 Bobbi Fleiss
Bobbi Fleiss Pierre Gressens
Pierre Gressens Helen B. Stolp
Helen B. Stolp- 1School of Health and Biomedical Sciences, RMIT University, Bundoora, VIC, Australia
- 2Université de Paris, NeuroDiderot, Inserm, Paris, France
- 3PremUP, Paris, France
- 4Centre for the Developing Brain, School of Biomedical Engineering and Imaging Sciences, King's College London, London, United Kingdom
- 5Comparative Biomedical Sciences, Royal Veterinary College, London, United Kingdom
Preterm-born infants frequently suffer from an array of neurological damage, collectively termed encephalopathy of prematurity (EoP). They also have an increased risk of presenting with a neurodevelopmental disorder (e.g., autism spectrum disorder; attention deficit hyperactivity disorder) later in life. It is hypothesized that it is the gray matter injury to the cortex, in addition to white matter injury, in EoP that is responsible for the altered behavior and cognition in these individuals. However, although it is established that gray matter injury occurs in infants following preterm birth, the exact nature of these changes is not fully elucidated. Here we will review the current state of knowledge in this field, amalgamating data from both clinical and preclinical studies. This will be placed in the context of normal processes of developmental biology and the known pathophysiology of neurodevelopmental disorders. Novel diagnostic and therapeutic tactics required integration of this information so that in the future we can combine mechanism-based approaches with patient stratification to ensure the most efficacious and cost-effective clinical practice.
Introduction
Preterm birth is defined as delivery before 37 completed weeks of gestation, and although the shorter the gestation, the higher risk of mortality and morbidity, even the late preterm-born infants are vulnerable to injury, including to the brain. The hallmarks of brain injury to the preterm born infant are: neuroinflammation, oligodendrocyte maturation arrest and hypomyelination, axonopathy, reduced fractional anisotropy and cortical volume determined by magnetic resonance imaging (MRI), and eventually, significant cognitive deficits (1). Collectively the brain damage associated with preterm birth is called encephalopathy of prematurity (EoP).
As long-term cognitive and behavioral consequences of preterm birth are increasingly recognized, neuropathological studies have focused on gray matter (GM), in addition to white matter (WM). It was initially thought that cortical GM injury only occurred in preterm infants in cases of very severe injury. Increased understanding of cortical development and more detailed post-mortem studies revealed that this not the case (2, 3). Over the past few years, work has increasingly indicated a widespread subtle neuronal injury in infants born preterm, that in some cases, such as interneuron deficits, may be independent of WM injury (4). Further than this, swathes of clinical and preclinical studies indicate that the cortical GM injury found in preterm infants significantly contributes to their increased risk of neurodevelopmental disorders (NNDs), such as autism spectrum disorder (ASD), attention deficit hyperactivity disorder (ADHD), and other learning and behavioral disorders.
The large and long running EPICURE (UK), EPIPAGE (France), and ELGAN (U.S.A.) studies have provided invaluable data on the incidence of neurological injury following very premature birth [e.g., (5–7)]. Together, these epidemiological studies confirm that preterm infants have a 25–30% incidence of neurological disorder, with as many as 40% of affected individuals having more than one diagnosable disorder (5–7). In all studies, incidence of cerebral palsy was 5–8% of preterm children, consistent between 2, 6, and 10 years of age (5, 6, 8). In addition, more than 40% of children at 2 years were below threshold for communication, motor, problem solving, and social skills (7), and 30% of children were diagnosed with cognitive impairment at 6 years of age (5), while at school age (10–11 years), 7–8% of preterm born children were diagnosed with ASD, 11% with ADHD, and 10% with emotional disorders, such as anxiety (8, 9). Using latent profile analysis in school age preterm born children (10 years of age), 25% of children were shown to have impaired executive functioning across a range of cognitive domains, while 41% of children fell into a “low-normal” category, where impairment was related to reasoning and working memory (10).
Environmental Contributors to EoP and Mechanisms of Injury
The maternal fetal membranes surrounding the amniotic cavity represent the boundaries of a sort of “black box,” inside which we struggle to know and understand the processes preceding preterm birth. This is due to technical difficulties in safely monitoring the biochemical processes ongoing in the uterine space. However, processes causing brain injury in the preterm born infants certainly begin before delivery, as indicated by a small study of brain functional connectivity in fetuses who went on to be born preterm (11) and an increasingly number of studies showing predictive biomarkers in maternal blood months before preterm birth (12–14). Numerous events and antenatal exposures have also been associated with preterm birth and EoP via epidemiological study and verified with preclinical studies. These include placental abruption or twin–twin transfusion, preeclampsia, or placental insufficiency (potentially contributing to a hypoxic-ischemic-like insult and/or intrauterine growth retardation) and, less commonly, complications linked to oligohydramnios and maternal substance abuse (15). A predominant role of hypoxia in EoP with no other complications (such as those described above) is not supported by clinical data (16). Chorioamnionitis, leading to a maternal–fetal inflammatory response, is a chief driver of the process of early parturition leading to preterm birth, demonstrated across clinical and preclinical studies (17–19). Maternal–fetal inflammatory response not only precipitates preterm birth, but a wealth of epidemiological and clinical studies have shown that, although it is often clinically silent, it is a major driver of EoP (20, 21) and its associated long-term neurological and behavioral/psychiatric deficits [see reviews (22, 23)]. While EoP can be initiated prenatally, there is evidence of continued disruption of the brain post-natally, which could be driven by a mixture of pre- and post-natal factors. For instance, Bouyssi-Kobar et al. (24) show reduced brain growth trajectories in preterm born infants compared to age-matched in utero controls that were associated with (antenatal) chorioamnionitis, as well as post-natal sepsis. Inflammatory drivers include pre- and post-natal events and conditions: chorioamnionitis, funisitis, early and late onset sepsis, and necrotizing enterocolitis. Other, non-inflammatory, post-natal contributors to EoP may include hyperoxia (25), as the ex utero environment is relatively high in oxygen compared to the in utero environment (26), and reduced exposure to maternal hormones, such as estrogen and other neuroactive precursors (27). While there has been little follow up on the estrogen hypothesis clinically (28), recent animal models have suggested a potential protective effect (4, 29, 30). That hyperoxia plays a role in EoP is also supported by animal studies (31–33).
Cellular Mediators of Brain Injury
How, specifically, do these perinatal events lead to EoP? In the case of maternal–fetal inflammation, systemic inflammation drives changes in the brain after either crossing directly through the endothelial cells making up the blood–brain barrier (BBB) or by stimulating, via receptors for cytokines, such as interleukin-1 (IL-1), production of pro-inflammatory molecules by the endothelial cells that are secreted into the brain parenchyma (34, 35). It is currently unclear whether these immune mediators act directly on neurons or have their actions performed via stimulated glial cells, such as microglia and astrocytes, though it is likely that both processes occur. Inflammation has been shown in the fetal brain to reduce neurogenesis in embryonic proliferative zones (36) or to increase neurogenesis in the SVZ and dentate gyrus (37), which has been linked to microglia activity in some cases [reviewed in (38–40)]. The timing of the exposure of neurons to inflammatory stimuli is undoubtedly critical. The exact timing of events impacting EoP is not clear, though it is likely that events occurring in the third trimester are most influential. Although the vast amount of proliferation is complete in the third trimester, there are specific cell types, interneurons of note, that are still being born and migrating in this period and that are increasingly demonstrated to be vulnerable in preterm born infants and animal models (41, 42). Susceptibility of processes, such as neuronal arborization and synapse formation may be even more important, given the intersect between injury events sensitizing to EoP and the developmental timetable of the brain (discussed in more detail below).
Over the past decade, the importance of microglial activation has been exhaustively demonstrated in human preterm-born infant post-mortem brain samples and in models of perinatal brain injury [reviewed in (43, 44)]. These studies have included experimental evidence that microglia are necessary for the evolution of injury in the developing brain (45, 46), but conversely, that microglia also play protective roles in perinatal brain injury (47, 48). Microglia are also activated by other modulators of brain injury, such as hypoxia or hypoxia-ischemia, as these events lead to cell injury and the release of damage-associated messenger proteins (DAMPS) and/or the production of toxic metabolites that also activate microglia directly (49, 50). Microglia establish territories in the developing brain from early in embryonic life and are intimately involved in the processes of brain building. Thus, microglia activation to an immune responsive state leads to EoP via a double hit, whereby there is production of toxic factors for neighboring neural cells and loss of normal microglial functions to shape axonal connectivity and synaptic elimination/function. This has been well-described in the WM (51–54). There is substantially less information on the specific effects of activated microglia on the GM, including synapses and interneurons, which are relevant to EoP. However, as increasing evidence shows that microglial dysfunction persists for weeks to years after insult (55–58), the importance of this phenomena may become more apparent with further study. It is also plausible that the GM and WM are differentially vulnerable, as microglia in these tissues have differing gene expression patterns in the basal state and after injury, based on studies in adults (59, 60). However, nothing is known of this difference in a model relevant to EoP. As such, although we can speculate that there may be specific soluble factors or regulators (micoRNAs, cytokines, etc.) released (possible via vesicles) from GM microglia that influence GM development in ways that would offer therapeutic avenues (61, 62), evidence is still required.
Reactive astrogliosis is also observed in some forms of human perinatal WM injury (63, 64) and is associated with deleterious effects mediated by agents, including hyaluronic acid (65), bone morphogenic protein (66), cycloxygenase-2 induction and associated prostaglandin E2 production (64), and endothelin-1 (67), which can impair oligodendrocyte precursor cell maturation. Clinical and experimental studies have shown a role for GFAP-positive astrocytes in WM injury in preterm born infants, but specifically in older preterm born infants [>32 weeks; (68, 69)] and during equivalent stages of rodent development [5 post-natal days plus; (70)]. In the GM, astrocytes increase in number with gestational age. Compared to the WM, GFAP positive cells represent a far smaller proportion of cells (<1%) (71) and the response of the populations a whole to injury is under-studied. In preterm-born infants, a small increase in the number of GM astrocytes was reported in a study of infants with cystic periventricular leukomalacia (cPVL; severe injury), but as the control group had a significantly lower gestational age, this effect did not survive correcting for development (72). However, studies in animals support the hypothesis that astrocytic dysfunction proceeds neuronal damage in at least some injury paradigms (73).
Developmental Events Susceptible to Injury in the Preterm Brain
When assessing how pre- and post-natal factors contribute to EoP, it is necessary to consider what developmental events happen during the preterm period that may be affected by preterm birth. The prenatal period most associated with EoP (from 23 to 32 weeks) is characterized by the final stages of neurogenesis in the human telencephalon, neuronal migration, differentiation and maturation, and the very early stages of cortical myelination. Neurogenesis peaks very early in gestation (8–12 weeks), but continues both in the ventricular zone of the dorsal cortex and within the ganglionic eminences for up to 29 weeks (41). The cortical plate forms around 11 weeks into gestation [reviewed in (74)] until shortly after the end of neurogenesis. Excitatory neurons primarily come from the progenitors in the ventricular zone of the dorsal cortex and migrate radially to the cortical plate [reviewed in (74)], while inhibitory cortical neurons derive from the ganglionic eminences and migrate tangentially to the cortical plate [reviewed in (75)]. Once neurons reach their final positions in the cortical plate, they start to mature—a process which includes extending dendritic arbors and forming synapses, detectable form 19–23 weeks gestation (76). At the same time, radial projections of the neural progenitors are lost, and tangential extension of subcortical and cortico-cortical projections continues. These processes continue through the prenatal and post-natal period of brain development, with an extensive period of synaptic modulation and pruning throughout the first year of life. Local electrical activity and connectivity between neurons can be detected early, undergoing numerous changes over development, and don't appear to find a mature state until early adolescence [reviewed by (77)]. Details of these events and many of the mechanisms underlying them are reviewed extensively in Molnar et al. (78) and Volpe (79). On top of these microstructural changes is a general increase in cortical thickness and a semi-stereotypical pattern of cortical folding, with primary sulci forming from 16 to 19 weeks gestation, and secondary and tertiary sulci formation starting from 32 to 36 weeks, respectively (80).
The real-time development of the brain, including the increasing complexity in the cortical structure, can be detected with non-invasive imaging methods, such as T1/T2 or diffusion-weighted MRI [(81–83); reviewed by (84)] and these techniques have allowed the detection of delayed or impaired cortical development in preterm born infants. However, given the relatively low resolution and integrated nature of diffusion MRI signal, interpreting the specific structural changes in relation to neurodevelopmental processes is difficult. Techniques are in development to scale match MRI and histological data [e.g., (85)], though changes in diffusion MRI are currently interpreted through comparison with standard histological measures. There is evidence that preterm birth can result in changes to all the processes of cortical development described above, including reduced progenitor proliferation, arborization, and myelination, as well as direct injury outcomes, such as cell death. The rest of this review will discuss a number of these in detail, including potential mechanisms of injury, overlap with neurodevelopmental disorders (NDDs), and potential therapeutics.
Macroscale Alterations in Cortical GM
Many elegant neuroimaging studies have begun to correlate both typical and pathological behavior with specific brain areas or functional readouts. Based on these, we know that many brain regions contribute to the diverse array of neurological disorders presented by preterm-born children. In particular, the important role of cortical dysfunction underlying these cognitive disorders (but not so prominently in motor disorders) is increasingly clear. Rathbone et al. (86), in their study of cortical growth (cerebral volume and cortical surface area) in the 20 weeks between birth and term-equivalent age in preterm infants, showed that slow rates of cortical growth correlated directly with neurocognitive ability at 2 and 6 years of age. In particular, impaired cortical surface area growth correlated with poorer scores in numerous features of executive function, learning, memory, and attention, as well as social ability. However, there was a clear lack of correlation between cortical growth and motor abilities (86). Numerous neuroimaging studies using different post-imaging assessment methods have shown reductions in cortical GM volumes in preterm infants, both in the preterm population as a whole (87–89) or specifically those with periventricular leukomalacia [PVL; (90, 91)] and in very preterm born children [assessed at 7 years; (92)] and adolescence [assessed at 15 years; (93, 94)]. Reductions in volume of the deep GM have also been reported (95, 96); changes in thalamic volume were found to be a predictor of reduced cortical GM volume and alterations in diffusivity within the thalamocortical networks (95) were found to correlate with cognitive performance at 2 years of age, though they only accounted for 11% of the variance (97).
Importantly, Bora et al. (98) showed that very preterm infants had a 13%-increased risk of inattention and hyperactivity behavior at school age (4, 6, and 9 years), which correlated with decreased GM volumes, particularly within the prefrontal region. Increased anxiety-like behavior has also been associated with preterm born infants with reduced GM volume (99). In a small study group, very preterm infants that went on to have a diagnosis of ASD were found to have increased incidence of cystic WM lesions, and reduced cerebellar volume, but no changes in GM volume (100). However, only eight children in the cohort (4.7%) were diagnosed with ASD by the age of 7 years, so the study may have been underpowered for detecting more subtle changes in cortical GM.
In animal models, mimicking changes in these cortical volume parameters is difficult, due to differences in the relative GM and WM volumes in experimental species and the differences in the size/complexity of the individual regions relative to one another. Sheep are used in studies of perinatal brain injury, with advantages regarding physiological and neurological similarities to preterm humans, including gray–white ratios [discussed in (101)]. Dean et al. (102) have studied intra-amniotic LPS in sheep, a paradigm able to cause cystic WM injury. This inflammatory exposure caused no obvious cell loss in the GM, but reduced cortical volume by ~18%. In further work in sheep models, Dean et al. (103) also showed a reduction in cortical GM volume after in utero hypoxia-ischemia, in which there was again no cortical cell loss. Interestingly, there was no early reduction in cortical volume (+7 days) but these became increasingly apparent with time after injury [starting at 2 weeks and at least up to 4 weeks; (103)]. In a mouse model of maternal immune activation using poly I:C, subtle decreases in GM volumes were observed throughout development (104), though changes in cortical volumes were not specifically reported.
In addition to alterations in GM volume, complex changes in cortical architecture have also been identified. For instance, Zhang et al. (92) determined that there was a decrease in cortical surface area and the gyrification index of 7-years-old following very preterm birth compared to term-born controls. Maps of cortical folding patterns in neonates suggest that preterm infants have fewer and shallower sulci than term equivalent controls (105). Data suggested that the lower gyrification index and cortical surface area in preterm-born neonates was likely to be due to a combination of altered in utero and post-natal growth, and it was a finding independent of reduced total brain volume (105, 106). Reduced cortical folding has also been associated with increased incidence of NDDs later in life (106), matching, at least partly with data from specific disorder cohorts (107–110), though the data are not substantial here, as existing studies are small.
Collectively, these data point to the possibility that alterations in cortical folding are driven by numerous age-specific microstructural changes. The theories behind cortical folding are many, and include processes such as the rate of neurogenesis, of tangential migration and neuronal arborization [reviewed by (111–113)]; currently no single one is sufficient to explain the biological underpinning to normal or abnormal cortical folding. Numerous aspects of the in utero environment and preterm injury have been associated with changes in cortical folding [reviewed by (114)], though the mechanism by which this injury is produced is still unclear. Recent work by Garcia et al. (115) has shown regional differences in cortical growth rate between post-menstrual age 30 and term equivalent age (based on 2–4 MRI scans over this period in preterm-born infants), which are disrupted in preterm-born infants with gross injury, such as intraventricular hemorrhage (IVH). Their work suggests that severe injury in preterm born infants may alter local cortical growth and subsequently cortical folding (115), supporting the hypothesis that cortical folding may result from mechanical instability as the GM grows faster than underlying WM (116). Alternatively, recent compelling evidence also shows that the extracellular matrix is essential in normal cortical folding (117), likely contributing to the mechanical tension within the brain. The link between these highly reductionist ex vivo studies and EoP is currently unclear, though they have suggested that hyaluronic acid can inhibit cortical folding (117), and increases in hyaluronic acid have been found within the preterm brain (118).
Gray Matter Neuropathology Associated With EoP
There are few neuropathological studies of GM injury in preterm-born infants, compared to the number of studies of WM injury. Complicating matters, due to the difficulties of defining appropriate controls, GM studies typically use evidence of WM injury as a starting point in the assessment of the GM, rather than searching for independent patterns of injury. We have created Table 1, which summarizes studies performed on human preterm-born, post-mortem tissue that have included GM analyses. From this, we can generalize that in studies of infants with severe and contemporaneously uncommon WM injury (cystic PVL), there are reductions in neuronal number and increased neuronal cell death (where assessed; 6/6 studies). However, in studies of infants predominantly with diffuse WM injury, global reductions in cell number are less frequently reported (1/5 studies), but interneurons seem to be a vulnerable subpopulation (present in 3/3 studies) and dysmaturation in cerebellar lamination are reported (1 study). Interestingly, there are complex subtle changes in interneurons in cases with non-cystic WM injury vs. controls (42) and when comparing very preterm infants to less preterm infants [irrespective of WM injury; (4)]. It is necessary to note that of the 12 studies identified, 6 of these were performed on archival tissue collected between 1993 and 2007 from the Department of Pathology at the Children's Hospital Boston. An additional observation study was not included in the table, as the data were expanded upon in a later study (127). It is not possible from the published details of the Boston group's work to determine whether cases in these studies have been reused. Thus, reports of cell death across regions in these studies may be interdependent, due to case severity in this center, and studies of other centers and in more contemporaneous cohorts are needed to determine the state of neuronal injury in preterm born infants more generally.
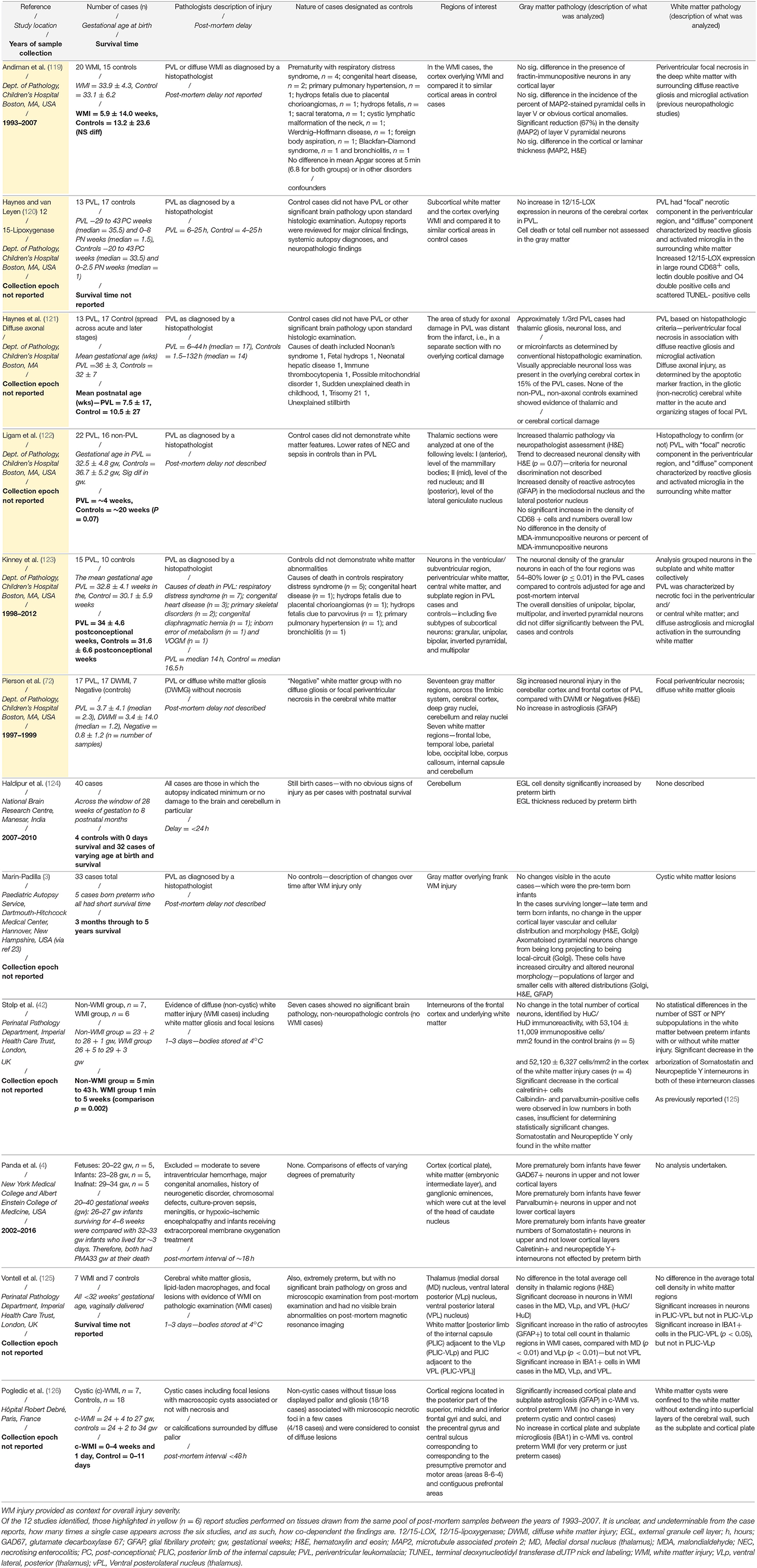
Table 1. Summary of post-mortem studies of preterm-born infants, including analysis of the GM, highlighting the case characteristics, regions of interest, and the GM and WM injuries described.
Animal Models of GM Pathology
Severe Injury
Severe brain injury, including cystic lesions and severe IVH, occurs in very few preterm-born infants [<5% cystic lesions, <5% IVH; (128)]. Historically, the proportion of infants with these forms of injury was much higher. It was also once considered that hypoxia-ischemia was the leading (possibly the predominant) cause of perinatal injury, including in preterm-born infants. Because of these historical trends and (now updated) ideas, much of the data that we have on GM injury in EoP is from animal models of gross clastic lesion (30–80% hemispheric ablation). This initial wave of data suggested that of the cortical layers, the subplate was most susceptible to hypoxic-ischemic injury at preterm equivalent ages (129), possibly due to its relatively early birth and maturation. However, subsequent studies agree that the extent of cortical injury is dependent on the severity of the insult, and all lower cortical layers have the capacity for cell loss after severe hypoxia-ischemia (130). In a study of partial uterine artery occlusion, modeling hypoxia-ischemia in the fetal sheep, immediate low level necrotic cell death was found throughout the deep and cortical GM, followed by extensive apoptosis in both the GM and WM at 3 h post-injury (131). Other studies of in utero hypoxia-ischemia in sheep have shown some increase in pyknotic cells and activated caspase-3 staining from 24 h up to 4 weeks in the caudate nucleus and subplate (132, 133), reduced NeuN and somatostatin positive neurons in the caudate and putamen (134), specific loss of glutamate decarboxylase interneurons (a marker of GABAergic neurons) and their perineuronal nets in the cortex (135), along with reduced arborization complexity and spine density in both the caudate, hippocampus, and cortex (103, 132–134, 136).
Moderate/Mild Injury
Improvements in antenatal and post-natal care, including the use of prenatal steroids and post-natal surfactants and improved respiratory support, have collectively led to the decrease in severe brain injury, so that now the vast majority of infants suffer from diffuse WM injury (118). This has inspired the revision/creation of animal models focused on modeling white matter dysmaturation. A number of these new(er) models providing insights into the role of contemporaneously relevant insults to GM injury are described. A landmark study in our understanding of the GM injury induced by preterm birth came from the team led by Sandra Rees (137), wherein they delivered baboons preterm and held them in a NICU environment for 2 weeks. This important study isolated the roles of prematurity itself from exogenous/precipitating factors (such as chorioamnionitis and sepsis). WM injury and hemorrhage were most common in preterm baboons, but there were significant pockets of necrosis in layer IV/V cortical neurons (4/16 cases) and in the head of the caudate (1/16 cases). One of the first attempts to nail down the cellular substrate of GM injury was the analysis of the effects of intrauterine hypoxia-ischemia on the fetal sheep (103). Although it can be debated whether hypoxia-ischemia is particularly relevant to the majority of cases of EoP (16), this team used cutting edge combinations of high-field MRI and detailed neuropathological assessments of cell number and structure to reveal novel insights into brain injury. The team found that overall reductions in GM volume were not precipitated by neuronal cell loss, but that there were frank changes in dendritic arborizations [length, number, intersections; (103)].
Sheep models of moderate inflammatory injury are also providing important information. Exposure to intra-amniotic inflammation prior to preterm birth, was used to reveal the evolution of in utero inflammatory brain injury (138). Interestingly, sheep were all exposed to inflammation on the same gestational day, but sub-groups were culled every 24 h providing a detailed time course of events. Cleave caspase-3 positive cells were increased in number in the hippocampus at 2, 4, 8, and 15 days following LPS exposure, and in the cortex (at 8 days only), along with increases in MAP2 staining in both GM regions. Strikingly, there was only moderate WM injury (few cleaved caspase-3 positive cells, microgliosis, and mild demyelination) suggesting that neurons may be vulnerable to injury in the absence of overt WM damage. Reduced cortical neurons were found in another study exposing the developing sheep to LPS (139), 7 days after a single LPS challenge. In these experiments there was no difference in either astrogliosis or microgliosis at the time point analyzed compared with the previous sheep study (138) in which microgliosis was present, but astrogliosis was not.
Rodent models are by far the most common for studying potential neuropathology of EoP. In a rat model of inflammatory exposure (maternal LPS exposure at the end of gestation), significant post-natal reduction in brain and body weight were observed, and a small increase in cell death in the striatum and germinal matrix (140). In a milder injury model of prolonged induction of systemic inflammation [using systemic IL-1β exposure; (52)], there was no gross body weight change, no evidence of caspase-3 positive dying cells or alteration in the number of NeuN-positive neurons in the neocortex. However, in this injury model, there were numerous changes in gene expression for synaptic and neurotransmission related genes (141). In this same animal model, and similar to data reported in human cases, a specific alteration in interneuron number was identified in the neocortex (42)—a finding supported by a number of other early-life inflammatory exposure models (142, 143) and preterm birth models (4). It is likely that the migration and differentiation of these cell populations is affected, though many studies show that injury reduces or repairs in adult mice, following early-life inflammation (42, 143). The important advantage of rodent models is the potential for behavioral testing, where aspects of human clinical disease can be recapitulated. In the inflammatory injury models just described, behavioral dysfunction has been reported, including reduced social interaction (143), short and long-term memory deficits (46, 52), attention-shifting deficits, and anxiety-like behavior (142). These behaviors are commonly found in preterm infants, as described above, and in other NDDs, thus supporting the face validity of these models. This is further supported by an extensive body of work showing reduction in GABAergic interneurons or expression of parvalbumin (as distinct from a reduction in cell number) in clinical ASD cases (144–146) and genetic models of NDDs (147–149).
EoP as a Synaptopathy
A synaptopathy is a disease or disorder caused by dysfunction of synapses. This dysfunction can arise due to a mutation in a gene encoding a synaptic-related protein, such as an ion channel, a neurotransmitter receptor, or a protein involved in neurotransmitter release; alternatively, a synaptopathy may be due to structural deficits in extension of neuronal arbors and synaptic process. Whether EoP can be defined as synaptopathy requires further study, but we suggest that this is likely to be an important part of the neuropathology of this condition. The changes in EoP of gross GM volume changes, variations in growth rate, and patterns of cortical folding discussed above all reflect a combination of microstructural deficits (150) and connectivity (97, 151, 152) including delayed acquisition of the default mode network, as assessed by MRI techniques. Additionally, there is the fact that EoP predisposes to strikingly increased odds of a diagnosis of a NDD that are clearly recognized as synaptopathies, such as: ASD, up to 17-fold increased rates (9, 153); attention deficit disorder, up to 2.5-fold increased rates (154, 155); epilepsy, up to 5-fold increased rates (156, 157); and decreases in IQ directly proportional to the severity of their preterm birth (158, 159).
Considering the developmental events happening during the period of preterm birth, it may be expected that alterations should be found in patterns of neuronal migration, time frames and degrees of arborization, axon extension, and synapse formation. On this subject, the recent study by Petrenko et al. (160), provides a number of important insights. In a highly reductionist model of selective neuronal apoptosis in layer 5 of the cortex, induced by diphtheria toxin (161), the authors showed a progressive loss of ~20% of neurons within the cortex over a 14-days period. While this degree of neuronal loss is unlikely to occur in EoP, the pre- and post-apoptosis changes to the brain have interesting correlates for the injury observed in EoP. Specifically, there was an associated increasing presence of astro- and microgliosis, retraction of dendritic arbors in dying neurons (days 3–5), and increased arborization (branch number and length) in the surviving neurons [day 14; (160)]. Alterations in dendritic arborization have been found in the GM in a number of experimental studies, many of which have already been referred to above [e.g., (103, 138)]. Additionally, a model of intrauterine growth restriction in pig, initiated at 100 days of pregnancy and assessed 22 days later, showed a loss of MAP2 staining in the parietal cortex and hippocampus, which was interpreted as disrupted somatodendritic neurites (162). Intrauterine growth restriction is an important contributor to poor perinatal outcomes, particularity in preterm born infants [see (163)]. Reduced dendritic branching and spine immaturity have also been reported in the CA1 region of hippocampus in a model of preterm birth in rabbit kits (30) and in the granular layer of the dentate gyrus in a maternal inflammatory activation (using i.p. poly I:C exposure) model in mice (164). These assessments are harder to perform in neuropathology on clinical samples, though reduced dendritic complexity (branch number and length) have been described for somatostatin and neuropeptide Y-positive neurons in the subcortical WM of preterm born infants with WM injury (42). Dendritic arborization, and relatedly, synapse formation [something also disrupted in these models; (42, 102, 165)], are essential developmental events for ensuring appropriate connectivity in the brain, and disruption in these processes have been implicated in a number of functional disorders of the brain (discussed below). The vulnerability of synapse structure in preterm born infants is clearly shown in a study that revealed a relationship between brain injury in preterm born infants and single nucleotide polymorphism (SNP) variants in the gene for the post-synaptic protein 95 [PSD-95, DLG4; (166)]. This work focused on a novel role for PSD-95 expressed specifically by microglia in early development in EoP, but the patient SNP data also suggest a wider vulnerability of synapse structure in preterm-born infants.
While evidence of EoP as a synaptopathy inevitable comes from neuropathological studies, our best capacity to clinical recognize disease, stratify patients for treatment, and monitor progress comes from neuroimaging. When relating in vivo imaging to pathology, the study by Petrenko et al. (160) suggested that (a) neurons loss could be detected by decrease in N-acetylaspartate and N-acetylaspartylglutamate and astrogliosis with reduced Glutamate/Glycine ratio, using magnetic resonance spectroscopy within 3 days of injury; and (b) diffusion MRI could also detect microstructural injury within 3 days of cell death induction, starting with increased water diffusivity (mean diffusivity) and extending to reduced factional anisotropy (FA) due to altered dendritic arrangement. Ball et al. (150) have shown a developmental decrease in FA in the cortex over the preterm period, with preterm born infants lagging behind term born infants in this maturational process, i.e., with a higher FA at term equivalent age. Modeling by Dean et al. (103) supports the idea that this increased FA value is due to delays in the normal dendritic arborization of the cortical neurons over this period. Vinall et al. (167), studied variation in diffusion MRI values between two scans in a cohort of very preterm infants. Their work showed that increased FA in the cortical GM at scan two was independently associated with reduced gestational age, birth weight, and slow weight gain. In addition, changes in FA were related to the second and third eigenvector direction, rather than the primary eigenvector direction. Collectively, these data imply that delays in cortical maturation were most likely driven by delays in neuronal process formation, or cell loss, and that cortical maturation was associated primarily with the phase of neonatal growth (167). Structural connectivity studies, typically based on the integrity of WM tracts using diffusion MRI, have shown a topographically dependent timetable of connectivity developing brain, which is impaired in the preterm brain (168, 169), and which is altered in nature over time, but persists in some form to adulthood (170). While these measures are not directly assessing cortical GM injury, it is likely that an interplay between WM and GM development occurs and that altered connectivity maps will reflect changes in GM development. These structural alterations are also likely to have functional consequences that reflect both local and global connectivity.
Interplay of Structural and Functional Deficits in EoP
Altered structural and functional connectivity can be identified in the brains of preterm infants at term equivalent age, using combined diffusion and functional MRI (171). Aside from studies testing passive function, including touch and auditory stimulation, the majority of functional MRI studies in preterm infants have investigated resting states. Collectively, these resting state studies suggest that there is modular organization of the connectivity of the preterm brain, as is seen in the mature brain, but that integration between networks is altered (172–174). In these studies, there is evidence for disruption in both cortico-cortical and cortico-subcortical networks (172, 174), and reduced connectivity between areas associated with motor function, cognition, language, and executive function (173).
The electroencephalogram (EEG) is a clinical tool that has been shown to have some potential to monitor and predict severity and outcome of EoP. EEG waveforms are immature in the preterm brain, but appear to have some characteristic changes that can be used as a biomarker, including seizures, EEG suppression, and mechanical delta brush activity (175–178). The rate of spontaneous activity transients on EEG in preterm born infants with or without GM-IVH, measured over the first 48 h of life, was associated with cortical GM volume growth, increased gyrification index, and increased FA in WM tracts (179). Additional studies of the association between early EEG and cortical growth have revealed very specific band frequency relationships and with spontaneous activity transients (SATs) (180). As we begin to understand the biological drivers of these events, it will provide further information of the structure function relationship of the EEG recordings. Whitehead et al. (181), using EEG, showed that gross injury initially disrupts signal recruitment from cortical circuits. Signal recruitment appears to eventually be reinstated following injury but remains different from individuals without gross injury. Importantly, EEG abnormalities assessed shortly after birth (a week to a month after birth) were able to predict both developmental delay and cerebral palsy at 18–24 months (182, 183).
In animal models, fMRI has not been used, but EEG has been used extensively in sheep models of in utero hypoxic ischemic injury (more closely modeling hypoxic-ischemic encephalopathy) and shows reduced maturation of the EEG signal over time, seizure susceptibility, and microscale epileptiform events in the latent phase (up to 7 h post-injury) prior to seizure onset that correlates with cell death (184, 185). Following intrauterine inflammatory exposure in fetal sheep, changes in developmental patterns in alpha and beta power (reduced) and delta power (increased) have also been reported (186). However, while there is widespread evidence of altered EEG parameters in both clinical and preclinical studies, it is not clear how well these changes related to the neuropathology and how predictive they are for outcome. This work is only just beginning in clinical populations [e.g., (181–183)], but in pre-clinical studies, a number of studies have found a disconnect between EEG results and activity and arousal (187) or neuropathology (188, 189). However, it should be noted that the pathology examined in the study by Galinsky et al. (189) was largely focused on WM, rather than GM, features, and therefore may provide a limited understanding the pathological correlates of EEG. Van den Heuij et al. (185), for instance, have reported improved EEG findings together with reduced cortical and deep GM damage following intrauterine artery occlusion in the fetal sheep. In rodent models, EEG studies are less common, due to the size of the post-natal brain. Using ex vivo multi-electrode arrays, Mordel et al. (190) showed that inflammation and hypoxia, alone or together, increased the excitability of cortical neurons, in a glutamate receptor dependent manner. Interestingly, this research group has also shown that inflammation-induced alterations in cortical neuron spontaneous burst activity subsequently results in an increase in apoptosis in the same cell population (191). The work of Mordel et al. (190) suggests that altered electrical activity in the cortex occurs only in the first few weeks after injury and that it recovers in adulthood. However, long-term alterations in spontaneous and mini-inhibitory post-synaptic currents, a more subtle measure of neuronal activity, was found specifically for parvalbumin-positive interneurons following fetal exposure to inflammation (142). Electrophysiological studies in the preterm sheep have shown altered excitability in subplate neurons (133), as well as reductions in intrinsic excitability, altered polarization dynamics and reduced long-term synaptic plasticity in the hippocampus, following hypoxia-ischemia and hypoxia alone (136).
The relationship with structure and function is complex and needs to be understood better at a (sub-)cellular level in the context of EoP. However, the study of Zaslavsky et al. (192) in iPSCs from ASD patients shows increased dendritic arborization and synaptic connectivity associated with a significant increase in sESPC frequency, supporting suggestions that altered neuronal morphology does change cellular function (rather than being compensated for in the function of the cellular communications pathway). This link between structure and function, the capacity for one to affect the other, and the plasticity for recovery is a particularly important point to consider when exploring new therapeutic targets, and optimal periods of treatment, for EoP and NDDs. This concept has recently been supported in a study of genetically encoded epilepsy, where timely treatment with Bumetanide altered long-term neuronal activity and network formation (193).
Potential Therapies for EoP
Gray Matter Targets
The most obvious change in the GM of preterm born infants are reductions in volumes on MRI, changes that persist with increasing age. These gross changes are likely mediated by limited but significant cell death, changes in sub-classes of interneurons, and, across neuronal classes, reductions in arborization and/or synaptic number. There are no therapies designed to target GM injury in the preterm specifically. Given that there are striking similarities between the GM changes in EoP and NDDs, it would seem appropriate to consider if any therapeutic candidates from the NDD field might have efficacy in EoP. Current pharmacological strategies for the treatment of ADHD focus on normalizing, but not repairing, disturbances in synaptic transmission and activity (194), and the same is the case for the various forms of epilepsy (195, 196). For ASD, therapy focuses on treating the symptoms of the disorder, such as risperidone, to reduce irritability via antagonism of central type 2 serotonergic (5-HT2) receptors and central dopamine D2 receptors (197). There are no therapies for ASD to treat the underlying deficits in social abilities. Other NDDs, such as intellectual disability and learning disorders (dyslexia and dyscalculia), together with ASD and ADHD, are successfully treated with behavioral interventions. It is believed that these therapies do rewire the brain (198), but whether they are capable of repair is not at all established.
A recent review of the drugs under investigation review for ASD highlighted that potential therapies fall into several clear classes—GABA/glutamate modulators, neuropeptides, immunologics, and dietary supplementation (199). The only therapies whose specific underlying premise is to permanently alter the structure of the brain are immunological therapies; although, neurotransmitter modulators given at the optimal stage of development may normalize aspects of structural and functional development—something that needs to be considered in future research. That immunological therapies might enable repair is based on the underlying idea that, in the brains of people with ASD, there is a persistent immunological dysfunction that itself is the cause of the core social deficits. As such, removing this dysfunction allows the brain to return to a normal structural and functional state. A very similar process of persisting and damaging inflammation is hypothesized to occur in the brain after perinatal brain injury (200) that evidence begins to accrue, which, in this context, it is also a valid therapeutic target (57, 201).
Another exploratory area of understanding and treating ASD and other NDDs is the gut–microbiome–brain axis (202, 203). Gene mutations associated with autism pathogenesis impair brain and gut function and contribute to core and comorbid symptoms reported in autism (204, 205). The gut and brain share cellular structures, molecular pathways and processes that likely cause shared vulnerability to processes leading to autism (203). For instance, gut and brain synaptic structure and function are similarly vulnerable to disturbances in structural proteins, such as neuroligins, post-synaptic density proteins, and Shanks (166, 206–210). An inexorable production of gut microbe-derived neuroactive metabolites influences gastrointestinal function, and these also traverse the BBB to exert potent effects on the brain (211–213). Importantly, microbiome-mediated gut and brain crosstalk even alters early brain development (214, 215) via dysbiosis, which impairs the function of the brain's chief “building managers” and resident immune cells—microglia. Microbe-derived metabolites also regulate the function of the BBB itself (216) demonstrating the integral nature of the microbiome-gut-brain axis in brain health. As such, research investigating factors modulating the gut–microbiome axis in NDDs may uncover novel mechanisms for treatment (217, 218).
Considering the options from the classes of drugs already being tested in models of EoP, we find that, despite many compounds being tested (with mixed results), most have not considered outcomes in the GM. There are some notable exceptions, such as MgSO4 pre-treatment in a rat model of preterm HI (modeling antenatal treatment in at-risk mothers), significantly reduced tissue loss in the hippocampus and striatum and were associated with reduced neurological injury score (219). MgSO4 has also been tested in a sheep model of perinatal asphyxia, reporting reduced seizure burden, but worse WM outcomes and no GM neuropathology (189). Clinically, MgSO4 has a number to treat of 54 (220), though due to the nature of pre-treatment of at risk individuals, the exact efficacy is difficult to determine; a Cochrane review of four trials of antenatal treatment of at-risk women showed no significant effect on mortality or neurological outcome (220). Erythropoietin in this environment has not been shown to be protective for qualitative WM or GM injury when administered as three doses of 25 μg/kg within the first 2 days of birth in preterm infants (221). This is despite positive GM outcomes in rodent (188, 222) and sheep models (223). Robinson et al. (222) showed that 2,000 U/kg erythropoietin (~17 μg/kg), administered post-natally following intrauterine occlusion, was beneficial for both WM and GM, ameliorating behavioral deficits in gait and social interaction and fractional anisotropy changes in the WM, hippocampus, and striatum. In their study of perinatal injury, hypoxia-ischemia in the post-natal day-3 rat, van de Looij et al. (188) showed that erythropoietin improved somatosensory-evoked potentials and diffusion parameters in the WM, when measured with MRI, but didn't prevent cortical tissue loss. Wassink et al. (223) assessed neuronal number and cell death in the caudate, showing a significant improvement with erythropoietin (5,000 IU loading dose, followed by 832 IU/h) in the preterm sheep, as well as reduced seizure burden. More positive data on erythropoietin have been found for WM injury [reviewed in (79, 224)], supporting the numerous on-going clinical trials for this drug; however, it is clear that additional therapeutic agents need to be tested for GM efficacy.
Links Between EoP and Neurodevelopmental Diseases
It has been unequivocally established that preterm born infants have increased rates of diagnosis for NDDs, including ASD, ADHD, and generalized learning disorders (5, 7–9, 225). It is also clear that, in the brains of people who suffered from EoP and those with NDDs (and from their matched preclinical models), there are a striking number of shared pathomechanisms. In this section, we will highlight key phenotypic, macrostructural, genetic, cellular, and sub-cellular processes shared with EoP and in cases of NDD. We will focus on the GM; but, we wish to highlight that for the WM these links between EoP and NDD are more established, such as shared deficits in corpus callosum structure in people after EoP and those with ASD and ADHD (226).
Recent work has assessed in detail the specific characteristics of behavioral disturbances in people born preterm with NDD, compared with people born at-term with an NDD [see reviews (227, 228)]. In general, in preterm vs. term NDD, the phenotypic presentations are similar. However, there are important differences. For instance, in people born at term, there is a higher rate of ADHD in males compared with females; but, this sex difference in not observed in people with ADHD who were born preterm (229). For ASD, a greater proportion of preterm (vs. term-born) males reported comorbidities (sleep apnea, seizure disorders, and ADHD) and people born preterm (particularly females) were more often non-verbal (230). Another recent small study of children with ASD demonstrated that, compared to term children, the preterm children had higher quality peer relationships and socioemotional reciprocity, but poorer non-verbal behaviors that regulate social communication (231). None of the current literature has indicated a problem with diagnosing those born preterm using the current diagnostic criteria. However, we speculate that just as autism has been expanded and refined into a complex spectrum of disorders that, in the future, ASD phenotypes specific to preterm born infants may be defined.
With increased MRI analyses of the GM in individuals with EoP, we begin to see a clear pattern of similarities in changes in brain structure in people with ASD and those born preterm—there are shared changes in the orbitofrontal regions, the amygdala, the basal ganglia, the hippocampus, and the cerebellum [reviewed in (227, 232)]. There is also a parallel with the altered cortical growth in preterm born infants and equivalent findings in ASD and ADHD patients. In MRI studies of ASD and ADHD, decreased GM volume has been associated with both conditions (233–237). In ASD, decreases have particularly been found in areas related to social behavior networks (233, 235, 237), while regions associated with inhibitory control (234) were changed in ADHD. In both cases, it is clear that patterns of GM deficits alter through the disease course (235, 238). Changes in the volume of GM in preterm infants/children/adolescents have been found in many of these regions [e.g., (91, 92, 239)], but are typically more widespread. Variation between studies has, of course, been reported, with not all studies finding cortical GM volume changes or associating them with neurological outcome. However, these are in the minority, and it has been suggested that these may be due to difficulties in accurately recognizing the gray-white matter boundary in the developing brain (240). Interestingly, in addition to this overlap in affected brain areas in both EoP and NDDs, MRI studies are also showing alterations in cortical networks [e.g., (236, 241)] in ASD and ADHD that warrant further exploration, and may come from as similar anatomical basis as in the EoP studies.
A newer avenue to link EoP and NDDs are genetic studies, such as genome-wide association studies (GWAS), copy number variant (CNV) studies, SNP, and haplotype studies, and these are revealing common risk factors. For instance, we have recently uncovered that an SNP in the gene for PSD95 is associated with poorer outcome for preterm born infants (166), mentioned above, as genetic variation in polymorphisms for PSD95 is a known risk factor for ASD (242). Common genetic variants and methylation patterns have been revealed in focused studies of people with ASD, with and without prior history of preterm birth (243). Changes uncovered by these targeted studies include tyrosine-protein kinase Met (MET), Neuregulin 3 (NRG3), and serotonin transporter (SLC6A4). A great deal can also be learned from comparing findings from studies of NDDs and studies of prematurity and EoP. For NDDs, there are numerous genes associated with synapse formation identified from GWAS studies including Shanks, Neuroregulin, Neurexin, and Contactins [reviewed by (244, 245)]. Many of these genes also associate with preterm birth or outcomes after preterm birth. Of note, neuregulin is found associated with infant outcome, with polymorphisms increasing mRNA levels in patients associated with better outcomes in babies born preterm (246).
A key vulnerable neuronal subpopulation in EoP is interneurons, although it is still unclear which populations are the most vulnerable at which time point and in what regions based on the human and preclinical studies (4, 42). Research into neuropathology in NDD, via post-mortem studies and animal models of NDD, also conclusively illustrates changes in interneurons (146). Quite strikingly, in a synaptic protein knockout model of ASD (PTEN KO), interneuron transplantation rescues social behavior deficits (247). This study also questions the established idea that interneuron deficits associate with NDDs due to negative effects on inhibitory circuit activation (248), as, although interneuron transplantation rescued the behavioral phenotype, there were no improvements in circuit function.
No discussion of the similarities between NDD and EoP could be complete with highlighting the shared common pathological process of neuroinflammation, which has, at its core, the aberrant activation of microglia. Indeed, across NDD and EoP models and human studies, evidence shows that microglial activities are altered [thoroughly reviewed in (22, 249–251)]. A chief function of microglia during development, but also throughout life, is regulation of connectivity via refinement of synaptic number [(252–254); and reviewed in (255–257)]. Based on all the evidence for the role of microglia and the presence of inflammation (both systemic and central) in EoP and NDD it is clear that microglia (and their effects on synapses and neurogenesis) are an important starting point in understanding GM pathology across NDDs and EoP and also a shared target for neurorepair.
We outlined above the reasons that EoP can be considered a synaptopathy, including genetic associations between injury severity and synaptic genes, connectivity deficits, and that preclinical studies show synaptic immaturity plus arborization deficits. These characteristics are also common among NDDs, and NDDs are clearly characterized as synaptopathies (258–261). For example, about half of the genes identified as candidate genes in people with ASD code synaptic proteins (262). Additionally, animal models of abnormal synaptic pruning induced by abnormal microglial function (227), or via genetic perturbation of synaptic structure (263), have cognitive and behavioral deficits reminiscent of NDDs. Thus, perhaps it is the collective change in these functional units of the neuron that give rise to the shared gross volumetric changes and pervasive behavioral problems in people with NDDs and due to EoP. Though it should also be said that a great many children and adults who were born preterm and who had EoP have typical neurodevelopmental profiles, potentially and interaction of genetics and environmental challenges in these case lead to structurally resilient synapses. There is clearly need for a better understanding of the vulnerabilities leading to NDDs and negative consequences after EoP.
Conclusions
Imaging and neuropathological studies indicate changes in GM are a subtle but substantial contributor to EoP. The full nature of this injury is probably only just being discovered and would benefit from more longitudinal MRI studies, with closer integration of both patient genetics data and neuropathology where possible. Given the link between GM injury and long-term cognitive and behavioral disorders, it is important to therapeutically target this injury, distinct from the WM injury aspects of EoP. In particular, while preterm birth and EoP increase the risk of NDD in later life, the current evidence suggests that preterm born infants may make up a specific subset of cases in these disorder spectrums and could benefit from a distinct treatment paradigm. In terms of what this therapeutic paradigm might look like, it is likely that a combination of ameliorating (e.g., anti-inflammatory or growth supporting) agents and restorative agents (e.g., drugs facilitating normal structural-functional development) will be required. If these treatments are delivered at optimal periods of brain development, it may be possible to limit the need for life-long symptom controlling medication. In this regard, it is necessary to focus more research on the synaptopathic aspects of EoP. Current research in this area is only the tip of the iceberg, particularly lacking in clinical studies, and increased understanding of the injury mechanisms and plasticity during the post-natal period may identify new therapeutic targets. Our great hope is actually that this proposed work becomes redundant. We hope that our highly skilled and motivated counterparts working on prediction and prevention of preterm birth have major breakthroughs. However, pragmatically, even major breakthroughs will take decades to make it across high and middle/low economic settings, meaning that millions more babies are going to need us to better understand the GM and its changes after EoP.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
The authors of this work are supported by grants from Sparks Children's Medical Research (15KCL05); King's Health Partners (R170506); the Medical Research Council (MR/K006355/1); Inserm, Université de Paris, Horizon 2020 Framework Program of the European Union (grant agreement no. 874721/PREMSTEM); Investissement d'Avenir (ANR-11-INBS-0011, NeurATRIS), Fondation de France; Fondation pour la Recherche sur le Cerveau, Fondation Grace de Monaco; Action Medical Research; the Australian National Health and Medical Research Council; and the Cerebral Palsy Alliance Research Foundation Australia. BF's research was also supported by a Vice Chancellor's Research Fellowship from RMIT University.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We wish to acknowledge the support of the Department of Perinatal Imaging & Health, King's College London. In addition, the authors acknowledge financial support from the National Institute for Health Research (NIHR) Biomedical Research Centre based at Guy's and St Thomas' NHS Foundation Trust and King's College London. The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health.
Abbreviations
ADHD, attention deficit hyperactivity disorder; ASD, autism spectrum disorder; EEG, electroencephalogram; EoP, encephalopathy of prematurity; GM, gray matter; FA, fractional anisotropy; IVH, intraventricular hemorrhage; MRI, magnetic resonance imaging; NDD, neurodevelopmental disorder; PVL, periventricular leukomalacia; WM, white matter.
References
1. Volpe JJ. The encephalopathy of prematurity–brain injury and impaired brain development inextricably intertwined. Semin Pediatr Neurol. (2009) 16:167–78. doi: 10.1016/j.spen.2009.09.005
2. Kostovic I, Judas M, Petanjek Z, Simic G. Ontogenesis of goal-directed behavior: anatomo-functional considerations. Int J Psychophysiol. (1995) 19:85–102. doi: 10.1016/0167-8760(94)00081-O
3. Marin-Padilla M. Developmental neuropathology and impact of perinatal brain damage. II: white matter lesions of the neocortex. J Neuropathol Exp Neurol. (1997) 56:219–35. doi: 10.1097/00005072-199703000-00001
4. Panda S, Dohare P, Jain S, Parikh N, Singla P, Mehdizadeh R, et al. Estrogen treatment reverses prematurity-induced disruption in cortical interneuron population. J Neurosci. (2018) 38:7378–91. doi: 10.1523/JNEUROSCI.0478-18.2018
5. Marlow N, Wolke D, Bracewell MA, Samara M, Group EPS. Neurologic and developmental disability at six years of age after extremely preterm birth. N Engl J Med. (2005) 352:9–19. doi: 10.1056/NEJMoa041367
6. O'Shea TM, Allred EN, Dammann O, Hirtz D, Kuban KC, Paneth N, et al. The ELGAN study of the brain and related disorders in extremely low gestational age newborns. Early Hum Dev. (2009) 85:719–25. doi: 10.1016/j.earlhumdev.2009.08.060
7. Pierrat V, Marchand-Martin L, Arnaud C, Kaminski M, Resche-Rigon M, Lebeaux C, et al. Neurodevelopmental outcome at 2 years for preterm children born at 22 to 34 weeks' gestation in France in 2011: EPIPAGE-2 cohort study. BMJ. (2017) 358:j3448. doi: 10.1136/bmj.j3448
8. Hirschberger RG, Kuban KCK, O'Shea TM, Joseph RM, Heeren T, Douglass LM, et al. Co-occurrence and severity of neurodevelopmental burden (cognitive impairment, cerebral palsy, autism spectrum disorder, and epilepsy) at age ten years in children born extremely preterm. Pediatr Neurol. (2018) 79:45–52. doi: 10.1016/j.pediatrneurol.2017.11.002
9. Johnson S, Hollis C, Kochhar P, Hennessy E, Wolke D, Marlow N. Psychiatric disorders in extremely preterm children: longitudinal finding at age 11 years in the EPICure study. J Am Acad Child Adolesc Psychiatry. (2010) 49:453–63 e451. doi: 10.1097/00004583-201005000-00006
10. Heeren T, Joseph RM, Allred EN, O'Shea TM, Leviton A, Kuban KCK. Cognitive functioning at the age of 10 years among children born extremely preterm: a latent profile approach. Pediatr Res. (2017) 82:614–9. doi: 10.1038/pr.2017.82
11. Thomason ME, Scheinost D, Manning JH, Grove LE, Hect J, Marshall N, et al. Weak functional connectivity in the human fetal brain prior to preterm birth. Sci Rep. (2017) 7:39286. doi: 10.1038/srep39286
12. Nunthapiwat S, Sekararithi R, Wanapirak C, Sirichotiyakul S, Tongprasert F, Srisupundit K, et al. Second trimester serum biomarker screen for fetal aneuploidies as a predictor of preterm delivery: a population-based study. Gynecol Obstet Invest. (2019) 84:326–33. doi: 10.1159/000495614
13. Souza RT, McKenzie EJ, Jones B, de Seymour JV, Thomas MM, Zarate E, et al. Trace biomarkers associated with spontaneous preterm birth from the maternal serum metabolome of asymptomatic nulliparous women–parallel case-control studies from the SCOPE cohort. Sci Rep. (2019) 9:13701. doi: 10.1038/s41598-019-50252-7
14. Leow SM, Di Quinzio MKW, Ng ZL, Grant C, Amitay T, Wei Y, et al. Preterm birth prediction in asymptomatic women at mid-gestation using a panel of novel protein biomarkers: the Prediction of PreTerm Labor (PPeTaL) study. Am J Obstet Gynecol. (2020) 2:100084. doi: 10.1016/j.ajogmf.2019.100084
15. Ananth CV, Vintzileos AM. Medically indicated preterm birth: recognizing the importance of the problem. Clin Perinatol. (2008) 35:53–67, viii. doi: 10.1016/j.clp.2007.11.001
16. Gilles F, Gressens P, Dammann O, Leviton A. Hypoxia-ischemia is not an antecedent of most preterm brain damage: the illusion of validity. Dev Med Child Neurol. (2018) 60:120–5. doi: 10.1111/dmcn.13483
17. Kaga N, Katsuki Y, Obata M, Shibutani Y. Repeated administration of low-dose lipopolysaccharide induces preterm delivery in mice: a model for human preterm parturition and for assessment of the therapeutic ability of drugs against preterm delivery. Am J Obstet Gynecol. (1996) 174:754–9. doi: 10.1016/S0002-9378(96)70460-X
18. Romero R, Gomez R, Chaiworapongsa T, Conoscenti G, Kim JC, Kim YM. The role of infection in preterm labour and delivery. Paediatr Perinat Epidemiol. (2001) 15:41–56. doi: 10.1046/j.1365-3016.2001.00007.x
19. Fidel P, Ghezzi F, Romero R, Chaiworapongsa T, Espinoza J, Cutright J, et al. The effect of antibiotic therapy on intrauterine infection-induced preterm parturition in rabbits. J Matern Fetal Neonat Med. (2003) 14:57–64. doi: 10.1080/jmf.14.1.57.64
20. Dammann O, Leviton A. Maternal intrauterine infection, cytokines, and brain damage in the preterm newborn. Pediatr Res. (1997) 42:1–8. doi: 10.1203/00006450-199707000-00001
21. Lau J, Magee F, Qiu Z, Hoube J, Von Dadelszen P, Lee SK. Chorioamnionitis with a fetal inflammatory response is associated with higher neonatal mortality, morbidity, and resource use than chorioamnionitis displaying a maternal inflammatory response only. Am J Obstet Gynecol. (2005) 193:708–13. doi: 10.1016/j.ajog.2005.01.017
22. Hagberg H, Mallard C, Ferriero DM, Vannucci SJ, Levison SW, Vexler ZS, et al. The role of inflammation in perinatal brain injury. Nat Rev Neurol. (2015) 11:192–208. doi: 10.1038/nrneurol.2015.13
23. Kuban KC, O'Shea TM, Allred EN, Fichorova RN, Heeren T, Paneth N, et al. The breadth and type of systemic inflammation and the risk of adverse neurological outcomes in extremely low gestation newborns. Pediatr Neurol. (2015) 52:42–8. doi: 10.1016/j.pediatrneurol.2014.10.005
24. Bouyssi-Kobar M, du Plessis AJ, McCarter R, Brossard-Racine M, Murnick J, Tinkleman L, et al. Third trimester brain growth in preterm infants compared with in utero healthy fetuses. Pediatrics. (2016) 138:e20161640. doi: 10.1542/peds.2016-1640
25. Wellmann S, Buhrer C, Schmitz T. Focal necrosis and disturbed myelination in the white matter of newborn infants: a tale of too much or too little oxygen. Front Pediatr. (2014) 2:143. doi: 10.3389/fped.2014.00143
26. Manley BJ, Owen LS, Hooper SB, Jacobs SE, Cheong JLY, Doyle LW, et al. Towards evidence-based resuscitation of the newborn infant. Lancet. (2017) 389:1639–48. doi: 10.1016/S0140-6736(17)30547-0
27. Shaw JC, Berry MJ, Dyson RM, Crombie GK, Hirst JJ, Palliser HK. Reduced neurosteroid exposure following preterm birth and its' contribution to neurological impairment: a novel avenue for preventative therapies. Front Physiol. (2019) 10:599. doi: 10.3389/fphys.2019.00599
28. Hunt R, Davis PG, Inder T. Replacement of estrogens and progestins to prevent morbidity and mortality in preterm infants. Cochrane Database Syst Rev. (2004) CD003848. doi: 10.1002/14651858.CD003848.pub2
29. Tibrewal M, Cheng B, Dohare P, Hu F, Mehdizadeh R, Wang P, et al. Disruption of interneuron neurogenesis in premature newborns and reversal with estrogen treatment. J Neurosci. (2018) 38:1100–13. doi: 10.1523/JNEUROSCI.1875-17.2017
30. Klebe D, Tibrewal M, Sharma DR, Vanaparthy R, Krishna S, Varghese M, et al. Reduced hippocampal dendrite branching, spine density and neurocognitive function in premature rabbits, and reversal with estrogen or TrkB agonist treatment. Cereb Cortex. (2019) 29:4932–47. doi: 10.1093/cercor/bhz033
31. Vottier G, Pham H, Pansiot J, Biran V, Gressens P, Charriaut-Marlangue C, et al. Deleterious effect of hyperoxia at birth on white matter damage in the newborn rat. Dev Neurosci. (2011) 33:261–9. doi: 10.1159/000327245
32. Schmitz T, Endesfelder S, Reinert MC, Klinker F, Muller S, Buhrer C, et al. Adolescent hyperactivity and impaired coordination after neonatal hyperoxia. Exp Neurol. (2012) 235:374–9. doi: 10.1016/j.expneurol.2012.03.002
33. Scheuer T, Brockmoller V, Blanco Knowlton M, Weitkamp JH, Ruhwedel T, Mueller S, et al. Oligodendroglial maldevelopment in the cerebellum after postnatal hyperoxia and its prevention by minocycline. Glia. (2015) 63:1825–39. doi: 10.1002/glia.22847
34. Banks WA, Kastin AJ, Broadwell RD. Passage of cytokines across the blood-brain barrier. Neuroimmunomodulation. (1995) 2:241–8. doi: 10.1159/000097202
35. Verma S, Nakaoke R, Dohgu S, Banks WA. Release of cytokines by brain endothelial cells: a polarized response to lipopolysaccharide. Brain Behav Immun. (2006) 20:449–55. doi: 10.1016/j.bbi.2005.10.005
36. Stolp HB, Turnquist C, Dziegielewska KM, Saunders NR, Anthony DC, Molnar Z. Reduced ventricular proliferation in the foetal cortex following maternal inflammation in the mouse. Brain. (2011) 134:3236–48. doi: 10.1093/brain/awr237
37. Pang Y, Dai X, Roller A, Carter K, Paul I, Bhatt AJ, et al. Early postnatal lipopolysaccharide exposure leads to enhanced neurogenesis and impaired communicative functions in rats. PLoS ONE. (2016) 11:e0164403. doi: 10.1371/journal.pone.0164403
39. Fan L-W, Pang Y. Dysregulation of neurogenesis by neuroinflammation: key differences in neurodevelopmental and neurological disorders. Neural Regen Res. (2017) 12:366–71. doi: 10.4103/1673-5374.202926
40. Prins JR, Eskandar S, Eggen BJL, Scherjon SA. Microglia, the missing link in maternal immune activation and fetal neurodevelopment; and a possible link in preeclampsia and disturbed neurodevelopment? J Reprod Immunol. (2018) 126:18–22. doi: 10.1016/j.jri.2018.01.004
41. Malik S, Vinukonda G, Vose LR, Diamond D, Bhimavarapu BB, Hu F, et al. Neurogenesis continues in the third trimester of pregnancy and is suppressed by premature birth. J Neurosci. (2013) 33:411–23. doi: 10.1523/JNEUROSCI.4445-12.2013
42. Stolp HB, Fleiss B, Arai Y, Supramaniam V, Vontell R, Birtles S, et al. Interneuron Development is disrupted in preterm brains with diffuse white matter injury: observations in mouse and human. Front Physiol. (2019) 10:955. doi: 10.3389/fphys.2019.00955
43. Mallard C, Tremblay ME, Vexler ZS. Microglia and neonatal brain injury. Neuroscience. (2019) 405:68–76. doi: 10.1016/j.neuroscience.2018.01.023
44. McNamara NB, Miron VE. Microglia in developing white matter and perinatal brain injury. Neurosci Lett. (2020) 714:134539. doi: 10.1016/j.neulet.2019.134539
45. Dommergues MA, Plaisant F, Verney C, Gressens P. Early microglial activation following neonatal excitotoxic brain damage in mice: a potential target for neuroprotection. Neuroscience. (2003) 121:619–28. doi: 10.1016/S0306-4522(03)00558-X
46. Van Steenwinckel J, Schang AL, Krishnan ML, Degos V, Delahaye-Duriez A, Bokobza C, et al. Decreased microglial Wnt/β-catenin signalling drives microglial pro-inflammatory activation in the developing brain. Brain. (2019) 142:3806–33. doi: 10.1093/brain/awz319
47. Faustino JV, Wang X, Johnson CE, Klibanov A, Derugin N, Wendland MF, et al. Microglial cells contribute to endogenous brain defenses after acute neonatal focal stroke. J Neurosci. (2011) 31:12992–3001. doi: 10.1523/JNEUROSCI.2102-11.2011
48. Fernández-López D, Faustino J, Klibanov AL, Derugin N, Blanchard E, Simon F, et al. Microglial cells prevent hemorrhage in neonatal focal arterial stroke. J Neurosci. (2016) 36:2881. doi: 10.1523/JNEUROSCI.0140-15.2016
49. Lafemina MJ, Sheldon RA, Ferriero DM. Acute hypoxia-ischemia results in hydrogen peroxide accumulation in neonatal but not adult mouse brain. Pediatr Res. (2006) 59:680–3. doi: 10.1203/01.pdr.0000214891.35363.6a
50. Gao Z, Tsirka SE. Animal models of MS reveal multiple roles of microglia in disease pathogenesis. Neurol Res Int. (2011) 2011:383087. doi: 10.1155/2011/383087
51. Pang Y, Campbell L, Zheng B, Fan L, Cai Z, Rhodes P. Lipopolysaccharide-activated microglia induce death of oligodendrocyte progenitor cells and impede their development. Neuroscience. (2010) 166:464–75. doi: 10.1016/j.neuroscience.2009.12.040
52. Favrais G, van de Looij Y, Fleiss B, Ramanantsoa N, Bonnin P, Stoltenburg-Didinger G, et al. Systemic inflammation disrupts the developmental program of white matter. Ann Neurol. (2011) 70:550–65. doi: 10.1002/ana.22489
53. Scafidi J, Hammond TR, Scafidi S, Ritter J, Jablonska B, Roncal M, et al. Intranasal epidermal growth factor treatment rescues neonatal brain injury. Nature. (2014) 506:230–4. doi: 10.1038/nature12880
54. Xie D, Shen F, He S, Chen M, Han Q, Fang M, et al. IL-1beta induces hypomyelination in the periventricular white matter through inhibition of oligodendrocyte progenitor cell maturation via FYN/MEK/ERK signaling pathway in septic neonatal rats. Glia. (2016) 64:583–602. doi: 10.1002/glia.22950
55. Ramlackhansingh AF, Brooks DJ, Greenwood RJ, Bose SK, Turkheimer FE, Kinnunen KM, et al. Inflammation after trauma: microglial activation and traumatic brain injury. Ann Neurol. (2011) 70:374–83. doi: 10.1002/ana.22455
56. Loane DJ, Kumar A, Stoica BA, Cabatbat R, Faden AI. Progressive neurodegeneration after experimental brain trauma: association with chronic microglial activation. J Neuropathol Exp Neurol. (2014) 73:14–29. doi: 10.1097/NEN.0000000000000021
57. Mattei D, Ivanov A, Ferrai C, Jordan P, Guneykaya D, Buonfiglioli A, et al. Maternal immune activation results in complex microglial transcriptome signature in the adult offspring that is reversed by minocycline treatment. Transl Psychiatry. (2017) 7:e1120. doi: 10.1038/tp.2017.80
58. Frost PS, Barros-Aragao F, da Silva RT, Venancio A, Matias I, Lyra ESNM, et al. Neonatal infection leads to increased susceptibility to Abeta oligomer-induced brain inflammation, synapse loss and cognitive impairment in mice. Cell Death Dis. (2019) 10:323. doi: 10.1038/s41419-019-1529-x
59. Lawson LJ, Perry VH, Dri P, Gordon S. Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience. (1990) 39:151–70. doi: 10.1016/0306-4522(90)90229-W
60. Hart AD, Wyttenbach A, Perry VH, Teeling JL. Age related changes in microglial phenotype vary between CNS regions: grey versus white matter differences. Brain Behav Immun. (2012) 26:754–65. doi: 10.1016/j.bbi.2011.11.006
61. Verderio C, Muzio L, Turola E, Bergami A, Novellino L, Ruffini F, et al. Myeloid microvesicles are a marker and therapeutic target for neuroinflammation. Ann Neurol. (2012) 72:610–24. doi: 10.1002/ana.23627
62. Lener T, Gimona M, Aigner L, Borger V, Buzas E, Camussi G, et al. Applying extracellular vesicles based therapeutics in clinical trials–an ISEV position paper. J Extracell Vesicles. (2015) 4:30087. doi: 10.3402/jev.v4.30087
63. Back SA, Miller SP. Brain injury in premature neonates: a primary cerebral dysmaturation disorder? Ann Neurol. (2014) 75:469–86. doi: 10.1002/ana.24132
64. Shiow LR, Favrais G, Schirmer L, Schang AL, Cipriani S, Andres C, et al. Reactive astrocyte COX2-PGE2 production inhibits oligodendrocyte maturation in neonatal white matter injury. Glia. (2017) 65:2024–37. doi: 10.1002/glia.23212
65. Preston M, Gong X, Su W, Matsumoto SG, Banine F, Winkler C, et al. Digestion products of the PH20 hyaluronidase inhibit remyelination. Ann Neurol. (2013) 73:266–80. doi: 10.1002/ana.23788
66. Wang Y, Cheng X, He Q, Zheng Y, Kim DH, Whittemore SR, et al. Astrocytes from the contused spinal cord inhibit oligodendrocyte differentiation of adult oligodendrocyte precursor cells by increasing the expression of bone morphogenetic proteins. J Neurosci. (2011) 31:6053. doi: 10.1523/JNEUROSCI.5524-09.2011
67. Hammond TR, Gadea A, Dupree J, Kerninon C, Nait-Oumesmar B, Aguirre A, et al. Astrocyte-Derived Endothelin-1 Inhibits Remyelination through Notch Activation. Neuron. (2014) 81:588–602. doi: 10.1016/j.neuron.2013.11.015
68. Gelot A, Villapol S, Billette de Villemeur T, Renolleau S, Charriaut-Marlangue C. Astrocytic demise in the developing rat and human brain after hypoxic-ischemic damage. Dev Neurosci. (2009) 31:459–70. doi: 10.1159/000232564
69. Verney C, Pogledic I, Biran V, Adle-Biassette H, Fallet-Bianco C, Gressens P. Microglial reaction in axonal crossroads is a hallmark of noncystic periventricular white matter injury in very preterm infants. J Neuropathol Exp Neurol. (2012) 71:251–64. doi: 10.1097/NEN.0b013e3182496429
70. Tahraoui SL, Marret S, Bodenant C, Leroux P, Dommergues MA, Evrard P, et al. Central role of microglia in neonatal excitotoxic lesions of the murine periventricular white matter. Brain Pathol. (2001) 11:56–71. doi: 10.1111/j.1750-3639.2001.tb00381.x
71. Roessmann U, Gambetti P. Astrocytes in the developing human brain. Acta Neuropathol. (1986) 70:308–13. doi: 10.1007/BF00686089
72. Pierson CR, Folkerth RD, Billiards SS, Trachtenberg FL, Drinkwater ME, Volpe JJ, et al. Gray matter injury associated with periventricular leukomalacia in the premature infant. Acta Neuropathol. (2007) 114:619–31. doi: 10.1007/s00401-007-0295-5
73. Sullivan SM, Bjorkman ST, Miller SM, Colditz PB, Pow DV. Structural remodeling of gray matter astrocytes in the neonatal pig brain after hypoxia/ischemia. Glia. (2010) 58:181–94. doi: 10.1002/glia.20911
74. Sidman RL, Rakic P. Neuronal migration, with special reference to developing human brain: a review. Brain Res. (1973) 62:1–35. doi: 10.1016/0006-8993(73)90617-3
75. Marin O. Cellular and molecular mechanisms controlling the migration of neocortical interneurons. Eur J Neurosci. (2013) 38:2019–29. doi: 10.1111/ejn.12225
76. Huttenlocher PR. Synaptic density in human frontal cortex–developmental changes and effects of aging. Brain Res. (1979) 163:195–205. doi: 10.1016/0006-8993(79)90349-4
77. Kilb W, Kirischuk S, Luhmann HJ. Electrical activity patterns and the functional maturation of the neocortex. Eur J Neurosci. (2011) 34:1677–86. doi: 10.1111/j.1460-9568.2011.07878.x
78. Molnar Z, Clowry GJ, Sestan N, Alzu'bi A, Bakken T, Hevner RF, et al. New insights into the development of the human cerebral cortex. J Anat. (2019) 235:432–51. doi: 10.1111/joa.13055
79. Volpe JJ. Dysmaturation of premature brain: importance, cellular mechanisms, and potential interventions. Pediatr Neurol. (2019) 95:42–66. doi: 10.1016/j.pediatrneurol.2019.02.016
80. Armstrong E, Schleicher A, Omran H, Curtis M, Zilles K. The ontogeny of human gyrification. Cereb Cortex. (1995) 5:56–63. doi: 10.1093/cercor/5.1.56
81. Neil JJ, Shiran SI, McKinstry RC, Schefft GL, Snyder AZ, Almli CR, et al. Normal brain in human newborns: apparent diffusion coefficient and diffusion anisotropy measured by using diffusion tensor MR imaging. Radiology. (1998) 209:57–66. doi: 10.1148/radiology.209.1.9769812
82. McKinstry RC, Mathur A, Miller JH, Ozcan A, Snyder AZ, Schefft GL, et al. Radial organization of developing preterm human cerebral cortex revealed by non-invasive water diffusion anisotropy MRI. Cereb Cortex. (2002) 12:1237–43. doi: 10.1093/cercor/12.12.1237
83. Batalle D, O'Muircheartaigh J, Makropoulos A, Kelly CJ, Dimitrova R, Hughes EJ, et al. Different patterns of cortical maturation before and after 38 weeks gestational age demonstrated by diffusion MRI in vivo. Neuroimage. (2019) 185:764–75. doi: 10.1016/j.neuroimage.2018.05.046
84. Lodygensky GA, Vasung L, Sizonenko SV, Huppi PS. Neuroimaging of cortical development and brain connectivity in human newborns and animal models. J Anat. (2010) 217:418–28. doi: 10.1111/j.1469-7580.2010.01280.x
85. Stolp HB, Ball G, So PW, Tournier JD, Jones M, Thornton C, et al. Voxel-wise comparisons of cellular microstructure and diffusion-MRI in mouse hippocampus using 3D bridging of optically-clear histology with neuroimaging data (3D-BOND). Sci Rep. (2018) 8:4011. doi: 10.1038/s41598-018-22295-9
86. Rathbone R, Counsell SJ, Kapellou O, Dyet L, Kennea N, Hajnal J, et al. Perinatal cortical growth and childhood neurocognitive abilities. Neurology. (2011) 77:1510–7. doi: 10.1212/WNL.0b013e318233b215
87. Boardman JP, Counsell SJ, Rueckert D, Kapellou O, Bhatia KK, Aljabar P, et al. Abnormal deep grey matter development following preterm birth detected using deformation-based morphometry. Neuroimage. (2006) 32:70–8. doi: 10.1016/j.neuroimage.2006.03.029
88. Ajayi-Obe M, Saeed N, Cowan FM, Rutherford MA, Edwards AD. Reduced development of cerebral cortex in extremely preterm infants. Lancet. (2000) 356:1162–3. doi: 10.1016/S0140-6736(00)02761-6
89. Makropoulos A, Aljabar P, Wright R, Huning B, Merchant N, Arichi T, et al. Regional growth and atlasing of the developing human brain. Neuroimage. (2016) 125:456–78. doi: 10.1016/j.neuroimage.2015.10.047
90. Inder TE, Huppi PS, Warfield S, Kikinis R, Zientara GP, Barnes PD, et al. Periventricular white matter injury in the premature infant is followed by reduced cerebral cortical gray matter volume at term. Ann Neurol. (1999) 46:755–60. doi: 10.1002/1531-8249(199911)46:5<755::AID-ANA11>3.0.CO
91. Zubiaurre-Elorza L, Soria-Pastor S, Junque C, Segarra D, Bargallo N, Mayolas N, et al. Gray matter volume decrements in preterm children with periventricular leukomalacia. Pediatr Res. (2011) 69:554–60. doi: 10.1203/PDR.0b013e3182182366
92. Zhang Y, Inder TE, Neil JJ, Dierker DL, Alexopoulos D, Anderson PJ, et al. Cortical structural abnormalities in very preterm children at 7 years of age. Neuroimage. (2015) 109:469–79. doi: 10.1016/j.neuroimage.2015.01.005
93. Nosarti C, Al-Asady MH, Frangou S, Stewart AL, Rifkin L, Murray RM. Adolescents who were born very preterm have decreased brain volumes. Brain. (2002) 125:1616–23. doi: 10.1093/brain/awf157
94. Nosarti C, Giouroukou E, Healy E, Rifkin L, Walshe M, Reichenberg A, et al. Grey and white matter distribution in very preterm adolescents mediates neurodevelopmental outcome. Brain. (2008) 131:205–17. doi: 10.1093/brain/awm282
95. Ball G, Boardman JP, Rueckert D, Aljabar P, Arichi T, Merchant N, et al. The effect of preterm birth on thalamic and cortical development. Cereb Cortex. (2012) 22:1016–24. doi: 10.1093/cercor/bhr176
96. Bjuland KJ, Rimol LM, Lohaugen GC, Skranes J. Brain volumes and cognitive function in very-low-birth-weight (VLBW) young adults. Eur J Paediatr Neurol. (2014) 18:578–90. doi: 10.1016/j.ejpn.2014.04.004
97. Ball G, Pazderova L, Chew A, Tusor N, Merchant N, Arichi T, et al. Thalamocortical connectivity predicts cognition in children born preterm. Cereb Cortex. (2015) 25:4310–8. doi: 10.1093/cercor/bhu331
98. Bora S, Pritchard VE, Chen Z, Inder TE, Woodward LJ. Neonatal cerebral morphometry and later risk of persistent inattention/hyperactivity in children born very preterm. J Child Psychol Psychiatry. (2014) 55:828–38. doi: 10.1111/jcpp.12200
99. Rogers CE, Barch DM, Sylvester CM, Pagliaccio D, Harms MP, Botteron KN, et al. Altered gray matter volume and school age anxiety in children born late preterm. J Pediatr. (2014) 165:928–35. doi: 10.1016/j.jpeds.2014.06.063
100. Ure AM, Treyvaud K, Thompson DK, Pascoe L, Roberts G, Lee KJ, et al. Neonatal brain abnormalities associated with autism spectrum disorder in children born very preterm. Autism Res. (2016) 9:543–52. doi: 10.1002/aur.1558
101. Jantzie LL, Robinson S. Preclinical models of encephalopathy of prematurity. Dev Neurosci. (2015) 37:277–88. doi: 10.1159/000371721
102. Dean JM, van de Looij Y, Sizonenko SV, Lodygensky GA, Lazeyras F, Bolouri H, et al. Delayed cortical impairment following lipopolysaccharide exposure in preterm fetal sheep. Ann Neurol. (2011) 70:846–56. doi: 10.1002/ana.22480
103. Dean JM, McClendon E, Hansen K, Azimi-Zonooz A, Chen K, Riddle A, et al. Prenatal cerebral ischemia disrupts MRI-defined cortical microstructure through disturbances in neuronal arborization. Sci Transl Med. (2013) 5:168ra167. doi: 10.1126/scitranslmed.3004669
104. Crum WR, Sawiak SJ, Chege W, Cooper JD, Williams SCR, Vernon AC. Evolution of structural abnormalities in the rat brain following in utero exposure to maternal immune activation: a longitudinal in vivo MRI study. Brain Behav Immun. (2017) 63:50–9. doi: 10.1016/j.bbi.2016.12.008
105. Engelhardt E, Inder TE, Alexopoulos D, Dierker DL, Hill J, Van Essen D, et al. Regional impairments of cortical folding in premature infants. Ann Neurol. (2015) 77:154–62. doi: 10.1002/ana.24313
106. Kersbergen KJ, Makropoulos A, Aljabar P, Groenendaal F, de Vries LS, Counsell SJ, et al. Longitudinal regional brain development and clinical risk factors in extremely preterm infants. J Pediatr. (2016) 178:93–100 e106. doi: 10.1016/j.jpeds.2016.08.024
107. Wolosin SM, Richardson ME, Hennessey JG, Denckla MB, Mostofsky SH. Abnormal cerebral cortex structure in children with ADHD. Hum Brain Mapp. (2009) 30:175–84. doi: 10.1002/hbm.20496
108. Kohli JS, Kinnear MK, Fong CH, Fishman I, Carper RA, Muller RA. Local cortical gyrification is increased in children with autism spectrum disorders, but decreases rapidly in adolescents. Cereb Cortex. (2019) 29:2412–23. doi: 10.1093/cercor/bhy111
109. Kohli JS, Kinnear MK, Martindale IA, Carper RA, Muller RA. Regionally decreased gyrification in middle-aged adults with autism spectrum disorders. Neurology. (2019) 93:e1900–5. doi: 10.1212/WNL.0000000000008478
110. Libero LE, Schaer M, Li DD, Amaral DG, Nordahl CW. A longitudinal study of local gyrification index in young boys with autism spectrum disorder. Cereb Cortex. (2019) 29:2575–87. doi: 10.1093/cercor/bhy126
111. Bayly PV, Taber LA, Kroenke CD. Mechanical forces in cerebral cortical folding: a review of measurements and models. J Mech Behav Biomed Mater. (2014) 29:568–81. doi: 10.1016/j.jmbbm.2013.02.018
112. Striedter GF, Srinivasan S, Monuki ES. Cortical folding: when, where, how, and why? Annu Rev Neurosci. (2015) 38:291–307. doi: 10.1146/annurev-neuro-071714-034128
113. Llinares-Benadero C, Borrell V. Deconstructing cortical folding: genetic, cellular and mechanical determinants. Nat Rev Neurosci. (2019) 20:161–76. doi: 10.1038/s41583-018-0112-2
114. Quezada S, Castillo-Melendez M, Walker DW, Tolcos M. Development of the cerebral cortex and the effect of the intrauterine environment. J Physiol. (2018) 596:5665–74. doi: 10.1113/JP277151
115. Garcia KE, Robinson EC, Alexopoulos D, Dierker DL, Glasser MF, Coalson TS, et al. Dynamic patterns of cortical expansion during folding of the preterm human brain. Proc Natl Acad Sci USA. (2018) 115:3156–61. doi: 10.1073/pnas.1715451115
116. Van Essen DC, Donahue CJ, Coalson TS, Kennedy H, Hayashi T, Glasser MF. Cerebral cortical folding, parcellation, and connectivity in humans, nonhuman primates, and mice. Proc Natl Acad Sci USA. (2019) 116:26173–80. doi: 10.1073/pnas.1902299116
117. Long KR, Newland B, Florio M, Kalebic N, Langen B, Kolterer A, et al. Extracellular matrix components HAPLN1, lumican, and collagen I cause hyaluronic acid-dependent folding of the developing human neocortex. Neuron. (2018) 99:702–19 e706. doi: 10.1016/j.neuron.2018.07.013
118. Buser JR, Maire J, Riddle A, Gong X, Nguyen T, Nelson K, et al. Arrested preoligodendrocyte maturation contributes to myelination failure in premature infants. Ann Neurol. (2012) 71:93–109. doi: 10.1002/ana.22627
119. Andiman SE, Haynes RL, Trachtenberg FL, Billiards SS, Folkerth RD, Volpe JJ, et al. The cerebral cortex overlying periventricular leukomalacia: analysis of pyramidal neurons. Brain Pathol. (2010) 20:803–14. doi: 10.1111/j.1750-3639.2010.00380.x
120. Haynes RL, van Leyen K. 12/15-lipoxygenase expression is increased in oligodendrocytes and microglia of periventricular leukomalacia. Dev Neurosci. (2013) 35:140–54. doi: 10.1159/000350230
121. Haynes RL, Billiards SS, Borenstein NS, Volpe JJ, Kinney HC. Diffuse axonal injury in periventricular leukomalacia as determined by apoptotic marker fractin. Pediatr Res. (2008) 63:656–61. doi: 10.1203/PDR.0b013e31816c825c
122. Ligam P, Haynes RL, Folkerth RD, Liu L, Yang M, Volpe JJ, et al. Thalamic damage in periventricular leukomalacia: novel pathologic observations relevant to cognitive deficits in survivors of prematurity. Pediatr Res. (2009) 65:524–9. doi: 10.1203/PDR.0b013e3181998baf
123. Kinney HC, Haynes RL, Xu G, Andiman SE, Folkerth RD, Sleeper LA, et al. Neuron deficit in the white matter and subplate in periventricular leukomalacia. Ann Neurol. (2012) 71:397–406. doi: 10.1002/ana.22612
124. Haldipur P, Bharti U, Alberti C, Sarkar C, Gulati G, Iyengar S, et al. Preterm delivery disrupts the developmental program of the cerebellum. PLoS ONE. (2011) 6:e23449. doi: 10.1371/journal.pone.0023449
125. Vontell R, Supramaniam V, Wyatt-Ashmead J, Gressens P, Rutherford M, Hagberg H, et al. Cellular mechanisms of toll-like receptor-3 activation in the thalamus are associated with white matter injury in the developing brain. J Neuropathol Exp Neurol. (2015) 74:273–85. doi: 10.1097/NEN.0000000000000172
126. Pogledic I, Kostovic I, Fallet-Bianco C, Adle-Biassette H, Gressens P, Verney C. Involvement of the subplate zone in preterm infants with periventricular white matter injury. Brain Pathol. (2014) 24:128–41. doi: 10.1111/bpa.12096
127. Haynes RL, Folkerth RD, Keefe RJ, Sung I, Swzeda LI, Rosenberg PA, et al. Nitrosative and oxidative injury to premyelinating oligodendrocytes in periventricular leukomalacia. J Neuropathol Exp Neurol. (2003) 62:441–50. doi: 10.1093/jnen/62.5.441
128. Ancel PY, Livinec F, Larroque B, Marret S, Arnaud C, Pierrat V, et al. Cerebral palsy among very preterm children in relation to gestational age and neonatal ultrasound abnormalities: the EPIPAGE cohort study. Pediatrics. (2006) 117:828–35. doi: 10.1542/peds.2005-0091
129. McQuillen PS, Sheldon RA, Shatz CJ, Ferriero DM. Selective vulnerability of subplate neurons after early neonatal hypoxia-ischemia. J Neurosci. (2003) 23:3308–15. doi: 10.1523/JNEUROSCI.23-08-03308.2003
130. Okusa C, Oeschger F, Ginet V, Wang WZ, Hoerder-Suabedissen A, Matsuyama T, et al. Subplate in a rat model of preterm hypoxia-ischemia. Ann Clin Transl Neurol. (2014) 1:679–91. doi: 10.1002/acn3.97
131. Goni-de-Cerio F, Alvarez A, Caballero A, Mielgo VE, Alvarez FJ, Rey-Santano MC, et al. Early cell death in the brain of fetal preterm lambs after hypoxic-ischemic injury. Brain Res. (2007) 1151:161–71. doi: 10.1016/j.brainres.2007.03.013
132. McClendon E, Chen K, Gong X, Sharifnia E, Hagen M, Cai V, et al. Prenatal cerebral ischemia triggers dysmaturation of caudate projection neurons. Ann Neurol. (2014) 75:508–24. doi: 10.1002/ana.24100
133. McClendon E, Shaver DC, Degener-O'Brien K, Gong X, Nguyen T, Hoerder-Suabedissen A, et al. Transient hypoxemia chronically disrupts maturation of preterm fetal ovine subplate neuron arborization and activity. J Neurosci. (2017) 37:11912–29. doi: 10.1523/JNEUROSCI.2396-17.2017
134. Ardalan M, Svedin P, Baburamani AA, Supramaniam VG, Ek J, Hagberg H, et al. Dysmaturation of somatostatin interneurons following umbilical cord occlusion in preterm fetal sheep. Front Physiol. (2019) 10:563. doi: 10.3389/fphys.2019.00563
135. Fowke TM, Galinsky R, Davidson JO, Wassink G, Karunasinghe RN, Prasad JD, et al. Loss of interneurons and disruption of perineuronal nets in the cerebral cortex following hypoxia-ischaemia in near-term fetal sheep. Sci Rep. (2018) 8:17686. doi: 10.1038/s41598-018-36083-y
136. McClendon E, Wang K, Degener-O'Brien K, Hagen MW, Gong X, Nguyen T, et al. Transient hypoxemia disrupts anatomical and functional maturation of preterm fetal ovine CA1 pyramidal neurons. J Neurosci. (2019) 39:7853–71. doi: 10.1523/JNEUROSCI.1364-19.2019
137. Dieni S, Inder T, Yoder B, Briscoe T, Camm E, Egan G, et al. The pattern of cerebral injury in a primate model of preterm birth and neonatal intensive care. J Neuropathol Exp Neurol. (2004) 63:1297–309. doi: 10.1093/jnen/63.12.1297
138. Gussenhoven R, Westerlaken RJJ, Ophelders D, Jobe AH, Kemp MW, Kallapur SG, et al. Chorioamnionitis, neuroinflammation, and injury: timing is key in the preterm ovine fetus. J Neuroinflammation. (2018) 15:113. doi: 10.1186/s12974-018-1149-x
139. Stojanovska V, Atik A, Nitsos I, Skiöld B, Barton SK, Zahra VA, et al. Effects of intrauterine inflammation on cortical gray matter of near-term lambs. Front Pediatr. (2018) 6:145. doi: 10.3389/fped.2018.00145
140. Rousset CI, Chalon S, Cantagrel S, Bodard S, Andres C, Gressens P, et al. Maternal exposure to LPS induces hypomyelination in the internal capsule and programmed cell death in the deep gray matter in newborn rats. Pediatr Res. (2006) 59:428–33. doi: 10.1203/01.pdr.0000199905.08848.55
141. Fleiss B, Tann CJ, Degos V, Sigaut S, Van Steenwinckel J, Schang AL, et al. Inflammation-induced sensitization of the brain in term infants. Dev Med Child Neurol. (2015) 57:17–28. doi: 10.1111/dmcn.12723
142. Canetta S, Bolkan S, Padilla-Coreano N, Song LJ, Sahn R, Harrison NL, et al. Maternal immune activation leads to selective functional deficits in offspring parvalbumin interneurons. Mol Psychiatry. (2016) 21:956–68. doi: 10.1038/mp.2015.222
143. Vasistha NA, Pardo-Navarro M, Gasthaus J, Weijers D, Muller MK, Garcia-Gonzalez D, et al. Maternal inflammation has a profound effect on cortical interneuron development in a stage and subtype-specific manner. Mol Psychiatry. (2019). doi: 10.1038/s41380-019-0539-5. [Epub ahead of print].
144. Zikopoulos B, Barbas H. Altered neural connectivity in excitatory and inhibitory cortical circuits in autism. Front Hum Neurosci. (2013) 7:609. doi: 10.3389/fnhum.2013.00609
145. Adorjan I, Ahmed B, Feher V, Torso M, Krug K, Esiri M, et al. Calretinin interneuron density in the caudate nucleus is lower in autism spectrum disorder. Brain. (2017) 140:2028–40. doi: 10.1093/brain/awx131
146. Lunden JW, Durens M, Phillips AW, Nestor MW. Cortical interneuron function in autism spectrum condition. Pediatr Res. (2019) 85:146–54. doi: 10.1038/s41390-018-0214-6
147. Yekhlef L, Breschi GL, Lagostena L, Russo G, Taverna S. Selective activation of parvalbumin- or somatostatin-expressing interneurons triggers epileptic seizurelike activity in mouse medial entorhinal cortex. J Neurophysiol. (2015) 113:1616–30. doi: 10.1152/jn.00841.2014
148. Lauber E, Filice F, Schwaller B. Dysregulation of parvalbumin expression in the Cntnap2−/− mouse model of autism spectrum disorder. Front Mol Neurosci. (2018) 11:262. doi: 10.3389/fnmol.2018.00262
149. Wiebe S, Nagpal A, Truong VT, Park J, Skalecka A, He AJ, et al. Inhibitory interneurons mediate autism-associated behaviors via 4E-BP2. Proc Natl Acad Sci USA. (2019) 116:18060–7. doi: 10.1073/pnas.1908126116
150. Ball G, Srinivasan L, Aljabar P, Counsell SJ, Durighel G, Hajnal JV, et al. Development of cortical microstructure in the preterm human brain. Proc Natl Acad Sci USA. (2013) 110:9541–6. doi: 10.1073/pnas.1301652110
151. Ball G, Boardman JP, Aljabar P, Pandit A, Arichi T, Merchant N, et al. The influence of preterm birth on the developing thalamocortical connectome. Cortex. (2013) 49:1711–21. doi: 10.1016/j.cortex.2012.07.006
152. Pandit AS, Robinson E, Aljabar P, Ball G, Gousias IS, Wang Z. Whole-brain mapping of structural connectivity in infants reveals altered connection strength associated with growth and preterm birth. Cereb Cortex. (2014) 24:2324–33. doi: 10.1093/cercor/bht086
153. Limperopoulos C, Bassan H, Sullivan NR, Soul JS, Robertson RL Jr, Moore M, et al. Positive screening for autism in ex-preterm infants: prevalence and risk factors. Pediatrics. (2008) 121:758–65. doi: 10.1542/peds.2007-2158
154. Delobel-Ayoub M, Arnaud C, White-Koning M, Casper C, Pierrat V, Garel M, et al. Behavioral problems and cognitive performance at 5 years of age after very preterm birth: the EPIPAGE study. Pediatrics. (2009) 123:1485–92. doi: 10.1542/peds.2008-1216
155. Lindstrom K, Lindblad F, Hjern A. Preterm birth and attention-deficit/hyperactivity disorder in schoolchildren. Pediatrics. (2011) 127:858–65. doi: 10.1542/peds.2010-1279
156. Hagberg B, Hagberg G, Olow I, von Wendt L. The changing panorama of cerebral palsy in Sweden. VII. Prevalence and origin in the birth year period 1987–90. Acta Paediatr. (1996) 85:954–60. doi: 10.1111/j.1651-2227.1996.tb14193.x
157. Crump C, Sundquist K, Winkleby MA, Sundquist J. Preterm birth and risk of epilepsy in Swedish adults. Neurology. (2011) 77:1376–82. doi: 10.1212/WNL.0b013e318231528f
158. Wood NS, Costeloe K, Gibson AT, Hennessy EM, Marlow N, Wilkinson AR, et al. The EPICure study: associations and antecedents of neurological and developmental disability at 30 months of age following extremely preterm birth. Arch Dis Child Fetal Neonatal Ed. (2005) 90:F134–140. doi: 10.1136/adc.2004.052407
159. Costeloe KL, Hennessy EM, Haider S, Stacey F, Marlow N, Draper ES. Short term outcomes after extreme preterm birth in England: comparison of two birth cohorts in 1995 and 2006 (the EPICure studies). BMJ. (2012) 345:e7976. doi: 10.1136/bmj.e7976
160. Petrenko V, van de Looij Y, Mihhailova J, Salmon P, Huppi PS, Sizonenko SV, et al. Multimodal MRI imaging of apoptosis-triggered microstructural alterations in the postnatal cerebral cortex. Cereb Cortex. (2018) 28:949–62. doi: 10.1093/cercor/bhw420
161. Petrenko V, Mihhailova J, Salmon P, Kiss JZ. Apoptotic neurons induce proliferative responses of progenitor cells in the postnatal neocortex. Exp Neurol. (2015) 273:126–37. doi: 10.1016/j.expneurol.2015.08.010
162. Kalanjati VP, Wixey JA, Miller SM, Colditz PB, Bjorkman ST. GABAA receptor expression and white matter disruption in intrauterine growth restricted piglets. Int J Dev Neurosci. (2017) 59:1–9. doi: 10.1016/j.ijdevneu.2017.02.004
163. Fleiss B, Wong F, Brownfoot F, Shearer IK, Baud O, Walker DW, et al. Knowledge gaps and emerging research areas in intrauterine growth restriction-associated brain injury. Front Endocrinol (Lausanne). (2019) 10:188. doi: 10.3389/fendo.2019.00188
164. Li WY, Chang YC, Lee LJH, Lee LJ. Prenatal infection affects the neuronal architecture and cognitive function in adult mice. Dev Neurosci. (2014) 36:359–70. doi: 10.1159/000362383
165. Balakrishnan B, Dai H, Janisse J, Romero R, Kannan S. Maternal endotoxin exposure results in abnormal neuronal architecture in the newborn rabbit. Dev Neurosci. (2013) 35:396–405. doi: 10.1159/000353156
166. Krishnan ML, Van Steenwinckel J, Schang AL, Yan J, Arnadottir J, Le Charpentier T, et al. Integrative genomics of microglia implicates DLG4 (PSD95) in the white matter development of preterm infants. Nat Commun. (2017) 8:428. doi: 10.1038/s41467-017-00422-w
167. Vinall J, Grunau RE, Brant R, Chau V, Poskitt KJ, Synnes AR, et al. Slower postnatal growth is associated with delayed cerebral cortical maturation in preterm newborns. Sci Transl Med. (2013) 5:168ra168. doi: 10.1126/scitranslmed.3004666
168. Pannek K, Hatzigeorgiou X, Colditz PB, Rose S. Assessment of structural connectivity in the preterm brain at term equivalent age using diffusion MRI and T2 relaxometry: a network-based analysis. PLoS ONE. (2013) 8:e68593. doi: 10.1371/journal.pone.0068593
169. Batalle D, Hughes EJ, Zhang H, Tournier JD, Tusor N, Aljabar P, et al. Early development of structural networks and the impact of prematurity on brain connectivity. Neuroimage. (2017) 149:379–92. doi: 10.1016/j.neuroimage.2017.01.065
170. Karolis VR, Froudist-Walsh S, Brittain PJ, Kroll J, Ball G, Edwards AD, et al. Reinforcement of the brain's rich-club architecture following early neurodevelopmental disruption caused by very preterm birth. Cereb Cortex. (2016) 26:1322–35. doi: 10.1093/cercor/bhv305
171. Arichi T, Counsell SJ, Allievi AG, Chew AT, Martinez-Biarge M, Mondi V, et al. The effects of hemorrhagic parenchymal infarction on the establishment of sensori-motor structural and functional connectivity in early infancy. Neuroradiology. (2014) 56:985–94. doi: 10.1007/s00234-014-1412-5
172. Ball G, Aljabar P, Arichi T, Tusor N, Cox D, Merchant N, et al. Machine-learning to characterise neonatal functional connectivity in the preterm brain. Neuroimage. (2016) 124:267–75. doi: 10.1016/j.neuroimage.2015.08.055
173. Gozdas E, Parikh NA, Merhar SL, Tkach JA, He L, Holland SK. Altered functional network connectivity in preterm infants: antecedents of cognitive and motor impairments? Brain Struct Funct. (2018) 223:3665–80. doi: 10.1007/s00429-018-1707-0
174. Bouyssi-Kobar M, De Asis-Cruz J, Murnick J, Chang T, Limperopoulos C. Altered functional brain network integration, segregation, and modularity in infants born very preterm at term-equivalent age. J Pediatr. (2019) 213:13–21 e11. doi: 10.1016/j.jpeds.2019.06.030
175. Ranasinghe S, Or G, Wang EY, Ievins A, McLean MA, Niell CM. Reduced cortical activity impairs development and plasticity after neonatal hypoxia ischemia. J Neurosci. (2015) 35:11946–59. doi: 10.1523/JNEUROSCI.2682-14.2015
176. Song J, Xu F, Wang L, Gao L, Guo J, Xia L, et al. Early amplitude-integrated electroencephalography predicts brain injury and neurological outcome in very preterm infants. Sci Rep. (2015) 5:13810. doi: 10.1038/srep13810
177. Pavlidis E, Lloyd RO, Boylan GB. EEG–a valuable biomarker of brain injury in preterm infants. Dev Neurosci. (2017) 39:23–35. doi: 10.1159/000456659
178. Whitehead K, Pressler R, Fabrizi L. Characteristics and clinical significance of delta brushes in the EEG of premature infants. Clin Neurophysiol Pract. (2017) 2:12–8. doi: 10.1016/j.cnp.2016.11.002
179. Tataranno ML, Claessens NHP, Moeskops P, Toet MC, Kersbergen KJ, Buonocore G, et al. Changes in brain morphology and microstructure in relation to early brain activity in extremely preterm infants. Pediatr Res. (2018) 83:834–42. doi: 10.1038/pr.2017.314
180. Benders MJ, Palmu K, Menache C, Borradori-Tolsa C, Lazeyras F, Sizonenko S, et al. Early brain activity relates to subsequent brain growth in premature infants. Cereb Cortex. (2015) 25:3014–24. doi: 10.1093/cercor/bhu097
181. Whitehead K, Jones L, Laudiano-Dray MP, Meek J, Fabrizi L. Altered cortical processing of somatosensory input in pre-term infants who had high-grade germinal matrix-intraventricular haemorrhage. Neuroimage Clin. (2020) 25:102095. doi: 10.1016/j.nicl.2019.102095
182. Hayashi-Kurahashi N, Kidokoro H, Kubota T, Maruyama K, Kato Y, Kato T, et al. EEG for predicting early neurodevelopment in preterm infants: an observational cohort study. Pediatrics. (2012) 130:e891–897. doi: 10.1542/peds.2012-1115
183. Reynolds LC, Pineda RG, Mathur A, Vavasseur C, Shah DK, Liao S, et al. Cerebral maturation on amplitude-integrated electroencephalography and perinatal exposures in preterm infants. Acta Paediatr. (2014) 103:e96–100. doi: 10.1111/apa.12485
184. Abbasi H, Drury PP, Lear CA, Gunn AJ, Davidson JO, Bennet L, et al. EEG sharp waves are a biomarker of striatal neuronal survival after hypoxia-ischemia in preterm fetal sheep. Sci Rep. (2018) 8:16312. doi: 10.1038/s41598-018-34654-7
185. van den Heuij LG, Fraser M, Miller SL, Jenkin G, Wallace EM, Davidson JO, et al. Delayed intranasal infusion of human amnion epithelial cells improves white matter maturation after asphyxia in preterm fetal sheep. J Cereb Blood Flow Metab. (2019) 39:223–9. doi: 10.1177/0271678X17729954
186. Keogh MJ, Bennet L, Drury PP, Booth LC, Mathai S, Naylor AS, et al. Subclinical exposure to low-dose endotoxin impairs EEG maturation in preterm fetal sheep. Am J Physiol Regul Integr Comp Physiol. (2012) 303:R270–8. doi: 10.1152/ajpregu.00216.2012
187. Plomgaard AM, Andersen AD, Petersen TH, van de Looij Y, Thymann T, Sangild PT, et al. Structural brain maturation differs between preterm and term piglets, whereas brain activity does not. Acta Paediatr. (2019) 108:637–44. doi: 10.1111/apa.14556
188. van de Looij Y, Chatagner A, Quairiaux C, Gruetter R, Hüppi PS, Sizonenko SV. Multi-modal assessment of long-term erythropoietin treatment after neonatal hypoxic-ischemic injury in rat brain. PLoS ONE. (2014) 9:e95643. doi: 10.1371/journal.pone.0095643
189. Galinsky R, Draghi V, Wassink G, Davidson JO, Drury PP, Lear CA, et al. Magnesium sulfate reduces EEG activity but is not neuroprotective after asphyxia in preterm fetal sheep. J Cereb Blood Flow Metab. (2017) 37:1362–73. doi: 10.1177/0271678X16655548
190. Mordel J, Sheikh A, Tsohataridis S, Kanold PO, Zehendner CM, Luhmann HJ. Mild systemic inflammation and moderate hypoxia transiently alter neuronal excitability in mouse somatosensory cortex. Neurobiol Dis. (2016) 88:29–43. doi: 10.1016/j.nbd.2015.12.019
191. Nimmervoll B, White R, Yang JW, An S, Henn C, Sun JJ, et al. LPS-induced microglial secretion of TNFalpha increases activity-dependent neuronal apoptosis in the neonatal cerebral cortex. Cereb Cortex. (2013) 23:1742–55. doi: 10.1093/cercor/bhs156
192. Zaslavsky K, Zhang WB, McCready FP, Rodrigues DC, Deneault E, Loo C, et al. (2019). SHANK2 mutations associated with autism spectrum disorder cause hyperconnectivity of human neurons. Nat Neurosci. 22:556–64. doi: 10.1038/s41593-019-0365-8
193. Marguet SL, Le-Schulte VT, Merseburg A, Neu A, Eichler R, Jakovcevski I, et al. Treatment during a vulnerable developmental period rescues a genetic epilepsy. Nat Med. (2015) 21:1436–44. doi: 10.1038/nm.3987
194. Mattingly GW, Wilson J, Rostain AL. A clinician's guide to ADHD treatment options. Postgrad Med. (2017) 129:657–66. doi: 10.1080/00325481.2017.1354648
195. Goldenberg MM. Overview of drugs used for epilepsy and seizures: etiology, diagnosis, and treatment. P T. (2010) 35:392–415.
196. De Crescenzo F, Cortese S, Adamo N, Janiri L. Pharmacological and non-pharmacological treatment of adults with ADHD: a meta-review. Evid Based Mental Health. (2017) 20:4. doi: 10.1136/eb-2016-102415
197. Heylen SL, Gelders YG. Risperidone, a new antipsychotic with serotonin 5-HT2 and dopamine D2 antagonistic properties. Clin Neuropharmacol. (1992) 15:180A−1A. doi: 10.1097/00002826-199201001-00095
198. Stavropoulos KK-M. Using neuroscience as an outcome measure for behavioral interventions in autism spectrum disorders (ASD): a review. Res Autism Spectr Disord. (2017) 35:62–73. doi: 10.1016/j.rasd.2017.01.001
199. Hong MP, Erickson CA. Investigational drugs in early-stage clinical trials for autism spectrum disorder. Expert Opin Invest Drugs. (2019) 28:709–18. doi: 10.1080/13543784.2019.1649656
200. Fleiss B, Gressens P. Tertiary mechanisms of brain damage: a new hope for treatment of cerebral palsy? Lancet Neurol. (2012) 11:556–66. doi: 10.1016/S1474-4422(12)70058-3
201. Han M, Zhang JC, Yao W, Yang C, Ishima T, Ren Q, et al. Intake of 7,8-dihydroxyflavone during juvenile and adolescent stages prevents onset of psychosis in adult offspring after maternal immune activation. Sci Rep. (2016) 6:36087. doi: 10.1038/srep36087
202. Iannone LF, Gomez-Eguilaz M, Citaro R, Russo E. The potential role of interventions impacting on gut-microbiota in epilepsy. Expert Rev Clin Pharmacol. (2020) 1:423–35. doi: 10.1080/17512433.2020.1759414
203. Hill-Yardin EL, McKeown SJ, Novarino G, Grabrucker AM. Extracerebral dysfunction in animal models of autism spectrum disorder. Adv Anat Embryol Cell Biol. (2017) 224:159–87. doi: 10.1007/978-3-319-52498-6_9
204. Hosie S, Ellis M, Swaminathan M, Ramalhosa F, Seger GO, Balasuriya GK, et al. Gastrointestinal dysfunction in patients and mice expressing the autism-associated R451C mutation in neuroligin-3. Autism Res. (2019) 12:1043–56. doi: 10.1002/aur.2127
205. Lasheras I, Seral P, Latorre E, Barroso E, Gracia-Garcia P, Santabarbara J. Microbiota and gut-brain axis dysfunction in autism spectrum disorder: evidence for functional gastrointestinal disorders. Asian J Psychiatr. (2020) 47:101874. doi: 10.1016/j.ajp.2019.101874
206. Sauer AK, Bockmann J, Steinestel K, Boeckers TM, Grabrucker AM. Altered intestinal morphology and microbiota composition in the autism spectrum disorders associated SHANK3 mouse model. Int J Mol Sci. (2019) 20:2134. doi: 10.3390/ijms20092134
207. Burrows EL, Laskaris L, Koyama L, Churilov L, Bornstein JC, Hill-Yardin EL, et al. A neuroligin-3 mutation implicated in autism causes abnormal aggression and increases repetitive behavior in mice. Mol Autism. (2015) 6:62. doi: 10.1186/s13229-015-0055-7
208. Hosie S, Malone DT, Liu S, Glass M, Adlard PA, Hannan AJ, et al. Altered amygdala excitation and CB1 receptor modulation of aggressive behavior in the neuroligin-3R451C mouse model of autism. Front Cell Neurosci. (2018) 12:234. doi: 10.3389/fncel.2018.00234
209. Matta SM, Hill-Yardin EL, Crack PJ. The influence of neuroinflammation in autism spectrum disorder. Brain Behav Immun. (2019) 79:75–90. doi: 10.1016/j.bbi.2019.04.037
210. Lee CYQ, Franks AE, Hill-Yardin EL. Autism-associated synaptic mutations impact the gut-brain axis in mice. Brain Behav Immun. (2020). doi: 10.1016/j.bbi.2020.05.072. [Epub ahead of print].
211. Hsiao EY, McBride SW, Hsien S, Sharon G, Hyde ER, McCue T, et al. Microbiota modulate behavioral and physiological abnormalities associated with neurodevelopmental disorders. Cell (2013) 155:1451–63. doi: 10.1016/j.cell.2013.11.024
212. Sharon G, Cruz NJ, Kang DW, Gandal MJ, Wang B, Kim YM, et al. Human gut microbiota from autism spectrum disorder promote behavioral symptoms in mice. Cell. (2019) 177:1600–18.e17. doi: 10.1016/j.cell.2019.05.004
213. Sgritta M, Dooling SW, Buffington SA, Momin EN, Francis MB, Britton RA, et al. Mechanisms underlying microbial-mediated changes in social behavior in mouse models of autism spectrum disorder. Neuron. (2019) 101:246–59.e6. doi: 10.1016/j.neuron.2018.11.018
214. Erny D, Hrabe de Angelis AL, Jaitin D, Wieghofer P, Staszewski O, David E, et al. Host microbiota constantly control maturation and function of microglia in the CNS. Nat Neurosci. (2015) 18:965–77. doi: 10.1038/nn.4030
215. Matcovitch-Natan O, Winter DR, Giladi A, Vargas Aguilar S, Spinrad A, Sarrazin S, et al. Microglia development follows a stepwise program to regulate brain homeostasis. Science. (2016) 353:aad8670. doi: 10.1126/science.aad8670
216. Hoyles L, Snelling T, Umlai U-K, Nicholson JK, Carding SR, Glen RC, et al. Microbiome-host systems interactions: protective effects of propionate upon the blood-brain barrier. Microbiome (2018) 6:55. doi: 10.1186/s40168-018-0439-y
217. Swiderski K, Bindon R, Trieu J, Naim T, Schokman S, Swaminathan M, et al. Spatiotemporal mapping reveals regional gastrointestinal dysfunction in MDX dystrophic mice ameliorated by oral L-arginine supplementation. J Neurogastroenterol Motil. (2019).
218. Nithianantharajah J, Balasuriya GK, Franks AE, Hill-Yardin EL. Using animal models to study the role of the gut–brain axis in autism. Curr Dev Disord Rep. (2017) 4:28–36. doi: 10.1007/s40474-017-0111-4
219. Koning G, Lyngfelt E, Svedin P, Leverin AL, Jinnai M, Gressens P, et al. Magnesium sulphate induces preconditioning in preterm rodent models of cerebral hypoxia-ischemia. Int J Dev Neurosci. (2018) 70:56–66. doi: 10.1016/j.ijdevneu.2018.01.002
220. Doyle LW, Crowther CA, Middleton P, Marret S. Magnesium sulphate for women at risk of preterm birth for neuroprotection of the fetus. Cochrane Database Syst Rev. (2007) 68:CD004661. doi: 10.1002/14651858.CD004661.pub2
221. Leuchter RH, Gui L, Poncet A, Hagmann C, Lodygensky GA, Martin E, et al. Association between early administration of high-dose erythropoietin in preterm infants and brain MRI abnormality at term-equivalent age. JAMA. (2014) 312:817–24. doi: 10.1001/jama.2014.9645
222. Robinson S, Corbett CJ, Winer JL, Chan LAS, Maxwell JR, Anstine CV, et al. Neonatal erythropoietin mitigates impaired gait, social interaction and diffusion tensor imaging abnormalities in a rat model of prenatal brain injury. Exp Neurol. (2018) 302:1–13. doi: 10.1016/j.expneurol.2017.12.010
223. Wassink G, Davidson JO, Dhillon SK, Fraser M, Galinsky R, Bennet L, et al. Partial white and grey matter protection with prolonged infusion of recombinant human erythropoietin after asphyxia in preterm fetal sheep. J Cereb Blood Flow Metab. (2017) 37:1080–94. doi: 10.1177/0271678X16650455
224. Fleiss B, Gressens P. Neuroprotection of the preterm brain. Handb Clin Neurol. (2019) 162:315–28. doi: 10.1016/B978-0-444-64029-1.00015-1
225. Sucksdorff M, Lehtonen L, Chudal R, Suominen A, Joelsson P, Gissler M, et al. Preterm birth and poor fetal growth as risk factors of attention-deficit/hyperactivity disorder. Pediatrics. (2015) 136:e599–608. doi: 10.1542/peds.2015-1043
226. Indredavik MS, Skranes JS, Vik T, Heyerdahl S, Romundstad P, Myhr GE, et al. Low-birth-weight adolescents: psychiatric symptoms and cerebral MRI abnormalities. Pediatr Neurol. (2005) 33:259–66. doi: 10.1016/j.pediatrneurol.2005.05.002
227. Bokobza C, Van Steenwinckel J, Mani S, Mezger V, Fleiss B, Gressens P. Neuroinflammation in preterm babies and autism spectrum disorders. Pediatr Res. (2019) 85:155–65. doi: 10.1038/s41390-018-0208-4
228. Fitzallen GC, Taylor HG, Bora S. What do we know about the preterm behavioural phenotype? A narrative review. Front Psychiatry. (2020) 11:154. doi: 10.3389/fpsyt.2020.00154
229. Elgen I, Sommerfelt K, Markestad T. Population based, controlled study of behavioural problems and psychiatric disorders in low birthweight children at 11 years of age. Arch Dis Child Fetal Neonatal Ed. (2002) 87:F128–32. doi: 10.1136/fn.87.2.F128
230. Bowers K, Wink LK, Pottenger A, McDougle CJ, Erickson C. Phenotypic differences in individuals with autism spectrum disorder born preterm and at term gestation. Autism. (2015) 19:758–63. doi: 10.1177/1362361314547366
231. Chen LW, Wang ST, Wang LW, Kao YC, Chu CL, Wu CC, et al. Behavioral characteristics of autism spectrum disorder in very preterm birth children. Mol Autism. (2019) 10:32. doi: 10.1186/s13229-019-0282-4
232. Batalle D, Edwards AD, O'Muircheartaigh J. Annual research review: not just a small adult brain: understanding later neurodevelopment through imaging the neonatal brain. J Child Psychol Psychiatry. (2017) 59:350–71. doi: 10.1111/jcpp.12838
233. McAlonan GM, Cheung V, Cheung C, Suckling J, Lam GY, Tai KS, et al. Mapping the brain in autism. A voxel-based MRI study of volumetric differences and intercorrelations in autism. Brain. (2005) 128:268–76. doi: 10.1093/brain/awh332
234. Batty MJ, Liddle EB, Pitiot A, Toro R, Groom MJ, Scerif G, et al. Cortical gray matter in attention-deficit/hyperactivity disorder: a structural magnetic resonance imaging study. J Am Acad Child Adolesc Psychiatry. (2010) 49:229–38. doi: 10.1097/00004583-201003000-00006
235. Greimel E, Nehrkorn B, Schulte-Ruther M, Fink GR, Nickl-Jockschat T, Herpertz-Dahlmann B, et al. Changes in grey matter development in autism spectrum disorder. Brain Struct Funct. (2013) 218:929–42. doi: 10.1007/s00429-012-0439-9
236. Griffiths KR, Grieve SM, Kohn MR, Clarke S, Williams LM, Korgaonkar MS. Altered gray matter organization in children and adolescents with ADHD: a structural covariance connectome study. Transl Psychiatry. (2016) 6:e947. doi: 10.1038/tp.2016.219
237. Sato W, Kochiyama T, Uono S, Yoshimura S, Kubota Y, Sawada R, et al. Reduced gray matter volume in the social brain network in adults with autism spectrum disorder. Front Hum Neurosci. (2017) 11:395. doi: 10.3389/fnhum.2017.00395
238. Nakao T, Radua J, Rubia K, Mataix-Cols D. Gray matter volume abnormalities in ADHD: voxel-based meta-analysis exploring the effects of age and stimulant medication. Am J Psychiatry. (2011) 168:1154–63. doi: 10.1176/appi.ajp.2011.11020281
239. Lawrence EJ, Froudist-Walsh S, Neilan R, Nam KW, Giampietro V, McGuire P, et al. Motor fMRI and cortical grey matter volume in adults born very preterm. Dev Cogn Neurosci. (2014) 10:1–9. doi: 10.1016/j.dcn.2014.06.002
240. Keunen K, Isgum I, van Kooij BJ, Anbeek P, van Haastert IC, Koopman-Esseboom C, et al. Brain volumes at term-equivalent age in preterm infants: imaging biomarkers for neurodevelopmental outcome through early school age. J Pediatr. (2016) 172:88–95. doi: 10.1016/j.jpeds.2015.12.023
241. Ecker C, Ronan L, Feng Y, Daly E, Murphy C, Ginestet CE, et al. Intrinsic gray-matter connectivity of the brain in adults with autism spectrum disorder. Proc Natl Acad Sci USA. (2013) 110:13222–7. doi: 10.1073/pnas.1221880110
242. Coley AA, Gao WJ. PSD95: a synaptic protein implicated in schizophrenia or autism? Prog Neuropsychopharmacol Biol Psychiatry. (2018) 82:187–94. doi: 10.1016/j.pnpbp.2017.11.016
243. Sajdel-Sulkowska EM, Makowska-Zubrycka M, Czarzasta K, Kasarello K, Aggarwal V, Bialy M, et al. Common genetic variants link the abnormalities in the gut-brain axis in prematurity and autism. Cerebellum. (2019) 18:255–65. doi: 10.1007/s12311-018-0970-1
244. Guang S, Pang N, Deng X, Yang L, He F, Wu L, et al. Synaptopathology involved in autism spectrum disorder. Front Cell Neurosci. (2018) 12:470. doi: 10.3389/fncel.2018.00470
245. Rylaarsdam L, Guemez-Gamboa A. Genetic causes and modifiers of autism spectrum disorder. Front Cell Neurosci. (2019) 13:385. doi: 10.3389/fncel.2019.00385
246. Hoffmann I, Bueter W, Zscheppang K, Brinkhaus MJ, Liese A, Riemke S, et al. Neuregulin-1, the fetal endothelium, and brain damage in preterm newborns. Brain Behav Immun. (2010) 24:784–91. doi: 10.1016/j.bbi.2009.08.012
247. Southwell DG, Seifikar H, Malik R, Lavi K, Vogt D, Rubenstein JL, et al. Interneuron transplantation rescues social behavior deficits without restoring wild-type physiology in a mouse model of autism with excessive synaptic inhibition. J Neurosci. (2020) 40:2215–27. doi: 10.1523/JNEUROSCI.1063-19.2019
248. Pizzarelli R, Cherubini E. Alterations of GABAergic signaling in autism spectrum disorders. Neural Plast. (2011) 2011:297153. doi: 10.1155/2011/297153
249. Bilbo SD, Smith SH, Schwarz JM. A lifespan approach to neuroinflammatory and cognitive disorders: a critical role for glia. J Neuroimmune Pharmacol. (2012) 7:24–41. doi: 10.1007/s11481-011-9299-y
250. Petrelli F, Pucci L, Bezzi P. Astrocytes and microglia and their potential link with autism spectrum disorders. Front Cell Neurosci. (2016) 10:21. doi: 10.3389/fncel.2016.00021
251. Vezzani A, Balosso S, Ravizza T. Neuroinflammatory pathways as treatment targets and biomarkers in epilepsy. Nat Rev Neurol. (2019) 15:459–72. doi: 10.1038/s41582-019-0217-x
252. Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, Panzanelli P, et al. Synaptic pruning by microglia is necessary for normal brain development. Science. (2011) 333:1456–8. doi: 10.1126/science.1202529
253. Zhan Y, Paolicelli RC, Sforazzini F, Weinhard L, Bolasco G, Pagani F, et al. Deficient neuron-microglia signaling results in impaired functional brain connectivity and social behavior. Nat Neurosci. (2014) 17:400–6. doi: 10.1038/nn.3641
254. Mallya AP, Wang HD, Lee HNR, Deutch AY. Microglial pruning of synapses in the prefrontal cortex during adolescence. Cereb Cortex. (2019) 29:1634–43. doi: 10.1093/cercor/bhy061
255. Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, Yamasaki R, et al. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron. (2012) 74:691–705. doi: 10.1016/j.neuron.2012.03.026
256. Bilimoria PM, Stevens B. Microglia function during brain development: new insights from animal models. Brain Res. (2015) 1617:7–17. doi: 10.1016/j.brainres.2014.11.032
257. Tay TL, Savage JC, Hui CW, Bisht K, Tremblay ME. Microglia across the lifespan: from origin to function in brain development, plasticity and cognition. J Physiol. (2017) 595:1929–45. doi: 10.1113/JP272134
258. Won H, Mah W, Kim E. Autism spectrum disorder causes, mechanisms, and treatments: focus on neuronal synapses. Front Mol Neurosci. (2013) 6:19. doi: 10.3389/fnmol.2013.00019
259. Ebrahimi-Fakhari D, Sahin M. Autism and the synapse: emerging mechanisms and mechanism-based therapies. Curr Opin Neurol. (2015) 28:91–102. doi: 10.1097/WCO.0000000000000186
260. Lepeta K, Lourenco MV, Schweitzer BC, Martino Adami PV, Banerjee P, Catuara-Solarz S, et al. Synaptopathies: synaptic dysfunction in neurological disorders–a review from students to students. J Neurochem. (2016) 138:785–805. doi: 10.1111/jnc.13713
261. Luo J, Norris RH, Gordon SL, Nithianantharajah J. Neurodevelopmental synaptopathies: insights from behaviour in rodent models of synapse gene mutations. Prog Neuropsychopharmacol Biol Psychiatry. (2018) 84:424–39. doi: 10.1016/j.pnpbp.2017.12.001
262. Delorme R, Ey E, Toro R, Leboyer M, Gillberg C, Bourgeron T. Progress toward treatments for synaptic defects in autism. Nat Med. (2013) 19:685–94. doi: 10.1038/nm.3193
Keywords: preterm, brain injury, development, inflammation, synaptopathy, MRI, functional activity, neuropathology
Citation: Fleiss B, Gressens P and Stolp HB (2020) Cortical Gray Matter Injury in Encephalopathy of Prematurity: Link to Neurodevelopmental Disorders. Front. Neurol. 11:575. doi: 10.3389/fneur.2020.00575
Received: 07 April 2020; Accepted: 19 May 2020;
Published: 14 July 2020.
Edited by:
Silvia Noemi Tenembaum, Garrahan Hospital, ArgentinaReviewed by:
Carlotta Spagnoli, Santa Maria Nuova Hospital, ItalyThalia Harmony, National Autonomous University of Mexico, Mexico
Copyright © 2020 Fleiss, Gressens and Stolp. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Helen B. Stolp, aHN0b2xwQHJ2Yy5hYy51aw==