Abstract
Pediatric epilepsy affects a large proportion of children, with a huge variability in seizure onset. Due to complicated etiology, wide range of associated comorbidities, and difficulty in obtaining clear physiological data from children, epilepsy management in pediatric patients often poses a critical challenge. Importantly, around 30% of these patients remain non-responsive to current anti-seizure drugs and develop a higher risk of developmental and cognitive delay and, in worse situations, premature death. One of the key treatment methods currently used for drug-resistant epilepsies is surgical resection of the epileptic foci. However, such patients often develop new epileptic foci post-surgery. This, in turn, enhances the need for recurrent invasive brain surgeries, impairing the overall quality of life in these children. Thus, mechanistic understanding of different types of pediatric epilepsy is critical to discovering more targeted molecular approach(es). For a long time, the occurrence of epilepsy was considered solely due to the abnormal functioning of single ion channels. However, in recent years, a huge number of genetic and non-genetic (environmental) factors have been associated with different types of pediatric epilepsy. Clinical diagnoses, coupled with a basic understanding of molecular and cellular mechanisms using different model systems, have been instrumental in unraveling new avenues for modern non-invasive targeted pharmacological therapies. Yet, the field has just started to evolve, and many challenges and contradictory hypotheses still exist. This comprehensive review discusses underlying developmental mechanisms associated with pediatric epilepsy. Specifically, we highlight how the PI3K-AKT–MTOR pathway acts as a critical node interconnecting the diverse mechanistic strategies, that may eventually help overcome the seizure burden in the future.
1 Introduction: epilepsy in children
Epilepsy is among the most common neurological disorders affecting people of all age groups (1, 2). As defined by the International League against Epilepsy (ILAE), epilepsy is one meeting any of the following conditions: (a) at least two unprovoked seizures occurring >24 h apart; (b) one unprovoked seizure and a probability of further seizures similar to the general recurrence risk after two unprovoked seizures; and (c) diagnosis of an epilepsy syndrome (3–5). Epileptic seizures are very distinct from just any other non-epileptic seizure event in the sense that epileptic seizures occur due to abnormal, excessive, or simultaneous activity of “neuronal populations” in the brain, which may or may not have any clinical manifestations. Epilepsy syndromes manifested in children are especially complicated, overlapping in nature, and multi-faceted to understand. Children with early-onset epilepsy are highly predisposed to developmental and cognitive delay and sensory-motor abnormalities since the critical period for many neurological functions lies in childhood (6, 7). Further, about 30% of patients suffering from childhood epilepsies are intractable to any regimen of current medications (8–10). The combination of associated comorbidities and drug resistance negatively influences the patient’s quality of life and is also potent in elevating their risk of premature death, creating a huge emotional burden on caregivers (11–13). Therefore, to fight this challenge and identify potential non-invasive therapeutic avenues, studying the underlying mechanisms driving pediatric epilepsy is critical.
Historically, epilepsy has always been considered a channel protein manifestation. However, advancements in genetic screens have identified diverse epilepsy-causing variants, ranging from mutations in cellular signaling pathways to the components of circadian rhythm (14–19). In this review, we will discuss the model systems that helped in identifying the molecular mechanisms underlying pediatric genetic epilepsies beyond channelopathies and how these models can be instrumental in discovering potential treatment strategies. Further, we strongly emphasize the ongoing challenges and controversies in the field. Certain common terminologies used throughout the text are defined in Table 1.
Table 1
| Term | Definition |
|---|---|
| Seizure | Transient symptoms due to abnormal excessive or simultaneous activity of a neuronal population in the brain, with or without any clinical manifestations |
| Epilepsy | A disease of the brain defined by any of the following conditions: (1) At least two unprovoked (or reflex) seizures occurring >24 h apart; (2) one unprovoked (or reflex) seizure and a probability of further seizures similar to the general recurrence risk (at least 60%) after two unprovoked seizures, occurring over the next 10 years; (3) diagnosis of an epilepsy syndrome. Epilepsy is resolved for individuals who have an age-dependent epilepsy syndrome but are now past the applicable age or those who have remained seizure-free for the last 10 years, with no antiseizure medicines for the last 5 years |
| Epileptogenesis | A process that includes mechanisms driving functional, structural, or network reorganization changes in the brain that may lead to the development of, or progression of, spontaneous seizures and epilepsy. |
| Status epilepticus | Status epilepticus occurs when a seizure lasts more than 5 min or when seizures occur very close together, and the person doesn’t recover consciousness between them. |
| Focal seizures | Originating within networks limited to one hemisphere. They may be discretely localized or more widely distributed. |
| Generalized seizures | Originating at some point within, and rapidly engaging, bilaterally distributed networks |
| Autonomic seizures | A distinct alteration of autonomic nervous system function involving cardiovascular, pupillary, gastrointestinal, sudomotor, vasomotor, and thermoregulatory functions |
| Tonic | A sustained increase in muscle contraction lasting a few seconds to minutes |
| Atonic | Sudden loss or diminution of muscle tone without apparent preceding myoclonic or tonic event lasting ~1–2 s, involving head, trunk, jaw, or limb musculature. |
| Clonic | Jerking, either symmetric or asymmetric, that is regularly repetitive and involves the same muscle groups |
| Myoclonic | Sudden, brief (<100 msec) involuntary single or multiple contraction(s) of muscles(s) or muscle groups of variable topography (axial, proximal limb, distal). Myoclonus is less repetitive and less sustained than clonus |
| Tonic–clonic | A sequence consisting of a tonic followed by a clonic phase |
| Generalized tonic–clonic (GTC) | Bilateral symmetric or sometimes asymmetric tonic contraction and then bilateral clonic contraction of somatic muscles, that are usually associated with autonomic phenomena and loss of awareness. These seizures engage networks in both hemispheres at the start of the seizure |
| Atypical absence seizures | An absence seizure with changes in tone that are more pronounced than in typical absence or the onset and/or cessation is not abrupt, often associated with slow, irregular, generalized spike–wave activity |
| Typical absence seizures | A sudden onset, interruption of ongoing activities, a blank stare, possibly a brief upward deviation of the eyes. Usually, the patient will be unresponsive when spoken to. Duration is a few seconds to half a minute, with very rapid recovery |
| Febrile seizures (FS) | Seizures occurring in pediatric patients between 6 and 60 months of age, triggered by fevers higher than 38°C (≥100.4°F), without any known underlying medical condition such as trauma, CNS infection, neurodevelopmental disorders, genetic mutation, afebrile seizures or predisposition to epilepsy. |
Common terminologies in the field of epilepsy [Adapted from Fisher et al. (3)].
2 Article types
This is a review article that comprehensively discusses different mechanistic aspects of pediatric epilepsy, using different model systems and finishing with newer avenues of prospective therapies and existing gaps. No specific databases have been used in this manuscript.
3 Current classification of pediatric epilepsy
Epilepsy is a multifactorial disorder, caused by genetic and/or environmental factors and is classified into different types, each with its own etiology, physiological properties, and onset (20). Differential diagnoses aid clinicians in classifying the type of epilepsy the patient is experiencing, which in turn may help ascertain the therapeutic strategy. Historically, epilepsy classification heavily relied on the clinical symptoms of the patients. However, this classification system has recently undergone a significant change from a symptom-based approach to a more sophisticated, multidimensional framework. A critical feature of this change is the integration of neuroimaging findings, genetic screen data, and etiology (21). The current classification system is a hierarchical three-level scheme (21). It begins with classifying a seizure type into focal, generalized, and of unknown onset/unclear origin, followed by classifying the epilepsy type, eventually categorizing as epilepsy syndromes. An epilepsy syndrome is defined as, “a characteristic cluster of clinical and electroencephalography (EEG) features, often supported by specific etiological findings” (4). It has a distinct set of comorbidities, etiology, and age of onset, and often has direct consequences for treatment and prognosis.
Pediatric epilepsy, defined in this review as those with seizure onset before 18 years of age, is generally classified into different types, mostly based on the age of seizure onset, set of comorbidities, and known etiologies (Figure 1). While there is significant progress in classifying pediatric epilepsy, certain limitations still exist. One of the persistent challenges is to define the boundaries of epilepsy syndromes. Multiple overlaps across categories and one type leading to or influencing the predisposition to other types of late-onset epilepsy are evident from clinical scenarios. For example, self-limited epilepsy is one of the most common and earliest types of epilepsy, accounting for about 25% of all pediatric epilepsy (22). Seizures are mostly focal in origin and subside within a few weeks or a few years after commencement, hence the name ‘self-limited’. Most patients with self-limited epilepsy respond to medication; seizures usually resolve by puberty but can occasionally occur up to 18 years of age. Interestingly, a proportion of these patients show a higher risk of developing developmental and epileptic encephalopathy (DEE) or genetic generalized epilepsy (GGE) (23–27). DEE is a broad umbrella term comprising many severe epileptic syndromes in children, each with its characteristic age of onset. DEE patients are characterized by the presence of developmental and cognitive impairment; both seizures and underlying etiology are suggested to contribute to these issues (28, 29). On the other hand, GGE is a broad term used for epilepsies with generalized seizures and genetic etiology identified through familial and twin studies (30). It constitutes 20–40% of all pediatric epilepsies (31). Considering the associated comorbidities and underlying genetic causes, the boundary between DEE and GGE often becomes diffused. Even the sub-syndromes within GGE (such as absence epilepsy and juvenile myoclonic epilepsy) show extensive overlap in terms of etiology despite having distinct electroclinical features. These overlapping phenomena make us hypothesize that different types of pediatric epilepsy generate a continuum of the disorder, considering the age of the patients, disease progression, and underlying etiology (Figure 2A).
Figure 1

Seizure onset for different pediatric epilepsies. Green blocks represent the age range in which different types of pediatric epilepsy show seizure onset. The first year of age (0–1 year) has been subdivided into 0 m, 3 m, 6 m, and 9 m of age (m, months). SeLNE, self-limited neonatal epilepsy; SeLIE, self-limited infantile epilepsy; SeLFNIE, self-limited familial neonatal-infantile epilepsy; SeLECTS, self-limited epilepsy with a centrotemporal spike; SeLEAS, self-limited epilepsy with autonomic seizures; COVE, childhood occipital visual epilepsy; POLE, photosensitive occipital lobe epilepsy; EIDDE, early infantile developmental and epileptic encephalopathy; EIMFS, epilepsy of infancy with migrating focal seizures; LGS, Lennox–Gastaut syndrome; DEE SWAS, developmental and/or epileptic encephalopathy with spike–wave activation in sleep; CAE, childhood absence epilepsy; JAE, juvenile absence epilepsy; JME, juvenile myoclonic epilepsy; GTCA, epilepsy with generalized tonic–clonic seizures alone; FS, febrile seizures; MTLE, mesial temporal lobe epilepsy.
Figure 2
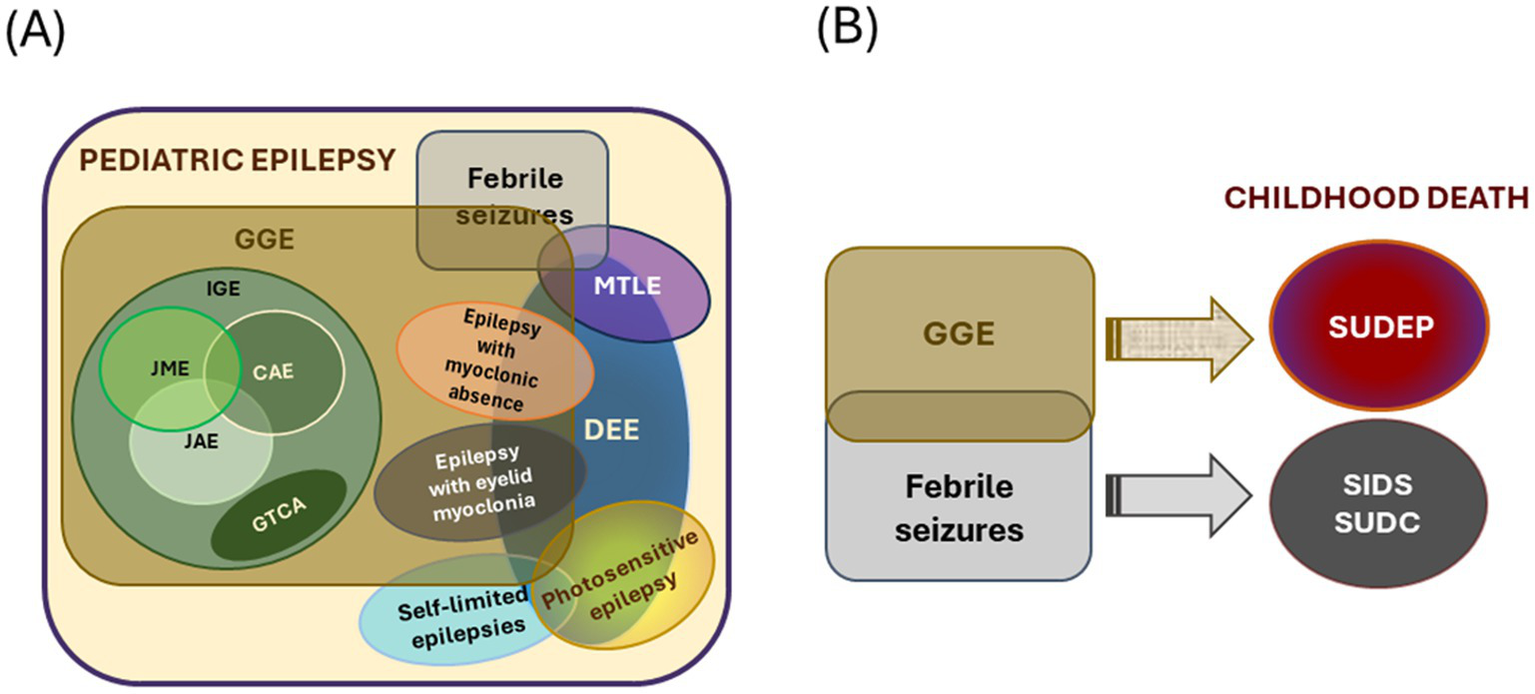
Types of pediatric epilepsies and connection to childhood death. (A) Schematic demonstrating overlapping features of different types of early-onset epilepsy. The overlapping sets mark the complexity of the scenario. (B) Genetic generalized epilepsies (GGE) and febrile seizures often make children predisposed to various premature death. The size of the circles/ovals is arbitrary and does not indicate the frequency of occurrence or other parameters. IGE, idiopathic generalized epilepsy; JME, juvenile myoclonic epilepsy; CAE, childhood absence epilepsy; JAE, juvenile absence epilepsy; GTCA, epilepsy with generalized tonic–clonic seizures alone; MTLE, mesial temporal lobe epilepsy; DEE, developmental and/or epileptic encephalopathy.
4 Current challenges in pediatric epilepsy
Apart from the diagnostic challenge due to diffused overlapping boundaries across different categories, one of the most critical challenges in the field of pediatric epilepsy is to combat drug-resistance. Drug-resistant epilepsy (DRE) is defined as “failure of adequate trials of two tolerated, appropriately chosen and used antiepileptic drug schedules (whether as monotherapies or in combination) to achieve sustained seizure freedom” (32). A large proportion of children suffering from early-onset epilepsy are non-responsive to current broad-spectrum medications that are largely strategized on certain channel proteins (11, 12). Surgical removal of dysplastic tissue is currently the only treatment against recurrent seizures in these patients (33). However, considering the complexity and exceptions, the chance of developing other epileptic foci in the future is quite high, thus triggering a vicious cycle of failure and recurrence of brain surgeries in these children (34). Parallelly, long-term anti-seizure drug therapy may negatively affect their cognitive development and increase the risk of premature death (35, 36). We elaborate on some of these clinical and mechanistic challenges in the following subsections.
4.1 Comorbidity with neurodevelopmental disorders
Up to 40% of cases of intractable pediatric epilepsy demonstrate significant association with brain malformations (32, 37). Such epilepsies mostly begin within the first year of life and are often accompanied by intellectual delay, motor impairment, and cognitive deficits (38). Magnetic resonance imaging (MRI) in such cases reveals blurring of white-grey matter demarcation, abnormal cortical thickening and folding, enlarged ventricles, and focal malformations of cortical development (MCD) (39, 40). Further, electroencephalograph (EEG) recordings demonstrate the presence of epileptiform discharges from the specific malformed or dysplastic region of the patient’s brain (41). However, it is still debatable whether brain malformation is the primary cause of epilepsy in such patients. Clinical evidence of extended epileptic foci exists beyond the dysplastic sites in the brain, suggesting that epileptogenesis can be dissociable from brain malformation (42).
Additionally, impairment of either voluntary or involuntary motor function is very common in epilepsy. Epilepsy and motor disorders can occur independently, harboring different underlying pathophysiology. However, despite having some distinct features, many motor disorders can also imitate epilepsy and vice versa (43). For instance, besides having other causative factors and comorbidities, disorders such as cerebral palsy, muscle dystonia, and ataxia show significant association with epilepsy (44). This is mostly because epilepsy affects the brain regions that control motor coordination, such as the frontal lobe, cortico-striatal connections, and basal ganglionic regions (45, 46). To add to complications, motor dysfunctions are often difficult to recognize at younger ages due to the developing state of the brain. It is seldom clear which problem precedes the other and hence, the complexity of diagnosing the etiology and eventual treatment increases in these patients.
4.2 Risk of mortality
Severe epileptic episodes or underlying neurological anomalies may be fatal for infants. The underlying mechanisms driving this epilepsy-related mortality have not yet been robustly explored, making this one of the most critical challenges in the field. Epilepsy that begins within the first year of life often predisposes patients to higher morbidity potential than children with late-onset epilepsy. More dramatically, children with malignant neonatal epilepsy are >12 times more likely to die than children with epilepsy onset at age ≥1 month (47). Some epilepsy syndromes affecting children at ≥1 year of age can have favorable prognoses. However, others result in medically refractory seizures, developmental delay/intellectual disability, and other neurologic handicaps (48, 49).
Sudden unexpected death in epilepsy (SUDEP) is the leading cause of mortality in children with epilepsy (50). SUDEP is defined as “a sudden, unexpected, witnessed or unwitnessed, non-traumatic, and non-drowning death in patients with epilepsy with or without evidence for a seizure, and excluding documented status epilepticus, in which postmortem examination does not reveal a structural or toxicologic cause of death” (51). Because most SUDEP incidents are unwitnessed, the exact sequence of events remains unknown. However, SUDEP nearly invariably happens after a generalized tonic–clonic (GTC) seizure (refer to Table 1) and is more common in patients with genetic epilepsies (52, 53). Additionally, patients with refractory epilepsy, early seizure-onset, intellectual disability, and male gender demonstrate increased risk (54, 55). Current pathophysiological mechanisms behind SUDEP are identified as an interplay among cardiac, respiratory, and autonomic nervous systems (56). Recent MRI studies have revealed reduced grey matter volume in the thalamus, frontal cortex, cerebellum, serotonin-producing neurons in the raphe nuclei and brainstem areas, and increased grey matter volume in the amygdala, hippocampus, cingulate areas in a significant proportion of SUDEP cases (57, 58). However, despite monitoring epileptic patients through clinical trials, the mechanism of SUDEP still remains unsubstantiated.
Beyond SUDEP, the other clinical categories leading to mortality in the pediatric population are sudden infant death syndrome (SIDS), and sudden unexplained death in childhood (SUDC). Although causation behind SIDS and SUDC is possibly multifactorial and largely unexplained, many reports suggest epilepsy as a potential contributing factor to predispose the child towards sudden death (Figure 2B).
SIDS is defined as “the sudden and unexpected death of an infant under the age of one year that remains unexplained after a thorough review of the clinical history and complete autopsy” (59). Some of the risk factors of SIDS include male gender, history of febrile seizures (FS), and preterm birth (60). Interestingly, like SUDEP and SUDC, many cases of SIDS show hippocampal abnormalities (61, 62). However, whether these abnormalities correlate to any underlying disorder or mortality in the patients is unclear. Interestingly, a significant proportion of these SIDS cases have shown underproduction of serotonin or downregulation of serotonin receptors (62–65). Some of these infants also demonstrated reduced GABA receptor binding (66). Whether these clinical features have any impact on the excitation/inhibition balance of the neurons is worth researching.
SUDC is defined as “the unexpected death of a child over 12 months of age which appears to be inexplicable even after a detailed case investigation” (67). SUDC usually occurs between 1 and 5 years of age, and it is the fifth leading cause of death in children. Unlike SUDEP, SUDC doesn’t occur after a seizure; however, patients with a history of FS have a higher vulnerability (68). A proportion of these patients also have hippocampal anomalies (69). Whilst clinical evidence suggests a relationship between hippocampal anomalies, FS history, and SUDC, the nature of this association is currently unclear.
Although there is awareness of SUDEP, SUDC, and SIDS, the etiology and mechanisms behind these sudden deaths are largely unknown. Most of the case studies are limited due to the spontaneity as well as the rarity of such cases, thus being difficult to model. A common link among SUDEP, SUDC, and SIDS could be the history of FS and that the brain regions in respiratory and circulatory regulation are involved and altered (57, 70). Indeed, abnormal breathing patterns, hypoxemia, and hypercapnia are more commonly observed, but the correlation is inconsistent (71–73).
4.3 Complex genetic etiology
Many genetic mutations are being identified in association with epilepsy; some important ones are listed in Table 2. Around 40–70% epilepsy cases show genetic etiology (74, 75). In certain epilepsy types, the genetic cause is well known, like SCN1A mutation in Dravet syndrome (28, 76). However, in most cases, the genetics are inferred based on the familial inheritance pattern. For instance, pediatric generalized epilepsies have a strong heritable nature (77), but the specific genes responsible for them are not fully known. Unfortunately, simple inheritance patterns are very rare in epilepsy. Pediatric epilepsy shows complex inheritance with varying expressivity patterns. It is often observed that mutations in the same gene can cause variable phenotypes in patients. This phenomenon is well characterized in patients with SCN1A mutations, which show a range of phenotypes from FS to Dravet syndrome (78). It is hypothesized that these phenotypes are dependent on mutation severity. Mild missense mutations in SCN1A are often identified in FS patients, while more severe loss-of-function mutations are associated with severe epilepsies such as Dravet syndrome (78, 79). Interestingly, phenotype variation is observed even in cases where the mutations are the same. For example, patients with mutations in the PI3K-AKT–MTOR pathway show a spectrum of developmental and epileptic phenotypes with varying severity (80). Mutations in genes coding for MAST (microtubule-associated serine/threonine) kinases (MAST1-4), upstream of the PI3K pathway, have recently been associated with developmental abnormalities, epileptic seizures, and cognitive impairment (81–83). Epilepsies related to MAST genes have also been categorized within DEE and GGE due to overlapping etiology, thus complicating the categorization. Conversely, there are multiple genes that are shown to be associated with the same epilepsy syndrome, as seen in the case of DEE (84–86). Further contradictions arise when genotype–phenotype cannot be correlated directly. For instance, the severity of phenotypes in patients with mutation in ciliary gene CDKL5 does not solely depend on the primary mutation, but also on the interplay between intrinsic and extrinsic factors (87). Such cases are difficult to treat and are often intractable.
Table 2
| Causative factor type | Causative factor | Type of epilepsy associated with | References |
|---|---|---|---|
| Signaling defects | kRAS | Temporal lobe epilepsy, DEE | (283, 284) |
| BRAF | Focal/Generalized epilepsy | (285) | |
| NF1 | Focal/Generalized epilepsy | (286, 287) | |
| PIK3CA, AKT3, PTEN, MTOR | Focal/Generalized epilepsy | (80, 109, 288) | |
| TSC1, TSC2 | Temporal lobe epilepsy | (289, 290) | |
| NPLR2, NPLR3 | Temporal lobe epilepsy | (291) | |
| DEPDC5 | Temporal lobe epilepsy | (292) | |
| Abnormal neural migration | RELN | Temporal lobe epilepsy | (123, 293) |
| DCX | Temporal lobe epilepsy | (198) | |
| LIS1 | Temporal lobe epilepsy | (294) | |
| Ciliopathy | EFHC1 | JME | (295) |
| CILK1 | JME | (223) | |
| CDKL5 | JME, DEE | (296, 297) | |
| Channelopathy | SCN1A | DEE, Dravet syndrome, MTLE | (76, 291, 298, 299) |
| SCN2A | SeLNE, SeLIE, DEE, Febrile seizures | (300–302) | |
| SCN8A | SeLIE, DEE, JAE | (303–305) | |
| GRIN2A | SeLECTS | (306) | |
| KCNT1 | EIMFS | (307) | |
| KCNQ2, KCNQ3 | SeLNE, DEE | (308–310) | |
| KCNJ10 | GGE, MTLE | (311, 312) | |
| CACNA1A | GGE, DEE | (313–315) | |
| CACNA1H | GGE | (316–318) | |
| HCN1 | Febrile seizure, DEE | (319, 320) | |
| GABRA1A, GABRG1 | Dravet syndrome, CAE, JME | (321–325) |
Summary of different genes and environmental factors implicated in pediatric epilepsies.
SeLNE, self-limited neonatal epilepsy; SeLIE, self-limited infantile epilepsy; SeLECTS, self-limited epilepsy with centrotemporal spike; DEE, developmental and/or epileptic encephalopathy; GGE, genetic generalized epilepsy; JME, juvenile myoclonic epilepsy, JAE, juvenile absence epilepsy, EIMFS, epilepsy of infancy with migrating focal seizures; CAE, childhood absence epilepsy.
The varying expressivity of epilepsy phenotype is hypothesized to be associated with genetic modifiers (88). Genetic modifiers are gene variants or non-coding single nucleotide polymorphisms (SNPs) present in a patient, in addition to the primary mutation, that are instrumental in modifying the phenotypic outcome in that patient. So, the final phenotype is determined by the close epigenetic, genetic, or functional interactions between these variants, which breaks the dogma of epilepsy being a monogenic disease. We think that identifying such novel epilepsy modifier genes can enhance our understanding of the underlying mechanisms.
5 Modeling epilepsy
Despite our understanding of the underlying causes and characteristics of various epilepsy syndromes, pediatric epilepsy remains a burden on the healthcare and economic sectors, mainly due to the predominance of drug-resistance and premature deaths (8–10). This emphasizes the importance of investigating the molecular and developmental mechanisms underlying early-onset epilepsies, which can eventually aid in designing targeted therapeutic strategies. To achieve this, epilepsy models that can closely recapitulate the human specific features are necessary. An ideal model should show construct validity (recapitulation of patient-specific etiology), face validity (replication of patient-specific phenotypes), and predictive validity (responding to treatments that can be effective in humans) (89). Although epilepsy has been modeled in various non-mammalian systems, such as Drosophila, C. elegans, and zebrafish, we consider rodents as a stronger candidate for being a model organism for epilepsy and neurodevelopmental disorders. This is because mice and humans show over 99% genetic homology, and more importantly, they show similarity in many stages of neurodevelopment and can undergo genetic manipulation to introduce patient-specific mutations for testing (90, 91). Currently, several mouse models are being used to study epilepsy; herein, we discuss different types of them. Broadly the mouse models of epilepsy can be categorized as induced models and genetic models.
5.1 Induced epilepsy models
An induced epilepsy model is where seizures are triggered in healthy mice by chemical or electrical stimulation of the brain (92). This is amongst the oldest strategies and is used extensively in current research as well. For chemical stimulation, an intracerebral or systemic injection of some of the neuroexcitatory drugs is used. Kainic acid is one of the first compounds used to recapitulate temporal lobe epilepsy (TLE), a common kind of focal epilepsy observed in both adults and adolescents (93, 94). It is a glutamate analog, and its administration causes neuronal depolarization, particularly in the mouse hippocampal region (95). However, patients with TLE also show neurological compromise in extrahippocampal regions (96, 97). Pilocarpine is often used to produce lesions in neocortical areas along with the hippocampus (98). Other compounds like pentylenetetrazol (PTZ), strychnine, N-methyl-D, L-aspartate, and penicillin are also used as convulsants to model seizures, and loosely epilepsy (92). On the other hand, electrical stimulation involves implanting electrodes in the region of interest and stimulating them to generate seizure outcomes (99). Less invasive methods, such as whole brain stimulation via trans-auricular or trans-corneal surface electrodes, are also available (100). It is important to note that most of the above-mentioned induced models exhibit acute seizures. Chronic seizures can be induced by kindling wherein mice exhibit spontaneous seizures after repeated electrical or chemical stimulation (101). Nevertheless, induced models generally do not validate the actual etiology of the disorder, especially with respect to a large proportion of pediatric epilepsy patients. In this regard, genetic models emerge as a more realistic tool to study underlying mechanisms.
5.2 Genetic epilepsy models
Advancements in gene editing technology have allowed researchers to integrate many patient-specific gain-of-function or loss-of-function genetic mutations in mice. Such models are very instrumental in understanding mechanisms as they have high construct validity. These models also provide a strong platform, allowing preclinical testing of small molecules. Some of the well-known mouse models of epilepsy include constitutive knockout of channel proteins such as SCN1A, SCN2A, SCN8A, KCNA1, GABRA1, KCNQ2, and many more (102–105). These models have not only helped us to understand the mechanistic aspect of these mutations but also the associated behavioral and developmental consequences. Parallelly, signaling pathways, especially the PI3K-AKT–MTOR pathway, are associated with pediatric epilepsies. Unlike most of the ion channels, the constitutive knockout of such pathway genes is often embryonically lethal (106–108). Furthermore, a large proportion of pediatric epilepsy patients exhibit spontaneous or de novo mutations in the pathway genes, where the phenotypic severity is often dependent on the developmental timing and regional extent of the mutation (109, 110). This further complicates the modeling design for such specific genetic variants since a constitutive deletion strategy may not mimic the actual scenario in such cases. To circumvent this problem, conditional strategies such as Cre-lox or FRT-Flp systems are used to induce the patient-specific mutations in specific cellular lineages, especially for genes associated with signaling pathways like PI3K-AKT–MTOR (111–115). The developmental timing of the genetic mutation can be more finely regulated by using inducible cre systems (116). Such strategies give more flexibility and allow researchers to spatially and temporally control their genetic modifications. Moreover, it allows partial manipulation of the genes that are important for early embryonic development and are fatal to mice if removed. While such conditional lines provide cell lineage and variant-specific modifications of the gene of interest, even finer focal genetic perturbations are possible in mice. One such example is a Pten mouse model, wherein an exogenous viral vector carrying Cre has been injected into the hippocampal area in Pten floxed background (117). By controlling the dose and injection site of the viral vector, one can create a mosaic loss of Pten in the region of interest. Another approach is to perform in utero electroporation (IUE), where a plasmid DNA targeting the gene of interest is injected in utero into the embryonic brain ventricles and then electroporated to the region of interest using a pair of electrodes. IUE strategy is widely implicated in inducing activating PI3K-AKT–MTOR pathway mutations in rodent systems (118–120). IUE strategy provides a deeper understanding of the function of a particular gene in a specific cellular population. These models are very relevant in studying developmental mechanisms underlying MCD-associated epilepsies or focal epilepsies, wherein patients often show somatic mosaicism (110, 121).
6 Mechanisms underlying epilepsy
Much of our current mechanistic understanding of pediatric epilepsy has come from studying model systems (mentioned in the previous section). Since the start of the epilepsy field, epilepsy has been always mechanistically attributed to mutations in ion channels and, in turn, alterations in neuronal excitability and inhibition (122). However, research over the past few decades has identified severe non-ion channel-associated genes being associated with pediatric epilepsy (109, 123–126). These studies highlighted novel mechanisms underlying epilepsy beyond ion channels. Herein we discuss some of these mechanisms.
6.1 Mechanisms involving ion channels: channelopathy
Several studies have associated either voltage- or ligand-gated ion channels with monogenic epilepsies, where the seizure onset coincides with the temporal expression of the affected channel during development (127–131). Mutations in ion channels are involved in different types of pediatric epilepsies, such as self-limiting epilepsies, generalized epilepsies, epileptic encephalopathies and FS (Table 2). Channel proteins and epilepsy by itself is a huge topic to cover; and has been discussed in vast lengths in several reviews and book chapters previously. In this review, we are only providing a brief overview of the mechanistic relevance of different channel mutations causing epilepsy; further elaboration is beyond the current focus of this review.
Voltage-gated sodium channels (NaV) allow voltage-dependent influx of Na+ ions to initiate neuronal depolarization; different genetic mutations in these channels can affect the functioning in varied ways (132). For instance, mutations in the inactivation gate domain of NaV causes the channel to close slowly or incompletely during the depolarization period, leading to an excessive influx of Na+ ions inside the neuron, making it intrinsically hyperexcitable (133, 134). In contrast, mutations that allow channels to recover faster from the inactivation state cause increased firing frequency. Some also lower the action potential threshold, causing neuronal hyperstimulation (133). NaV loss-of-function mutations found in inhibitory interneurons, in turn, can result in disinhibition in neural networks (Figure 3) (135). Again, voltage-gated potassium channels (KV) are critical for neuronal repolarization and bringing the membrane to the resting state. KV mutations can alter the electromechanical coupling in the channel, preventing it from sensing voltage (136); this results in repetitive neuronal firing and altering excitation/inhibition balance. Apart from the propagation of action potential, voltage-gated ion channels, such as voltage-gated calcium channels, trigger neurotransmitter release from presynaptic boutons. Genetic variants in a family of voltage-gated calcium channels are also associated with epilepsy (137, 138).
Figure 3

Network excitation in healthy and epileptic brain. (A) In a healthy brain, excitatory neurons (ENs) generate a balanced output due to feedback/feedforward inhibition from interneurons (INs). (B) In epileptic patients with channel mutations, this inhibition is reduced due to hypoexcitability or impaired action potential propagation in interneurons, or (C) ENs become inherently hyperexcitable or have a lower threshold for action potentials. These events can occur independently or together, leading to network hyperexcitation. ENs, excitatory neurons; INs, interneurons; IPSP, inhibitory postsynaptic potential; GOF, a gain of function; LOF, loss of function; NaV, voltage-gated sodium channel; KV, voltage-gated potassium channel.
Ligand-gated ion channels are typically found on the postsynaptic membranes or dendritic spines and play a vital role in signal reception. Mutations in these channels alter the ligand binding affinity and are implicated in epileptogenesis (139). For instance, mutant acetylcholine receptors get activated and remain open at abnormally lower levels of acetylcholine, causing hyperexcitability (140). Some mutations also impair the assembly or reduce current flow through ion channels, potentially increasing the net microcircuit excitability (141, 142). Not all mutations lead to altered biophysical properties of a channel. Some biophysically silent mutations keep the structural configuration undisturbed. However, their subcellular localization, expression levels, or affinity to the cytoplasmic interactor proteins may alter, disrupting membrane properties. Indeed, this feature is very well characterized in NMDA and AMPA receptors, which play important roles in long-term potentiation (LTP) and long-term depression (LTD) (143, 144). Patients harboring mutations in these receptor genes develop seizures, along with developmental and cognitive comorbidities (145–147). Similarly, there are molecules which aid in physical connection of synapses, primarily belonging to the neurexin-neuroligin family. Mutations in both have been associated with epilepsy (148, 149). Expression of neurexin-neuroligin proteins was reported to be increased in epileptic patients (150). Interestingly, this family of proteins was originally associated with neuropsychiatric conditions like autism spectrum disorder (ASD) (151). However, its newly identified role in epilepsy may provide insights into uncovering shared mechanisms between epilepsy and ASD.
Most of these mechanistic insights gained impetus from studying structural-biochemical properties and electrophysiological features of the channel proteins in various model systems, such as cell lines, oocytes expressing channel variants, and artificial modeling which are far away from the actual human condition (141, 142, 152). Unfortunately, studying isolated molecules or cells does not reveal the full picture, considering epilepsy is a network phenomenon. Another major caveat was that most channel-related research has an extreme bias toward neuronal activity, neglecting the potential roles of other neural cell types. Research over the past few decades has proven the importance of glia in modulating synaptic modulation and transmission by uptaking/redistributing ions, glucose, and water molecules at the synapse (153–155). In fact, patients with mesial temporal lobe epilepsy (MTLE) often carry mutations in inwardly rectifying potassium channels (KCNJ10) and water channels (AQP4), that are abundantly expressed in astrocytes (156–158). Our understanding of neuron-astrocyte interaction and its association with epilepsy currently remains limited. However, this association is gaining more and more relevance toward developing novel anti-epilepsy strategies.
6.2 Mechanisms involving proliferation and maturation of neural cells
Besides channelopathies, multiple gene variants in critical signaling pathways, specifically PI3K-AKT–MTOR and RAS–RAF–ERK pathways, have been identified as epileptogenic (159, 160). These pathways are highly conserved across evolution and interact with each other to promote critical processes like cell growth, proliferation, differentiation, and apoptosis, as well as the generation of different neural cell types and synapses (161–168) (Figure 4). Mutations in one such pathway often impair the regulation of other pathways, causing variable consequences downstream. Since global homozygous deletion in the components of PI3K-AKT–MTOR and RAS–RAF–ERK pathways mostly caused embryonic lethality in animal models, recent studies have used brain-specific conditional genetic deletion via either in utero electroporation or recombination techniques to study the effects. Genetic null mutants of Depdc5, Pten, NF1, Tsc1, and Tsc2 and in utero electroporation models for Akt, Kras, Braf, and Rheb recapitulate the range of clinical phenotypes, either fully or partially (119, 169–176). Here, we emphasize a few of these models which aided in understanding the mechanism behind pediatric epilepsy. Pten is one of the negative regulators of the PI3K-AKT–MTOR pathway, and its selective removal from the murine hippocampus resulted in spontaneous seizures (177). This model has also shown increased activation of the MTOR pathway. Further, gain-of-function mutations in PI3K or loss-of-function mutations in TSC1/TSC2 in the initial stages of brain development (radial glial cells, or RGCs) also resulted in seizures in the early stages of life (111, 178–180). Many chemically induced epilepsy models also show alteration in the PI3K-AKT–MTOR pathway (181). Mechanistically, Pten, Pik3ca, Tsc1, and Tsc2 mouse models have demonstrated that pathway hyperactivation results in increased cell proliferation, cellular hypertrophy, dendritic hypertrophy, and aberrant axonal growth (111, 171, 173, 182). Besides these cell-autonomous changes, the affected cell influences the development and function of the adjacent non-mutated cells (118, 183). Similar mechanisms have been identified in mouse models harboring mutations in the RAS pathway (176, 184). We hypothesize that the mutations in these critical signaling pathways not only alter the synaptic properties but also alter the numbers and diversity of the excitatory/inhibitory neurons in the neural network, leading to network hyperexcitability. Moreover, these mutants also modeled the coexisting MCDs such as megalencephaly, hydrocephalus, and hippocampal/cortical dysplasia along with epilepsy, thus accurately mimicking the clinical scenario (111, 171, 173). This strongly suggests that disruption of neurodevelopmental processes is central to both MCDs and epileptogenesis. Besides cell growth and proliferation, MTOR activity critically maintains cellular autophagy and vesicular trafficking, thus, in turn, regulating neurotransmitter release, synaptic recycling, and mitochondrial homeostasis (185). Indeed, reduced cellular autophagy is reported in conditional knockout models for Pten and Tsc1 (186). However, it remains disputed whether it is a cause or an effect (187, 188). Translation and surface expression of many ion channels are also dynamically regulated by PI3K-AKT–MTOR and RAS–RAF–ERK pathways (189). Pathway misregulation can alter the surface expression of these proteins and, in turn, cellular excitability, making this a plausible explanation for connecting signaling pathways to epilepsy.
Figure 4
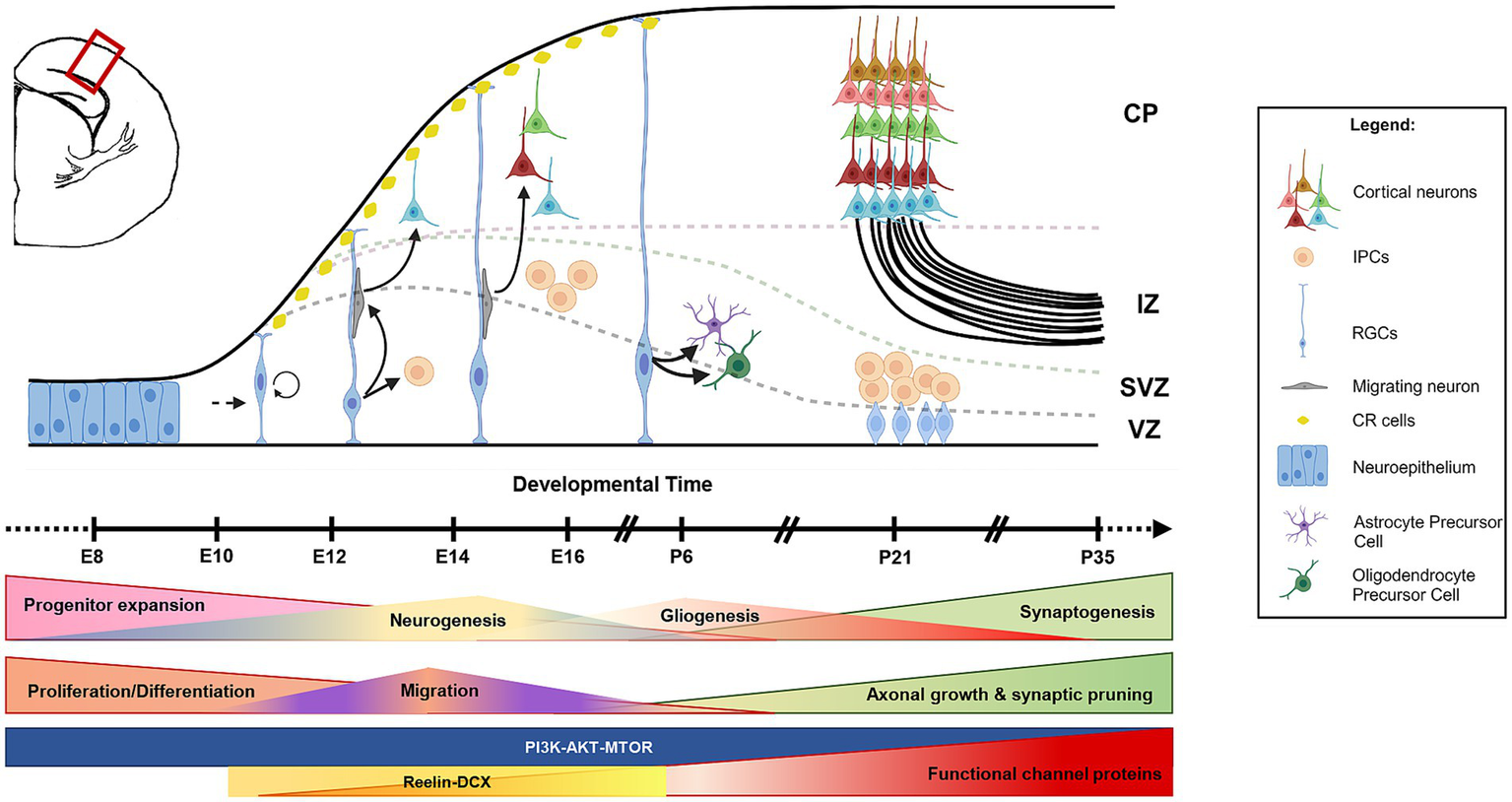
Cascade of neurogenesis and gliogenesis in developing mouse brain. The schematic of a coronal hemi-section of a developing mouse cortex shows that the initial phase of brain development involves the expansion of the progenitor pool (neuroepithelial cells and RGCs). This is followed by the formation of neurons (neurogenesis) and glia (gliogenesis) either directly (from RGCs) or indirectly (from IPCs). Newly formed neurons migrate toward the pial surface with the help of radial glial projections and occupy in an inside-out fashion (late-born neurons occupy upper layers, and early-born neurons occupy deep layers). CR cells regulate this radial migration process. Neurogenesis and gliogenesis are followed by synaptogenesis and functional network formation. RGCs, radial glial cells; IPCs, intermediate progenitor cells; CR cells, Cajal–Retzius cells; CP, cortical plate; IZ, intermediate zone; SVZ, subventricular zone; VZ, ventricular zone; E, embryonic; P, postnatal.
The above-mentioned mechanisms suggest that suppression of this pathway using MTOR inhibitors should rescue the phenotype. Unfortunately, use of inhibitors such as rapamycin and its analogs showed limited success in curbing epilepsy in both mouse models as well as patients (190, 191). An instrumental study using the clinically relevant Pik3ca genetic models demonstrated that activating Pik3ca mutation causes intrinsic neuronal hyperactivity in mice, which is, in turn, acutely suppressible by PI3K and/or AKT but not MTOR inhibition (192). These findings suggest that MTOR hyperactivation is not always the sole direct reason behind these epilepsies and possibly explain the reason behind the partial or complete failure of rapamycin analogs and other MTOR inhibitors in treating epilepsies of all kinds. The mouse models also revealed that the underlying mechanism behind the emergence of epilepsy in such pathway-related mutations traces back to abnormal neurogenesis and gliogenesis (111, 159, 174, 175, 192). This re-emphasizes the importance of early developmental processes in determining brain circuitry and expands epileptic mechanisms beyond channelopathies. As we will see in the upcoming sections different components of these pathways are involved in different intracellular signaling beyond MTOR-driven cell growth.
6.3 Mechanisms involving neural migration
Neural migration to specific zonal layers of the brain is crucial for making appropriate axonal connections and synaptic maturation (Figure 4). Many genes are involved in regulating these processes, such as RELN, DCX, LIS1, ARX, TUB1A, and FLNA. Mutations in these migration-related genes often result in lamination defects and epilepsy in humans (193–197). Here, we focus on reelin (RELN) and doublecortin (DCX), whose functions with respect to neural migration and epilepsy are more elucidated. DCX is an X-linked microtubule-associated protein; patients with DCX mutations have structural cortical malformations, often associated with epilepsy (194). Specifically, these patients are mosaic for DCX mutations, such that a proportion of neurons migrate successfully in the cortex, while the mutant ones fail and accumulate in subcortical regions (193, 198). This abnormal neuronal localization impacts the formation of functional networks and consequently leads to epileptogenesis. Interestingly, studies on resected human brain samples demonstrated fewer Reelin+ and DCX+ cells in MTLE-hippocampal sclerosis patients and chemically-induced seizure models (199–202). Rodent models of clinically relevant DCX mutations displayed impaired migration of hippocampal granule cells and spontaneous seizures (203, 204). A double-null model for DCX and Doublecortin-like kinase 1 was epileptic and exhibited more severe migration abnormalities in cortical projection neurons and inhibitory interneurons (205, 206). Similarly, Reelin is involved in neural migration, especially during the early period of embryonic neurogenesis and hippocampus formation. At this stage, Reelin is expressed in Cajal–Retzius cells that play instrumental roles in the inside-out layer formation of the neocortex as well as in hippocampal lamination. Indeed, focal malformations in cortex and hippocampal lamination defects are observed in patients with RELN mutations (207, 208). These patients also exhibit intractable epilepsy (209). Similar phenotypes were also observed in the Reeler mouse mutant (210). Later in development, reelin is also expressed in inhibitory GABAergic interneurons, which are important for regulating network excitability as well as synaptic maturation of hippocampal granule cells (211–213). Reduction in the interneuron number due to RELN mutation is hypothesized to cause network hyperexcitability (202, 214). However, this hypothesis is debatable as many clinical reports lack proper age-matched controls. In a recent retrospective pediatric brain study, no change was observed in the Reelin+ cell number in the hippocampus of epileptic human brains as compared to that of age-matched controls (215). Moreover, variability in immunohistochemistry results is common due to differential post-processing time for human brains (216, 217). On the other hand, commonly used rodent models, like kainic acid-induced rat seizure model, are complex to analyze; kainic acid itself induces death of hippocampal neurons. To complicate further, Reelin is known to be an upstream regulator of PI3K and RAS pathways (218). So, alteration in the reelin expression may have consequences with respect to cell proliferation and maturation; but these associations are yet to be proven. Taking together, it remains to be determined whether neuronal migration defects directly lead to epilepsy.
6.4 Mechanisms involving genesis and function of cilia: ciliopathy
Ciliary genes have recently been associated with epilepsy. There are two types of cilia present in the brain: primary/nonmotile cilia and secondary/motile cilia. Primary cilium is a specialized organelle found in almost all neural cells that senses and reacts to most of the signals and external environmental cues (219, 220). These cilia are rich in signaling receptors and ion channels, which are essential for brain growth and function (219). Therefore, it is no surprise that mutations in the genes involved in ciliary maturation and function cause neurodevelopmental disorders including epilepsy. Clinical studies have reported that a significant proportion of patients with juvenile myoclonic epilepsy (JME) harbor mutations in the genes involved in primary cilia formation, like CILK1, EFHC1, and CDKL5, while reduced number of primary cilia was identified in surgically resected brain samples of focal cortical dysplasia (FCD) patients (220–225). Such clinical findings hint at the role of primary cilium in epileptogenesis (Figure 5). However, the precise mechanistic relation between primary cilia dysfunction and epilepsy is yet to be known. Animal models indicate the importance of primary cilium in neural cell proliferation, differentiation, migration, and synaptogenesis (226, 227). Considering that primary cilia are like signal sensors for a cell, their loss possibly makes neurons insensitive to external cues such as neuromodulators and causes epilepsy (Figure 5). However, a few contradictory reports challenge this hypothesis. A mouse model harboring a patient-specific variant in CILK1 was epileptogenic, while another study on that identical variant failed to identify any epileptic behavior in mice (223, 228). Such contradictory results prevent us from confirming the direct correlation between ciliopathies and epilepsy. Nevertheless, the field of primary cilia is gaining momentum in the field of epilepsy. Very recent high-resolution electron microscopy data from human brain slices have shown that primary cilia are diverse in their shape, size, and microtubule architecture depending on the cell type and brain regions, which in turn can diversify the signaling competencies (229). This structural range of primary cilia provides each neuron or glial cell with a unique barcode of access to the surrounding neural network which influences the overall network excitability. Hence, primary cilia have now been considered an integral component of the synaptic signaling and neural connectome.
Figure 5
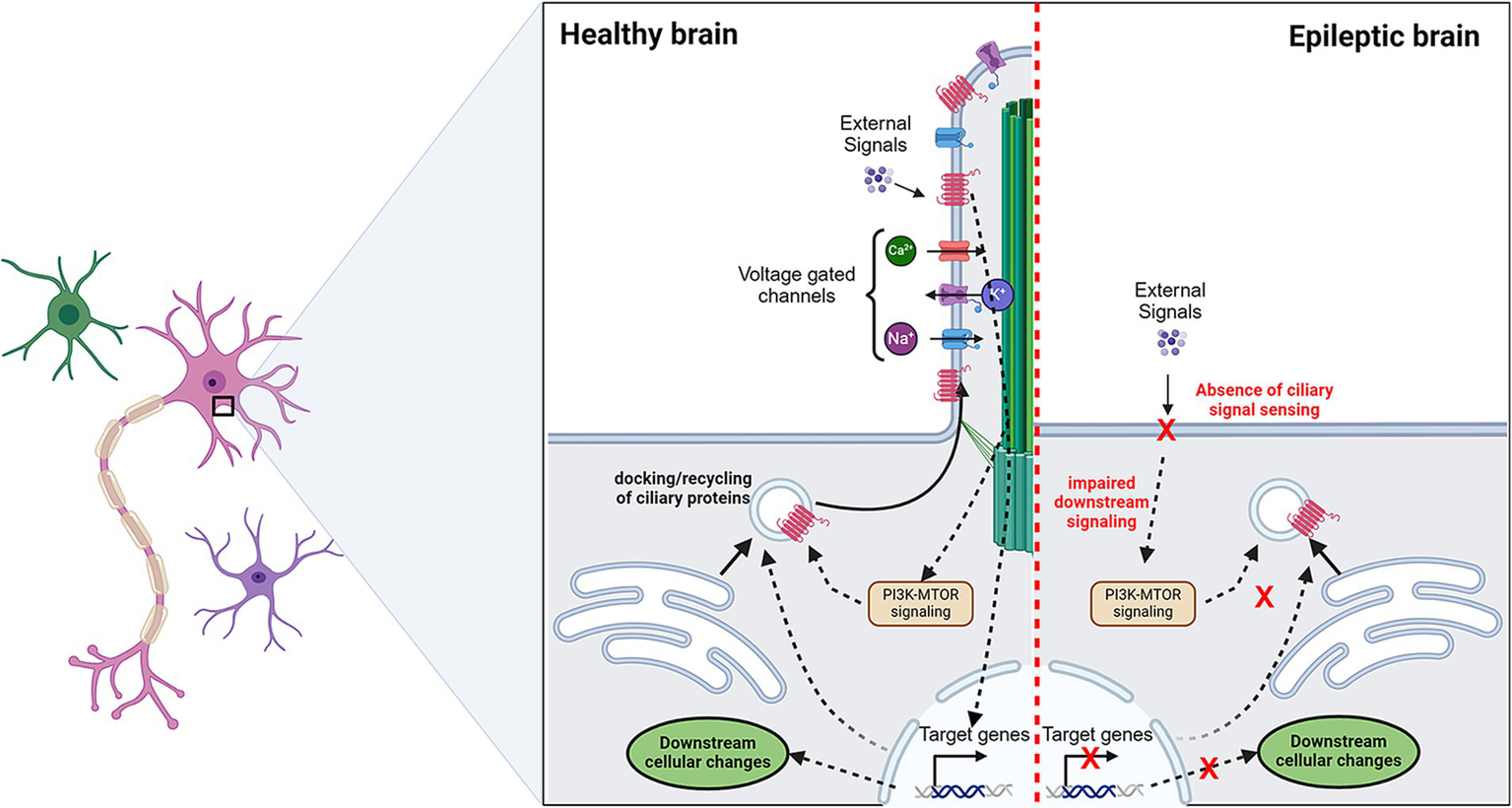
Potential role of primary cilia in epilepsy. Primary cilia sense the presence of extracellular cues via signaling receptors. This leads to a cellular cascade that may lead to activation/inactivation of a set of genes. These transcriptional changes can alter the docking or recycling of surface proteins which include signaling receptors and ion channels. In epilepsy, the absence of primary cilia is observed in a proportion of cells. This makes cells insensitive to “cilia-mediated signal sensing.” Making aberrant regulation of ion channels which may tweak the excitation/inhibition balance. Dotted arrows indicate the proposed pathways.
Even less information is known about motile cilia dysfunction and epilepsy. Motile cilia, present on brain ependymal cells, play an integral role in the circulation of cerebrospinal fluid. Recent studies indirectly suggest that blockage of fluid flow underlies epilepsy development (19, 230). This disrupted flow of cerebrospinal fluid is attributed to impaired development and function of ependymal cilia. However, the ciliopathy field is still in its infancy due to the cilium’s high structural complexity and diverse functional range based on cell type and developmental stage. Interestingly, the PI3K-AKT–MTOR pathway is very central to this process as well. Crosstalk between the PI3K-AKT–MTOR pathway and cilia is evident, with defects in the pathway function having adverse effects on the cilium length or even the development of ependymal cells (231–233). However, no direct role of MTOR-related ciliopathy in epilepsy has yet been established. Nonetheless, it opens a new avenue for understanding epilepsy and related neurodevelopmental disorders.
6.5 Epilepsy and association with sleep and body clock
Beyond direct mechanisms through ion channels or neurotransmitters, novel anti-epilepsy therapies can also be developed by studying body homeostasis and body cycles, including sleep and wakefulness. Epilepsy and sleep have been bidirectionally associated with each other for centuries (Figure 6). In other words, epilepsy can cause sleep problems in patients while sleep deprivation may trigger certain types of epilepsy (234, 235). Especially children with epilepsy have been documented for poor sleep quality, increased nocturnal awakenings, early morning awakenings, difficulty in falling asleep, and/or excessive daytime sleepiness. Certain epileptic seizures occur consistently during specific stages of sleep–wake cycles, suggesting a strong correlation between the two. Even with respect to the risk of mortality, most cases of SUDEP and SIDS occur during sleep, with SIDS specifically linked to the rapid eye movement (REM) sleep stage (56, 236, 237). In other cases, increased synchronous neuronal firing during non-REM (NREM) sleep may make the brain more susceptible to seizure activity, while inhibition of thalamocortical synchrony during REM lowers the epileptiform brain activities (238, 239). This association is often taken as an advantage to diagnose seizure activities. For instance, sleep deprivation is used before EEG recording, both in epilepsy models and pediatric patients, to evoke heightened neuronal activity (111, 240).
Figure 6
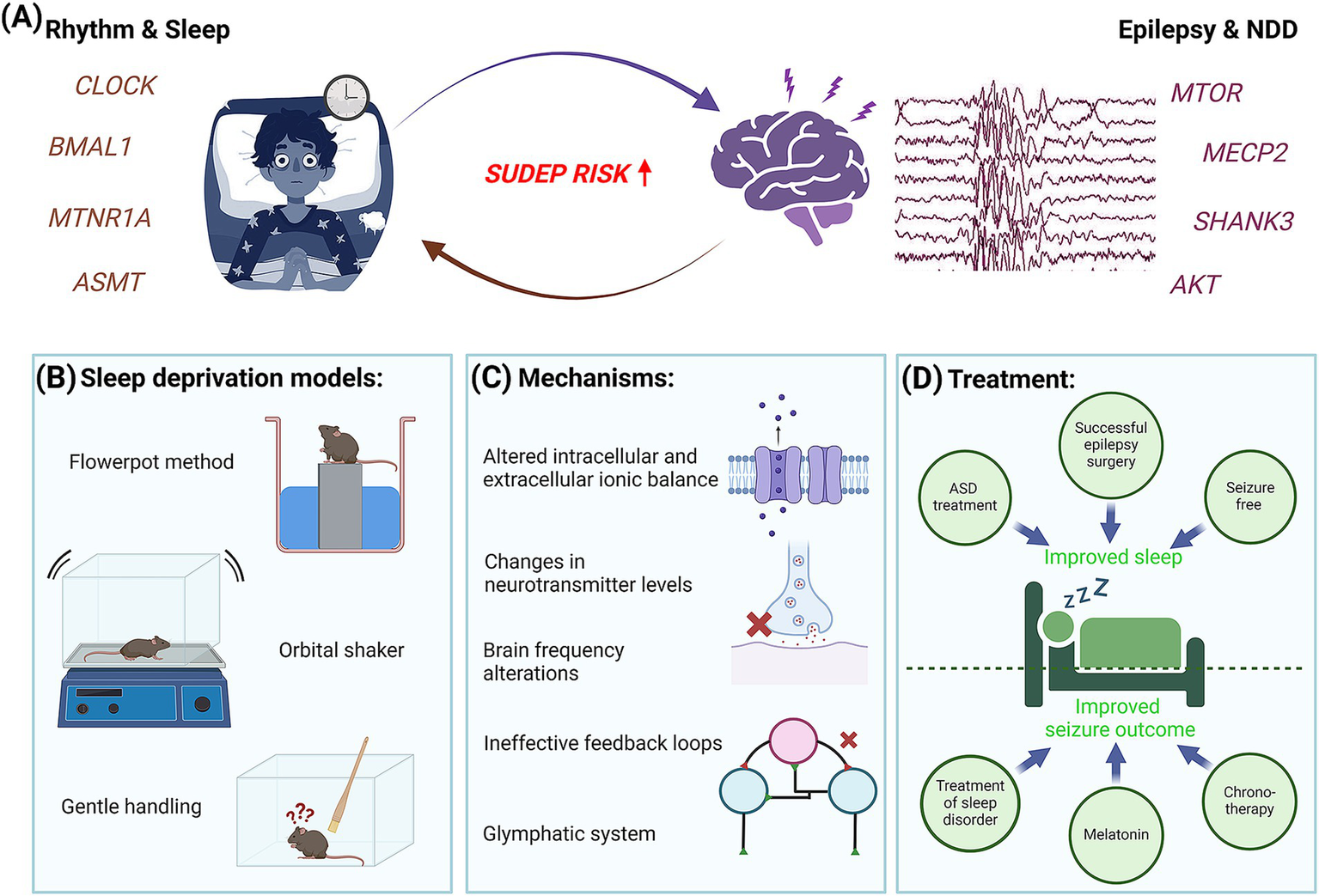
Interconnection of epilepsy with body rhythms and sleep. (A) Summary diagram representing the close association between sleep deprivation and epilepsy. Schematic demonstrates a patient suffering from either sleep deprivation or epilepsy triggers a higher risk of SUDEP. (B) Sleep deprivation in rodents is induced by changing the environment or application of stress. (C) Some of the common molecular and physiological mechanisms underlying sleep deprivation and epilepsy are shown. (D) Close association of sleep deprivation and epilepsy introduces a novel therapeutic angle.
The underlying reason behind this interesting connection may be aligned to changes in neurotransmitter levels, intracellular and extracellular ionic balance, alterations in the duration of sleep stages and in brain wave frequencies, ineffective feedback loops, and other possibilities (241, 242). Some reports also link sleep disruption and pediatric epilepsy through stress and induced neuroinflammation, marked by altered expression of inflammatory mediators and glial activation (243, 244). Further, the glymphatic system, a recently discovered waste clearance system of the central nervous system, is being considered as a potential mechanism connecting sleep and many neurological disorders including epilepsy, such that its dysfunction may account for the common association between disturbed sleep or sleep deprivation and increased seizure risk (245–247). This phenomenon, in turn, can critically disrupt normal brain development and cognition, more prominently if it begins at early stages in neonatal and pediatric populations (241). Although recent studies are trying to understand the effect of current anti-seizure drugs on the glymphatic system (248), there are a lot of open questions that remain unaddressed.
Curbing sleep issues is proving a good way to attenuate the severity of seizure episodes in children, thus becoming potential avenues for anti-epilepsy therapies. Certain anti-epileptic drugs have analgesic effects that help to alleviate sleep quality in some pediatric patients. Administration of melatonin, an endogenous hormone that inhibits brain excitability and induces balance in sleep–wake cycles and circadian rhythms, is found to be neuroprotective and anti-convulsant in nature in both patients and mouse seizure models (242, 249–251). The mechanism is based on the activation of two high-affinity G protein-coupled melatonin receptors, MT1 and MT2, which provide beneficial effects on regulating the circadian and sleep pathways without the baggage of side effects seen due to regular use of sleep medications (252). Slow-release melatonin and its agonists are currently being considered as sources of novel, efficacious therapeutics against certain types of epilepsies and sleep disorders (242, 252, 253). Parallelly, core circadian proteins, BMAL1 and CLOCK, have been shown to influence excitability and seizure threshold by regulating the PI3K-AKT–MTOR pathway (243, 254–257). Studies also reveal a feedback loop where MTOR activates BMAL1 via phosphorylation (254). Epilepsies associated with PI3K-AKT–MTOR pathway-related brain malformations also show a dysregulation of the expression of core circadian genes, thus making them excellent therapeutic targets for treating this specific type of epilepsies (258, 259). Consequently, the therapeutic possibilities for optogenetics and chronotherapy are actively being considered to treat seizures, especially those that are known to have a circadian sleep component as an underlying mechanism. Finally, given the considerable proportion of DRE in children, we feel an obvious treatment strategy would be toward advancements in personalized anti-epileptic treatment paradigms, using the time-dependent information relevant to the individual patient. This information may include times of day that witness the greatest occurrence of seizures of the highest levels of epileptogenicity in relation to sleep, wakefulness, and other body rhythms (242, 260).
7 Limitations of epilepsy models and novel advancements
Despite using diverse model systems, researchers continue to face challenges ranging from proper disease recapitulation to discovering novel therapeutics. The aforementioned induced epilepsy models are currently used by large screening consortia, such as the Epilepsy Therapy Screening Program, due to the ease and high throughput capacity (261). However, some of these inducible agents are known to cause neuronal loss and behavioral changes in mice. Moreover, chemical or electric stimulation is not a true cause of epilepsy in patients, restricting our capacity to extrapolate the findings in a human context. Conversely, genetic models recapitulate the genetic etiology and physiology of epilepsy to a much greater extent. Unfortunately, these models also face major challenges. One such challenge is the uneven frequency of spontaneous seizures, causing difficulty in assessing the output. The seizure onset in such models can also be different from that of actual patients. Besides seizure rarity, many models also exhibit absence or non-convulsive seizures, which are generally difficult to detect. In contrast to the models with low seizure frequency, there are models that suffer from severe and fatal seizures. These animals die after one or a few seizures, making them not useful for long-term epilepsy research, but may be beneficial for understanding SUDEP. Finally, genetic models face serious challenges to recapitulate the varying expressivity of epilepsy as seen in patients. As mentioned in Section 4, mutations in the same gene can cause a range of phenotypes in patients. This signifies the need to study gene interactors and modifiers in model systems. Recently, attempts have been made to investigate these connections by introducing the same mutation into mice of different strains (262–264). These studies have reported that the onset and frequency of seizures vary depending on the inbred strain used. We consider the introduction of both primary and modifier mutations in models as a useful alternative strategy to mitigate the challenge and advance our understanding of genetic modifiers.
Further, rodents cannot recapitulate certain genetic and developmental traits unique to humans regardless of the similarities (265). Due to this, not all aspects of human epilepsy can be accurately modeled in rodents, leading to unsuccessful preclinical trials. To overcome this, researchers have recently started using patient-derived induced pluripotent stem cells (iPSCs), brain organoids, and assembloids to generate disease-associated cell types for understanding the pathophysiology of certain diseases (266, 267). However, genetic variability among different iPSC lines and failure in developing the complete brain still limit the usefulness of such models (268, 269). Combining in vivo rodent models and in vitro human models seems a more efficient strategy to shed light on epilepsy. Recently, human-mouse chimeric brain models, or humanized mice, have been generated wherein patient-derived iPSCs are engrafted in different regions of the brain (270). This allows the integration of human neural cell development and function in vivo, thus aiding the enhancement of our understanding of human brain development and epilepsy.
In addition to the limitation of having accurate model systems, a major challenge in epilepsy management is pharmacoresistance. Despite proper diagnosis and drug treatment, seizures often tend to stay unresolved. Unfortunately, little is known about what causes this drug resistance. Alteration of the blood–brain barrier or drug targets due to SNPs, environmental influence, genetic background, and associated comorbidities are some of the hypothetical mechanisms underlying DRE (271, 272); however, nothing is yet proven preclinically or clinically (273). Some novel therapeutic approaches, including vagus nerve stimulation (VNS), responsive stimulation, and deep brain stimulation, involve invasive surgical intervention (274–276). Although these strategies have shown some success in recent times, their invasive nature limits their application in pediatric patients. On the other hand, an increase in extracellular serotonin (5-HT) levels is reported to inhibit various seizure types (277). Complementarily, patients with genetic or acquired 5-HT defects are more susceptible to SUDEP (278). In fact, stimulation of 5-HT receptors beneficially influences certain preclinical epilepsy models (279). Clinically, rapamycin and its analogs are currently the only pathway-related medications utilized to treat epilepsy noninvasively (280, 281). Unfortunately, these drugs work on a specific cohort of tuberous sclerosis patients, harboring rare mutations in TSC1/TSC2, resulting in overactivating the PI3K-AKT–MTOR pathway (159, 282). Recently, a preclinical study has shown that targeting different upstream components of the PI3K-AKT–MTOR pathway but not MTOR can acutely treat epilepsy, suggesting that epilepsy is more than channelopathies and TORopathies (192). This study also suggested that developing epilepsy may have distinct acute and chronic mechanisms that can differentially respond to the administered cocktail of anti-seizure medications. Together, such studies highlight the need to further explore different mechanisms underlying pediatric epilepsy and to develop a patient-specific therapeutic approach.
8 Discussion
In this review, we have discussed established and evolving cellular and molecular mechanisms underlying the development of pediatric epilepsy, emphasizing the use of diverse types of models and their pros and cons. Perspectives regarding potential reasons behind refractory epilepsy, along with challenges and contradictions in the field, are also brought forward.
Novel epilepsy-related gene variants are progressively being identified. However, lack of adequate sample size, heterogeneity in the cohort, diagnostic biases, and differential genetic and geographical backgrounds often lead to confusion in understanding the disease manifestation across patient groups. For a long time, the cause of epilepsy was considered monogenic, primarily based on defects in single ion channel proteins. However, it has become evident that epilepsy is not merely a channelopathy. Instead, it is multigenic, often involving molecules instrumental in cell cycle and signaling, ciliogenesis, and biological rhythms. Among all epilepsies, we opine that pediatric epilepsies are possibly the most difficult ones to interpret and treat for different reasons. First, a child may suffer from multiple overlapping epilepsy types, either simultaneously or sequentially, as they age. Next, pediatric patients often suffer from one or more developmental comorbidities; this may confuse the diagnoses as the boundary between cause and effect becomes blurred in such cases. Further, causative de novo point mutations can be difficult to identify in a newborn. Moreover, the same gene can be associated with different types of epilepsy with variable onsets. Worst of all, pediatric patients with very early-onset epilepsies are non-verbal, extremely mobile, and cannot emote what they are experiencing, resulting in added complications and delayed detection of the disorder. Parallelly, invasive brain surgeries on a few weeks-to-month-old patients are life-threatening and painful. In such situations, recent advancements in EEG, high-resolution brain imaging, genetic screening, and detection strategies provide hope. Our review has highlighted the evolving complexity of pediatric epilepsy, its close connection with neurodevelopment and cognition, as well as the associated challenges in the field. We also put forward the concept that pediatric epilepsy is a part of the neurodevelopmental disorder continuum.
Finally, it is interesting to note that there is a deep underlying connection between epileptogenesis and the PI3K-AKT–MTOR signaling pathway (Figure 7). Be it the cause or the effect, the development of epilepsy is always correlated to the overactivated PI3K-AKT–MTOR pathway. As mentioned in different sections of the review, this signaling is responsible for various developmental processes, such as proliferation, neural differentiation, and formation of ion channels and cilia, as well as helps in the regulation of the circadian body clock and sleep rhythms. Disruption of any of these cellular processes can also perturb the pathway functioning as a feedback mechanism, further triggering epileptogenesis. Activating mutations of the pathway themselves are known to cause a spectrum of neurodevelopmental disorders, including epilepsy. These phenomena highlight common nodes in the form of small-molecule targets, that can be utilized in developing potential therapeutic strategies. With the advent of these new concepts and tools in the epilepsy field, we are presently at an exciting juncture to circumvent the current bottleneck of drug resistance in children and reduce the need for invasive surgeries in order to provide a better quality of life in future patients with pediatric epilepsy.
Figure 7
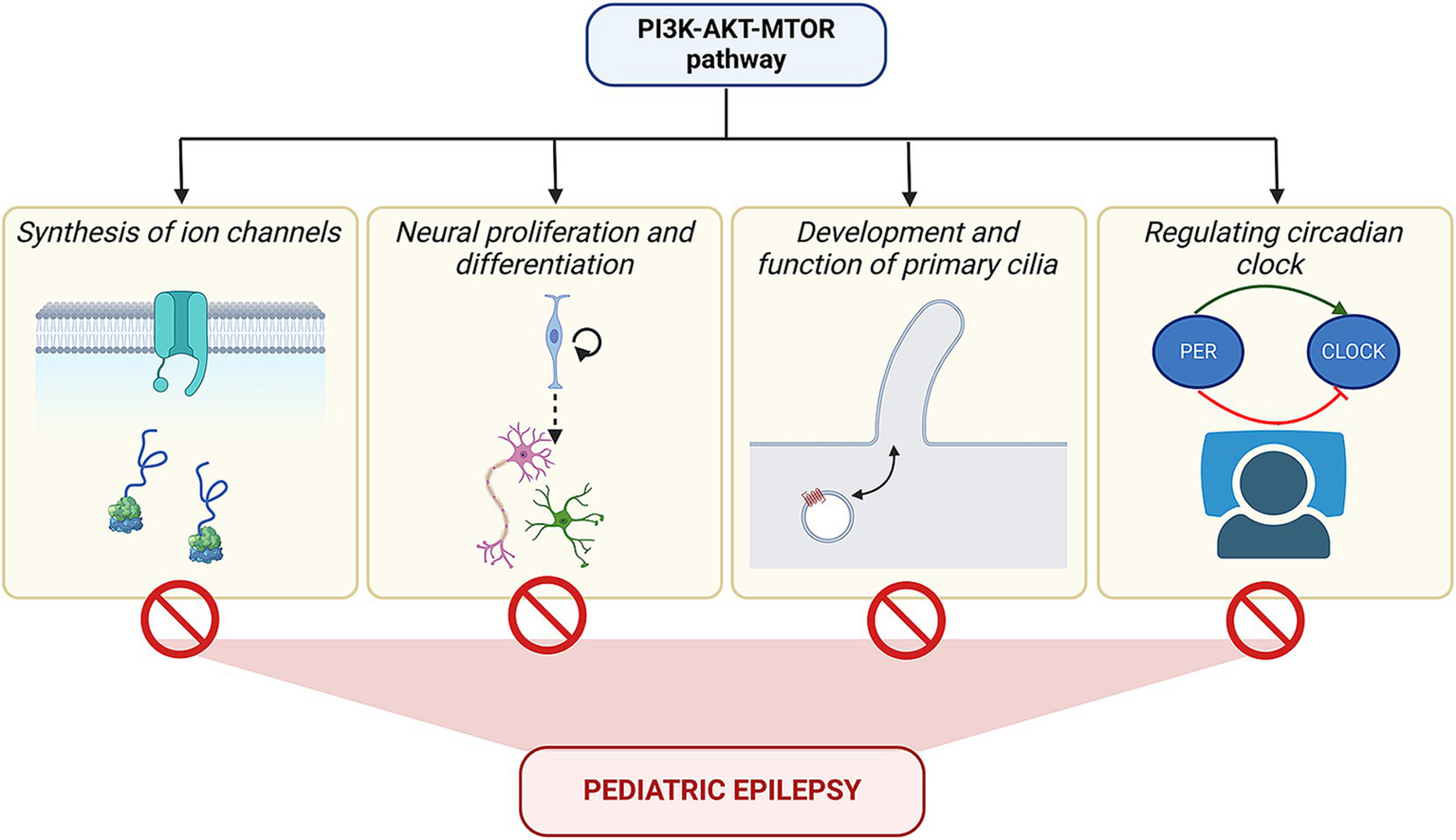
Converging influence of PI3K-AKT–MTOR pathway on epilepsy. The figure summarizes the varied roles of the PI3K-AKT–MTOR pathway in regulating channel protein synthesis, cell migration, formation of primary cilia, and molecular circuit of the circadian clock, besides its function in cell proliferation and differentiation. Mutations in the pathway components can alter these above-mentioned processes, which, in turn, may lead to epilepsy.
Statements
Author contributions
VL: Conceptualization, Investigation, Validation, Writing – original draft, Writing – review & editing, Visualization. AR: Conceptualization, Investigation, Validation, Visualization, Writing – original draft, Writing – review & editing, Funding acquisition, Project administration, Supervision.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was funded by the Ramalingaswami Re-entry Fellowship (D.O. No. BT/HRD/35/02/2006) from the Department of Biotechnology (DBT), India (AR), a research donation from M-CM Foundation, USA (AR), and the intramural funding from the Jawaharlal Nehru Centre for Advanced Scientific Research, India (AR).
Acknowledgments
We thank Shatabdi Choudhury and Aman Sharma for their technical support. Schematics in Figures 3, 5–7 have been created using the BioRender software (https://www.biorender.com/).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Gen AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1.
Hirtz D Thurman DJ Gwinn-Hardy K Mohamed M Chaudhuri AR Zalutsky R . How common are the “common” neurologic disorders?Neurology. (2007) 68:326–37. doi: 10.1212/01.wnl.0000252807.38124.a3
2.
Dalic L Cook MJ . Managing drug-resistant epilepsy: challenges and solutions. Neuropsychiatr Dis Treat. (2016) 12:2605–16. doi: 10.2147/NDT.S84852
3.
Fisher RS Cross JH D’Souza C French JA Haut SR Higurashi N et al . Instruction manual for the ILAE 2017 operational classification of seizure types. Epilepsia. (2017) 58:531–42. doi: 10.1111/epi.13671
4.
Fisher RS Cross JH French JA Higurashi N Hirsch E Jansen FE et al . Operational classification of seizure types by the International League Against Epilepsy: Position Paper of the ILAE Commission for Classification and Terminology. Epilepsia. (2017) 58:522–30. doi: 10.1111/epi.13670
5.
Pressler RM Cilio MR Mizrahi EM Moshé SL Nunes ML Plouin P et al . The ILAE classification of seizures and the epilepsies: Modification for seizures in the neonate. Position paper by the ILAE Task Force on Neonatal Seizures. Epilepsia. (2021) 62:615–28. doi: 10.1111/epi.16815
6.
Nickels KC Zaccariello MJ Hamiwka LD Wirrell EC . Cognitive and neurodevelopmental comorbidities in paediatric epilepsy. Nat Rev Neurol. (2016) 12:465–76. doi: 10.1038/nrneurol.2016.98
7.
Sorg AL von Kries R Borggraefe I . Cognitive disorders in childhood epilepsy: a comparative longitudinal study using administrative healthcare data. J Neurol. (2022) 269:3789–99. doi: 10.1007/s00415-022-11008-y
8.
Lin CH Chou IC Hong SY . Genetic factors and the risk of drug-resistant epilepsy in young children with epilepsy and neurodevelopment disability: A prospective study and updated meta-analysis. Medicine. (2021) 100:e25277. doi: 10.1097/MD.0000000000025277
9.
Kumar G . Evaluation and management of drug resistant epilepsy in children. Curr Probl Pediatr Adolesc Health Care. (2021) 51:101035. doi: 10.1016/j.cppeds.2021.101035
10.
Barker-Haliski M Steve WH . Validated animal models for antiseizure drug (ASD) discovery: Advantages and potential pitfalls in ASD screening. Neuropharmacology. (2020) 167:107750. doi: 10.1016/j.neuropharm.2019.107750
11.
Sperling MR Harris A Nei M Liporace JD O’Connor MJ . Mortality after epilepsy surgery. Epilepsia. (2005) 46:49–53. doi: 10.1111/j.1528-1167.2005.00410.x
12.
Sillanpää M Jalava M Kaleva O Shinnar S . Long-term prognosis of seizures with onset in childhood. N Engl J Med. (1998) 338:1715–22. doi: 10.1056/NEJM199806113382402
13.
Saad K Eldaly EH Abdelall HM Abdelgabaar NM Zaki DM Dailah HG et al . Contemporary Insights into Intractable Epilepsy in Children. J Pharm Bioallied Sci. (2024) 16:S909–11. doi: 10.4103/jpbs.jpbs_1075_23
14.
Wang J Lin ZJ Liu L Xu HQ Shi YW Yi YH et al . Epilepsy-associated genes. Seizure. (2017) 44:11–20. doi: 10.1016/j.seizure.2016.11.030
15.
Noebels JL . The biology of epilepsy genes. Annu Rev Neurosci. (2003) 26:599–625. doi: 10.1146/annurev.neuro.26.010302.081210
16.
Speed D Hoggart C Petrovski S Tachmazidou I Coffey A Jorgensen A et al . A genome-wide association study and biological pathway analysis of epilepsy prognosis in a prospective cohort of newly treated epilepsy. Hum Mol Genet. (2014) 23:247–58. doi: 10.1093/hmg/ddt403
17.
Bozzi Y Dunleavy M Henshall DC . Cell signaling underlying epileptic behavior. Front Behav Neurosci. (2011) 5:45. doi: 10.3389/fnbeh.2011.00045
18.
Liu S Trupiano MX Simon J Guo J Anton ES . The essential role of primary cilia in cerebral cortical development and disorders. Curr Top Dev Biol. (2021) 142:99–146. doi: 10.1016/bs.ctdb.2020.11.003
19.
Faubel RJ Santos Canellas VS Gaesser J Beluk NH Feinstein TN Wang Y et al . Flow blockage disrupts cilia-driven fluid transport in the epileptic brain. Acta Neuropathol. (2022) 144:691–706. doi: 10.1007/s00401-022-02463-y
20.
Vezzani A Fujinami RS White HS Preux PM Blümcke I Sander JW et al . Infections, inflammation and epilepsy. Acta Neuropathol. (2016) 131:211–34. doi: 10.1007/s00401-015-1481-5
21.
Scheffer IE Berkovic S Capovilla G Connolly MB French J Guilhoto L et al . ILAE classification of the epilepsies: Position paper of the ILAE Commission for Classification and Terminology. Epilepsia. (2017) 58:512–21. doi: 10.1111/epi.13709
22.
Panayiotopoulos CP Michael M Sanders S Valeta T Koutroumanidis M . Benign childhood focal epilepsies: assessment of established and newly recognized syndromes. Brain. (2008) 131:2264–86. doi: 10.1093/brain/awn162
23.
Taylor I Berkovic SF Kivity S Scheffer IE . Benign occipital epilepsies of childhood: clinical features and genetics. Brain. (2008) 131:2287–94. doi: 10.1093/brain/awn138
24.
Caraballo RH Sologuestua A Grañana N Adi JN Cersósimo RO Mazza E et al . Idiopathic occipital and absence epilepsies appearing in the same children. Pediatr Neurol. (2004) 30:24–8. doi: 10.1016/S0887-8994(03)00409-0
25.
Verrotti A D’Alonzo R Rinaldi VE Casciato S D’Aniello A Di Gennaro G . Childhood absence epilepsy and benign epilepsy with centro-temporal spikes: a narrative review analysis. World J Pediatr. (2017) 13:106–11. doi: 10.1007/s12519-017-0006-9
26.
Garcia-Ramos C Jackson DC Lin JJ Dabbs K Jones JE Hsu DA et al . Cognition and brain development in children with benign epilepsy with centrotemporal spikes. Epilepsia. (2015) 56:1615–22. doi: 10.1111/epi.13125
27.
Halász P Szũcs A . Self-limited childhood epilepsies are disorders of the perisylvian communication system, carrying the risk of progress to epileptic encephalopathies-Critical review. Front Neurol. (2023) 14:1092244. doi: 10.3389/fneur.2023.1092244
28.
Zuberi SM Wirrell E Yozawitz E Wilmshurst JM Specchio N Riney K et al . ILAE classification and definition of epilepsy syndromes with onset in neonates and infants: Position statement by the ILAE Task Force on Nosology and Definitions. Epilepsia. (2022) 63:1349–97. doi: 10.1111/epi.17239
29.
Raga S Specchio N Rheims S Wilmshurst JM . Developmental and epileptic encephalopathies: recognition and approaches to care. Epileptic Disord. (2021) 23:40–52. doi: 10.1684/epd.2021.1244
30.
Helbig I . Genetic Causes of Generalized Epilepsies. Semin Neurol. (2015) 35:288–92. doi: 10.1055/s-0035-1552922
31.
Verducci C Friedman D Donner E Devinsky O . Genetic generalized and focal epilepsy prevalence in the North American SUDEP Registry. Neurology. (2020) 94:e1757–63. doi: 10.1212/WNL.0000000000009295
32.
Kwan P Arzimanoglou A Berg AT Brodie MJ Allen Hauser W Mathern G et al . Definition of drug resistant epilepsy: consensus proposal by the ad hoc Task Force of the ILAE Commission on Therapeutic Strategies. Epilepsia. (2010) 51:1069–77. doi: 10.1111/j.1528-1167.2009.02397.x
33.
Sperling MR O’Connor MJ Saykin AJ Plummer C . Temporal lobectomy for refractory epilepsy. JAMA. (1996) 276:470–5. doi: 10.1001/jama.1996.03540060046034
34.
Sisodiya SM . Surgery for malformations of cortical development causing epilepsy. Brain. (2000) 123:1075–91. doi: 10.1093/brain/123.6.1075
35.
Laxer KD Trinka E Hirsch LJ Cendes F Langfitt J Delanty N et al . The consequences of refractory epilepsy and its treatment. Epilepsy Behav. (2014) 37:59–70. doi: 10.1016/j.yebeh.2014.05.031
36.
Kalilani L Sun X Pelgrims B Noack-Rink M Villanueva V . The epidemiology of drug-resistant epilepsy: A systematic review and meta-analysis. Epilepsia. (2018) 59:2179–93. doi: 10.1111/epi.14596
37.
Barkovich AJ Guerrini R Kuzniecky RI Jackson GD Dobyns WB . A developmental and genetic classification for malformations of cortical development: update 2012. Brain. (2012) 135:1348–69. doi: 10.1093/brain/aws019
38.
Berg AT Tarquinio D Koh S . Early Life Epilepsies are a Comorbidity of Developmental Brain Disorders. Semin Pediatr Neurol. (2017) 24:251–63. doi: 10.1016/j.spen.2017.10.008
39.
Liu W An D Xiao J Li J Hao N Zhou D . Malformations of cortical development and epilepsy: A cohort of 150 patients in western China. Seizure. (2015) 32:92–9. doi: 10.1016/j.seizure.2015.09.009
40.
Barkovich AJ Dobyns WB Guerrini R . Malformations of cortical development and epilepsy. Cold Spring Harb Perspect Med. (2015) 5:a022392. doi: 10.1101/cshperspect.a022392
41.
Blackmon K . Structural MRI biomarkers of shared pathogenesis in autism spectrum disorder and epilepsy. Epilepsy Behav. (2015) 47:172–82. doi: 10.1016/j.yebeh.2015.02.017
42.
Jehi L . The Epileptogenic Zone: Concept and Definition. Epilepsy Curr. (2018) 18:12–6. doi: 10.5698/1535-7597.18.1.12
43.
Freitas ME Ruiz-Lopez M Dalmau J Erro R Privitera M Andrade D et al . Seizures and movement disorders: phenomenology, diagnostic challenges and therapeutic approaches. J Neurol Neurosurg Psychiatry. (2019) 90:920–8. doi: 10.1136/jnnp-2018-320039
44.
Cooper MS Mackay MT Dagia C Fahey MC Howell KB Reddihough D et al . Epilepsy syndromes in cerebral palsy: varied, evolving and mostly self-limited. Brain. (2023) 146:587–99. doi: 10.1093/brain/awac274
45.
Papandreou A Danti FR Spaull R Leuzzi V Mctague A Kurian MA . The expanding spectrum of movement disorders in genetic epilepsies. Dev Med Child Neurol. (2020) 62:178–91. doi: 10.1111/dmcn.14407
46.
David CV Mac Allister WS . Fine motor impairment in children with epilepsy: Relations with seizure severity and lateralizing value. Epilepsy Behav. (2022) 127:108518. doi: 10.1016/j.yebeh.2021.108518
47.
Moseley BD Wirrell EC Wong-Kisiel LC Nickels K . Early onset epilepsy is associated with increased mortality: a population-based study. Epilepsy Res. (2013) 105:410–4. doi: 10.1016/j.eplepsyres.2013.03.002
48.
Nunes ML Martins MP Barea BM Wainberg RC da CJC . Neurological outcome of newborns with neonatal seizures: a cohort study in a tertiary university hospital. Arq Neuropsiquiatr. (2008) 66:168–74. doi: 10.1590/S0004-282X2008000200005
49.
van der Heide MJ Roze E van der Veere CN Ter Horst HJ Brouwer OF Bos AF . Long-term neurological outcome of term-born children treated with two or more anti-epileptic drugs during the neonatal period. Early Hum Dev. (2012) 88:33–8. doi: 10.1016/j.earlhumdev.2011.06.012
50.
Tomson T Nashef L Ryvlin P . Sudden unexpected death in epilepsy: current knowledge and future directions. Lancet Neurol. (2008) 7:1021–31. doi: 10.1016/S1474-4422(08)70202-3
51.
Nashef L So EL Ryvlin P Tomson T . Unifying the definitions of sudden unexpected death in epilepsy. Epilepsia. (2012) 53:227–33. doi: 10.1111/j.1528-1167.2011.03358.x
52.
DeGiorgio CM Markovic D Mazumder R Moseley BD . Ranking the Leading Risk Factors for Sudden Unexpected Death in Epilepsy. Front Neurol. (2017) 8:473. doi: 10.3389/fneur.2017.00473
53.
Bagnall RD Crompton DE Semsarian C . Genetic Basis of Sudden Unexpected Death in Epilepsy. Front Neurol. (2017) 8:348. doi: 10.3389/fneur.2017.00348
54.
Téllez-Zenteno JF Ronquillo LH Wiebe S . Sudden unexpected death in epilepsy: evidence-based analysis of incidence and risk factors. Epilepsy Res. (2005) 65:101–15. doi: 10.1016/j.eplepsyres.2005.05.004
55.
Hesdorffer DC Tomson T Benn E Sander JW Nilsson L Langan Y et al . Combined analysis of risk factors for SUDEP. Epilepsia. (2011) 52:1150–9. doi: 10.1111/j.1528-1167.2010.02952.x
56.
Wicker E Cole JW . Sudden Unexpected Death in Epilepsy (SUDEP): A Review of Risk Factors and Possible Interventions in Children. J Pediatr Pharmacol Ther. (2021) 26:556–64. doi: 10.5863/1551-6776-26.6.556
57.
Allen LA Vos SB Kumar R Ogren JA Harper RK Winston GP et al . Cerebellar, limbic, and midbrain volume alterations in sudden unexpected death in epilepsy. Epilepsia. (2019) 60:718–29. doi: 10.1111/epi.14689
58.
Wandschneider B Koepp M Scott C Micallef C Balestrini S Sisodiya SM et al . Structural imaging biomarkers of sudden unexpected death in epilepsy. Brain. (2015) 138:2907–19. doi: 10.1093/brain/awv233
59.
Willinger M James LS Catz C . Defining the sudden infant death syndrome (SIDS): deliberations of an expert panel convened by the National Institute of Child Health and Human Development. Pediatr Pathol. (1991) 11:677–84. doi: 10.3109/15513819109065465
60.
Hoffman HJ Damus K Hillman L Krongrad E . Risk factors for SIDS. Results of the National Institute of Child Health and Human Development SIDS Cooperative Epidemiological Study. Ann N Y Acad Sci. (1988) 533:13–30. doi: 10.1111/j.1749-6632.1988.tb37230.x
61.
Kon FC Vázquez RZ Lang A Cohen MC . Hippocampal abnormalities and seizures: a 16-year single center review of sudden unexpected death in childhood, sudden unexpected death in epilepsy and SIDS. Forensic Sci Med Pathol. (2020) 16:423–34. doi: 10.1007/s12024-020-00268-7
62.
Haynes RL Kinney HC Haas EA Duncan JR Riehs M Trachtenberg F et al . Medullary Serotonergic Binding Deficits and Hippocampal Abnormalities in Sudden Infant Death Syndrome: One or Two Entities?Front Pediatr. (2021) 9:762017. doi: 10.3389/fped.2021.762017
63.
Duncan JR Paterson DS Hoffman JM Mokler DJ Borenstein NS Belliveau RA et al . Brainstem serotonergic deficiency in sudden infant death syndrome. JAMA. (2010) 303:430–7. doi: 10.1001/jama.2010.45
64.
Kinney HC Randall LL Sleeper LA Willinger M Belliveau RA Zec N et al . Serotonergic brainstem abnormalities in Northern Plains Indians with the sudden infant death syndrome. J Neuropathol Exp Neurol. (2003) 62:1178–91. doi: 10.1093/jnen/62.11.1178
65.
Bright FM Byard RW Vink R Paterson DS . Medullary Serotonin Neuron Abnormalities in an Australian Cohort of Sudden Infant Death Syndrome. J Neuropathol Exp Neurol. (2017) 76:864–73. doi: 10.1093/jnen/nlx071
66.
Broadbelt KG Paterson DS Belliveau RA Trachtenberg FL Haas EA Stanley C et al . Decreased GABAA receptor binding in the medullary serotonergic system in the sudden infant death syndrome. J Neuropathol Exp Neurol. (2011) 70:799–810. doi: 10.1097/NEN.0b013e31822c09bc
67.
Krous HF Chadwick AE Crandall L Nadeau-Manning JM . Sudden unexpected death in childhood: a report of 50 cases. Pediatr Dev Pathol. (2005) 8:307–19. doi: 10.1007/s10024-005-1155-8
68.
Hesdorffer DC Crandall LA Friedman D Devinsky O . Sudden unexplained death in childhood: A comparison of cases with and without a febrile seizure history. Epilepsia. (2015) 56:1294–300. doi: 10.1111/epi.13066
69.
Hefti MM Cryan JB Haas EA Chadwick AE Crandall LA Trachtenberg FL et al . Hippocampal malformation associated with sudden death in early childhood: a neuropathologic study: Part 2 of the investigations of The San Diego SUDC Research Project. Forensic Sci Med Pathol. (2016) 12:14–25. doi: 10.1007/s12024-015-9731-3
70.
Whatley BP Winston JS Allen LA Vos SB Jha A Scott CA et al . Distinct Patterns of Brain Metabolism in Patients at Risk of Sudden Unexpected Death in Epilepsy. Front Neurol. (2021) 12:623358. doi: 10.3389/fneur.2021.623358
71.
Kinney HC Cryan JB Haynes RL Paterson DS Haas EA Mena OJ et al . Dentate gyrus abnormalities in sudden unexplained death in infants: morphological marker of underlying brain vulnerability. Acta Neuropathol. (2015) 129:65–80. doi: 10.1007/s00401-014-1357-0
72.
Ryvlin P Nashef L Lhatoo SD Bateman LM Bird J Bleasel A et al . Incidence and mechanisms of cardiorespiratory arrests in epilepsy monitoring units (MORTEMUS): a retrospective study. Lancet Neurol. (2013) 12:966–77. doi: 10.1016/S1474-4422(13)70214-X
73.
Hunt CE . Abnormal hypercarbic and hypoxic sleep arousal responses in Near-Miss SIDS infants. Pediatr Res. (1981) 15:1462–4. doi: 10.1203/00006450-198111000-00015
74.
Thakran S Guin D Singh P Singh P Kukal S Rawat C et al . Genetic Landscape of Common Epilepsies: Advancing towards Precision in Treatment. Int J Mol Sci [Internet]. (2020) 21:7784. doi: 10.3390/ijms21207784
75.
Sokka A Olsen P Kirjavainen J Harju M Keski-Nisula L Räisänen S et al . Etiology, syndrome diagnosis, and cognition in childhood-onset epilepsy: A population-based study. Epilepsia Open. (2017) 2:76–83. doi: 10.1002/epi4.12036
76.
Sun H Zhang Y Liu X Ma X Yang Z Qin J et al . Analysis of SCN1A mutation and parental origin in patients with Dravet syndrome. J Hum Genet. (2010) 55:421–7. doi: 10.1038/jhg.2010.39
77.
Wang X Rao X Zhang J Gan J . Genetic mechanisms in generalized epilepsies. Acta Epileptologica. (2023) 5:1–13. doi: 10.1186/s42494-023-00118-3
78.
Choohan MA Ahmed I Syed SK Naeem M . SCN1A gene mutation: A rising cause of human epilepsy syndrome. Journal of Contemporary Pharmacy. (2022) 6:32–40. doi: 10.56770/jcp2022615
79.
Zhang D Liu X Deng X . Genetic basis of pediatric epilepsy syndromes. Exp Ther Med. (2017) 13:2129–33. doi: 10.3892/etm.2017.4267
80.
Pirozzi F Berkseth M Shear R Gonzalez L Timms AE Sulc J et al . Profiling PI3K-AKT-MTOR variants in focal brain malformations reveals new insights for diagnostic care. Brain. (2022) 145:925–38. doi: 10.1093/brain/awab376
81.
Spinelli E Christensen KR Bryant E Schneider A Rakotomamonjy J Muir AM et al . Pathogenic MAST3 Variants in the STK Domain Are Associated with Epilepsy. Ann Neurol. (2021) 90:274–84. doi: 10.1002/ana.26147
82.
Tripathy R Leca I van Dijk T Weiss J van Bon BW Sergaki MC et al . Mutations in MAST1 Cause Mega-Corpus-Callosum Syndrome with Cerebellar Hypoplasia and Cortical Malformations. Neuron. (2018) 100:1354–68.e5. doi: 10.1016/j.neuron.2018.10.044
83.
Landoulsi Z Laatar F Noé E Mrabet S Ben Djebara M Achaz G et al . Clinical and genetic study of Tunisian families with genetic generalized epilepsy: contribution of CACNA1H and MAST4 genes. Neurogenetics. (2018) 19:165–78. doi: 10.1007/s10048-018-0550-z
84.
Dibbens LM Mullen S Helbig I Mefford HC Bayly MA Bellows S et al . Familial and sporadic 15q13.3 microdeletions in idiopathic generalized epilepsy: precedent for disorders with complex inheritance. Hum Mol Genet. (2009) 18:3626–31. doi: 10.1093/hmg/ddp311
85.
Ottman R . Analysis of genetically complex epilepsies. Epilepsia. (2005) 46:7–14. doi: 10.1111/j.1528-1167.2005.00350.x
86.
Oliver KL Ellis CA Scheffer IE Ganesan S Leu C Sadleir LG et al . Common risk variants for epilepsy are enriched in families previously targeted for rare monogenic variant discovery. EBioMedicine. (2022) 81:104079. doi: 10.1016/j.ebiom.2022.104079
87.
Dell’Isola GB Fattorusso A Pisani F Mastrangelo M Cordelli DM Pavone P et al . CDKL5 deficiency-related neurodevelopmental disorders: a multi-center cohort study in Italy. J Neurol. (2024) 271:5368–7649. doi: 10.1007/s00415-024-12653-1
88.
de Lange IM Mulder F van’t Slot R ACM S MJA v K Nijman IJ et al . Modifier genes in SCN1A-related epilepsy syndromes. Mol Genet Genomic Med. (2020) 8:e 1103. doi: 10.1002/mgg3.1103
89.
Tadenev ALD Burgess RW . Model validity for preclinical studies in precision medicine: precisely how precise do we need to be?Mamm Genome. (2019) 30:111–22. doi: 10.1007/s00335-019-09798-0
90.
Nakajima R Hagihara H Miyakawa T . Similarities of developmental gene expression changes in the brain between human and experimental animals: rhesus monkey, mouse, Zebrafish, and Drosophila. Mol Brain. (2021) 14:135. doi: 10.1186/s13041-021-00840-4
91.
Wong HHW Chou CYC Watt AJ Sjöström PJ . Comparing mouse and human brains. eLife. (2023) 12:12. doi: 10.7554/eLife.90017
92.
Kandratavicius L Balista PA Lopes-Aguiar C Ruggiero RN Umeoka EH Garcia-Cairasco N et al . Animal models of epilepsy: use and limitations. Neuropsychiatr Dis Treat. (2014) 10:1693–705. doi: 10.2147/NDT.S50371
93.
Bourgeois BF . Temporal lobe epilepsy in infants and children. Brain Dev. (1998) 20:135–41. doi: 10.1016/S0387-7604(98)00010-2
94.
Riney K Bogacz A Somerville E Hirsch E Nabbout R Scheffer IE et al . International League Against Epilepsy classification and definition of epilepsy syndromes with onset at a variable age: position statement by the ILAE Task Force on Nosology and Definitions. Epilepsia. (2022) 63:1443–74. doi: 10.1111/epi.17240
95.
Nadler JV Perry BW Cotman CW . Intraventricular kainic acid preferentially destroys hippocampal pyramidal cells. Nature. (1978) 271:676–7. doi: 10.1038/271676a0
96.
Bonilha L Edwards JC Kinsman SL Morgan PS Fridriksson J Rorden C et al . Extrahippocampal gray matter loss and hippocampal deafferentation in patients with temporal lobe epilepsy. Epilepsia. (2010) 51:519–28. doi: 10.1111/j.1528-1167.2009.02506.x
97.
Bonilha L Alessio A Rorden C Baylis G Damasceno BP Min LL et al . Extrahippocampal gray matter atrophy and memory impairment in patients with medial temporal lobe epilepsy. Hum Brain Mapp. (2007) 28:1376–90. doi: 10.1002/hbm.20373
98.
da SAV Regondi MC Cipelletti B Frassoni C Cavalheiro EA Spreafico R . Neocortical and hippocampal changes after multiple pilocarpine-induced status epilepticus in rats. Epilepsia. (2005) 46:636–42. doi: 10.1111/j.1528-1167.2005.31604.x
99.
Rolston JD Desai SA Laxpati NG Gross RE . Electrical stimulation for epilepsy: experimental approaches. Neurosurg Clin N Am. (2011) 22:425–42, v. doi: 10.1016/j.nec.2011.07.010
100.
Wang Y Wei P Yan F Luo Y Zhao G . Animal models of epilepsy: A phenotype-oriented review. Aging Dis. (2022) 13:215–31. doi: 10.14336/AD.2021.0723
101.
Goddard GV McIntyre DC Leech CK . A permanent change in brain function resulting from daily electrical stimulation. Exp Neurol. (1969) 25:295–330. doi: 10.1016/0014-4886(69)90128-9
102.
Kaneko K Currin CB Goff KM Wengert ER Somarowthu A Vogels TP et al . Developmentally regulated impairment of parvalbumin interneuron synaptic transmission in an experimental model of Dravet syndrome. Cell Rep. (2022) 38:110580. doi: 10.1016/j.celrep.2022.110580
103.
Wong JC Grieco SF Dutt K Chen L Thelin JT Inglis GAS et al . Autistic-like behavior, spontaneous seizures, and increased neuronal excitability in a Scn 8a mouse model. Neuropsychopharmacology. (2021) 46:2011–20. doi: 10.1038/s41386-021-00985-9
104.
Gautier NM Glasscock E . Spontaneous seizures in Kcna 1-null mice lacking voltage-gated Kv1.1 channels activate Fos expression in select limbic circuits. J Neurochem. (2015) 135:157–64. doi: 10.1111/jnc.13206
105.
Zhang Q Forster-Gibson C Bercovici E Bernardo A Ding F Shen W et al . Epilepsy plus blindness in microdeletion of GABRA1 and GABRG2 in mouse and human. Exp Neurol. (2023) 369:114537. doi: 10.1016/j.expneurol.2023.114537
106.
Hare LM Schwarz Q Wiszniak S Gurung R Montgomery KG Mitchell CA et al . Heterozygous expression of the oncogenic Pik 3ca (H1047R) mutation during murine development results in fatal embryonic and extraembryonic defects. Dev Biol. (2015) 404:14–26. doi: 10.1016/j.ydbio.2015.04.022
107.
Kobayashi T Minowa O Sugitani Y Takai S Mitani H Kobayashi E et al . A germ-line Tsc 1 mutation causes tumor development and embryonic lethality that are similar, but not identical to, those caused by Tsc 2 mutation in mice. Proc Natl Acad Sci USA. (2001) 98:8762–7. doi: 10.1073/pnas.151033798
108.
Gangloff YG Mueller M Dann SG Svoboda P Sticker M Spetz JF et al . Disruption of the mouse mTOR gene leads to early postimplantation lethality and prohibits embryonic stem cell development. Mol Cell Biol. (2004) 24:9508–16. doi: 10.1128/MCB.24.21.9508-9516.2004
109.
Jansen LA Mirzaa GM Ishak GE O’Roak BJ Hiatt JB Roden WH et al . PI3K/AKT pathway mutations cause a spectrum of brain malformations from megalencephaly to focal cortical dysplasia. Brain. (2015) 138:1613–28. doi: 10.1093/brain/awv045
110.
Kim J Park SM Koh HY Ko A Kang HC Chang WS et al . Threshold of somatic mosaicism leading to brain dysfunction with focal epilepsy. Brain. (2024) 147:2983–90. doi: 10.1093/brain/awae190
111.
Roy A Skibo J Kalume F Ni J Rankin S Lu Y et al . Mouse models of human PIK3CA-related brain overgrowth have acutely treatable epilepsy. eLife. (2015) 4:4. doi: 10.7554/eLife.12703
112.
Abs E Goorden SMI Schreiber J Overwater IE Hoogeveen-Westerveld M Bruinsma CF et al . TORC1-dependent epilepsy caused by acute biallelic Tsc 1 deletion in adult mice. Ann Neurol. (2013) 74:569–79. doi: 10.1002/ana.23943
113.
Backman SA Stambolic V Suzuki A Haight J Elia A Pretorius J et al . Deletion of Pten in mouse brain causes seizures, ataxia and defects in soma size resembling Lhermitte-Duclos disease. Nat Genet. (2001) 29:396–403. doi: 10.1038/ng782
114.
Wong M Bordey A Danzer SC . MTOR in acquired and genetic models of epilepsy In: NoebelsJLAvoliMRogawskiMAVezzaniADelgado-EscuetaAV, editors. Jasper’s Basic Mechanisms of the Epilepsies. Oxford University Press: New York (2024). 45–74.
115.
Tsien JZ . Cre-lox neurogenetics: 20 years of versatile applications in brain research and counting …. Front Genet. (2016) 7:19. doi: 10.3389/fgene.2016.00019
116.
Feil S Valtcheva N Feil R . Inducible Cre mice. Methods Mol Biol. (2009) 530:343–63. doi: 10.1007/978-1-59745-471-1_18
117.
Yonan JM Chen KD Baram TZ Steward O . PTEN deletion in the adult dentate gyrus induces epilepsy. Neurobiol Dis. (2024) 203:106736. doi: 10.1016/j.nbd.2024.106736
118.
Zhong S Zhao Z Xie W Cai Y Zhang Y Ding J et al . GABAergic interneuron and neurotransmission are mTOR-dependently disturbed in experimental focal cortical dysplasia. Mol Neurobiol. (2021) 58:156–69. doi: 10.1007/s12035-020-02086-y
119.
Baek ST Copeland B Yun EJ Kwon SK Guemez-Gamboa A Schaffer AE et al . An AKT3-FOXG1-reelin network underlies defective migration in human focal malformations of cortical development. Nat Med. (2015) 21:1445–54. doi: 10.1038/nm.3982
120.
Proietti Onori M Koene LMC Schäfer CB Nellist M de Brito van Velze M Gao Z et al . RHEB/mTOR hyperactivity causes cortical malformations and epileptic seizures through increased axonal connectivity. PLoS Biol. (2021) 19:e3001279. doi: 10.1371/journal.pbio.3001279
121.
D’Gama AM Poduri A . Brain somatic mosaicism in epilepsy: Bringing results back to the clinic. Neurobiol Dis. (2023) 181:106104. doi: 10.1016/j.nbd.2023.106104
122.
Shao LR Habela CW Stafstrom CE . Pediatric Epilepsy Mechanisms: Expanding the Paradigm of Excitation/Inhibition Imbalance. Children. (2019) 6:23. doi: 10.3390/children6020023
123.
Dazzo E Fanciulli M Serioli E Minervini G Pulitano P Binelli S et al . Heterozygous reelin mutations cause autosomal-dominant lateral temporal epilepsy. Am J Hum Genet. (2015) 96:992–1000. doi: 10.1016/j.ajhg.2015.04.020
124.
Suzuki T Delgado-Escueta AV Aguan K Alonso ME Shi J Hara Y et al . Mutations in EFHC1 cause juvenile myoclonic epilepsy. Nat Genet. (2004) 36:842–9. doi: 10.1038/ng1393
125.
Liu YD Ma MY Hu XB Yan H Zhang YK Yang HX et al . Brain proteomic profiling in intractable epilepsy caused by TSC1 truncating mutations: A small sample study. Front Neurol. (2020) 11:475. doi: 10.3389/fneur.2020.00475
126.
Re CJ Batterman AI Gerstner JR Buono RJ Ferraro TN . The molecular genetic interaction between circadian rhythms and susceptibility to seizures and epilepsy. Front Neurol. (2020) 11:520. doi: 10.3389/fneur.2020.00520
127.
George AL Jr . Inherited Channelopathies Associated with Epilepsy. Epilepsy Curr. (2004) 4:65–70. doi: 10.1111/j.1535-7597.2004.42010.x
128.
Guerrini R Balestrini S Wirrell EC Walker MC . Monogenic epilepsies: Disease mechanisms, clinical phenotypes, and targeted therapies. Neurology. (2021) 97:817–31. doi: 10.1212/WNL.0000000000012744
129.
Ng ACH Chahine M Scantlebury MH Appendino JP . Channelopathies in epilepsy: an overview of clinical presentations, pathogenic mechanisms, and therapeutic insights. J Neurol. (2024) 271:3063–94. doi: 10.1007/s00415-024-12352-x
130.
Viswanathan LG Alapati S Nagappa M Mundlamuri R Kenchaiah R Asranna A et al . Phenotypic features of epilepsy due to sodium channelopathies - A single center experience from India. J Neurosci Rural Pract. (2023) 14:603–9. doi: 10.25259/JNRP_329_2023
131.
Mulley JC Scheffer IE Petrou S Berkovic SF . Channelopathies as a genetic cause of epilepsy. Curr Opin Neurol. (2003) 16:171–6. doi: 10.1097/00019052-200304000-00009
132.
Wang J Ou SW Wang YJ . Distribution and function of voltage-gated sodium channels in the nervous system. Channels. (2017) 11:534–54. doi: 10.1080/19336950.2017.1380758
133.
Wengert ER Patel MK . The Role of the Persistent Sodium Current in Epilepsy. Epilepsy Curr. (2021) 21:40–7. doi: 10.1177/1535759720973978
134.
Berecki G Bryson A Terhag J Maljevic S Gazina EV Hill SL et al . SCN1A gain of function in early infantile encephalopathy. Ann Neurol. (2019) 85:514–25. doi: 10.1002/ana.25438
135.
Bryson A Petrou S . SCN1A channelopathies: Navigating from genotype to neural circuit dysfunction. Front Neurol. (2023) 14:1173460. doi: 10.3389/fneur.2023.1173460
136.
Yang ND Kanyo R Zhao L Li J Kang PW Dou AK et al . Electro-mechanical coupling of KCNQ channels is a target of epilepsy-associated mutations and retigabine. Sci Adv. (2022) 8:eabo3625. doi: 10.1126/sciadv.abo3625
137.
Khosravani H Zamponi GW . Voltage-gated calcium channels and idiopathic generalized epilepsies. Physiol Rev. (2006) 86:941–66. doi: 10.1152/physrev.00002.2006
138.
Hering S Berjukow S Sokolov S Marksteiner R Weiss RG Kraus R et al . Molecular determinants of inactivation in voltage-gated Ca2+ channels. J Physiol. (2000) 528:237–49. doi: 10.1111/j.1469-7793.2000.t01-1-00237.x
139.
Lerche H Shah M Beck H Noebels J Johnston D Vincent A . Ion channels in genetic and acquired forms of epilepsy. J Physiol. (2013) 591:753–64. doi: 10.1113/jphysiol.2012.240606
140.
Indurthi DC Qudah T Liao VW Ahring PK Lewis TM Balle T et al . Revisiting autosomal dominant nocturnal frontal lobe epilepsy (ADNFLE) mutations in the nicotinic acetylcholine receptor reveal an increase in efficacy regardless of stochiometry. Pharmacol Res. (2019) 139:215–27. doi: 10.1016/j.phrs.2018.11.031
141.
Syed P Durisic N Harvey RJ Sah P Lynch JW . Effects of GABAA Receptor α3 Subunit Epilepsy Mutations on Inhibitory Synaptic Signaling. Front Mol Neurosci. (2020) 13:13. doi: 10.3389/fnmol.2020.602559
142.
Hernandez CC Zhang Y Hu N Shen D Shen W Liu X et al . GABA A Receptor Coupling Junction and Pore GABRB3 Mutations are Linked to Early-Onset Epileptic Encephalopathy. Sci Rep. (2017) 7:15903. doi: 10.1038/s41598-017-16010-3
143.
Egbenya DL Hussain S Lai YC Xia J Anderson AE Davanger S . Changes in synaptic AMPA receptor concentration and composition in chronic temporal lobe epilepsy. Mol Cell Neurosci. (2018) 92:93–103. doi: 10.1016/j.mcn.2018.07.004
144.
Rahimi-Madiseh M Lorigooini Z Boroujeni SN Taji M Amini-Khoei H . The Role of the NMDA Receptor in the Anticonvulsant Effect of Ellagic Acid in Pentylenetetrazole-Induced Seizures in Male Mice. Behav Neurol. (2022) 2022:1–6. doi: 10.1155/2022/9015842
145.
Oyrer J Maljevic S Scheffer IE Berkovic SF Petrou S Reid CA . Ion Channels in Genetic Epilepsy: From Genes and Mechanisms to Disease-Targeted Therapies. Pharmacol Rev. (2018) 70:142–73. doi: 10.1124/pr.117.014456
146.
Salpietro V Dixon CL Guo H Bello OD Vandrovcova J Efthymiou S et al . AMPA receptor Glu A2 subunit defects are a cause of neurodevelopmental disorders. Nat Commun. (2019) 10:3094. doi: 10.1038/s41467-019-10910-w
147.
Xiang Wei W Jiang Y Yuan H . De Novo Mutations and Rare Variants Occurring in NMDA Receptors. Curr Opin Physio. (2018) 2:27–35. doi: 10.1016/j.cophys.2017.12.013
148.
Zhao S Yang L Liang L Zheng Y Wang Y Wang D . A child with febrile and atypical absence seizures caused by a NLGN2 variant. Seizure. (2023) 111:36–8. doi: 10.1016/j.seizure.2023.07.014
149.
Pavone P Pappalardo XG Parano C Falsaperla R Corsello A Parano E et al . NRXN1-related disorders, attempt to better define clinical assessment. Open Med (Warsz). (2024) 19:20240979. doi: 10.1515/med-2024-0979
150.
Fang M Wei J-L Tang B Liu J Chen L Tang Z-H et al . Neuroligin-1 knockdown suppresses seizure activity by regulating neuronal hyperexcitability. Mol Neurobiol. (2016) 53:270–84. doi: 10.1007/s12035-014-8999-8
151.
Südhof TC . Neuroligins and neurexins link synaptic function to cognitive disease. Nature. (2008) 455:903–11. doi: 10.1038/nature07456
152.
Hinckley CA Zhu Z Chu JH Gubbels C Danker T Cherry JJ et al . Functional evaluation of epilepsy-associated KCNT1 variants in multiple cellular systems reveals a predominant gain of function impact on channel properties. Epilepsia. (2023) 64:2126–36. doi: 10.1111/epi.17648
153.
Heuser K Szokol K Taubøll E . The role of glial cells in epilepsy. Tidsskr Nor Laegeforen. (2014) 134:37–41. doi: 10.4045/tidsskr.12.1344
154.
Nagelhus EA Horio Y Inanobe A Fujita A Haug FM Nielsen S et al . Immunogold evidence suggests that coupling of K+ siphoning and water transport in rat retinal Müller cells is mediated by a coenrichment of Kir 4.1 and AQP4 in specific membrane domains. Glia. (1999) 26:47–54. doi: 10.1002/(SICI)1098-1136(199903)26:1<47::AID-GLIA5>3.0.CO;2-5
155.
Stogsdill JA Eroglu C . The interplay between neurons and glia in synapse development and plasticity. Curr Opin Neurobiol. (2017) 42:1–8. doi: 10.1016/j.conb.2016.09.016
156.
Heuser K Eid T Lauritzen F Thoren AE Vindedal GF Taubøll E et al . Loss of perivascular Kir 4.1 potassium channels in the sclerotic hippocampus of patients with mesial temporal lobe epilepsy. J Neuropathol Exp Neurol. (2012) 71:814–25. doi: 10.1097/NEN.0b013e318267b5af
157.
Binder DK Yao X Zador Z Sick TJ Verkman AS Manley GT . Increased seizure duration and slowed potassium kinetics in mice lacking aquaporin-4 water channels. Glia. (2006) 53:631–6. doi: 10.1002/glia.20318
158.
Eid T Lee TSW Thomas MJ Amiry-Moghaddam M Bjørnsen LP Spencer DD et al . Loss of perivascular aquaporin 4 may underlie deficient water and K+ homeostasis in the human epileptogenic hippocampus. Proc Natl Acad Sci USA. (2005) 102:1193–8. doi: 10.1073/pnas.0409308102
159.
Nguyen LH Bordey A . Convergent and Divergent Mechanisms of Epileptogenesis in mTORopathies. Front Neuroanat. (2021) 15:664695. doi: 10.3389/fnana.2021.664695
160.
Sran S Bedrosian TA . RAS pathway: The new frontier of brain mosaicism in epilepsy. Neurobiol Dis. (2023) 180:106074. doi: 10.1016/j.nbd.2023.106074
161.
Represa A . Why Malformations of Cortical Development Cause Epilepsy. Front Neurosci. (2019) 13:250. doi: 10.3389/fnins.2019.00250
162.
Porter BE . Neurogenesis and epilepsy in the developing brain. Epilepsia. (2008) 49:50–4. doi: 10.1111/j.1528-1167.2008.01637.x
163.
Zhou YD Lee S Jin Z Wright M Smith SEP Anderson MP . Arrested maturation of excitatory synapses in autosomal dominant lateral temporal lobe epilepsy. Nat Med. (2009) 15:1208–14. doi: 10.1038/nm.2019
164.
Ammothumkandy A Ravina K Wolseley V Tartt AN Yu PN Corona L et al . Altered adult neurogenesis and gliogenesis in patients with mesial temporal lobe epilepsy. Nat Neurosci. (2022) 25:493–503. doi: 10.1038/s41593-022-01044-2
165.
Wang L Zhou K Fu Z Yu D Huang H Zang X et al . Brain Development and Akt Signaling: the Crossroads of Signaling Pathway and Neurodevelopmental Diseases. J Mol Neurosci. (2017) 61:379–84. doi: 10.1007/s12031-016-0872-y
166.
Iroegbu JD Ijomone OK Femi-Akinlosotu OM Ijomone OM . ERK/MAPK signalling in the developing brain: Perturbations and consequences. Neurosci Biobehav Rev. (2021) 131:792–805. doi: 10.1016/j.neubiorev.2021.10.009
167.
Zhong J . RAS and downstream RAF-MEK and PI3K-AKT signaling in neuronal development, function and dysfunction. Biol Chem. (2016) 397:215–22. doi: 10.1515/hsz-2015-0270
168.
Wong M Crino PB . MTOR and epileptogenesis in developmental brain malformations In: JLNoebelsMAvoliMARogawskiAVezzaniAVDelgado-Escueta, editors. Jasper’s Basic Mechanisms of the Epilepsies. 4th Edn. Oxford University Press (2012). 835–44.
169.
Yuskaitis CJ Jones BM Wolfson RL Super CE Dhamne SC Rotenberg A et al . A mouse model of DEPDC5-related epilepsy: Neuronal loss of Depdc 5 causes dysplastic and ectopic neurons, increased mTOR signaling, and seizure susceptibility. Neurobiol Dis. (2018) 111:91–101. doi: 10.1016/j.nbd.2017.12.010
170.
Kassai H Sugaya Y Noda S Nakao K Maeda T Kano M et al . Selective activation of mTORC1 signaling recapitulates microcephaly, tuberous sclerosis, and neurodegenerative diseases. Cell Rep. (2014) 7:1626–39. doi: 10.1016/j.celrep.2014.04.048
171.
Kwon CH Luikart BW Powell CM Zhou J Matheny SA Zhang W et al . Pten regulates neuronal arborization and social interaction in mice. Neuron. (2006) 50:377–88. doi: 10.1016/j.neuron.2006.03.023
172.
Sanchez-Ortiz E Cho W Nazarenko I Mo W Chen J Parada LF . NF1 regulation of RAS/ERK signaling is required for appropriate granule neuron progenitor expansion and migration in cerebellar development. Genes Dev. (2014) 28:2407–20. doi: 10.1101/gad.246603.114
173.
Meikle L Talos DM Onda H Pollizzi K Rotenberg A Sahin M et al . A mouse model of tuberous sclerosis: neuronal loss of Tsc 1 causes dysplastic and ectopic neurons, reduced myelination, seizure activity, and limited survival. J Neurosci. (2007) 27:5546–58. doi: 10.1523/JNEUROSCI.5540-06.2007
174.
Zhang L Huang T Teaw S Nguyen LH Hsieh LS Gong X et al . Filamin A inhibition reduces seizure activity in a mouse model of focal cortical malformations. Sci Transl Med. (2020) 12:12 (531). doi: 10.1126/scitranslmed.aay0289
175.
Kim YE Kim YS Lee HE So KH Choe Y Suh BC et al . Reversibility and developmental neuropathology of linear nevus sebaceous syndrome caused by dysregulation of the RAS pathway. Cell Rep. (2023) 42:112003. doi: 10.1016/j.celrep.2023.112003
176.
Koh HY Kim SH Jang J Kim H Han S Lim JS et al . BRAF somatic mutation contributes to intrinsic epileptogenicity in pediatric brain tumors. Nat Med. (2018) 24:1662–8. doi: 10.1038/s41591-018-0172-x
177.
Williams MR DeSpenza T Jr Li M Gulledge AT Luikart BW . Hyperactivity of newborn Pten knock-out neurons results from increased excitatory synaptic drive. J Neurosci. (2015) 35:943–59. doi: 10.1523/JNEUROSCI.3144-14.2015
178.
Uhlmann EJ Wong M Baldwin RL Bajenaru ML Onda H Kwiatkowski DJ et al . Astrocyte-specific TSC1 conditional knockout mice exhibit abnormal neuronal organization and seizures. Ann Neurol. (2002) 52:285–96. doi: 10.1002/ana.10283
179.
Goto J Talos DM Klein P Qin W Chekaluk YI Anderl S et al . Regulable neural progenitor-specific Tsc 1 loss yields giant cells with organellar dysfunction in a model of tuberous sclerosis complex. Proc Natl Acad Sci USA. (2011) 108:E1070–9. doi: 10.1073/pnas.1106454108
180.
Magri L Cominelli M Cambiaghi M Cursi M Leocani L Minicucci F et al . Timing of mTOR activation affects tuberous sclerosis complex neuropathology in mouse models. Dis Model Mech. (2013) 6:1185–97. doi: 10.1242/dmm.012096
181.
Macias M Blazejczyk M Kazmierska P Caban B Skalecka A Tarkowski B et al . Spatiotemporal characterization of mTOR kinase activity following kainic acid induced status epilepticus and analysis of rat brain response to chronic rapamycin treatment. PLoS One. (2013) 8:e64455. doi: 10.1371/journal.pone.0064455
182.
Benthall KN Ong SL Bateup HS . Corticostriatal transmission is selectively enhanced in striatonigral neurons with postnatal loss of Tsc 1. Cell Rep. (2018) 23:3197–208. doi: 10.1016/j.celrep.2018.05.037
183.
Koh HY Jang J Ju SH Kim R Cho GB Kim DS et al . Non-cell autonomous epileptogenesis in focal cortical dysplasia. Ann Neurol. (2021) 90:285–99. doi: 10.1002/ana.26149
184.
Nateri AS Raivich G Gebhardt C Da Costa C Naumann H Vreugdenhil M et al . ERK activation causes epilepsy by stimulating NMDA receptor activity. EMBO J. (2007) 26:4891–901. doi: 10.1038/sj.emboj.7601911
185.
Limanaqi F Biagioni F Busceti CL Fabrizi C Frati A Fornai F . mTOR-Related Cell-Clearing Systems in Epileptic Seizures, an Update. Int J Mol Sci [Internet]. (2020) 21:1642. doi: 10.3390/ijms21051642
186.
McMahon J Huang X Yang J Komatsu M Yue Z Qian J et al . Impaired autophagy in neurons after disinhibition of mammalian target of rapamycin and its contribution to epileptogenesis. J Neurosci. (2012) 32:15704–14. doi: 10.1523/JNEUROSCI.2392-12.2012
187.
Shacka JJ Lu J Xie ZL Uchiyama Y Roth KA Zhang J . Kainic acid induces early and transient autophagic stress in mouse hippocampus. Neurosci Lett. (2007) 414:57–60. doi: 10.1016/j.neulet.2006.12.025
188.
Rami A Benz AP Niquet J Langhagen A . Axonal Accumulation of Lysosomal-Associated Membrane Protein 1 (LAMP1) Accompanying Alterations of Autophagy Dynamics in the Rat Hippocampus Upon Seizure-Induced Injury. Neurochem Res. (2016) 41:53–63. doi: 10.1007/s11064-015-1704-0
189.
Niere F Raab-Graham KF . mTORC1 Is a Local, Postsynaptic Voltage Sensor Regulated by Positive and Negative Feedback Pathways. Front Cell Neurosci. (2017) 11:152. doi: 10.3389/fncel.2017.00152
190.
Sliwa A Plucinska G Bednarczyk J Lukasiuk K . Post-treatment with rapamycin does not prevent epileptogenesis in the amygdala stimulation model of temporal lobe epilepsy. Neurosci Lett. (2012) 509:105–9. doi: 10.1016/j.neulet.2011.12.051
191.
Hartman AL Santos P Dolce A Hardwick JM . The mTOR inhibitor rapamycin has limited acute anticonvulsant effects in mice. PLoS One. (2012) 7:e45156. doi: 10.1371/journal.pone.0045156
192.
Roy A Han VZ Bard AM Wehle DT Smith SEP Ramirez JM et al . Non-synaptic Cell-Autonomous Mechanisms Underlie Neuronal Hyperactivity in a Genetic Model of PIK3CA-Driven Intractable Epilepsy. Front Mol Neurosci. (2021) 14:772847. doi: 10.3389/fnmol.2021.772847
193.
Bozzi Y Casarosa S Caleo M . Epilepsy as a neurodevelopmental disorder. Front Psych. (2012) 3:19. doi: 10.3389/fpsyt.2012.00019
194.
Mahmud R . Subcortical Band Heterotopia Presented With Refractory Epilepsy and Reversible Aphasia. Cureus. (2021) 13:e16990. doi: 10.7759/cureus.16990
195.
Dazzo E Nobile C . Epilepsy-causing Reelin mutations result in impaired secretion and intracellular degradation of mutant proteins. Hum Mol Genet. (2022) 31:665–73. doi: 10.1093/hmg/ddab271
196.
Schröter J Popp B Brennenstuhl H Döring JH Donze SH Bijlsma EK et al . Complementing the phenotypical spectrum of TUBA1A tubulinopathy and its role in early-onset epilepsies. Eur J Hum Genet. (2022) 30:298–306. doi: 10.1038/s41431-021-01027-0
197.
Kaur S Ghuman MS Devarajan LJ . A pediatric epilepsy classic: “Double cortex” syndrome. J Pediatr Neurosci. (2015) 10:125–6. doi: 10.4103/1817-1745.159201
198.
Tsai MH Kuo PW Myers CT Li SW Lin WC Fu TY et al . A novel DCX missense mutation in a family with X-linked lissencephaly and subcortical band heterotopia syndrome inherited from a low-level somatic mosaic mother: Genetic and functional studies. Eur J Paediatr Neurol. (2016) 20:788–94. doi: 10.1016/j.ejpn.2016.05.010
199.
Haas CA Dudeck O Kirsch M Huszka C Kann G Pollak S et al . Role for reelin in the development of granule cell dispersion in temporal lobe epilepsy. J Neurosci. (2002) 22:5797–802. doi: 10.1523/JNEUROSCI.22-14-05797.2002
200.
D’Alessio L Konopka H López EM Seoane E Consalvo D Oddo S et al . Doublecortin (DCX) immunoreactivity in hippocampus of chronic refractory temporal lobe epilepsy patients with hippocampal sclerosis. Seizure. (2010) 19:567–72. doi: 10.1016/j.seizure.2010.09.004
201.
Weiss KH Johanssen C Tielsch A Herz J Deller T Frotscher M et al . Malformation of the radial glial scaffold in the dentate gyrus of reeler mice, scrambler mice, and Apo ER2/VLDLR-deficient mice. J Comp Neurol. (2003) 460:56–65. doi: 10.1002/cne.10644
202.
Duveau V Madhusudan A Caleo M Knuesel I Fritschy JM . Impaired reelin processing and secretion by Cajal-Retzius cells contributes to granule cell dispersion in a mouse model of temporal lobe epilepsy. Hippocampus. (2011) 21:935–44. doi: 10.1002/hipo.20793
203.
Nosten-Bertrand M Kappeler C Dinocourt C Denis C Germain J Phan Dinh Tuy F et al . Epilepsy in Dcx knockout mice associated with discrete lamination defects and enhanced excitability in the hippocampus. PLoS One. (2008) 3:e 2473. doi: 10.1371/journal.pone.0002473
204.
Corbo JC Deuel TA Long JM LaPorte P Tsai E Wynshaw-Boris A et al . Doublecortin Is Required in Mice for Lamination of the Hippocampus But Not the Neocortex. J Neurosci. (2002) 22:7548–57. doi: 10.1523/JNEUROSCI.22-17-07548.2002
205.
Deuel TAS Liu JS Corbo JC Yoo SY Rorke-Adams LB Walsh CA . Genetic interactions between doublecortin and doublecortin-like kinase in neuronal migration and axon outgrowth. Neuron. (2006) 49:41–53. doi: 10.1016/j.neuron.2005.10.038
206.
Lapray D Popova IY Kindler J Jorquera I Becq H Manent JB et al . Spontaneous epileptic manifestations in a DCX knockdown model of human double cortex. Cereb Cortex. (2010) 20:2694–701. doi: 10.1093/cercor/bhq014
207.
Crino P . New RELN Mutation Associated with Lissencephaly and Epilepsy. Epilepsy Curr. (2001) 1:72–3. doi: 10.1046/j.1535-7597.2001.00017.x
208.
Machado RA Benjumea-Cuartas V Zapata Berruecos JF Agudelo-Flóres PM Salazar-Peláez LM . Reelin, tau phosphorylation and psychiatric complications in patients with hippocampal sclerosis and structural abnormalities in temporal lobe epilepsy. Epilepsy Behav. (2019) 96:192–9. doi: 10.1016/j.yebeh.2019.04.052
209.
Leifeld J Förster E Reiss G Hamad MIK . Considering the Role of Extracellular Matrix Molecules, in Particular Reelin, in Granule Cell Dispersion Related to Temporal Lobe Epilepsy. Front Cell Dev Biol. (2022) 10:917575. doi: 10.3389/fcell.2022.917575
210.
D’Arcangelo G . Reelin mouse mutants as models of cortical development disorders. Epilepsy Behav. (2006) 8:81–90. doi: 10.1016/j.yebeh.2005.09.005
211.
Warm D Schroer J Sinning A . Gabaergic Interneurons in Early Brain Development: Conducting and Orchestrated by Cortical Network Activity. Front Mol Neurosci. (2021) 14:807969. doi: 10.3389/fnmol.2021.807969
212.
Pallotto M Deprez F . Regulation of adult neurogenesis by GABAergic transmission: signaling beyond GABAA-receptors. Front Cell Neurosci. (2014) 8:166. doi: 10.3389/fncel.2014.00166
213.
Vílchez-Acosta A Manso Y Cárdenas A Elias-Tersa A Martínez-Losa M Pascual M et al . Specific contribution of Reelin expressed by Cajal-Retzius cells or GABAergic interneurons to cortical lamination. Proc Natl Acad Sci USA. (2022) 119:e2120079119. doi: 10.1073/pnas.2120079119
214.
Haas CA Frotscher M . Reelin deficiency causes granule cell dispersion in epilepsy. Exp Brain Res. (2010) 200:141–9. doi: 10.1007/s00221-009-1948-5
215.
Roy A Millen KJ Kapur RP . Hippocampal granule cell dispersion: a non-specific finding in pediatric patients with no history of seizures. Acta Neuropathol Commun. (2020) 8:54. doi: 10.1186/s40478-020-00928-3
216.
Gallardo-Caballero M Rodríguez-Moreno CB Álvarez-Méndez L Terreros-Roncal J Flor-García M Moreno-Jiménez EP et al . Prolonged fixation and post-mortem delay impede the study of adult neurogenesis in mice. Commun Biol. (2023) 6:978. doi: 10.1038/s42003-023-05367-z
217.
Ma W Zeighami Y Dadar M . Differential effects of prolonged post-fixation on immunohistochemical and histochemical staining for postmortem human brains. Bio Rxiv. (2024). 18. doi: 10.1101/2024.05.13.593904
218.
Jossin Y Goffinet AM . Reelin signals through phosphatidylinositol 3-kinase and Akt to control cortical development and through mTor to regulate dendritic growth. Mol Cell Biol. (2007) 27:7113–24. doi: 10.1128/MCB.00928-07
219.
Green JA Mykytyn K . Neuronal primary cilia: an underappreciated signaling and sensory organelle in the brain. Neuropsychopharmacology. (2014) 39:244–5. doi: 10.1038/npp.2013.203
220.
Park SM Jang HJ Lee JH . Roles of Primary Cilia in the Developing Brain. Front Cell Neurosci. (2019) 13:218. doi: 10.3389/fncel.2019.00218
221.
Fehr S Wilson M Downs J Williams S Murgia A Sartori S et al . The CDKL5 disorder is an independent clinical entity associated with early-onset encephalopathy. Eur J Hum Genet. (2013) 21:266–73. doi: 10.1038/ejhg.2012.156
222.
Bahi-Buisson N Nectoux J Rosas-Vargas H Milh M Boddaert N Girard B et al . Key clinical features to identify girls with CDKL5 mutations. Brain. (2008) 131:2647–61. doi: 10.1093/brain/awn197
223.
Bailey JN de Nijs L Bai D Suzuki T Miyamoto H Tanaka M et al . Variant Intestinal-Cell Kinase in Juvenile Myoclonic Epilepsy. N Engl J Med. (2018) 378:1018–28. doi: 10.1056/NEJMoa1700175
224.
Medina MT Suzuki T Alonso ME Durón RM Martínez-Juárez IE Bailey JN et al . Novel mutations in Myoclonin 1/EFHC1 in sporadic and familial juvenile myoclonic epilepsy. Neurology. (2008) 70:2137–44. doi: 10.1212/01.wnl.0000313149.73035.99
225.
Park SM Lim JS Ramakrishina S Kim SH Kim WK Lee J et al . Brain Somatic Mutations in MTOR Disrupt Neuronal Ciliogenesis. Leading to Focal Cortical Dyslamination Neuron. (2018) 99:83–97.e7. doi: 10.1016/j.neuron.2018.05.039
226.
Han YG Alvarez-Buylla A . Role of primary cilia in brain development and cancer. Curr Opin Neurobiol. (2010) 20:58–67. doi: 10.1016/j.conb.2009.12.002
227.
Chizhikov VV Davenport J Zhang Q Shih EK Cabello OA Fuchs JL et al . Cilia proteins control cerebellar morphogenesis by promoting expansion of the granule progenitor pool. J Neurosci. (2007) 27:9780–9. doi: 10.1523/JNEUROSCI.5586-06.2007
228.
Salvati KA Mason AJ Gailey CD Wang EJ Fu Z Beenhakker MP . Mice Harboring a Non-Functional CILK1/ICK Allele Fail to Model the Epileptic Phenotype in Patients Carrying Variant CILK1/ICK. Int J Mol Sci [Internet]. (2021) 22:8875. doi: 10.3390/ijms22168875
229.
Wu JY Cho SJ Descant K Li PH Shapson-Coe A Januszewski M et al . Mapping of neuronal and glial primary cilia contactome and connectome in the human cerebral cortex. Neuron. (2024) 112:41–55.e3. doi: 10.1016/j.neuron.2023.09.032
230.
Suzuki T Inoue I Yamakawa K . Epilepsy protein Efhc 1/myoclonin 1 is expressed in cells with motile cilia but not in neurons or mitotic apparatuses in brain. Sci Rep. (2020) 10:22076. doi: 10.1038/s41598-020-79202-4
231.
Conduit SE Pearce W Bhamra A Bilanges B Bozal-Basterra L Foukas LC et al . A class I PI3K signalling network regulates primary cilia disassembly in normal physiology and disease. Nat Commun. (2024) 15:7181. doi: 10.1038/s41467-024-51354-1
232.
Prosseda PP Dannewitz Prosseda S Tran M Liton PB Sun Y . Crosstalk between the mTOR pathway and primary cilia in human diseases. Curr Top Dev Biol. (2023) 155:1–37. doi: 10.1016/bs.ctdb.2023.09.004
233.
Roy A Murphy RM Deng M Mac Donald JW Bammler TK Aldinger KA et al . PI3K-Yap activity drives cortical gyrification and hydrocephalus in mice. eLife. (2019) 8:8. doi: 10.7554/eLife.45961
234.
Krutoshinskaya Y Coulehan K Pushchinska G Spiegel R . The Reciprocal Relationship between Sleep and Epilepsy. J Pers Med. (2024) 14:118. doi: 10.3390/jpm14010118
235.
Latreille V Schiller K Peter-Derex L Frauscher B . Does epileptic activity impair sleep-related memory consolidation in epilepsy? A critical and systematic review. J Clin Sleep Med. (2022) 18:2481–95. doi: 10.5664/jcsm.10166
236.
Ali A Wu S Issa NP Rose S Towle VL Warnke P et al . Association of sleep with sudden unexpected death in epilepsy. Epilepsy Behav. (2017) 76:1–6. doi: 10.1016/j.yebeh.2017.08.021
237.
Duncan JR Byard RW . (eds.). SIDS Sudden infant and early childhood death: The past, the present and the future. Adelaide (AU): University of Adelaide Press. (2018). Available from: https://www.ncbi.nlm.nih.gov/books/NBK513384/.
238.
Halász P Bódizs R Ujma PP Fabó D Szűcs A . Strong relationship between NREM sleep, epilepsy and plastic functions - A conceptual review on the neurophysiology background. Epilepsy Res. (2019) 150:95–105. doi: 10.1016/j.eplepsyres.2018.11.008
239.
Chokroverty S . Sleep Disorders Medicine: Basic Science, Technical Considerations and Clinical Aspects. Springer Nature (2017). 1269 p.
240.
Shahar E Genizi J Ravid S Schif A . The complementary value of sleep-deprived EEG in childhood onset epilepsy. Eur J Paediatr Neurol. (2010) 14:308–12. doi: 10.1016/j.ejpn.2009.08.001
241.
Roliz AH Kothare S . The Relationship Between Sleep, Epilepsy, and Development: a Review. Curr Neurol Neurosci Rep. (2023) 23:469–77. doi: 10.1007/s11910-023-01284-0
242.
Nobili L Frauscher B Eriksson S Gibbs SA Halasz P Lambert I et al . Sleep and epilepsy: A snapshot of knowledge and future research lines. J Sleep Res. (2022) 31:e13622. doi: 10.1111/jsr.13622
243.
Bonilla-Jaime H Zeleke H Rojas A Espinosa-Garcia C . Sleep disruption worsens seizures: Neuroinflammation as a potential mechanistic link. Int J Mol Sci. (2021) 22:12531. doi: 10.3390/ijms222212531
244.
Espinosa-Garcia C Zeleke H Rojas A . Impact of stress on epilepsy: Focus on neuroinflammation-A mini review. Int J Mol Sci. (2021) 22:4061. doi: 10.3390/ijms22084061
245.
Lee DA Park BS Ko J Park SH Park JH Kim IH et al . Glymphatic system function in patients with newly diagnosed focal epilepsy. Brain Behav. (2022) 12:e2504. doi: 10.1002/brb3.2504
246.
Christensen J Yamakawa GR Shultz SR Mychasiuk R . Is the glymphatic system the missing link between sleep impairments and neurological disorders? Examining the implications and uncertainties. Prog Neurobiol. (2021) 198:101917. doi: 10.1016/j.pneurobio.2020.101917
247.
Anzai Y Minoshima S . Why We Need to Sleep: Glymphatic Pathway and Neurodegenerative Disease. Radiology. (2021) 300:669–70. doi: 10.1148/radiol.2021211140
248.
Kim ST Kim SE Lee DA Lee HJ Park KM . Anti-seizure medication response and the glymphatic system in patients with focal epilepsy. Eur J Neurol. (2024) 31:e16097. doi: 10.1111/ene.16097
249.
Gupta M Aneja S Kohli K . Add-on melatonin improves sleep behavior in children with epilepsy: randomized, double-blind, placebo-controlled trial. J Child Neurol. (2005) 20:112–5. doi: 10.1177/08830738050200020501
250.
Guerrero JM Reiter RJ . Melatonin-immune system relationships. Curr Top Med Chem. (2002) 2:167–79. doi: 10.2174/1568026023394335
251.
Costa-Lotufo LV de F FMM ISP L de Oliveira AA Nascimento VS VMS d B et al . Attenuating effects of melatonin on pilocarpine-induced seizures in rats. Comp Biochem Physiol C Toxicol Pharmacol. (2002) 131:521–9. doi: 10.1016/S1532-0456(02)00037-6
252.
Liu J Clough SJ Hutchinson AJ Adamah-Biassi EB Popovska-Gorevski M Dubocovich ML . MT1 and MT2 Melatonin Receptors: A Therapeutic Perspective. Annu Rev Pharmacol Toxicol. (2016) 56:361–83. doi: 10.1146/annurev-pharmtox-010814-124742
253.
Maghbooli M Alyan Najaf Abadi S Malek Mahmoudi G Molseghi MH . Effect of add-on melatonin on seizure outcomes and quality of sleep in epilepsy with idiopathic generalized tonic-clonic seizures alone in adult patients: Cross-sectional, randomized, double-blind, placebo-controlled clinical trial. Brain Behav. (2023) 13:e2860. doi: 10.1002/brb3.2860
254.
Lipton JO Yuan ED Boyle LM Ebrahimi-Fakhari D Kwiatkowski E Nathan A et al . The Circadian Protein BMAL1 Regulates Translation in Response to S6K1-Mediated Phosphorylation. Cell. (2015) 161:1138–51. doi: 10.1016/j.cell.2015.04.002
255.
Gerstner JR Smith GG Lenz O Perron IJ Buono RJ Ferraro TN . BMAL1 controls the diurnal rhythm and set point for electrical seizure threshold in mice. Front Syst Neurosci. (2014) 8:121. doi: 10.3389/fnsys.2014.00121
256.
Li P Fu X Smith NA Ziobro J Curiel J Tenga MJ et al . Loss of CLOCK Results in Dysfunction of Brain Circuits Underlying Focal Epilepsy. Neuron. (2017) 96:387–401.e6. doi: 10.1016/j.neuron.2017.09.044
257.
Hodges SL Lugo JN . Therapeutic role of targeting mTOR signaling and neuroinflammation in epilepsy. Epilepsy Res. (2020) 161:106282. doi: 10.1016/j.eplepsyres.2020.106282
258.
Jin B Aung T Geng Y Wang S . Epilepsy and Its Interaction With Sleep and Circadian Rhythm. Front Neurol. (2020) 11:327. doi: 10.3389/fneur.2020.00327
259.
Ramanathan C Kathale ND Liu D Lee C Freeman DA Hogenesch JB et al . mTOR signaling regulates central and peripheral circadian clock function. PLoS Genet. (2018) 14:e1007369. doi: 10.1371/journal.pgen.1007369
260.
Ramgopal S Thome-Souza S Loddenkemper T . Chronopharmacology of anti-convulsive therapy. Curr Neurol Neurosci Rep. (2013) 13:339. doi: 10.1007/s11910-013-0339-2
261.
Wilcox KS West PJ Metcalf CS . The current approach of the Epilepsy Therapy Screening Program contract site for identifying improved therapies for the treatment of pharmacoresistant seizures in epilepsy. Neuropharmacology. (2020) 166:107811. doi: 10.1016/j.neuropharm.2019.107811
262.
Bergren SK Chen S Galecki A Kearney JA . Genetic modifiers affecting severity of epilepsy caused by mutation of sodium channel Scn 2a. Mamm Genome. (2005) 16:683–90. doi: 10.1007/s00335-005-0049-4
263.
Miller AR Hawkins NA McCollom CE Kearney JA . Mapping genetic modifiers of survival in a mouse model of Dravet syndrome. Genes Brain Behav. (2014) 13:163–72. doi: 10.1111/gbb.12099
264.
Hawkins NA Calhoun JD Huffman AM Kearney JA . Gene expression profiling in a mouse model of Dravet syndrome. Exp Neurol. (2019) 311:247–56. doi: 10.1016/j.expneurol.2018.10.010
265.
Florio M Huttner WB . Neural progenitors, neurogenesis and the evolution of the neocortex. Development. (2014) 141:2182–94. doi: 10.1242/dev.090571
266.
Zhao X Bhattacharyya A . Human Models Are Needed for Studying Human Neurodevelopmental Disorders. Am J Hum Genet. (2018) 103:829–57. doi: 10.1016/j.ajhg.2018.10.009
267.
Simkin D Kiskinis E . Modeling Pediatric Epilepsy Through iPSC-Based Technologies. Epilepsy Curr. (2018) 18:240–5. doi: 10.5698/1535-7597.18.4.240
268.
Andrews MG Kriegstein AR . Challenges of Organoid Research. Annu Rev Neurosci. (2022) 45:23–39. doi: 10.1146/annurev-neuro-111020-090812
269.
Kim J Koo BK Knoblich JA . Human organoids: model systems for human biology and medicine. Nat Rev Mol Cell Biol. (2020) 21:571–84. doi: 10.1038/s41580-020-0259-3
270.
Zhao Y Liu K Wang Y Ma Y Guo W Shi C . Human-mouse chimeric brain models constructed from iPSC-derived brain cells: Applications and challenges. Exp Neurol. (2024) 379:114848. doi: 10.1016/j.expneurol.2024.114848
271.
Kwan P Schachter SC Brodie MJ . Drug-resistant epilepsy. N Engl J Med. (2011) 365:919–26. doi: 10.1056/NEJMra1004418
272.
Tang F Hartz AMS Bauer B . Drug-Resistant Epilepsy: Multiple Hypotheses. Few Answers Front Neurol. (2017) 8:301. doi: 10.3389/fneur.2017.00301
273.
Löscher W Potschka H Sisodiya SM Vezzani A . Drug Resistance in Epilepsy: Clinical Impact, Potential Mechanisms, and New Innovative Treatment Options. Pharmacol Rev. (2020) 72:606–38. doi: 10.1124/pr.120.019539
274.
Lim MJR Fong KY Zheng Y Chua CYK Miny S Lin JB et al . Vagus nerve stimulation for treatment of drug-resistant epilepsy: a systematic review and meta-analysis. Neurosurg Rev. (2022) 45:2361–73. doi: 10.1007/s10143-022-01757-9
275.
Li MCH Cook MJ . Deep brain stimulation for drug-resistant epilepsy. Epilepsia. (2018) 59:273–90. doi: 10.1111/epi.13964
276.
Fisher B Des Marteau JA Koontz EH Wilks SJ Melamed SE . Responsive Vagus Nerve Stimulation for Drug Resistant Epilepsy: A Review of New Features and Practical Guidance for Advanced Practice Providers. Front Neurol. (2020) 11:610379. doi: 10.3389/fneur.2020.610379
277.
Richerson GB Buchanan GF . The serotonin axis: Shared mechanisms in seizures, depression, and SUDEP. Epilepsia. (2011) 52:28–38. doi: 10.1111/j.1528-1167.2010.02908.x
278.
Petrucci AN Joyal KG Purnell BS Buchanan GF . Serotonin and sudden unexpected death in epilepsy. Exp Neurol. (2020) 325:113145. doi: 10.1016/j.expneurol.2019.113145
279.
Sourbron J Lagae L . Serotonin receptors in epilepsy: Novel treatment targets?Epilepsia Open. (2022) 7:231–46. doi: 10.1002/epi4.12580
280.
Crino PB . The mTOR signalling cascade: paving new roads to cure neurological disease. Nat Rev Neurol. (2016) 12:379–92. doi: 10.1038/nrneurol.2016.81
281.
Kim JK Lee JH . Mechanistic Target of Rapamycin Pathway in Epileptic Disorders. J Korean Neurosurg Soc. (2019) 62:272–87. doi: 10.3340/jkns.2019.0027
282.
Tehrani F Khosroshahi N Keihani Doust Z Dabiran S Zarkesh MR . The efficacy and safety of rapamycin in children with tuberous sclerosis: A cross-sectional study. Iran J Child Neurol. (2023) 17:19–29. doi: 10.22037/ijcn.v17i2.36243
283.
Chang CA Perrier R Kurek KC Estrada-Veras J Lehman A Yip S et al . Novel findings and expansion of phenotype in a mosaic RASopathy caused by somatic KRAS variants. Am J Med Genet A. (2021) 185:2829–45. doi: 10.1002/ajmg.a.62356
284.
Pepi C de Palma L Trivisano M Pietrafusa N Lepri FR Diociaiuti A et al . The Role of KRAS Mutations in Cortical Malformation and Epilepsy Surgery: A Novel Report of Nevus Sebaceous Syndrome and Review of the Literature. Brain Sci. (2021) 11:793. doi: 10.3390/brainsci11060793
285.
Battaglia DI Gambardella ML Veltri S Contaldo I Chillemi G Veredice C et al . Epilepsy and BRAF Mutations: Phenotypes, Natural History and Genotype-Phenotype Correlations. Genes (Basel). (2021) 12:1316. doi: 10.3390/genes12091316
286.
Pecoraro A Arehart E Gallentine W Radtke R Smith E Pizoli C et al . Epilepsy in neurofibromatosis type 1. Epilepsy Behav. (2017) 73:137–41. doi: 10.1016/j.yebeh.2017.05.011
287.
Khair A Kaur G Falchek S . Epilepsy Characteristics in Children With Neurofibromatosis type 1, What We Have Learnt From a Tertiary Center Five Years’ Experience (P12-5.005). Neurology. (2022) 98. doi: 10.1212/WNL.98.18_supplement.1027
288.
Lim JS Kim WI Kang HC Kim SH Park AH Park EK et al . Brain somatic mutations in MTOR cause focal cortical dysplasia type II leading to intractable epilepsy. Nat Med. (2015) 21:395–400. doi: 10.1038/nm.3824
289.
Almobarak S Almuhaizea M Abukhaled M Alyamani S Dabbagh O Chedrawi A et al . Tuberous Sclerosis Complex: Clinical Spectrum and Epilepsy: A Retrospective Chart Review Study. Transl Neurosci. (2018) 9:154–60. doi: 10.1515/tnsci-2018-0023
290.
Ding Y Zhou Y Yu L Zhang L Zhou S Wang Y et al . Correlation between epilepsy and genotype: A large retrospective tuberous sclerosis complex cohort. Seizure. (2021) 91:273–7. doi: 10.1016/j.seizure.2021.06.036
291.
Harris RV Oliver KL Perucca P Striano P Labate A Riva A et al . Familial Mesial Temporal Lobe Epilepsy: Clinical Spectrum and Genetic Evidence for a Polygenic Architecture. Ann Neurol. (2023) 94:825–35. doi: 10.1002/ana.26765
292.
Dibbens LM de Vries B Donatello S Heron SE Hodgson BL Chintawar S et al . Mutations in DEPDC5 cause familial focal epilepsy with variable foci. Nat Genet. (2013) 45:546–51. doi: 10.1038/ng.2599
293.
Michelucci R Pulitano P Di Bonaventura C Binelli S Luisi C Pasini E et al . The clinical phenotype of autosomal dominant lateral temporal lobe epilepsy related to reelin mutations. Epilepsy Behav. (2017) 68:103–7. doi: 10.1016/j.yebeh.2016.12.003
294.
Gilliam FG Ssentongo P Sather M Kawasawa YI . Case Report: PAFAH1B1 Mutation and Posterior Band Heterotopia With Focal Temporal Lobe Epilepsy Treated by Responsive Neurostimulation. Front Neurol. (2021) 12:779113. doi: 10.3389/fneur.2021.779113
295.
Thounaojam R Langbang L Itisham K Sobhani R Srivastava S Ramanujam B et al . EFHC1 mutation in Indian juvenile myoclonic epilepsy patient. Epilepsia Open. (2017) 2:84–9. doi: 10.1002/epi4.12037
296.
Bahi-Buisson N Kaminska A Boddaert N Rio M Afenjar A Gérard M et al . The three stages of epilepsy in patients with CDKL5 mutations. Epilepsia. (2008) 49:1027–37. doi: 10.1111/j.1528-1167.2007.01520.x
297.
Kobayashi Y Tohyama J Takahashi Y Goto T Haginoya K Inoue T et al . Clinical manifestations and epilepsy treatment in Japanese patients with pathogenic CDKL5 variants. Brain Dev. (2021) 43:505–14. doi: 10.1016/j.braindev.2020.12.006
298.
Gowda VK Amoghimath R Battina M Shivappa SK Benakappa N . Case Series of Early SCN1A-Related Developmental and Epileptic Encephalopathies. J Pediatr Neurosci. (2021) 16:212–7. doi: 10.4103/jpn.JPN_99_20
299.
Clatot J Parthasarathy S Cohen S McKee JL Massey S Somarowthu A et al . SCN1A gain-of-function mutation causing an early onset epileptic encephalopathy. Epilepsia. (2023) 64:1318–30. doi: 10.1111/epi.17444
300.
Heron SE Crossland KM Andermann E Phillips HA Hall AJ Bleasel A et al . Sodium-channel defects in benign familial neonatal-infantile seizures. Lancet. (2002) 360:851–2. doi: 10.1016/S0140-6736(02)09968-3
301.
Sugawara T Tsurubuchi Y Agarwala KL Ito M Fukuma G Mazaki-Miyazaki E et al . A missense mutation of the Na+ channel αII subunit gene Nav1.2 in a patient with febrile and afebrile seizures causes channel dysfunction. Proc Natl Acad Sci. (2001) 98:6384–9. doi: 10.1073/pnas.111065098
302.
Nakamura K Kato M Osaka H Yamashita S Nakagawa E Haginoya K et al . Clinical spectrum of SCN2A mutations expanding to Ohtahara syndrome. Neurology. (2013) 81:992–8. doi: 10.1212/WNL.0b013e3182a43e57
303.
Cutts A Savoie H Hammer MF Schreiber J Grayson C Luzon C et al . Clinical characteristics and treatment experience of individuals with SCN8A developmental and epileptic encephalopathy (SCN8A-DEE): Findings from an online caregiver survey. Seizure. (2022) 97:50–7. doi: 10.1016/j.seizure.2022.03.008
304.
Fatema K Rahman MM Faruk O . SCN8A Mutation in Infantile Epileptic Encephalopathy: Report of Two Cases. J Epilepsy Res. (2019) 9:147–51. doi: 10.14581/jer.19017
305.
Gardella E Becker F Møller RS Schubert J Lemke JR Larsen LHG et al . Benign infantile seizures and paroxysmal dyskinesia caused by an SCN8A mutation. Ann Neurol. (2016) 79:428–36. doi: 10.1002/ana.24580
306.
Lemke JR Lal D Reinthaler EM Steiner I Nothnagel M Alber M et al . Mutations in GRIN2A cause idiopathic focal epilepsy with rolandic spikes. Nat Genet. (2013) 45:1067–72. doi: 10.1038/ng.2728
307.
Barcia G Fleming MR Deligniere A Gazula VR Brown MR Langouet M et al . De novo gain-of-function KCNT1 channel mutations cause malignant migrating partial seizures of infancy. Nat Genet. (2012) 44:1255–9. doi: 10.1038/ng.2441
308.
Soldovieri MV Boutry-Kryza N Milh M Doummar D Heron B Bourel E et al . Novel KCNQ2 and KCNQ3 mutations in a large cohort of families with benign neonatal epilepsy: first evidence for an altered channel regulation by syntaxin-1A. Hum Mutat. (2014) 35:356–67. doi: 10.1002/humu.22500
309.
Chokvithaya S Caengprasath N Buasong A Jantasuwan S Santawong K Leela-Adisorn N et al . Nine patients with KCNQ2-related neonatal seizures and functional studies of two missense variants. Sci Rep. (2023) 13:3328. doi: 10.1038/s41598-023-29924-y
310.
Ye J Tang S Miao P Gong Z Shu Q Feng J et al . Clinical analysis and functional characterization of KCNQ2-related developmental and epileptic encephalopathy. Front Mol Neurosci. (2023) 16:1205265. doi: 10.3389/fnmol.2023.1205265
311.
Guo Y Yan KP Qu Q Qu J Chen ZG Song T et al . Common variants of KCNJ10 are associated with susceptibility and anti-epileptic drug resistance in Chinese genetic generalized epilepsies. PLoS One. (2015) 10:e0124896. doi: 10.1371/journal.pone.0124896
312.
Sicca F Imbrici P D’Adamo MC Moro F Bonatti F Brovedani P et al . Autism with seizures and intellectual disability: possible causative role of gain-of-function of the inwardly-rectifying K+ channel Kir 4.1. Neurobiol Dis. (2011) 43:239–47. doi: 10.1016/j.nbd.2011.03.016
313.
Reinson K Õiglane-Shlik E Talvik I Vaher U Õunapuu A Ennok M et al . Biallelic CACNA1A mutations cause early onset epileptic encephalopathy with progressive cerebral, cerebellar, and optic nerve atrophy. Am J Med Genet A. (2016) 170:2173–6. doi: 10.1002/ajmg.a.37678
314.
Epi 4K Consortium . De Novo Mutations in SLC1A2 and CACNA1A Are Important Causes of Epileptic Encephalopathies. Am J Hum Genet. (2016) 99:287–98. doi: 10.1016/j.ajhg.2016.06.003
315.
Li XL Li ZJ Liang XY Liu DT Jiang M Gao LD et al . CACNA1A Mutations Associated With Epilepsies and Their Molecular Sub-Regional Implications. Front Mol Neurosci. (2022) 15:860662. doi: 10.3389/fnmol.2022.860662
316.
Chen Y Lu J Pan H Zhang Y Wu H Xu K et al . Association between genetic variation of CACNA1H and childhood absence epilepsy. Ann Neurol. (2003) 54:239–43. doi: 10.1002/ana.10607
317.
Splawski I Yoo DS Stotz SC Cherry A Clapham DE Keating MT . CACNA1H mutations in autism spectrum disorders. J Biol Chem. (2006) 281:22085–91. doi: 10.1074/jbc.M603316200
318.
Chourasia N Ossó-Rivera H Ghosh A Von Allmen G Koenig MK . Expanding the Phenotypic Spectrum of CACNA1H Mutations. Pediatr Neurol. (2019) 93:50–5. doi: 10.1016/j.pediatrneurol.2018.11.017
319.
Marini C Porro A Rastetter A Dalle C Rivolta I Bauer D et al . HCN1 mutation spectrum: from neonatal epileptic encephalopathy to benign generalized epilepsy and beyond. Brain. (2018) 141:3160–78. doi: 10.1093/brain/awy263
320.
Nava C Dalle C Rastetter A Striano P de Kovel CGF Nabbout R et al . De novo mutations in HCN1 cause early infantile epileptic encephalopathy. Nat Genet. (2014) 46:640–5. doi: 10.1038/ng.2952
321.
Johannesen K Marini C Pfeffer S Møller RS Dorn T Niturad CE et al . Phenotypic spectrum of GABRA1: From generalized epilepsies to severe epileptic encephalopathies. Neurology. (2016) 87:1140–51. doi: 10.1212/WNL.0000000000003087
322.
Fonte J Videira G Chorão R Freitas J Carrilho I Freixo JP et al . Familial occipital lobe epilepsy associated with GABAA receptor variants. Seizure. (2023) 112:139–42. doi: 10.1016/j.seizure.2023.10.003
323.
Carvill GL Weckhuysen S McMahon JM Hartmann C Møller RS Hjalgrim H et al . GABRA1 and STXBP1: novel genetic causes of Dravet syndrome. Neurology. (2014) 82:1245–53. doi: 10.1212/WNL.0000000000000291
324.
Cossette P Liu L Brisebois K Dong H Lortie A Vanasse M et al . Mutation of GABRA1 in an autosomal dominant form of juvenile myoclonic epilepsy. Nat Genet. (2002) 31:184–9. doi: 10.1038/ng885
325.
Kang JQ Macdonald RL . Molecular Pathogenic Basis for GABRG2 Mutations Associated With a Spectrum of Epilepsy Syndromes, From Generalized Absence Epilepsy to Dravet Syndrome. JAMA Neurol. (2016) 73:1009–16. doi: 10.1001/jamaneurol.2016.0449
Summary
Keywords
epilepsy – abnormalities, pediatric epilepsy, SUDEP, neurodevelopment, drug resistance, Cilia, sleep and circadian rhythm, PI3K-AKT–MTOR pathway
Citation
Lolam V and Roy A (2025) Developmental mechanisms underlying pediatric epilepsy. Front. Neurol. 16:1586947. doi: 10.3389/fneur.2025.1586947
Received
03 April 2025
Accepted
16 May 2025
Published
03 June 2025
Volume
16 - 2025
Edited by
Piero Pavone, University of Catania, Italy
Reviewed by
Lorenzo Pavone, University of Catania, Italy
Sotaro Kanai, University of California, Los Angeles, United States
Updates
Copyright
© 2025 Lolam and Roy.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Achira Roy, achira.roy@jncasr.ac.in
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.