 David S. Liebeskind1*
David S. Liebeskind1* Jason D. Hinman1
Jason D. Hinman1 Naoki Kaneko1Hiroaki Kitajima1Tristan Honda1
Naoki Kaneko1Hiroaki Kitajima1Tristan Honda1 Adam H. De Havenon2Edward Feldmann3
Adam H. De Havenon2Edward Feldmann3 Raul G. Nogueira4
Raul G. Nogueira4 Shyam Prabhakaran5
Shyam Prabhakaran5 Jose G. Romano6Peter W. Callas7David J. Schneider8
Jose G. Romano6Peter W. Callas7David J. Schneider8- 1Department of Neurology, Neurovascular Imaging Research Core and UCLA Stroke Center, University of California, Los Angeles, Los Angeles, CA, United States
- 2Department of Neurology, University of Utah, Salt Lake City, UT, United States
- 3Department of Neurology, The University of Massachusetts Medical School-Baystate, Springfield, MA, United States
- 4Department of Neurology, Marcus Stroke & Neuroscience Center, Emory University School of Medicine, Atlanta, GA, United States
- 5Department of Neurology, The University of Chicago, Chicago, IL, United States
- 6Department of Neurology, University of Miami, Miami, FL, United States
- 7Department of Biostatistics, University of Vermont, Burlington, VT, United States
- 8Department of Medicine, Cardiovascular Research Institute, University of Vermont, Burlington, VT, United States
Intracranial atherosclerotic disease (ICAD) has been characterized by the degree of arterial stenosis and downstream hypoperfusion, yet microscopic derangements of endothelial shear stress at the luminal wall may be key determinants of plaque growth, vascular remodeling and thrombosis that culminate in recurrent stroke. Platelet interactions have similarly been a principal focus of treatment, however, the mechanistic basis of anti-platelet strategies is largely extrapolated rather than directly investigated in ICAD. Platelet FcγRIIa expression has been identified as a potent risk factor in cardiovascular disease, as elevated expression markedly increases the risk of recurrent events. Differential activation of the platelet FcγRIIa receptor may also explain the variable response of individual patients to anti-platelet medications. We review existing data on endothelial shear stress and potential interactions with the platelet FcγRIIa receptor that may alter the evolving impact of ICAD, based on local pathophysiology at the site of arterial stenosis. Current methods for quantification of endothelial shear stress and platelet activation are described, including tools that may be readily adapted to the clinical realm for further understanding of ICAD.
Introduction
Intracranial atherosclerotic disease (ICAD) is the most common cause of stroke worldwide (1, 2). The devastating consequences of ICAD reflect racial, sex and ethnic disparities, impact a broad age group and lack strategies for prevention (3). Overwhelming recurrent risk amounts to an excessive burden of disease and public health priority (4). ICAD engenders a ~12.5% rate of recurrent clinical strokes within 1 year (5, 6). The impact of “silent” strokes, evident only on surveillance imaging, may be even greater when one considers cognitive or other impairment.
Recurrent ischemic stroke due to ICAD is extremely common despite treatment with anti-platelet medications. Heterogeneity of the arterial architecture and associated blood flow changes in ICAD-related stenoses result in different patterns of wall shear stress (WSS) from one individual to the next. Such wall shear stress can be readily quantified with computational fluid dynamics (CFD) from non-invasive CT angiography (CTA), routinely acquired in patients with minor stroke or transient ischemic attack (TIA) due to ICAD. These shear stress changes in blood flow promote platelet aggregation and thereby alter the response to anti-platelet therapy. Additionally, greater platelet FcγRIIa expression increases platelet reactivity and promotes thrombosis when platelets are exposed to increased shear stress. In coronary artery disease (CAD), greater platelet expression of FcγRIIa identifies patients at greater risk of recurrent cardiovascular events, including stroke. Numerous mechanisms have been invoked in the recurrence of ischemia in ICAD, yet focused research on the pathophysiology of shear stress and platelet activation has not been evaluated to explain the high rate of imaging evidence and clinical strokes following minor stroke or TIA due to ICAD. Given the shared pathology of coronary artery disease and ICAD, the data suggest that individual differences in CFD-derived WSS and platelet FcγRIIa expression may inform a precision medicine strategy to prevent recurrent stroke.
Shear-Induced Platelet Aggregation in ICAD
More than 25 years ago, stroke research underscored the pathophysiology of shear-induced platelet aggregation (7–9). In vitro studies showed a protective effect of thienopyridines (e.g., clopidogrel), creating parallel approaches to ICAD and CAD, based on anti-platelet effects. These studies revealed that aspirin has limited effect on platelet aggregation, modified largely by local hemodynamics, forming the rationale for dual anti-platelet therapy (DAPT) in ICAD and CAD. Distinct zones in the region of arterial narrowing or stenosis and immediately downstream in the post-stenotic segment influence platelet activation, modulated by shear stress. As in Figure 1, wall shear stress (WSS, calculated as ts) increases as blood flows tangentially to the arterial wall of the narrowed lumen or stenosis, measured by the residual radius. As blood flow volume asymmetrically exits the stenosis, flow vortices create oscillating gradients in both direction and intensity of WSS. High shear stress and the oscillatory shear index (OSI) can be measured with CTA techniques and are closely linked to platelet activity (10–13).
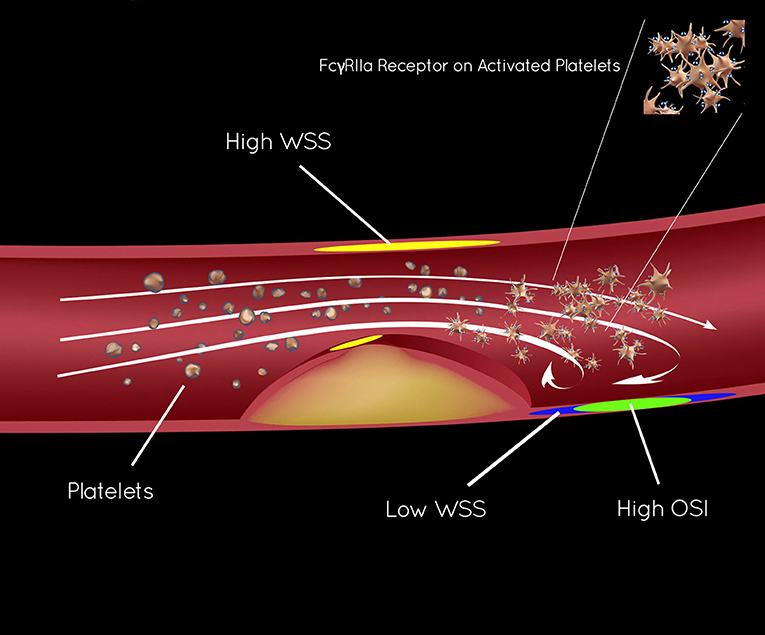
Figure 1. Endothelial shear stress in ICAD and activation of platelet FcγRIIa.
Platelet Reactivity and Platelet Expression of FCγRIIA
Increased platelet reactivity has identified patients with minor stroke or TIA who are at greater risk of recurrent stroke (14). Similarly, increased platelet reactivity has consistently identified patients with CAD who are at greater risk of subsequent cardiovascular events (15–17). Two large clinical trials in CAD failed to demonstrate that currently available non-specific platelet function tests can be used to guide treatment (18, 19). Intra-individual variability in platelet function over time is substantial and likely to be a major contributor to the failure (20–22). Because of the failure of platelet function tests to effectively guide treatment in patients with CAD, it is unlikely that existing platelet function tests will be able to guide individualized care in ICAD.
FcγRIIa is a member of the Fc family of proteins that is expressed on the surface of platelets and amplifies platelet activation (23, 24). The Schneider Lab has pioneered the platelet biology of FcγRIIa and established it as a potential marker of risk for secondary thrombotic events in circulatory disorders. FcyRIIa amplifies activation of platelets in response to any stimulus or agonist. Importantly, platelet FcγRIIa expression amplifies thrombosis in the setting of shear forces (25). In a single center study, we found that high platelet FcγRIIa expression (≥11,000/platelet) is associated with a greater risk (odds ratio > 4) of myocardial infarction (MI), stroke and death (26). Platelet FcγRIIa expression does not require activation of platelets and does not exhibit the magnitude of intra-individual variability seen with platelet function tests (27). The emphasis on platelet activation directly focuses our stroke prevention efforts in ICAD where anti-platelets have been paramount and shear-induced platelet aggregation pivotal. FcγRIIa may identify those at high or low risk of recurrent stroke and serve as an effective tool to guide precision medicine in ICAD.
FCγRIIA as a Marker of Platelet Reactivity and Risk of Cardiovascular Events
FcγRIIa was identified as a low-affinity receptor for the fragment constant (Fc) portion of immunoglobulin (Ig) G (28, 29). FcγRIIa markedly enhances thrombus formation when platelets are perfused over a collagen-coated flow chamber under conditions of arterial and venous shear (30). Phosphorylation of FcγRIIa amplifies the activation of platelets (23, 24). We demonstrated that platelets with more FcγRIIa exhibited greater activation in response to sub-maximal concentrations of multiple agonists (31). FcγRIIa may therefore be a novel biomarker capable of identifying patients with increased platelet reactivity. A prospective trial was designed to determine the prognostic implications of platelet FcγRIIa expression (26). Patients (n = 197) were enrolled shortly before discharge from hospitalization for myocardial infarction (MI, both ST elevation and non-ST elevation were included). All patients were treated with aspirin (81 mg) and treatment with clopidogrel (~64%) and ticagrelor (~36%) was balanced in patients with high and low platelet expression of FcγRIIa (26). Clinical characteristics were well-balanced with the exception of older age, diabetes, and prior revascularization being more prominent in the high expression group. Patients with platelet expression of FcγRIIa ≥11,000 had a greater risk of heart attack, stroke, and death that became apparent after 6 months. Cox regression analysis was performed and platelet expression of FcγRIIa was the sole covariate (hazard ratio 3.9, p = 0.035) associated with freedom from MI, stroke, and death. The sensitivity of high expression to identify patients with cardiovascular events was 0.82 (95% confidence intervals 0.57 to 0.92) and the specificity was 0.51 (95% confidence intervals 0.43 to 0.58). Cardiovascular events (heart attack, stroke, and death) were uncommon (8% of all patients experienced an event). The negative predictive value of low platelet expression of FcγRIIa was 0.97 (95% confidence intervals 0.89 to 0.98). Based on preliminary retrospective studies it has been hypothesized that a threshold of 11,000 molecules of FcγRIIa/platelet may identify high and low risk of subsequent cardiovascular events. Analysis of patients with heart attack confirmed that this threshold discriminated high and low risk most efficiently (26). As platelet expression of FcγRIIa is a continuous variable, a larger study will be required to address whether the relationship between cardiovascular events and FcγRIIa expression is continuous.
Defining Platelet Activity and Anti-Platelet Strategies in Secondary Stroke Prevention
Anti-platelet therapies have been the mainstay of secondary stroke prevention for decades. In ICAD, “best medical therapy” is currently defined as DAPT with aspirin and clopidogrel for 90 days after stroke or TIA as in the Stenting and Aggressive Medical Management for Preventing Recurrent Stroke in Intracranial Stenosis (SAMMPRIS) trial (6). Determining platelet activity, defining long-term “best medical therapy” and establishing criteria for “failure” of anti-platelet strategies remain unaddressed. Extensive variation exists in combinations of anti-platelet strategies used and platelet activity monitoring remains a quandary. The measures in Table 1 are used sporadically, imparting bias without systematically assaying platelet activity, offering a role for FcγRIIa.
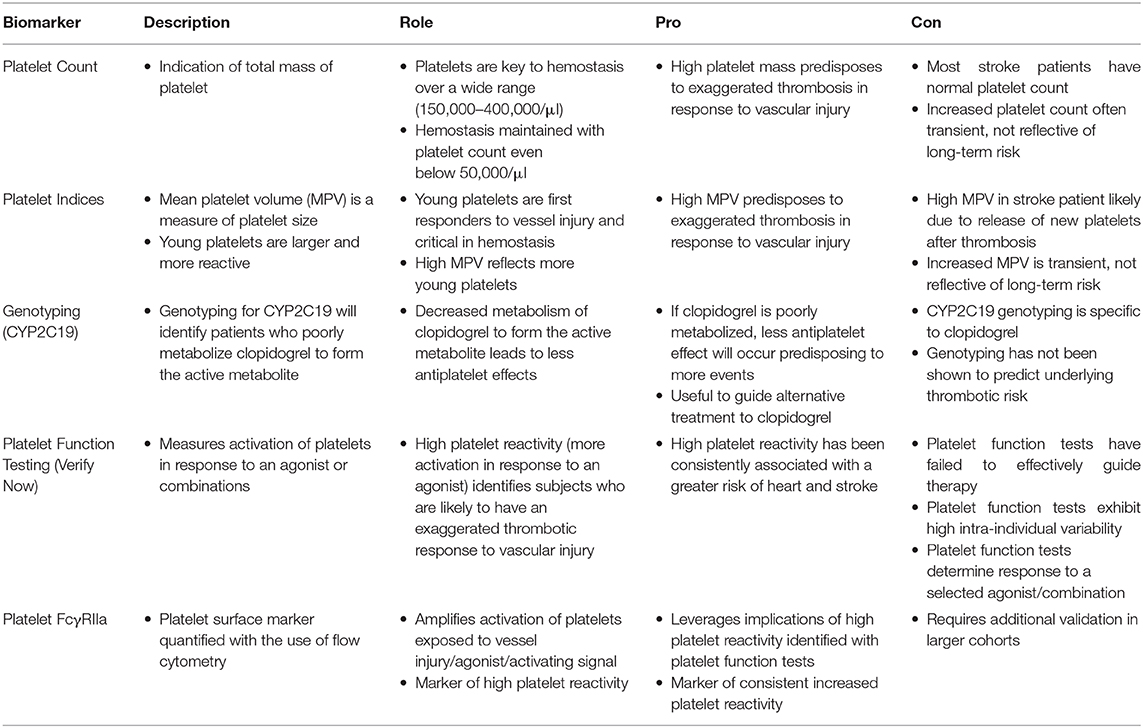
Table 1. Platelet assays and potential use in anti-platelet stroke prevention strategies.
Arterial Hemodynamics of ICAD With CTA Computational Fluid Dynamics (CFD)
For more than a decade, routinely acquired, non-invasive CTA has been used to generate CFD measures of arterial hemodynamics in the coronary and cerebral circulations. CTA CFD has measured fractional flow reserve (FFR), elevated wall shear stress associated with arterial stenoses and post-stenotic flow aberrations, including focal areas of atherogenic low shear stress. In ICAD, almost all cases are treated with medical therapy with very few undergoing endovascular revascularization or alteration of the arterial lesion. As a result, CTA CFD can be used to characterize the local arterial hemodynamics that may predict future events. Our group has pioneered the use of CFD to quantify specific arterial hemodynamic parameters in ICAD for more than a decade (32–40). Our collaborative efforts with investigators in Beijing and Hong Kong have yielded insight on WSS in ICAD stenoses and subsequent clinical events. In a multicenter study of 245 patients (median age = 61 years, 63.7% men) we demonstrated the pivotal prognostic implication of high WSS in the stenosis (35). Stroke in the territory (SIT) occurred in 20 (8.2%) patients, mostly with multiple infarcts in the borderzone and/or cortical regions. In multivariate Cox regression, high WSS ratio (WSSR) of stenotic WSS to pre-stenotic WSS was independently associated with SIT (adjusted HR = 3.05, p = 0.014). These data suggest that high WSS will predict recurrent stroke, yet many other instrumental variables were not captured in that study, including post-stenotic shear force. In our most recent shear stress and endothelial pathophysiology study, we are investigating post-stenotic foci of low shear stress as a nidus for specific endothelial genotype expression, laden with atherogenic and pro-thrombotic stimuli (41). Our collaborative work integrating CFD of ICAD with microfluidic and endothelial expertise has analyzed the CTA subset acquired in the SAMMPRIS trial, showing low shear stress in the post-stenotic segment due to flow vortices proving our ability to extract and define WSS ratios at various arterial lesion sites. These retrospective analyses of SAMMPRIS are limited in ability to prove recurrent stroke due to post-stenotic low shear stress, particularly as they lack systematic MRI follow up to discern interval ischemic injury. These studies focus on only 70–99% stenoses of the proximal MCA and other potentially critical variables regarding platelet biology, anti-platelet treatment, and platelet resistance or response were not collected. In vitro work on shear-induced platelet aggregation strongly suggests that not just elevated WSS, but immediate downstream fluctuations in shear stress are instrumental. We have used the OSI in the post-stenotic segment to calculate, map and quantify this influential variable on in silico models of ICAD with CTA CFD.
Discussion
Poor understanding of ICAD pathophysiology has been a critical barrier to progress in the field of stroke prevention. Targeting specific mechanisms of recurrent ischemia may enable clinicians to match diagnostic findings of ICAD in a given patient with the most effective therapies. Such strategies have been limited due to gaps in clinical trial design, dearth of observational studies, simplistic definitions of ICAD lesion type, empiric use of “best medical therapy,” choice of endpoints and failure to maximally leverage patient-level information from diagnostic imaging. ICAD trials increasingly focus on the most severe (70–99%) stenosis, yet almost half of ischemic strokes due to ICAD occur in milder (50–69%) lesions (42). We have previously shown that hemodynamics in ICAD are pivotal for risk stratification. Dual anti-platelet treatment (DAPT) is often used for variable durations after stroke without recognizing individual anti-platelet response or effects.
It may be possible to tackle these weaknesses using precise individual platelet biology, arterial hemodynamics of shear force across a spectrum of ICAD lesions to ascertain effect on both clinical and imaging ischemic endpoints in a multicenter cohort study. Preliminary data have established that increased platelet FcγRIIa expression is a stable measure of increased platelet reactivity. FcγRIIa can now be quantified for comparison across centers. Unlike platelet function tests, platelet FcγRIIa expression is not substantially affected by assay conditions. Finally, FcγRIIa ≥ 11,000/platelet identifies patients with ~4-fold greater risk of MI, stroke and death. Using the baseline CTA routinely acquired in our recently completed MyRIAD study, we calculated the time averaged WSS in the ICAD lesion, WSS ratio and post-stenotic OSI under pulsatile flow conditions. This preliminary research enabled us to develop standard methodology for quantification of these variables in circumferential bands of the stenosis and equivalent length post-stenosis. A larger study may extend these findings to ICAD patients, show key interplay between platelet FcγRIIa expression and WSS, providing these markers as a basis to guide individualized ICAD stroke prevention.
We have been ardently detailing a vision for precision medicine approaches to stroke and ICAD for many years now (43–56). We have described the potential of imaging features, novel assays and individual clinical characteristics of patients with acute stroke and chronic ICAD to identify therapeutic opportunities based on an n of 1. At a population level, we have advocated for innovative statistical methods such as clustering to discern key predictors of not just risk, but also of propensity for benefit with specific therapeutics. In retrospective analyses, we have leveraged clustering and principal component analyses to reclassify and stratify patient subsets at heightened risk of recurrent events in the datasets of past stroke randomized, controlled trials (44, 49, 57). We developed a novel approach to validate CTA CFD values of WSS in stenoses in ICAD with precision 3D cerebrovascular models, including data from the landmark SAMMPRIS trial. In other collaborations, we have separately studied the potential impact of elevated WSS on stroke recurrence in ICAD and conducted an observational multicenter study on mechanisms of recurrent stroke in ICAD. It has been demonstrated that greater platelet FcγRIIa expression increases the activation of platelets in response to agonists and shear stress. These synergies now enable investigation of how the interaction of anti-platelet therapies with individual platelet expression of FcγRIIa and WSS calculated from patient-specific CTA CFD may explain recurrent ischemia after minor stroke or TIA due to ICAD. The culmination of parallel work on shear stress-induced platelet activation in ICAD leverages preliminary data on FcγRIIa, CTA CFD of WSS and precision medicine analytics in stroke.
Author Contributions
DL conceived and designed the manuscript, analyzed and interpreted the data, handled funding and supervision, drafted the manuscript, and made critical revision of the manuscript for important intellectual content. JH, NK, HK, TH, AD, EF, RN, SP, PC, and DS analyzed and interpreted the data and made critical revision of the manuscript for important intellectual content. All authors contributed to the article and approved the submitted version.
Funding
Funding includes support from NIH-NINDS R01NS112799, Shear Stress and Endothelial Pathophysiology in Intracranial Atherosclerosis.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Banerjee C, Chimowitz MI. Stroke caused by atherosclerosis of the major intracranial arteries. Circ Res. (2017) 120:502–13. doi: 10.1161/CIRCRESAHA.116.308441
2. Chimowitz MI, Lynn MJ, Howlett-Smith H, Stern BJ, Hertzberg VS, Frankel MR, et al. Comparison of warfarin and aspirin for symptomatic intracranial arterial stenosis. N Engl J Med. (2005) 352:1305–16. doi: 10.1056/NEJMoa043033
3. Sacco RL, Kargman DE, Gu Q, Zamanillo MC. Race-ethnicity and determinants of intracranial atherosclerotic cerebral infarction. The Northern Manhattan Stroke Study. Stroke. (1995) 26:14–20. doi: 10.1161/01.STR.26.1.14
4. Hurford R, Wolters FJ, Li L, Lau KK, Küker W, Rothwell PM. Prevalence, predictors, and prognosis of symptomatic intracranial stenosis in patients with transient ischaemic attack or minor stroke: a population-based cohort study. Lancet Neurol. (2020) 19:413–21. doi: 10.1016/S1474-4422(20)30079-X
5. Zaidat OO, Fitzsimmons BF, Woodward BK, Wang Z, Killer-Oberpfalzer M, Wakhloo A, et al. Effect of a balloon-expandable intracranial stent vs medical therapy on risk of stroke in patients with symptomatic intracranial stenosis: the VISSIT randomized clinical trial. JAMA. (2015) 313:1240–8. doi: 10.1001/jama.2015.1693
6. Chimowitz MI, Lynn MJ, Derdeyn CP, Turan TN, Fiorella D, Lane BF, et al. Stenting versus aggressive medical therapy for intracranial arterial stenosis. N Engl J Med. (2011) 365:993–1003. doi: 10.1056/NEJMoa1105335
7. Rana A, Westein E, Niego B, Hagemeyer CE. Shear-dependent platelet aggregation: mechanisms and therapeutic opportunities. Front Cardiovasc Med. (2019) 6:141. doi: 10.3389/fcvm.2019.00141
8. Konstantopoulos K, Grotta JC, Sills C, Wu KK, Hellums JD. Shear-induced platelet aggregation in normal subjects and stroke patients. Thromb Haemost. (1995) 74:1329–34. doi: 10.1055/s-0038-1649935
9. Uchiyama S, Yamazaki M, Maruyama S, Handa M, Ikeda Y, Fukuyama M, et al. Shear-induced platelet aggregation in cerebral ischemia. Stroke. (1994) 25:1547–51. doi: 10.1161/01.STR.25.8.1547
10. Hoogendoorn A, Kok AM, Hartman EMJ, de Nisco G, Casadonte L, Chiastra C, et al. Multidirectional wall shear stress promotes advanced coronary plaque development: comparing five shear stress metrics. Cardiovasc Res. (2019) 116:1136–46. doi: 10.1093/cvr/cvz212
11. Peiffer V, Sherwin SJ, Weinberg PD. Does low and oscillatory wall shear stress correlate spatially with early atherosclerosis? A systematic review. Cardiovasc Res. (2013) 99:242–50. doi: 10.1093/cvr/cvt044
12. Pinto SIS, Campos JBLM. Numerical study of wall shear stress-based descriptors in the human left coronary artery. Comput Methods Biomech Biomed Eng. (2016) 19:1443–55. doi: 10.1080/10255842.2016.1149575
13. Hoi Y, Zhou YQ, Zhang X, Henkelman RM, Steinman DA. Correlation between local hemodynamics and lesion distribution in a novel aortic regurgitation murine model of atherosclerosis. Ann Biomed Eng. (2011) 39:1414–22. doi: 10.1007/s10439-011-0255-z
14. Breet NJ, van Werkum JW, Bouman HJ, Kelder JC, Ruven HJ, Bal ET, et al. Comparison of platelet function tests in predicting clinical outcome in patients undergoing coronary stent implantation. JAMA. (2010) 303:754–62. doi: 10.1001/jama.2010.181
15. Wisman PP, Roest M, Asselbergs FW, de Groot PG, Moll FL, van der Graaf Y, et al. Platelet-reactivity tests identify patients at risk of secondary cardiovascular events: a systematic review and meta-analysis. J Thromb Haemost. (2014) 12:736–47. doi: 10.1111/jth.12538
16. Reny JL, Fontana P, Hochholzer W, Neumann FJ, Ten Berg J, Janssen PW, et al. Vascular risk levels affect the predictive value of platelet reactivity for the occurrence of MACE in patients on clopidogrel. Systematic review and meta-analysis of individual patient data. Thromb Haemost. (2016) 115:844–55. doi: 10.1160/TH15-09-0742
17. Fiolaki A, Katsanos AH, Kyritsis AP, Papadaki S, Kosmidou M, Moschonas IC, et al. High on treatment platelet reactivity to aspirin and clopidogrel in ischemic stroke: a systematic review and meta-analysis. J Neurol Sci. (2017) 376:112–6. doi: 10.1016/j.jns.2017.03.010
18. Price MJ, Berger PB, Teirstein PS, Tanguay JF, Angiolillo DJ, Spriggs D, et al. Standard- vs high-dose clopidogrel based on platelet function testing after percutaneous coronary intervention: the GRAVITAS randomized trial. JAMA. (2011) 305:1097–105. doi: 10.1001/jama.2011.290
19. Collet JP, Cuisset T, Rangé G, Cayla G, Elhadad S, Pouillot C, et al. Bedside monitoring to adjust antiplatelet therapy for coronary stenting. N Engl J Med. (2012) 367:2100–9. doi: 10.1056/NEJMoa1209979
20. Frelinger AL, 3rd, Bhatt DL, Lee RD, Mulford DJ, Wu J, Nudurupati S, et al. Clopidogrel pharmacokinetics and pharmacodynamics vary widely despite exclusion or control of polymorphisms (CYP2C19, ABCB1, PON1), noncompliance, diet, smoking, co-medications (including proton pump inhibitors), and pre-existent variability in platelet function. J Am Coll Cardiol. (2013) 61:872–9. doi: 10.1016/j.jacc.2012.11.040
21. Hochholzer W, Ruff CT, Mesa RA, Mattimore JF, Cyr JF, Lei L, et al. Variability of individual platelet reactivity over time in patients treated with clopidogrel: insights from the ELEVATE-TIMI 56 trial. J Am Coll Cardiol. (2014) 64:361–8. doi: 10.1016/j.jacc.2014.03.051
22. Nührenberg TG, Stratz C, Leggewie S, Hochholzer W, Valina CM, Gick M, et al. Temporal variability in the antiplatelet effects of clopidogrel and aspirin after elective drug-eluting stent implantation. An ADAPT-DES substudy. Thromb Haemost. (2015) 114:1020–7. doi: 10.1160/TH15-03-0257
23. Boylan B, Gao C, Rathore V, Gill JC, Newman DK, Newman PJ. Identification of FcgammaRIIa as the ITAM-bearing receptor mediating alphaIIbbeta3 outside-in integrin signaling in human platelets. Blood. (2008) 112:2780–6. doi: 10.1182/blood-2008-02-142125
24. Lova P, Paganini S, Sinigaglia F, Balduini C, Torti M. A Gi-dependent pathway is required for activation of the small GTPase Rap1B in human platelets. J Biol Chem. (2002) 277:12009–15. doi: 10.1074/jbc.M111803200
25. Chen J, Dong JF, Sun C, Bergeron A, McBride L, Pillai M, et al. Platelet FcgammaRIIA His131Arg polymorphism and platelet function: antibodies to platelet-bound fibrinogen induce platelet activation. J Thromb Haemost. (2003) 1:355–62. doi: 10.1046/j.1538-7836.2003.00054.x
26. Schneider DJ, McMahon SR, Chava S, Taatjes-Sommer HS, Meagher S, Ehle GL, et al. FcγRIIa: a new cardiovascular risk marker. J Am Coll Cardiol. (2018) 72:237–8. doi: 10.1016/j.jacc.2018.04.046
27. McMahon SR, Chava S, Taatjes-Sommer HS, Meagher S, Brummel-Ziedins KE, Schneider DJ. Variation in platelet expression of FcγRIIa after myocardial infarction. J Thromb Thrombolysis. (2019) 48:88–94. doi: 10.1007/s11239-019-01852-7
28. Karas SP, Rosse WF, Kurlander RJ. Characterization of the IgG-Fc receptor on human platelets. Blood. (1982) 60:1277–82. doi: 10.1182/blood.V60.6.1277.1277
29. Arman M, Krauel K. Human platelet IgG Fc receptor FcγRIIA in immunity and thrombosis. J Thromb Haemost. (2015) 13:893–908. doi: 10.1111/jth.12905
30. Zhi H, Rauova L, Hayes V, Gao C, Boylan B, Newman DK, et al. Cooperative integrin/ITAM signaling in platelets enhances thrombus formation in vitro and in vivo. Blood. (2013) 121:1858–67. doi: 10.1182/blood-2012-07-443325
31. Serrano FA, El-Shahawy M, Solomon RJ, Sobel BE, Schneider DJ. Increased platelet expression of FcGammaRIIa and its potential impact on platelet reactivity in patients with end stage renal disease. Thromb J. (2007) 5:7. doi: 10.1186/1477-9560-5-7
32. Ip HL, Liebeskind DS. The future of ischemic stroke: flow from prehospital neuroprotection to definitive reperfusion. Interv Neurol. (2014) 2:105–17. doi: 10.1159/000357164
33. Lan L, Leng X, Ip V, Soo Y, Abrigo J, Liu H, et al. Sustaining cerebral perfusion in intracranial atherosclerotic stenosis: the roles of antegrade residual flow and leptomeningeal collateral flow. J Cereb Blood Flow Metab. (2020) 40:126–34. doi: 10.1177/0271678X18805209
34. Lan L, Liu H, Ip V, Soo Y, Abrigo J, Fan F, et al. Regional high wall shear stress associated with stenosis regression in symptomatic intracranial atherosclerotic disease. Stroke. (2020) 51:3064–73. doi: 10.1161/STROKEAHA.120.030615
35. Leng X, Lan L, Ip HL, Abrigo J, Scalzo F, Liu H, et al. Hemodynamics and stroke risk in intracranial atherosclerotic disease. Ann Neurol. (2019) 85:752–64. doi: 10.1002/ana.25456
36. Leng X, Lan L, Ip HL, Fan F, Ma SH, Ma K, et al. Translesional pressure gradient and leptomeningeal collateral status in symptomatic middle cerebral artery stenosis. Eur J Neurol. (2018) 25:404–10. doi: 10.1111/ene.13521
37. Leng X, Lan L, Ip VHL, Liu H, Abrigo J, Liebeskind DS, et al. Noninvasive fractional flow in intracranial atherosclerotic stenosis: reproducibility, limitations, and perspectives. J Neurol Sci. (2017) 381:150–2. doi: 10.1016/j.jns.2017.08.3239
38. Leng X, Scalzo F, Ip HL, Johnson M, Fong AK, Fan FS, et al. Computational fluid dynamics modeling of symptomatic intracranial atherosclerosis may predict risk of stroke recurrence. PLoS ONE. (2014) 9:e97531. doi: 10.1371/journal.pone.0097531
39. Leung TWH, Fan SY, Ip HL, Lau AYL, Siu DYW, Dai EYL, et al. Effects of collateral circulation on haemodynamic flow status in intracranial artery stenosis depicted by computational fluid dynamics. Hong Kong Med J. (2019) 25(Suppl. 5):18–21.
40. Nam HS, Scalzo F, Leng X, Ip HL, Lee HS, Fan F, et al. Hemodynamic impact of systolic blood pressure and hematocrit calculated by computational fluid dynamics in patients with intracranial atherosclerosis. J Neuroimaging. (2016) 26:331–8. doi: 10.1111/jon.12314
41. Amaya R, Cancel LM, Tarbell JM. Interaction between the Stress Phase Angle (SPA) and the Oscillatory Shear Index (OSI) affects endothelial cell gene expression. PLoS ONE. (2016) 11:e0166569. doi: 10.1371/journal.pone.0166569
42. Feldmann E, Wilterdink JL, Kosinski A, Lynn M, Chimowitz MI, Sarafin J, et al. The Stroke Outcomes and Neuroimaging of Intracranial Atherosclerosis (SONIA) trial. Neurology. (2007) 68:2099–106. doi: 10.1212/01.wnl.0000261488.05906.c1
43. Feldmann E, Liebeskind DS. Developing precision stroke imaging. Front Neurol. (2014) 5:29. doi: 10.3389/fneur.2014.00029
44. Hinman JD, Rost NS, Leung TW, Montaner J, Muir KW, Brown S, et al. Principles of precision medicine in stroke. J Neurol Neurosurg Psychiatry. (2017) 88:54–61. doi: 10.1136/jnnp-2016-314587
45. Liebeskind DS. Mapping the collaterome for precision cerebrovascular health: theranostics in the continuum of stroke and dementia. J Cereb Blood Flow Metab. (2018) 38:1449–60. doi: 10.1177/0271678X17711625
46. Liebeskind DS. Commentary: theranostics for stroke: precision medicine is about tailoring therapy to the mechanism of ischemia. Neurol India. (2018) 66:526–8. doi: 10.4103/0028-3886.227260
47. Liebeskind DS. Big data for a big problem: precision medicine of stroke in neurocritical care. Crit Care Med. (2018) 46:1189–91. doi: 10.1097/CCM.0000000000003165
48. Liebeskind DS. Editorial commentary: beyond the guidelines to expertise in precision stroke medicine. Trends Cardiovasc Med. (2017) 27:67–8. doi: 10.1016/j.tcm.2016.08.001
49. Liebeskind DS. Crowdsourcing precision cerebrovascular health: imaging and cloud seeding a million brains initiative. Front Med. (2016) 3:62. doi: 10.3389/fmed.2016.00062
50. Liebeskind DS. Innovative interventional and imaging registries: precision medicine in cerebrovascular disorders. Interv Neurol. (2015) 4:5–17. doi: 10.1159/000438773
51. Liebeskind DS, Derdeyn CP, Wechsler LR, Consortium SX. STAIR X: emerging considerations in developing and evaluating new stroke therapies. Stroke. (2018) 49:2241–7. doi: 10.1161/STROKEAHA.118.021424
52. Liebeskind DS, Feldmann E. Imaging of cerebrovascular disorders: precision medicine and the collaterome. Ann N Y Acad Sci. (2016) 1366:40–8. doi: 10.1111/nyas.12765
53. Liebeskind DS, Feldmann E. Data considerations in ischemic stroke trials. Neurol Res. (2014) 36:423–6. doi: 10.1179/1743132814Y.0000000357
54. Liebeskind DS, Malhotra K, Hinman JD. Imaging as the nidus of precision cerebrovascular health: a million brains initiative. JAMA Neurol. (2017) 74:257–8. doi: 10.1001/jamaneurol.2016.4896
55. Liebeskind DS, Woolf GW, Shuaib A, Collaterals C. Collaterals 2016: translating the collaterome around the globe. Int J Stroke. (2017) 12:338–42. doi: 10.1177/1747493017701942
56. Saber H, Somai M, Rajah GB, Scalzo F, Liebeskind DS. Predictive analytics and machine learning in stroke and neurovascular medicine. Neurol Res. (2019) 41:681–90. doi: 10.1080/01616412.2019.1609159
Keywords: intracranial atherosclerosis, stroke, shear stress, FcγRIIa receptor, platelet activation and reactivity
Citation: Liebeskind DS, Hinman JD, Kaneko N, Kitajima H, Honda T, De Havenon AH, Feldmann E, Nogueira RG, Prabhakaran S, Romano JG, Callas PW and Schneider DJ (2021) Endothelial Shear Stress and Platelet FcγRIIa Expression in Intracranial Atherosclerotic Disease. Front. Neurol. 12:646309. doi: 10.3389/fneur.2021.646309
Received: 26 December 2020; Accepted: 05 February 2021;
Published: 25 February 2021.
Edited by:
Osama O. Zaidat, Northeast Ohio Medical University, United StatesReviewed by:
Rabia Zubair, St. Vincent Mercy Medical Center, United StatesJong-Won Chung, Sungkyunkwan University School of Medicine, South Korea
Luis Antonio Sanchez Iglesias, Neurosciences Institute, United States
Copyright © 2021 Liebeskind, Hinman, Kaneko, Kitajima, Honda, De Havenon, Feldmann, Nogueira, Prabhakaran, Romano, Callas and Schneider. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: David S. Liebeskind, ZGF2aWRsaWViZXNraW5kQHlhaG9vLmNvbQ==