 Kenneth C. Gorson
Kenneth C. Gorson- Department of Neurology, St Elizabeth’s Medical Center, Tufts University School of Medicine, Boston, MA, United States
Guillain-Barré syndrome (GBS) is a rare, frequently postinfectious neuromuscular emergency and the leading cause of acute paralytic neuropathy worldwide. GBS incidence varies considerably across geographic regions, owing predominantly to different infectious exposures. In GBS, antecedent infection leads to production of immunoglobulin G and immunoglobulin M antibodies that cross-react with the myelin sheath and axons of peripheral nerves. These antibodies activate the classical complement pathway, which plays a key role in peripheral nerve injury regardless of autoantibody binding to myelin or axons as a target. The heterogeneous clinical presentation and progression of GBS symptoms have long been attributed to binary axonal and demyelinating neurophysiologic classifications; however, evolving evidence indicates that these pathophysiologic processes overlap. Intravenous immunoglobulin and plasma exchange, the current standard-of-care therapies in GBS, both reduce autoantibody levels and complement activation, thereby aiming to address this convergence of pathophysiology. However, these therapies only partially decrease antibody levels and complement activity and require extended courses of treatment (5 days for intravenous immunoglobulin and 7–14 days for plasma exchange), limiting their effectiveness in addressing acute neuronal damage during the active phase of disease. Given its evolutionary role in antibody binding and activating the classical complement pathway, the complement component C1q has been proposed as a therapeutic target in GBS. The clinical trial program of the C1q inhibitor ANX005, including placebo-controlled, double-blind phase 1b and phase 3 trials in GBS, provides insight into the pathophysiology of GBS and the efficacy of C1q inhibition regardless of neurophysiologic classification or geographic location.
1 Introduction
Guillain-Barré syndrome (GBS) is a neuromuscular emergency and the most common and severe acute paralytic neuropathy worldwide (1, 2). The global incidence of GBS ranges from 0.30 to 6.08 cases per 100,000 population depending on geography, age, exposures to infections, and other risk factors (3, 4). The incidence is especially high in low- and middle-income countries, including Bangladesh and the Philippines (5, 6).
In most cases, GBS occurs following an antecedent infection in a previously healthy patient. The disease often begins with muscle weakness that rapidly progresses over a few days and, in up to 30% of patients, can lead to respiratory failure requiring mechanical ventilation and intensive care admission (2, 7, 8). Patients may also present with sensory symptoms, such as pain and paresthesia, as well autonomic dysfunction that may result in life-threatening hemodynamic instability or arrhythmias (1, 2). Mortality rates range from 3 to 10% (1, 9–11) and are highest among those who require mechanical ventilation (12, 13). Although most deaths in GBS occur in the first 6 months after disease onset, mortality risk may be increased for several years beyond the acute phase of illness and does not appear to be influenced by current treatments (1, 14, 15).
Many patients with GBS who achieve good functional recovery following the acute phase of disease may still experience prolonged pain and fatigue (2). The prevalence of residual pain and other sensory deficits varies from 33% to 82% depending on symptom location, muscle group (for those with residual chronic muscle pain), and subtype of sensory deficit (e.g., paresthesia, dysesthesias, radicular or neuropathic pain, allodynia) (16–20). Sensory deficits are associated with disruptions in quality of life, while pain intensity correlates with disability, strength impairment, and fatigue (16, 19). As with disease incidence, mortality and other outcomes in GBS may vary regionally, based on extrinsic factors, including access to health care resources (6, 9, 21, 22).
GBS is a prototypical postinfectious autoimmune disorder (2, 23), and Campylobacter jejuni is the most prevalent antecedent infection worldwide (24, 25). The prevailing evidence strongly supports molecular mimicry as the pathogenic mechanism of disease (26, 27). Antibodies that are generated against C jejuni or other infectious agents cross-react with antigens in peripheral nerve axons and myelin (27, 28). Serum immunoglobulin (Ig) M and IgG antibodies against gangliosides or glycolipid complexes are detected in up to 92% of patients with GBS during the acute, active phase of disease, and their levels subsequently decline in the following weeks (27, 29–31). Regardless of the antecedent infection, the cross-reactive autoantibodies engage the classical complement pathway, which drives the peripheral nerve injury and neuroinflammation underlying disease pathology (23, 27, 28, 32–34).
The heterogeneous clinical presentation and progression of symptoms in GBS may lead to diagnostic delay during the active phase of the disease, further underscoring the need for a treatment that rapidly and completely inhibits key pathophysiologic components of disease once the diagnosis is confirmed.
2 Pathophysiology of GBS
2.1 Reappraisal of the distinction between axonal and demyelinating mechanisms in GBS
The variability in prognosis and long-term recovery in GBS has historically been attributed to the specific site of peripheral nerve damage, as characterized by nerve conduction studies (NCSs). In this classification, the primary targets of immune-mediated attack are either the axonal components of peripheral nerves, leading to acute motor axonal neuropathy (AMAN), or the myelin sheath (produced by Schwann cells), resulting in acute inflammatory demyelinating polyneuropathy (AIDP). AMAN is often associated with more severe disease (27, 35). The relative frequencies of each NCS subtype vary across geographic regions, potentially due to differing infectious exposures or genetic susceptibilities (27, 36).
Despite the dichotomized NCS classifications, the diagnostic criteria for GBS are identical for AMAN and AIDP, and neither classification dictates treatment strategies or prognosis (37–43). While earlier studies suggested that patients with AMAN experience a worse prognosis compared with those with AIDP (12, 27), more recent research indicates that other factors such as baseline severity of weakness, as measured by Medical Research Council (MRC) scale sum score, and serum and cerebrospinal fluid (CSF) neurofilament light chain (NfL) and neurofilament heavy chain levels during active disease are more relevant in predicting outcome in patients with GBS (12, 44–48). Acutely elevated serum NfL levels reflect the level of axonal damage in early disease and are elevated in both the axonal and demyelinating NCS subtypes, indicating varying degrees of axonal involvement, even in those patients with a primarily demyelinating presentation (45).
Axonal loss has been demonstrated in AIDP as well as AMAN and is the main determinant of long-term disability (35, 48, 49). Ultimately, electrodiagnostic studies often reveal a mixed pattern of demyelination and axonal damage or yield inconclusive results. In a retrospective study of patients with very early-stage GBS who had initial NCSs performed within 4 days of symptom onset, classification was possible in only 20% of patients. Specifically, 40% showed a mixed pattern of demyelination and axonal degeneration on NCSs, and 33% showed equivocal results (50). Similarly, in a study of patients with AIDP assessed within 10 days of symptom onset, NCSs often showed nonspecific abnormalities (51). Furthermore, the classification of electrodiagnostic findings in GBS appears to vary based on which NCS criteria are used. In an analysis of 1,137 NCSs from the first 1,500 patients enrolled in the International GBS Outcome Study, only 68% were classified identically according to the Hadden and Rajabally criteria, which are among the most commonly used NCS criteria in GBS research (52). NCS abnormalities are typically not confirmed via pathologic material (i.e., nerve biopsy), further complicating these seemingly artificial distinctions.
Given these limitations, a reappraisal of the oversimplified classification of GBS into axonal and demyelinating forms is warranted from diagnostic, pathophysiologic, and prognostic standpoints. The evolving understanding of common pathways in GBS pathophysiology further challenges the application of rigid electrophysiologic classifications when considering current and future treatment options. Regardless of NCS findings, GBS represents a neuromuscular emergency (1, 2, 39, 43), in which rapid, effective treatment is required to prevent acute and ongoing damage to axons and mitigate neuroinflammation (2, 8, 42, 53). As research advances in GBS and treatment becomes further personalized, pathophysiological differences between AMAN and AIDP which are clinically relevant may be uncovered or targeted with new treatments. However, the convergence of axonal loss in both NCS classifications also suggests that a unified, targeted treatment approach that addresses complement-mediated nerve damage driven by autoantibodies against either myelin or axons can be effective for all patients with GBS.
2.2 Revelation of key mechanistic characteristics in GBS based on current standard-of-care therapies
Intravenous immunoglobulin (IVIg) and plasma exchange (PE) have been the standard treatments for GBS since their introduction in the 1980s (53–55). Both are believed to function by reducing circulating autoantibody levels and decreasing complement activity (27, 35, 56–59). IVIg is used in most patients with GBS, given its relative ease of administration and accessibility (54, 58, 60). However, evidence suggests that IVIg and PE are equivalent in efficacy (35, 42, 53, 55, 60, 61), consistent with their overlapping mechanisms and the core pathophysiology of GBS. Both IVIg and PE require a prolonged treatment cycle, typically spanning 5 days for IVIg and 7 to 14 days for PE for a full course of therapy (35, 42, 53, 62–64), and neither fully eliminates autoantibodies or halts complement activity (65–69).
Despite treatment with IVIg or PE, along with optimal supportive care including mechanical ventilation, a substantial proportion of patients with GBS experience persistent symptoms (1, 2, 58). Resolution of the disease is rare in the first week of treatment (9), and up to 50% of patients treated with IVIg or PE show no improvement on the GBS Disability Scale (GBS-DS) at 4 weeks (70). Even with time, only a minority of patients with GBS achieve a GBS-DS score of 0, indicating a return to pre-disease levels of function or being healthy (15, 17, 18, 71). Moreover, even among those who regain functional abilities, including restored ambulation, many continue to face challenges with walking, experience limitations in activities of daily living, including work and hobbies, and report suboptimal health-related quality of life (2, 72–77).
Global expert consensus emphasizes the importance of early treatment to achieve maximum therapeutic benefits and avert irreversible axonal damage in this rapidly progressive disease (1, 2, 27, 42, 53, 78, 79). A recent study involving 136 IVIg-treated patients found that shorter time to treatment initiation was associated with better outcomes regardless of NCS pattern, with the ideal treatment window being within the first 2 weeks (79). Early treatment becomes even more critical given that IVIg and PE tend to act slowly and require several days to complete a full course of therapy.
Although as many as a third of patients with GBS receive additional treatment after the initial intervention, owing to persistent deterioration or lack of improvement after first-line therapy (60, 80), current evidence does not support this practice. There is no proven benefit to combining or sequentially administering IVIg and PE (42, 58, 62, 70, 81), and repeated dosing of either therapy has not demonstrated superior outcomes relative to single dosing (82–84). For instance, a second dose of IVIg did not provide additional benefit over placebo and was associated with a higher frequency of serious adverse events in patients with severe GBS who deteriorated 1 week after their first course of IVIg (84). Similarly, increasing the number of PE sessions from 4 to 6 did not yield better results in patients with severe GBS (82).
Ultimately, findings from studies evaluating alternative regimens for IVIg and PE suggest that the full therapeutic potential of these therapies in GBS has been realized, highlighting a substantial unmet medical need for therapies capable of rapidly preventing acute and ongoing nerve damage and axonal loss. Moreover, it appears that the treatment window in GBS is relatively short, necessitating an intervention that acts rapidly to prevent further deterioration that limits the ability to recover.
2.3 Central role of classical complement activation in GBS pathophysiology
Complement represents a distinct pathway in GBS that mediates rapid tissue damage and destruction and inflammation (23, 27, 32, 33). When the pathway is engaged on the cell surface, it activates within seconds to coat the surface with complement activation products; these products cause direct membrane damage or destruction and recruit inflammatory macrophages that destroy tissue (1, 27, 33, 34, 36, 85). Anaphylatoxins released during classical complement activation drive neuroinflammation by activating a number of inflammatory cell types, including macrophages (33, 34, 86, 87). This mechanism is consistent with the rapid course of disease in GBS that can trigger pain, paralysis, and even death within a period of days (8, 11). In a large cohort of 567 patients with GBS from multiple clinical trials, 80% of patients reached clinical nadir (i.e., maximum disability or muscle weakness) within 2 weeks following the onset of weakness, and 97% reached their nadir within 4 weeks (88).
The complement system functions similarly in all individuals, independent of their genetic or ethnic background, because it is an evolutionarily conserved mechanism designed to recognize and eliminate pathogens by using pattern recognition (34, 89). The high conservation of complement components C1q, C2, C3, C4, and C5-C9 and their activities across species underscores the essential and nonredundant roles of these universal processes in ensuring rapid immune responses (90). In GBS, binding of C1q to antibodies bound to peripheral nerves activates the classical complement pathway directly on the nerve surface, mediating a cascade of events that culminates in neuroinflammation and nerve damage and destruction (Figure 1) (23, 27, 32–34, 91–94). Evidence also suggests that classical complement activation underlies nerve injury in GBS regardless of axonal or demyelinating pathophysiology (27, 28, 33, 36, 85). Given the early role of complement activation in the pathophysiology of GBS, complement inhibition is a potentially effective target for halting significant axonal damage in the acute phase of the disease and for optimizing long-term function.
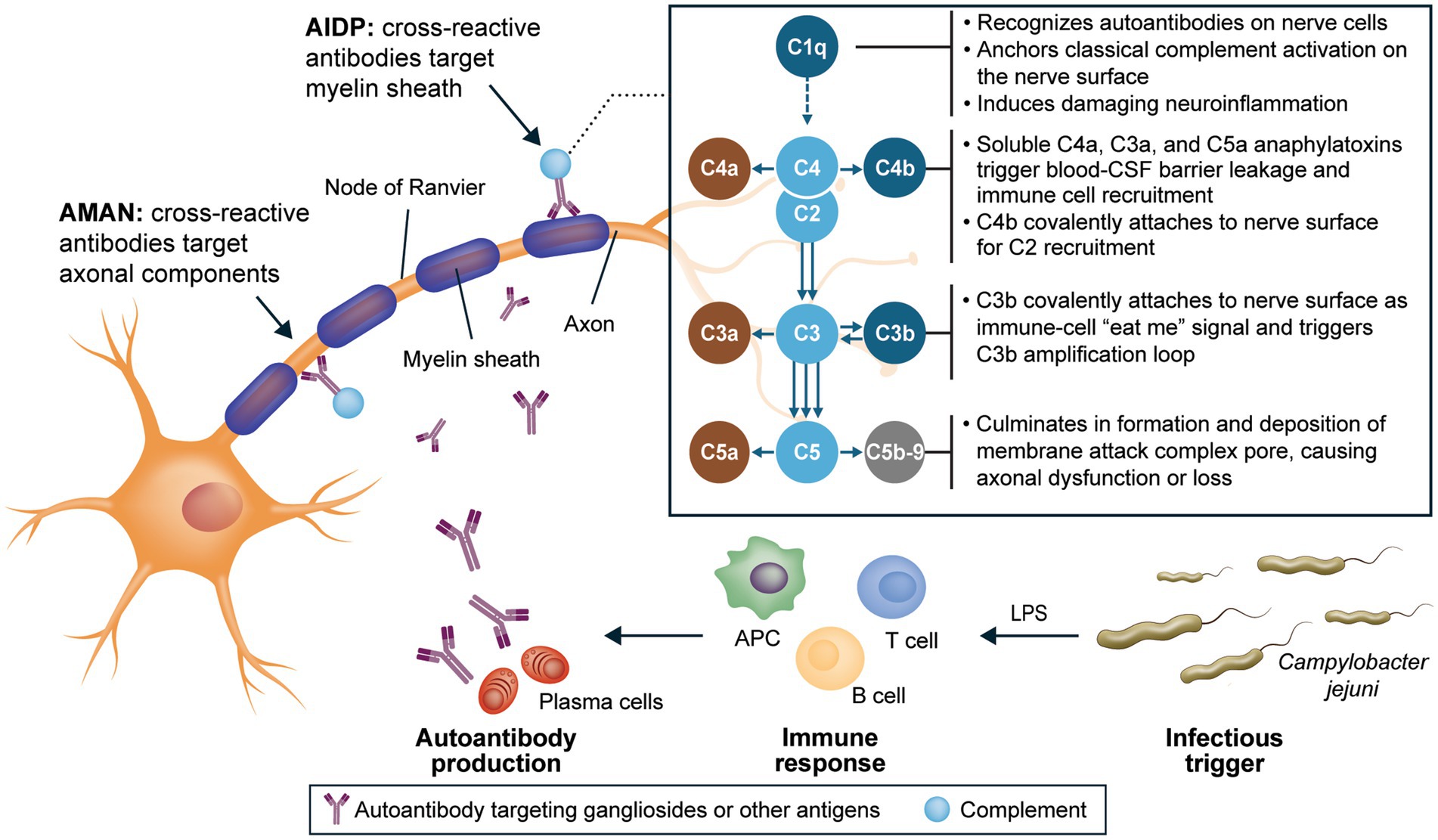
Figure 1. Integral role of C1q in neuroinflammation and nerve damage in GBS (23, 27, 32–34, 91–94). AIDP, acute inflammatory demyelinating polyneuropathy; AMAN, acute motor axonal neuropathy; APC, antigen-presenting cell; C, complement component; CSF, cerebrospinal fluid; GBS, Guillain-Barré syndrome; LPS, lipopolysaccharide.
3 Insights from a C1q inhibitor clinical development program in GBS
ANX005, a humanized monoclonal antibody, is designed to inhibit C1q selectively, rapidly, and fully, thus preventing initiation of the classical complement cascade (32). Blocking C1q at the start of the classical complement pathway inhibits tissue inflammation and damage driven by downstream complement components, including C3a and C5a (soluble anaphylatoxins that are highly proinflammatory), C4b and C3b (cell surface phagocytosis signals), and C5b-9 (which causes direct membrane damage; Figure 1) (32–34, 86, 87). Furthermore, due to the nature of the complement system with C1q as its initiating molecule, the biological mechanisms of ANX005 are expected to be consistent regardless of a patient’s race, ethnicity, or geographic location. The ANX005 development program in GBS consists of 2 placebo-controlled, double-blind clinical trials, a phase 1b trial and a phase 3 trial (NCT04701164), designed to evaluate the safety and efficacy of a single dose of ANX005 as a treatment for GBS. The phase 1b trial was conducted in Bangladesh, and the phase 3 trial was conducted in Bangladesh and the Philippines.
Among patients in the phase 1b trial with antibodies against the GM1 ganglioside in CSF (including patients with either AMAN or AIDP), complement deposition on GM1-coated plates was rapidly reduced in those treated with a single dose of ANX005 vs. placebo, consistent with complete inhibition of C1q (95). An analysis of data from this study also provided valuable insight into the coexistence of axonal damage and demyelination in GBS pathogenesis (96). Patients exhibited elevated levels of biomarkers of both axonal damage (NfL) (48) and demyelination (CSF sphingomyelin) (97) regardless of the NCS pattern. Peak NfL levels inversely correlated with MRC scores, and patients with lower NfL levels at baseline were more likely to show improvement on GBS-DS score at week 8, a consistent observation regardless of axonal or demyelinating physiology. Furthermore, and also independent of NCS findings, NfL level was the most important predictor of GBS-DS score at week 8 based on random forest modeling of multiple prognostic biomarkers and clinical characteristics, followed by CSF sphingomyelin level. The results of this study support a common pathophysiologic pathway across both axonal and demyelinating NCS patterns and suggest the effectiveness of complement inhibition in all patients with GBS (96).
In the subsequent phase 3 trial, patients who received a single 30-mg/kg ANX005 infusion showed complete inhibition of complement on the first day of administration and experienced rapid and sustained improvements in function and health status compared with those receiving placebo. Significant response was seen as early as week 1 in MRC sum score change from baseline (p < 0.0001) compared to placebo, and ANX005 30-mg/kg–treated participants were able to discontinue mechanical ventilation 28 days earlier and walk independently 31 days sooner than participants receiving placebo. This response was maintained, with a greater percentage in the ANX005 30-mg/kg group returning to a GBS-DS score of 0 at 26 weeks (22% vs. 9% in the placebo group; odds ratio, 4.1; 95% CI, 1.42–12.04; p = 0.0092). Significant response was maintained in patients with AMAN and AIDP and in subgroups with baseline MRC sum score and NfL levels typical of approximately 80% of patients with GBS in the United States and Europe. Hence, the results of this study are expected to be generalizable to patients in those regions. ANX005 was also well tolerated, with infusion-related reactions being the most common adverse events (98).
4 Discussion
GBS stands as the most prevalent and severe acute paralytic neuropathy globally, presenting significant challenges in diagnosis, treatment, and long-term management. The heterogeneous clinical manifestations and variable prognosis of this disease are rooted in its complex pathophysiology, primarily driven by autoantibody-mediated complement activation that results in both axonal damage and demyelination. Standard therapies, namely IVIg and PE, have been instrumental in the management of GBS; however, they are constrained by their delayed effect, prolonged treatment courses, and failure to consistently prevent residual symptoms such as pain, fatigue, and functional disabilities. Moreover, the critical window for effective intervention in GBS is notably short, underscoring the urgent need for treatments that act rapidly to halt disease progression and mitigate irreversible nerve damage.
Recent advancements in the understanding of GBS pathophysiology have highlighted the central role of classical complement activation in driving neuroinflammation and nerve damage and destruction, regardless of NCS pattern. This recognition has paved the way for targeted therapeutic approaches, such as the development of ANX005, a humanized monoclonal antibody designed to inhibit C1q selectively. Clinical trials have demonstrated that a single dose of ANX005 not only effectively suppressed complement activation but also yielded significant and sustained improvements in functional outcomes in patients with GBS, independent of NCS subtype. These findings validate the hypothesis that complement inhibition can serve as a unifying therapeutic strategy, addressing the underlying mechanisms common to all patients with GBS.
The success of ANX005 in clinical trials emphasizes the necessity of moving beyond traditional binary classifications of GBS and adopting a more nuanced understanding of its pathogenesis. By targeting universal pathogenic pathways, ANX005 offers a promising solution to the unmet medical need for rapid and comprehensive treatments that can prevent both acute and ongoing nerve damage. However, new treatments may also come with other limitations, such as limited initial accessibility and clinical experience.
While IVIg and PE have established a foundation for GBS management, their limitations highlight the pressing demand for novel therapies that can deliver faster and more complete therapeutic benefits. The development and validation of complement inhibitors like ANX005 represent a significant breakthrough, offering hope for improved outcomes and enhanced quality of life for patients afflicted by this debilitating neuromuscular emergency. As our understanding of GBS continues to evolve, embracing targeted treatments that address its core pathophysiologic mechanisms will be pivotal in transforming the landscape of GBS care and patient recovery.
Data availability statement
The original contributions presented in the commentary are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
Author contributions
KG: Conceptualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. Medical writing support for the development of this manuscript was provided by Precision AQ and funded by Annexon Biosciences.
Acknowledgments
Medical writing assistance with the development and revision of the manuscript under the direction of the authors was provided by Lara Primak, MD, Theresa Leichner, PhD, of Precision AQ, and Michael Yeakey of Annexon Biosciences, with financial support from Annexon Biosciences and in compliance with international Good Publication Practice guidelines.
Conflict of interest
KG has provided consulting activities for Annexon Biosciences, Janssen, Sanofi, and argenx and served as chair of the data safety monitoring board for the phase 3 trial of ANX005.
The author declares that this study and article received funding from Annexon Biosciences. The funder had the following involvement in the study: study design, collection, analysis, interpretation of data, the writing of this article, and the decision to submit it for publication.
Generative AI statement
The author declares that no Gen AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Leonhard, SE, Mandarakas, MR, Gondim, FAA, Bateman, K, Ferreira, MLB, Cornblath, DR, et al. Diagnosis and management of Guillain-Barré syndrome in ten steps. Nat Rev Neurol. (2019) 15:671–83. doi: 10.1038/s41582-019-0250-9
2. Willison, HJ, Jacobs, BC, and van Doorn, PA. Guillain-Barré syndrome. Lancet. (2016) 388:717–27. doi: 10.1016/S0140-6736(16)00339-1
3. Bragazzi, NL, Kolahi, AA, Nejadghaderi, SA, Lochner, P, Brigo, F, Naldi, A, et al. Global, regional, and national burden of Guillain-Barré syndrome and its underlying causes from 1990 to 2019. J Neuroinflammation. (2021) 18:264. doi: 10.1186/s12974-021-02319-4
4. Wachira, VK, Farinasso, CM, Silva, RB, Peixoto, HM, and de Oliveira, MRF. Incidence of Guillain-Barré syndrome in the world between 1985 and 2020: a systematic review. Glob Epidemiol. (2023) 5:100098. doi: 10.1016/j.gloepi.2023.100098
5. Co, AP, and Laxamana, LC. Guillain-Barre syndrome ensuing dengue fever in a filipino male: a case report. J Clin Images Med Case Rep. (2023) 4:2618. doi: 10.52768/2766-7820/2618
6. Papri, N, Islam, Z, Leonhard, SE, Mohammad, QD, Endtz, HP, and Jacobs, BC. Guillain-Barré syndrome in low-income and middle-income countries: challenges and prospects. Nat Rev Neurol. (2021) 17:285–96. doi: 10.1038/s41582-021-00467-y
7. Shang, P, Feng, J, Wu, W, and Zhang, HL. Intensive care and treatment of severe Guillain-Barré syndrome. Front Pharmacol. (2021) 12:608130. doi: 10.3389/fphar.2021.608130
8. Leonhard, SE, Papri, N, Querol, L, Rinaldi, S, Shahrizaila, N, and Jacobs, BC. Guillain–Barré syndrome. Nat Rev Dis Primers. (2024) 10:97. doi: 10.1038/s41572-024-00580-4
9. Doets, AY, Verboon, C, van den Berg, B, Harbo, T, Cornblath, DR, Willison, HJ, et al. Regional variation of Guillain-Barré syndrome. Brain. (2018) 141:2866–77. doi: 10.1093/brain/awy232
10. Dourado, ME, Félix, RH, da Silva, WK, Queiroz, JW, and Jeronimo, SM. Clinical characteristics of Guillain-Barré syndrome in a tropical country: a Brazilian experience. Acta Neurol Scand. (2012) 125:47–53. doi: 10.1111/j.1600-0404.2011.01503.x
11. van den Berg, B, Bunschoten, C, van Doorn, PA, and Jacobs, BC. Mortality in Guillain-Barre syndrome. Neurology. (2013) 80:1650–4. doi: 10.1212/WNL.0b013e3182904fcc
12. Grelowska, M, Logoń, K, and Dziadkowiak, E. Prognostic factors associated with worse outcomes in patients with GBS: a systematic review. Adv Clin Exp Med. (2024) 34:457–67. doi: 10.17219/acem/186949
13. Netto, AB, Taly, AB, Kulkarni, GB, Rao, UG, and Rao, S. Mortality in mechanically ventilated patients of Guillain Barré syndrome. Ann Indian Acad Neurol. (2011) 14:262–6. doi: 10.4103/0972-2327.91942
14. Levison, LS, Thomsen, RW, and Andersen, H. Increased mortality following Guillain-Barré syndrome: a population-based cohort study. Eur J Neurol. (2022) 29:1145–54. doi: 10.1111/ene.15204
15. Wang, Y, Lang, W, Zhang, Y, Ma, X, Zhou, C, and Zhang, HL. Long-term prognosis of Guillain-Barré syndrome not determined by treatment options? Oncotarget. (2017) 8:79991–80001. doi: 10.18632/oncotarget.20620
16. Bernsen, RA, Jager, AE, Schmitz, PI, and van der Meché, FG. Long-term sensory deficit after Guillain-Barré syndrome. J Neurol. (2001) 248:483–6. doi: 10.1007/s004150170157
17. Estublier, B, Colineaux, H, Arnaud, C, Cintas, P, Baudou, E, Chaix, Y, et al. Long-term outcomes of paediatric Guillain-Barré syndrome. Dev Med Child Neurol. (2024) 66:176–86. doi: 10.1111/dmcn.15693
18. Martic, V, Bozovic, I, Berisavac, I, Basta, I, Peric, S, Babic, M, et al. Three-year follow-up study in patients with Guillain-Barré syndrome. Can J Neurol Sci. (2018) 45:269–74. doi: 10.1017/cjn.2018.12
19. Ruts, L, Drenthen, J, Jongen, JL, Hop, WC, Visser, GH, Jacobs, BC, et al. Pain in Guillain-Barre syndrome: a long-term follow-up study. Neurology. (2010) 75:1439–47. doi: 10.1212/WNL.0b013e3181f88345
20. Uz, FB, Uz, C, and Karaahmet, OZ. Three-year follow-up outcomes of adult patients with Guillain-Barré syndrome after rehabilitation. Malawi Med J. (2023) 35:156–62. doi: 10.4314/mmj.v35i3.4
21. Bayu, HT, Demilie, AE, Molla, MT, Kumie, FT, and Endeshaw, AS. Mortality and its predictors among patients with Guillain-Barré syndrome in the intensive care unit of a low-income country, Ethiopia: a multicenter retrospective cohort study. Front Neurol. (2024) 15:1484661. doi: 10.3389/fneur.2024.1484661
22. Ishaque, T, Islam, MB, Ara, G, Endtz, HP, Mohammad, QD, Jacobs, BC, et al. High mortality from Guillain-Barré syndrome in Bangladesh. J Peripher Nerv Syst. (2017) 22:121–6. doi: 10.1111/jns.12215
23. Laman, JD, Huizinga, R, Boons, GJ, and Jacobs, BC. Guillain-Barré syndrome: expanding the concept of molecular mimicry. Trends Immunol. (2022) 43:296–308. doi: 10.1016/j.it.2022.02.003
24. Leonhard, SE, Tan, CY, van der Eijk, AA, Reisin, RR, Franken, SC, Huizinga, R, et al. Antecedent infections in Guillain-Barré syndrome in endemic areas of arbovirus transmission: a multinational case-control study. J Peripher Nerv Syst. (2021) 26:449–60. doi: 10.1111/jns.12469
25. Leonhard, SE, van der Eijk, AA, Andersen, H, Antonini, G, Arends, S, Attarian, S, et al. An international perspective on preceding infections in Guillain-Barré syndrome: the IGOS-1000 cohort. Neurology. (2022) 99:e1299–313. doi: 10.1212/WNL.0000000000200885
26. Stein, RA. Campylobacter jejuni and postinfectious autoimmune diseases: a proof of concept in glycobiology. ACS Infect Dis. (2022) 8:1981–91. doi: 10.1021/acsinfecdis.2c00397
27. van den Berg, B, Walgaard, C, Drenthen, J, Fokke, C, Jacobs, BC, and van Doorn, PA. Guillain-Barré syndrome: pathogenesis, diagnosis, treatment and prognosis. Nat Rev Neurol. (2014) 10:469–82. doi: 10.1038/nrneurol.2014.121
28. Hagen, KM, and Ousman, SS. The neuroimmunology of Guillain-Barré syndrome and the potential role of an aging immune system. Front Aging Neurosci. (2021) 12:613628. doi: 10.3389/fnagi.2020.613628
29. Halstead, SK, Kalna, G, Islam, MB, Jahan, I, Mohammad, QD, Jacobs, BC, et al. Microarray screening of Guillain-Barré syndrome sera for antibodies to glycolipid complexes. Neurol Neuroimmunol Neuroinflamm. (2016) 3:e284. doi: 10.1212/NXI.0000000000000284
30. Rinaldi, S, Brennan, KM, Kalna, G, Walgaard, C, van Doorn, P, Jacobs, BC, et al. Antibodies to heteromeric glycolipid complexes in Guillain-Barré syndrome. PLoS One. (2013) 8:e82337. doi: 10.1371/journal.pone.0082337
31. Thomma, RCM, Halstead, SK, de Koning, LC, Wiegers, E, Gourlay, DS, Tio-Gillen, AP, et al. Large-scale profiling of antibody reactivity to glycolipids in patients with Guillain-Barré syndrome. Brain. (2025). doi: 10.1093/brain/awaf102 [Epub ahead of print].
32. Lansita, JA, Mease, KM, Qiu, H, Yednock, T, Sankaranarayanan, S, and Kramer, S. Nonclinical development of ANX005: a humanized anti-C1q antibody for treatment of autoimmune and neurodegenerative diseases. Int J Toxicol. (2017) 36:449–62. doi: 10.1177/1091581817740873
33. McGonigal, R, Cunningham, ME, Yao, D, Barrie, JA, Sankaranarayanan, S, Fewou, SN, et al. C1q-targeted inhibition of the classical complement pathway prevents injury in a novel mouse model of acute motor axonal neuropathy. Acta Neuropathol Commun. (2016) 4:23. doi: 10.1186/s40478-016-0291-x
34. Janeway, CA, Travers, P, Walport, M, and Shlomchik, MJ. The complement system and innate immunity. Immunobiology: The immune system in health and disease. 5th ed. New York, NY: Garland Science (2001).
35. Shahrizaila, N, Lehmann, HC, and Kuwabara, S. Guillain-Barré syndrome. Lancet. (2021) 397:1214–28. doi: 10.1016/S0140-6736(21)00517-1
36. Hafer-Macko, C, Hsieh, ST, Li, CY, Ho, TW, Sheikh, K, Cornblath, DR, et al. Acute motor axonal neuropathy: an antibody-mediated attack on axolemma. Ann Neurol. (1996) 40:635–44. doi: 10.1002/ana.410400414
37. Asbury, AK, and Cornblath, DR. Assessment of current diagnostic criteria for Guillain-Barré syndrome. Ann Neurol. (1990) 27:S21–4. doi: 10.1002/ana.410270707
38. Hiraga, A, Mori, M, Ogawara, K, Kojima, S, Kanesaka, T, Misawa, S, et al. Recovery patterns and long term prognosis for axonal Guillain-Barré syndrome. J Neurol Neurosurg Psychiatry. (2005) 76:719–22. doi: 10.1136/jnnp.2004.051136
39. Konuşkan, B, Okuyaz, Ç, Taşdelen, B, Kurul, SH, and Anlar, B. Turkish childhood Guillan-Barre syndrome study group. Electrophysiological subtypes and prognostic factors of childhood Guillain-Barré syndrome. Noro Psikiyatr Ars. (2018) 55:199–204. doi: 10.5152/npa.2017.16996
40. Nagasawa, K, Kuwabara, S, Misawa, S, Fujii, K, Tanabe, Y, Yuki, N, et al. Electrophysiological subtypes and prognosis of childhood Guillain–Barré syndrome in Japan. Muscle Nerve. (2006) 33:766–70. doi: 10.1002/mus.20520
41. Tian, J, Cao, C, Li, T, Zhang, K, Li, P, Liu, Y, et al. Electrophysiological subtypes and prognostic factors of Guillain-Barre syndrome in northern China. Front Neurol. (2019) 10:714. doi: 10.3389/fneur.2019.00714
42. van Doorn, PA, Van den Bergh, PYK, Hadden, RDM, Avau, B, Vankrunkelsven, P, Attarian, S, et al. European academy of neurology/peripheral nerve society guideline on diagnosis and treatment of Guillain-Barré syndrome. Eur J Neurol. (2023) 30:3646–74. doi: 10.1111/ene.16073
43. Ye, Y, Wang, K, Den, F, and Xing, Y. Electrophysiological subtypes and prognosis of Guillain-Barré syndrome in northeastern China. Muscle Nerve. (2013) 47:68–71. doi: 10.1002/mus.23477
44. Khedr, EM, Mohamed, MZ, and Shehab, MMM. The early clinical and laboratory predictors of GBS outcome: hospital-based study, Assiut University, upper Egypt. Egypt J Neurol Psychiatry Neurosurg. (2023) 59:45. doi: 10.1186/s41983-023-00646-2
45. van Tilburg, SJ, Teunissen, CE, Maas, CCHM, Thomma, RCM, Walgaard, C, Heijst, H, et al. Dynamics and prognostic value of serum neurofilament light chain in Guillain-Barré syndrome. EBioMedicine. (2024) 102:105072. doi: 10.1016/j.ebiom.2024.105072
46. Axelsson, M, Sjögren, M, Andersen, O, Blennow, K, Zetterberg, H, and Lycke, J. Neurofilament light protein levels in cerebrospinal fluid predict long-term disability of Guillain-Barré syndrome: a pilot study. Acta Neurol Scand. (2018) 138:143–50. doi: 10.1111/ane.12927
47. Dujmovic, I, Lunn, MP, Reilly, MM, and Petzold, A. Serial cerebrospinal fluid neurofilament heavy chain levels in severe Guillain-Barré syndrome. Muscle Nerve. (2013) 48:132–4. doi: 10.1002/mus.23752
48. Martín-Aguilar, L, Camps-Renom, P, Lleixà, C, Pascual-Goñi, E, Díaz-Manera, J, Rojas-García, R, et al. Serum neurofilament light chain predicts long-term prognosis in Guillain-Barré syndrome patients. J Neurol Neurosurg Psychiatry. (2021) 92:70–7. doi: 10.1136/jnnp-2020-323899
49. Berciano, J. Inflammatory oedema of nerve trunks may be pathogenic in very early Guillain-Barré syndrome. Acta Neurol Belg. (2020) 120:1061–5. doi: 10.1007/s13760-020-01413-3
50. Berciano, J, Orizaola, P, Gallardo, E, Pelayo-Negro, AL, Sánchez-Juan, P, Infante, J, et al. Very early Guillain-Barré syndrome: a clinical-electrophysiological and ultrasonographic study. Clin Neurophysiol Pract. (2019) 5:1–9. doi: 10.1016/j.cnp.2019.11.003
51. Vucic, S, Cairns, KD, Black, KR, Chong, PS, and Cros, D. Neurophysiologic findings in early acute inflammatory demyelinating polyradiculoneuropathy. Clin Neurophysiol. (2004) 115:2329–35. doi: 10.1016/j.clinph.2004.05.009
52. Arends, S, Drenthen, J, de Koning, L, van den Bergh, P, Hadden, RDM, Kuwabara, S, et al. Electrodiagnostic subtyping in Guillain-Barré syndrome patients in the international Guillain-Barré outcome study. Eur J Neurol. (2024) 31:e16335. doi: 10.1111/ene.16335
53. Yao, J, Zhou, R, Liu, Y, and Lu, Z. Progress in Guillain-Barré syndrome immunotherapy—a narrative review of new strategies in recent years. Hum Vaccin Immunother. (2023) 19:2215153. doi: 10.1080/21645515.2023.2215153
54. Goodfellow, JA, and Willison, HJ. Guillain-Barré syndrome: a century of progress. Nat Rev Neurol. (2016) 12:723–31. doi: 10.1038/nrneurol.2016.172
55. Zaki, HA, Iftikhar, H, Najam, M, Masood, M, Al-Marri, NDR, Elgassim, MAM, et al. Plasma exchange (PE) versus intravenous immunoglobulin (IVIG) for the treatment of Guillain-Barré syndrome (GBS) in patients with severe symptoms: a systematic review and meta-analysis. eNeurologicalSci. (2023) 31:100468. doi: 10.1016/j.ensci.2023.100468
56. Dalakas, MC. Mechanisms of action of IVIg and therapeutic considerations in the treatment of acute and chronic demyelinating neuropathies. Neurology. (2002) 59:S13–21. doi: 10.1212/wnl.59.12_suppl_6.s13
57. Hughes, RA, Swan, AV, and van Doorn, PA. Intravenous immunoglobulin for Guillain-Barré syndrome. Cochrane Database Syst Rev. (2014) 2019:CD002063. doi: 10.1002/14651858.CD002063.pub6
58. Liu, S, Dong, C, and Ubogu, EE. Immunotherapy of Guillain-Barré syndrome. Hum Vaccin Immunother. (2018) 14:1–12. doi: 10.1080/21645515.2018.1493415
59. Velikova, T, Sekulovski, M, Bogdanova, S, Vasilev, G, Peshevska-Sekulovska, M, Miteva, D, et al. Intravenous immunoglobulins as immunomodulators in autoimmune diseases and reproductive medicine. Antibodies (Basel). (2023) 12:20. doi: 10.3390/antib12010020
60. Rath, J, Zulehner, G, Schober, B, Grisold, A, Krenn, M, Cetin, H, et al. Real-world treatment of adult patients with Guillain-Barré syndrome over the last two decades. Sci Rep. (2021) 11:19170. doi: 10.1038/s41598-021-98501-y
61. American Academy of Neurology. AAN guideline summary for clinicians. Immunotherapy for Guillain-Barré Syndrome. Available online at: https://www.aan.com/Guidelines/home/GetGuidelineContent/115 (Accessed May 7, 2025).
62. Plasma Exchange/Sandoglobulin Guillain-Barré Syndrome Trial Group. Randomised trial of plasma exchange, intravenous immunoglobulin, and combined treatments in Guillain-Barré syndrome. Lancet. (1997) 349:225–30.
63. The Guillain-Barré Syndrome Study Group. Plasmapheresis and acute Guillain-Barré syndrome. Neurology. (1985) 35:1096–104. doi: 10.1212/WNL.35.8.1096
64. van der Meché, FG, and Schmitz, PIThe Dutch Guillain-Barré Study Group. A randomized trial comparing intravenous immune globulin and plasma exchange in Guillain-Barré syndrome. N Engl J Med. (1992) 326:1123–9. doi: 10.1056/NEJM199204233261705
65. Baines, AC, and Brodsky, RA. Complementopathies. Blood Rev. (2017) 31:213–23. doi: 10.1016/j.blre.2017.02.003
66. Bleeker, WK, Teeling, JL, and Hack, CE. Accelerated autoantibody clearance by intravenous immunoglobulin therapy: studies in experimental models to determine the magnitude and time course of the effect. Blood. (2001) 98:3136–42. doi: 10.1182/blood.v98.10.3136
67. Hansen, RJ, and Balthasar, JP. Intravenous immunoglobulin mediates an increase in anti-platelet antibody clearance via the FcRn receptor. Thromb Haemost. (2002) 88:898–9. doi: 10.1055/s-0037-1613331
68. Lehmann, HC, Hartung, HP, Hetzel, GR, Stüve, O, and Kieseier, BC. Plasma exchange in neuroimmunological disorders: part 1: rationale and treatment of inflammatory central nervous system disorders. Arch Neurol. (2006) 63:930–5. doi: 10.1001/archneur.63.7.930
69. Piepers, S, Jansen, MD, Cats, EA, van Sorge, NM, van den Berg, LH, and van der Pol, WL. IVIg inhibits classical pathway activity and anti-GM1 IgM-mediated complement deposition in MMN. J Neuroimmunol. (2010) 229:256–62. doi: 10.1016/j.jneuroim.2010.08.023
70. Verboon, C, van Doorn, PA, and Jacobs, BC. Treatment dilemmas in Guillain-Barré syndrome. J Neurol Neurosurg Psychiatry. (2017) 88:346–52. doi: 10.1136/jnnp-2016-314862
71. Dhar, R, Stitt, L, and Hahn, AF. The morbidity and outcome of patients with Guillain-Barré syndrome admitted to the intensive care unit. J Neurol Sci. (2008) 264:121–8. doi: 10.1016/j.jns.2007.08.005
72. Bersano, A, Carpo, M, Allaria, S, Franciotta, D, Citterio, A, and Nobile-Orazio, E. Long term disability and social status change after Guillain-Barré syndrome. J Neurol. (2006) 253:214–8. doi: 10.1007/s00415-005-0958-x
73. Darweesh, SK, Polinder, S, Mulder, MJ, Baena, CP, van Leeuwen, N, Franco, OH, et al. Health-related quality of life in Guillain-Barré syndrome patients: a systematic review. J Peripher Nerv Syst. (2014) 19:24–35. doi: 10.1111/jns5.12051
74. Dornonville de la Cour, C, and Jakobsen, J. Residual neuropathy in long-term population-based follow-up of Guillain-Barré syndrome. Neurology. (2005) 64:246–53. doi: 10.1212/01.WNL.0000149521.65474.83
75. Gupta, A, Taly, AB, Srivastava, A, and Murali, T. Guillain-Barre syndrome–rehabilitation outcome, residual deficits and requirement of lower limb orthosis for locomotion at 1 year follow-up. Disabil Rehabil. (2010) 32:1897–902. doi: 10.3109/09638281003734474
76. Khan, F, Pallant, JF, Ng, L, and Bhasker, A. Factors associated with long-term functional outcomes and psychological sequelae in Guillain-Barre syndrome. J Neurol. (2010) 257:2024–31. doi: 10.1007/s00415-010-5653-x
77. Witsch, J, Galldiks, N, Bender, A, Kollmar, R, Bösel, J, Hobohm, C, et al. Long-term outcome in patients with Guillain-Barré syndrome requiring mechanical ventilation. J Neurol. (2013) 260:1367–74. doi: 10.1007/s00415-012-6806-x
78. Meena, AK, Khadilkar, SV, and Murthy, JM. Treatment guidelines for Guillain-Barré syndrome. Ann Indian Acad Neurol. (2011) 14:S73–81. doi: 10.4103/0972-2327.83087
79. Min, YG, Hong, YH, Rajabally, YA, and Sung, JJ. Timing of intravenous immunoglobulin treatment and outcome in Guillain-Barré syndrome: is time nerve? Muscle Nerve. (2024) 70:1215–22. doi: 10.1002/mus.28271
80. Verboon, C, Doets, AY, Galassi, G, Davidson, A, Waheed, W, Péréon, Y, et al. Current treatment practice of Guillain-Barré syndrome. Neurology. (2019) 93:e59–76. doi: 10.1212/WNL.0000000000007719
81. Oczko-Walker, M, Manousakis, G, Wang, S, Malter, JS, and Waclawik, AJ. Plasma exchange after initial intravenous immunoglobulin treatment in Guillain-Barré syndrome: critical reassessment of effectiveness and cost-efficiency. J Clin Neuromuscul Dis. (2010) 12:55–61. doi: 10.1097/CND.0b013e3181f3dbbf
82. The French cooperative group on plasma exchange in Guillain-Barré syndrome. Appropriate number of plasma exchanges in Guillain-Barré syndrome. Ann Neurol. (1997) 41:298–306. doi: 10.1002/ana.410410304
83. Verboon, C, van den Berg, B, Cornblath, DR, Venema, E, Gorson, KC, Lunn, MP, et al. Original research: second IVIg course in Guillain-Barré syndrome with poor prognosis: the non-randomised ISID study. J Neurol Neurosurg Psychiatry. (2020) 91:113–21. doi: 10.1136/jnnp-2019-321496
84. Walgaard, C, Jacobs, BC, Lingsma, HF, Steyerberg, EW, van den Berg, B, Doets, AY, et al. Second intravenous immunoglobulin dose in patients with Guillain-Barré syndrome with poor prognosis (SID-GBS): a double-blind, randomised, placebo-controlled trial. Lancet Neurol. (2021) 20:275–83. doi: 10.1016/S1474-4422(20)30494-4
85. Hafer-Macko, CE, Sheikh, KA, Li, CY, Ho, TW, Cornblath, DR, McKhann, GM, et al. Immune attack on the Schwann cell surface in acute inflammatory demyelinating polyneuropathy. Ann Neurol. (1996) 39:625–35. doi: 10.1002/ana.410390512
86. Klos, A, Tenner, AJ, Johswich, KO, Ager, RR, Reis, ES, and Köhl, J. The role of the anaphylatoxins in health and disease. Mol Immunol. (2009) 46:2753–66. doi: 10.1016/j.molimm.2009.04.027
87. Schanzenbacher, J, Köhl, J, and Karsten, CM. Anaphylatoxins spark the flame in early autoimmunity. Front Immunol. (2022) 13:958392. doi: 10.3389/fimmu.2022.958392
88. Fokke, C, van den Berg, B, Drenthen, J, Walgaard, C, van Doorn, PA, and Jacobs, BC. Diagnosis of Guillain-Barré syndrome and validation of Brighton criteria. Brain. (2014) 137:33–43. doi: 10.1093/brain/awt285
89. Reis, ES, Mastellos, DC, Hajishengallis, G, and Lambris, JD. New insights into the immune functions of complement. Nat Rev Immunol. (2019) 19:503–16. doi: 10.1038/s41577-019-0168-x
90. Dodds, AW, and Matsushita, M. The phylogeny of the complement system and the origins of the classical pathway. Immunobiology. (2007) 212:233–43. doi: 10.1016/j.imbio.2006.11.009
91. Elward, K, and Gasque, P. "Eat me" and "don't eat me" signals govern the innate immune response and tissue repair in the CNS: emphasis on the critical role of the complement system. Mol Immunol. (2003) 40:85–94. doi: 10.1016/s0161-5890(03)00109-3
92. Propson, NE, Roy, ER, Litvinchuk, A, Köhl, J, and Zheng, H. Endothelial C3a receptor mediates vascular inflammation and blood-brain barrier permeability during aging. J Clin Invest. (2021) 131:e140966. doi: 10.1172/JCI140966
93. Rajabally, YA. Immunoglobulin and monoclonal antibody therapies in Guillain-Barré syndrome. Neurotherapeutics. (2022) 19:885–96. doi: 10.1007/s13311-022-01253-4
94. Orsini, F, De Blasio, D, Zangari, R, Zanier, ER, and De Simoni, MG. Versatility of the complement system in neuroinflammation, neurodegeneration and brain homeostasis. Front Cell Neurosci. (2014) 8:380. doi: 10.3389/fncel.2014.00380
95. Suri, P, Sankaranarayanan, S, Cahir-McFarland, E, Kroon, HA, Islam, Z, and Yednock, T. Intravenous Administration of ANX005, an anti-C1q therapy, reduced CSF antibody-driven complement activity in early-stage Guillain-Barré syndrome. Oral presentation at: the 2021 Peripheral Nerve Society (PNS) Annual Meeting; June 12–13 and June 25–27 (2021)
96. Kroon, HA, Paliwal, P, Kanani, K, Humphriss, E, Islam, Z, and Mohammad, QD. Beyond binary: biomarkers reveal the coexistence of acute motor axonal neuropathy (AMAN) and acute inflammatory demyelinating polyneuropathy (AIDP) in Guillain-Barré syndrome. Poster presented at: The 2024 American Academy of Neurology annual meeting, Denver, Colorado, USA; (2024).
97. Capodivento, G, De Michelis, C, Carpo, M, Fancellu, R, Schirinzi, E, Severi, D, et al. CSF sphingomyelin: a new biomarker of demyelination in the diagnosis and management of CIDP and GBS. J Neurol Neurosurg Psychiatry. (2021) 92:303–10. doi: 10.1136/jnnp-2020-324445
98. Kroon, HA, Islam, Z, Hoehn, B, Humphriss, E, Lin, P, Morrison, G, et al. GBS-02: a phase 3 study to evaluate efficacy, safety, pharmacokinetics, and pharmacodynamics of ANX005 in patients with Guillain-Barré syndrome (GBS). Poster presented at: The 2024 neuromuscular study group annual scientific meeting; Tarrytown, New York, USA; (2024)
Keywords: Guillain-Barré syndrome, classical complement, C1q, pathophysiology, acute motor axonal neuropathy (AMAN), acute inflammatory demyelinating polyneuropathy (AIDP), ANX005, tanruprubart
Citation: Gorson KC (2025) Evolving understanding of Guillain-Barré syndrome pathophysiology and the central role of the classical complement pathway in axonal injury. Front. Neurol. 16:1572949. doi: 10.3389/fneur.2025.1572949
Edited by:
Mamede De Carvalho, University of Lisbon, PortugalReviewed by:
Hongliang Zhang, National Natural Science Foundation of China, ChinaJustin Kwan, National Institutes of Health (NIH), United States
Copyright © 2025 Gorson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kenneth C. Gorson, a2VuZ29yc29uQGNvbWNhc3QubmV0