Abstract
Krabbe disease (KD), also known as globoid cell leukodystrophy, is a rare autosomal recessive condition caused by mutations in the galactocerebrosidase (GALC) gene. KD is more common in infants and young children than in adults. We reported the case of an adult-onset KD presenting with progressive myoclonic epilepsy (PME) and cortical lesions mimicking mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes (MELAS) syndrome. The whole-exome sequencing (WES) identified a pathogenic homozygous missense mutation of the GALC gene. Parents of the patient were heterozygous for the mutation. The clinical, electrophysiological, and radiological data of the patient were retrospectively analyzed. The patient was a 24-year-old woman presenting with generalized seizures, progressive cognitive decline, psychiatric symptoms, gait ataxia, and action-induced myoclonus. The brain magnetic resonance imaging (MRI) revealed a right occipital cortical ribbon sign without any other damage. This single case expands the clinical phenotypes of adult-onset KD.
Introduction
Krabbe disease (KD) is an autosomal recessive condition initially reported by the Danish neuropathologist Knud Krabbe (1). It is now classified as a sphingolipidosis, a lipid storage disorder caused by the deficiency of an enzyme that is required for the catabolism of lipids that contain ceramide. Mutations in the galactocerebrosidase (GALC) gene on chromosome 14q31 cause a deficiency of an enzyme called galactosylceramidase, resulting in an abnormal and toxic accumulation of psychosine in oligodendrocytes. The clinical hallmarks of KD include the demyelination and gliosis of multinuclear macrophages (globoid cells) in the white matter. The most common form of KD usually begins before the age of 1 year. Initial signs and symptoms typically include irritability, feeding difficulties, developmental delay, and seizures. Brain magnetic resonance imaging (MRI) is characterized by aberrant signals in the basal ganglia, cerebellum, corpus callosum, and demyelination of the white matter in posterior regions. Due to the severity of the condition, infants with KD rarely survive beyond the age of 2 years. Less commonly, KD begins in childhood, adolescence, or adulthood (2). Individuals with late-onset KD may survive many years after the condition begins and are identified by progressive spastic paraplegia and gait abnormalities. In the late-onset KD, brain MRI abnormalities implicated the bilateral pyramidal tracts, the posterior ventricle, parietal-occipital white matter, and the corpus callosum (3).
In this study, we described a Chinese patient with adult-onset KD and a homozygous missense mutation in the GALC gene, characterized by progressive myoclonic epilepsy and an asymmetric occipital cortical ribbon sign. The patient was previously misdiagnosed as having mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes (MELAS) syndrome. This case report describes new clinical and radiological manifestations of adult-onset KD.
Case presentation
In August 2019, a 22-year-old Chinese woman born from consanguineous parents (cousins) developed generalized tonic-clonic seizures (GTCS). There was no family history of relevant disorders. In early life, her physical and intellectual growth was unremarkable. The GTCS was treated at the local hospital. At home, the patient discontinued several anti-seizure drugs, such as valproate, clonazepam, and oxcarbazepine. The patient's family noticed limited responsiveness to external stimuli, with decreased production and fluidity of speech. The patient's behaviors became childlike with no apparent reason. The patient started to exhibit action- or posture-induced myoclonus involving the trunk and extremities, particularly the legs. She experienced difficulty in walking and intolerance to physical exercise. In January 2020, she complained several episodes of GTCS. The frequency of seizures decreased when she started taking levetiracetam and lamotrigine. To determine the epileptic cause, she was referred to our hospital in October 2021.
At the time of admission, the physical examination showed horizontal nystagmus, mild dysarthria, gait ataxia, hypotonia and hyperreflexia in all limbs, bilateral pyramidal signs, and pes cavus. The finger-to-nose and rapid alternating movement tests were abnormal. Her head and limbs showed shaking-like involuntary movements while walking or standing. Orientation in space and time was normal but attention, calculation, and comprehension were impaired. She did not cooperate during the sensory testing.
Laboratory tests and cerebrospinal fluid evaluation were normal, including the screening for autoimmune encephalitis and paraneoplastic syndromes-related antibodies. On ultrasound examination, there was no visceromegaly or evident masses. The optical coherence tomography fundus images were normal. The reduced motor nerve conduction velocity, compound muscle action potential amplitude of median nerves, and the sensory conduction velocity of peroneal and sural nerves suggested the presence of a demyelinating peripheral neuropathy. The interictal electroencephalogram (EEG) showed severe generalized slowing waves (Figure 1). Brain MRI exhibited a right occipital cortical ribbon sign on T2-weighted and fluid-attenuated inversion recovery (FLAIR) sequences, with global brain atrophy and ventricular enlargement. Diffusion-weighted imaging (DWI) and susceptibility-weighted imaging (SWI) sequences revealed no other abnormalities (Figure 2).
Figure 1
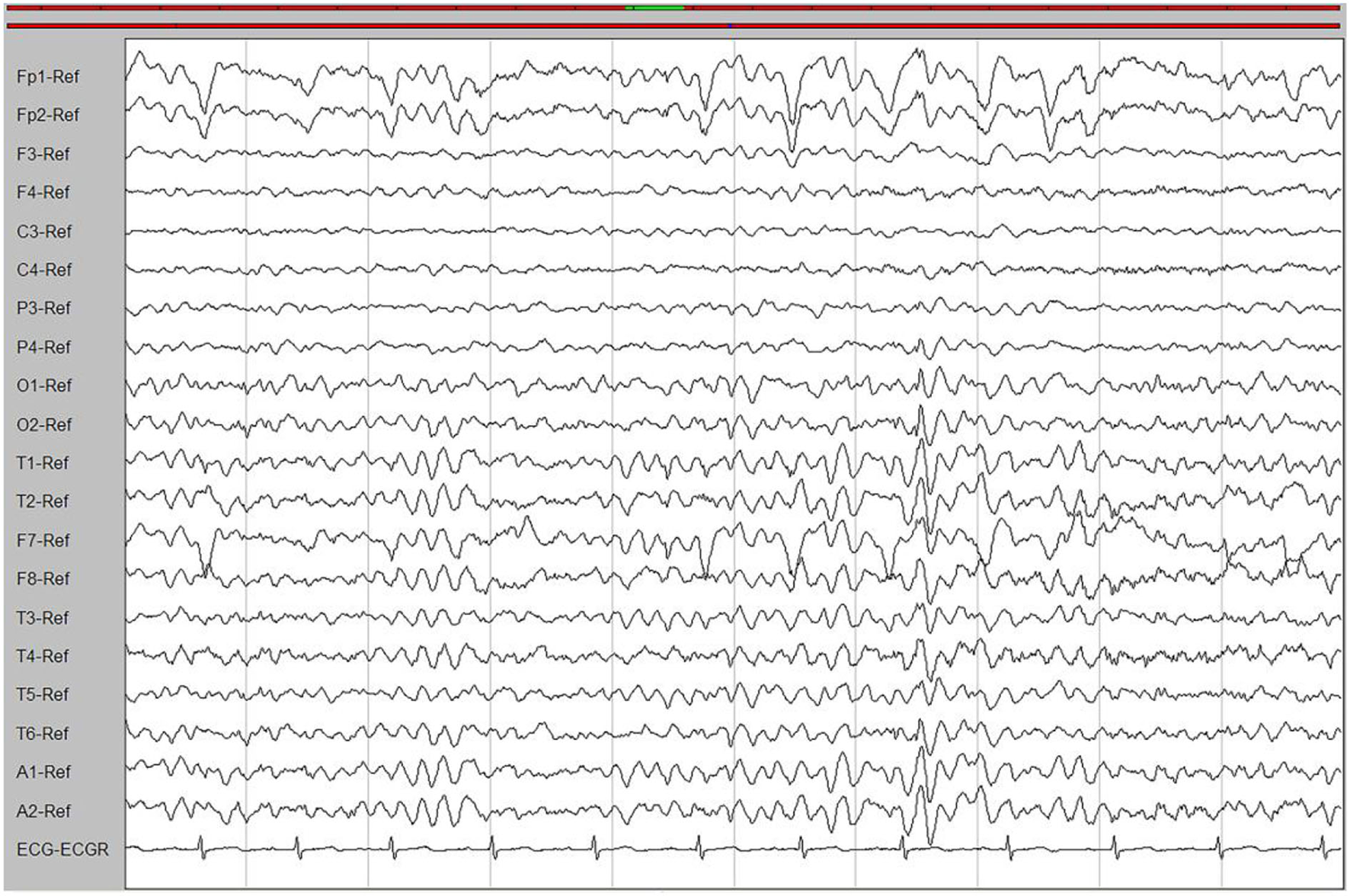
An interictal electroencephalogram (EEG). The EEG showed generalized slowing waves (20–40 μV) and theta waves (6–7 Hz).
Figure 2
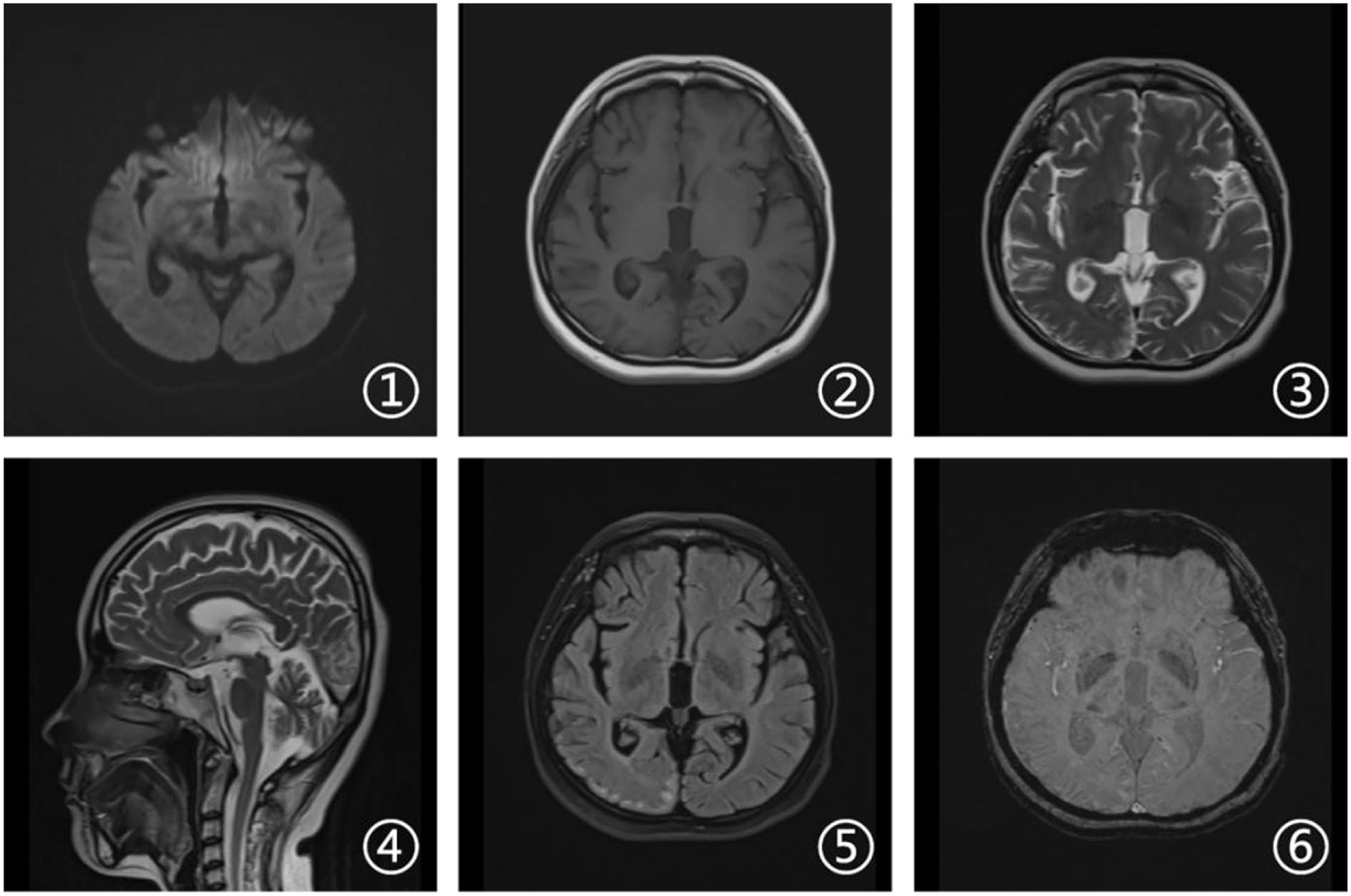
Brain magnetic resonance imaging (MRI). Axial diffusion-weighted imaging (DWI), T1-weighted, and susceptibility-weighted imaging (SWI) sequences (1, 2, and 6) revealed no other abnormalities. Axial T2-weighted and fluid attenuated inversion recovery (FLAIR) sequences (3 and 5) showed a right occipital cortical hyperintense signal. Sagittal T2-weighted scan (4) showed global brain atrophy and ventricular enlargement with occipital cortical hyperintense signal.
There were other manifestations of psychiatric, cognitive, or movement disorders. Although antibodies were negative for autoimmune encephalitis, we used high-dose glucocorticoid therapy (500 mg/day for 5 days). The treatment, however, was ineffective. Meanwhile, we diagnosed a progressive myoclonic epilepsy (PME). Based on clinical presentations and MRI findings, we suspected probable MELAS syndrome or atypical autosomal-recessive cerebellar ataxia (ARCA). The whole-exome sequencing (WES) analysis identified pathogenic homozygous missense mutations of the GALC gene, c.1901T>C (p.Leu634Ser). The Sanger sequencing confirmed that these mutations were inherited from her parents (Figure 3). Then, we diagnosed an adult-onset KD. Serum GALC enzyme activity was found to be ~3.3 nmol/17 h/mg protein (normal range > 12.7 nmol/17 h/mg protein), providing additional support for the clinical diagnosis. There are currently no approved treatments for KD. Adult-onset KD might benefit from hematopoietic stem cell transplantation, as described in a single case report (4). The family refused the treatment due to the high risk of failure.
Figure 3
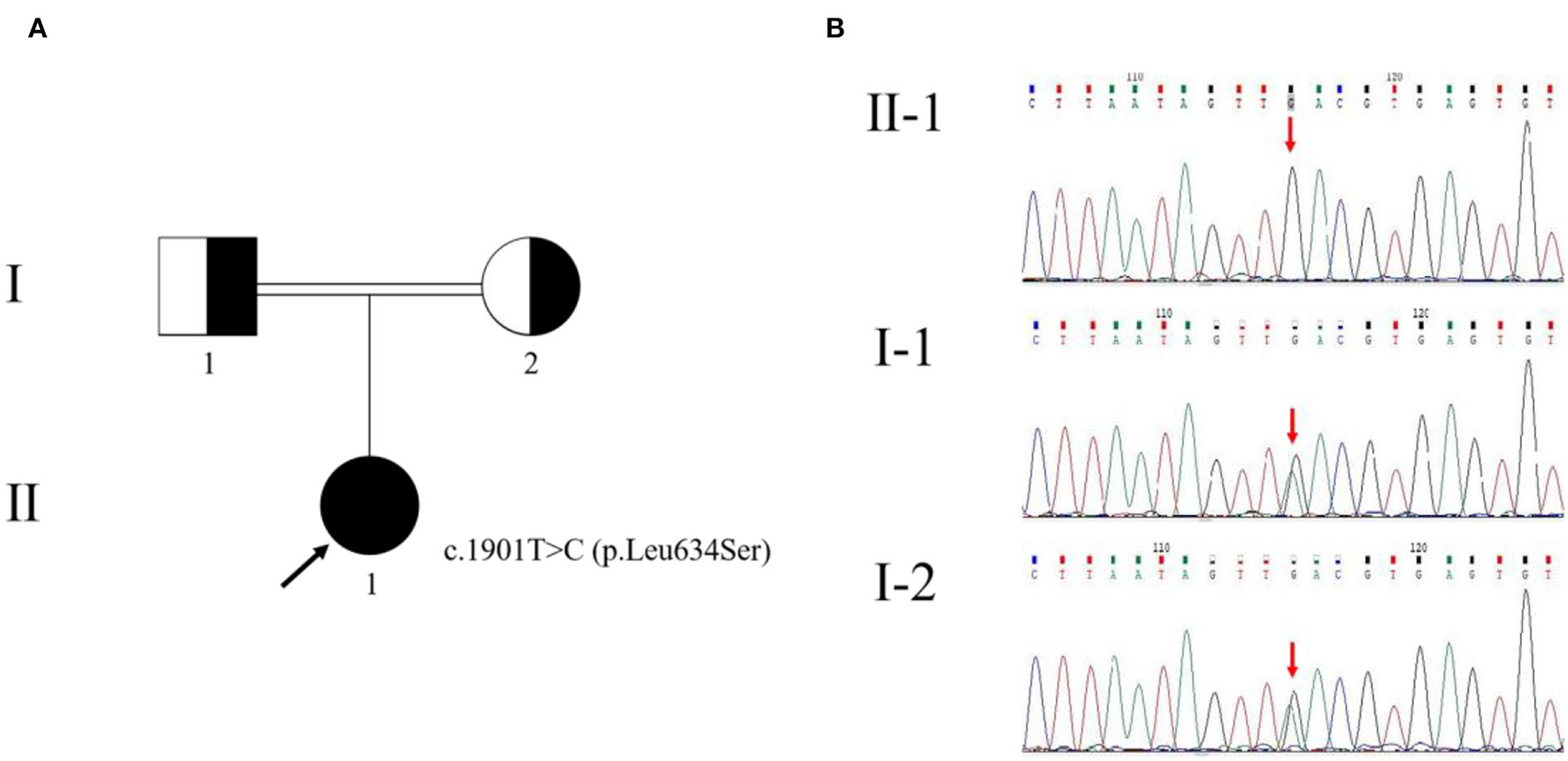
Pedigree of the family with Krabbe disease (A) and genetic evaluation of GALC(B). The pedigree chart shows that the patient's father (I-1) and mother (I-2) were cousins. The proband (II-1) is indicated by a black arrow. The genetic evaluation showed that the c.1901T>C (p.Leu634Ser) mutation of the proband was inherited from her parents.
A few months later, the patient experienced new GTCS with a general deterioration of neurological function, such as persistent confusion, cataphasia, and insomnia. Her family reported a transient gaze paresis of the right side, with involuntary twitch-like movements affecting her right upper limb and mouth. The EEG findings showed numerous irregular left-sided delta waves mixed with a large number of sharp waves, spikes, and wave discharges (Figure 4). Brain MRI showed a damage to the left cerebral hemisphere (Figure 5). Magnetic resonance spectroscopy (MRS) found an increased peak of choline, a decreased peak of N-acetylaspartate and an inverted peak of lipid-lactate in bilateral basal ganglia (Figure 6). She was treated with levetiracetam (1,500 mg/day) and lamotrigine (200 mg/day). She gradually returned to consciousness, the frequency of abnormal involuntary movements decreased.
Figure 4
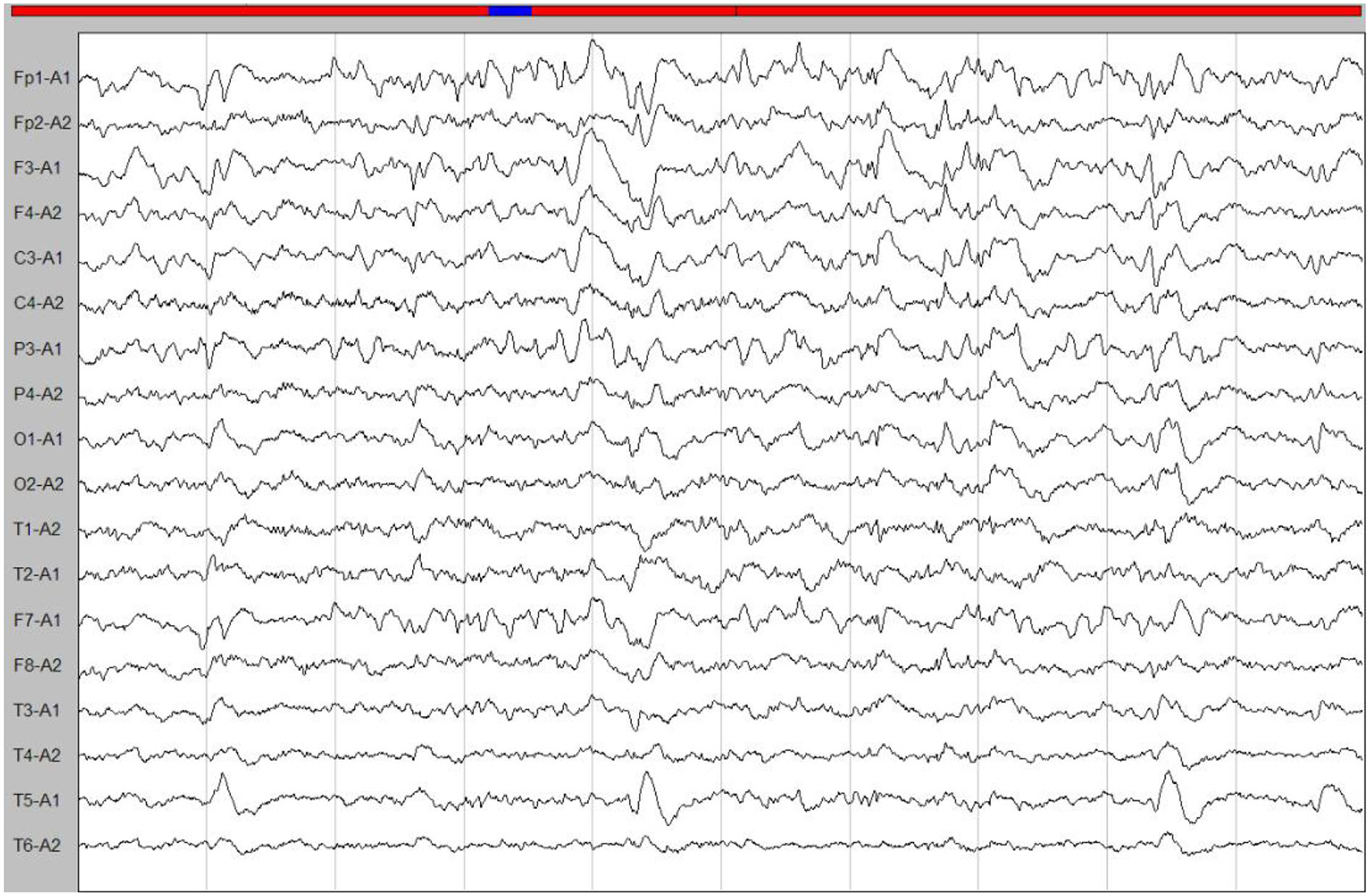
An ictal EEG. The EEG showed left-sided abnormalities with numerous delta waves, including slow waves mixed with sharp waves, spikes, and wave discharges.
Figure 5
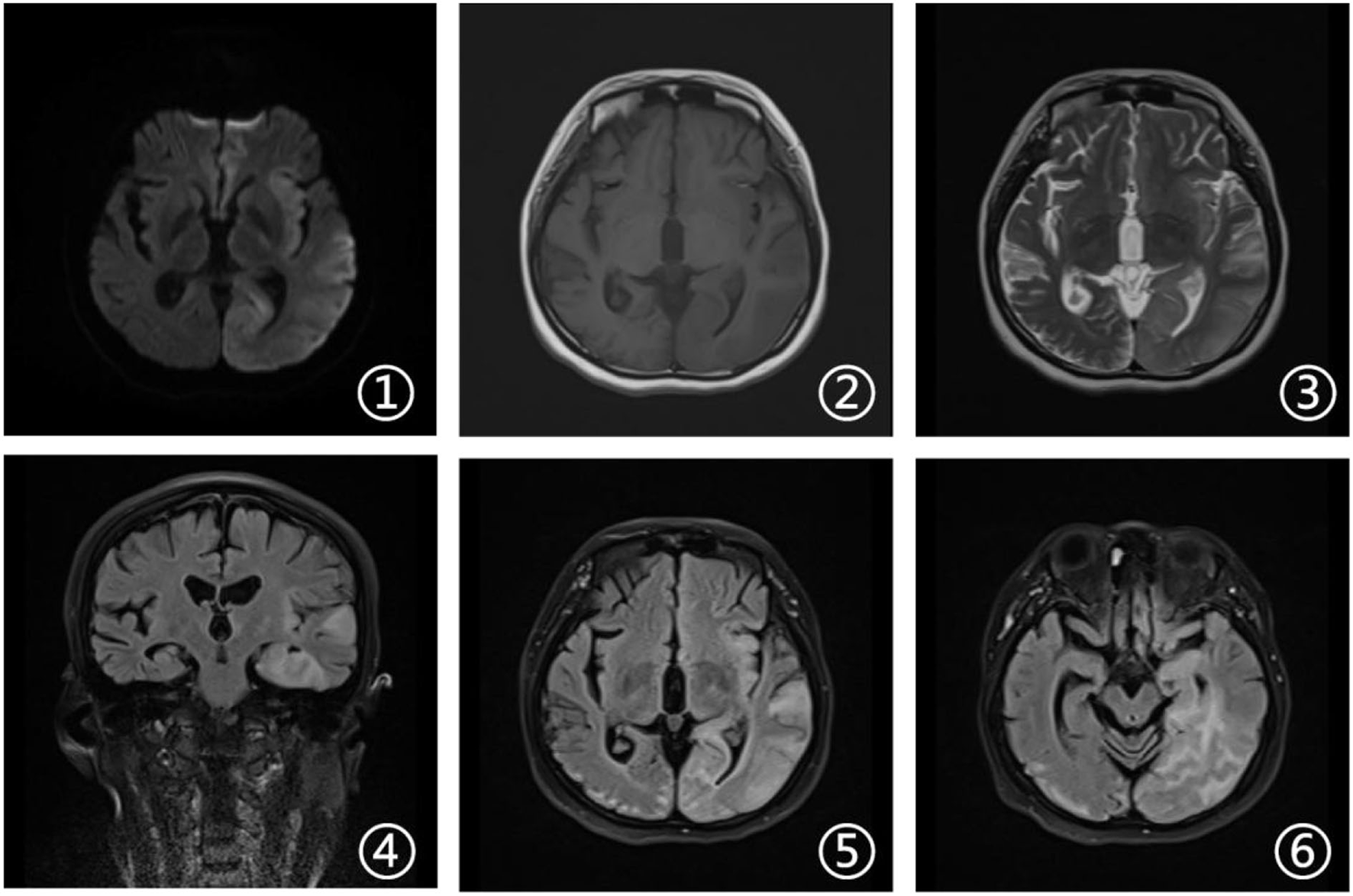
Brain magnetic resonance imaging. Axial DWI, T2-weighted and FLAIR sequences (1, 3, 5, and 6) showed damage in the left cerebral hemisphere covering the frontal, temporal, and occipital lobes. Axial T1-weighted sequences (2) revealed hypointense signal in corresponding areas. Coronal FLAIR MRI (4) revealed high signal changes in the hippocampal area and the left temporal cortex.
Figure 6
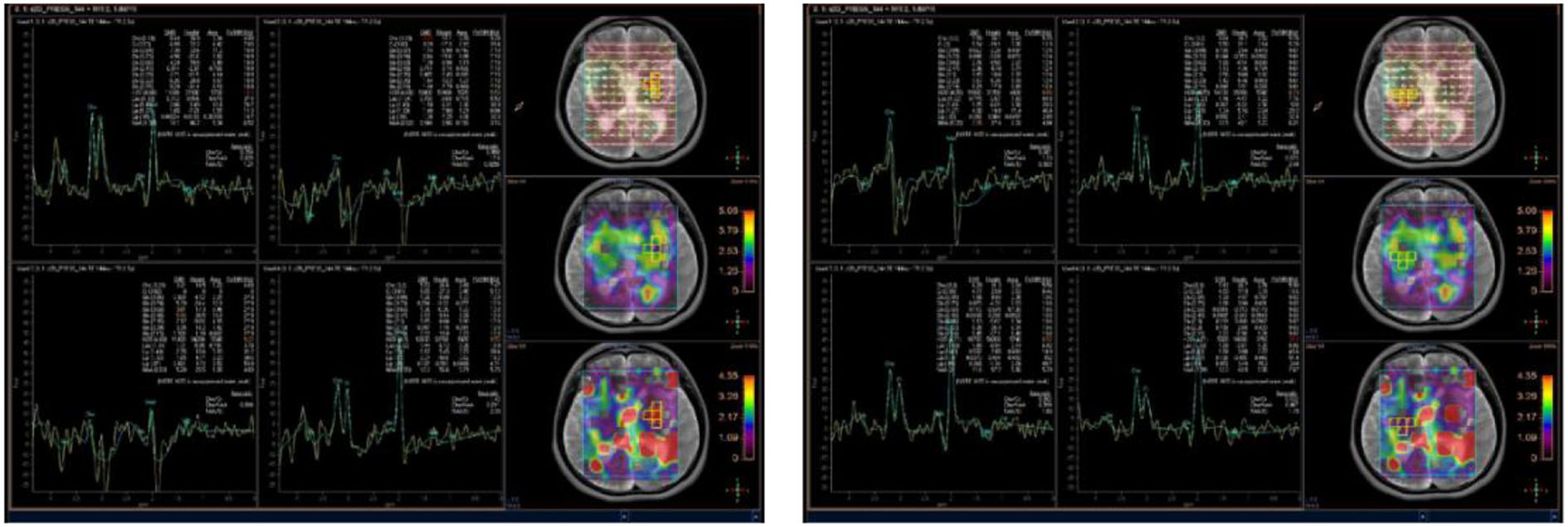
Brain magnetic resonance spectroscopy (MRS) showed an increased peak of choline, a decreased peak of N-acetylaspartate, and an inverted peak of lipid-lactate around bilateral basal ganglia.
Discussion
We described an adult case of KD with atypical clinical and neuroimaging characteristics. We reported for the first time an adult-onset KD presenting PME, a rare epileptic syndrome most commonly inherited in a recessive manner. PME usually appears in late childhood or adolescence with myoclonus, multiple seizure types, cerebellar symptoms, and a progressive degeneration of neurological function (5). The most common causes of PME are lysosomal storage disorders (e.g., neuronal ceroid-lipofuscinoses, Gaucher disease, and sialidosis), mitochondrial disorders (e.g., myoclonic epilepsy with ragged red fibers), spinocerebellar ataxia, Lafora disease, and Unverricht-Lundborg disease (6). Delays in diagnosis are common due to the lack of disease awareness, non-specific clinical presentation, and limited access to diagnostic testing resources in some areas. Our case was early misdiagnosed as an autoimmune encephalitis and incorrectly treated with glucocorticoids. When epilepsy or autoimmune encephalitis is suspected but patients do not respond well to immunotherapies or show no radiological improvements, there is a possibility of misdiagnosis. Only four provinces and cities in China have implemented newborn screening for lysosomal storage disorders. In addition, the test of enzyme is very common in pediatric departments, while it tends to be ignored in adult neurology, which may lead to the underestimation of the late-onset KD prevalence. Therefore, more attention should be directed to adult-onset genetic disorders, especially when common causes have been excluded.
In neurological disorders, an imaging examination is an essential component in the differential diagnosis. In our patient, the MRI findings lead to diagnostic pitfalls. The main imaging feature of KD is leukoencephalopathy, but our patient revealed no widespread supra- or infratentorial white matter signals. The corticospinal tracts, regarded as the most frequent areas involved in adult-onset KD, were not damaged. On initial inspection, the occipital lobe showed a unilateral cortical ribbon sign. When combined with clinical symptoms, the cortical ribbon sign is highly suggestive of mitochondrial disorders. When the cortical ribbon sign is present, multiple autoimmune or metabolic encephalitis, or sporadic Creutzfeldt-Jakob disease should be considered in the differential diagnosis. MELAS syndrome is a relatively common mitochondrial disorder characterized by stroke-like episodes involving the cerebral cortex and diagnosed by genetic testing. We detected no mutations in mitochondrial or nuclear DNA, but we found a homozygous missense variant carrying a c.1901T>C (p.Leu634Ser) mutation in the GALC gene. So far, more than 300 mutations of the GALC gene have been found in patients with KD. The identified pathogenic variant indicated that thymidine at the 1901st position of the coding region of GALC changed to a methylated cytosine, leading to an amino acid substitution (leucine to serine, the 634th amino acid). Her parents were phenotypically normal with a heterozygous GALC gene missense variant. According to the GnomAD database, the frequency of this variant is 0.006 in the East Asian population. This variant was predicted to be “probably damaging” by PolyPhen-2 and “deleterious” by the Sorting Intolerant from Tolerant (SIFT). Previous research demonstrated that this missense mutation inhibited protein synthesis in vitro. Of interest, all patients carrying this variant were associated with a late-onset and mild form of KD. They all were Asian people, particularly from Japan (7), China (8), and Korea (Table 1). Unlike previous reported cases with a relatively benign phenotype, our patient was associated with a more rapid course and worse outcomes (9).
Table 1
| References | Patient no. | Sex/age at onset | Origin | Phenotype | Family history | Genotype | Clinical symptoms | Brain MRI |
|---|---|---|---|---|---|---|---|---|
| Furuya et al. (14) | 1 | N/20y | Japanese | Adult | None | p.[L618S]; [IVS6 + 5G-A] | Slowly progressive spastic paraplegia | T2WI showed high signal in the white matter and pyramidal tracts |
| Satoh et al. (15) | 2 | F/38y | Japanese | Adult | None | p.[L618S]; [L618S] | Slowly progressive spastic paraparesis and diminished vibration sense | T2WI showed symmetric high signal in the bilateral frontoparietal white matter |
| Xu et al. (16) | 3 | N/8m | Japanese | Late-infantile | None | p.[P302A]; [L618S] | N | N |
| Hossain et al. (17) | 4 | N/11m | Japanese | Late-infantile | None | c.[1719dupT]; p.[L618S] | N | N |
| 5 | N/14m | Japanese | Late-infantile | None | c.[1719dupT]; p.[L618S] | N | N | |
| 6 | N/2y | Japanese | Late-infantile | None | p.[L618S]; [?] | N | N | |
| 7 | N/3y | Japanese | Late-infantile | None | c.[P302A]; p.[L618S] | N | N | |
| 8 | N/14y | Japanese | Juvenile | None | p.[L618S]; [?] | N | N | |
| 9 | N/35y | Japanese | Adult | None | p.[L618S]; [G646A] | N | N | |
| 10 | N/56y | Japanese | Adult | None | p.[L618S]; [?] | N | N | |
| Orsini et al. (18) | 11 | M/6m | American | Early-infantile | N | p.[L618S]; [L618S] | N | N |
| 12 | F/13m | American | Late-infantile | N | p.[R38W]; [L618S] | N | N | |
| Yoshimura et al. (7) | 13 | F/11m | Japanese | Late-infantile | None | c.635_646 delinsCTC; [p.L618S] | Developmental regression and spastic quadriplegia | Predominant corticospinal tract involvement |
| Lim et al. (19) | 14 | M/12y | Korean | Juvenile | None | p.[K563*]; [L634S] | Slowly progressive spastic paraplegia | T2WI and FLAIR showed high signal in the precentral motor cortex, corona radiata, posterior limb of the internal capsule, cerebral peduncle of the bilateral pyramidal tracts and optic radiation |
| Zhang et al. (9) | 15 | M/20y | Chinese | Adult | None | p.[L634S]; [L634X] | Acute hemiplegia | selective pyramidal tract involvement with restricted water diffusion on DWI |
| 16 | M/>20y | Chinese | Adult | None | p.[L634S]; [L634S] | None | Hyperintensities of the bilateral pyramidal tracts at the centrum semiovale level and visual radiations | |
| Bascou et al. (20) | 17 | N/2y | American | Late-infantile | N | p.[L634S]; [Arg127Cys] | Abnormal gait characterized by poor balance, ataxia, a wide base, and decreased trunk rotation | Increased T2 signal in the periventricular white matter |
| Zhao et al. (8) | 18 | F/10m | Chinese | Late-infantile | N | p.[F350V]; [L634S] | Motor regression, language development delay, hearing and vision impairment | N |
| 19 | M/ <2y | Chinese | Late-infantile | N | p.[R129X]; [L634S] | Motor regression | N | |
| 20 | M/8y | Chinese | Juvenile | N | p.[L634S]; [?] | Walking and vision impairment | N | |
| 21 | M/ <5y | Chinese | Juvenile | N | p.[P318L]; [L634S] | Left limb movement disorder | N | |
| 22 | F/20y | Chinese | Adult | N | p.[L634S]; [?] | Psychomotor regression, aphasia | N | |
| Irahara-Miyana et al. (21) | 23 | N/>6m | Japanese | Late-infantile | N | p.[L634S]; [?] | N | N |
| Zhang et al. (22) | 24 | F/25y | Chinese | Adult | None | p.[L634S]; [I250T] | Slowly progressive gait disturbance and mild to moderate mental retardation | Corticospinal tract and parieto-occipital white matter involvement |
| 25 | F/14y | Chinese | Juvenile | None | p.[L634S]; [L95fs] | Impairment of visual acuity and Slowly progressive gait disturbance | Corticospinal tract and parieto-occipital white matter involvement | |
| Zhou et al. (23) | 26 | F/22y | Chinese | Adult | Consanguineous | p.[L634S]; [L634S] | Generalized convulsions and cognitive decline | Brain atrophy, no abnormal signals |
| Xie et al. (24) | 27 | M/12 | Chinese | Juvenile | N | p.[L634S]; [Q441X] | Shuffling gait, spastic paraplegia, and ataxia | No visible abnormality |
| 28 | M/26 | Chinese | Adult | N | p.[L634S]; [V681M] | Spastic paraplegia | No visible abnormality | |
| Meng et al. (25) | 29 | M/40y | Chinese | Adult | None | p.[L634S]; [Y335X] | Paralytic paraplegia | Demyelination of parts of the white matter |
| Bascou et al. (12) | 30 | M/5y | American | Juvenile | N | p.[R396W]; [L634S] | Vision reduced | Confluent hyperintensities in the periventricular region with posterior distribution and involvement of the optic radiation |
| Zhang et al. (26) | 31 | M/20y | Chinese | Adult | None | p.[L634S]; [L634X] | Slowly progressive spastic paraplegia | Hyperintensities of the cortico-spinal tracts |
| 32 | M/43y | Chinese | Adult | None | p.[L634S]; [R290C] | Slowly progressive spastic paraplegia | Hyperintensities of the cortico-spinal tracts | |
| 33 | M/46y | Chinese | Adult | None | p.[S200X]; [L634S] | Slowly progressive spastic paraplegia | Hyperintensities of the cortico-spinal tracts | |
| He et al. (27) | 34 | F/40y | Chinese | Adult | None | p.[G553E]; [L634S] | Slowly progressive spastic paraplegia | White matter hyperintensities along bilateral optic radiations |
The clinical and radiological characteristics of Krabbe disease (KD) patients with p.L634S (also known was L618S) mutation previously reported.
F, female; M, male; N, not described in the studies; FLAIR, fluid attenuated inversion recovery; T2WI, T2-weighted images; DWI, diffusion-weighted imaging; “?”: indicates no mutation was found in the second allele or not identified. The * symbol indicates the stop codon, according the HGVS.
The GALC gene dysfunction lowers the activity of the GALC enzyme, which catalyzes the hydrolysis of galactose from several glycosphingolipids (10). Psychosine, a substrate of the GALC enzyme, is a biomarker for infant KD. In the late-onset KD, the diagnosis is supported by a low activity of the GALC enzyme (0–5% of normal activity) in leukocytes. The age of onset and severity of lysosomal storage disorders are often related to the activities of enzymes involved in the disease, with individuals with some enzymatic activity having a higher age of occurrence. In KD, the loss of enzymatic activity does not always predict the development of the disease (11). We found a relatively low activity of the GALC enzyme (about 25.98% of normal activity) in leukocytes, suggesting that KD patients with the p.Leu634Ser mutation may exhibit a broader range of GALC activities. The severity of phenotype may not be necessarily represented by very low enzymatic activity. It is possible that different activities of the GALC enzyme coexist between peripheral blood and the central nervous system. The activity measured in leukocytes may not accurately reflect the activity necessary to maintain an appropriate myelination in neural tissues.
Bascou et al. suggested that vision loss could be the first indicator of late-onset KD due to a p.Leu634Ser variant (12). Our patient was able to see and count fingers but the visual acuity was not tested because of cognitive impairment. The loss of GALC activity can cause oligodendrocyte toxicity, demyelination, and poor remyelination in the brain and peripheral nerves (13). Electrophysiological findings and high arches in the feet supported the presence of a peripheral neuropathy. Previously, a restricted diffusion on DWI suggested an acute aggravation of KD in a few individuals (9). In our patient, we found a limited high signal on DWI in the interictal stage and a high signal in the acute period. DWI appears to be useful in assessing disease progression in people with KD.
We reported the case of a single patient with adult-onset KD, PME, and a cortical ribbon sign. At the beginning, the clinical and neuroimaging characteristics were strongly suggestive of MELAS syndrome. This study could help extending genetic and clinical aspects of adult-onset KD. We acknowledge that our study investigated only a single individual. Skin or muscle samples, if available, could better inform the specificities of adult-onset KD.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Statements
Data availability statement
The datasets presented in this article are not readily available because of ethical and privacy restrictions. Requests to access the datasets should be directed to the corresponding author/s.
Ethics statement
The Medical Research Ethics Committee of the Affiliated Hospital of the Neurology Institute of Anhui University of Chinese Medicine provided the formal approval to this study. The patient and her family provided a written informed consent for the publication of this case report.
Author contributions
YW and S-yW wrote the manuscript with input from all authors. All authors contributed to data acquisition and analysis. All authors contributed to the article and approved the submitted version.
Acknowledgments
We thank the patient and her family for placing their trust in us. We also acknowledge TopEdit LLC for linguistic editing and proofreading during the preparation of this manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1.
Compston A . From the archives. Brain. (2013) 136:2649–51. 10.1093/brain/awt232
2.
Komatsuzaki S Zielonka M Mountford WK Kölker S Hoffmann GF Garbade SF et al . Clinical characteristics of 248 patients with Krabbe disease: quantitative natural history modeling based on published cases. Genet Med. (2019) 21:2208–15. 10.1038/s41436-019-0480-7
3.
Cousyn L Law-Ye B Pyatigorskaya N Debs R Froissart R Piraud M et al . Brain MRI features and scoring of leukodystrophy in adult-onset Krabbe disease. Neurology. (2019) 93:e647–52. 10.1212/WNL.0000000000007943
4.
Laule C Vavasour IM Shahinfard E Mädler B Zhang J Li DK et al . Hematopoietic stem cell transplantation in late-onset krabbe disease: no evidence of worsening demyelination and axonal loss 4 years post-allograft. J Neuroimag. (2018) 28:252–5. 10.1111/jon.12502
5.
Genton P Striano P Minassian BA . The history of progressive myoclonus epilepsies. Epileptic Disorders. (2016) 18:3–10. 10.1684/epd.2016.0834
6.
Kravljanac R Vucetic Tadic B Djordjevic M Lalic T Kravljanac D Cerovic I . The improvement in diagnosis and epilepsy managing in children with progressive myoclonus epilepsy during the last decade—a tertiary center experience in cohort of 51 patients. Epilepsy Behav. (2020) 113:107456. 10.1016/j.yebeh.2020.107456
7.
Yoshimura A Kibe T Irahara K Sakai N Yokochi K . Predominant corticospinal tract involvement in a late infant with Krabbe disease. Jpn Clin Med. (2016) 7:JCM.S40470. 10.4137/JCM.S40470
8.
Zhao S Zhan X Wang Y Ye J Han L Qiu W et al . Large-scale study of clinical and biochemical characteristics of Chinese patients diagnosed with Krabbe disease. Clin Genet. (2018) 93:248–54. 10.1111/cge.13071
9.
Zhang T Yan C Ji K Lin P Chi L Zhao X et al . Adult-onset Krabbe disease in two generations of a Chinese family. Ann Transl Med. (2018) 6:174–174. 10.21037/atm.2018.04.30
10.
Bradbury AM Bongarzone ER Sands MS . Krabbe disease: new hope for an old disease. Neurosci Lett. (2021) 752:135841. 10.1016/j.neulet.2021.135841
11.
Jalal K Carter R Yan L Barczykowski A Duffner PK . Does galactocerebrosidase activity predict Krabbe phenotype?Pediatr Neurol. (2012) 47:324–9. 10.1016/j.pediatrneurol.2012.07.003
12.
Bascou NA Beltran-Quintero ML Escolar ML . Pathogenic variants in GALC gene correlate with late onset krabbe disease and vision loss: case series and review of literature. Front Neurol. (2020) 11:563724. 10.3389/fneur.2020.563724
13.
Tagliapietra M Crescenzo F Castellotti B Gellera C Polo D Cavallaro T et al . Peripheral nerve enlargement on nerve ultrasound parallels neuropathological changes in adult-onset Krabbe disease. Muscle Nerve. (2021) 63:E33–5. 10.1002/mus.27175
14.
Furuya H Kukita YJ Nagano S Sakai Y Yamashita Y Fukuyama H et al . Adult onset globoid cell leukodystrophy (Krabbe disease): analysis of galactosylceramidase cDNA from four Japanese patients. Hum Genet. (1997) 100:450–6. 10.1007/s004390050532
15.
Satoh JI Tokumoto H Kurohara K Yukitake M Matsui M Kuroda Y et al . Adult-onset Krabbe disease with homozygous T1853C mutation in the galactocerebrosidase gene. Unusual MRI findings of corticospinal tract demyelination. Neurology. (1997) 49:1392–9. 10.1212/WNL.49.5.1392
16.
Xu C Sakai N Taniike M Inui K Ozono K . Six novel mutations detected in the GALC gene in 17 Japanese patients with Krabbe disease, and new genotype–phenotype correlation. J Hum Genet. (2006) 51:548–54. 10.1007/s10038-006-0396-3
17.
Hossain MA Otomo T Saito S Ohno K Sakuraba H Hamada Y et al . Late-onset Krabbe disease is predominant in Japan and its mutant precursor protein undergoes more effective processing than the infantile-onset form. Gene. (2014) 534:144–54. 10.1016/j.gene.2013.11.003
18.
Orsini JJ Kay DM Saavedra-Matiz CA Wenger DA Duffner PK Erbe RW et al . Newborn screening for Krabbe disease in New York State: the first eight years' experience. Genet Med. (2016) 18:239–48. 10.1038/gim.2015.211
19.
Lim SM Choi BO Oh SI Choi WJ Oh KW Nahm M et al . Patient fibroblasts-derived induced neurons demonstrate autonomous neuronal defects in adult-onset Krabbe disease. Oncotarget. (2016) 7:74496–509. 10.18632/oncotarget.12812
20.
Bascou N DeRenzo A Poe MD Escolar ML A . prospective natural history study of Krabbe disease in a patient cohort with onset between 6 months and 3 years of life. Orphanet J Rare Dis. (2018) 13:126. 10.1186/s13023-018-0872-9
21.
Irahara-Miyana K Enokizono T Ozono K Sakai N . Exonic deletions in GALC are frequent in Japanese globoid-cell leukodystrophy patients. Hum Genome Var. (2018) 5:28. 10.1038/s41439-018-0027-5
22.
Zhang C Liu Z Dong H . Two Cases of Female Chinese Adult-Onset Krabbe Disease with One Novel Mutation and a Review of Literature. J Mol Neurosci. (2021) 71:1185–92. 10.1007/s12031-020-01742-1
23.
Xia Z Wenwen Y Xianfeng Y Panpan H Xiaoqun Z Zhongwu S . Adult-onset Krabbe disease due to a homozygous GALC mutation without abnormal signals on an MRI in a consanguineous family: a case report. Mol Genet Genomic Med. (2020) 8:e1407. 10.1002/mgg3.1407
24.
Xie JJ Ni W Wei Q Ma H Bai G Shen Y et al . New clinical characteristics and novel pathogenic variants of patients with hereditary leukodystrophies. CNS Neurosci Ther. (2020) 26:567–75. 10.1111/cns.13284
25.
Meng X Li Y Lian Y Li Y Du L Xie N et al . A new compound heterozygous mutation in adult-onset Krabbe disease. Int J Neurosci. (2020) 130:1267–71. 10.1080/00207454.2020.1731504
26.
Zhang T Yan C Liu Y Cao L Ji K Li D et al . Late-Onset Leukodystrophy Mimicking Hereditary Spastic Paraplegia without Diffuse Leukodystrophy on Neuroimaging. Neuropsychiatr Dis Treat. (2021) 17:1451–8. 10.2147/NDT.S296424
27.
He Z Pang X Bai J Wang H Feng F Du R et al . A novel GALC gene mutation associated with adult-onset Krabbe disease: a case report. Neurocase. (2022) 4:1–6. 10.1080/13554794.2022.2083518
Summary
Keywords
adult-onset Krabbe disease, brain MRI, GALC gene, MELAS syndrome, progressive myoclonic epilepsy
Citation
Wang Y, Wang S-y, Li K, Zhu Y-l, Xia K, Sun D-d, Ai W-l, Fu X-m, Ye Q-r, Li J and Chen H-z (2022) Adult-onset Krabbe disease presenting with progressive myoclonic epilepsy and asymmetric occipital lesions: A case report. Front. Neurol. 13:1010150. doi: 10.3389/fneur.2022.1010150
Received
02 August 2022
Accepted
30 September 2022
Published
21 October 2022
Volume
13 - 2022
Edited by
Huifang Shang, Sichuan University, China
Reviewed by
Atsushi Ishii, Fukuoka Sanno Hospital, Japan; Yanping Wei, Peking Union Medical College Hospital, China
Updates
Copyright
© 2022 Wang, Wang, Li, Zhu, Xia, Sun, Ai, Fu, Ye, Li and Chen.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kai Li anhuitcmlikai@163.com
†These authors share first authorship
This article was submitted to Neurogenetics, a section of the journal Frontiers in Neurology
‡ORCID: Yu Wang orcid.org/0000-0002-8098-2491
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.