 Yuri Seo1†
Yuri Seo1† Tae Young Kim2†
Tae Young Kim2† Dongju Won3
Dongju Won3 Saeam Shin3
Saeam Shin3 Jong Rak Choi3,4
Jong Rak Choi3,4 Seung-Tae Lee3,4
Seung-Tae Lee3,4 Byung Joo Lee5Hyun Taek Lim5,6Sueng-Han Han7*‡Jinu Han2*‡
Byung Joo Lee5Hyun Taek Lim5,6Sueng-Han Han7*‡Jinu Han2*‡- 1Department of Ophthalmology, Institute of Vision Research, Yongin Severance Hospital, Yonsei University College of Medicine, Yongin, South Korea
- 2Department of Ophthalmology, Institute of Vision Research, Gangnam Severance Hospital, Yonsei University College of Medicine, Seoul, South Korea
- 3Department of Laboratory Medicine, Yonsei University College of Medicine, Seoul, South Korea
- 4Dxome Co., Ltd., Seongnam-si, South Korea
- 5Department of Ophthalmology, Asan Medical Center, University of Ulsan College of Medicine, Seoul, South Korea
- 6Seoul Orthopia Eye Clinic, Seoul, South Korea
- 7Department of Ophthalmology, Institute of Vision Research, Severance Hospital, Yonsei University College of Medicine, Seoul, South Korea
Aims: To evaluate the clinical characteristics and causative genetic variants in autosomal optic atrophy diagnosed using next-generation sequencing (NGS).
Methods: A cohort of 57 unrelated families affected with bilateral optic atrophy were recruited from two university-based tertiary referral hospitals from May 2016 to April 2022. Genetic variants were detected using a target enrichment panel consisting of 429 or 595 genes and known deep intronic variants associated with inherited eye diseases, exome sequencing, or genome sequencing. The results of detailed clinical examinations, disease-causing variants, and clinical diagnoses were analyzed.
Results: Among the 57 probands, 33 (57.9%) were men, and the median age at genetic testing was 19.1 years (interquartile range, 7.6–42.5 years). We identified 22 likely causative variants in 18 families and corresponding diagnostic yields of 31.6% (95% confidence interval, 21.0–44.5%). The diagnostic rate of NGS was higher in patients with infantile or early childhood onset optic atrophy than in those with late-onset or unknown optic atrophy (18/39, 46.2% vs. 0/18, 0%, P < 0.001). Among the 22 variants, 15 were novel in our cohort. The OPA1 variants (n = 7) were found to be the major genetic causes, followed by the NR2F1 variant (n = 4). The causative variants in PTPN23, TMEM126A, NBAS, and WFS1 genes were identified in 4 probands with a recessive form of optic atrophy.
Conclusions: Based on the results of diagnostic NGS for optic atrophy, the causative variant could be detected in 31.6% of patients. Our study also demonstrated that NGS is unlikely to help identify molecular causes in late-onset unexplained optic atrophy.
Introduction
Inherited optic atrophy is the major underlying etiology of inherited visual impairment (1). This condition is the end result of injury to either the retinal ganglion cells, retinal nerve fiber layer, optic nerve, optic chiasm, or optic tract, and it is caused by various factors such as hereditary, metabolic, radiation-induced, malnutrition-induced, toxic, ischemic, inflammatory, or infiltrative lesions. In patients with optic atrophy, the pertinent history and ophthalmological findings (onset of symptoms, optic disc appearance, pattern of visual field loss, and color vision deficiency) may suggest specific causes of optic atrophy, but not all cases are readily classifiable (2). Defining the cause of optic atrophy in children is more difficult since complete ophthalmological assessments and recording clinical history are sometimes challenging.
To successfully diagnose this condition, first, a thorough gestational, prenatal, and neonatal history is essential because periventricular leukomalacia, intraventricular hemorrhage, or hydrocephalus can cause transsynaptic retrograde degeneration of the optic nerve (3). In addition, a previous history of head trauma, encephalitis, meningitis, malnutrition, or medications (e.g., vigabatrin and ethambutol) should be specifically elicited (2, 4). If no causes of optic atrophy are found in the history or clinical investigation, hereditary causes should be investigated. While dominant optic atrophy and Leber hereditary optic neuropathy (LHON) are the predominant forms of hereditary optic neuropathy (5), other rare diseases such as Bosch-Boonstra-Schaff optic atrophy syndrome (BBSOAS), SSBP1 or RTN4IP1 optic neuropathy, or Costeff syndrome should also be considered in the differential diagnosis (6). Patients with hereditary optic atrophy exhibit either isolated optic nerve dysfunction or accompanying syndromic features such as auditory, neurologic, or systemic abnormalities (7). Therefore, the recognition of accompanying signs and symptoms may aid in the diagnosis.
In recent years, next-generation sequencing (NGS) technology has transitioned from research to clinical use and is being used for inherited retinal diseases, congenital cataracts, and infantile nystagmus syndrome (8–10). Moreover, the use of genetic testing in diagnosis has been gaining more attention due to the recent studies on gene therapies for inherited optic neuropathies (11). Importantly, a few studies have also investigated the use of NGS on patients with hereditary optic atrophy (12–14). However, its clinical utility for diagnosing these patients remains largely unknown. Therefore, herein, the genotypic and phenotypic characteristics of optic atrophy diagnosed with NGS were analyzed in patients with suspected hereditary optic atrophy over the past 6 years in two tertiary referral centers.
Materials and methods
Recruitment and selection of patients with hereditary optic atrophy
The study cohort consisted of 57 unrelated, consecutively sampled patients with suspected hereditary optic atrophy who were undergoing further investigation and were recruited from two university-based tertiary referral hospitals: the Asan Medical Center, Seoul, South Korea and Severance Hospital, Seoul, South Korea between March 2016 and March 2022. All patients had an insidious onset of optic atrophy. Patients who were previously diagnosed with known neurodegenerative disorders commonly associated with optic atrophy such as Leigh syndrome; myoclonic epilepsy with ragged-red fibers; mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes; neuropathy, ataxia, and retinitis pigmentosa; cerebellar ataxia, areflexia, pes cavus, optic atrophy, and sensorineural hearing loss syndrome; Canavan disease; Charcot-Marie-Tooth disease; metachromatic leukodystrophy; metabolic syndromes such as 3-methylglutaconic aciduria or maple syrup urine disease; or Pelizaeus-Merzbacher disease were excluded from the study. Careful history-taking was done to exclude patients with risk factors for any non-genetic causes of optic atrophy such as a history of prematurity (e.g., periventricular leukomalacia and hypoxic ischemic encephalopathy), severe malnutrition, infectious diseases, a previous history of cancer, demyelinating diseases, non-arteritic anterior ischemic optic neuropathy, head trauma, or drug-induced or toxic optic neuropathy (e.g., ethambutol, linezolid or heavy metals).
In this study, all the patients had an insidious onset of bilateral generalized or temporal optic atrophy, and none of the patients displayed band optic atrophy or superior segmental optic atrophy. Sanger sequencing for mitochondrial ND1, ND4, and ND6 genes associated with LHON was also previously conducted in all patients except those with congenital or infantile-onset optic atrophy. No structural abnormalities explaining the bilateral optic atrophy were identified in the previous brain magnetic resonance imaging (MRI) or computerized tomography scans.
All patients underwent ophthalmologic examination, which included the measurement of visual acuity and slit-lamp and fundus examinations. If applicable, a color vision test, spectral domain optical coherence tomography, and automated visual field tests were also performed. Peripheral blood samples from the patients were also collected for genetic analysis. Informed written consents were obtained from all the patients. This study was approved by the institutional review board of Gangnam Severance Hospital, Seoul, South Korea (3-2020-0063) and adhered to the tenets of the Declaration of Helsinki.
NGS analysis
NGS analysis was primarily performed using the customized NGS panel analysis with 429 or 595 targeted genes (Supplementary Tables 1–3) or exome sequencing (ES). Secondarily, exome sequencing (ES) or genome sequencing (GS) were proposed as a diagnostic option for patients when targeted panel sequencing failed to identify the causative variants. With the consent of the patients, target enrichment for targeted panel was performed with a molecular inversion probe-based capture method using a customized target enrichment kit (Dxome, South Korea). ES was performed using either the xGen Exome Research Panel v1 (Integrated DNA technologies, Coralville, IA, USA) or Twist Human Core Exome kit (Twist Bioscience, San Francisco, CA, USA). If the identification was unsuccessful following ES, GS was suggested using the TruSeq Nano DNA sample prep kit (Illumina, San Diego, CA, USA). Pooled libraries were sequenced using NextSeq 550 for the targeted sequencing panel and NovaSeq6000 (Illumina, San Diego, CA, USA) for ES or GS. NGS data analysis was performed primarily through our custom pipeline (9, 15). The interpretation of variants was done according to the 5-tier classification system recommended by the American College of Medical Genetics and Genomics and the Association for Molecular Pathology using a step-by-step approach (Supplementary Figure 1) (16). The systematic approaches for variant classification have also been described in Supplementary Methods.
Results
Clinical characteristics
Among the 57 unrelated Korean patients, 33 patients (57.9%) were male, and the median age of the patients at the time of genetic testing was 19.1 years (interquartile range [IQR], 7.6–42.5 years, Supplementary Figure 2 in the Supplement). The median best-corrected visual acuity was 0.30 (IQR, 0.15–0.56) in the right eye and 0.40 (IQR, 0.13–0.85) in the left eye (logMAR). Seventeen patients (29.8%) had systemic features such as facial dysmorphism, intellectual disability, juvenile-onset diabetes mellitus, cerebellar atrophy, or developmental delay, while the remaining forty patients only had non-syndromic isolated dominant optic atrophy. The onset of optic atrophy was either infantile (n = 14), early childhood (n = 25), late-onset (n = 13), or unknown (n = 5). Seven probands had family histories of optic atrophy. Among these, dominant inheritance was noted in Six patients and autosomal recessive pattern of inheritance in one patient. Sixteen patients (28.1%) had various types of nystagmus including infantile, manifest latent, gaze-evoked, or spasmus nutans-like nystagmus. The clinical features of the patients are summarized in Table 1 and Supplementary Table 4.
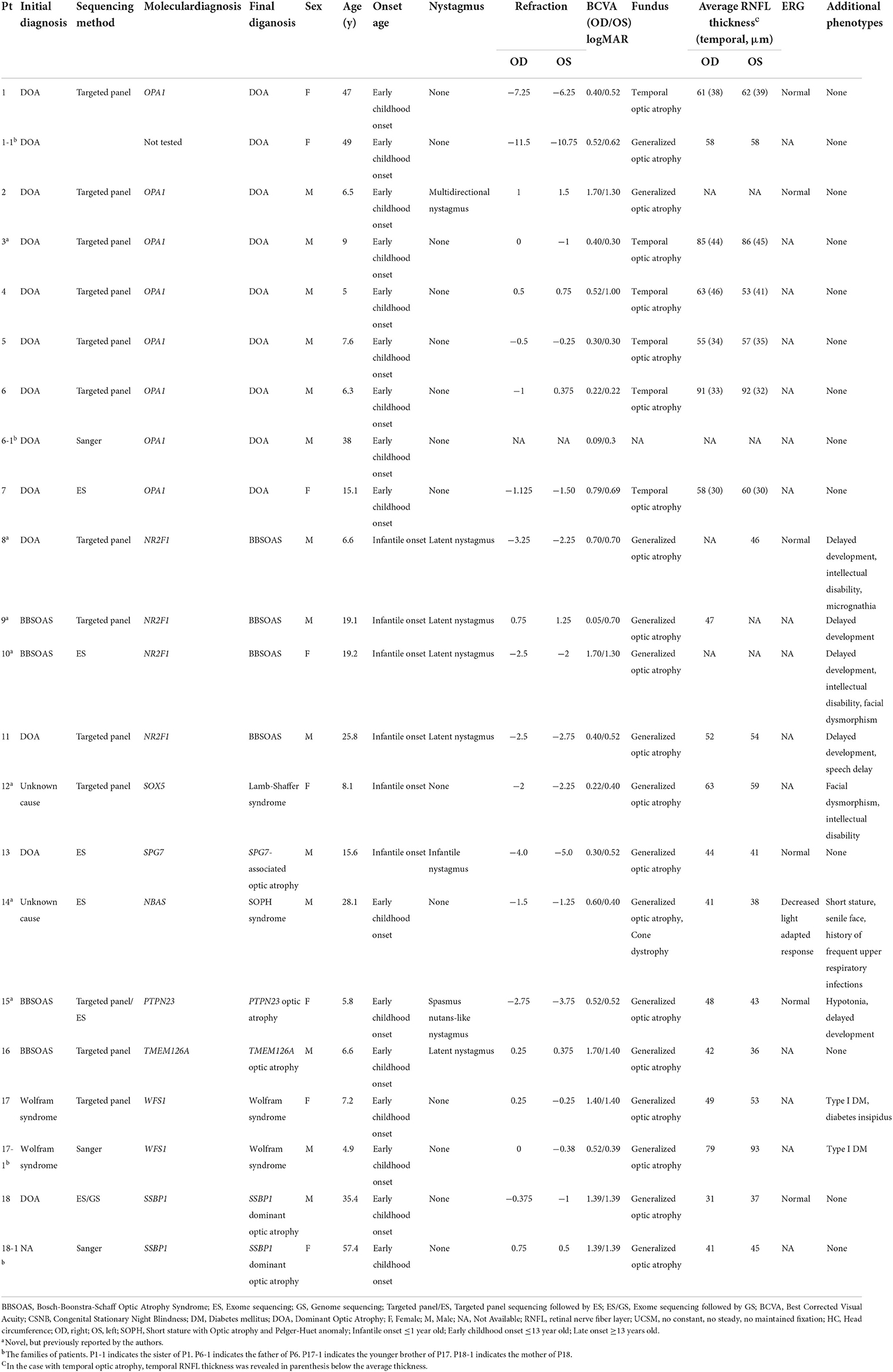
Table 1. The clinical features of 18 patients with hereditary optic atrophy and their families.
NGS results and genetic findings
A total of 18 patients received molecular diagnoses after NGS, while 39 patients remained as unsolved cases, corresponding to a molecular detection rate of 31.6% (95% confidence interval, 21.0–44.5%) (Figure 1). Among the 18 solved cases, 12 cases (66.7%) were diagnosed using targeted panel sequencing and the remaining 6 cases (33.3%) were solved using ES only (n = 4), targeted panel sequencing followed by ES (n = 1) or ES followed by GS (n = 1). For the 39 patients with unsolved cases, either targeted panel sequencing (n = 32) or ES (n = 7) was performed without further testing.
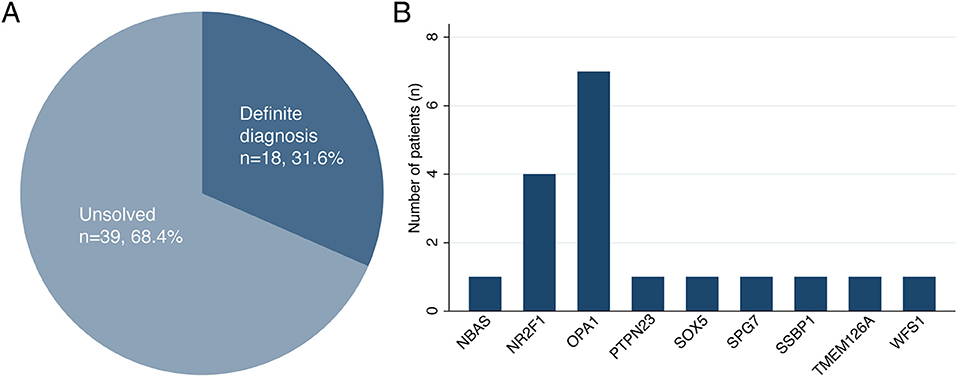
Figure 1. (A,B) The diagnostic rate of next-generation sequencing and genetic distribution of causative variants with hereditary optic atrophy.
Furthermore, a total of 22 disease-causing variants were identified, 15 of which were novel (Table 2; Supplementary Table 5). Among the 39 patients with unsolved cases, one patient was found to have only one likely pathogenic variant in his/her recessive genes (Supplementary Table 6; Supplementary Figure 3. The causative variants were identified in all seven out of the seven patients with a familial history of optic neuropathy (100%), a diagnostic rate higher than in sporadic cases (22.0%) (P < 0.001, Fisher's exact test). Moreover, there was no difference in the diagnostic rate of NGS between the syndromic and non-syndromic cases (47.1 vs. 25.0%, P = 0.101). There was no significant difference in the diagnosis rate according to the presence or absence of nystagmus (43.8 vs. 26.8%, P = 0.217). The diagnostic rate of NGS was much higher in the patients with infantile or early childhood onset optic atrophy than in the patients with late-onset or unknown optic atrophy (18/39, 46.2 vs. 0/18, 0%, P < 0.001).
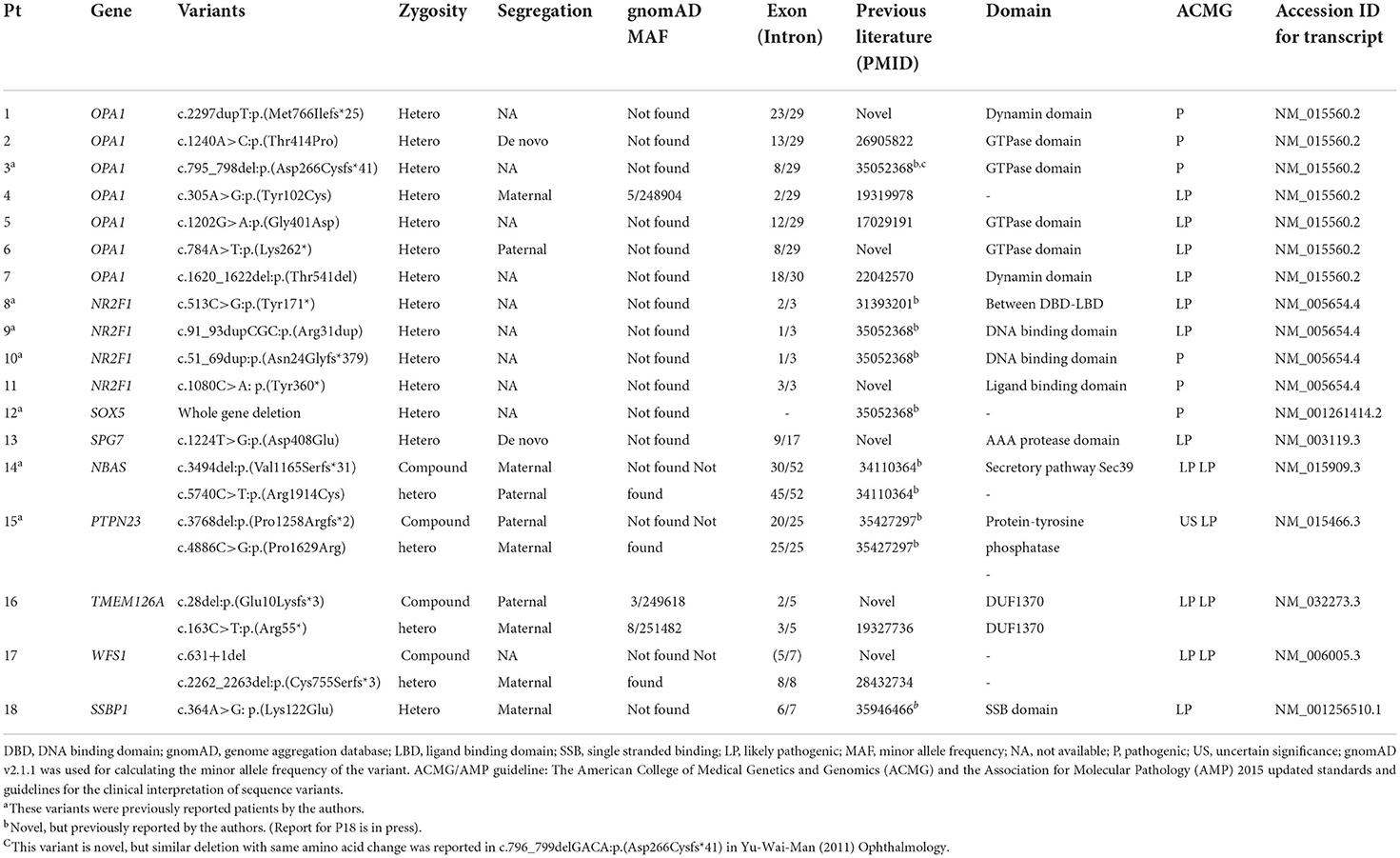
Table 2. Likely causative variants identified in patients with hereditary optic atrophy.
The most frequently mutated genes were OPA1 (n = 7) and NR2F1 (n = 4). The variants in SOX5 (n = 1), SPG7 (n = 1), and SSBP1 (n = 1) genes were responsible for cases of other dominantly inherited optic atrophies. Among the cases of recessive optic atrophy, there was one case of short stature, optic nerve atrophy, Pelger-Huet anomaly (SOPH syndrome) caused by NBAS variants, one case of PTPN23 optic atrophy syndrome, one case of TMEM126A optic atrophy, and one case of Wolfram syndrome. The clinical phenotypes and genotype results of the 18 patients with definite diagnoses are summarized in Tables 1, 2. The initial clinical diagnoses were revised in eight patients (14%) after performing NGS.
Genotype-phenotype correlations
OPA1 dominant optic atrophy
All seven cases of optic atrophy with variants in OPA1 were non-syndromic. P2 exhibited a visual acuity of 1.70 in the right eye and 1.30 in the left eye (logMAR). Furthermore, optical coherence tomography imaging confirmed the presence of severe thinning of the peripapillary retinal nerve fiber layer in all quadrants (Supplementary Figure 4). This patient had been diagnosed with multidirectional nystagmus at 17 months of age. Targeted NGS on this patient revealed a de novo c.1240A>C:p.(Thr414Pro) variant in the OPA1 gene. A previous study reported of a patient with the same variant who had a visual acuity of 1.52 in the right eye and 2.0 in the left eye (logMAR) (17). Notably, missense OPA1 variants located within the GTPase catalytic domain are more likely to cause severe phenotypes than variants resulting in haploinsufficiency (6, 18).
Lamb–Shaffer syndrome
P12 was an 8-year-old female patient presenting with dissociated vertical deviation and inferior oblique muscle overaction. On fundus examination, fundus photography and spectral-domain optical coherence tomography showed bilateral diffuse optic atrophy in both eyes (Figures 2A,B, right eye is shown). Visual acuity was 0.22 in the right eye and 0.40 in the left eye (logMAR). P12 exhibited no definite delayed development or nystagmus, but mild facial dysmorphic features and hirsutism in the philtrum area were noted. Deletion of chromosome 12p12 was suspected based on bioinformatics analysis using CopywriteR program (Figure 2C, red arrow), and array comparative genomic hybridization confirmed 12p12.2p12.1 deletion (hg19:chr12:20286266-25154015, Figure 2D, red arrow). Optic atrophy was considered as a clinical feature of SOX5 gene deletion, known as Lamb–Shaffer syndrome. This chromosomal deletion encompasses several disease-associated genes such as ABCC9, GYS2, LDHB, PDE3A, PYROXD1, SLCO1B1, SLCO1B3, and SOX5. Among these gene, only ABCC9, PDE3A, and SOX5 were inherited as autosomal dominant. In dosage sensitivity curation in ClinGen, only SOX5 had sufficient evidence of haploinsufficiency score: 3. The other genes were not determined yet. All reported variants in PDE3A gene, which causes hypertension and brachydactyly syndrome, were missense variants, and blood pressure, fingers and toes were normal. Because pathogenic variants in ABCC9 gene had been known to cause dilated cardiomyopathy or atrial fibrillation, regular cardiac function check-up was recommended for the patient.
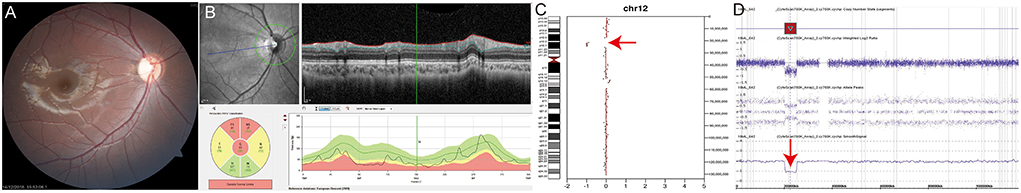
Figure 2. (P12) Chromosomal copy number variations analysis using off-target reads reveals SOX5 deletion in Lamb–Shaffer syndrome. An 8-year-old female patient shows strabismus and hirsutism in the philtrum area. Best corrected visual acuity is 0.22 in the right eye and 0.40 in the left eye (logMAR). (A,B) Fundus photography and spectral-domain optical coherence tomography show optic nerve atrophy in both eyes (right eye was shown). (C) Deletion of the chromosome 12p12 is suspected by bioinformatics analysis using CopywriteR program (red arrow). (D) Array comparative genomic hybridization confirms 12p12.2p12.1 deletion (red arrow). Optic atrophy is thought to be related to SOX5 gene deletion. Regular cardiac function check-up is recommended because ABCC9 gene deletion is found by array comparative genomic hybridization.
TMEM126 optic atrophy
P16 was a 6-year-old male patient with bilateral optic atrophy and low visual acuity. His best corrected visual acuity was 1.70 in the right eye and 1.40 in the left eye (logMAR). He had a mild degree of delayed development and latent nystagmus, but had normal intelligence. The patient's initial clinical diagnosis was BBSOAS. The targeted NGS showed the presence of the novel compound heterozygous TMEM126A c.28del:p.(Glu10Lysfs*3) / c.163C>T:p.(Arg55*) variants (Figure 3A). Based on this, the patient's clinical diagnosis was revised to TMEM126A optic atrophy. Diffuse optic atrophy was observed on fundus photography (Figures 3B,C), and profound loss of peripapillary retinal nerve fiber layer was detected on spectral-domain optical coherence tomography (Figures 3D,E). The location of this novel variant is indicated in a schematic representation of the TMEM126A protein comparing previously reported variants (Figure 3F).
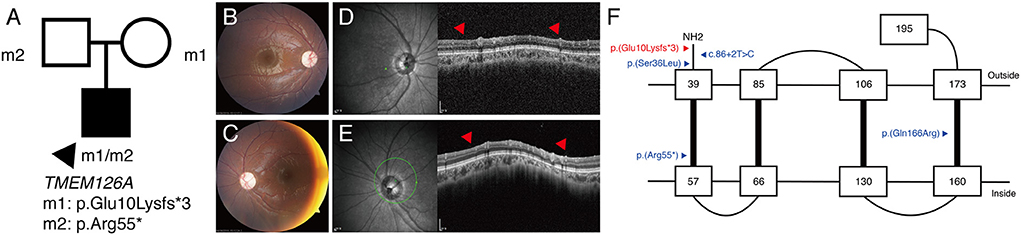
Figure 3. (P16) TMEM126A optic atrophy. The best-corrected visual acuity is 1.70 in the right eye and 1.40 in the left eye (logMAR). (A) Segregation analysis revealing the presence of compound heterozygous TMEM126A variants. (B,C) Fundus photographs showing diffuse optic atrophy (D,E) Profound loss of peripapillary retinal nerve fiber layer is detected in both eyes (red arrowhead). (F) Schematic representation of the TMEM126A protein and previously reported variants (blue). The red color indicates the novel variant in this study.
Wolfram syndrome
P17 was a 7-year-old female patient who was brought to the emergency room with a complaint of severe fatigue. Examination revealed diabetic ketoacidosis, which was her first diagnosis of diabetes. She was diagnosed with optic atrophy 2 years ago and had never been genetically evaluated. Her best corrected visual acuity was 1.4 in both eyes (logMAR) and showed no definite nystagmus. She was diagnosed with type 1 diabetes mellitus, but no definite developmental delay or intellectual disability was noted. The segregation analysis revealed compound heterozygous c.631+1del/c.2262_2263del:p.(Cys755Serfs3) variants. Therefore, she was diagnosed with Wolfram syndrome. These variants were also found in her brother (Figure 4A). The c.631+1del variant was identified as a novel variant (Supplementary Figure 5). Generalized optic atrophy was shown on fundus photography and optical coherence tomography (Figures 4B–E). An audiometry test showed normal hearing function (Figure 4F).
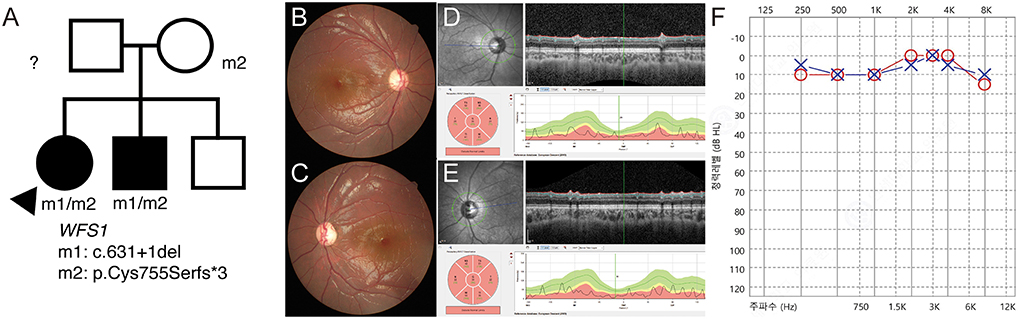
Figure 4. (P17) Targeted next-generation sequencing identify WFS1 variants in optic atrophy with diabetic ketoacidosis. Clinical diagnosis before genetic testing is Wolfram syndrome. Best corrected visual acuity is 1.40 logMAR in both eyes. (A) The segregation analysis shows compound heterozygous c.631+1del/c.2262_2263del:p.(Cys755Serfs*3) variants. These variants are also detected in her brother. The paternal sample is not available. (B,C) Fundus photographs showing generalized diffuse optic atrophy. (D,E) The optical coherence tomography revealing generalized retinal nerve thinning. (F) Audiometry is normal at the age of 7 years.
The clinical features of the BBSOAS in P11 is presented in Supplementary Figure 6. In early childhood, P11 presented delayed speech that was corrected with rehabilitation. At the age of 25-years old, P11 revealed normal intelligence and no other neurological symptoms. Fundus photographs revealed diffuse RNFL thinning. BCVA was 0.4 in the right eye and 0.52 in the left eye (logMAR). Targeted sequencing identified c.1080C>A:p.(Tyr360*) variant in NR2F1.
The clinical information of SOPH syndrome and PTPN23 optic atrophy were described in our previous study (19, 20). P8, P9, and P10 were reported in our previous study (1, 21).
Discussion
The molecular diagnostic yield for hereditary optic atrophy is relatively low compared to that for other hereditary eye diseases such as inherited retinal dystrophy or infantile nystagmus syndrome (diagnostic rate: 75–90%) (1, 9). Previous studies reported various diagnostic rates for hereditary optic atrophy ranging from 20.2–40% (12–14). Our study demonstrated that NGS identified the causative variants in 31.6% of the patients with suspected hereditary optic atrophy. This result is comparable to that of previous studies. In addition, variants in OPA1 were observed to be major causes of optic atrophy in our cohort, accounting for 38.9% of our solved cases. BBSOAS was also found to be a common cause of optic atrophy, accounting for 22.2% of our solved cases. A previous study reported that 78.9% of solved cases in their study were caused mainly by OPA1 gene variants followed by WFS1 gene variants (12). Yu-Wai-Man et al. reported that screening the OPA1 and OPA3 genes allowed for the detection of pathogenic variants in 27 (14.4%) of the 188 probands in their study, a finding which is consistent with our study (22). No OPA3 variants were found in our cohort.
Unexplained insidious onset of optic atrophy in adulthood might result from genetic defects. A previous study reported that the age of onset of isolated optic atrophy was 20–50 years in AFG3L2 and SPG7 dominant optic atrophy. However, our study failed to identify the responsible genetic causes for unexplained adult-onset optic atrophies. This can be interpreted in various ways. A recent study found dominant MEIF1 variants as a cause for late-onset optic neuropathy in two unrelated patients (23). Therefore, other novel genes, including MEIF1, might be the cause of late-onset optic neuropathy in our cohort. Second, non-coding pathogenic variants or structural variants can be missed, and those were misclassified as unsolved. Third, unknown cause of late-onset optic atrophy can be resulted from toxic, medication-induced, or environmental causes which patients did not recognize previously.
The SOX5 gene encodes the member of the SOX (SRY-related high mobility group-box) gene family that is related to the regulation of embryonic development such as neurogenesis and skeletogenesis (24). Haploinsufficiency of SOX5 causes Lamb–Shaffer syndrome, clinically characterized with developmental delay, speech delay, and behavioral disturbances (25). Ophthalmic features, such as strabismus, optic nerve atrophy, amblyopia, and cortical visual impairment have been frequently reported (26, 27). In our patient, mild intellectual disability and facial dysmorphism was noted, which have been known as the most common symptoms of Lamb–Shaffer syndrome (28). The large deletion also included ABCC9 gene, and pathogenic variant in this gene is known to cause cardiomyopathy, excess hair growth, and intellectual disability named as Cantú syndrome. Most reported pathogenic variant in ABCC9 gene was missense, and null variants had been reported to be associated with dilated cardiomyopathy. Therefore, regular monitoring of cardiac function is needed in this patient.
TMEM126A is a gene that encodes an assembly factor for the ND4-module of mitochondrial complex I (29, 30), in which variants of this gene cause non-syndromic autosomal-recessive optic atrophy (31). The TMEM126A protein is located in the mitochondrial cristae, along with the OPA1 protein. Notably, our case is the very first case of TMEM126A optic atrophy reported in East Asian patients. The c.163C>T/p:(Arg55*) variant has been well known as a founder variant in North African descent (31, 32). The minor allele frequencies of the novel c.28del:p.(Glu10Lysfs*3) variant were reported to be 3/18358 in East Asians in the genome aggregation database (gnomAD) and 0.0002 in the 4.7K ToMMo (Tohoku Medical Megabank Organization) Japanese database (33). Therefore, we expect more cases with TMEM126A optic atrophy to be discovered among East Asian patients.
Wolfram syndrome has clinically been termed as “DIDMOAD” (diabetes insipidus, diabetes mellitus, optic atrophy, and deafness), and is usually inherited with an autosomal recessive pattern. WFS1 encodes wolframin which is a transmembrane protein localized to the endoplasmic reticulum and is highly expressed in pancreatic beta cells and neuronal cells (34). The functional loss of wolframin causes the aggregation of misfolded protein in the endoplasmic reticulum, resulting in retinal ganglion cell death. Diabetes tends to appear as the first sign of Wolfram syndrome, followed by optic atrophy in the early twenties (35). Optic atrophy presented in about 84.4% of patients with Wolfram syndrome and the decline in visual acuity progressed to 20/200 or worse within 8 years from the onset of disease (36). However, P17 presented with optic atrophy as the first symptom in early childhood and was diagnosed with diabetes 2 years later. Her younger brother also had the same history of being diagnosed with optic atrophy first and then later with diabetes. Therefore, if isolated optic atrophy is diagnosed in early childhood and a genetic cause is strongly suspected, genetic evaluation of the patient is strongly recommended and regular check-up is necessary.
This study had several limitations. First, the study was limited by its retrospective design and all the patients in this study were of Korean ethnicity. Thus, it should be noted that other genetic backgrounds may have different profiles of gene variants. Although our targeted panel included 595 genes and deep intronic c.713-1075C>G in the WFS1 gene, other deep intronic variants (Supplementary Table 7), secondary mitochondrial DNA variations or copy number variations could have been missed. Second, our panel also did not include the PTPN23 gene, which was recently reported to be involved in hereditary optic atrophy (37). However, our targeted panel captures the poor coverage region efficiently in the NR2F1 exon 1 (Supplementary Figure 7). Third, our panel did not include DNAJC30 gene that is recently known as a cause of autosomal recessive LHON (38). We conducted exome re-analysis in 17 patients, but no candidate variants were found in DNAJC30 gene. In addition, LHON sequencing was not performed in patients with congenital or infantile-onset optic atrophy. As childhood onset LHON had been reported (39, 40), this could be one of limitations of the study. Fourth, segregation analyses could not be performed for all the patients. Fifth, we did not perform functional studies to corroborate the pathogenicity of the novel variants identified in this study. Also, the minor allele frequency of c.305A>G OPA1 variant was relatively high (5/248904), and unaffected mother of the proband also had this variant. Although previous studies reported this variant as disease-causing and non-penetrance was well characterized in OPA1-dominant optic atrophy (41, 42), this variant might be questionable in pathogenicity. Lastly, although we attempted to carefully exclude non-genetic causes of optic neuropathies, there may have been other non-genetic causes of optic atrophy that were missed in the unsolved patients.
This study demonstrated that NGS can be used for the diagnosis of patients with hereditary optic atrophy, with 31.6% of patients in our cohort having a definite diagnosis. This study emphasized that examining genotype-phenotype correlations and family segregation analyses are important in interpreting genetic variations in patients with hereditary optic atrophy. Accurate molecular diagnosis will enable ophthalmologists to conduct genetic counseling for future family planning and patient-specific diagnostic workups, including diabetes screening, auditory function tests, and cardiac evaluation. Given that optic atrophy has various causes, the careful collection of patient history, recognition of syndromic features, appropriate brain imaging, and laboratory investigations should be considered first to avoid unnecessary genetic investigations (Supplementary Figure 8).
Data availability statement
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding author. Requests to access the datasets should be directed to JH, amludWhhbkB5dWhzLmFj.
Ethics statement
This study was approved by the institutional review board of Gangnam Severance Hospital, Seoul, South Korea (3-2020-0063) and adhered to the tenets of the Declaration of Helsinki. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin.
Author contributions
JH and S-HH contributed to conception and design of the study. YS and TK organized the database. YS and JH performed the statistical analysis and wrote the first draft of the manuscript. DW, SS, JC, S-TL, HL, and S-HH wrote sections of the manuscript. BL acquired and analyzed the data. All authors contributed to manuscript revision, read, and approved the submitted version.
Funding
This study was supported by a Faculty Research Grant of Yonsei University College of Medicine (6-2017-0044) (6-2020-0146) and National Research Foundation of Korea (NRF) grant funded by the Korean Government (MSIT) (No. 2020R1C1C1007965).
Conflict of interest
JC and S-TL were employed by Dxome Co., Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2022.978532/full#supplementary-material
Abbreviations
NGS, next-generation sequencing; ES, exome sequencing; GS, genome sequencing; BBSOAS, Bosch-Boonstra-Schaff optic atrophy syndrome.
References
1. Moon D, Park HW, Surl D, Won D, Lee ST, Shin S, et al. Precision medicine through next-generation sequencing in inherited eye diseases in a Korean Cohort. Genes. (2021) 13:27. doi: 10.3390/genes13010027
2. Brodsky MC. Optic atrophy in children. Pediatric Neuro-Ophthalmology. Berlin: Springer (2016). p. 569–95.
3. Rudanko SL, Fellman V, Laatikainen L. Visual Impairment in Children Born Prematurely from 1972 through 1989. Ophthalmology. (2003) 110:1639–45. doi: 10.1016/S0161-6420(03)00498-6
4. Lee AG, Chau FY, Golnik KC, Kardon RH, Wall M. The diagnostic yield of the evaluation for isolated unexplained optic atrophy. Ophthalmology. (2005) 112:757–9. doi: 10.1016/j.ophtha.2004.12.009
5. Newman NJ. Hereditary optic neuropathies: from the mitochondria to the optic nerve. Am J Ophthalmol. (2005) 140:517–23. doi: 10.1016/j.ajo.2005.03.017
6. Lenaers G, Neutzner A, Le Dantec Y, Jüschke C, Xiao T, Decembrini S, et al. Dominant optic atrophy: culprit mitochondria in the optic nerve. Prog Retin Eye Res. (2020) 83:100935. doi: 10.1016/j.preteyeres.2020.100935
7. Yu-Wai-Man P, Griffiths PG, Gorman GS, Lourenco CM, Wright AF, Auer-Grumbach M, et al. Multi-system neurological disease is common in patients with opa1 mutations. Brain. (2010) 133:771–86. doi: 10.1093/brain/awq007
8. Gillespie RL, O'Sullivan J, Ashworth J, Bhaskar S, Williams S, Biswas S, et al. Personalized diagnosis and management of congenital cataract by next-generation sequencing. Ophthalmology. (2014) 121:2124-37.e1–2. doi: 10.1016/j.ophtha.2014.06.006
9. Rim JH, Lee ST, Gee HY, Lee BJ, Choi JR, Park HW, et al. Accuracy of next-generation sequencing for molecular diagnosis in patients with infantile nystagmus syndrome. JAMA Ophthalmol. (2017) 135:1376–85. doi: 10.1001/jamaophthalmol.2017.4859
10. Stark Z, Dolman L, Manolio TA, Ozenberger B, Hill SL, Caulfied MJ, et al. Integrating genomics into healthcare: a global responsibility. Am J Hum Genet. (2019) 104:13–20. doi: 10.1016/j.ajhg.2018.11.014
11. Newman NJ, Yu-Wai-Man P, Carelli V, Moster ML, Biousse V, Vignal-Clermont C, et al. Efficacy and safety of intravitreal gene therapy for leber hereditary optic neuropathy treated within 6 months of disease onset. Ophthalmology. (2021) 128:649–60. doi: 10.1016/j.ophtha.2020.12.012
12. Chen AT, Brady L, Bulman DE, Sundaram ANE, Rodriguez AR, Margolin E, et al. An evaluation of genetic causes and environmental risks for bilateral optic atrophy. PLoS ONE. (2019) 14:e0225656. doi: 10.1371/journal.pone.0225656
13. Li JK Li W, Gao FJ, Qu SF, Hu FY, Zhang SH, et al. Mutation screening of mtdna combined targeted exon sequencing in a cohort with suspected hereditary optic neuropathy. Transl Vis Sci Technol. (2020) 9:11. doi: 10.1167/tvst.9.8.11
14. Charif M, Bris C, Goudenege D, Desquiret-Dumas V, Colin E, Ziegler A, et al. Use of next-generation sequencing for the molecular diagnosis of 1,102 patients with a autosomal optic neuropathy. Front Neurol. (2021) 12:602979. doi: 10.3389/fneur.2021.602979
15. Lee J, Jeong H, Won D, Shin S, Lee ST, Choi JR, et al. Noncanonical splice site and deep intronic Frmd7 variants activate cryptic exons in X-linked infantile nystagmus. Transl Vis Sci Technol. (2022) 11:25. doi: 10.1167/tvst.11.6.25
16. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
17. Namba K, Mutai H, Takiguchi Y, Yagi H, Okuyama T, Oba S, et al. Molecular impairment mechanisms of novel opa1 mutations predicted by molecular modeling in patients with autosomal dominant optic atrophy and auditory neuropathy spectrum disorder. Otol Neurotol. (2016) 37:394–402. doi: 10.1097/MAO.0000000000000978
18. Yu-Wai-Man P, Trenell MI, Hollingsworth KG, Griffiths PG, Chinnery PF. Opa1 mutations impair mitochondrial function in both pure and complicated dominant optic atrophy. Brain. (2011) 134(Pt 4):e164. doi: 10.1093/brain/awq288
19. Seo Y, Kim SS, Han J. Short stature with optic atrophy and cone dystrophy. JAMA Ophthalmol. (2021) 139:910–1. doi: 10.1001/jamaophthalmol.2020.5777
20. Seo Y, Kim TY, Won D, Choi JR, Seo GH, Lee ST, et al. Ptpn23 neurodevelopmental disorder presenting with optic atrophy and spasmus nutans-like nystagmus. J Neuroophthalmol. (2022). doi: 10.1097/WNO.0000000000001582
21. Park SE, Lee JS, Lee ST, Kim HY, Han SH, Han J. Targeted panel sequencing identifies a novel Nr2f1 mutations in a patient with bosch-boonstra-schaaf optic atrophy syndrome. Ophthalmic Genet. (2019) 40:359–61. doi: 10.1080/13816810.2019.1650074
22. Yu-Wai-Man P, Shankar SP, Biousse V, Miller NR, Bean LJ, Coffee B, et al. Genetic screening for Opa1 and Opa3 mutations in patients with suspected inherited optic neuropathies. Ophthalmology. (2011) 118:558–63. doi: 10.1016/j.ophtha.2010.07.029
23. Charif M, Wong YC, Kim S, Guichet A, Vignal C, Zanlonghi X, et al. Dominant mutations in Mief1 affect mitochondrial dynamics and cause a singular late onset optic neuropathy. Mol Neurodegener. (2021) 16:12. doi: 10.1186/s13024-021-00431-w
24. Kwan KY, Lam MM, Krsnik Z, Kawasawa YI, Lefebvre V, Sestan N. Sox5 postmitotically regulates migration, postmigratory differentiation, and projections of subplate and deep-layer neocortical neurons. Proc Natl Acad Sci U S A. (2008) 105:16021–6. doi: 10.1073/pnas.0806791105
25. Lamb AN, Rosenfeld JA, Neill NJ, Talkowski ME, Blumenthal I, Girirajan S, et al. Haploinsufficiency of Sox5 at 12p121 is associated with developmental delays with prominent language delay, behavior problems, and mild dysmorphic features. Hum Mutat. (2012) 33:728–40. doi: 10.1002/humu.22037
26. Nesbitt A, Bhoj EJ, McDonald Gibson K, Yu Z, Denenberg E, Sarmady M, et al. Exome sequencing expands the mechanism of Sox5-Associated intellectual disability: a case presentation with review of sox-related disorders. Am J Med Genet A. (2015) 167a:2548–54. doi: 10.1002/ajmg.a.37221
27. Bosch DG, Boonstra FN, de Leeuw N, Pfundt R, Nillesen WM, de Ligt J, et al. Novel genetic causes for cerebral visual impairment. Eur J Hum Genet. (2016) 24:660–5. doi: 10.1038/ejhg.2015.186
28. Schanze I, Schanze D, Bacino CA, Douzgou S, Kerr B, Zenker M. Haploinsufficiency of Sox5, a member of the sox (Sry-Related Hmg-Box) family of transcription factors is a cause of intellectual disability. Eur J Med Genet. (2013) 56:108–13. doi: 10.1016/j.ejmg.2012.11.001
29. D'Angelo L, Astro E, De Luise M, Kurelac I, Umesh-Ganesh N, Ding S, et al. Ndufs3 depletion permits Complex I maturation and reveals Tmem126a/Opa7 as an assembly factor binding the Nd4-module intermediate. Cell Rep. (2021) 35:109002. doi: 10.1016/j.celrep.2021.109002
30. Formosa LE, Reljic B, Sharpe AJ, Hock DH, Muellner-Wong L, Stroud DA, et al. Optic atrophy-associated Tmem126a is an assembly factor for the Nd4-Module of mitochondrial complex I. Proc Natl Acad Sci U S A. (2021) 118:e2019665118. doi: 10.1073/pnas.2019665118
31. Hanein S, Perrault I, Roche O, Gerber S, Khadom N, Rio M, et al. Tmem126a, encoding a mitochondrial protein, is mutated in autosomal-recessive nonsyndromic optic atrophy. Am J Hum Genet. (2009) 84:493–8. doi: 10.1016/j.ajhg.2009.03.003
32. La Morgia C, Caporali L, Tagliavini F, Palombo F, Carbonelli M, Liguori R, et al. First Tmem126a missense mutation in an italian proband with optic atrophy and deafness. Neurol Genet. (2019) 5:e329. doi: 10.1212/NXG.0000000000000329
33. Tadaka S, Katsuoka F, Ueki M, Kojima K, Makino S, Saito S, et al. 3.5kjpnv2: an allele frequency panel of 3552 Japanese individuals including the X chromosome. Hum Genome Var. (2019) 6:28. doi: 10.1038/s41439-019-0059-5
34. Schmidt-Kastner R, Kreczmanski P, Preising M, Diederen R, Schmitz C, Reis D, et al. Expression of the diabetes risk gene wolframin (Wfs1) in the human retina. Exp Eye Res. (2009) 89:568–74. doi: 10.1016/j.exer.2009.05.007
35. de Heredia ML, Clèries R, Nunes V. Genotypic classification of patients with wolfram syndrome: insights into the natural history of the disease and correlation with phenotype. Genet Med. (2013) 15:497–506. doi: 10.1038/gim.2012.180
36. Barrett TG, Bundey SE, Fielder AR, Good PA. Optic atrophy in wolfram (Didmoad) syndrome. Eye. (1997) 11:882–8. doi: 10.1038/eye.1997.226
37. Jurkute N, Leu C, Pogoda HM, Arno G, Robson AG, Nürnberg G, et al. Ssbp1 mutations in dominant optic atrophy with variable retinal degeneration. Ann Neurol. (2019) 86:368–83. doi: 10.1002/ana.25550
38. Stenton SL, Sheremet NL, Catarino CB, Andreeva NA, Assouline Z, Barboni P, et al. Impaired complex I repair causes recessive leber's hereditary optic neuropathy. J Clin Invest. (2021) 131:e138267. doi: 10.1172/JCI138267
39. Barboni P, Savini G, Valentino ML, La Morgia C, Bellusci C, De Negri AM, et al. Leber's hereditary optic neuropathy with childhood onset. Invest Ophthalmol Vis Sci. (2006) 47:5303–9. doi: 10.1167/iovs.06-0520
40. Majander A, Bowman R, Poulton J, Antcliff RJ, Reddy MA, Michaelides M, et al. Childhood-onset leber hereditary optic neuropathy. Br J Ophthalmol. (2017) 101:1505–9. doi: 10.1136/bjophthalmol-2016-310072
41. Ferré M, Bonneau D, Milea D, Chevrollier A, Verny C, Dollfus H, et al. Molecular screening of 980 cases of suspected hereditary optic neuropathy with a report on 77 novel opa1 mutations. Hum Mutat. (2009) 30:E692–705. doi: 10.1002/humu.21025
Keywords: hereditary optic atrophy, inherited optic neuropathy, OPA1, TMEM126A, SOX5
Citation: Seo Y, Kim TY, Won D, Shin S, Choi JR, Lee S-T, Lee BJ, Lim HT, Han S-H and Han J (2022) Genetic spectrum and characteristics of autosomal optic neuropathy in Korean: Use of next-generation sequencing in suspected hereditary optic atrophy. Front. Neurol. 13:978532. doi: 10.3389/fneur.2022.978532
Received: 26 June 2022; Accepted: 29 July 2022;
Published: 22 August 2022.
Edited by:
Chiara La Morgia, IRCCS Institute of Neurological Sciences of Bologna (ISNB), ItalyReviewed by:
Bernd Wissinger, Centre for Ophthalmology, University Clinics Tuebingen, GermanyLeonardo Caporali, IRCCS Institute of Neurological Sciences of Bologna (ISNB), Italy
Valerio Carelli, University of Bologna, Italy
Copyright © 2022 Seo, Kim, Won, Shin, Choi, Lee, Lee, Lim, Han and Han. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jinu Han, amludWhhbkB5dWhzLmFj; Sueng-Han Han, c2hoYW4yMjJAeXVocy5hYw==
†These authors have contributed equally to this work and share first authorship
‡These authors have contributed equally to this work