Abstract
Tauopathies are both clinical and pathological heterogeneous disorders characterized by neuronal and/or glial accumulation of misfolded tau protein. It is now well understood that every pathologic tauopathy may present with various clinical phenotypes based on the primary site of involvement and the spread and distribution of the pathology in the nervous system making clinicopathological correlation more and more challenging. The clinical spectrum of tauopathies includes syndromes with a strong association with an underlying primary tauopathy, including Richardson syndrome (RS), corticobasal syndrome (CBS), non-fluent agrammatic primary progressive aphasia (nfaPPA)/apraxia of speech, pure akinesia with gait freezing (PAGF), and behavioral variant frontotemporal dementia (bvFTD), or weak association with an underlying primary tauopathy, including Parkinsonian syndrome, late-onset cerebellar ataxia, primary lateral sclerosis, semantic variant PPA (svPPA), and amnestic syndrome. Here, we discuss clinical syndromes associated with various primary tauopathies and their distinguishing clinical features and new biomarkers becoming available to improve in vivo diagnosis. Although the typical phenotypic clinical presentations lead us to suspect specific underlying pathologies, it is still challenging to differentiate pathology accurately based on clinical findings due to large phenotypic overlaps. Larger pathology-confirmed studies to validate the use of different biomarkers and prospective longitudinal cohorts evaluating detailed clinical, biofluid, and imaging protocols in subjects presenting with heterogenous phenotypes reflecting a variety of suspected underlying pathologies are fundamental for a better understanding of the clinicopathological correlations.
Introduction
Tauopathies are both clinical and pathological heterogenous disorders characterized by neuronal and/or glial accumulation of misfolded tau protein. Since the first appearance of the name “tau protein” in the literature (1), many studies have been conducted to explain its structure and function. A larger and expanding body of literature focuses on untangling the process of tau accumulation in the neural tissue. Tau is a microtubule-associated protein translated from the MAPT gene on chromosome 17q21 which is predominantly expressed in the brain and the skeletal muscles (2). Alternate splicing of three exons of the MAPT gene produces two sets of isoforms with either three or four microtubule-binding domains, namely 3R and 4R isoforms (3). Based on the ratio of the 3R to 4R isoforms in the pathologic aggregates, tauopathies could be pathologically classified as 4R, 3R, or mixed 3R and 4R. Table 1 shows the pathologic spectrum of tauopathies based on 3R:4R ratio in tau aggregates. Although cryo-electron microscopic (cryo-EM) evaluation of tau filaments in tauopathies led to a new classification based on the structure of tau filaments (16), the clinical significance of this classification is not yet known. There is also a distinction between primary vs. secondary tauopathies based on the tauopathy being the predominant or merely a co-pathology. In fact, even in many secondary tauopathies, tau is the most important co-pathology and a critical player in the process of neurodegeneration (17, 18). It is now well understood that every pathologic tauopathy may present with various clinical phenotypes based on the primary site of involvement and the spread and distribution of the pathology in the nervous system (19). Thus, clinicopathological correlation is becoming more and more challenging and defining clinical diagnostic criteria for pathologic disorders more nuanced and sometimes almost impossible.
Table 1
| 4R tauopathies | 3R tauopathy | Mixed 3R/4R tauopathy | |||||
|---|---|---|---|---|---|---|---|
| Lesion | PSP (4, 5) | CBD (5, 6) | GGT (7, 8) | AGD (9, 10) | ARTAG (11, 12) | PiD (13, 14) | PART (15) |
| Neuronal loss/gliosis | Frontal / paracentral Globus pallidus Subthalamic nucleus Substantia nigra Brainstem tegmentum Red nucleus Midbrain tectum Dentate nucleus |
Asymmetric peri-rolandic areas, superior frontal and parietal Substantia nigra |
Frontotemporal (type I, III) Motor cortex / corticospinal tract (types II, III) |
Ambient gyrus medial temporal structures (CA1) |
Subpial, subependymal, perivascular in basal or lobar regions | Frontal, anterior temporal, medial temporal Subcortical/ white matter degeneration |
Medial temporal Hippocampal formation (CA2) |
| Neuronal pathology/inclusions | Dense argyrophilic “globose” tangles | Disperse globose inclusions (less argyrophilic mostly consisted of pre-tangles) Abundant achromatic /ballooned neurons |
Diffuse granular /thick cord-like /round horseshoe-shaped neuronal cytoplasmic inclusions | Comma-shaped argyrophilic grains Cytoplasmic pre-tangles Ballooned neurons |
Strongly argyrophilic neuronal cytoplasmic inclusions (Pick bodies) Pick cells (ballooned achromatic neurons) |
Intracellular neurofibrillary tangles Extracellular “ghost” tangles | |
| Glial pathology/ inclusions | Tufted astrocytes (highly argyrophilic dense NFTs in soma) | Astrocytic plaques (less argyrophilic NFTs in processes) | Globular oligodendroglial / astrocytic inclusions Non-argyrophilic tufted astrocytes-like inclusions | Bush-like astrocytes | Thorn-shaped, granular or fuzzy astrocytes | “Ramified” astrocytes | |
| Threads / coiled bodies | Coiled bodies Threads | Coiled bodies Widespread threads |
Coiled bodies | Coiled bodies | |||
Pathologic spectrum of primary tauopathies and their defining hallmarks.
AGD, argyrophilic grain disease; ARTAG, aging related tau astrogliopathy; CA, cornu Ammonis; CBD, corticobasal degeneration; PSP, progressive supranuclear palsy; GGT, globular glial tauopathy; NFT, neurofibrillary tangle; PART, primary age related tauopathy; PiD, Pick's disease.
The pathologic spectrum of primary tauopathies include progressive supranuclear palsy (PSP), corticobasal degeneration (CBD), Pick's disease (PiD), globular glial tauopathy (GGT), argyrophilic grain disease (AGD), primary age-related tauopathy (PART), and aging-related tau astrogliopathy (ARTAG). On the other hand, the clinical spectrum of tauopathies include some major syndromes strongly associated with an underlying primary tauopathy and a number of other syndromes with lower specificity. The first group includes PSP or Richardson syndrome (RS), corticobasal syndrome (CBS), non-fluent agrammatic primary progressive aphasia (nfaPPA)/apraxia of speech, pure akinesia with gait freezing (PAGF), and behavioral variant frontotemporal dementia (bvFTD). The second group with less specificity includes Parkinsonian syndrome, late onset cerebellar ataxia, primary lateral sclerosis, semantic variant PPA (svPPA), and amnestic syndrome of the hippocampal type. Figure 1 shows the clinicopathologic correlation in the tauopathy spectrum. Of all clinical variants, RS has the most consistent correlation with an underlying tau, more specifically 4R tau, pathology (Figure 1). Here, we summarize main clinical syndromes associated with various primary tauopathies.
Figure 1
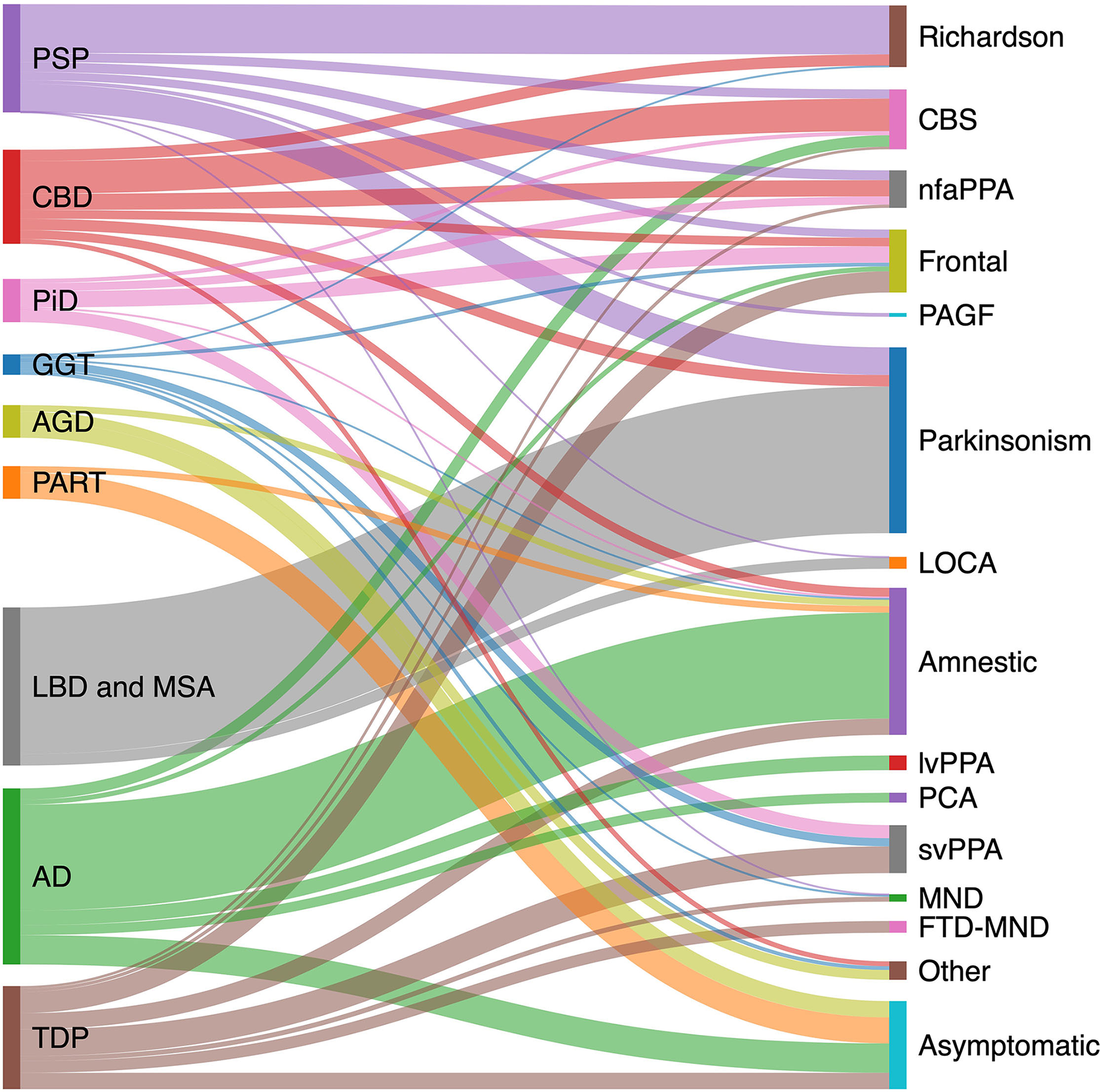
Clinicopathological correlation in tauopathies spectrum. Pathologic diagnoses are shown on the left and clinical syndromes on the right. Degree to which various pathologies contribute to a specific clinical phenotype is estimated based on available pathology-confirmed studies, including: PSP (20–23); CBD (23–27); AD (28–31); PiD (32); LBD (33); GGT (34); TDP (35–37); AGD (38); and PART (15, 39, 40). AD, Alzheimer's disease; AGD, argyrophilic grain disease; CBD, corticobasal degeneration; CBS corticobasal syndrome; FTD-MND, frontotemporal dementia-motor neuron disease; GGT, globular glial tauopathy; LBD, Lewy body disease; LOCA, late onset cerebellar ataxia; lvPPA logopenic variant primary progressive aphasia; MND, motor neuron disease; nfaPPA, non-fluent agrammatic primary progressive aphasia; PAGF, progressive akinesia and gait freezing; PART, primary age-related tauopathy; PCA, posterior cortical atrophy; PiD, Pick's disease; PSP, progressive supranuclear palsy; svPPA, semantic variant primary progressive aphasia; TDP, transactive response DNA binding protein 43 kDa pathology.
Clinical Syndromes With Strong Association to a Primary Tauopathy
Richardson Syndrome
Progressive supranuclear palsy (PSP) syndrome was first described by Steele, Richardson, and Olszewski in 1964 as a distinct clinicopathological entity (41). Although typical PSP pathology with tufted astrocytes (Table 1) is the most common underlying pathology of this clinical syndrome (PSP-RS), a small subset of patients with an underlying CBD pathology might present with RS (CBD-RS) and there are rare reports of GGT presenting with a PSP syndrome (Figure 1) (34, 42). Original description of RS included nine patients presenting with early and prominent supranuclear ophthalmoplegia, severe dysarthria, axial rigidity with extensor posturing of the neck, mild dementia, and pseudobulbar palsy with subsequent progression to a bedridden state and death in 5 to 7 years (41). Patients also showed gait problems with frequent falls and unsteadiness which originally were attributed by authors to the gaze palsy and truncal rigidity or truncal apraxia. However, postural instability was later acknowledged as the major cause of gait impairment in these patients (43) and as the second clinical hallmark of RS, along with supranuclear gaze palsy (44, 45).
Before recognition of the various clinical phenotypes, the prevalence of PSP has been estimated to be about 5–8/100,000 (46–48). This figure in fact might better reflect the prevalence of PSP-RS which accounts for about 40–50% of patients with PSP (20, 21). In a more recent study by Stang et al. (49), the new MDS-PSP criteria for PSP was used which led to an estimated PSP incidence of 2.6 per 100,000. Median age of the onset of RS is about 63 years (50). Survival has not changed since the first description of RS.
Clinical Characteristics
Ocular manifestations in RS are diverse and include eye movement, fixation, lid function, and pupillary abnormalities (51). Supranuclear ophthalmoplegia starts with subtle slowing and decreased accuracy of vertical, especially downward saccades which is better demonstrated on optokinetic nystagmus (OKN) testing (52). This will later be followed by decreased saccadic amplitude and eventual involvement of horizontal saccades (53) and smooth pursuit system (54) leading to complete ophthalmoplegia. Fixation impairment with frequent high amplitude square wave jerks (55, 56) is a differentiating features of RS from similar parkinsonian syndromes especially CBS (56). Decreased blink rate, lid retraction, blepharospasm, and eyelid opening apraxia are other ocular manifestations of RS.
Postural instability leading to frequent falls is a common feature among various Parkinsonian disorders. However, it is most severe and occurs the earliest in the course of RS, in the first 3 years and more specifically in the first year of disease onset (45), compared to other Parkinsonian syndromes and other PSP variants (57). On stance, patients with RS assume a wide base and tend to drift backwards (44, 58) and postural reflexes are impaired on the pull test. During gait initiation, the center of pressure changes in opposite direction in these patients compared to patients with Parkinson's disease (PD) and healthy controls (59). Walking in RS is markedly slow with decreased cadence and has been described as “reckless” or like a “drunken sailor,” emphasizing unsteadiness and increased variability. Increased double support time and the wide base are compensatory strategies to improve balance (60); however, despite these strategies, patients with RS are very prone to falls following minor internal or external perturbations. In addition to the aforementioned abnormalities, other factors, such as vertical supranuclear gaze palsy, bradykinesia, axial rigidity, and cognitive impairment have been proposed as contributors to the fall risk. However, a large recent study showed that vertical supranuclear gaze palsy and cognitive impairment were not independent predictors of fall risk while markers of disease duration and severity, such as horizontal gaze palsy, were independent predictors of fall risk (43). Freezing of gait presents in 20% of patients in the first year of symptom onset and increases with disease duration (61).
Parkinsonism in RS presents as axial-predominant symmetric bradykinesia, rigidity, and less frequently tremor, usually of low amplitude postural/kinetic or mixed type (62). While at the time of diagnosis over 75 and 40% of patients with RS present symptoms of bradykinesia and axial rigidity, respectively, nearly all show these signs throughout disease course, and only 10–20% show tremor (22, 63). This is a distinguishing feature of RS from another PSP variant, PSP-Parkinsonism (PSP-P) in which tremor is present in more than half of the patients (22).
In patients with RS, dysarthria, of hypokinetic or mixed hypokinetic-spastic nature, occurs earlier and in a higher number of patients than dysphagia, although both, dysarthria and dysphagia are more frequent and more severe in RS than in other parkinsonian syndromes (64, 65). At least one third of patients have speech impairment at the time of diagnosis that leads to unintelligible speech in about 6 years and eventually involves nearly all patients (22, 66). Significant oropharyngeal dysphagia develops in about 3 to 4 years, although it might occur during the first 3 years which is an indicator of poor prognosis considering that aspiration pneumonia is the most common cause of death in these patients (67, 68). Feeding tube placement is considered after seven years of symptom onset for many patients (66).
Mild non-amnestic cognitive impairment is frequently encountered in the first 3 years and has a high risk for conversion to frontal/subcortical dementia. Older age at disease onset is the major risk factor for the development of dementia. The incidence rate of dementia in RS is estimated to be about 241 per 1,000 patients per year which is 3 times greater than that of PD (69). Cognitive changes in RS have classically been defined as a prototypic subcortical dementia with slowed central processing speed and pronounced executive dysfunction (70–72). More recent studies, separating different variants of PSP, found that phonemic verbal fluency and global cognitive performance are consistently and more severely impaired in RS (73, 74) compared to other PSP variants (75, 76). Insight is also characteristically impaired (77). Behavioral changes, mainly in the frontotemporal spectrum, are among common features of RS and in fact many patients with RS fulfill the possible bvFTD criteria (78). Social cognition, including both basic emotion recognition and more complex theory of mind functions are impaired in patients with RS. However, these patients do not usually present socially inappropriate behavior like patients with bvFTD (79). Neuropsychiatric changes are common, especially apathy and less commonly depression. Apathy affects about 80–90 % of patients with RS while depression is more common in CBS than RS (80). Sleep and eating-related disorders are probably the next common neuropsychiatric symptoms. Other behavioral symptoms with less frequency include disinhibition, aggressive behavior, irritability, and anxiety (81). In terms of impulsivity and disinhibition, a behavioral/cognitive and a motor aspect could be considered. The cognitive aspect is reflected, for example, in the Tower test. In addition to overall lower scores due to executive/planning dysfunction, patients with PSP-RS present higher number of rule-violation errors compared to other types of dementias and healthy subjects which is a marker of disinhibition. Possin et al. (82) found that, after controlling for general cognitive performance, the number of rule violation errors was correlated with the right lateral prefrontal cortex atrophy in these patients. For the motor aspect, one might think of the reckless gait, such as rocket sign and sitting en bloc as motor presentations of impulsivity. Hallucination and delusions are very rare in RS and if present are indicative of either co-pathology or medication side-effects (63, 83–85). Environmental dependency syndrome, defined as excessive dependence on environmental cues (86), which includes symptoms, such as imitation behaviors, palilalia, echolalia, echopraxia, utilization behavior, and palmar and visual grasping are also frequently encountered in patients with PSP (87, 88). Parietal lobe disinhibition due to frontal lobe and/or frontoparietal network dysfunction is believed to be the underlying cause of environmental dependency syndrome.
Diagnostic Criteria
The National Institute of Neurological Disorders and Stroke and Society for PSP, Inc. (NINDS-SPSP) criteria of PSP was developed in 1996, before the recognition of the majority of the clinical phenotypes of PSP pathology (45). These criteria include slowing of vertical saccades/supranuclear gaze palsy and postural instability with falls in the first year as possible and probable PSP. The NINDS-SPSP criteria have in fact high specificity for the diagnosis of the RS but moderate sensitivity (89, 90). The need for higher sensitivity and recognition of all new clinical phenotypes led to the development of the Movement Disorders Society criteria for PSP (MDS-PSP) (91) (Table 2). These criteria define three levels of certainty: suggestive, possible, and probable for each clinical phenotype to allow for the early diagnosis of patients with PSP. Wide application of the criteria revealed that the MDS-PSP criteria unexpectedly allowed for a single patient to fulfill criteria for multiple phenotypes at a time (94), which led to later development of the multiple application extinction (MAX) rules (95). Despite the new addition, which resulted in more complexity (96–98), problems remain with differentiating PSP-Parkinsonism variants from RS and to accurately diagnose PSP-Speech/language phenotype (99–102). Moreover, specificity of the criteria, especially in early stages, remains low (103).
Table 2
| Clinical syndrome | ||||||
|---|---|---|---|---|---|---|
| Criteria set | RS | CBS | nfaPPA | bvFTD | PAGF | Tauopathies with Parkinsonism |
| MDS-PSP criteria 1 | ||||||
| Probable | VSGP or SVS + Repeated falls or fall on pull test in first 3 years | VSGP or SVS + ≥ 3 of the following: • Apathy • Bradyphrenia • Dysexecutive syndrome • Reduced phonemic verbal fluency • Impulsivity, disinhibition, or perseveration |
VSGP or SVS + Progressive gait freezing (Sudden, transient motor blocks/start hesitation, no/mild parkinsonism, levodopa resistant) in first 3 years | VSGP or SVS + one of: • Axial predominant, levodopa resistant bradykinesia and rigidity • Parkinsonism that is asymmetrical/with tremor/levodopa responsive |
||
| Possible | SVS + >2 steps backward on pull test in first 3 years | VSGP or SVS + Limb rigidity or akinesia or myoclonus + ≥1 cortical sign: • Orobuccal/limb apraxia • Cortical sensory deficit • Alien limb phenomena |
VSGP or SVS + nfaPPA or PAOS | Progressive gait freezing in first 3 years | ||
| Suggestive | Frequent mSWJs + Fall or >2 steps backward on pull test in first 3 years | Limb rigidity or akinesia or myoclonus + ≥1 cortical sign: • Orobuccal/limb apraxia • Cortical sensory deficit • Alien limb phenomena |
nfaPPA or Progressive AOS | Frequent mSWJs or >2 steps backward on pull test in first 3 years + ≥3 of the following: • Apathy • Bradyphrenia • Dysexecutive syndrome • Reduced phonemic verbal fluency • Impulsivity, disinhibition, or perseveration |
Axial predominant, levodopa resistant bradykinesia and rigidity or Parkinsonism that is asymmetrical/with tremor/levodopa responsive + one of: • Frequent mSWJs • Fall or >2 steps backward on pull test in first 3 years • s.o. PSP-SL • s.o. PSP-F • Levodopa resistant • Hypokinetic, spastic dysarthria • Dysphagia • Photophobia |
|
| Armstrong CBD criteria | ||||||
| Probable | ≥3 of: • Axial or symmetric limb rigidity or akinesia • Postural instability/falls • Urinary incontinence • Behavioral changes • VSGP/SVS |
Asymmetric presentation of ≥2 cortical + ≥2 movement signs: Cortical signs: • Orobuccal/limb apraxia • Cortical sensory deficit • Alien limb phenomena Movement signs: • Limb rigidity • Limb akinesia • Limb myoclonus Exclusionary criteria: • Positive CSF, PET, or genetic AD biomarkers2 • Evidence of: LBD3/MSA4/ALS5/svPPA or nfaPPA • Structural lesion suggestive of focal cause • Granulin mutation or reduced plasma progranulin levels • TDP-43 mutations • FUS mutations ≥1 movement sign + ≥1 cortical sign Meeting no exclusionary criteria |
Effortful, agrammatic speech + ≥1 of: • Impaired grammar/sentence comprehension with relatively preserved single word comprehension • Groping, distorted speech production (AOS) |
≥2 of: • Executive dysfunction • Behavioral or personality changes • Visuospatial deficits |
||
| Rascovsky bvFTD criteria 6 | ||||||
| Possible | Presence in the first 3 years of ≥3 of these symptoms: • Behavioral disinhibition7 • Apathy or inertia8 • Loss of sympathy or empathy9 • Perseverative, stereotyped or compulsive/ritualistic behavior10 • Hyperorality and dietary changes11 • Neuropsychological profile12 |
|||||
| Probable | All of below: • Meets criteria for possible bvFTD • Significant functional decline • Imaging results consistent with bvFTD, ≥ 1 of: • Frontal and/or anterior temporal atrophy on MRI or CT • Frontal and/or anterior temporal hypoperfusion or hypometabolism on PET or SPECT |
|||||
| Definite | Meets criteria for possible or probable bvFTD + • Histopathological evidence of FTLD on biopsy or at post-mortem OR • Presence of a known pathogenic mutation |
|||||
| Gorno-Tempini PPA criteria 13 | ||||||
| nfaPPA | svPPA | lvPPA | ||||
| Clinical | At least one core feature: • Agrammatism • Effortful, halting speech with inconsistent speech sound errors and distortions (apraxia of speech) + ≥2 of: • Impaired comprehension of syntactically complex sentences • Spared single-word comprehension • Spared object knowledge |
Both of the following core features: • Impaired confrontation naming • Impaired single-word comprehension + ≥3 of: • Impaired object knowledge, particularly for low frequency or low-familiarity items • Surface dyslexia or dysgraphia • Spared repetition • Spared speech production (grammar and motor) |
Both of the following core features: • Impaired single-word retrieval in spontaneous speech and naming • Impaired repetition of sentences and phrases + ≥3 of: • Speech (phonologic) errors in spontaneous speech and naming • Spared single-word comprehension and object knowledge • Spared motor speech • Absence of frank agrammatism |
|||
| nfaPPA | svPPA | lvPPA | ||||
| Imaging supported | • Clinical diagnosis of nfaPPA (as above) + ≥1 of: • Predominant left posterior fronto-insular atrophy on MRI • Predominant left posterior fronto-insular hypoperfusion or hypometabolism on SPECT or PET |
• Clinical diagnosis of svPPA (as above) + ≥1 of: • Predominant anterior temporal lobe atrophy • Predominant anterior temporal hypoperfusion or hypometabolism on SPECT or PET |
• Clinical diagnosis of lvPPA (as above) + ≥1 of: • Predominant left posterior perisylvian or parietal atrophy on MRI • Predominant left posterior perisylvian or parietal hypoperfusion or hypometabolism on SPECT or PET |
|||
| nfaPPA | svPPA | lvPPA | ||||
| Definite | Clinical diagnosis of nfaPPA (as above) + ≥1 of: • Histopathologic evidence of a specific neurodegenerative pathology (e.g., FTLD-tau, FTLD-TDP, AD, other) • Presence of a known pathogenic mutation |
Clinical diagnosis of svPPA (as above) + ≥1 of: • Histopathologic evidence of a specific neurodegenerative pathology (e.g., FTLD-tau, FTLD-TDP, AD, other) • Presence of a known pathogenic mutation |
Clinical diagnosis of lvPPA (as above) + ≥1 of: • Histopathologic evidence of a specific neurodegenerative pathology (AD, FTLD-tau, FTLD-TDP, other) • Presence of a known pathogenic mutation |
|||
Standardized clinical diagnostic criteria of phenotypes related to primary tauopathies based on Movement Disorders Society Progressive Supranuclear Palsy (MDS-PSP) criteria, (91), Armstrong corticobasal degeneration (CBD) (24) criteria, Gorno-Tempini Primary Progressive Aphasia (PPA) criteria (92), and Rascovsky behavioral variant Frontotemporal Dementia (bvFTD) criteria (93).
AD, Alzheimer's disease; ALS, amyotrophic lateral sclerosis; AOS, apraxia of speech; bvFTD, behavioral variant frontotemporal dementia; CBD, corticobasal degeneration; CBS, corticobasal syndrome; CSF, cerebrospinal fluid; CT, computed tomography; FTLD, frontotemporal lobar degeneration; FUS, fused in sarcoma; LBD, Lewy body disease; lvPPA, logopenic variant primary progressive aphasia; MRI, magnetic resonance imaging; MSA, multiple system atrophy; mSWJs, macro-square wave jerks; nfaPPA, non-fluent agrammatic primary progressive aphasia; PAGF, progressive akinesia and gait freezing; PET, positron emission tomography; PSP, progressive supranuclear palsy; PSP-F, frontal variant of progressive supranuclear palsy; PSP-SL, speech-language variant of progressive supranuclear palsy; RS, Richardson syndrome; s.o., suggestive of; SPECT, single photon emission computed tomography; svPPA, semantic variant primary progressive aphasia; SVS, slow vertical saccades; TDP-43, transactive response DNA binding protein 43 kDa; VSGP, vertical supranuclear gaze palsy.
Exclusionary criteria for the MDS-PSP criteria include clinical, imaging, laboratory, and genetic markers of any PSP-mimics or differential diagnoses including AD, PD, other atypical parkinsonian disorders, motor neuron disease, vascular or other structural brain lesions, autoimmune encephalitis, metabolic encephalopathies, prion disease, sensory deficit, vestibular dysfunction, severe spasticity, lower motor neuron syndrome, leukoencephalopathy, normal pressure or obstructive hydrocephalus, Wilson's disease, Niemann-Pick disease type C, hypoparathyroidism, Neuroacanthocytosis, Neurosyphilis, Whipple's disease, MAPT, and other genetic mutations mimicking PSP clinically.
Laboratory findings strongly suggestive of AD such as low CSF Aβ42 to tau ratio or positive 11C–Pittsburgh compound B PET; or genetic mutation suggesting AD (e.g., presenilin, amyloid precursor protein).
Classic 4-Hz Parkinson disease resting tremor, excellent and sustained levodopa response, or hallucinations.
Dysautonomia or prominent cerebellar signs.
Presence of both upper and lower motor neuron signs.
Exclusion criteria: Pattern of deficits is better accounted for by other non-degenerative nervous system or medical disorders/Behavioral disturbance is better accounted for by a psychiatric diagnosis/Biomarkers strongly indicative of Alzheimer's disease or other neurodegenerative process.
At least one of: Socially inappropriate behavior/Loss of manners or decorum/Impulsive, rash, or careless actions.
At least one of: Apathy/Inertia.
At least one of: Diminished response to other people's needs and feelings/Diminished social interest, interrelatedness or personal warmth.
At least one of: Simple repetitive movements/Complex, compulsive or ritualistic behaviors/Stereotypy of speech.
At least one of: Altered food preferences/Binge eating, increased consumption of alcohol or cigarettes/Oral exploration or consumption of inedible objects.
All of: Deficits in executive tasks/Relative sparing of episodic memory/Relative sparing of visuospatial skills.
Inclusion criteria: most prominent clinical feature is difficulty with language; these deficits are the principal cause of impaired daily living activities; aphasia should be the most prominent deficit at symptom onset and for the initial phases of the disease. Exclusion criteria: none of these criteria apply: pattern of deficits is better accounted for by other non-degenerative nervous system or medical disorders; cognitive disturbance is better accounted for by a psychiatric diagnosis; prominent initial episodic memory, visual memory, and visuoperceptual impairments; prominent, initial behavioral disturbance.
Differential Diagnosis
Corticobasal degeneration-Richardson syndrome (CBD-RS) presents with a similar clinical picture as described above and clinical distinction from PSP-RS is sometimes very challenging. Few studies compared clinical features of the two tauopathies. Behavioral abnormalities and urinary incontinence were significantly associated with CBD-RS compared to PSP-RS in one autopsy-confirmed study (104). Among early features, dysarthria was more frequent in PSP-RS while memory complaints were more common in patients with CBD-RS. It is suggested that the presence of CBS-specific clinical features (such as increased saccadic latency, cortical sensory loss, action myoclonus, limb or orobuccal apraxia, and limb dystonia or rigidity, especially in an asymmetric pattern) in a patient with RS is probably indicative of an underlying CBD pathology, but these features may occur relatively late (105, 106). Integration of biomarkers into current diagnostic criteria might be a necessary next step to improve the diagnostic accuracy. We will discuss recent advancements in the field of biomarkers in a later section.
Corticobasal Syndrome
In 1967, Rebeiz et al. (107) described three patients with progressive myoclonus, dystonia, gaze limitation, akinesia and monolimbic rigidity, speech disorder, and limb apraxia. Frontoparietal atrophy with special type of neuronal achromasia was evident in pathologic examination. It was initially called “corticodentatonigral degeneration with neuronal achromasia.” Since cerebellar features were not part of the clinical syndrome, its terminology changed to corticobasal ganglionic degeneration and later to CBD (108). Presently, CBD is considered a pathologic entity with hyperphosphorylated 4R-tau accumulation in astrocytes forming the astrocytic plaques which is the hallmark of pathology (109). A constellation of symptoms originally thought to be specific for CBD pathology is currently called corticobasal syndrome (CBS). Incidence of CBS is estimated to be 0.4 per 100,000 (49) which is less common than RS as an atypical parkinsonism. Mean age at disease onset is about 64 years. It seems to be a little more common in females and has a mean survival of about 6.5 years (24).
Clinical Characteristics
Corticobasal syndrome symptoms include orobuccal and/or limb apraxia, cortical sensory loss, alien limb phenomena, asymmetrical limb rigidity/akinesia, limb myoclonus, and limb dystonia. The first three symptoms are classified as cortical and the other three as motor symptoms.
Although parkinsonism is typically asymmetric (24), there are reports of symmetric parkinsonism (110). A minority of parkinsonian syndromes in CBS is levodopa responsive. Tremor is not common and is usually postural or action induced with a jerky myoclonic-like quality (24). About 40% of patients suffer from dystonia which is usually asymmetric and is more common in upper limbs. Lower limb dystonia, blepharospasm, and cervical dystonia is not common in CBS. Dystonia is usually associated with myoclonus and presents during the first 2 years of disease onset (109). Myoclonus is usually of the cortical reflex type (111) and has been proposed to be the result of lack of inhibitory input from the sensory cortex (112).
Apraxia is the clinical hallmark of CBS. Although it was believed that apraxia is also specific to CBS, it can be seen in other parkinsonian syndromes especially RS but is not as severe (113, 114). The alien-limb syndrome usually associates with apraxia, but these symptoms are unrelated (115). An alien limb is associated with a sense that the limb is foreign and acts on its own, which is not characteristic of apraxia (115). There are two alien-limb syndromes. In the sensory or posterior type, the patient believes that the limb is not their own and levitation may be present (116). In the anterior or motor type, the patient shows utilization-dependent limb activities which may result in inter-manual conflict (117).
Diagnostic Criteria
Based on the most recent clinical CBD criteria by Armstrong et al. (24), asymmetric presentation of a combination of at least two of these cortical and at least two of these motor symptoms leads to the diagnosis of probable CBD-CBS while the presence of one cortical plus one motor symptom suggests the diagnosis of possible CBD-CBS. It is important to note that exclusion criteria exist for both categories and include the evidence of biomarkers to exclude Alzheimer's disease [AD; such as amyloid positron emission tomography (PET) or tau and amyloid in cerebrospinal fluid (CSF)], structural lesion suggestive of focal causes, granulin mutation or reduced plasma progranulin levels, TDP-43 mutations, fused in sarcoma (FUS) mutations, evidence of Lewy body disease (LBD), multiple system atrophy (MSA), amyotrophic lateral sclerosis, or svPPA (24).
Differential Diagnosis
Although CBS is the most common clinical phenotype of CBD (37%), there are other clinical phenotypes that CBD can present with, including RS (23%), frontal behavioral-spatial (13%), amnestic syndrome of the hippocampal type (8%), nfaPPA (5%), and other rare presentations including PD-like or mixed phenotypes (Figure 1) (24). On the other hand, CBS phenotype may be the predominant clinical picture of various pathologies other than CBD in addition to PSP (PSP-CBS), including PiD (PiD-CBS), Alzheimer's disease pathology (AD-CBS), transactive response DNA binding protein 43 kDa (TDP-43) (TDP-CBS), and rarely LBD, GGT, vascular pathology, or even sporadic Creutzfeldt- Jacob disease (sCJD) (25, 32, 118–121).
There are few studies comparing CBD-CBS and PSP-CBS. In a small clinicopathologic study of 11 cases, eyelid opening apraxia, early falls, and predominant downgaze abnormalities were more frequent in patients with PSP-CBS while cortical sensory loss and limb clumsiness or alien limb were more common in CBD-CBS (106).
A number of studies compared AD-CBS to CBD-CBS (122–129) and found that in AD-CBS myoclonus, memory impairment, dressing apraxia, visuospatial neglect, and hallucinations were more common while in CBD-CBS dystonia, falls, tremor, and early frontal behavioral symptoms were more frequent and MMSE scores were higher at the disease onset. Boyd et al. (129), in an interesting pathology-confirmed study, found that the spatial subset of a visuospatial battery, more specifically the cube analysis test, was significantly impaired in patients with AD-CBS compared to CBD-CBS. This finding is consistent with the preferential involvement of the dorsal stream in parietal lobes by Alzheimer's disease (AD) pathology in AD-CBS cases.
TDP-43 proteinopathy Mackenzie type A [Sampathu type 3 (130)] may present with CBS as one of its clinical phenotypes which also include bvFTD with or without motor neuron disease (MND) and nfaPPA (26, 131, 132). In TDP-CBS, the related underlying genetic predisposition, if present, is one of the progranulin gene variants (133, 134). There are no reports comparing the clinical features of CBS with underlying TDP-43, LBD, or GGT to CBD-CBS. Vascular pathology and sCJD-CBS could be diagnosed based on the relevant MRI findings and a very rapid course of less than 12 months in case of sCJD (118, 120).
It is very challenging to determine the underlying pathology of CBS solely based on clinical grounds. However, if the Armstrong CBD exclusionary criteria that includes the use of biomarkers to exclude AD (using CSF or PET biomarkers), TDP-43, or FUS are applied, the set of diagnostic criteria will be very specific. Unfortunately, attempts to validate the diagnostic criteria of CBD-CBS failed to do so because none of the investigators used biomarkers as exclusionary criteria (135, 136). This again underlines the importance of integrating biomarkers to the clinical criteria.
Non-fluent/Agrammatic Primary Progressive Aphasia
The non-fluent agrammatic variant of primary progressive aphasia (nfaPPA) is very specific for an underlying tauopathy. CBD is the most common tau pathology (44%) followed by PSP (24%), and PiD (16%). Non-tauopathy nfaPPA is usually associated with TDP-43 type A or AD pathologies (Figure 1) (27). A minority of nfaPPA cases have a genetic etiology, commonly in association with either GRN or C9orf72 but only rarely with the MAPT gene (137–139). Incidence of nfaPPA is estimated about 0.9 per 100,000 per year based on a recent population-based Italian study (140). NfaPPA is usually accompanied with apraxia of speech, in more than half of the cases, although, the two syndromes may occur in isolation (141, 142). Based on the current clinical criteria, the two disorders are merged under the term nfaPPA. However, it is now understood that the two disorders are separate entities although they often co-occur.
Clinical Characteristics
Clinical hallmark of nfaPPA is agrammatism and effortful, distorted speech. Agrammatism is a a primary disorder of the language system and the term, primary agrammatic aphasia is used when it occurs as an isolated neurodegenerative process (143). Agrammatism is primarily a disorder of language and symptoms include moderate to severe decrease in the fluency or verbal output, a telegraphic speech with frequent loss of articles and conjunctions. As a part of agrammatism, understanding of syntactically complex sentences is particularly impaired. Early in the disease course, writing might be spared (144). Apraxia of speech is essentially a motor speech disorder in which motor programs necessary for the production of phonetically and prosodically normal speech are impaired (145). A pure, progressive, neurodegenerative apraxia of speech without involvement of the language system is then called primary progressive apraxia of speech. Symptoms include mispronunciation of words, speech sound distortion, visible or audible groping to find the suitable movements to articulate, false start, restart, omission, or addition of distorted sounds, all in an effortful and deliberately slow rate. Milder degrees of dysarthria may accompany both agrammatism and apraxia of speech (143, 146). Since agrammatism and apraxia of speech both affect speech output and effortful, nonfluent speech is a feature of both disorders, differentiation between agrammatism and apraxia of speech might be challenging. However, in contrast to agrammatism, speech sound production errors and sound distortion is seen in patients with apraxia of speech. In addition, syntax and comprehension of syntactically complex sentences is preserved in isolated forms.
Diagnostic Criteria
Either agrammatism or apraxia of speech could be the core criteria which defines probable nfaPPA in combination with at least two of these three symptoms: impaired comprehension of syntactically complex sentences, spared object knowledge, and spared single-word comprehension (Table 2) (92). The current PPA criteria have been challenged for being non-specific in differentiating between logopenic and nfaPPA variants. There are clinical suggestions for the improvement of its specificity and several new biomarkers can also be helpful to differentiate AD and logopenic variant from patients with a nfaPPA tauopathy (147).
Differential Diagnosis
Although by definition, speech/language features should be the most prominent deficit on symptom onset and in the first few years, other clinical symptoms usually accompany nfaPPA/apraxia of speech after 2–3 years of disease onset. Especially, those with an underlying PSP (PSP-SL) or CBD (CBD-nfaPPA) pathology will probably develop parkinsonism, gait disturbance, limb apraxia, dystonia, and sometimes vertical supranuclear gaze palsy/slow vertical saccades. Other features that might predict an underlying tauopathy, far more commonly a 4R tauopathy, include apraxia of speech as the first presentation, higher general cognitive performance, presence of behavioral impairment, and early occurrence of mixed/hypokinetic dysarthria (27, 148). Early motor speech disorder has specifically been linked to a PSP pathology (149). A pathology-based study showed that among patients fulfilling the criteria for PSP-SL, those with probable PSP-SL are more likely to have an underlying PSP pathology, while those with suggestive-of-PSP-SL are more consistently associated with a CBD pathology (101). In a recent study, survival was about 6 years for patients presenting with primary progressive apraxia of speech, around 5 years for agrammatic aphasia, and about 4 years for combined apraxia of speech and agrammatism (150).
Frontal Behavioral/Dysexecutive Syndrome
About 30% of patients presenting with bvFTD suffer from some form of tauopathy, almost equally distributed between PiD, CBD, and PSP. More than 50% are due to TDP-43 proteinopathies and about 13% have an AD pathology (Figure 1) (35). The typical clinical picture of bvFTD, which is dominated by PiD, TDP-43, and FUS pathologies, includes disinhibition, apathy, loss of sympathy/empathy, perseveration or compulsive/ritualistic behavior, hyperorality, and executive dysfunction with relatively spared episodic memory and visuospatial domain (93). The presence of at least 3 of the above 6 features in addition to frontal/anterior temporal atrophy or hypometabolism on brain imaging in a setting of progressive functional decline defines probable bvFTD (93).
In a large pathology-based study (35), language dysfunction, depression, and myoclonus were more common in patients presenting with a frontotemporal syndrome with an underlying AD compared to all other pathologies while falls, dysphagia, dysarthria, mental rigidity, aggression, apathy, and lack of insight, were against an AD pathology. Recently, the term behavioral variant AD (bvAD) was introduced by Ossenkoppele et al. (151) in their systematic review and meta-analysis of bvAD vs. bvFTD. Ossenkoppele et al. found that behavioral profile is generally milder in bvAD; the patients have fewer compulsive behaviors and less hyperorality but more agitation, delusions, and hallucinations, and more severe memory and/or executive dysfunction compared to bvFTD. These findings were incorporated in the research criteria for bvAD (151). Fronto-behavioral AD, frontal variant AD, and frontal/dysexecutive AD are other terms that have been used to describe the population of patients with a frontal-dominant dementia syndrome and an underlying AD pathology.
It has been suggested that accompanying symptoms of MND or semantic dementia are indicative of TDP-43 pathology, type B and C, respectively (26, 36). Moreover, executive dysfunction occurred earlier and was more severe in fronto-behavioral AD cases (35, 152). Features against an underlying tauopathy include earlier age of onset, delusions, visual misperceptions, and signs of MND. In contrast, binge eating was in favor of bvFTD tauopathy (35, 153). Loss of empathy and compulsive behavior were less common in frontal variant PSP (PSP-F) or CBD-frontal behavioral-spatial syndrome (CBD-FBS) compared to PiD or bvFTD with non-tauopathies but dysarthria and dysphagia were more frequent in PSP-F (35). In another study, patients with fronto-behavioral AD were less likely to have personality changes but frequently showed memory impairment. Personality change was significantly higher in tauopathies presenting with bvFTD compared to non-tauopathies (154). The MDS-PSP criteria for PSP-F requires at least three of the following symptoms: apathy, bradyphrenia, dysexecutive syndrome, reduced phonemic verbal fluency and impulsivity, disinhibition, or perseveration, in addition to vertical supranuclear gaze palsy/slow vertical saccades (91). However, vertical supranuclear gaze palsy/slow vertical saccades occurs in <20% of patients during the first 4 years of symptom onset which increases diagnostic latency in cortical variants of PSP (21, 155). Misdiagnosis as psychiatric disorders is another common source of diagnostic delay in patients presenting with bvFTD syndrome (156).
Globular glial tauopathy presents with bvFTD syndrome in 39 and 27% of type I and III cases, respectively (34). Most common genetic causes of bvFTD syndrome include C9orf72 repeat expansions and GRN and MAPT mutations. MAPT mutations cause tauopathies that present in about 45% of cases with a bvFTD syndrome followed by unspecified dementia (34.6%), parkinsonian syndrome (4.9%), RS (4.2%), amnestic syndrome (3%), CBS (1.8%), nfaPPA (1.8%), and svPPA (1.8%) phenotypes (157). GRN mutations and C9orf72 repeat expansions present with the similar range of clinical syndromes; however, PPA syndromes are more common with GRN mutations and motor neuron disease syndromes are more common with C9orf72 repeat expansions (157). Pro301Leu MAPT mutation is the most common cause of genetic tauopathy followed by IVS10 + 16C>T, Arg406Trp, and Asn279Lys. Parkinsonian symptoms, sometimes mimicking RS and/or CBS, are common later in the course of MAPT mutations presenting with bvFTD or other syndromes (158). Although penetrance is almost 100%, age of onset is highly variable ranging from the first decade to 82 years (157) and there is both inter- and intra-familial variability in clinical presentation and symptoms.
Progressive Akinesia and Gait Freezing (PAGF)
Progressive supranuclear palsy-progressive akinesia and gait freezing (PSP-PAGF) is the only tauopathy presenting with this clinical syndrome. It was first reported in 1980 from Japan with subsequent pathologic confirmation of underlying PSP (159, 160). The clinical picture is dominated by the freezing of gait with frequent start hesitation and blocking, but freezing could also involve other movements, such as speech and writing (161). Axial rigidity might be present; however, typical parkinsonian features, such as limb rigidity or bradykinesia and tremor are lacking, and symptoms do not respond to levodopa. Frequent falls follow severe gait freezing. Speech becomes hypophonic and writing is rapid micrographic. Hypomimia is also a frequently reported symptom (161–163). Typical PSP features, such as impaired postural reflexes, vertical supranuclear gaze palsy/slow vertical saccades, eyelid opening apraxia, dysarthria, dysphagia, apathy, and cognitive frontal impairment are delayed by at least 5 years and usually 8–10 years (161, 162, 164). Some patients may never develop vertical supranuclear gaze palsy/slow vertical saccades or dementia during life (163, 165). Other etiologies, such as vascular white matter disease (166) and carbon monoxide poisoning (167) have been ascribed to PAGF syndrome. However, their courses are different from neurodegenerative diseases and other clinical features, especially more apparent parkinsonism and pyramidal signs, differentiate these disorders from PSP-PAGF. A case of PAGF with subsequent progression to a corticobasal syndrome has also been reported but pathologic diagnosis was lacking in this report (168).
Clinical Syndromes With Less Association to a Primary Tauopathy
Parkinsonian Syndrome
Progressive supranuclear palsy-Parkinsonism (PSP-P) is the second most common PSP phenotype, accounting for about 37% of PSP cases (20, 22). PD is the most common alternative diagnosis when a patient with PSP first approaches a neurologist (20). PSP-P is the most common tauopathy that closely resembles PD, presenting with asymmetric akineto-rigid parkinsonism, rest tremor, and relatively good levodopa response (169). Williams et al. (22) first introduced this phenotype. The mean age of the onset of PSP-P was similar to PSP-RS in Williams' series. However, other studies reported higher age of onset of about 68 years (21). This phenotype is considered as one of the brainstem restricted forms of PSP with survival of about 9–13 years (22, 169, 170), longer than RS but shorter than PD, so it is crucial to differentiate the three disorders. It is important to emphasize that even at the same baseline disease severity, patients with PSP-P progress at a slower pace than patients with RS (171). It is, however, difficult to diagnose PSP-P early in the disease course and diagnosis is often delayed by up to 10 years of symptom onset because characteristic PSP features, vertical supranuclear gaze palsy, and postural instability, occur relatively late in patients with PSP-P (21).
Postural instability usually does not occur in the first 2–3 years of symptom onset in PSP-P and vertical supranuclear gaze palsy/slow vertical saccades has a latency of about 5 years in these patients while parkinsonism occurs in the first year (99, 169). In about 40–80% of patients, parkinsonism starts asymmetrically, 50% of patients present with tremor, and about 50–70% of patients have moderate to good response to levodopa early in the disease course (169, 172). While some studies found no difference in apathy or depression between PSP-P and RS (76, 173), apathy was more frequent in PSP-P in one study (75) and in PSP-R in another (174), but further studies are needed. Early cognitive decline is more common in RS than PSP-P (76, 172) and generally, neuropsychiatric and cognitive symptoms are more severe in both PSP variants compared to PD (76). On the other hand, PD-specific features, such as autonomic dysfunction and levodopa-induced dyskinesia are not usually seen in patients with PSP-P (169).
Late Onset Cerebellar Ataxia Syndrome (LOCA)
A cerebellar ataxic syndrome is very unspecific for tau pathology since a lot of genetic, autoimmune, degenerative, and many other structural disorders are present with this syndrome. PSP-Cerebellar (PSP-C) is the only rare tauopathy that presents with a cerebellar syndrome. It was first proposed as a PSP phenotype when retrospective evaluation of a Japanese autopsy series with pathological diagnosis of PSP revealed that some cases had a primary and predominant syndrome of cerebellar ataxia before the presence of PSP-specific features. There was abundant tau pathology involving Purkinje cells and more severe tau pathology in dentate nucleus (175). Prospective autopsy-confirmed cases were reported thereafter (176, 177). Some of these patients had been diagnosed with a form of spinocerebellar ataxia. The closest neurodegenerative differential diagnosis is multiple system atrophy-cerebellar (MSA-C) which could be differentiated because their relatively younger age of onset, early onset of more severe autonomic disturbances, lower rates of falls, and vertical supranuclear gaze palsy/slow vertical saccades, present hot-cross-bun sign on brain MRI (178–180). In recent reviews of 16 PSP-C cases, of which 12 were pathology-proven, the mean age of onset was 65 (range 57–73) years, mean disease duration was about 6 years, and symptoms were mostly restricted to a cerebellar ataxia syndrome for about 2 years (181). Interestingly, some patients never developed vertical supranuclear gaze palsy.
Primary Lateral Sclerosis
A few patients presented have been reported with pyramidal symptoms including spastic gait and speech indicative of primary lateral sclerosis (PLS) but the pathological diagnosis was PSP in the absence of any PLS-related pathologies, such as TDP-43 or FUS proteinopathies (182, 183). These findings suggest that PSP-PLS might be a rare PSP phenotype.
Prior to these reports, a retrospective study of 12 autopsied cases with prominent upper motor neuron as well as extrapyramidal symptoms revealed atypical PSP-like pathological changes, with Gallyas-negative tufted astrocytes and minimal involvement of subcortical nuclei typically involved with PSP, but with significant degeneration of the corticospinal tract due to 4R tau pathology (184). Subsequent studies led to further characterization of the distinct pathological changes and identified them as a new pathological entity named globular glial tauopathy (GGT) (7, 185). The GGT has been divided pathologically into three subtypes: type I, involving frontotemporal areas and causing frontotemporal dementia (FTD); type II, which involves motor cortex and the descending corticospinal tracts causing MND; and type III that is a combination of the first two (FTD-MND). In all types, extrapyramidal symptoms might be a part of the clinical picture (7). Thus, PLS syndrome is associated with GGT types II or III and mostly in combination with other symptoms and sometimes other pathologies (34, 42). Recent cryo-EM studies suggest that GGT type III might be a distinct pathological entity since its tau filament structure does not twist, unlike the other types (16).
Other Primary Progressive Aphasias
Semantic variant PPA (svPPA) is a clinical syndrome tightly associated with an underlying TDP-43 (mostly type C) pathology. However, <15% of patients may have an underlying tauopathy of PiD or GGT type and about similar proportion are due to AD pathology (27, 186–188). Bilateral involvement of the anteromedial and inferior temporal areas more severely on the dominant side could be appreciated on the structural or functional neuroimaging of svPPA (189). Based on the most current clinical criteria, the core clinical features required for the diagnosis of svPPA include deficits in both confrontation naming and single word comprehension as a result of progressive defect in semantic memory (92). Motor aspects of speech, fluency, and grammar are characteristically spared, and patient might even be verbose; however, the speech content lacks meaning. More generic category names might be used instead of specific names and filler words are used more frequently. Surface dyslexia and dysgraphia might also be present. Behavioral features are not uncommon in svPPA; however, they are more commonly encountered in the right-sided semantic dementia. Extrapyramidal motor features when present in the course of svPPA are consistent with an underlying tau rather than TDP pathology (27) while a temporoparietal pattern of brain atrophy on imaging are probably indicative of underlying AD (190).
Logopenic variant PPA (lvPPA) is believed to be associated with an underlying AD pathology in most of the cases (27, 191, 192). Non-AD pathologies presenting with lvPPA include TDP-43 type A, LBD, CJD, and rarely tauopathies, such as CBD, PSP, and PiD (141, 188). Core clinical features of lvPPA include impairment in single-word retrieval and repetition of sentences and phrases which are probably due to a malfunctioning phonological loop of verbal working memory (192). Recently, Mesulam et al. (147) argued that the current clinical criteria for lvPPA are unspecific and allow for misclassification of other variants, especially nfaPPA, into logopenic category. Considering that the rate of amyloid beta biomarker positivity is the highest in lvPPA compared to other PPAs; evaluation of these biomarkers might be useful in differentiating the two variants (188).
Amnestic Syndrome of the Hippocampal Type
Alzheimer's disease is the most common secondary tauopathy. As a mixed or pure pathology, it is the major cause of an amnestic syndrome in about 56–90% of cases (28–30). Other pathologies that may present with amnestic syndrome include limbic-predominant age-related TDP-43 encephalopathy (LATE), non-AD tauopathies, LBD, vascular, CJD, and mixed pathologies (29). Non-AD tauopathy categories presenting with amnestic syndrome include AGD, primary age-related tauopathy (PART), CBD, GGT, and PiD.
Argyrophilic grain disease, a 4R tauopathy first introduced in 1987 (10), has been associated with many psychiatric symptoms including emotional instability, delusions, hallucinations, depression, anxiety, and personality changes (193, 194). Although an Alzheimer-like dementia seems to be the dominant clinical presentation of this age-related tauopathy, in a large pathological study, about 59% of the cases with an AGD pathology had no cognitive impairment (38). AGD has a prevalence of 5–15% in community-based autopsy studies and usually co-occurs with other neurodegenerative disorders, especially AD and other age-related pathologies including PART and hippocampal sclerosis (38, 195). Therefore, it is challenging to attribute cognitive decline to AGD alone but preferential involvement of the limbic and hippocampal structures in AGD supports its contribution to cognitive decline (195, 196).
Primary age-related tauopathy is a mixed 3R/4R tauopathy commonly found in the brains of cognitively normal individuals in the form of neurofibrillary tangles restricted to the medial temporal lobes and structures typically involved in AD but with only minimal neocortical involvement. Amyloid beta plaques are either absent (definite PART) or mild (possible PART) (15). When symptomatic, PART usually presents with a slowly progressive cognitive decline which starts at a higher age and progresses at a slower pace compared to AD, and when associated with AD pathology, hastens cognitive decline (39, 40).
Pick's disease constitutes the postmortem diagnosis of a minority of amnestic syndrome cases in autopsy series, ranging from 4 to 10% (28, 31). Early occurrence of frontal behavioral symptoms have been reported in these patients as a distinguishing clinical feature (197).
Compared to typical AD, CBD-amnestic patients are younger and several features including hyperreflexia, gait abnormalities, parkinsonism, dystonia, and asymmetric motor/sensory symptoms are significantly more frequent in these patients early in the disease course. Falls, urinary incontinence, and ocular movement abnormalities are distinguishing clinical features in later stages (198).
Memory impairment is the predominant clinical syndrome in <10% of patients with GGT (34). AD-mimicking presentation has rarely been reported in association with this tauopathy (199).
Asymptomatic Tauopathies
Aging-related tau astrogliopathy (ARTAG) is an essentially asymptomatic 4R tauopathy that exclusively involves astrocytes in various cortical and subcortical brain areas in individuals older than 60 years (11). ARTAG is frequently encountered as a single pathology in individuals with no neurologic symptoms or as a co-pathology along with many neurodegenerative disorders (11). In a large community-based autopsy series, 38% of brains over 75 years of age contained cortical ARTAG (200). Although, parkinsonism, aphasia, or cognitive decline have been reported in association with this tauopathy, clinical significance of ARTAG is yet to be determined (201, 202).
Incidental finding of mild sub-threshold stages of many tauopathies have been reported in autopsy series. Incidental PSP has been reported in 2.5–6.9% of community-based autopsy series (203–205) and the rate of incidental CBD was about 0.23% in a Japanese forensic autopsy series (206). AGD and PART, as stated above, usually present as asymptomatic tauopathies in autopsy studies.
Biomarkers to Differentiate Tauopathies Sharing a Clinical Phenotype
Accurate in vivo diagnosis of the underlying pathology is yet an unsolved issue facing all therapeutic clinical trials of tauopathies. Clinical diagnostic criteria are currently used both in clinical and research settings as the basis for diagnosing various tauopathies from one another and from non-tauopathy disorders. However, clinical heterogeneity and huge phenotypic overlap of different tauopathies, as discussed above, necessitates the development and inclusion of biomarkers into the clinical diagnostic criteria of tauopathies in the near future to improve their diagnostic accuracy, as previously tested in diagnostic criteria of AD (207, 208).
In the field of diagnosis, biomarkers could be helpful in (1) the early diagnosis of the clinical phenotype when the clinical picture of a tauopathy is not yet complete to fit in a specific clinical criteria; (2) confirming a syndromic diagnosis when clinical findings are conflicting or a patient fulfills the criteria for multiple phenotypes; (3) molecular diagnosis of the underlying etiology/proteinopathy; and (4) as exclusionary criteria.
Structural MRI measures can supplement clinical findings to confirm a given phenotypic diagnosis or possibly can be used for the early detection of clinical syndromes. However, considering the scarcity of evidence from pathology-confirmed studies, not only the correlation of specific conventional or quantitative MRI measures with specific pathologic diagnoses is not yet known, but structural imaging cannot differentiate the underlying pathology. PET and fluid biomarkers are rapidly evolving to address this purpose. On the other hand, frequent presence of associated pathologies is a major hinder that prevents molecular diagnoses to rest on one biomarker alone. Therefore, a combination of various biomarkers might be necessary for more accurate diagnosis.
Structural MRI
Midbrain and superior cerebellar peduncle atrophy and their derivatives, such as magnetic resonance parkinsonian index (MRPI) and MRPI 2.0, are hallmark MRI findings of RS which can distinguish this clinical syndrome from healthy controls, other atypical parkinsonian syndromes, and PD (209–211). However, these indices cannot differentiate between RS due to various pathologies (e.g., PSP-RS from CBD-RS) (212).
In a longitudinal study, all patients with a clinical diagnosis of PSP-P and an abnormal MRPI developed vertical supranuclear gaze palsy during 4 years of follow up (213). In another study, 73% of patients with undifferentiated parkinsonism who had abnormal MRPI fulfilled possible or probable PSP criteria up to 4 years after MRI acquisition (214). A recent study evaluated brainstem atrophy measures including midbrain area, midbrain/pons ratio, and MRPI across eight PSP variants who were clinically diagnosed using the MDS-PSP criteria. Results showed no difference between brainstem and cortical variants, but of note, possible PSP-SL patients had less midbrain atrophy while PSP-CBS and PSP-F patients had highly abnormal indices. Among 26% of the cases in this study who underwent an autopsy, one third had non-PSP pathology dominated by SL phenotype (215). These studies, although mostly lacking pathologic confirmation, show that midbrain/superior cerebellar peduncle atrophy and MRPI might be useful in the early diagnosis of RS or other probable/possible variants by improving the sensitivity of the clinical criteria in the first visit.
In a conventional MRI prominent atrophy, peri-rolandic cortices in an asymmetric pattern was thought to be specific to CBS (216, 217), although this is again not specific to any underlying pathology (218, 219). Voxel-based morphometry (VBM) studies confirmed these findings and showed atrophy involving primary and supplementary motor and parietal, especially superior parietal lobule, cortical areas prominently in the side contralateral to the symptomatic limb; these changes are probably helpful in differentiating CBS from other parkinsonian phenotypes especially PD, MSA-Parkinsonism, and RS (220–223). In a comparative VBM MRI study of CBS due to four common pathologies of CBD, PSP, AD, and TDP-43, Whitwell et al. (224) found that more widespread cortical atrophy is indicative of an underlying AD or TDP-43 while more focal atrophy of premotor and supplemental motor area is seen with CBD and PSP .
In nfaPPA, due to tauopathies, MRI atrophy was found on the left posterior frontal cortex especially the opercular part of the inferior frontal gyrus, premotor and supplementary motor areas, and striatum along with severe frontal white matter atrophy, while in nfaPPA, due to TDP-43, white matter was spared (148). More specifically, the proportion of gray matter to white matter atrophy was found to be higher in CBD-nfaPPA compared to PSP-SL which showed more severe contraction of the white matter running between the striatum, premotor, and prefrontal areas (149). More focal involvement of the motor, premotor, and supplementary motor areas have been reported in the primary progressive apraxia of speech (225).
Patients with bvFTD irrespective of the underlying pathology share atrophy in the anterior cingulate, fronto-insula, striatum, and amygdala areas (35). PiD-bvFTD shows more widespread atrophy of the right frontal and anterior temporal lobes while CBD and PSP had less anterior temporal atrophy (35). Marked atrophy of bilateral temporoparietal regions with minimal frontal atrophy is indicative of an underlying AD (152). Parietal atrophy is also more severe in TDP-43 pathology compared to PiD or CBD (226), and the accompanying midbrain atrophy is indicative of an underlying PSP (227).
A recent study evaluated the pattern of covarying brain atrophy and clinical features across a mixed sample of clinically diagnosed patients including RS, CBS, bvFTD, and PPA variants. It showed that these syndromes exist as a continuous spectrum of structural imaging and clinical features, in the three dimensions of behavior, movement, and language, rather than discrete entities. Overlap is common and increases with time (228).
Positron Emission Tomography
Tau PET tracers are viewed as the future of in vivo pathological diagnosis of tauopathies. Flortaucipir ([18F]AV- 1451), was the first of its kind to be approved by the FDA on May 2020 for in vivo diagnosis of tau pathology in AD (229). Despite having high sensitivity and specificity for higher stages of AD pathology (230), flortaucipir showed much lower, but still significant, affinity to non-AD tau fibrils and, in this regard, significant off-target binding that is seen with this tracer (231) is a greater challenge in non-AD tau imaging. Although flortaucipir binds with reduced affinity to pathologic tau in non-AD tauopathies, such as PSP and CBD (232–234), it is not specific to a single type of tau fibril (235). Nevertheless, tracer uptake in probable CBD-CBS was distinct compared to AD-CBS which showed more diffuse uptake especially in the temporal and parietal areas (234, 236). These findings can be used in the differentiation of the underlying pathology in CBS. In a more recent study on patients with PSP of various clinical phenotypes, Whitwell et al. (19) found that flortaucipir uptake was correlated with areas of expected involvement and MRI volume loss and in some autopsied cases with postmortem tau burden. In this study, putamen and globus pallidus uptake was the highest with PAGF and parkinsonism variants, and supplementary motor and precentral cortices with speech/language variant.
An earlier first generation tracer, [11C]PBB3 and its derivatives, [18F]PBB3 and [18F]-PM-PBB3 ([18F]APN-1607), are also among tracers with acceptable non-AD tau binding affinity with some off-target binding in the basal ganglia, thalamus, and choroid plexus but not to monoamine oxidase (MAO) (237). In a recent study comparing clinically diagnosed patients with PSP to healthy controls, Endo et al. (238) found that [11C]PBB3 binding to frontoparietal white matter is correlated with disease severity in patients with PSP. Still another study showed differential [11C]PBB3 binding, reflecting areas typically involved with tau pathology, in clinically diagnosed PSP, CBS, and AD cases (239). However, significant [11C]PBB3 binding to alpha-synuclein and TDP-43 has been reported in other studies (240, 241).
The newer [18F] APN-1607 was recently tested in patients clinically diagnosed with PSP and MSA and the results showed that tracer uptake in the brainstem structures including the subthalamic nucleus, midbrain, substantia nigra, red nucleus, pontine base, and raphe nuclei was associated with disease severity in PSP. Moreover, the basal ganglia and the midbrain uptake could differentiate PSP from MSA (242). Trials are ongoing for the evaluation of [18F] APN-1607 in a larger sample of various parkinsonian disorders including PSP and CBD (NCT04557865, ClinicalTrials.gov).
Many second generation tau tracers have improved the affinity and reduced the off-target binding but remained mainly AD-specific (237). More recently, [18F]-PI-2620 was introduced as a new second generation tau tracer with better affinity to PSP, CBD, and PiD but still with relatively much higher affinity for AD (243). Clinical studies, however, were not able to find a consistent binding to non-AD tau. Moreover, there was no postmortem correlation to tau pathology in a CBD case (244). However, in another study using a dynamic technique, [18F]-PI-2620 was able to differentiate PSP-RS from healthy controls, PD, MSA, and non-RS PSP (245) and in a follow-up study, an improved imaging technique was proposed (246). Palleis et al. (247) compared patients with CBS having underlying AD, based on amyloid-PET imaging, and non-AD tau and suggested that higher dorsolateral prefrontal cortical binding of this tracer may differentiate AD-CBS from tau-CBS. None of the first or second-generation tau tracers is able to differentiate between various 3R or 4R tauopathies.
Cryo-EM ultrastructural evaluation of tau fibrils studies show that among tauopathies, CBD, AGD, and ARTAG fibrils have more structural resemblance (four-layered fold), while PSP fibrils are similar to those of GGT (three-layered fold), and PART and AD fibrils are identical (16). This suggests that different tau tracers might be needed for the diagnosis of CBD and PSP. A new 4R-specific tau tracer, [18F]CBD-2115, has been developed by Lindberg et al. on 2021 which showed higher affinity for PSP-specific 4R tau compared to flortaucipir in preclinical evaluations (248). Clinical studies are yet to be done.
Cerebrospinal Fluid and Serum Biomarkers
Cerebrospinal fluid levels of total tau, phosphorylated tau 181 (p-tau181), and amyloid beta 1-42 (Aβ42) have been studied for more than 30 years for the diagnosis of AD pathology (249), especially as an early diagnostic tool and eventually incorporated into the diagnostic criteria of AD in 2018 (207). Wagshal et al. (250) showed that CSF p-tau181 and total tau are decreased in RS (250) in sharp contrast to what occurs in AD. In another study, Lleo et al. (251) showed that FTLD-tau generally have lower total and p-tau levels compared to AD, but patients with sporadic FTLD-TDP had even lower p-tau181 compared to PSP for which they could be used as a diagnostic marker. This was confirmed by a recent study using pathologically-confirmed cases and automated laboratory techniques (252). These investigators found in this relatively large sample of AD and FTLD cases including tauopathies and TDP-43opathies that CSF Aβ42/Aβ40 or p-tau/Aβ42 ratios diagnose not only primary AD but also AD co-pathology in PSP and FTLD-tau. The investigators also found decreased p-tau/total tau and increased CSF neurofilament light chain (NfL) in FTLD-TDP compared to FTLD-tau, in line with prior studies (253), and that the FTLD (PSP) had low CSF total tau.
Thijssen et al. (254, 255) found that serum p-tau217 outperformed serum p-tau181 in differentiating AD from FTLD and healthy controls, while both markers were correlated with their CSF level, Aβ PET results, and underlying AD pathology. Previous clinical studies demonstrated that CSF total tau is increased in CBS compared to PSP or other atypical parkinsonism (256, 257); however, this could be due to underlying AD or at least co-pathology.
Considering that post-translational modifications of tau protein are different in PSP and AD (258, 259) and other tauopathies, more studies are needed to characterize PSP- and CBD-specific tau strains in CSF and specific assays are then necessary to detect these strains.
In PSP, serum NfL as a non-specific biomarker of neuronal injury is associated with disease severity and survival (260, 261) and high CSF NfL and p-tau181 predict prognosis (262). Moreover, high NfL levels in CSF or serum can differentiate atypical parkinsonian disorders from PD (263, 264). MicroRNAs and proteomic studies are currently being investigated in search of pathology-specific CSF or serum candidate markers to differentiate tauopathies (265, 266).
Tau real-time quaking induced conversion (RT-QuIC) assays are probably the most sensitive and specific single laboratory techniques for the determination of underlying pathology in every tauopathy-related clinical syndrome. Tau RT-QuIC assays for the detection of 3R and mixed 3R/4R seeds of PiD, AD, and chronic traumatic encephalopathy have already been developed (267). Recently, the range of tau RT-QuIC assays was extended to detect 4R seeds of PSP and CBD (268).
Conclusion
Major advances in molecular biology, cryo-EM, and detailed clinical and pathologic characterization have allowed to disentangle various primary tauopathies, as well as their association with an heterogenous spectrum of phenotypic clinical presentations. Classifying the tauopathies according to the similarities of their strains using cryo-EM further supports the notion that PSP and CBD are different disorders. While this structural classification may allow to identify more specific biomarkers, it is unclear if it may allow to better understand their etiopathogenesis.
The typical phenotypic clinical presentations (RS, CBS, nfaPPA, fronto-behavioral/dysexecutive syndrome, and PAGF) lead us to suspect specific underlying pathologies, but it is still challenging to clinically differentiate PSP and CBD accurately due to their phenotypic overlap. Moreover, at present, only the RS presentation accurately predicts an underlying 4R tau pathology, usually PSP. On the other hand, it would also be difficult to differentiate CBD-CBS vs. AD-CBS without using specific AD biomarkers. However, the inclusion of exclusionary biomarkers required by the CBD clinical diagnostic criteria allows their distinction, which is critical for subjects' participation in therapeutic trials to slow disease progression and into future effective therapeutic interventions. This approach could improve the accuracy of the MDS-PSP criteria to diagnose PSP-CBS.
The discussed promising CSF biomarkers (Aβ42, Aβ42/Aβ40, total tau, p-tau 217, p-tau/Aβ42, NfL, RT- QuIC 3R, and 4R tau) and others (e.g., alpha-synuclein and future second generation tau PET ligands) will need to be validated in larger samples of pathologically confirmed cases. It would be relevant that these cases would have been prospectively and longitudinally evaluated in a cohort of subjects presenting with heterogenous phenotypes reflecting a variety of suspected underlying pathologies using detailed clinical, biofluid and imaging protocols. While we mostly discussed the use of biofluids and tau PET in view of the available literature, the role of cutaneous biopsies and other approaches will also need to be further investigated.
It remains to be understood how early these biomarkers could be identified to accurately diagnose these disorders. Of similar importance is to decrease the cost of assays and imaging to allow their use to diagnose subjects around the world. Improving the accuracy of prodromal and early clinical diagnostic criteria by inclusion of more sensitive and specific biomarkers, will allow to determine more efficiently which are the best therapeutic approaches to stop the progression of these disorders.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Statements
Author contributions
NO and AS prepared the first draft of the manuscript. IL wrote sections of the manuscript and provided review and critique to the content. All authors contributed to the article and approved the submitted version.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest. IL is the Field Editor of Frontiers in Neurology however was not involved in the review of this manuscript.
References
1.
Weingarten MD Lockwood AH Hwo SY Kirschner MW . A Protein Factor essential for microtubule assembly. Proc Natl Acad Sci U S A. (1975) 72:1858–62. 10.1073/pnas.72.5.1858
2.
The Human Protein Atlas (2021) . Available online at: https://www.proteinatlas.org/ENSG00000186868-MAPT. [cited 2021 Nov 29].
3.
Goedert M Spillantini MG Jakes R Rutherford D Crowther RA . Multiple isoforms of human microtubule-associated protein tau: sequences and localization in neurofibrillary tangles of Alzheimer's disease. Neuron. (1989) 3:519–26. 10.1016/0896-6273(89)90210-9
4.
Hauw JJ Daniel SE Dickson D Horoupian DS Jellinger K Lantos PL et al . Preliminary ninds neuropathologic criteria for Steele-Richardson-Olszewski Syndrome (Progressive Supranuclear Palsy). Neurology. (1994) 44:2015–9. 10.1212/WNL.44.11.2015
5.
Yoshida M . Astrocytic inclusions in progressive supranuclear palsy and corticobasal degeneration. Neuropathology. (2014) 34:555–70. 10.1111/neup.12143
6.
Dickson DW Bergeron C Chin SS Duyckaerts C Horoupian D Ikeda K et al . office of rare diseases neuropathologic criteria for corticobasal degeneration. J Neuropathol Exp Neurol. (2002) 61:935–46. 10.1093/jnen/61.11.935
7.
Ahmed Z Bigio EH Budka H Dickson DW Ferrer I Ghetti B et al . Globular Glial Tauopathies (Ggt): consensus recommendations. Acta Neuropathol. (2013) 126:537–44. 10.1007/s00401-013-1171-0
8.
Tanaka H Toyoshima Y Kawakatsu S Kobayashi R Yokota O Terada S et al . Morphological characterisation of glial and neuronal tau pathology in globular glial tauopathy (Types II and III). Neuropathol Appl Neurobiol. (2020) 46:344–58. 10.1111/nan.12581
9.
Saito Y Ruberu NN Sawabe M Arai T Tanaka N Kakuta Y et al . Staging of argyrophilic grains: an age-associated tauopathy. J Neuropathol Exp Neurol. (2004) 63:911–8. 10.1093/jnen/63.9.911
10.
Braak H Braak E . Argyrophilic grains: characteristic pathology of cerebral cortex in cases of adult onset dementia without alzheimer changes. Neurosci Lett. (1987) 76:124–7. 10.1016/0304-3940(87)90204-7
11.
Kovacs GG Ferrer I Grinberg LT Alafuzoff I Attems J Budka H et al . Aging-Related Tau Astrogliopathy (Artag): harmonized evaluation strategy. Acta Neuropathol. (2016) 131:87–102. 10.1007/s00401-015-1509-x
12.
Das S . Aging-related tau astrogliopathy: a brief review. Clin Neuropathol. (2020) 39:245–9. 10.5414/NP301264
13.
Probst A Tolnay M Langui D Goedert M Spillantini MG . Pick's disease: Hyperphosphorylated tau protein segregates to the somatoaxonal compartment. Acta Neuropathol. (1996) 92:588–96. 10.1007/s004010050565
14.
Dickson DW . Neuropathology of Pick's disease. Neurology. (2001) 56:S16–20. 10.1212/WNL.56.suppl_4.S16
15.
Crary JF Trojanowski JQ Schneider JA Abisambra JF Abner EL Alafuzoff I et al . Primary age-related tauopathy (Part): A common pathology associated with human aging. Acta Neuropathol. (2014) 128:755–66. 10.1007/s00401-014-1349-0
16.
Shi Y Zhang W Yang Y Murzin AG Falcon B Kotecha A et al . Structure-based classification of tauopathies. Nature. (2021) 598:359–63. 10.1038/s41586-021-03911-7
17.
Gao YL Wang N Sun FR Cao XP Zhang W Yu JT . Tau in neurodegenerative disease. Ann Transl Med. (2018) 6:175. 10.21037/atm.2018.04.23
18.
Do Carmo S Spillantini MG Cuello AC . Editorial: tau pathology in neurological disorders. Front Neurol. (2021) 12:754669. 10.3389/fneur.2021.754669
19.
Whitwell JL Tosakulwong N Botha H Ali F Clark HM Duffy JR et al . Brain volume and flortaucipir analysis of progressive supranuclear palsy clinical variants. Neuroimage Clin. (2020) 25:102152. 10.1016/j.nicl.2019.102152
20.
Morgan JC Ye X Mellor JA Golden KJ Zamudio J Chiodo LA et al . Disease course and treatment patterns in progressive supranuclear Palsy: a real-world study. J Neurol Sci. (2021) 421:117293. 10.1016/j.jns.2020.117293
21.
Respondek G Stamelou M Kurz C Ferguson LW Rajput A Chiu WZ et al . The phenotypic spectrum of progressive supranuclear palsy: a retrospective multicenter study of 100 definite cases. Mov Disord. (2014) 29:1758–66. 10.1002/mds.26054
22.
Williams DR de Silva R Paviour DC Pittman A Watt HC Kilford L et al . Characteristics of two distinct clinical phenotypes in pathologically proven progressive supranuclear palsy: Richardson's Syndrome and Psp-Parkinsonism. Brain. (2005) 128(Pt 6):1247–58. 10.1093/brain/awh488
23.
Respondek G Grimm MJ Piot I Arzberger T Compta Y Englund E et al . Validation of the movement disorder society criteria for the diagnosis of 4-repeat tauopathies. Mov Disord. (2020) 35:171–6. 10.1002/mds.27872
24.
Armstrong MJ Litvan I Lang AE Bak TH Bhatia KP Borroni B et al . Criteria for the Diagnosis of Corticobasal Degeneration. Neurology. (2013) 80:496–503. 10.1212/WNL.0b013e31827f0fd1
25.
Parmera JB Rodriguez RD Studart Neto A Nitrini R Brucki SMD . Corticobasal syndrome: a diagnostic conundrum. Dement Neuropsychol. (2016) 10:267–75. 10.1590/s1980-5764-2016dn1004003
26.
Rohrer JD Geser F Zhou J Gennatas ED Sidhu M Trojanowski JQ et al . Tdp-43 subtypes are associated with distinct atrophy patterns in frontotemporal dementia. Neurology. (2010) 75:2204–11. 10.1212/WNL.0b013e318202038c
27.
Spinelli EG Mandelli ML Miller ZA Santos-Santos MA Wilson SM Agosta F et al . Typical and atypical pathology in primary progressive aphasia variants. Ann Neurol. (2017) 81:430–43. 10.1002/ana.24885
28.
Mendez MF Mastri AR Sung JH Frey WH . Clinically diagnosed alzheimer disease: neuropathologic findings in 650 cases alzheimer. Dis Assoc Disord. (1992) 6:35–43. 10.1097/00002093-199205000-00004
29.
Bertoux M Cassagnaud P Lebouvier T Lebert F Sarazin M Le Ber I et al . Does amnesia specifically predict alzheimer's pathology? A neuropathological study. Neurobiol Aging. (2020) 95:123–30. 10.1016/j.neurobiolaging.2020.07.011
30.
Gauthreaux K Bonnett TA Besser LM Brenowitz WD Teylan M Mock C et al . Concordance of clinical alzheimer diagnosis and neuropathological features at autopsy. J Neuropathol Exp Neurol. (2020) 79:465–73. 10.1093/jnen/nlaa014
31.
Risse SC Raskind MA Nochlin D Sumi SM Lampe TH Bird TD et al . Neuropathological findings in patients with clinical diagnoses of probable alzheimer's disease. Am J Psychiatry. (1990) 147:168–72.
32.
Choudhury P Scharf EL Paolini MA Graff-Radford J Alden EC Machulda MM et al . Pick's disease: clinicopathologic characterization of 21 cases. J Neurol. (2020) 267:2697–704. 10.1007/s00415-020-09927-9
33.
Koga S Cheshire WP Tipton PW Driver-Dunckley ED Wszolek ZK Uitti RJ et al . Clinical features of autopsy-confirmed multiple system atrophy in the mayo clinic Florida Brain Bank. Parkinsonism Relat Disord. (2021) 89:155–61. 10.1016/j.parkreldis.2021.07.007
34.
Forrest SL Kril JJ Kovacs GG . Association between globular glial tauopathies and frontotemporal dementia-expanding the spectrum of gliocentric disorders: a review. JAMA Neurol. (2021) 78:1004–14. 10.1001/jamaneurol.2021.1813
35.
Perry DC Brown JA Possin KL Datta S Trujillo A Radke A et al . Clinicopathological correlations in behavioural variant frontotemporal dementia. Brain. (2017) 140:3329–45. 10.1093/brain/awx254
36.
Rohrer JD Lashley T Schott JM Warren JE Mead S Isaacs AM et al . Clinical and neuroanatomical signatures of tissue pathology in frontotemporal lobar degeneration. Brain. (2011) 134(Pt 9):2565-81. 10.1093/brain/awr198
37.
Nelson PT Dickson DW Trojanowski JQ Jack CR Boyle PA Arfanakis K et al . Limbic-predominant Age-Related Tdp-43 Encephalopathy (Late): Consensus Working Group Report. Brain. (2019) 142:1503–27. 10.1093/brain/awz099
38.
Rodriguez RD Suemoto CK Molina M Nascimento CF Leite RE de Lucena Ferretti-Rebustini RE et al . Argyrophilic grain disease: demographics, clinical, and neuropathological features from a large autopsy study. J Neuropathol Exp Neurol. (2016) 75:628–35. 10.1093/jnen/nlw034
39.
Hickman RA Flowers XE Wisniewski T . Primary Age-Related Tauopathy (Part): addressing the spectrum of neuronal tauopathic changes in the aging brain. Curr Neurol Neurosci Rep. (2020) 20:39. 10.1007/s11910-020-01063-1
40.
Teylan M Mock C Gauthreaux K Chen YC Chan KCG Hassenstab J et al . Cognitive trajectory in mild cognitive impairment due to primary age-related tauopathy. Brain. (2020) 143:611–21. 10.1093/brain/awz403
41.
Steele JC Richardson JC Olszewski J . Progressive supranuclear palsy. A heterogeneous degeneration involving the brain stem, basal ganglia and cerebellum with vertical gaze and pseudobulbar palsy, nuchal dystonia and dementia. Arch Neurol. (1964) 10:333–59. 10.1001/archneur.1964.00460160003001
42.
Liu AJ Chang JE Naasan G Boxer AL Miller BL Spina S . Progressive supranuclear palsy and primary lateral sclerosis secondary to globular glial tauopathy: a case report and a practical theoretical framework for the clinical prediction of this rare pathological entity. Neurocase. (2020) 26:91–7. 10.1080/13554794.2020.1732427
43.
Bluett B Litvan I Cheng S Juncos J Riley DE Standaert DG et al . Understanding falls in progressive supranuclear palsy. Parkinsonism Relat Disord. (2017) 35:75–81. 10.1016/j.parkreldis.2016.12.009
44.
Brusa A Mancardi GL Bugiani O . Progressive supranuclear palsy 1979: an overview. Ital J Neurol Sci. (1980) 1:205–22. 10.1007/BF02336701
45.
Litvan I Agid Y Calne D Campbell G Dubois B Duvoisin RC et al . Clinical research criteria for the diagnosis of progressive supranuclear palsy (Steele-Richardson-Olszewski Syndrome): report of the Ninds-Spsp International Workshop. Neurology. (1996) 47:1–9. 10.1212/WNL.47.1.1
46.
Nath U Ben-Shlomo Y Thomson RG Morris HR Wood NW Lees AJ et al . The prevalence of progressive supranuclear palsy (Steele-Richardson-Olszewski Syndrome) in the Uk. Brain. (2001) 124(Pt 7):1438-49. 10.1093/brain/124.7.1438
47.
Golbe LI . The epidemiology of progressive supranuclear palsy. Handb Clin Neurol. (2008) 89:457–9. 10.1016/S0072-9752(07)01242-0
48.
Fleury V Brindel P Nicastro N Burkhard PR . Descriptive Epidemiology of Parkinsonism in the Canton of Geneva, Switzerland. Parkinsonism Relat Disord. (2018) 54:30–9. 10.1016/j.parkreldis.2018.03.030
49.
Stang CD Turcano P Mielke MM Josephs KA Bower JH Ahlskog JE et al . Incidence and trends of progressive supranuclear palsy and corticobasal syndrome: a population-based study. J Parkinsons Dis. (2020) 10:179–84. 10.3233/JPD-191744
50.
Golbe LI Davis PH Schoenberg BS Duvoisin RC . Prevalence and natural history of progressive supranuclear palsy. Neurology. (1988) 38:1031–4. 10.1212/WNL.38.7.1031
51.
Phokaewvarangkul O Bhidayasiri R . How to spot ocular abnormalities in progressive supranuclear palsy? A practical review transl. Neurodegener. (2019) 8:20. 10.1186/s40035-019-0160-1
52.
Garbutt S Riley DE Kumar AN Han Y Harwood MR Leigh RJ . Abnormalities of Optokinetic Nystagmus in progressive supranuclear palsy. J Neurol Neurosurg Psychiatry. (2004) 75:1386–94. 10.1136/jnnp.2003.027367
53.
Terao Y Fukuda H Shirota Y Yugeta A Yoshioka M Suzuki M et al . Deterioration of horizontal saccades in progressive supranuclear palsy. Clin Neurophysiol. (2013) 124:354–63. 10.1016/j.clinph.2012.07.008
54.
Malessa S Gaymard B Rivaud S Cervera P Hirsch E Verny M et al . Role of pontine nuclei damage in smooth pursuit impairment of progressive supranuclear palsy: a clinical-pathologic study. Neurology. (1994) 44:716–21. 10.1212/WNL.44.4.716
55.
Troost BT Daroff RB . The ocular motor defects in progressive supranuclear palsy. Ann Neurol. (1977) 2:397–403. 10.1002/ana.410020509
56.
Rivaud-Pechoux S Vidailhet M Gallouedec G Litvan I Gaymard B Pierrot-Deseilligny C . Longitudinal ocular motor study in corticobasal degeneration and progressive supranuclear palsy. Neurology. (2000) 54:1029–32. 10.1212/WNL.54.5.1029
57.
Picillo M Ricciardi C Tepedino MF Abate F Cuoco S Carotenuto I et al . Gait analysis in progressive supranuclear palsy phenotypes. Front Neurol. (2021) 12:674495. 10.3389/fneur.2021.674495
58.
Litvan I . Progressive supranuclear palsy revisited. Acta Neurol Scand. (1998) 98:73–84. 10.1111/j.1600-0404.1998.tb01723.x
59.
Amano S Skinner JW Lee HK Stegemoller EL Hack N Akbar U et al . Discriminating features of gait performance in progressive supranuclear palsy. Parkinsonism Relat Disord. (2015) 21:888–93. 10.1016/j.parkreldis.2015.05.017
60.
Egerton T Williams DR Iansek R . Comparison of gait in progressive supranuclear palsy, parkinson's disease and healthy older adults. BMC Neurol. (2012) 12:116. 10.1186/1471-2377-12-116
61.
Rezvanian S Litvan I Standaert D Jankovic J Reich SG Hall D et al . Understanding the relationship between freezing of gait and other progressive supranuclear palsy features. Parkinsonism Relat Disord. (2020) 78:56–60. 10.1016/j.parkreldis.2020.07.009
62.
Fujioka S Algom AA Murray ME Sanchez-Contreras MY Tacik P Tsuboi Y et al . Tremor in progressive supranuclear palsy. Parkinsonism Relat Disord. (2016) 27:93–7. 10.1016/j.parkreldis.2016.03.015
63.
Litvan I Mangone CA McKee A Verny M Parsa A Jellinger K et al . Natural history of progressive supranuclear palsy (Steele-Richardson-Olszewski Syndrome) and clinical predictors of survival: a clinicopathological study. J Neurol Neurosurg Psychiatry. (1996) 60:615–20. 10.1136/jnnp.60.6.615
64.
Rusz J Bonnet C Klempir J Tykalova T Baborova E Novotny M et al . Speech disorders reflect differing pathophysiology in parkinson's disease, progressive supranuclear palsy and multiple system atrophy. J Neurol. (2015) 262:992–1001. 10.1007/s00415-015-7671-1
65.
Clark HM Utianski RL Ali F Botha H Whitwell JL Josephs KA . Motor speech disorders and communication limitations in progressive supranuclear palsy. Am J Speech Lang Pathol. (2021) 30(3S):1361–72. 10.1044/2020_AJSLP-20-00126
66.
Goetz CG Leurgans S Lang AE Litvan I . Progression of gait, speech and swallowing deficits in progressive supranuclear palsy. Neurology. (2003) 60:917–22. 10.1212/01.WNL.0000052686.97625.27
67.
Litvan I Sastry N Sonies BC . Characterizing swallowing abnormalities in progressive supranuclear palsy. Neurology. (1997) 48:1654–62. 10.1212/WNL.48.6.1654
68.
dell'Aquila C Zoccolella S Cardinali V de Mari M Iliceto G Tartaglione B et al . Predictors of survival in a series of clinically diagnosed progressive supranuclear palsy patients. Parkinsonism Relat Disord. (2013) 19:980-5. 10.1016/j.parkreldis.2013.06.014
69.
Pilotto A Gazzina S Benussi A Manes M. Dell'Era V Cristillo V et al . Mild cognitive impairment and progression to dementia in progressive supranuclear palsy. Neurodegener Dis. (2017) 17:286–91. 10.1159/000479110
70.
Albert ML Feldman RG Willis AL . The 'Subcortical Dementia' of progressive supranuclear palsy. J Neurol Neurosurg Psychiatry. (1974) 37:121–30. 10.1136/jnnp.37.2.121
71.
Litvan I . Cognitive disturbances in progressive supranuclear palsy. J Neural Transm Suppl. (1994) 42:69–78. 10.1007/978-3-7091-6641-3_6
72.
Gerstenecker A Mast B Duff K Ferman TJ Litvan I Group E-PS . Executive dysfunction is the primary cognitive impairment in progressive supranuclear palsy. Arch Clin Neuropsychol. (2013) 28:104–13. 10.1093/arclin/acs098
73.
Foley JA Niven EH Abrahams S Cipolotti L . Phonemic fluency quantity and quality: comparing patients with psp, parkinson's disease and focal frontal and subcortical lesions. Neuropsychologia. (2021) 153:107772. 10.1016/j.neuropsychologia.2021.107772
74.
Luca A Nicoletti A Donzuso G Terravecchia C Cicero CE D'Agate C et al . Phonemic verbal fluency and midbrain atrophy in progressive supranuclear palsy. J Alzheimers Dis. (2021) 80:1669–74. 10.3233/JAD-210023
75.
Pellicano C Assogna F Cellupica N Piras F Pierantozzi M Stefani A et al . Neuropsychiatric and cognitive profile of early richardson's syndrome, progressive supranuclear palsy-parkinsonism and parkinson's disease. Parkinsonism Relat Disord. (2017) 45:50–6. 10.1016/j.parkreldis.2017.10.002
76.
Horta-Barba A Pagonabarraga J Martinez-Horta S Busteed L Pascual-Sedano B Illan-Gala I et al . Cognitive and behavioral profile of progressive supranuclear palsy and its phenotypes. J Neurol. (2021) 268:3400–8. 10.1007/s00415-021-10511-y
77.
O'Keeffe FM Murray B Coen RF Dockree PM Bellgrove MA Garavan H et al . Loss of insight in frontotemporal dementia, corticobasal degeneration and progressive supranuclear palsy. Brain. (2007) 130(Pt 3):753–64. 10.1093/brain/awl367
78.
Kobylecki C Jones M Thompson JC Richardson AM Neary D Mann DM et al . Cognitive-behavioural features of progressive supranuclear palsy syndrome overlap with frontotemporal dementia. J Neurol. (2015) 262:916–22. 10.1007/s00415-015-7657-z
79.
Ghosh BC Calder AJ Peers PV Lawrence AD Acosta-Cabronero J Pereira JM et al . Social cognitive deficits and their neural correlates in progressive supranuclear palsy. Brain. (2012) 135(Pt 7):2089–102. 10.1093/brain/aws128
80.
Litvan I Cummings JL Mega M . Neuropsychiatric features of corticobasal degeneration. J Neurol, Neurosurg Psych. (1998) 65:717–21. 10.1136/jnnp.65.5.717
81.
Gerstenecker A Duff K Mast B Litvan I Group E-PS . Behavioral abnormalities in progressive supranuclear palsy. Psychiatry Res. (2013) 210:1205–10. 10.1016/j.psychres.2013.08.045
82.
Possin KL Brambati SM Rosen HJ Johnson JK Pa J Weiner MW et al . Rule violation errors are associated with right lateral prefrontal cortex atrophy in neurodegenerative disease. J Int Neuropsychol Soc. (2009) 15:354–64. 10.1017/S135561770909050X
83.
Belvisi D Berardelli I Suppa A Fabbrini A Pasquini M Pompili M et al . Neuropsychiatric disturbances in atypical parkinsonian disorders. Neuropsychiatr Dis Treat. (2018) 14:2643–56. 10.2147/NDT.S178263
84.
Kompoliti K Goetz CG Litvan I Jellinger K Verny M . Pharmacological therapy in progressive supranuclear palsy. Arch Neurol. (1998) 55:1099–102. 10.1001/archneur.55.8.1099
85.
Aarsland D Litvan I Larsen JP . Neuropsychiatric symptoms of patients with progressive supranuclear palsy and parkinson's disease. J Neuropsychiatry Clin Neurosci. (2001) 13:42–9. 10.1176/jnp.13.1.42
86.
Lhermitte F . Human autonomy and the frontal lobes. part II: patient behavior in complex and social situations: the “Environmental Dependency Syndrome.” Ann Neurol. (1986) 19:335–43. 10.1002/ana.410190405
87.
Lagarde J Valabregue R Corvol JC Le Ber I Colliot O Vidailhet M et al . The clinical and anatomical heterogeneity of environmental dependency phenomena. J Neurol. (2013) 260:2262–70. 10.1007/s00415-013-6976-1
88.
Ghika J Tennis M Growdon J Hoffman E Johnson K . Environment-driven responses in progressive supranuclear palsy. J Neurol Sci. (1995) 130:104–11. 10.1016/0022-510X(95)00015-T
89.
Litvan I Agid Y Jankovic J Goetz C Brandel JP Lai EC et al . Accuracy of clinical criteria for the diagnosis of progressive supranuclear palsy (Steele-Richardson-Olszewski Syndrome). Neurology. (1996) 46:922–30. 10.1212/WNL.46.4.922
90.
Ali F Martin PR Botha H Ahlskog JE Bower JH Masumoto JY et al . Sensitivity and specificity of diagnostic criteria for progressive supranuclear palsy. Movement Disorders. (2019) 34:1144–53. 10.1002/mds.27619
91.
Hoglinger GU Respondek G Stamelou M Kurz C Josephs KA Lang AE et al . Clinical diagnosis of progressive supranuclear palsy: the movement disorder society criteria. Mov Disord. (2017) 32:853–64. 10.1002/mds.26987
92.
Gorno-Tempini ML Hillis AE Weintraub S Kertesz A Mendez M Cappa SF et al . Classification of primary progressive aphasia and its variants. Neurology. (2011) 76:1006–14. 10.1212/WNL.0b013e31821103e6
93.
Rascovsky K Hodges JR Knopman D Mendez MF Kramer JH Neuhaus J et al . Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain. (2011) 134(Pt 9):2456-77. 10.1093/brain/awr179
94.
Picillo M Erro R Cuoco S Tepedino MF Manara R Pellecchia MT et al . Mds Psp criteria in real-life clinical setting: motor and cognitive characterization of subtypes. Movement Disorders. (2018) 33:1361–5. 10.1002/mds.27408
95.
Grimm MJ Respondek G Stamelou M Arzberger T Ferguson L Gelpi E et al . How to apply the movement disorder society criteria for diagnosis of progressive supranuclear palsy. Mov Disord. (2019) 34:1228–32. 10.1002/mds.27666
96.
Iankova V Respondek G Saranza G Painous C Camara A Compta Y et al . Video-tutorial for the movement disorder society criteria for progressive supranuclear palsy. Parkinsonism Relat Disord. (2020) 78:200–3. 10.1016/j.parkreldis.2020.06.030
97.
Ogawa T Hatano T Oyama G Takanashi M Taniguchi D Hattori N . Graphic summary of movement disorders society criteria for progressive supranuclear palsy and multiple allocations extinction rules. Mov Disord Clin Pract. (2020) 7:240–2. 10.1002/mdc3.12894
98.
Cozma L Avasilichioaei M Dima N Popescu BO . Of criteria and men-diagnosing atypical parkinsonism: towards an algorithmic approach. Brain Sci. (2021) 11:695. 10.3390/brainsci11060695
99.
Shoeibi A Litvan I Juncos JL Riley DE Bordelon Y Standaert D et al . Are the International Parkinson Disease and Movement Disorder Society Progressive Supranuclear Palsy (Ipmds-Psp) diagnostic criteria accurate enough to differentiate common psp phenotypes?Parkinsonism Relat Disord. (2019). 10.1016/j.parkreldis.2019.10.012
100.
Frank A Peikert K Linn J Brandt MD Hermann A . Mds Criteria for the diagnosis of progressive supranuclear palsy overemphasize richardson syndrome. Ann Clin Transl Neurol. (2020) 7:1702–7. 10.1002/acn3.51065
101.
Hokelekli FO Duffy JR Clark HM Utianski RL Botha H Ali F et al . Autopsy validation of progressive supranuclear palsy-predominant speech/language disorder criteria. Mov Disord. (2022) 37:213–8. 10.1002/mds.28822
102.
Sanchez-Ruiz de. Gordoa J Zelaya V Tellechea-Aramburo P Acha B Roldan M Lopez-Molina C et al . Is the phenotype designation by Psp-Mds criteria stable throughout the disease course and consistent with tau distribution?Front Neurol. (2022) 13:827338. 10.3389/fneur.2022.827338
103.
Ali F Botha H Whitwell JL Josephs KA . Utility of the movement disorders society criteria for progressive supranuclear palsy in clinical practice: clinical utility of Psp criteria. Mov Disord Clin Pract. (2019). 10.1002/mdc3.12807
104.
Kouri N Murray ME Hassan A Rademakers R Uitti RJ Boeve BF et al . Neuropathological features of corticobasal degeneration presenting as corticobasal syndrome or richardson syndrome. Brain. (2011) 134(Pt 11):3264–75. 10.1093/brain/awr234
105.
Litvan I Grimes DA Lang AE Jankovic J McKee A Verny M et al . clinical features differentiating patients with postmortem confirmed progressive supranuclear palsy and corticobasal degeneration. J Neurol. (1999) 246 (Suppl 2):II1–5. 10.1007/BF03161075
106.
Ling H O'Sullivan SS Holton JL Revesz T Massey LA Williams DR et al . Does corticobasal degeneration exist? A clinicopathological re-evaluation. Brain. (2010) 133(Pt 7):2045–57. 10.1093/brain/awq123
107.
Rebeiz JJ Kolodny EH Richardson EP Jr . Corticodentatonigral degeneration with neuronal achromasia: a progressive disorder of late adult life. Trans Am Neurol Assoc. (1967) 92:23–6.
108.
Gibb WR Luthert PJ Marsden CD . Corticobasal degeneration. Brain. (1989) 112 (Pt 5):1171-92. 10.1093/brain/112.5.1171
109.
Constantinides VC Paraskevas GP Paraskevas PG Stefanis L Kapaki E . Corticobasal degeneration and corticobasal syndrome: a review. Clin Park Relat Disord. (2019) 1:66–71. 10.1016/j.prdoa.2019.08.005
110.
Hassan A Whitwell JL Boeve BF Jack CR Jr Parisi JE Dickson DW et al . Symmetric corticobasal degeneration (S-Cbd). Parkinsonism Relat Disord. (2010) 16:208–14. 10.1016/j.parkreldis.2009.11.013
111.
Brunt ER van Weerden TW Pruim J Lakke JW . Unique myoclonic pattern in corticobasal degeneration. Mov Disord. (1995) 10:132–42. 10.1002/mds.870100203
112.
Lu CS Ikeda A Terada K Mima T Nagamine T Fukuyama H et al . Electrophysiological studies of early stage corticobasal degeneration. Mov Disord. (1998) 13:140–6. 10.1002/mds.870130126
113.
Leiguarda RC Pramstaller PP Merello M Starkstein S Lees AJ Marsden CD . Apraxia in parkinson's disease, progressive supranuclear palsy, multiple system atrophy and neuroleptic-induced parkinsonism. Brain. (1997) 120 (Pt 1):75–90. 10.1093/brain/120.1.75
114.
Pharr V Uttl B Stark M Litvan I Fantie B Grafman J . Comparison of apraxia in corticobasal degeneration and progressive supranuclear palsy. Neurology. (2001) 56:957–63. 10.1212/WNL.56.7.957
115.
Lewis-Smith DJ Wolpe N Ghosh BCP Rowe JB . Alien limb in the corticobasal syndrome: phenomenological characteristics and relationship to apraxia. J Neurol. (2020) 267:1147–57. 10.1007/s00415-019-09672-8
116.
Bundick T Jr. Spinella M . Subjective experience, involuntary movement, and posterior alien hand syndrome. J Neurol Neurosurg Psychiatry. (2000) 68:83–5. 10.1136/jnnp.68.1.83
117.
Aboitiz F Carrasco X Schröter C Zaidel D Zaidel E Lavados M . The alien hand syndrome: classification of forms reported and discussion of a new condition. Neurol Sci. (2003) 24:252–7. 10.1007/s10072-003-0149-4
118.
Lee W Simpson M Ling H McLean C Collins S Williams DR . Characterising the uncommon corticobasal syndrome presentation of sporadic creutzfeldt-jakob disease. Parkinsonism Relat Disord. (2013) 19:81–5. 10.1016/j.parkreldis.2012.07.010
119.
Kasanuki K Josephs KA Ferman TJ Murray ME Koga S Konno T et al . Diffuse lewy body disease manifesting as corticobasal syndrome: a rare form of lewy body disease. Neurology. (2018) 91:e268–e79. 10.1212/WNL.0000000000005828
120.
Koga S Roemer SF Kasanuki K Dickson DW . Cerebrovascular pathology presenting as corticobasal syndrome: an autopsy case series of “Vascular Cbs”. Parkinsonism Relat Disord. (2019) 68:79–84. 10.1016/j.parkreldis.2019.09.001
121.
Ichinose K Watanabe M Mizutani S Tanizawa T Uchihara T Fujigasaki H . An autopsy case of corticobasal syndrome with pure diffuse lewy body disease. Neurocase. (2021) 27:231–7. 10.1080/13554794.2021.1921220
122.
Murray R Neumann M Forman MS Farmer J Massimo L Rice A et al . Cognitive and motor assessment in autopsy-proven corticobasal degeneration. Neurology. (2007) 68:1274–83. 10.1212/01.wnl.0000259519.78480.c3
123.
Hu WT Rippon GW Boeve BF Knopman DS Petersen RC Parisi JE et al . Alzheimer's disease and corticobasal degeneration presenting as corticobasal syndrome. Mov Disord. (2009) 24:1375–9. 10.1002/mds.22574
124.
Shelley BP Hodges JR Kipps CM Xuereb JH Bak TH . Is the Pathology of corticobasal syndrome predictable in life?Mov Disord. (2009) 24:1593–9. 10.1002/mds.22558
125.
Hassan A Whitwell JL Josephs KA . The corticobasal syndrome-alzheimer's disease conundrum. Expert Rev Neurother. (2011) 11:1569–78. 10.1586/ern.11.153
126.
Lee SE Rabinovici GD Mayo MC Wilson SM Seeley WW DeArmond SJ et al . Clinicopathological correlations in corticobasal degeneration. Ann Neurol. (2011) 70:327–40. 10.1002/ana.22424
127.
Jung NY Lee JH Lee YM Shin JH Shin MJ Lee MJ et al . Early stage memory impairment, visual hallucinations, and myoclonus combined with temporal lobe atrophy predict alzheimer's disease pathology in corticobasal syndrome. Neurocase. (2018) 24:145–50. 10.1080/13554794.2018.1494290
128.
Sakae N Josephs KA Litvan I Murray ME Duara R Uitti RJ et al . Clinicopathologic subtype of alzheimer's disease presenting as corticobasal syndrome. Alzheimers Dement. (2019) 15:1218–28. 10.1016/j.jalz.2019.04.011
129.
Boyd CD Tierney M Wassermann EM Spina S Oblak AL Ghetti B et al . Visuoperception test predicts pathologic diagnosis of alzheimer disease in corticobasal syndrome. Neurology. (2014) 83:510–9. 10.1212/WNL.0000000000000667
130.
Sampathu DM Neumann M Kwong LK Chou TT Micsenyi M Truax A et al . Pathological heterogeneity of frontotemporal lobar degeneration with ubiquitin-positive inclusions delineated by ubiquitin immunohistochemistry and novel monoclonal antibodies. Am J Pathol. (2006) 169:1343–52. 10.2353/ajpath.2006.060438
131.
Mackenzie IR Neumann M Baborie A Sampathu DM Du Plessis D Jaros E et al . A harmonized classification system for Ftld-Tdp pathology. Acta Neuropathol. (2011) 122:111–3. 10.1007/s00401-011-0845-8
132.
de Boer EMJ Orie VK Williams T Baker MR De Oliveira HM Polvikoski T et al . Tdp-43 proteinopathies: a new wave of neurodegenerative diseases. J Neurol Neurosurg Psychiatry. (2020) 92:86–95. 10.1136/jnnp-2020-322983
133.
Tartaglia MC Sidhu M Laluz V Racine C Rabinovici GD Creighton K et al . Sporadic corticobasal syndrome due to Ftld-Tdp. Acta Neuropathol. (2010) 119:365–74. 10.1007/s00401-009-0605-1
134.
Dopper EG Seelaar H Chiu WZ de Koning I van Minkelen R Baker MC et al . Symmetrical corticobasal syndrome caused by a novel C314dup progranulin mutation. J Mol Neurosci. (2011) 45:354–8. 10.1007/s12031-011-9626-z
135.
Alexander SK Rittman T Xuereb JH Bak TH Hodges JR Rowe JB . Validation of the new consensus criteria for the diagnosis of corticobasal degeneration. J Neurol Neurosurg Psychiatry. (2014) 85:925–9. 10.1136/jnnp-2013-307035
136.
Ouchi H Toyoshima Y Tada M Oyake M Aida I Tomita I et al . Pathology and sensitivity of current clinical criteria in corticobasal syndrome. Mov Disord. (2014) 29:238–44. 10.1002/mds.25746
137.
Saracino D Geraudie A Remes AM Ferrieux S Nogues-Lassiaille M Bottani S et al . Primary progressive aphasias associated with C9orf72 expansions: another side of the story. Cortex. (2021) 145:145–59. 10.1016/j.cortex.2021.09.005
138.
Van Mossevelde S van der Zee J Gijselinck I Engelborghs S Sieben A Van Langenhove T et al . Clinical features of Tbk1 carriers compared with C9orf72, Grn and non-mutation carriers in a Belgian Cohort. Brain. (2016) 139(Pt 2):452–67. 10.1093/brain/awv358
139.
Rohrer JD . The genetics of primary progressive aphasia. Aphasiology. (2014) 28:941–7. 10.1080/02687038.2014.911242
140.
Logroscino G Piccininni M Binetti G Zecca C Turrone R Capozzo R et al . Incidence of frontotemporal lobar degeneration in Italy: the Salento-Brescia registry study. Neurology. (2019) 92:e2355–e63. 10.1212/WNL.0000000000007498
141.
Harris JM Gall C Thompson JC Richardson AM Neary D du Plessis D et al . Classification and pathology of primary progressive aphasia. Neurology. (2013) 81:1832–9. 10.1212/01.wnl.0000436070.28137.7b
142.
Wicklund MR Duffy JR Strand EA Machulda MM Whitwell JL Josephs KA . Quantitative application of the primary progressive aphasia consensus criteria. Neurology. (2014) 82:1119–26. 10.1212/WNL.0000000000000261
143.
Duffy JR Utianski RL Josephs KA . Primary progressive apraxia of speech: from recognition to diagnosis and care. Aphasiology. (2021) 35:560–91. 10.1080/02687038.2020.1787732
144.
Grossman M . The non-fluent/agrammatic variant of primary progressive aphasia. Lancet Neurol. (2012) 11:545–55. 10.1016/S1474-4422(12)70099-6
145.
Duffy JR . Motor Speech Disorders : Substrates, Differential Diagnosis, and Management. St Louis, Missouri: Elsevier. (2020).
146.
Marshall CR Hardy CJD Volkmer A Russell LL Bond RL Fletcher PD et al . Primary progressive aphasia: a clinical approach. J Neurol. (2018) 265:1474–90. 10.1007/s00415-018-8762-6
147.
Mesulam M Coventry C Bigio EH Geula C Thompson C Bonakdarpour B et al . Nosology of primary progressive aphasia and the neuropathology of language. Adv Exp Med Biol. (2021) 1281:33–49. 10.1007/978-3-030-51140-1_3
148.
Caso F Mandelli ML Henry M Gesierich B Bettcher BM Ogar J et al . In vivo signatures of nonfluent/agrammatic primary progressive aphasia caused by Ftld pathology. Neurology. (2014) 82:239–47. 10.1212/WNL.0000000000000031
149.
Santos-Santos MA Mandelli ML Binney RJ Ogar J Wilson SM Henry ML et al . Features of patients with nonfluent/agrammatic primary progressive aphasia with underlying progressive supranuclear palsy pathology or corticobasal degeneration. JAMA Neurol. (2016) 73:733–42. 10.1001/jamaneurol.2016.0412
150.
Whitwell JL Martin P Duffy JR Clark HM Utianski RL Botha H et al . Survival analysis in primary progressive apraxia of speech and agrammatic aphasia. Neurol Clin Pract. (2021) 11:249–55. 10.1212/CPJ.0000000000000919
151.
Ossenkoppele R Singleton EH Groot C Dijkstra AA Eikelboom WS Seeley WW et al . Research criteria for the behavioral variant of alzheimer disease: a systematic review and meta-analysis. JAMA Neurol. (2022) 79:48–60. 10.1001/jamaneurol.2021.4417
152.
Ossenkoppele R Pijnenburg YA Perry DC Cohn-Sheehy BI Scheltens NM Vogel JW et al . The behavioural/dysexecutive variant of alzheimer's disease: clinical, neuroimaging and pathological features. Brain. (2015) 138(Pt 9):2732–49. 10.1093/brain/awv191
153.
Kobayashi Z Arai T Kawakami I Yokota O Hosokawa M Oshima K et al . Clinical features of the behavioural variant of frontotemporal dementia that are useful for predicting underlying pathological subtypes of frontotemporal lobar degeneration. Psychogeriatrics. (2018) 18:307–12. 10.1111/psyg.12334
154.
Mendez MF Joshi A Tassniyom K Teng E Shapira JS . Clinicopathologic differences among patients with behavioral variant frontotemporal dementia. Neurology. (2013) 80:561–8. 10.1212/WNL.0b013e3182815547
155.
Street D Malpetti M Rittman T Ghosh BCP Murley AG Coyle-Gilchrist I et al . Clinical progression of progressive supranuclear palsy: impact of trials bias and phenotype variants. Brain Commun. (2021) 3:fcab206. 10.1093/braincomms/fcab206
156.
Zapata-Restrepo L Rivas J Miranda C Miller BL Ibanez A Allen IE et al . The psychiatric misdiagnosis of behavioral variant frontotemporal dementia in a Colombian sample. Front Neurol. (2021) 12:729381. 10.3389/fneur.2021.729381
157.
Moore KM Nicholas J Grossman M McMillan CT Irwin DJ Massimo L et al . Age at symptom onset and death and disease duration in genetic frontotemporal dementia: an International Retrospective Cohort Study. The Lancet Neurology. (2020) 19:145–56. 10.1016/S1474-4422(19)30394-1
158.
Boeve BF Rosen H . Clinical and neuroimaging aspects of familial frontotemporal lobar degeneration associated with Mapt and Grn mutations. Adv Exp Med Biol. (2021) 1281:77–92. 10.1007/978-3-030-51140-1_6
159.
Imai H Narabayashi H Sakata E . “Pure Akinesia” and the later added supranuclear ophthalmoplegia. Adv Neurol. (1987) 45:207–12.
160.
Matsuo H Takashima H Kishikawa M Kinoshita I Mori M Tsujihata M et al . Pure Akinesia: An Atypical Manifestation Of Progressive Supranuclear Palsy. J Neurol Neurosurg Psychiatry. (1991) 54:397–400. 10.1136/jnnp.54.5.397
161.
Williams DR Holton JL Strand K Revesz T Lees AJ . Pure akinesia with gait freezing: a third clinical phenotype of progressive supranuclear palsy. Mov Disord. (2007) 22:2235–41. 10.1002/mds.21698
162.
Ahmed Z Josephs KA Gonzalez J DelleDonne A Dickson DW . Clinical and neuropathologic features of progressive supranuclear palsy with severe Pallido-Nigro-Luysial degeneration and axonal dystrophy. Brain. (2008) 131(Pt 2):460–72. 10.1093/brain/awm301
163.
Owens E Josephs KA Savica R Hassan A Klassen B Bower J et al . The Clinical spectrum and natural history of pure akinesia with gait freezing. J Neurol. (2016) 263:2419–23. 10.1007/s00415-016-8278-x
164.
Lee YC Williams DR Anderson JFI . Prospective characterization of cognitive function in typical and 'brainstem predominant'progressive supranuclear palsy Phenotypes. J Mov Disord. (2018) 11:72–7. 10.14802/jmd.17067
165.
Elkouzi A Bit-Ivan EN Elble RJ . Pure akinesia with gait freezing: a clinicopathologic study. J Clin Mov Disord. (2017) 4:15. 10.1186/s40734-017-0063-1
166.
Baezner H Hennerici M . From Trepidant abasia to motor network failure–gait disorders as a consequence of subcortical vascular encephalopathy (Sve): review of historical and contemporary concepts. J Neurol Sci. (2005) 229–30:81–8. 10.1016/j.jns.2004.11.008
167.
Lee MS Lyoo CH Choi YH . Primary progressive freezing gait in a patient with co-induced parkinsonism. Mov Disord. (2010) 25:1513–5. 10.1002/mds.23124
168.
Factor SA Higgins DS Qian J . Primary progressive freezing gait: a syndrome with many causes. Neurology. (2006) 66:411–4. 10.1212/01.wnl.0000196469.52995.ab
169.
Williams DR Lees AJ . What features improve the accuracy of the clinical diagnosis of progressive supranuclear palsy-parkinsonism (Psp-P)?Mov Disord. (2010) 25:357–62. 10.1002/mds.22977
170.
Jecmenica-Lukic M Petrovic IN Pekmezovic T Kostic VS . Clinical outcomes of two main variants of progressive supranuclear palsy and multiple system atrophy: a prospective natural history study. J Neurol. (2014) 261:1575–83. 10.1007/s00415-014-7384-x
171.
Shoeibi A Litvan I Tolosa E Ser TD Lee E Investigators T . Progression of two progressive supranuclear palsy phenotypes with comparable initial disability. Parkinsonism Relat Disord. (2019) 66:87–93. 10.1016/j.parkreldis.2019.07.010
172.
Srulijes K Mallien G Bauer S Dietzel E Groger A Ebersbach G et al . In vivo comparison of Richardson's syndrome and progressive supranuclear palsy-parkinsonism. J Neural Transm. (2011) 118:1191–7. 10.1007/s00702-010-0563-8
173.
Bower SM Weigand SD Ali F Clark HM Botha H Stierwalt JA et al . Depression and apathy across different variants of progressive supranuclear palsy. Mov Disord Clin Pract. (2022) 9:212–7. 10.1002/mdc3.13396
174.
Kok ZQ Murley AG Rittman T Rowe J Passamonti L . Co-occurrence of apathy and impulsivity in progressive supranuclear palsy. Mov Disord Clin Pract. (2021) 8:1225–33. 10.1002/mdc3.13339
175.
Kanazawa M Shimohata T Toyoshima Y Tada M Kakita A Morita T et al . Cerebellar involvement in progressive supranuclear palsy: a clinicopathological study. Mov Disord. (2009) 24:1312–8. 10.1002/mds.22583
176.
Kanazawa M Shimohata T Endo K Koike R Takahashi H Nishizawa M et al . Serial Mri study in a patient with progressive supranuclear palsy with cerebellar ataxia. Parkinsonism Relat Disord. (2012) 18:677–9. 10.1016/j.parkreldis.2011.11.011
177.
Iwasaki Y Mori K Ito M Tatsumi S Mimuro M Yoshida M . An Autopsied Case of Progressive Supranuclear Palsy Presenting with Cerebellar Ataxia and Severe Cerebellar Involvement. Neuropathology. (2013) 33:561–7. 10.1111/neup.12012
178.
Kanazawa M Tada M Onodera O Takahashi H Nishizawa M Shimohata T . Early clinical features of patients with progressive supranuclear palsy with predominant cerebellar ataxia. Parkinsonism Relat Disord. (2013) 19:1149–51. 10.1016/j.parkreldis.2013.07.019
179.
Koga S Aoki N Uitti RJ van Gerpen JA Cheshire WP Josephs KA et al . When Dlb, Pd, and Psp Masquerade as Msa: an autopsy study of 134 patients. Neurology. (2015) 85:404–12. 10.1212/WNL.0000000000001807
180.
Koga S Josephs KA Ogaki K Labbe C Uitti RJ Graff-Radford N et al . Cerebellar ataxia in progressive supranuclear palsy: an autopsy study of Psp-C. Mov Disord. (2016) 31:653–62. 10.1002/mds.26499
181.
Ando S Kanazawa M Onodera O . Progressive supranuclear palsy with predominant cerebellar ataxia. J Mov Disord. (2020) 13:20–6. 10.14802/jmd.19061
182.
Nagao S Yokota O Nanba R Takata H Haraguchi T Ishizu H et al . Progressive supranuclear palsy presenting as primary lateral sclerosis but lacking parkinsonism, gaze palsy, aphasia, or dementia. J Neurol Sci. (2012) 323:147–53. 10.1016/j.jns.2012.09.005
183.
King A Curran O Al-Sarraj S . Atypical progressive supranuclear palsy presenting as primary lateral sclerosis. J Neurol Sci. (2013) 329:69. 10.1016/j.jns.2013.03.015
184.
Josephs KA Katsuse O Beccano-Kelly DA Lin WL Uitti RJ Fujino Y et al . Atypical progressive supranuclear palsy with corticospinal tract degeneration. J Neuropathol Exp Neurol. (2006) 65:396–405. 10.1097/01.jnen.0000218446.38158.61
185.
Ahmed Z Doherty KM Silveira-Moriyama L Bandopadhyay R Lashley T Mamais A et al . Globular Glial Tauopathies (Ggt) presenting with motor neuron disease or frontotemporal dementia: an emerging group of 4-repeat tauopathies. Acta Neuropathol. (2011) 122:415–28. 10.1007/s00401-011-0857-4
186.
Graff-Radford J Josephs KA Parisi JE Dickson DW Giannini C Boeve BF . Globular glial tauopathy presenting as semantic variant primary progressive aphasia. JAMA Neurol. (2016) 73:123–5. 10.1001/jamaneurol.2015.2711
187.
Harris JM Jones M . Pathology in primary progressive aphasia syndromes. Curr Neurol Neurosci Rep. (2014) 14:466. 10.1007/s11910-014-0466-4
188.
Bergeron D Gorno-Tempini ML Rabinovici GD Santos-Santos MA Seeley W Miller BL et al . Prevalence of amyloid-beta pathology in distinct variants of primary progressive aphasia. Ann Neurol. (2018) 84:729–40. 10.1002/ana.25333
189.
Chan D Fox NC Scahill RI Crum WR Whitwell JL Leschziner G et al . Patterns of temporal lobe atrophy in semantic dementia and alzheimer's disease. Ann Neurol. (2001) 49:433–42. 10.1002/ana.92
190.
Josephs KA Whitwell JL Duffy JR Vanvoorst WA Strand EA Hu WT et al . Progressive aphasia secondary to alzheimer disease Vs Ftld pathology. Neurology. (2008) 70:25–34. 10.1212/01.wnl.0000287073.12737.35
191.
Gorno-Tempini ML Brambati SM Ginex V Ogar J Dronkers NF Marcone A et al . The logopenic/phonological variant of primary progressive aphasia. Neurology. (2008) 71:1227–34. 10.1212/01.wnl.0000320506.79811.da
192.
Giannini LAA Irwin DJ McMillan CT Ash S Rascovsky K Wolk DA et al . Clinical marker for alzheimer disease pathology in logopenic primary progressive aphasia. Neurology. (2017) 88:2276–84. 10.1212/WNL.0000000000004034
193.
Togo T Isojima D Akatsu H Suzuki K Uchikado H Katsuse O et al . Clinical features of argyrophilic grain disease: a retrospective survey of cases with neuropsychiatric symptoms. Am J Geriatr Psychiatry. (2005) 13:1083–91. 10.1097/00019442-200512000-00008
194.
Nagao S Yokota O Ikeda C Takeda N Ishizu H Kuroda S et al . Argyrophilic grain disease as a neurodegenerative substrate in late-onset schizophrenia and delusional disorders. Eur Arch Psychiatry Clin Neurosci. (2014) 264:317–31. 10.1007/s00406-013-0472-6
195.
Jicha GA Petersen RC Knopman DS Boeve BF Smith GE Geda YE et al . Argyrophilic grain disease in demented subjects presenting initially with amnestic mild cognitive impairment. J Neuropathol Exp Neurol. (2006) 65:602–9. 10.1097/01.jnen.0000225312.11858.57
196.
Jellinger KA . Dementia with grains (Argyrophilic Grain Disease). Brain Pathol. (1998) 8:377–86. 10.1111/j.1750-3639.1998.tb00161.x
197.
Toller G Zitser J Sukhanov P Grant H Miller BL Kramer JH et al . Clinical, neuroimaging, and neuropathological characterization of a patient with alzheimer's disease syndrome due to Pick's pathology. Neurocase. (2021) 28:19–28. 10.1080/13554794.2021.1936072
198.
Day GS Lim TS Hassenstab J Goate AM Grant EA Roe CM et al . Differentiating cognitive impairment due to corticobasal degeneration and alzheimer disease. Neurology. (2017) 88:1273–81. 10.1212/WNL.0000000000003770
199.
SantaCruz KS Rottunda SJ Meints JP Bearer EL Bigio EH McCarten JR et al . Case of globular glial tauopathy presenting clinically as alzheimer disease. Alzheimer Dis Assoc Disord. (2015) 29:82–4. 10.1097/WAD.0b013e318298e531
200.
Forrest SL Kril JJ Wagner S Honigschnabl S Reiner A Fischer P et al . Chronic Traumatic Encephalopathy (Cte) is absent from a European community-based aging cohort while cortical Aging-Related Tau Astrogliopathy (Artag) is highly prevalent. J Neuropathol Exp Neurol. (2019) 78:398–405. 10.1093/jnen/nlz017
201.
Munoz DG Woulfe J Kertesz A . Argyrophilic thorny astrocyte clusters in association with alzheimer's disease pathology in possible primary progressive aphasia. Acta Neuropathol. (2007) 114:347–57. 10.1007/s00401-007-0266-x
202.
Kovacs GG Milenkovic I Wohrer A Hoftberger R Gelpi E Haberler C et al . Non-alzheimer neurodegenerative pathologies and their combinations are more frequent than commonly believed in the elderly brain: a community-based autopsy series. Acta Neuropathol. (2013) 126:365–84. 10.1007/s00401-013-1157-y
203.
Evidente VG Adler CH Sabbagh MN Connor DJ Hentz JG Caviness JN et al . Neuropathological findings of Psp in the elderly without clinical Psp: possible incidental Psp?Parkinsonism Relat Disord. (2011) 17:365–71. 10.1016/j.parkreldis.2011.02.017
204.
Dugger BN Hentz JG Adler CH Sabbagh MN Shill HA Jacobson S et al . Clinicopathological outcomes of prospectively followed normal elderly brain bank volunteers. J Neuropathol Exp Neurol. (2014) 73:244–52. 10.1097/NEN.0000000000000046
205.
Nogami A Yamazaki M Saito Y Hatsuta H Sakiyama Y Takao M et al . Early stage of progressive supranuclear palsy: a neuropathological study of 324 consecutive autopsy cases. J Nippon Med Sch. (2015) 82:266–73. 10.1272/jnms.82.266
206.
Nishida N Yoshida K Hata Y Arai Y Kinoshita K . Pathological features of preclinical or early clinical stages of corticobasal degeneration: a comparison with advanced cases. Neuropathol Appl Neurobiol. (2015) 41:893–905. 10.1111/nan.12229
207.
Jack CR Jr. Bennett DA Blennow K Carrillo MC Dunn B Haeberlein SB et al . Nia-Aa research framework: toward a biological definition of alzheimer's disease. Alzheimers Dement. (2018) 14:535–62. 10.1016/j.jalz.2018.02.018
208.
Dubois B Villain N Frisoni GB Rabinovici GD Sabbagh M Cappa S et al . Clinical diagnosis of alzheimer's disease: recommendations of the international working group. Lancet Neurol. (2021) 20:484–96. 10.1016/S1474-4422(21)00066-1
209.
Nicoletti G Caligiuri ME Cherubini A Morelli M Novellino F Arabia G et al . A fully automated, atlas-based approach for superior cerebellar peduncle evaluation in progressive supranuclear palsy phenotypes. AJNR Am J Neuroradiol. (2017) 38:523–30. 10.3174/ajnr.A5048
210.
Quattrone A Morelli M Nigro S Quattrone A Vescio B Arabia G et al . A new Mr imaging index for differentiation of progressive supranuclear palsy-parkinsonism from parkinson's disease. Parkinsonism Relat Disord. (2018) 54:3–8. 10.1016/j.parkreldis.2018.07.016
211.
Albrecht F Bisenius S Neumann J Whitwell J Schroeter ML . Atrophy in midbrain & cerebral/cerebellar pedunculi is characteristic for progressive supranuclear palsy - a double-validation whole-brain meta-analysis. Neuroimage Clin. (2019) 22:101722. 10.1016/j.nicl.2019.101722
212.
Whitwell JL Jack CR Jr. Parisi JE Gunter JL Weigand SD Boeve BF et al . Midbrain atrophy is not a biomarker of progressive supranuclear palsy pathology. Eur J Neurol. (2013) 20:1417–22. 10.1111/ene.12212
213.
Quattrone A Morelli M Williams DR Vescio B Arabia G Nigro S et al . Mr parkinsonism index predicts vertical supranuclear gaze palsy in patients with Psp-Parkinsonism. Neurology. (2016) 87:1266–73. 10.1212/WNL.0000000000003125
214.
Morelli M Arabia G Novellino F Salsone M Giofre L Condino F et al . Mri Measurements predict Psp in unclassifiable parkinsonisms: a cohort study. Neurology. (2011) 77:1042–7. 10.1212/WNL.0b013e31822e55d0
215.
Grijalva RM Pham NTT Huang Q Martin PR Ali F Clark HM et al . Brainstem biomarkers of clinical variant and pathology in progressive supranuclear palsy. Mov Disord. (2021) 37:702–12. 10.1002/mds.28901
216.
Grisoli M Fetoni V Savoiardo M Girotti F Bruzzone MG . Mri in Corticobasal Degeneration. Eur J Neurol. (1995) 2:547–52. 10.1111/j.1468-1331.1995.tb00172.x
217.
Soliveri P Monza D Paridi D Radice D Grisoli M Testa D et al . Cognitive and Magnetic Resonance Imaging Aspects of Corticobasal Degeneration and Progressive Supranuclear Palsy. Neurology. (1999) 53:502–7. 10.1212/WNL.53.3.502
218.
Josephs KA Tang-Wai DF Edland SD Knopman DS Dickson DW Parisi JE et al . Correlation between antemortem magnetic resonance imaging findings and pathologically confirmed corticobasal degeneration. Arch Neurol. (2004) 61:1881–4. 10.1001/archneur.61.12.1881
219.
Chand P Grafman J Dickson D Ishizawa K Litvan I . Alzheimer's disease presenting as corticobasal syndrome. Mov Disord. (2006) 21:2018–22. 10.1002/mds.21055
220.
Boxer AL Geschwind MD Belfor N Gorno-Tempini ML Schauer GF Miller BL et al . Patterns of brain atrophy that differentiate corticobasal degeneration syndrome from progressive supranuclear palsy. Arch Neurol. (2006) 63:81–6. 10.1001/archneur.63.1.81
221.
Josephs KA Whitwell JL Dickson DW Boeve BF Knopman DS Petersen RC et al . Voxel-based morphometry in autopsy proven Psp and Cbd. Neurobiol Aging. (2008) 29:280–9. 10.1016/j.neurobiolaging.2006.09.019
222.
Yu F Barron DS Tantiwongkosi B Fox P . Patterns of gray matter atrophy in atypical parkinsonism syndromes: a Vbm meta-analysis. Brain Behav. (2015) 5:e00329. 10.1002/brb3.329
223.
Ferrea S Junker FB Hartmann CJ Dinkelbach L Eickhoff SB Moldovan AS et al . Brain volume patterns in corticobasal syndrome vs. idiopathic parkinson's disease. J Neuroimaging. (2022). 10.1111/jon.12971
224.
Whitwell JL Jack CR Jr. Boeve BF Parisi JE Ahlskog JE Drubach DA et al . Imaging correlates of pathology in corticobasal syndrome. Neurology. (2010) 75:1879–87. 10.1212/WNL.0b013e3181feb2e8
225.
Tetzloff KA Duffy JR Clark HM Strand EA Machulda MM Schwarz CG et al . Longitudinal structural and molecular neuroimaging in agrammatic primary progressive aphasia. Brain. (2018) 141:302–17. 10.1093/brain/awx293
226.
Whitwell JL Jack CR Jr. Parisi JE Knopman DS Boeve BF Petersen RC et al . Imaging signatures of molecular pathology in behavioral variant frontotemporal dementia. J Mol Neurosci. (2011) 45:372–8. 10.1007/s12031-011-9533-3
227.
Hassan A Parisi JE Josephs KA . Autopsy-proven progressive supranuclear palsy presenting as behavioral variant frontotemporal dementia. Neurocase. (2012) 18:478–88. 10.1080/13554794.2011.627345
228.
Murley AG Coyle-Gilchrist I Rouse MA Jones PS Li W Wiggins J et al . Redefining the multidimensional clinical phenotypes of frontotemporal lobar degeneration syndromes. Brain. (2020) 143:1555–71. 10.1093/brain/awaa097
229.
FDAA . FDA Approves First Drug to Image Tau Pathology in Patients Being Evaluated for Alzheimer's Disease: Food and Drug Administration. (2020). Available online at: https://www.fda.gov/news-events/press-announcements/fda-approves-first-drug-image-tau-pathology-patients-being-evaluated-alzheimers-disease.
230.
Fleisher AS Pontecorvo MJ Devous MD Sr. Lu M Arora AK Truocchio SP et al . Positron emission tomography imaging with [18f]flortaucipir and postmortem assessment of alzheimer disease neuropathologic changes. JAMA Neurol. (2020) 77:829–39. 10.1001/jamaneurol.2020.0528
231.
Marquie M Normandin MD Vanderburg CR Costantino IM Bien EA Rycyna LG et al . Validating novel tau positron emission tomography tracer [F-18]-Av-1451 (T807) on postmortem brain tissue. Ann Neurol. (2015) 78:787–800. 10.1002/ana.24517
232.
Passamonti L Vazquez Rodriguez P Hong YT Allinson KS Williamson D Borchert RJ et al . 18f-Av-1451 positron emission tomography in alzheimer's disease and progressive supranuclear palsy. Brain. (2017) 140:781–91. 10.1093/brain/aww340
233.
Schonhaut DR McMillan CT Spina S Dickerson BC Siderowf A Devous MD. Sr. et al . F-flortaucipir tau positron emission tomography distinguishes established progressive supranuclear palsy from controls and parkinson disease: a multicenter study. Ann Neurol. (2017) 82:622–34. 10.1002/ana.25060
234.
Smith R Schöll M Widner H van Westen D Svenningsson P Hägerström D et al . In vivo retention of F-Av-1451 in corticobasal syndrome. Neurology. (2017) 89:845–53. 10.1212/WNL.0000000000004264
235.
Josephs K Tosakulwong N Weigand S Buciuc M Lowe V Dickson D et al . Relationship between F-flortaucipir uptake and histologic lesion types in 4-repeat tauopathies. J Nucl Med. (2021). 10.2967/jnumed.121.262685
236.
Goodheart AE Locascio JJ Samore WR Collins JA Brickhouse M Schultz A et al . 18f-Av-1451 positron emission tomography in neuropathological substrates of corticobasal syndrome. Brain. (2021) 144:266–77. 10.1093/brain/awaa383
237.
Li Y Liu T Cui M . Recent development in selective tau tracers for pet imaging in the brain. Chin Chem Lett. (2022) 33:3339–48. 10.1016/j.cclet.2022.03.024
238.
Endo H Shimada H Sahara N Ono M Koga S Kitamura S et al . In vivo binding of a tau imaging probe, [ C]Pbb3, in patients with progressive supranuclear palsy. Mov Disord. (2019) 34:744–54. 10.1002/mds.27643
239.
Schroter N Blazhenets G Frings L Barkhausen C Jost WH Weiller C et al . Tau imaging in the 4-repeat-tauopathies progressive supranuclear palsy and corticobasal syndrome: a 11c-pyridinyl-butadienyl-benzothiazole 3 pet pilot study. Clin Nucl Med. (2020) 45:283–7. 10.1097/RLU.0000000000002949
240.
Koga S Ono M Sahara N Higuchi M Dickson DW . Fluorescence and autoradiographic evaluation of tau pet ligand Pbb3 to Alpha-Synuclein Pathology. Mov Disord. (2017) 32:884–92. 10.1002/mds.27013
241.
Perez-Soriano A Arena JE Dinelle K Miao Q McKenzie J Neilson N et al . Pbb3 imaging in parkinsonian disorders: evidence for binding to tau and other proteins. Mov Disord. (2017) 32:1016–24. 10.1002/mds.27029
242.
Li L Liu FT Li M Lu JY Sun YM Liang X et al . Clinical Utility of F-Apn-1607 Tau Pet imaging in patients with progressive supranuclear palsy. Mov Disord. (2021) 36:2314–23. 10.1002/mds.28672
243.
Kroth H Oden F Molette J Schieferstein H Capotosti F Mueller A et al . Discovery and preclinical characterization of [F]Pi-2620, a next-generation tau pet tracer for the assessment of tau pathology in alzheimer's disease and other tauopathies. Eur J Nucl Med Mol Imaging. (2019) 46:2178–89. 10.1007/s00259-019-04397-2
244.
Tezuka T Takahata K Seki M Tabuchi H Momota Y Shiraiwa M et al . Evaluation of [F]Pi-2620, a second-generation selective tau tracer, for assessing four-repeat tauopathies. Brain Commun. (2021) 3:fcab190. 10.1093/braincomms/fcab190
245.
Brendel M Barthel H van Eimeren T Marek K Beyer L Song M et al . Assessment of 18f-Pi-2620 as a biomarker in progressive supranuclear palsy. JAMA Neurol. (2020) 77:1408–19. 10.1001/jamaneurol.2020.2526
246.
Song M Scheifele M Barthel H van Eimeren T Beyer L Marek K et al . Feasibility of short imaging protocols for [F]Pi-2620 Tau-Pet in progressive supranuclear palsy. Eur J Nucl Med Mol Imaging. (2021) 48:3872–85. 10.1007/s00259-021-05391-3
247.
Palleis C Brendel M Finze A Weidinger E Bötzel K Danek A et al . Cortical [ F]Pi-2620 Binding differentiates corticobasal syndrome subtypes. Mov Disord. (2021) 36:2104–15. 10.1002/mds.28624
248.
Lindberg A Knight AC Sohn D Rakos L Tong J Radelet A et al . Radiosynthesis, in vitro and in vivo evaluation of [f]cbd-2115 as a first-in-Class Radiotracer for Imaging 4r-tauopathies. ACS Chem Neurosci. (2021) 12:596–602. 10.1021/acschemneuro.0c00801
249.
Vandermeeren M Mercken M Vanmechelen E Six J van de Voorde A Martin JJ et al . Detection of tau proteins in normal and alzheimer's disease cerebrospinal fluid with a sensitive sandwich enzyme-linked immunosorbent assay. J Neurochem. (1993) 61:1828–34. 10.1111/j.1471-4159.1993.tb09823.x
250.
Wagshal D Sankaranarayanan S Guss V Hall T Berisha F Lobach I et al . Divergent Csf tau alterations in two common tauopathies: alzheimer's disease and progressive supranuclear palsy. J Neurol Neurosurg Psychiatry. (2015) 86:244–50. 10.1136/jnnp-2014-308004
251.
Lleo A Irwin DJ Illan-Gala I McMillan CT Wolk DA Lee EB et al . A 2-step cerebrospinal algorithm for the selection of frontotemporal lobar degeneration subtypes. JAMA Neurol. (2018) 75:738–45. 10.1001/jamaneurol.2018.0118
252.
Mattsson-Carlgren N Grinberg LT Boxer A Ossenkoppele R Jonsson M Seeley W et al . Cerebrospinal fluid biomarkers in autopsy-confirmed alzheimer disease and frontotemporal lobar degeneration. Neurology. (2022) 98:e1137–e50. 10.1212/WNL.0000000000200040
253.
Abu-Rumeileh S Mometto N Bartoletti-Stella A Polischi B Oppi F Poda R et al . Cerebrospinal fluid biomarkers in patients with frontotemporal dementia spectrum: a single-center study. J Alzheimers Dis. (2018) 66:551–63. 10.3233/JAD-180409
254.
Thijssen EH La Joie R Wolf A Strom A Wang P Iaccarino L et al . Diagnostic value of plasma phosphorylated tau181 in alzheimer's disease and frontotemporal lobar degeneration. Nat Med. (2020) 26:387–97. 10.1038/s41591-020-0762-2
255.
Thijssen EH La Joie R Strom A Fonseca C Iaccarino L Wolf A et al . Plasma phosphorylated tau 217 and phosphorylated tau 181 as biomarkers in alzheimer's disease and frontotemporal lobar degeneration: a retrospective diagnostic performance study. Lancet Neurol. (2021) 20:739–52. 10.1016/S1474-4422(21)00214-3
256.
Aerts MB Esselink RA Bloem BR Verbeek MM . Cerebrospinal Fluid Tau And Phosphorylated Tau Protein Are Elevated In Corticobasal Syndrome. Mov Disord. (2011) 26:169–73. 10.1002/mds.23341
257.
Hall S Ohrfelt A Constantinescu R Andreasson U Surova Y Bostrom F et al . Accuracy of a panel of 5 cerebrospinal fluid biomarkers in the differential diagnosis of patients with dementia and/or parkinsonian disorders. Arch Neurol. (2012) 69:1445–52. 10.1001/archneurol.2012.1654
258.
Shoeibi A Olfati N Litvan I . Preclinical, Phase I, and Phase Ii investigational clinical trials for treatment of progressive supranuclear palsy. Expert Opin Investig Drugs. (2018) 27:349–61. 10.1080/13543784.2018.1460356
259.
Cicognola C Brinkmalm G Wahlgren J Portelius E Gobom J Cullen NC et al . Novel Tau fragments in cerebrospinal fluid: relation to tangle pathology and cognitive decline in alzheimer's disease. Acta Neuropathol. (2019) 137:279–96. 10.1007/s00401-018-1948-2
260.
Rojas JC Karydas A Bang J Tsai RM Blennow K Liman V et al . Plasma neurofilament light chain predicts progression in progressive supranuclear palsy. Ann Clin Transl Neurol. (2016) 3:216–25. 10.1002/acn3.290
261.
Donker Kaat L Meeter LH Chiu WZ Melhem S Boon AJW Blennow K et al . Serum neurofilament light chain in progressive supranuclear palsy. Parkinsonism Relat Disord. (2018) 56:98–101. 10.1016/j.parkreldis.2018.06.018
262.
Rojas JC Bang J Lobach IV Tsai RM Rabinovici GD Miller BL et al . Csf Neurofilament light chain and phosphorylated tau 181 predict disease progression in Psp. Neurology. (2018) 90:e273–e81. 10.1212/WNL.0000000000004859
263.
Constantinescu R Rosengren L Johnels B Zetterberg H Holmberg B . Consecutive analyses of cerebrospinal fluid axonal and glial markers in parkinson's disease and atypical parkinsonian disorders. Parkinsonism Relat Disord. (2010) 16:142–5. 10.1016/j.parkreldis.2009.07.007
264.
Hansson O Janelidze S Hall S Magdalinou N Lees AJ Andreasson U et al . Blood-Based Nfl: a biomarker for differential diagnosis of parkinsonian disorder. Neurology. (2017) 88:930–7. 10.1212/WNL.0000000000003680
265.
Paslawski W Bergström S Zhang X Remnestål J He Y Boxer A et al . Cerebrospinal fluid proteins altered in corticobasal degeneration. Mov Disord. (2021) 36:1278–80. 10.1002/mds.28543
266.
Bougea A . Microrna as candidate biomarkers in atypical parkinsonian syndromes: systematic literature review. Medicina. (2022) 58:483. 10.3390/medicina58040483
267.
Metrick MA 2nd, Ferreira NDC Saijo E Kraus A Newell K Zanusso G et al . A single ultrasensitive assay for detection and discrimination of tau aggregates of alzheimer and pick diseases. Acta Neuropathol Commun. (2020) 8:22. 10.1186/s40478-020-0887-z
268.
Saijo E Metrick MA. 2nd, Koga S Parchi P Litvan I Spina S et al . 4-repeat tau seeds and templating subtypes as brain and Csf biomarkers of frontotemporal lobar degeneration Acta Neuropathol. (2020) 139:63–77. 10.1007/s00401-019-02080-2
Summary
Keywords
tauopathy, movement, clinical, progressive supranuclear palsy, corticobasal, neurodegenerative, frontotemporal dementia, primary progressive aphasia
Citation
Olfati N, Shoeibi A and Litvan I (2022) Clinical Spectrum of Tauopathies. Front. Neurol. 13:944806. doi: 10.3389/fneur.2022.944806
Received
15 May 2022
Accepted
20 June 2022
Published
14 July 2022
Volume
13 - 2022
Edited by
Huifang Shang, Sichuan University, China
Reviewed by
Alexander Murley, University of Cambridge, United Kingdom; Emilia J. Sitek, Medical University of Gdansk, Poland; Donna Clark Tippett, Johns Hopkins Medicine, United States
Updates
Copyright
© 2022 Olfati, Shoeibi and Litvan.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Irene Litvan ilitvan@health.ucsd.edu
This article was submitted to Movement Disorders, a section of the journal Frontiers in Neurology
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.