 Subit Barua
Subit Barua Salomon Kuizon
Salomon Kuizon W. Ted Brown2
W. Ted Brown2 Mohammed A. Junaid
Mohammed A. Junaid- 1Departments of Developmental Biochemistry, New York State Institute for Basic Research in Developmental Disabilities, Staten Island, NY, USA
- 2Human Genetics, New York State Institute for Basic Research in Developmental Disabilities, Staten Island, NY, USA
It is becoming increasingly more evident that lifestyle, environmental factors, and maternal nutrition during gestation can influence the epigenome of the developing fetus and thus modulate the physiological outcome. Variations in the intake of maternal nutrients affecting one-carbon metabolism may influence brain development and exert long-term effects on the health of the progeny. In this study, we investigated whether supplementation with high maternal folic acid during gestation alters DNA methylation and gene expression in the cerebellum of mouse offspring. We used reduced representation bisulfite sequencing to analyze the DNA methylation profile at the single-base resolution level. The genome-wide DNA methylation analysis revealed that supplementation with higher maternal folic acid resulted in distinct methylation patterns (P < 0.05) of CpG and non-CpG sites in the cerebellum of offspring. Such variations of methylation and gene expression in the cerebellum of offspring were highly sex-specific, including several genes of the neuronal pathways. These findings demonstrate that alterations in the level of maternal folic acid during gestation can influence methylation and gene expression in the cerebellum of offspring. Such changes in the offspring epigenome may alter neurodevelopment and influence the functional outcome of neurologic and psychiatric diseases.
Introduction
DNA reprogramming is essential for early embryonic development; around the time of implantation, de novo methylation is initiated in embryonic cells and is required for complete embryonic development (Li et al., 1992; Lei et al., 1996; Okano et al., 1999; Dean et al., 2001). In mammals, cytosine methylation is highly prevalent at CpG islands that modulate the chromatin structure and binding of transcriptional factors at promoter regions (Jaenisch and Bird, 2003). Thus, alterations in DNA methylation and epigenetic modification that occur during a specific window of gestational development can dysregulate gene expression and are associated with many diseases (Oberlander et al., 2008; Stein et al., 2009; Suter et al., 2010; Gore et al., 2011; Li et al., 2013; Perkins et al., 2013; West et al., 2013; Vanhees et al., 2014). Studies in humans and animal models have shown that variations in intake of the maternal nutrients involved in one-carbon metabolism, including folic acid (FA), during pregnancy can induce persistent changes in the offspring's epigenome and modulate various physiological outcomes (Cooney et al., 2002; Bean et al., 2011; Boeke et al., 2012; Greenop et al., 2014; O'Neill et al., 2014; Barua and Junaid, 2015). Our earlier studies in a mouse model had shown that exposure to high FA supplementation during gestation causes widespread changes in the methylation and gene expression in the cerebral hemisphere of the offspring (Barua et al., 2014b). Moreover, such exposure during gestation and the post-weaning period resulted in moderate changes in behavior (Barua et al., 2014a).
Over the past decades, several studies have shown that the cerebellum (CB) plays a significant role in coordination to motor functions and is involved in various cognitive processes, including perception, attention, and emotional behavior (Leiner et al., 1991; Martin et al., 2003; Schmahmann and Caplan, 2006). Recent studies with post-mortem brain samples have shown widespread aberrant methylation and gene expression in the CB of psychotic patients (Chen et al., 2014). Studies with a mouse model and in human post-mortem CB of individuals with autism have shown altered patterns of DNA methylation (Shpyleva et al., 2014), and indeed, cerebellar abnormalities have been reported in more than 95% of post-mortem examinations of individuals with autism (Marzban et al., 2014). To investigate whether higher supplementation with a methyl diet during gestation impacts the cerebellar development of offspring, in this study, we tested the hypothesis that higher folic acid supplementation during gestation can alter the methylation and gene expression in the CB of offspring.
Materials and Methods
Animals and Experimental Design
All animal experiments were performed in accordance with protocols reviewed and approved by the Institute for Basic Research Institutional Animal Care and Use Committee in conformity with the NIH Guide for Care and Use of Laboratory Animals (NIH publication No. 86-23, revised 1985). One week prior to mating and throughout gestation, adult 8–10 week-old C57BL/6 J female mice were fed a custom AIN-93G amino acid–based diet (Research Diet, Inc., North Brunswick, NJ), having either low maternal folic acid (LMFA), at 0.4 mg/kg (n = 12), or high maternal folic acid (HMFA), at 4 mg/kg (n = 12). These levels of FA supplementation were chosen in this study, as women with a prior history of complicated and neural tube defect (NTD)–affected pregnancy are recommended to take 10-fold higher FA (4 mg/day) in comparison to other pregnant women (400–800 μg/day). FA at the 0.4 mg/kg diet level is necessary for a normal healthy litter, whereas FA at the 4 mg/kg diet level is 10 times higher.
Tissue Collection and Processing
At post-natal day one (P1), pups from different dams were sacrificed, and CB tissues were dissected. The numbers of tissues collected from the LMFA group were: male pups, n = 15, and female pups n = 15. The numbers of tissues collected from the HMFA were: male pups n = 15, and female pups, n = 15. Tissues were immediately stored at −70°C until further use. From these, tissues were distributed for subsequent DNA/RNA and protein analysis.
DNA Extraction
CB tissues were extracted from P1 pups and pooled (n = 3/gender/group, each from an independent dam). DNA was extracted with the Epicenter MasterPure DNA purification kit (Epicenter Biotechnologies, Madison, WI, USA) by following the manufacturer's instructions, and concentration was measured by using a NanoDrop ND-1000 (Thermo Scientific, Wilmington, DE, USA).
Library Construction, Sequence Alignments, and Data Analysis
Library construction, sequence alignments, and data analysis were performed by following the detailed protocol previously described (Barua et al., 2014b). Libraries were prepared from 200 to 500 ng of genomic DNA after sequential digestion with 60 units of TaqI and 30 units of MspI (New England Biolabs, Ipswich, MA, USA), and sequencing was performed on an Illumina HiSeq genome analyzer. Sequence reads from bisulfite-treated EpiQuest libraries were identified using standard Illumina base-calling software and then analyzed using a Zymo Research proprietary analysis pipeline, which is written in Python. Bismark (http://www.bioinformatics.babraham.ac.uk/projects/bismark/) was the alignment software in the analysis pipeline. Index files were constructed by bismark_genome_preparation command using the entire reference genome. –non_directional and all the other default parameters were applied for running Bismark. Filled-in nucleotides were trimmed off when doing methylation calling. The number of reads reporting a C was divided by the total number of reads reporting a C or T was used to estimate the methylation level of sample cytosine. For each CpG site, Fisher's exact test was performed, which covered at least five reads. Moreover, for each CpG promoter, gene body and CpG island annotations were added. The total numbers of reads that were taken into account for each CpG site are given in Tables 2, 3, and Tables S1–S4 (column total CpG). All the procedures above were carried out in the Zymo Epigentic Core Services (Zymo Research, Irvine, CA). Sequence data has been deposited at the Sequence Read Archive (accession number SRX1608467) in the National Center for Biotechnology Information (NCBI).
Quantitative Real-Time Polymerase Chain Reaction (qPCR) Analysis
Total RNA was extracted (n = 12, segregated by gender) by lysing the cells with Trizol reagent (Life Technologies, Inc., Carlsbad, CA) and was further purified by Qiagen RNeasy kit (Qiagen, Valencia, CA), according to the manufacturer's protocol as described earlier (Barua et al., 2014c). For each sample, on-column DNase digestion was performed to remove any DNA contamination. Quantitative RT-PCR was performed with the One-Step iScript kit (BioRad, Hercules, CA) or the Two-step kit (Affymetrix, Santa Clara, CA) by following the manufacturer's instructions. Hprt1 was used as the endogenous control, and the relative expression was calculated using the Pfaffl method. For each gene, the mRNA expression was measured from n = 3/gender/group, each from an independent dam. Each reaction was run in triplicates, and only the expression of those genes that exhibited the same directional changes in independent pools from at least two independent animals was considered significant. The statistical difference between samples was determined by Student's t-test by using Prism Software (GraphPad, San Diego, CA); values are presented as means ± standard deviation. Primers used for qRT-PCR are listed in Table S5.
Western Blot Analyses
Total cell lysates (n = 16) from the CB of male (M) and female (F) pups were prepared from both the HMFA and the LMFA groups. For each of these two groups, four male CBs (each male pup from an independent dam) and four female CBs (each female pup from an independent dam), for a total eight pups were analyzed. Western blot analyses were done as previously described (Barua et al., 2014c). Targeted proteins were detected by incubating with primary antibodies overnight at 4°C (dilutions for anti-GAD1 1:500 and anti-PARK2 1:200) followed by the secondary antibody coupled with horseradish peroxidase. Densitometric evaluation of the bands was calculated by using ImageJ software (NIH) and was normalized to the densities of β-actin staining as housekeeping control. The average calculated ratios of target protein to β-actin are presented.
Results
Global DNA Methylation Patterns of the Offspring's CB from HMFA
The statistics of the mapping of the methylation profiles of pups CB's of pups from mothers supplemented with LMFA and HMFA are given in Table 1. The ratio of mapped reads to total reads ranged from 40 to 46.34% in male pups and from 56.87 to 61.33% in female pups from the LMFA and HMFA groups, respectively, with an average depth of CpG coverage (12x–13x) in males and (10x–16x) in females. The bisulfate conversion rates were approximately 98% for all the samples. Analysis of the global methylation profile revealed that 19% of the CpG sites were differentially methylated in pups from the HMFA group in comparison to that of the LMFA group in both male and female pups (n = 40.376 for male and n = 44.974 for female). Moreover, the majority of differentially methylated regions (DMRs) were in the intergenic or introns, whereas 19–20% were in exons, and 8–9% were in the promoter regions in the CpG-island sequence (Figures S1A,B). Similar to the CpG sites, the distribution of DMRs in the non-CpG sites revealed that the majority of DMRs were in the intergenic or introns, whereas 9–10% in exons, and 15–23% in the promoter regions (Figure S2). The distribution of methylation ratios and the Pearson's correlation coefficient for the corresponding CpG and non-CpG sites of male and female pups are shown in Figures S3, S4. The hexbin plot (Figures S5–S7) shows the distribution of overlapped CpG and non-CpG sites (P < 0.05) in male and female pups as a result of HMFA. The result of global DNA methylation profiling indicated that HMFA altered the methylation pattern of the epigenome of the offspring's CB.
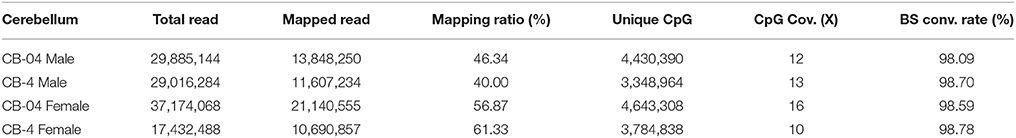
Table 1. Descriptive statistics of the mapping of methylation profile of pups' cerebellums from mothers supplemented with FA at 0.4 (LMFA) and 4 mg/kg (HMFA).
Maternal FA Alters DNA Methylation Status of Several Genes in the CpG and Non-CpG Sites in Offspring's CB
DNA methylation analysis at the single base level revealed that HMFA resulted in significant alterations in the methylation level of several genes. The alterations of methylation level were found in both CpG and non-CpG (CHG, CHH) sites throughout the entire genome in both male and female pups from HMFA group (Figures S1, S2). Such alterations were evident both in promoter and gene body regions and resulted in either hyper-methylation or hypo-methylation (P ≤ 0.05) in pups supplemented with HMFA. Multiple testing corrections revealed that HMFA resulted in hyper-methylation (Table 2) of several genes that modulate neuronal pathways in male pups (Atp1a1, Kcnq4, Bre, Scnn1a, Celsr3, Kcnk10), whereas the methylation of a gene of the neurexin gene family (Nrxn2) was hyper-methylated in female pups. Further analysis of methylation data revealed significant hypo-methylation (Table 3) of several genes in both male and female pups from the HMFA group. Several genes related to intellectual disability (Dcaf17, Myst4, Park2, Rbfox1) were found to be hypo-methylated in male pups, and genes (Pfn1, Cntnap1, Drp2) related to normal function of the nervous system were hypo-methylated in female pups from the HMFA group. Transcriptional factors in male pups (Gtf2i, Nr3c2) and in female pups (Foxl2, Rfx1) were hyper-methylated, whereas transcriptional factor Rn45s was hypo-methylated in female pups. Of note, the methylation level of sidekick cell adhesion molecule 1 (Sdk1) was hyper-methylated in both male and female pups from the HMFA group.
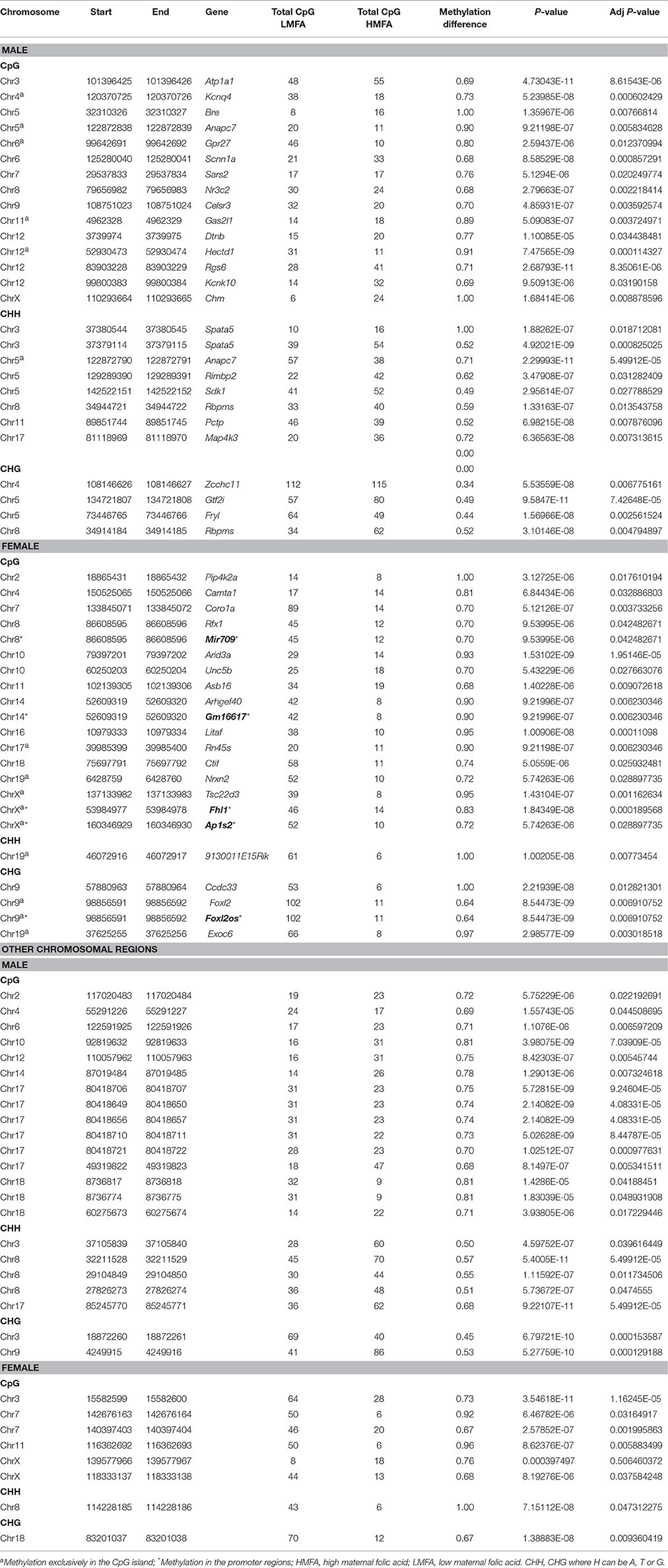
Table 2. List of hypermethylated CpG/CHH/CHG sites in the gene body/promoter/other chromosomal region of genes from high maternal folic acid diet that were significantly altered after multiple testing corrections.
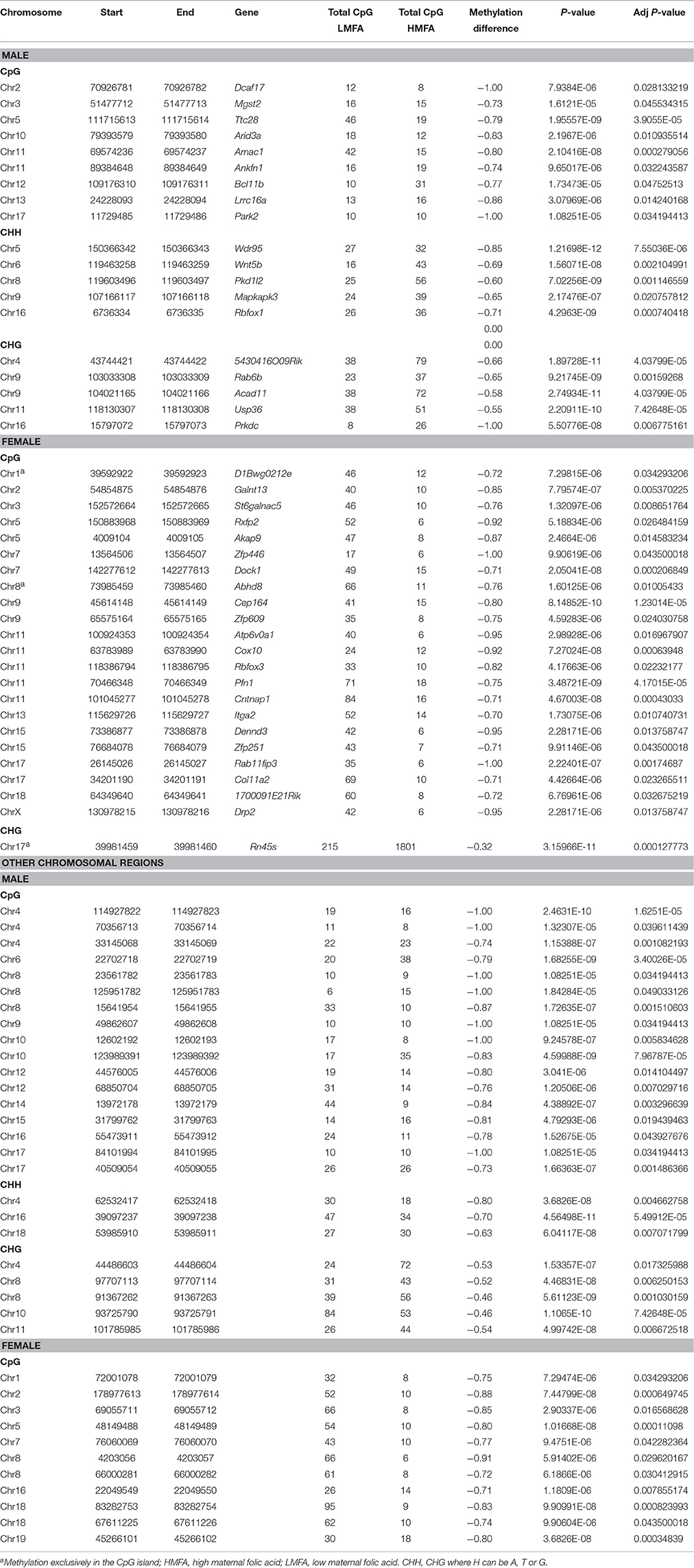
Table 3. List of hypomethylated CpG/CHH/CHG sites in the gene body/promoter/other chromosomal region of genes from high maternal folic acid diet that were significantly altered after multiple testing corrections.
Further analysis of all P ≤ 0.05 differential methylation sites without correction for multiple testing identified several genes that were hyper-methylated or hypo-methylated in male and female pups, in both the CpG and non-CpG sites (Tables S1–S4). The methylation level of genes associated with autism spectrum disorder (Plxna4, Arid1b, Kdm4c, Runx1, Accn1, Aff2, Chd9, Cntnap2, Grip1, Grin2b, and Mid1); imprinted genes (Peg12, Tsix); transcriptional factors (Ebf2, Lmx1b, Runx3, Sox13, and Mef2a) that modulate neurogenesis; and genes related to neurodevelopment (Grik4, Ntrk2, Sgk1, Cacna1a, Gabrg3, Erbb3, and Gfra1) were found to be altered in CB of both male and female pups from the HMFA group. Our findings suggest that maternal diet during gestation, specifically HMFA, can modulate the methylation profile of several genes, including those involved in neural development in the gene bodies and the promoter, CpG, and non-CpG sites in the CB of pups' brains.
Maternal FA Alters Expression of Several Differentially Methylated Genes in Offspring's CB
To extend our findings, we then analyzed whether HMFA induced changes in the overall methylation profile in offspring's CB correlates with the alterations in gene expression. Quantitative RT-PCR analysis of several genes that showed differential methylation (P ≤ 0.05) exhibited variations in expression in pups from the HMFA group. Genes in male pups from the HMFA group that exhibited significant hyper-methylation after multiple testing corrections in CpG sites (Atp1a1, Bre, Celsr3, Kcnq4) and Gtf2i in CHG sites did not exhibit any changes in expression level compared to pups from the LMFA group (Figure 1A). In contrast, expression of Kcnk10, a gene that encodes protein from the potassium channel family and is hyper-methylated in CpG sites, was significantly down-regulated in male pups from the HMFA group (Figure 1A). In female pups from the HMFA group, genes that exhibited hyper-methylation in CpG sites after multiple corrections (Arid3a, Nrxn2, Unc5b) exhibited no significant change in expression, whereas the expression of Coro1a was up-regulated significantly (Figure 1B). Similarly, analysis of expression of genes that were hypo-methylated at CpG sites by HMFA revealed significant up-regulation in expression of Arid3a in male pups whereas the expression of several other genes in male (Dcaf17, Park2, Rbfox1) and in female (Col11a2, Cox10, Drp2, Itga2, Pfn1, and Rxfp2) pups did not exhibit any significant changes (Figures 1C,D).
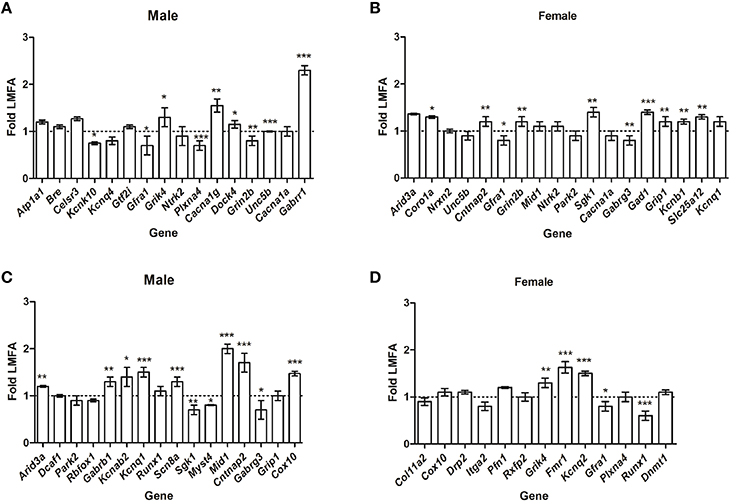
Figure 1. Relative expression of the genes that exhibited hyper-methylation (A,B) and hypo-methylation (C,D). The results were normalized to Hprt transcript expression and were expressed as relative values in comparison with corresponding transcripts from low maternal folic acid (LMFA). Results represent mean ± standard deviation (SD); asterisks denote statistically significant change (*P < 0.05, **P < 0.01, ***P < 0.001).
To further reveal the impact of maternal FA, we next assessed whether HMFA-induced changes of methylation (P ≤ 0.05, without corrections) alter the expression of genes related to neuronal pathways (Figures 1A–D). Our results showed significant down-regulation in the expression of genes (Gfra1, Plxna4, and Grin2b), up-regulation in the expression of genes (Grik4, Cacna1g, Dock4, Unc5b, and Gabrr1), and no changes in the expression level of genes (Ntrk2, Cacna1a) in male pups that exhibited hyper-methylation at CpG or non-CpG sites (Figure 1A). In female pups from the HMFA group, several genes that were hyper-methylated at CpG or non-CpG sites also exhibited changes in expression level (Figure 1B).
Although the expression of some genes (Gfra1, Gabrg3) was down-regulated and of other genes (Cntnap2, Grin2b, Sgk1, Gad1, Grip1, Kcnb1, and Slc25a12) was up-regulated, the expression of some other genes (Mid1, Ntrk2, Park2, Cacna1a, Park2, and Kcnq1) was unaltered. Similarly, expression analysis of genes that exhibited hypo-methylation (P ≤ 0.05, without corrections) in CpG and non-CpG sites revealed down-regulation in the expression of genes in male (Sgk1, Myst4, Gabrg3) and in female (Gfra1, Runx1) pups (Figures 1C,D). HMFA resulted in up-regulation in the expression of some genes in male (Gabrb1, Kcnab2, Kcnq1, Scn8a, Mid1, Cntnap2, Cox10) and in female (Grik4, Fmr1, Kcnq2) pups, whereas the expression of other genes in male (Runx1, Grip1) and in female (Plxna4, Dnmt1) pups remained unaltered (Figures 1C,D).
Maternal FA Down-Regulates Expression of Gad1p in Offspring's CB
Next, we analyzed whether the expression of two genes (Gad1, hyper-methylated at CpG in female pups and Park2, hyper- and hypo-methylated at both CpG and non-CpG sites in male and female pups) that play a role in neuronal pathways including autism are altered at the protein level. Western blot analysis and densitometric quantification of the protein levels revealed that the expression of Gad1p (Figure 2A) in male pups remains unaltered; however, the expression of Gad1p in female pups from the HMFA group was significantly reduced (Figure 2B). In contrast, the expression of Park2p remained unchanged in both male and female pups from the HMFA group (Figures 3A,B). These results show that HMFA during gestation can modulate the expression of genes in offspring's CB and that such impact can be gender-specific.
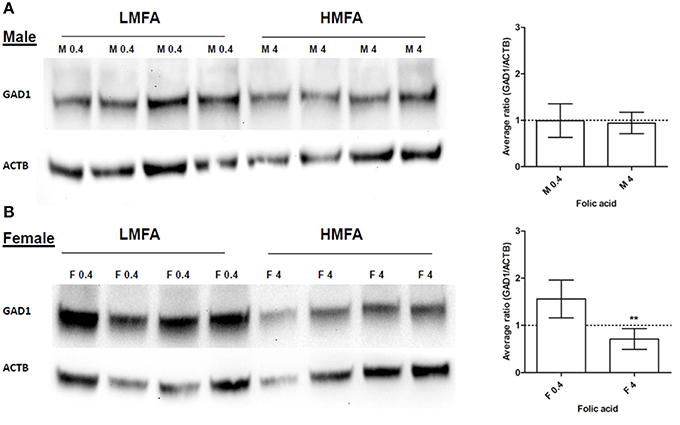
Figure 2. Western blot analysis showing the expression of Gad1p in the cerebellum of (A) male (M) and (B) female (F) pups from LMFA (0.4 mg/kg diet) and HMFA (4 mg/kg diet) groups. The left panel shows one representative blot, and the right panel shows the mean densitometric evaluation. The error bar ± SD represents the inter-variability among independent samples (n = 4). Asterisks denote statistically significant change (**P < 0.05) by unpaired T-test.
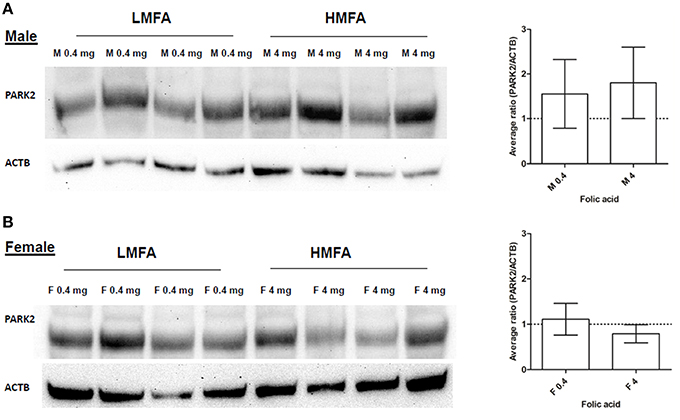
Figure 3. Western blot analysis showing the expression of Park2p in the CB of (A) male and (B) female pups from LMFA (0.4 mg/kg diet) and HMFA (4 mg/kg diet). The left panel represents one representative blot, and the right panel shows the mean densitometric evaluation. The error bar ± SD represents the inter-variability among independent samples (n = 4).
Maternal FA Modulates Sex-Specific Alterations in Expression of Genes in the Offspring's CB
To gain insight into whether HMFA-induced changes in methylation profiles modulate sex-specific alterations in the expression of genes, we analyzed the expression of several genes that exhibited alterations in methylation in the opposite sex. In male pups, the expression of the genes Coro1a, Gad1, Kcnb1, and Fmr1 (which exhibited changes in methylation and gene expression in female pups) was not altered (Figure 4A); in contrast, the expression of Drp2, Itga2, and Pfn1 was altered in male pups from the HMFA group, although it exhibited no alterations in methylation profile in comparison to the LMFA group. However, the expression of the genes Nrxn2, Col11a2, Dnmt1, and Rxfp2 was unaltered; while Slc25a12, and Kcnq2 was altered in pups of both genders from the HMFA group. Similarly, in female pups, the expression of genes (Figure 4B) Kcnk10, Cacna1g, Dock4, Gabrr1, Gabrb1, Kcnab2, Scn8a, and Myst4 (which exhibited changes in methylation and gene expression in male pups) was not altered, in contrast to the expression of Gtf2i being altered in female pups from the HMFA group, although it exhibited no alterations in methylation profile in comparison to the LFMA group. However, expression of the genes Atp1a1, Bre, Celsr3, Kcnq4, Dcaf1, and Rbfox1 was unaltered in pups of both genders from the HMFA group.
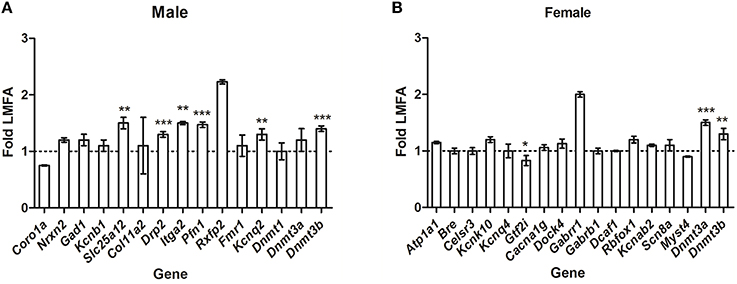
Figure 4. Quantitative real time reverse transcription-polymerase chain reaction (qRT-PCR) showing relative expression of the transcripts of genes in (A) male pups and (B) female pups from the HMFA group that exhibited no alterations in the methylation profile in promoter and gene body in the cerebellum compared with the LMFA. The results were normalized to Hprt transcript expression and were expressed as relative values in comparison with corresponding transcripts from LMFA. Results represent mean ± standard deviation (SD); asterisks denote statistically significant change (*P < 0.05, **P < 0.01, ***P < 0.001).
Discussion
DNA methylation in the early embryo has long been associated with the maintenance of genomic integrity, with its crucial role in modulating gene expression and genomic imprinting (Farthing et al., 2008; Messerschmidt et al., 2014). Epigenetic alterations of effectors in the developing brains of mammals can impair normal development and result in neurodevelopmental disorders (Schanen, 2006; LaSalle and Yasui, 2009; LaSalle, 2011). In this study, we performed genome-wide methylation analysis to test the hypothesis that exposure to HMFA during gestation induces epigenetic changes in the mouse offspring's CB and contributes to alterations in gene expression. We found distinct alterations in the methylation profile in The CB of offspring from mothers that received HMFA in comparison to LMFA. Such changes were widespread throughout the genome and were evident in both promoter and intragenic regions. These data establish that in addition to its role in preventing NTD's, FA during gestation can induce epigenetic changes in the brain.
As a pteroylmonoglutamate, FA cannot breach the blood brain barrier, with only methylfolate appearing in the cerebrospinal fluid (CSF). After FA is consumed, metabolically it is chemically reduced to tetrahydrofolate by the enzyme dihydrofolate reductase (DHFR) in the liver, where it collects a formyl group, which is reduced to methyl to form 5-methyltetrahydrofolic acid (Barua et al., 2014c). However, excess supplementation with FA may overwhelm this process and possibly saturate the DHFR activity. Perhaps the benefit of high doses of FA may be limited by saturation of DHFR, as studies have shown that DHFR activity in human liver is extremely slow and variable (Bailey and Ayling, 2009). We speculate that mechanistically one possibility is that excess FA in blood may compete to bind with the folate receptor and inhibit methyl-tetrahydrofoalte transport, resulting in changes in brain homeostasis (Smith et al., 2008). Alternatively, elevated methylfolate via maternal metabolism may concentrate against a gradient in the CSF by the choroid plexus and thus interfere with the regulatory functions of the brain. Because the developing brain is potentially vulnerable to maternal nutrient availability (Antonow-Schlorke et al., 2011), such exposure to HMFA may impact the brain function of offspring with alterations in methylation and gene expression, as is evident from the data of our study.
To date, much progress has been made to characterize the dynamic changes of DNA methylation in CpG sites and its role in functional outcome. However, the research to elucidate the precise mechanism by which methylation in non-CpG sites affects gene expression and its role in functional outcomes is still in its infancy. Studies have shown that Dnmts have sequence specificity and can influence the methylation in non-CpG sites (Ramsahoye et al., 2000; Gowher and Jeltsch, 2001; Mund et al., 2004). Similar to our previous findings (Barua et al., 2014b), the data generated from this study suggest that alterations in FA intake during gestation can modulate the methylation profile of both CpG and non-CpG sites in the brains of offspring. Our results revealed that HMFA increases the expression levels of Dnmt3b in male pups and of Dnmt3a, Dnmt3b in female pups (Figure 4). Such evidence supports the idea that HMFA may increase the activity of Dnmts which can modulate neuronal differentiation (Luo et al., 2013); perhaps neuronal cells have a higher variation in DNA methylation than non-neuronal cells (Zhang et al., 2010; Melka et al., 2014). This study further establishes the theory of variations in methylation of neuronal cells, with distinct methylation patterns of CpG and non-CpG sites throughout the genome, including promoters and intergenic and intragenic regions in the CB of pups from the HMFA group in comparison to the LMFA group. This finding is in agreement with our previous reports and suggests that HMFA during gestation can induce unbiased distribution of methylation in the brains of offspring. Such differences in the methylation pattern in the intragenic region may have different correlations to the expression of transcriptional levels, as is evident from our study. Indeed, studies in mammalian cells and human brains correlated changes in methylation of intragenic regions with both up-regulation and down-regulation of transcripts (Ball et al., 2009; Rauch et al., 2009; Maunakea et al., 2010).
Our data also highlighted that several genes that play a role in neuronal pathways exhibited alterations in expression in the CB of offspring. For example, pups from HMFA exhibited significant changes in the expression of several voltage-dependent ion channels (Cacna1g, Scna8a, Kcnk10, Kcnab2, Kcnq1, Kcnq2), which may result in modulation in neuronal synaptic transmission and excitability (Vacher et al., 2008).
Similarly, several genes (Grik4, Gabrr1, Gabrg3, Gabrb1, and Gad1) involved in GABAergic (gamma-aminobutyric acid) or glutamatergic synaptic transmissions exhibited alterations in expression in pups from HMFA. Studies have reported that GABAergic interneurons play a central role in the modulation of excitatory output in the brain and have been documented with a wide variety of psychiatric diseases, including mood disorders, schizophrenia and autism (Fatemi and Folsom, 2014). Biochemical studies have revealed that the expressions of GAD65/67 are decreased in the CB of subjects with developmental disorders such as autism and schizophrenia (Bullock et al., 2008; Blatt and Fatemi, 2011). In the current study, we found that HMFA decreased the expression of Gad1p in female pups, where the expression in the male pups remains unaltered.
Such alterations in the expression of Gad1p may induce alterations in learning and social behavior, as studies with Gad67-deficient mice have been reported to exhibit behavioral abnormalities and reduced sociability (Sandhu et al., 2014; Zhang et al., 2014). Indeed, our previous studies showed that gestational and post-weaning HMFA resulted in changes in behavior, with an increase in ultrasonic vocalization as neonates (Barua et al., 2014a). Such findings indicate that maternal nutritional status, specifically variations in the methyl diet, can induce changes in DNA methylation and thus modulate the expression of genes involved in neuronal pathways and may impact the behavioral outcomes.
Off note, neuronal cells are found to exhibit more inter-individual variations compare to non-neuronal cells (Iwamoto et al., 2011). Moreover, studies with post-mortem brain samples from patient with major depression have shown significant cell specific epigenetic variation in between brain regions (Guintivano et al., 2013). One of the limitations of our current study is that we used the whole cerebellum from pups for methylation and expression analysis; thus some of the alterations of DNA methylation profile may results due to cellular heterogeneity.
Previous studies have reported sexual dimorphism and asymmetry in animal and human cerebellum (Ramirez and Jimenez, 2002; Fan et al., 2010). It is also reported that perinatal exposure to chemical and physical perturbations impact differential neurodevelopment and behavioral effects in males and females (Nguon et al., 2005); other studies with mouse placenta have shown that maternal diet impacts gene expression differentially in males and females (Mao et al., 2010; Gabory et al., 2012). Our data also shows a significant bias between genders in methylation and expression of genes in the offspring CB as a result of HMFA. This finding is congruent with our previous report that HMFA induces gender-specific methylation changes in the brains of offspring (Barua et al., 2014b). We speculate that such differential methylation could results from alterations in the uterine environment because of excess FA, and biased sensitivity to nutritional perturbations resulted from gender specific distribution of specific receptors and methylation of imprinted genes.
Conclusions
Given the role of maternal FA in the methylation pathway, it remains an open questions how such epigenetic modifications impact the brain and behavior of offspring. Our findings support the idea that epigenetic variations may have distinct sex biased functional consequences to certain neuropsychiatric disorders. It further highlights the relevance of studying both sexes in both experimental model and clinical studies to study the epigenetic impact of maternal diet.
In summary, a key finding of this study is that FA during gestation, aside from its role in preventing NTDs, can induce alterations in the methylation of several genes in both CpG and non-CpG regions in the offspring's CB, and such alterations in methylation are gene- and sex- specific. Such changes in DNA methylation during gestation can induce alterations in gene regulatory structures, and given the role the CB in regulation of higher order functions, including motor function and cognition (Schmahmann, 2004; Tavano et al., 2007), such changes may modulate the functional outcome of neurologic and psychiatric diseases.
Author Contributions
WB and MJ conceived the experiments; SB, WB, and MJ designed the experiments; SB, SK, and MJ performed the experiments; SB and MJ analyzed the data; SB, SK, WB, and MJ contributed reagents/materials/analysis tools; SB wrote the paper, and WB and MJ critically revised the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer SK and handling Editor declared their shared affiliation, and the handling Editor states that the process nevertheless met the standards of a fair and objective review.
Acknowledgments
Financial support from the March of Dimes Foundation (12-FY12-170) and the New York State Office for People with Developmental Disabilities is gratefully acknowledged. We acknowledge Ms. Maureen Marlow for help with editorial corrections with the manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fnins.2016.00168
Figure S1. Distribution of differentially methylated sites in CpG island sequences. (A) Male; low maternal folic acid (LMFA) vs. high maternal folic acid (HMFA). (B) Female LMFA vs. HMFA.
Figure S2. Distribution of differentially methylated sites in non-CpG (CHG/CHH) sites of cerebellum. (A) Male low maternal folic acid (LMFA) vs. high maternal folic acid (HMFA). (B) Female LMFA vs. HMFA.
Figure S3. Scatter plot representing the distribution of the methylation ratio for corresponding sites (A) CpG, (B) CHG, and (C) CHH of LMFA vs. HMFA of male pups. Pearson's correlation coefficient is denoted in the center of each scatter plot.
Figure S4. Scatter plot representing the distribution of the methylation ratio for corresponding sites (A) CpG, (B) CHG, and (C) CHH of LMFA vs. HMFA of female pups. Pearson's correlation coefficient is denoted in the center of each scatter plot.
Figures S5–S7. Hexbin plot representing the overlapped sites in the CpG (n = 2715), CHG (n = 111), and CHH (n = 192) regions between male and female pups from the HMFA group in comparison with the LMFA group from all the significant (P < 0.05) differential methylation sites. Each dot on hexbin plot is one of the overlapped sites. The colors blue, green, yellow and red represent the dot density from lower to higher order, in accordance with the prevalence of the overlapping sites.
Table S1. Genes in male offspring from high maternal folic acid (HMFA) diet, which exhibited hyper methylation (P < 0.05) compared with genes in male offspring from low maternal folic acid (LMFA) in CpG/CHG/CHH contexts.
Table S2. Genes in male offspring from high maternal folic acid (HMFA) diet, which exhibited hypo methylation (P < 0.05) compared with genes in male offspring from low maternal folic acid (LMFA) in CpG/CHG/CHH contexts.
Table S3. Genes in male offspring from high maternal folic acid (HMFA) diet, which exhibited hyper methylation (P < 0.05) compared with genes in female offspring from low maternal folic acid (LMFA) in CpG/CHG/CHH contexts.
Table S4. Genes in male offspring from high maternal folic acid (HMFA) diet, which exhibited hypo methylation (P < 0.05) compared with genes in female offspring from low maternal folic acid (LMFA) in CpG/CHG/CHH contexts.
Table S5. List of primers used in this study.
Abbreviations
FA, Folic acid; NTDs, Neural tube defects; LMFA, Low maternal folic acid; HMFA, High maternal folic acid; 5 mC, 5-methylcytosine; CB, Cerebellum.
References
Antonow-Schlorke, I., Schwab, M., Cox, L. A., Li, C., Stuchlik, K., Witte, O. W., et al. (2011). Vulnerability of the fetal primate brain to moderate reduction in maternal global nutrient availability. Proc. Natl. Acad. Sci. U.S.A. 108, 3011–3016. doi: 10.1073/pnas.1009838108
Bailey, S. W., and Ayling, J. E. (2009). The extremely slow and variable activity of dihydrofolate reductase in human liver and its implications for high folic acid intake. Proc. Natl. Acad. Sci. U.S.A. 106, 15424–15429. doi: 10.1073/pnas.0902072106
Ball, M. P., Li, J. B., Gao, Y., Lee, J. H., LeProust, E. M., Park, I. H., et al. (2009). Targeted and genome-scale strategies reveal gene-body methylation signatures in human cells. Nat. Biotechnol. 27, 361–368. doi: 10.1038/nbt.1533
Barua, S., Chadman, K. K., Kuizon, S., Buenaventura, D., Stapley, N. W., Ruocco, F., et al. (2014a). Increasing maternal or post-weaning folic acid alters gene expression and moderately changes behavior in the offspring. PLoS ONE 9:e101674. doi: 10.1371/journal.pone.0101674
Barua, S., and Junaid, M. A. (2015). Lifestyle, pregnancy and epigenetic effects. Epigenomics 7, 85–102. doi: 10.2217/epi.14.71
Barua, S., Kuizon, S., Chadman, K. K., Flory, M. J., Brown, W. T., and Junaid, M. A. (2014b). Single-base resolution of mouse offspring brain methylome reveals epigenome modifications caused by gestational folic acid. Epigenetics. Chromatin. 7:3. doi: 10.1186/1756-8935-7-3
Barua, S., Kuizon, S., and Junaid, M. A. (2014c). Folic acid supplementation in pregnancy and implications in health and disease. J. Biomed. Sci. 21:77. doi: 10.1186/s12929-014-0077-z
Bean, L. J., Allen, E. G., Tinker, S. W., Hollis, N. D., Locke, A. E., Druschel, C., et al. (2011). Lack of maternal folic acid supplementation is associated with heart defects in Down syndrome: a report from the National Down Syndrome Project. Birth Defects Res. A Clin. Mol. Teratol. 91, 885–893. doi: 10.1002/bdra.22848
Blatt, G. J., and Fatemi, S. H. (2011). Alterations in GABAergic biomarkers in the autism brain: research findings and clinical implications. Anat. Rec. 294, 1646–1652. doi: 10.1002/ar.21252
Boeke, C. E., Baccarelli, A., Kleinman, K. P., Burris, H. H., Litonjua, A. A., Rifas-Shiman, S. L., et al. (2012). Gestational intake of methyl donors and global LINE-1 DNA methylation in maternal and cord blood: prospective results from a folate-replete population. Epigenetics 7, 253–260. doi: 10.4161/epi.7.3.19082
Bullock, W. M., Cardon, K., Bustillo, J., Roberts, R. C., and Perrone-Bizzozero, N. I. (2008). Altered expression of genes involved in GABAergic transmission and neuromodulation of granule cell activity in the cerebellum of schizophrenia patients. Am. J. Psychiatry 165, 1594–1603. doi: 10.1176/appi.ajp.2008.07121845
Chen, C., Zhang, C., Cheng, L., Reilly, J. L., Bishop, J. R., Sweeney, J. A., et al. (2014). Correlation between DNA methylation and gene expression in the brains of patients with bipolar disorder and schizophrenia. Bipolar. Disord. 16, 790–799. doi: 10.1111/bdi.12255
Cooney, C. A., Dave, A. A., and Wolff, G. L. (2002). Maternal methyl supplements in mice affect epigenetic variation and DNA methylation of offspring. J. Nutr. 132, 2393S–2400S.
Dean, W., Santos, F., Stojkovic, M., Zakhartchenko, V., Walter, J., Wolf, E., et al. (2001). Conservation of methylation reprogramming in mammalian development: aberrant reprogramming in cloned embryos. Proc. Natl. Acad. Sci. U.S.A. 98, 13734–13738. doi: 10.1073/pnas.241522698
Fan, L., Tang, Y., Sun, B., Gong, G., Chen, Z. J., Lin, X., et al. (2010). Sexual dimorphism and asymmetry in human cerebellum: an MRI-based morphometric study. Brain Res. 1353, 60–73. doi: 10.1016/j.brainres.2010.07.031
Farthing, C. R., Ficz, G., Ng, R. K., Chan, C. F., Andrews, S., Dean, W., et al. (2008). Global mapping of DNA methylation in mouse promoters reveals epigenetic reprogramming of pluripotency genes. PLoS. Genet. 4:e1000116. doi: 10.1371/journal.pgen.1000116
Fatemi, S. H., and Folsom, T. D. (2014). GABA receptor subunit distribution and FMRP-mGluR5 signaling abnormalities in the cerebellum of subjects with schizophrenia, mood disorders, and autism. Schizophr. Res. 167, 42–56. doi: 10.1016/j.schres.2014.10.010
Gabory, A., Ferry, L., Fajardy, I., Jouneau, L., Gothie, J. D., Vige, A., et al. (2012). Maternal diets trigger sex-specific divergent trajectories of gene expression and epigenetic systems in mouse placenta. PLoS ONE 7:e47986. doi: 10.1371/journal.pone.0047986
Gore, A. C., Walker, D. M., Zama, A. M., Armenti, A. E., and Uzumcu, M. (2011). Early life exposure to endocrine-disrupting chemicals causes lifelong molecular reprogramming of the hypothalamus and premature reproductive aging. Mol. Endocrinol. 25, 2157–2168. doi: 10.1210/me.2011-1210
Gowher, H., and Jeltsch, A. (2001). Enzymatic properties of recombinant Dnmt3a DNA methyltransferase from mouse: the enzyme modifies DNA in a non-processive manner and also methylates non-CpG [correction of non-CpA] sites. J. Mol. Biol. 309, 1201–1208. doi: 10.1006/jmbi.2001.4710
Greenop, K. R., Miller, M., de Klerk, N. H., Scott, R. J., Attia, J., Ashton, L. J., et al. (2014). Maternal dietary intake of folate and vitamins b6 and B12 during pregnancy and risk of childhood brain tumors. Nutr. Cancer 66, 800–809. doi: 10.1080/01635581.2014.916326
Guintivano, J., Aryee, M. J., and Kaminsky, Z. A. (2013). A cell epigenotype specific model for the correction of brain cellular heterogeneity bias and its application to age, brain region and major depression. Epigenetics 8, 290–302. doi: 10.4161/epi.23924
Iwamoto, K., Bundo, M., Ueda, J., Oldham, M. C., Ukai, W., Hashimoto, E., et al. (2011). Neurons show distinctive DNA methylation profile and higher interindividual variations compared with non-neurons. Genome Res. 21, 688–696. doi: 10.1101/gr.112755.110
Jaenisch, R., and Bird, A. (2003). Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat. Genet. 33(suppl.), 245–254. doi: 10.1038/ng1089
LaSalle, J. M. (2011). A genomic point-of-view on environmental factors influencing the human brain methylome. Epigenetics 6, 862–869. doi: 10.4161/epi.6.7.16353
LaSalle, J. M., and Yasui, D. H. (2009). Evolving role of MeCP2 in Rett syndrome and autism. Epigenomics 1, 119–130. doi: 10.2217/epi.09.13
Lei, H., Oh, S. P., Okano, M., Juttermann, R., Goss, K. A., Jaenisch, R., et al. (1996). De novo DNA cytosine methyltransferase activities in mouse embryonic stem cells. Development 122, 3195–3205.
Leiner, H. C., Leiner, A. L., and Dow, R. S. (1991). The human cerebro-cerebellar system: its computing, cognitive, and language skills. Behav. Brain Res. 44, 113–128. doi: 10.1016/S0166-4328(05)80016-6
Li, C. C., Young, P. E., Maloney, C. A., Eaton, S. A., Cowley, M. J., Buckland, M. E., et al. (2013). Maternal obesity and diabetes induces latent metabolic defects and widespread epigenetic changes in isogenic mice. Epigenetics 8, 602–611. doi: 10.4161/epi.24656
Li, E., Bestor, T. H., and Jaenisch, R. (1992). Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell 69, 915–926. doi: 10.1016/0092-8674(92)90611-F
Luo, S., Zhang, X., Yu, M., Yan, H., Liu, H., Wilson, J. X., et al. (2013). Folic acid acts through DNA methyltransferases to induce the differentiation of neural stem cells into neurons. Cell Biochem. Biophys. 66, 559–566. doi: 10.1007/s12013-012-9503-6
Mao, J., Zhang, X., Sieli, P. T., Falduto, M. T., Torres, K. E., and Rosenfeld, C. S. (2010). Contrasting effects of different maternal diets on sexually dimorphic gene expression in the murine placenta. Proc. Natl. Acad. Sci. U.S.A. 107, 5557–5562. doi: 10.1073/pnas.1000440107
Martin, L. A., Goldowitz, D., and Mittleman, G. (2003). The cerebellum and spatial ability: dissection of motor and cognitive components with a mouse model system. Eur. J. Neurosci. 18, 2002–2010. doi: 10.1046/j.1460-9568.2003.02921.x
Marzban, H., Del Bigio, M. R., Alizadeh, J., Ghavami, S., Zachariah, R. M., and Rastegar, M. (2014). Cellular commitment in the developing cerebellum. Front. Cell Neurosci. 8:450. doi: 10.3389/fncel.2014.00450
Maunakea, A. K., Nagarajan, R. P., Bilenky, M., Ballinger, T. J., D'souza, C., Fouse, S. D., et al. (2010). Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature 466, 253–257. doi: 10.1038/nature09165
Melka, M. G., Laufer, B. I., McDonald, P., Castellani, C. A., Rajakumar, N., O'Reilly, R., et al. (2014). The effects of olanzapine on genome-wide DNA methylation in the hippocampus and cerebellum. Clin. Epigenet. 6:1. doi: 10.1186/1868-7083-6-1
Messerschmidt, D. M., Knowles, B. B., and Solter, D. (2014). DNA methylation dynamics during epigenetic reprogramming in the germline and preimplantation embryos. Genes Dev. 28, 812–828. doi: 10.1101/gad.234294.113
Mund, C., Musch, T., Strodicke, M., Assmann, B., Li, E., and Lyko, F. (2004). Comparative analysis of DNA methylation patterns in transgenic Drosophila overexpressing mouse DNA methyltransferases. Biochem. J. 378, 763–768. doi: 10.1042/bj20031567
Nguon, K., Ladd, B., Baxter, M. G., and Sajdel-Sulkowska, E. M. (2005). Sexual dimorphism in cerebellar structure, function, and response to environmental perturbations. Prog. Brain Res. 148, 341–351. doi: 10.1016/S0079-6123(04)48027-3
Oberlander, T. F., Weinberg, J., Papsdorf, M., Grunau, R., Misri, S., and Devlin, A. M. (2008). Prenatal exposure to maternal depression, neonatal methylation of human glucocorticoid receptor gene (NR3C1) and infant cortisol stress responses. Epigenetics 3, 97–106. doi: 10.4161/epi.3.2.6034
Okano, M., Bell, D. W., Haber, D. A., and Li, E. (1999). DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 99, 247–257. doi: 10.1016/S0092-8674(00)81656-6
O'Neill, R. J., Vrana, P. B., and Rosenfeld, C. S. (2014). Maternal methyl supplemented diets and effects on offspring health. Front. Genet. 5:289. doi: 10.3389/fgene.2014.00289
Perkins, A., Lehmann, C., Lawrence, R. C., and Kelly, S. J. (2013). Alcohol exposure during development: impact on the epigenome. Int. J. Dev. Neurosci. 31, 391–397. doi: 10.1016/j.ijdevneu.2013.03.010
Ramirez, O., and Jimenez, E. (2002). Sexual dimorphism in rat cerebrum and cerebellum: different patterns of catalytically active creatine kinase isoenzymes during postnatal development and aging. Int. J. Dev. Neurosci. 20, 627–639. doi: 10.1016/S0736-5748(02)00102-8
Ramsahoye, B. H., Biniszkiewicz, D., Lyko, F., Clark, V., Bird, A. P., and Jaenisch, R. (2000). Non-CpG methylation is prevalent in embryonic stem cells and may be mediated by DNA methyltransferase 3a. Proc. Natl. Acad. Sci. U.S.A. 97, 5237–5242. doi: 10.1073/pnas.97.10.5237
Rauch, T. A., Wu, X., Zhong, X., Riggs, A. D., and Pfeifer, G. P. (2009). A human B cell methylome at 100-base pair resolution. Proc. Natl. Acad. Sci. U.S.A. 106, 671–678. doi: 10.1073/pnas.0812399106
Sandhu, K. V., Lang, D., Muller, B., Nullmeier, S., Yanagawa, Y., Schwegler, H., et al. (2014). Glutamic acid decarboxylase 67 haplodeficiency impairs social behavior in mice. Genes Brain Behav. 13, 439–450. doi: 10.1111/gbb.12131
Schanen, N. C. (2006). Epigenetics of autism spectrum disorders. Hum. Mol. Genet. 15 Spec No 2, R138–R150. doi: 10.1093/hmg/ddl213
Schmahmann, J. D. (2004). Disorders of the cerebellum: ataxia, dysmetria of thought, and the cerebellar cognitive affective syndrome. J. Neuropsychiatry Clin. Neurosci. 16, 367–378. doi: 10.1176/jnp.16.3.367
Schmahmann, J. D., and Caplan, D. (2006). Cognition, emotion and the cerebellum. Brain 129, 290–292. doi: 10.1093/brain/awh729
Shpyleva, S., Ivanovsky, S., de Conti, A., Melnyk, S., Tryndyak, V., Beland, F. A., et al. (2014). Cerebellar oxidative DNA damage and altered DNA methylation in the BTBR T+tf/J mouse model of autism and similarities with human post mortem cerebellum. PLoS ONE 9:e113712. doi: 10.1371/journal.pone.0113712
Smith, A. D., Kim, Y. I., and Refsum, H. (2008). Is folic acid good for everyone? Am. J. Clin. Nutr. 87, 517–533.
Stein, A. D., Pierik, F. H., Verrips, G. H., Susser, E. S., and Lumey, L. H. (2009). Maternal exposure to the Dutch famine before conception and during pregnancy: quality of life and depressive symptoms in adult offspring. Epidemiology 20, 909–915. doi: 10.1097/EDE.0b013e3181b5f227
Suter, M., Abramovici, A., and Aagaard-Tillery, K. (2010). Genetic and epigenetic influences associated with intrauterine growth restriction due to in utero tobacco exposure. Pediatr. Endocrinol. Rev. 8, 94–102.
Tavano, A., Grasso, R., Gagliardi, C., Triulzi, F., Bresolin, N., Fabbro, F., et al. (2007). Disorders of cognitive and affective development in cerebellar malformations. Brain 130, 2646–2660. doi: 10.1093/brain/awm201
Vacher, H., Mohapatra, D. P., and Trimmer, J. S. (2008). Localization and targeting of voltage-dependent ion channels in mammalian central neurons. Physiol. Rev. 88, 1407–1447. doi: 10.1152/physrev.00002.2008
Vanhees, K., Vonhogen, I. G., van Schooten, F. J., and Godschalk, R. W. (2014). You are what you eat, and so are your children: the impact of micronutrients on the epigenetic programming of offspring. Cell Mol. Life Sci. 71, 271–285. doi: 10.1007/s00018-013-1427-9
West, N. A., Kechris, K., and Dabelea, D. (2013). Exposure to maternal diabetes in utero and DNA methylation patterns in the offspring. Immunometabolism 1, 1–9. doi: 10.2478/immun-2013-0001
Zhang, D., Cheng, L., Badner, J. A., Chen, C., Chen, Q., Luo, W., et al. (2010). Genetic control of individual differences in gene-specific methylation in human brain. Am. J. Hum. Genet. 86, 411–419. doi: 10.1016/j.ajhg.2010.02.005
Keywords: folic acid, DNA methylation, gestational development, brain development, cerebellum, psychiatric disease
Citation: Barua S, Kuizon S, Brown WT and Junaid MA (2016) DNA Methylation Profiling at Single-Base Resolution Reveals Gestational Folic Acid Supplementation Influences the Epigenome of Mouse Offspring Cerebellum. Front. Neurosci. 10:168. doi: 10.3389/fnins.2016.00168
Received: 20 January 2016; Accepted: 04 April 2016;
Published: 03 May 2016.
Edited by:
Shaida A. Andrabi, Johns Hopkins University School of Medicine, USAReviewed by:
Jorgina Satrustegui, Universidad Autónoma de Madrid, SpainMaría A. García, Universidad de Concepción, Chile
Sung Ung Kang, Johns Hopkins University School of Medicine, USA
Copyright © 2016 Barua, Kuizon, Brown and Junaid. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mohammed A. Junaid, bW9oYW1tZWQuanVuYWlkQG9wd2RkLm55Lmdvdg==
†Present Address: Subit Barua, Department of Pathology and Cell Biology, College of Physicians and Surgeons, Columbia University, New York, USA