 Farhana Akter1,2,3*†
Farhana Akter1,2,3*† Xinming Yu1†
Xinming Yu1† Xingping Qin3
Xingping Qin3 Shun Yao4Parisa Nikrouz5
Shun Yao4Parisa Nikrouz5 Yasir Ahmed Syed6,7
Yasir Ahmed Syed6,7 Mark Kotter1
Mark Kotter1- 1Department of Clinical Neuroscience, University of Cambridge, Cambridge, United Kingdom
- 2Faculty of Arts and Sciences, Harvard University, Cambridge, MA, United States
- 3Massachusetts General Hospital Cancer Center, Harvard Medical School, Boston, MA, United States
- 4Department of Neurosurgery, Brigham and Women's Hospital, Harvard Medical School, Boston, MA, United States
- 5Maidstone and Tunbridge Wells Trust, Maidstone, United Kingdom
- 6Neuroscience and Mental Health Research Institute (NMHRI), Cathays, United Kingdom
- 7School of Bioscience, Cardiff University, The Sir Martin Evans Building, Cardiff, United Kingdom
Background: Degenerative cervical myelopathy (DCM), also known as cervical spondylotic myelopathy is the leading cause of spinal cord compression in adults. The mainstay of treatment is surgical decompression, which leads to partial recovery of symptoms, however, long term prognosis of the condition remains poor. Despite advances in treatment methods, the underlying pathobiology is not well-known. A better understanding of the disease is therefore required for the development of treatments to improve outcomes following surgery.
Objective: To systematically evaluate the pathophysiology of DCM and the mechanism underlying recovery following decompression.
Methods: A total of 13,808 published articles were identified in our systematic search of electronic databases (PUBMED, WEB OF SCIENCE). A total of 51 studies investigating the secondary injury mechanisms of DCM or physiology of recovery in animal models of disease underwent comprehensive review.
Results: Forty-seven studies addressed the pathophysiology of DCM. Majority of the studies demonstrated evidence of neuronal loss following spinal cord compression. A number of studies provided further details of structural changes in neurons such as myelin damage and axon degeneration. The mechanisms of injury to cells included direct apoptosis and increased inflammation. Only four papers investigated the pathobiological changes that occur in spinal cords following decompression. One study demonstrated evidence of axonal plasticity following decompressive surgery. Another study demonstrated ischaemic-reperfusion injury following decompression, however this phenomenon was worse when decompression was delayed.
Conclusions: In preclinical studies, the pathophysiology of DCM has been poorly studied and a number of questions remain unanswered. The physiological changes seen in the decompressed spinal cord has not been widely investigated and it is paramount that researchers investigate the decompressed spinal cord further to enable the development of therapeutic tools, to enhance recovery following surgery.
Introduction
Degenerative cervical myelopathy (DCM), previously known as cervical spondylotic myelopathy (CSM) is a degenerative condition of the spine leading to mechanical compression of the spinal cord. Despite the prevalence of this condition, the pathogenesis underlying mechanical stress induced injury is poorly understood. Interestingly, to complicate matters further, the degree of compression of the spinal cord is not related to the severity of cord compression (Boden et al., 1990; Lebl et al., 2011). DCM can occur due to both static and dynamic stressors. The former is related to stenosis of the developmental canal, intervertebral disc bulging, and hypertrophy of the ligamentum flavum. Dynamic stressors include invagination of the ligamentum flavum (Nishida et al., 2012). Histopathological analysis of DCM suggests that the disease is mainly confined to the white matter tracts with evidence of wallerian degeneration of motor axons in the lateral corticospinal tract (McMinn, 1998), leading to clinical symptoms such as spastic gait. Patients who suffer with symptoms of poor sensation, proprioception defects or sphincter disturbance typically have degeneration of the central gray matter and posterior column (Lunsford et al., 1980). The underlying mechanism of injury includes cell death, increased inflammation and myelin damage. However, the exact pathophysiological mechanisms of DCM are not fully understood, thereby limiting our ability to develop therapeutics that can be used to improve prognosis. Most patients with DCM are treated with surgery, which can halt disease progression. However, many patients relapse and the long term outcomes are poor. In this review, we discuss the findings of our systematic analysis of the available literature to enable us to understand the pathobiology of this condition during compression and also the cellular and structural changes that take place following decompression.
Objectives
To systematically review the literature on the pathophysiology of DCM and physiology of recovery following surgical decompression.
Research questions:
What is the pathophysiology of DCM?
What are the physiological and pathological changes following decompression?
Methods
An electronic search of the literature was performed by two reviewers (F.A and X.Y) to identify all studies, on the pathophysiology of DCM.
The MEDLINE (January 1955 to February 2019) AND Web Of Science (January 1900 to December 2019) databases were searched by using a sensitive search strategy (Leenaars et al., 2012) that combined medical subject headings and terms with free text words in Ovid; these terms were the following:
[(“cervical spondylotic myelopathy” OR “cervical myelopathy” OR “spinal cord compression” OR “spinal cord decompression” OR “degenerative cervical myelopathy”)] AND (“pathogenesis” OR “pathology” OR “pathobiology” OR “biology” OR “inflammation” OR “apoptosis” OR “ischemia” OR “demyelination” OR “axon loss”).
Two reviewers (F.A and X.Y) independently assessed the titles and key words of all eligible citations to determine if the studies met our inclusion criteria. If the content of a study was not obvious from the title and key words, the abstract was retrieved and evaluated by both reviewers for eligibility. In the second step, abstracts of articles that were eligible for inclusion in the study were reviewed independently. We also screened the reference lists of the included papers for further studies. Finally, the original studies of the selected articles were evaluated independently (F.A and X.Y). At any stage, disagreements were discussed and resolved in a consensus meeting with the senior author before the next step could be performed.
Inclusion Criteria (See Table 1)
This systematic review includes papers, which investigated the pathophysiology of DCM in pre-clinical models. Specifically, we included studies, which reported on the cellular and the mechanisms of injury seen in DCM. We included studies involving both in vitro models and in vivo models.
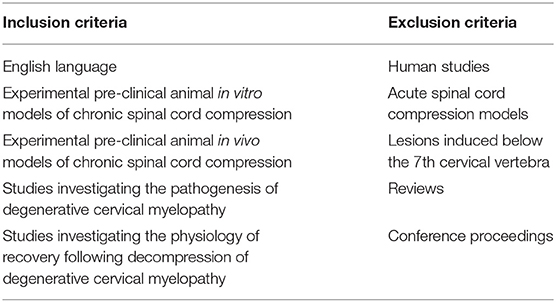
Table 1. Inclusion and exclusion criteria.
Excluded were studies, which looked at acute spinal cord injury or compression, reviews, and spinal cord injuries induced at levels other than the cervical region. We also excluded single case reports and conference proceedings. Articles written in languages other than English were excluded, however we did not restrict our search to any particular geographical region.
Data Extraction
Data extraction and quality assessment was performed independently by the two investigators using “The Animals in Research: Reporting in vivo Experiments (ARRIVE)” Guidelines. The design and methodology of individual studies were extracted including the type of study, sample size, experimental groups, animal housing, and length of follow-up. Species of animal as well as age, strain, weight, sex, and genetic modification factors were extracted. These results were entered into an Excel spreadsheet and studies were assessed for heterogeneity and quality.
Assessment of Risk of Bias and Study Quality
Risk of bias was assessed by two authors. Discrepancies between authors were resolved by discussion with the senior author (MK) until consensus was achieved. The Systematic Review Center for Laboratory animal Experimentation (SYRCLE) risk of bias tool, which includes domains for selection bias (sequence generation, baseline characteristics, allocation concealment), performance bias (random housing and blinding), detection bias (random outcome assessment and blinding), attrition bias, reporting bias, and other biases was used. Studies were subsequently graded as “low,” “moderate,” or “high” according to quality. This grading method was designed by the authors to provide an overall quality rating following an assessment of the overall methodology of the study. Studies were graded as of “high” quality, when two or more methods were used to cross validate any findings and thus increasing the confidence that the stated conclusions of the paper were more likely to be a true effect. If a study was conducted using a robust methodology but not appropriately quantified or cross validated, it was downgraded to “moderate.” Studies were further downgraded to “low” if there were significant and unexplained variability in results or conclusions were made using simple observational methods without any cross-validation. There is a considerable amount of subjectivity using this method and thus any differing conclusions made by the primary authors were discussed with the senior author to help achieve a consensus regarding the quality of the paper.
Results
For our research questions, we included 51 studies in our analysis using our search strategy (Figure 1). Our initial search produced 13,808 possible publications for review for these three key questions. After title and abstract review, we excluded 13,700 publications, the majority of which did not discuss DCM or were review studies. We found 15 additional publications from the references in-text, giving a total of 108 publications for full-text review. After full-text review, 57 were excluded for the following reasons: human studies (n = 8), compression in thoracic level (n = 12), compression in lumbar region (n = 1), not in English language (n = 4), paper did not investigate the pathophysiology of DCM or physiology of recovery (n = 32). Fifty-one studies were subsequently included in the quantitative analysis. The full evidence summary for these included studies can be found in Tables S1–S3. We used our grading method grade the quality of evidence found (Table 2).
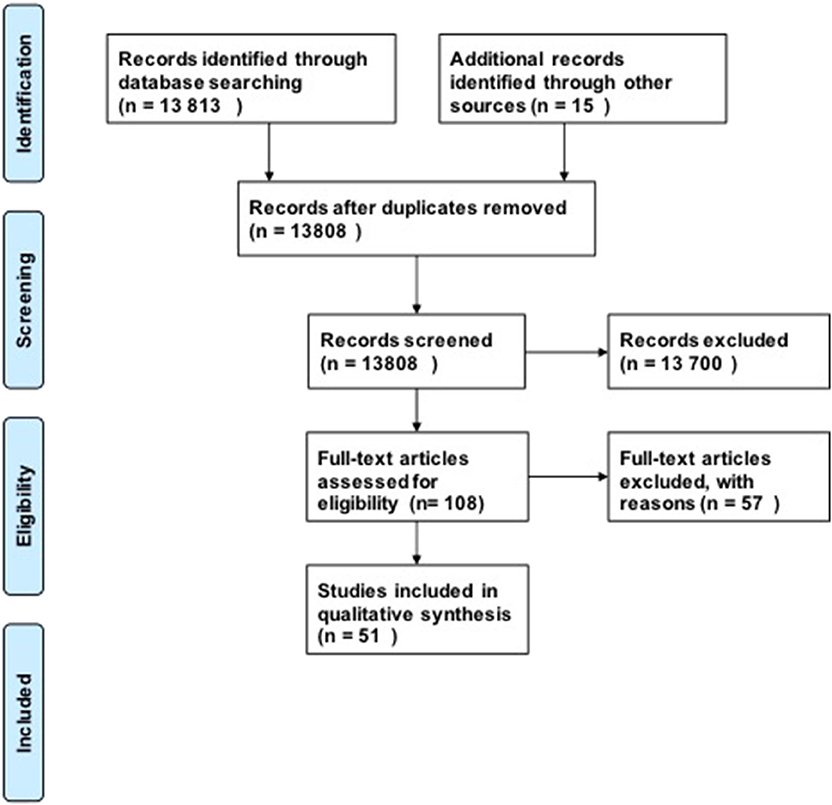
Figure 1. PRISMA Flow diagram.
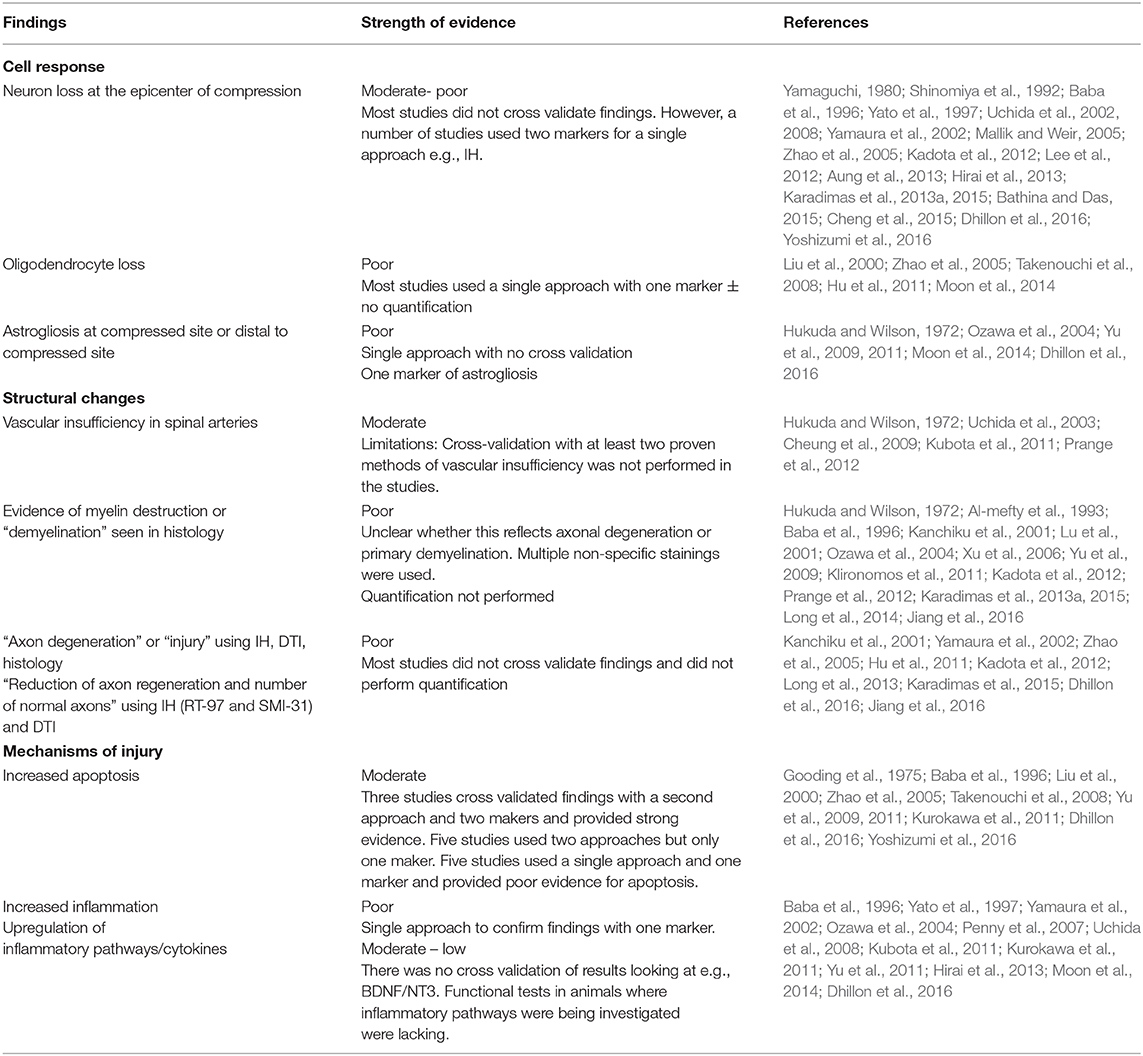
Table 2. Summary of pathophysiological changes following chronic spinal cord compression.
Discussion
Pathophysiology of DCM
Fifty-one studies addressed the pathophysiology of DCM (Table 2; Tables S1, S2; Figure 2) or physiological changes following decompression (Table 2; Table S3; Figures 3, 4). The most common mechanisms involved in the pathophysiology of compression include cell apoptosis, vascular changes, inflammatory responses, axon degeneration and myelin changes (Figure 2). Fourteen papers studied the extrinsic apoptosis pathway. Fourteen papers studied the role of inflammation, of which 10 papers explicitly assessed the microglial response following chronic cord compression. Eight groups studied the vascular changes seen following chronic compression of the spinal cord. Sixteen assessed myelin changes after chronic compression injury. Ten studies investigated axon degeneration in DCM pathogenesis. Twenty-one papers investigated the compressive effects on neurons. Seven papers investigated presence of astrogliosis in DCM. Six studies demonstrated loss of oligodendrocytes following chronic cord compression. Four papers investigated the physiological changes following decompressive surgery (Figure 3).
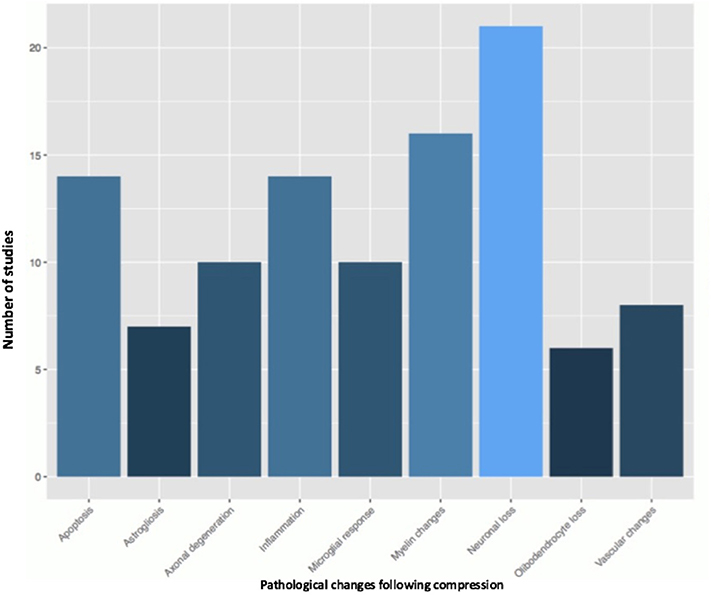
Figure 2. Pathological changes found in the chronically compressed cervical spinal cord.
Figure 3. Pathological changes in the decompressed spinal cord.
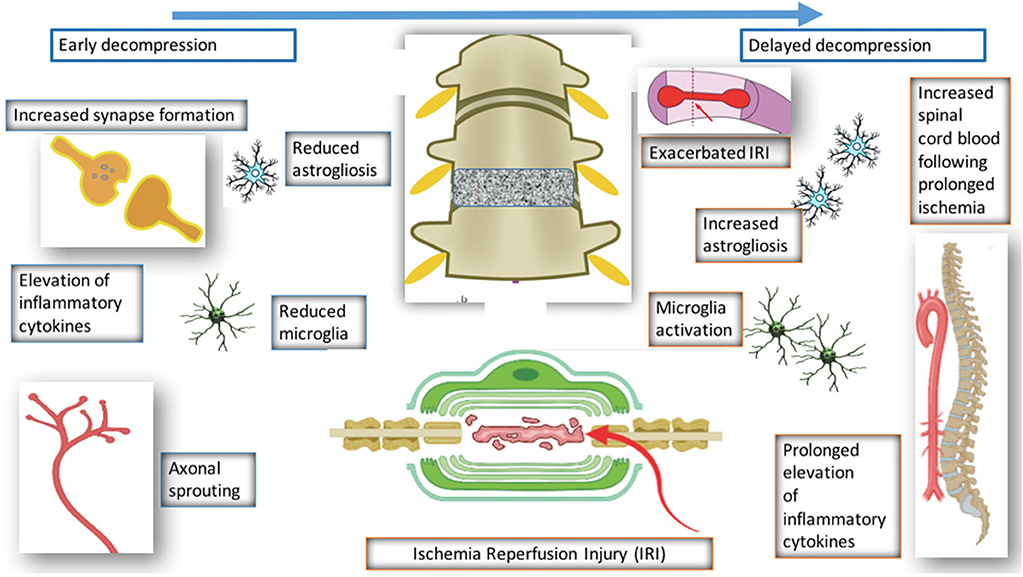
Figure 4. Pathological changes following decompression.
Structural Changes
Structural damage in pre-clinical DCM models has not been fully investigated. A number of histological studies describe structural changes in the compressed spinal cord, which include vacuolar degeneration (Gooding et al., 1975; Al-mefty et al., 1993; Klironomos et al., 2011; Long et al., 2014; Jiang et al., 2016), interstitial oedema (Al-mefty et al., 1993), mitochondrial oedema (Long et al., 2013), cytoplasmic reduction (Aung et al., 2013) and cavity formation. Novel imaging techniques such as diffusion tensor imaging (DTI) with evidence of decreased diffusivity and fractional anisotropy at the compressed site suggesting reduced microstructural integrity (Yu et al., 2011).
Cellular Changes
Neuron loss
It has been well-documented in post-mortem studies that spinal cord compression leads to neuronal and axonal degeneration in the anterior horn (Yu et al., 2011), with evidence of loss of interneurons (Ogino et al., 1983) and lower motor (Ito et al., 1996). Animal studies also demonstrate loss of neurons in the gray matter of the ventral horns (Kanchiku et al., 2001; Kim et al., 2004; Klironomos et al., 2011; Dhillon et al., 2016; Jiang et al., 2016; Yoshizumi et al., 2016). Compression occupation rate is thought to be an important factor in neuronal survival, with a level >30% reducing levels of cholinergic neurons (Yu et al., 2011) and large motor neurons (Yato et al., 1997). In contrast to acute spinal cord injury, DCM patients often have preserved respiratory function. In one elegant study, it was shown that an excitatory neural network in the mid-cervical region is responsible for maintenance of breathing in DCM providing insight into how excitatory interneurons could be used to modulate respiratory function in acute spinal cord injury (Satkunendrarajah et al., 2018). Neuron loss can occur due to a number of factors, including apoptosis, inflammation or ischemia. Neuron loss appears to be mediated by various innate immune responses with evidence of increased levels of pro-inflammatory cytokines and macrophages (Al-mefty et al., 1993). The role of the adaptive immune system, if any, has not been investigated in this model. Moreover, the exact interplay between the different modes of injury is also not fully understood.
Axon degeneration
Chronic cord progression leads to axonal destruction at the site of compression (Kubota et al., 2011) and at sites cranial and caudal to the compression site (Prange et al., 2012). Neuroaxonal changes in compressed cords can be studied by immunoreactivity (IR) for amyloid precursor protein (APP) and phosphorylated neurofilaments (SMI312, SMI31, SMI32). Chronic compression is associated with increased APP at the compressed site (Dhillon et al., 2016). APP is transported by axoplasmic flow and accumulates when there are cytoskeletal defects (Dhillon et al., 2016). However, the mechanism underlying this is not well-known.
There is evidence of both retrograde degeneration and Wallerian degeneration of axons (Karadimas et al., 2013b) and this leads to irreversible damage to structural components of axons such as neurofilaments (Wang et al., 2012). Phosphorylated neurofilaments such as SMI-31 are found in normal axons and can therefore be used as a marker of healthy axons. In chronically compressed cords, there is evidence of a reduction in SMI-31–positive fibers (Takano et al., 2013). There is evidence of widespread axonal damage in the lateral and posterior funiculi (Kanchiku et al., 2001) and slight axonal damage in the anterior funiculi of compressed cords in rabbits (Ozawa et al., 2004). Histological analysis reveals evidence of Wallerian degeneration in the lateral funiculi at sites caudal to the compressed region in sheep (Penny et al., 2007). Early studies in dogs show slight degrees of demyelination in the ventral funiculi and the dorsal part of the lateral funiculi (Harkey et al., 1995). To further assess neuronal damage, one study investigated the serotonergic (5-HT) axons of the descending raphespinal tract in the spinal cord and found that compression was associated with a significant loss of 5HT- positive axons at the center of compression (Dhillon et al., 2016).
Oligodendrocyte loss and demyelination
Oligodendrocytes are supporting cells of the central nervous system (CNS) involved in myelination of axons. Primary loss of oligodendrocytes therefore leads to denuded axons, which display impaired axon conductance (Franklin and Ffrench-Constant, 2008). Six studies demonstrated apoptosis of oligodendrocytes in chronic compression models (Liu et al., 2000; Zhao et al., 2005; Takenouchi et al., 2008; Inukai et al., 2009; Uchida et al., 2012; Moon et al., 2014). This was predominantly found in the twy (tiptoe walking—Yoshimura) mouse model, an autosomal recessive mutant model which leads to calcification of the C1-C2 region and compression of the spinal cord. However, these studies typically only confirmed oligodendrocyte loss using one technique with little evidence of quantitative analysis. Many studies used techniques such as Luxol Fast Blue (LFB) staining to demonstrate myelin sheath changes, however electron microscopy, considered the gold standard in demonstrating evidence of nude axons and thereby demyelination was not performed (Franklin and Ffrench-Constant, 2008). A number of studies cross-validated their data using SEP by measuring the amplitude giving useful information about axon density and latency, which is prolonged following demyelination (Mallik and Weir, 2005), however these should be performed in addition to electron microscopy.
Axonal degeneration leads to secondary demyelination, with consequential loss of oligodendrocytes. More rigorous attempts are mandated in order to differentiate between primary and secondary demyelination in DCM models.
Astrogliosis
Astrogliosis is a reactive process that occurs in response to damage to nearby structures such as neurons. It can be typically demonstrated using immunohistochemistry and the presence of glial fibrillary acidic protein (GFAP). Chronically compressed cords appear to be associated with increased expression of GFAP in a number of different models (Hukuda and Wilson, 1972; Harkey et al., 1995; Yu et al., 2009; Moon et al., 2014). However, one study found evidence of astrogliosis not in the compressed region, but in areas proximal and distal to the maximal compressed site (Long et al., 2014). This may occur to protect intact regions of the cord from further damage and separate the damaged region from normal regions. The exact timing when the studies were performed following compression may also have an impact on the different results obtained. This is important to determine as prolonged astrogliosis may have detrimental effects. An understanding of molecular aberrations underlying astrogliosis has also not been investigated fully in these models. Further studies are required to quantify the loss of astrocytes with multiples markers and at different time points following compression.
Mechanisms of Injury
Apoptosis
Apoptosis is a form of programmed cell death found in both acute spinal cord injury and in chronically compressed spinal cords. It is possible that the mechanical stress associated with chronic cord compression directly leads to activation of pro-apoptotic factors, and this has been hypothesized in the case of traumatic brain injury (Ng and Lee, 2019). Alternatively, apoptosis may occur as a consequence of other destructive processes, such as inflammation or hypoxic damage. Apoptosis can be mediated by various cell signaling processes involving cell surface death receptors such as those from the tumor necrosis factor receptor (TNFR) family (Inukai et al., 2009; Uchida et al., 2012; Takano et al., 2014). Apoptosis can also be activated by intrinsic pathways initiated by mitochondrial damage and are characterized by the presence of apoptotic proteins such as Bcl-2 (Yu et al., 2011). Our analysis reveals an important role of the extrinsic pathway in apoptosis of cells in DCM. A number of studies have shown evidence of increased apoptosis mediated by the Fas pathway (Moon et al., 2014) activating caspase (Yu et al., 2011) which can be used as a therapeutic target. However, a time course analysis of apoptosis appears to be lacking in the current studies. Furthermore, other mediators of cellular apoptosis such as oxidants have also not been investigated.
Vascular changes
Changes in vascularity is commonly seen following acute spinal cord injuries (Figley et al., 2014) and appears to play a significant role in DCM. Microvascular swelling has been described following compression (Yamaguchi, 1980; Jiang et al., 2016), and proliferation of arterioles has also been described in one study (Al-mefty et al., 1993). Venous thickening and sinusoidal swelling has been described in three papers (Penny et al., 2007; Wang et al., 2012; Yamamoto et al., 2014). Vascular dysfunction was demonstrated in both the early studies performed in dogs and in more recent studies. Damage to the vasculature can lead to myelin damage (Gooding et al., 1975), glial fibrosis and necrosis (Ng and Lee, 2019). Early studies however were limited by small samples and limited quantitative analysis. Modern imaging techniques have attempted to visualize the exact vascular damage that occurs in DCM. Chronic cord compression can lead to damage to anterior spinal artery (ASA) (Long et al., 2014; Cheng et al., 2015) and ARA (Kurokawa et al., 2011). Vascular dysfunction appears to be associated with reduced blood flow to the spinal cord (Franklin and Ffrench-Constant, 2008; Kurokawa et al., 2011) and this can be demonstrated with various methods such as the microsphere technique which allows blood flow measurement using a fluorescence microsphere and avoids directly manipulating the spinal cord. However, injection of the microsphere in small animals can itself cause haemodynamic abnormalities (Prinzen and Bassingthwaighte, 2000). Other methods include the hydrogen clearance method, used by Al-mefty et al. (1993) and Harkey et al. (1995). This is an adaptation of the Fick principle where blood is linearly related to the local hydrogen concentration. Hydrogen clearance can be detected in any tissue where an electrode can be placed. Unlike microsphere techniques it allows repeated measurements of blood flow. However, the hydrogen clearance method often manipulates the spinal cord, which can affect the blood flow measurement. If there is a disruption of the gray matter/white matter interface after compression, this method cannot be used to determine blood flow in the two areas (Stalberg et al., 1998).
Hypoxia
Hypoxic changes in DCM have not been widely investigated. Tanabe et al. (2011), demonstrated increase in HIF-1α, a transcriptional regulator of oxygen homeostasis in the Twy/Twy model. However, their findings were not substantiated by investigation of other hypoxia induced factors, angiogenesis markers or the presence of reactive oxygen species. Furthermore, HIF-1α activates transcription of genes encoding various other factors including vascular endothelial growth factor (VEGF) (Ramakrishnan et al., 2014) and this needs to be investigated in relation to hypoxia in the DCM model.
Inflammation
A number of studies have shown evidence of increased inflammatory cells such as microglia in DCM (Moon et al., 2014), which increased with worsening cord compression (Hirai et al., 2013). Activated microglia in the form of the pro-inflammatory cells expressing markers such as TNF-α and CD86, are present in chronically compressed spinal cords (Ng and Lee, 2019). The role of the anti-inflammatory microglia is less clear with some studies showing no significant differences between control and compressed cords (Takano et al., 2013), whilst other studies showing these are present (Hirai et al., 2013) perhaps as a mechanism of recovery. Against, the time course when the activated microglia appear are not fully clear and this may help us determine the precise role of microglia activation patterns. Various other cytokines are also increased in compressed cords such as Th1 cytokines (Hirai et al., 2013) and increased TNFR1, CD95, and p75NTR due to increased chemokine signals (Takano et al., 2013). Inflammatory signaling pathways such as the NF-κB pathway is also upregulated in DCM (Karadimas et al., 2013a).
Changes in the extracellular matrix
The extracellular matrix (ECM) surrounding neurons in the form of perineuronal nets is composed of hyaluronan, proteoglycans and tenascin. Hyaluronan is released into the extracellular space and subsequently localizes around astrocytes, oligodendrocytes and neuron cell bodies (Eggli et al., 1992; Struve et al., 2005). The Hyaluronan tetrasaccharide (HA4), (hyaluronan degradation product), is known to have regenerative properties (Torigoe et al., 2011), and in a rat model of DCM appears to increase following 3 weeks of compression (Wang et al., 2012). This is associated with increased cellular inhibitor of apoptosis protein-2 (cIAP2) as well as a gradual reduction in apoptosis. There was also increased BDNF and VEGF expression at 4 weeks post-compression compared with the control group likely induced by astrocytes to aid in repair of neurons and oligodendrocytes. These results however have not been fully replicated in other studies, with some showing evidence of reduced BDNF levels in compressed cords (Uchida et al., 2003). Matrix metalloproteinases (MMP) are associated with dysfunction of the blood-brain barrier and increased levels of MMP in DCM has been described in one paper (Karadimas et al., 2013a).
Mechanisms of Recovery Following Decompression
Surgical decompression of the compressed spinal cord leads to partial recovery in patients and preclinical models of DCM. However, the mechanism underlying this recovery has seldom been investigated. Four studies have investigated the decompressed state of DCM (Harkey et al., 1995; Karadimas et al., 2015; Dhillon et al., 2016). Harkey et al. (1995) experimented on 12 dogs, which were compressed with an anterior screw device, followed by decompression of six dogs. The decompressed group demonstrated neurological improvement; however, there were no significant differences in SEP amplitudes and latencies, spinal cord blood flow, or MRI imaging characteristics between the compressed and decompressed groups. Histopathology was carried out on spinal cord tissue from the decompressed group, and this showed variable changes including necrosis and cavitation. Despite the use of higher order species, there were several limitations of the study such as the lack of quantitative data. Furthermore, majority of the data is descriptive and there no evidence of any systematic approaches to the study.
Dhillon et al. (2016) performed a study on 15 rats, which were randomly and equally allocated to control, compressed and decompressed groups. Evidence of axonal plasticity was observed following decompression. In the compressed group, increased levels of axon injury was observed using the marker APP, together with decreased numbers of serotonergic axons. Following decompression, there was increased GAP-43, which indicates increased axon growth rates, and increased 5HT+ axons and 5HT+/synaptophysin were also demonstrated, providing evidence for axonal sprouting.
Karadimas et al. (2015) demonstrated evidence of ischemia-reperfusion injury post decompression in a rat model, and suggested that this could be one of the reasons why some patients don't fully recover following decompression surgery. They demonstrated that decompression-induced ischemia-reperfusion injury however could be prevented by the administration of Riluzole and this can lead to better long term neurological outcomes compared to the group that received decompression surgery alone.
Vidal et al. (2017) investigated the consequences of delayed surgical management in DCM. Decompression is associated with various changes such as reduced microglia activation and reduced astrogliosis. However, when decompression is delayed, there is increased astrogliosis, microglia activation, and reduced spinal blood cord flow. Decompression itself is associated with ischemia reperfusion and elevation of inflammatory cytokines, however if decompression is delayed, this is prolonged and leads to worse neurological outcome. This highlights the importance of a therapeutic window in DCM and also the need to operate early with or without the use of any medical adjuncts.
Signaling Pathways and Therapeutic Strategies Explored for Promoting Recovery in DCM
Therapeutic signaling pathways which have been used to promote recovery in DCM include the brain derived neutrophic factor (BDNF) signaling pathway (Uchida et al., 2003; Xu et al., 2006; Hirai et al., 2013), Neurotrophin-3 (NT-3) pathway (Uchida et al., 1998, 2008; Hirai et al., 2013), Nuclear factor–[kappa] B (NF-[kappa]B) pathway (Karadimas et al., 2013a), Mitogen Activated Protein Kinase (MAPK) (Takenouchi et al., 2008) and phosphodiesterase (PDE)/cylic AMP pathway (Yamamoto et al., 2014). Other therapeutic agents used include prostaglandins (Kurokawa et al., 2011), granulocyte-colony stimulating factor (G-CSF) (Yoshizumi et al., 2016) and Riluzole (Karadimas et al., 2013a).
Brain-derived neurotrophic factor (BDNF)
BDNF is a member of the neurotrophin family and is important for survival of neurons (Bathina and Das, 2015). BDNF signaling is elicited after it binds to TrkB receptor. Chronic cord compression appears to reduce BDNF at the compressed site in the Twy/Twy mice model (Uchida et al., 2003). There is an increase at rostral and caudal sites suggesting that in the compressed model there is an attempt to promote regeneration. However other studies have demonstrated that with an increase in duration of compression, there is increased BDNF intensity (Hirai et al., 2013). Xu et al. (2006) showed that administration of BDNF can increase the number of neurons at the compressed spinal cord. It appears from the above studies that the response of BDNF in compressed cords is not fully characterized. A full understanding of its response at different times during compression and decompression is warranted.
Neurotrophin
Neurotrophin (NT-3) are a family of growth factors involved in neuron survival and axonal regeneration (Lu et al., 2001). Compression of the spinal cord appears to downregulate the levels of NT-3 (Uchida et al., 1998, 2003) at the epicenter and also downregulates the levels of its receptor, trKC (Uchida et al., 2003). However, there is upregulation rostral to the compressed site (Uchida et al., 1998, 2003) and caudal to the compressed site (Uchida et al., 2003). Targeted retrograde gene delivery of NT-3 has been shown to reduces loss of neurons seen in compressed cords and improves their morphology (Takenouchi et al., 2008). Targeted retrograde gene delivery of NT-3 therefore may be used to promote neuronal survival in compressed spinal cords.
Mitogen-activated protein kinase (MAPK)
The MAPK signaling pathway is an intracellular signaling system that controls many basic cellular functions, including apoptosis (Chang and Karin, 2001), which induces optimal stress responses. It encompasses a core signaling module, which includes MAPK kinase kinase (MAPKKK), MAPK kinase, and MAPK (Chang and Karin, 2001). One of the MAPKKKs is apoptosis signal related kinase 1 (ASK1), which is activated by reactive oxygen species and tumor necrosis factor (TNF)-[alpha] (Long et al., 2013). Other stress-activated MAPK pathways include JNK and p38. All three pathways were upregulated in chronically compressed cords and located in apoptotic cells (Takenouchi et al., 2008). Similar dysregulation of MAPK has been found in mechanically-induced apoptosis of chondrocytes (Kong et al., 2013) supporting the notion that inhibitors of ASK1-JNK/-p38-caspase pathway mediated apoptosis may be a therapeutic target in DCM.
Phosphodiesterase (PDE)/cyclic AMP
The phosphodiesterase (PDE)/cyclic AMP pathway has been investigated in one study. Phosphodiesterase are enzymes that degrade the phosphodiester bond in the secondary messengers, cAMP and cGMP. PDE inhibitors inhibit degradation of these messengers and thereby enhance their effects, in part via modulation of MAPK. The PDE3 inhibitor Cilastazol has been shown to inhibit platelet aggregation and acts as a vasodilator by inhibiting activation of myosin light chain kinase, which contracts smooth muscle cells. Cilastazol has been shown to improve neurological function, reduce neuron loss and apoptosis in a rat model of DCM (Yamamoto et al., 2014).
Nuclear factor–[kappa] B (NF-[kappa]B)
Nuclear factor–[kappa] B (NF-[kappa]B) are transcriptional factors, composed of the Rel family of proteins including RelA (p65), NF-[kappa]B1 (p50), and NF-[kappa]B2 (p52) (Christian et al., 2016). The most common form is the NF-[kappa]B1-RelA that contains the p65 and p50 proteins. Resting NF-Kb in the cytoplasm becomes activated after phosphorylation of its inhibitory component, followed by ubiquitination, and degradation. The NF-[kappa]B then translocate to the nucleus and binds to target genes involved in various functions (Christian et al., 2016). The role of NF-KB has been investigated in one study in rabbits (Karadimas et al., 2013a). Chronic compression of the spinal cord leads to increased levels of nuclear and cytoplasm p50 and p60. However, the exact effects of activated NF-Kb in DCM is not known. The role of these transcriptional factors in the recovery of DCM has not been investigated.
Prostaglandins
Prostaglandins are lipid compounds found in most tissues with diverse functions. A prostaglandin PGE1 derivative Limaprost has been shown to reduce neuron loss seen in the rat DCM model (Kurokawa et al., 2011). Limaprost is an anti-platelet agent and a vasodilator and it is thus possible it played a vasodilatory role in the DCM model. However, how the drug prevented neuron loss was not clear (Kurokawa et al., 2011).
Granulocyte colony stimulating factor (G-CSF)
Granulocyte colony stimulating factor (G-CSF) is a 19.6-kD glycoprotein that has neuroprotective effects in experimental spinal cord injury. It can induce proliferation of oligodendrocyte precursors, inhibit apoptotic cell death, and suppress inflammatory cytokines (Kadota et al., 2012). G-CSF has been shown in one study to reduce the decline in motor function and loss of motor neurons in a rat model of DCM (Yoshizumi et al., 2016). G-CSF was also shown to restore motor function with the effectiveness observed at approximately 8 weeks. The authors suggested that surgical decompression may be considered during that period to enhance the benefits of the drug.
Riluzole
Riluzole is a sodium glutamate antagonist, which appears to be neuroprotective in acute and chronic injuries of the CNS. Riluzole decreases NMDA receptor phosphorylation in astrocytes and can reduce microglia activation in the chronically compressed spinal cord (Moon et al., 2014). In the decompressed spinal cord, there is evidence for ischemia-reperfusion injury, which may be responsible for initial post-operative neurological decline after decompression surgery. This process can be attenuated using Riluzole, which appears to reduce oxidative DNA damage in the spinal cord (Karadimas et al., 2015).
Autophagy
Autophagy is a mechanism controlling the turnover of organelles and proteins in cells. Autophagy can act to prevent apoptosis and therefore could be exploited therapeutically. One study investigated the neuroprotective role of autophagy in DCM. Tanabe et al. (2011) demonstrated increased expression of autophagy markers in compressed spinal cords and this was associated with a reduction in neuronal apoptosis.
Conclusions and Recommendations
In this systematic review we were able to delineate some of the core pathophysiological processes that have been described in DCM. Our main findings however demonstrate that this condition is not well-studied, and numerous key questions remain regarding the pathobiology of this condition thereby hindering progress in developing therapeutics to improve outcomes. We have highlighted the key processes involved in this condition such as axonal degeneration, apoptosis of cells, and vascular dysfunction. However, how these process lead to cellular and molecular changes in the condition has not been well-studied. We have demonstrated the lack of understanding of the changes that occur following decompression. In addition to early surgery, there is a therapeutic window in which outcomes can be improved. Further studies investigating the mechanisms of injury and recovery in the decompressed spinal cord and how these can be modulated for patient benefit is warranted.
Recommendations
We recommend the following based on the results of our systematic review
1. Define standardized protocols for pre-clinical DCM models, including outcome measures that are relevant to the clinical setting.
2. Delineate the intrinsic and extrinsic mechanisms responsible for axonal degeneration and cell death in DCM, and in particular how mechanical stress translates into cellular injury.
3. In human patients the extent of cord compression correlates poorly with neurological function. It is therefore important to determine mechanisms, genes and pathways that are able to modulate the neurological decline caused by chronic spinal cord compression.
4. Investigate whether intra-cellular repair mechanisms such as autophagy can prevent neuronal and axonal loss, halt disease progression or promote outcomes in DCM.
5. Investigate the role of key cellular elements of the CNS in the decline of neurology caused by cord compression and the recovery following decompression, including the role of oligodendrocytes, astrocytes, microglia, pericytes, etc.
6. Investigate the role of inflammation, and in particular the innate immune response, cytokine profiles and signaling during compression and following decompression.
Data Availability Statement
All datasets generated for this study are included in the article/supplementary material.
Author Contributions
FA, XY, and MK: manuscript design, data collection, analysis, drafting of manuscript, editing of manuscript. XQ and SY: analysis, drafting of manuscript, editing of manuscript. PN and YS: drafting of manuscript, editing of manuscript.
Funding
We gratefully acknowledge support by the Cambridge NIHR Brain Injury MedTech Cooperative. MK was funded by a NIHR Clinician Scientist Award CS-2015-15-023.
Disclaimer
This report is independent research arising from a Clinician Scientist Award, CS-2015-15-023, supported by the National Institute for Health Research. The views expressed in this publication are those of the authors and not necessarily those of the NHS, the National Institute for Health Research or the Department of Health.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnins.2020.00138/full#supplementary-material
References
Al-mefty, O., Harkey, H. L., Marawai, I., Haines, D. E., Peeler, D. F., Wilner, H. I., et al. (1993). Experimental chronic compressive cervical myelopathy. J. Neurosurg. 79, 550–561. doi: 10.3171/jns.1993.79.4.0550
Aung, W. Y., Mar, S., and Benzinger, T. L. (2013). Diffusion tensor MRI as a biomarker in axonal and myelin damage. Imaging Med. 5, 427–440. doi: 10.2217/iim.13.49
Baba, H., Maezawa, Y., Imura, S., Kawahara, N., Nakahashi, K., and Tomita, K. (1996). Quantitative analysis of the spinal cord motoneuron under chronic compression: an experimental observation in the mouse. J. Neurol. 243, 109–116. doi: 10.1007/BF02443999
Bathina, S., and Das, U. N. (2015). Brain-derived neurotrophic factor and its clinical implications. Arch. Med. Sci. 11, 1164–1178. doi: 10.5114/aoms.2015.56342
Boden, S. D., McCowin, P. R., Davis, D. O., Dina, T. S., Mark, A. S., and Wiesel, S. (1990). Abnormal magnetic-resonance scans of the cervical spine in asymptomatic subjects. A prospective investigation. J. Bone Joint Surg. Am. 72, 1178–1184. doi: 10.2106/00004623-199072080-00008
Chang, L., and Karin, M. (2001). Mammalian MAP kinase signalling cascades. Nature. 410, 37–40. doi: 10.1038/35065000
Cheng, X., Long, H., Chen, W., Xu, J., Huang, Y., and Li, F. (2015). Three-dimensional alteration of cervical anterior spinal artery and anterior radicular artery in rat model of chronic spinal cord compression by micro-CT. Neurosci Lett. 606, 106–112. doi: 10.1016/j.neulet.2015.08.050
Cheung, M. M., Li, D. T., Hui, E. S., Fan, S., Ding, A. Y., Hu, Y., et al. (2009). In vivo diffusion tensor imaging of chronic spinal cord compression in rat model. Conf. Proc. IEEE Eng. Med. Biol. Soc. 2009, 2715–2718. doi: 10.1109/IEMBS.2009.5333389
Christian, F., Smith, E. L., and Carmody, R. J. (2016). The Regulation of NF-κB Subunits by Phosphorylation. Cells 18:5. doi: 10.3390/cells5010012
Dhillon, R. S., Parker, J., Syed, Y. A., Edgley, S., Young, A., Fawcett, J. W., et al. (2016). Axonal plasticity underpins the functional recovery following surgical decompression in a rat model of cervical spondylotic myelopathy. Acta Neuropathol. Commun. 4:89. doi: 10.1186/s40478-016-0359-7
Eggli, P. S., Lucocq, J., Ott, P., Graber, W., and van der Zypen, E. (1992). Ultrastructural localization of hyaluronan in myelin sheaths of the rat central and rat and human peripheral nervous systems using hyaluronan-binding protein-gold and link protein-gold. Neuroscience 48, 737–744. doi: 10.1016/0306-4522(92)90417-Z
Figley, S. A., Khosravi, R., Legasto, J. M., Tseng, Y. F., and Fehlings, M. G. (2014). Characterization of vascular disruption and blood-spinal cord barrier permeability following traumatic spinal cord injury. J. Neurotrauma 31, 541–552. doi: 10.1089/neu.2013.3034
Franklin, R. J. M., and Ffrench-Constant, C. (2008). Remyelination in the CNS: from biology to therapy. Nat. Rev. Neurosci. 9, 839–855. doi: 10.1038/nrn2480
Gooding, M. R., Wilson, C. B., and Hoff, J. T. (1975). Experimental cervical myelopathy- effects of ischemia and compression of canine cervical spinal cord. J. Neurosurg. 43, 9–17. doi: 10.3171/jns.1975.43.1.0009
Harkey, H. L., Al-Mefty, O., Marawi, I., Peeler, D. F., Haines, D. E., and Alexander, L. F. (1995). Experimental chronic compressive cervical myelopathy: effects of decompression. J. Neurosurg. 83, 336–341. doi: 10.3171/jns.1995.83.2.0336
Hirai, T., Uchida, K., Nakajima, H., et al. (2013). The prevalence and phenotype of activated microglia/macrophages within the spinal cord of the hyperostotic mouse (Twy/Twy) changes in response to chronic progressive spinal cord compression: implications for human cervical compressive myelopathy. PLoS ONE 8:e64528. doi: 10.1371/journal.pone.0064528
Hu, Y., Wen, C. Y., Li, T. H., Cheung, M. M., Wu, E. X., and Luk, K. D. (2011). Somatosensory-evoked potentials as an indicator for the extent of ultrastructural damage of the spinal cord after chronic compressive injuries in a rat model. Clin. Neurophysiol. 122, 1440–1447. doi: 10.1016/j.clinph.2010.12.051
Hukuda, S., and Wilson, C. B. (1972). Experimental cervical myelopathy- effects of compression and ischemia on canine cervical cord. J. Neurosurg. 37:631. doi: 10.3171/jns.1972.37.6.0631
Inukai, T., Uchida, K., Nakajima, H., Yayama, T., Kobayashi, S., Mwaka, E., et al. (2009). Tumor necrosis factor-alpha and its receptors contribute to apoptosis of oligodendrocytes in the spinal cord of spinal hyperostotic mouse (Twy/Twy) sustaining chronic mechanical compression. Spine 34, 2848–2857. doi: 10.1097/BRS.0b013e3181b0d078
Ito, T., Oyanagi, K., Takahashi, H., Takahashi, H. E., and Ikuta, F. (1996). Cervical spondylotic myelopathy. Clinicopathologic study on the progression pattern and thin myelinated fibers of the lesions of seven patients examined during complete autopsy. Spine 21, 827–833. doi: 10.1097/00007632-199604010-00010
Jiang, H., Wang, J., Xu, B., Yang, H., and Zhu, Q. (2016). A model of acute central cervical spinal cord injury syndrome combined with chronic injury in goats. Eur. Spine J. 26, 56–63. doi: 10.1007/s00586-016-4573-6
Kadota, R., Koda, M., Kawabe, J., Hashimoto, M., Nishio, Y., Mannoji, C., et al. (2012). Granulocyte colony-stimulating factor (G-CSF) protects oligodendrocyte and promotes hindlimb functional recovery after spinal cord injury in rats. PLoS ONE 7:e50391. doi: 10.1371/journal.pone.0050391
Kanchiku, T., Taguchi, T., Kaneko, K., Yonemura, H., Kawai, S., and Gondo, T. (2001). A new rabbit model for the study on cervical compressive myelopathy. J. Orthop. Res. 19, 605–613. doi: 10.1016/S0736-0266(00)00058-9
Karadimas, S. K., Klironomos, G., Papachristou, D. J., Papanikolaou, S., Papadaki, E., and Gatzounis, G. (2013a). Immunohistochemical profile of NF-kappaB/p50, NF-kappaB/p65, MMP-9, MMP-2, and u-PA in experimental cervical spondylotic myelopathy. Spine 38, 4–10. doi: 10.1097/BRS.0b013e318261ea6f
Karadimas, S. K., Laliberte, A. M., Tetreault K Ching, Y. S., Arnold, P., Foltz, W. D., et al. (2015). Riluzole blocks perioperative ischemia-reperfusion injury and enhances postdecompression outcomes in cervical spondylotic myelopathy. Sci. Transl. Med. 7:316ra194 doi: 10.1126/scitranslmed.aac6524
Karadimas, S. K., Moon, E. S., Yu, W. R., Satkunendrarajah, K., Kallitsis, J. K., Gatzounis, G., et al. (2013b). A novel experimental model of cervical spondylotic myelopathy (CSM) to facilitate translational research. Neurobiol. Dis. 54, 43–58. doi: 10.1016/j.nbd.2013.02.013
Kim, P., Haisa, T., Kawamoto, T., Kirino, T., and Wakai, S. (2004). Delayed myelopathy induced by chronic compression in the rat spinal cord. Ann. Neurol. 55, 503–511. doi: 10.1002/ana.20018
Klironomos, G., Karadimas, S., Mavrakis, A., et al. (2011). New experimental rabbit animal model for cervical spondylotic myelopathy. Spinal Cord. 49, 1097–1102. doi: 10.1038/sc.2011.71
Kong, D., Zheng, T., Zhang, M., Wang, D., Du, S., Li, X., et al. (2013). Static mechanical stress induces apoptosis in rat endplate chondrocytes through MAPK and mitochondria-dependent caspase activation signaling pathways. PLoS ONE 8:e69403. doi: 10.1371/journal.pone.0069403
Kubota, M., Kobayashi, S., Nonoyama, T., Shimada, S., Takeno, K., Miyazaki, T., et al. (2011). Development of a chronic cervical cord compression model in rat: changes in the neurological behaviors and radiological and pathological findings. J. Neurotrauma. 28, 459–467. doi: 10.1089/neu.2010.1610
Kurokawa, R., Murata, H., Ogino, M., Ueki, K., and Kim, P. (2011). Altered blood flow distribution in the rat spinal cord under chronic compression. Spine 36, 1006–1009. doi: 10.1097/BRS.0b013e3181eaf33d
Lebl, D. R., Hughes, A., Cammisa, F. P., and O'Leary, P. F. (2011). Cervical spondylotic myelopathy: pathophysiology, clinical presentation, and treatment. HSS J. 7, 170–178. doi: 10.1007/s11420-011-9208-1
Lee, J., Satkunendrarajah, K., and Fehlings, M. G. (2012). Development and characterization of a novel rat model of cervical spondylotic myelopathy: the impact of chronic cord compression on clinical, neuroanatomical, and neurophysiological outcomes. J. Neurotrauma 29, 1012–1027. doi: 10.1089/neu.2010.1709
Leenaars, M., Hooijmans, C. R., van Veggel, N., ter Riet, G., Leeflang, M., Hooft, L., et al. (2012). A step-by-step guide to systematically identify all relevant animal studies. Lab. Anim. 46, 24–31. doi: 10.1258/la.2011.011087
Liu, H., Nishitoh, H., Ichijo, H., and Kyriakis, J. M. (2000). Activation of apoptosis signal-regulating kinase 1 (ASK1) by tumor necrosis factor receptor-associated factor 2 requires prior dissociation of the ASK1 inhibitor thioredoxin. Mol. Cell Biol. 20, 2198–2208. doi: 10.1128/MCB.20.6.2198-2208.2000
Long, H. Q., Li, G. S., Lin, E. J., Xie, W. H., Chen, W. L., Luk, K. D., et al. (2013). Is the speed of chronic compression an important factor for chronic spinal cord injury rat model? Neurosci. Lett. 545, 75–80. doi: 10.1016/j.neulet.2013.04.024
Long, H. Q., Xie, W. H., Chen, W. L., Xie, W. L., Xu, J. H., and Hu, Y. (2014). Value of micro-CT for monitoring spinal microvascular changes after chronic spinal cord compression. Int. J. Mol. Sci. 15, 12061–12073. doi: 10.3390/ijms150712061
Lu, B., Pang, P. T., and Woo, N. H. (2001). The yin and yang of neurotrophin action. Nat. Rev. Neurosci. (2005) 6, 603–614. doi: 10.1038/nrn1726
Lunsford, L. D., Bissonette, D. J., and Zorub, D. S. (1980). Anterior surgery for cervical disc disease. Part 2: Treatment of cervical spondylotic myelopathy in 32 cases. J. Neurosurg. 53, 12–19. doi: 10.3171/jns.1980.53.1.0012
Ma, L., Zhang, D., Chen, W., Shen, Y., Zhang, Y., Ding, W., et al. (2014). Correlation between magnetic resonance T2 image signal intensity ratio and cell apoptosis in a rabbit spinal cord cervical myelopathy model. Chin. Med. J. 127, 305–313. doi: 10.3760/cma.j.issn.0366-6999.20132385
Mallik, A., and Weir, A. I. (2005). Nerve conduction studies: essentials and pitfalls in practice. J. Neurol. Neurosurg. Psychiatry. 76(Suppl 2), ii23–31. doi: 10.1136/jnnp.2005.069138
Moon, E. S., Karadimas, S. K., Yu, W.-R., Austin, J. W., and Fehlings, M. G. (2014). Riluzole attenuates neuropathic pain and enhances functional recovery in a rodent model of cervical spondylotic myelopathy. Neurobiol. Dis. 62, 394–406. doi: 10.1016/j.nbd.2013.10.020
Ng, S. Y., and Lee, A. Y. W. (2019). Traumatic brain injuries: pathophysiology and potential therapeutic targets. Front. Cell Neurosci. 13:528. doi: 10.3389/fncel.2019.00528
Nishida, N., Kato, Y., Imajo, Y., Kawano, S., and Taguchi, T. (2012). Biomechanical analysis of cervical spondylotic myelopathy: The influence of dynamic factors and morphometry of the spinal cord. J. Spinal Cord Med. 35, 256–261. doi: 10.1179/2045772312Y.0000000024
Ogino, H. K., Tada, K., Okada, K., Yonenobu, K., Yamamoto, T., Ono, K., et al. (1983). Canal diameter, anteroposterior compression ratio, and spondylotic myelopathy of the cervical spine. Spine. 8, 1–15 doi: 10.1097/00007632-198301000-00001
Ozawa, H., Jian Wu, Z., Tanaka, Y., and Kokubun, S. (2004). Morphologic change and astrocyte response to unilateral spinal cord compression in rabbits. J. Neurotrauma 21, 944–955. doi: 10.1089/0897715041526159
Penny, C., Macrae, A., Hagen, R., et al. (2007). Compressive cervical myelopathy in young Texel and Beltex sheep. J. Vet. Intern. Med. 21, 322–327. doi: 10.1111/j.1939-1676.2007.tb02967.x
Prange, T., Carr, E. A., Stick, J. A., Garcia-Pereira, F. L., Patterson, J. S., and Derksen, F. J. (2012). Cervical vertebral canal endoscopy in a horse with cervical vertebral stenotic myelopathy. Equine Vet. J. 44, 116–119. doi: 10.1111/j.2042-3306.2011.00395.x
Prinzen, F. W., and Bassingthwaighte, J. B. (2000). Blood flow distributions by microsphere deposition methods. Cardiovasc. Res. 45, 13–21. doi: 10.1016/S0008-6363(99)00252-7
Ramakrishnan, S., Anand, V., and Roy, S. (2014). Vascular endothelial growth factor signaling in hypoxia and inflammation. J. Neuroimmune Pharmacol. 9, 142–160. doi: 10.1007/s11481-014-9531-7
Satkunendrarajah, K., Karadimas, S. K., Laliberte, A. M., Montandon, G., and Fehlings, M. G. (2018). Cervical excitatory neurons sustain breathing after spinal cord injury. Nature 562, 419–422. doi: 10.1038/s41586-018-0595-z
Shinomiya, K., Mutoh, N., and Furuya, K. (1992). Study of experimental cervical spondylotic myelopathy. Spine 17(10 Suppl), S383–S387. doi: 10.1097/00007632-199210001-00007
Stalberg, E., Sharma, H., and Olsson, Y. (1998). Spinal Cord Monitoring. Basic Principles, Regeneration, Pathophysiology, and Clinical Aspects. Vienna: Springer. doi: 10.1007/978-3-7091-6464-8
Struve, J., Maher, P. C., Li, Y. Q., Kinney, S., Fehlings, M. G., Kuntz, C., et al. (2005). Disruption of the hyaluronan-based extracellular matrix in spinal cord promotes astrocyte proliferation. Glia 52, 16–24. doi: 10.1002/glia.20215
Takano, M., Kawabata, S., Komaki, Y., Shibata, S., Hikishima, K., Toyama, Y., et al. (2014). Inflammatory cascades mediate synapse elimination in spinal cord compression. J. Neuroinflammation 11:40. doi: 10.1186/1742-2094-11-40
Takano, M., Kodak, Y., Hikishama, K., Konomi, T., Fujiyoshi, K., Tsuji, O., et al. (2013). In vivo tracing of neural tracts in tiptoe walking Yoshimura mice by diffusion tensor tractography. Spine. 38, E66–72. doi: 10.1097/BRS.0b013e31827aacc2
Takenouchi, T., Setoguchi, T., Yone, K., and Komiya, S. (2008). Expression of apoptosis signal-regulating kinase 1 in mouse spinal cord under chronic mechanical compression: possible involvement of the stress-activated mitogen-activated protein kinase pathways in spinal cord cell apoptosis. Spine 33, 1943–1950. doi: 10.1097/BRS.0b013e3181822ed7
Tanabe, F., Yone, K., Kawabata, N., et al. (2011). Accumulation of p62 in degenerated spinal cord under chronic mechanical compression: functional analysis of p62 and autophagy in hypoxic neuronal cells. Autophagy 7, 1462–1471. doi: 10.4161/auto.7.12.17892
Torigoe, K., Tanaka, H. F., Ohkochi, H., Miyasaka, M., Yamanokuchi, H., Yoshidad, K., et al. (2011). Hyaluronan tetrasaccharide promotes regeneration of peripheral nerve: in vivo analysis by film model method. Brain Res. 1385, 87–92. doi: 10.1016/j.brainres.2011.02.020
Uchida, K., Baba, H., Maezawa, Y., Furukawa, S., Furusawa, N., and Imura, S. (1998). Histological investigation of spinal cord lesions in the spinal hyperostotic mouse (Twy/Twy): morphological changes in anterior horn cells and immunoreactivity to neurotropic factors. J. Neurol. 245, 781–793. doi: 10.1007/s004150050287
Uchida, K., Baba, H., Maezawa, Y., Furukawa, S., Omiya, M., Kokubo, Y., et al. (2003). Increased expression of neurotrophins and their receptors in the mechanically compressed spinal cord of the spinal hyperostotic mouse (twy/twy). Acta Neuropathol. 106, 29–36. doi: 10.1007/s00401-003-0691-4
Uchida, K., Baba, H., Maezawa, Y., and Kubota, C. (2002). Progressive changes in neurofilament proteins and growth-associated protein-43 immunoreactivities at the site of cervical spinal cord compression in spinal hyperostotic mice. Spine 27, 480–486 doi: 10.1097/00007632-200203010-00008
Uchida, K., Nakajima, H., Inukai, T., Takamura, T., Kobayashi, S., Furukawa, S., et al. (2008). Adenovirus-mediated retrograde transfer of neurotrophin-3 gene enhances survival of anterior horn neurons of twy/twy mice with chronic mechanical compression of the spinal cord. J. Neurosci. Res. 86, 1789–1800. doi: 10.1002/jnr.21627
Uchida, K., Nakajima, H., Watanabe, S., Yayama, T., Inukai, T., et al. (2012). Apoptosis of neurons and oligodendrocytes in the spinal cord of spinal hyperostotic mouse (Twy/Twy): possible pathomechanism of human cervical compressive myelopathy. Eur. Spine J. 21, 490–497. doi: 10.1007/s00586-011-2025-x
Vidal, P. M., Karadimas, S. K., Ulndreaj, A., Laliberte, A. M., Tetreault, L., Forner, S., et al. (2017). Delayed decompression exacerbates ischemia-reperfusion injury in cervical compressive myelopathy. JCI Insight. 2:92512. doi: 10.1172/jci.insight.92512
Wang, H., Wu, M., Zhan, C., Ma, E., Yang, M., Yang, X., et al. (2012). Neurofilament proteins in axonal regeneration and neurodegenerative diseases. Neural Regen. Res. 7, 620–626.
Xu, K., Uchida, K., Nakajima, H., Kobayashi, S., and Baba, H. (2006). Targeted retrograde transfection of adenovirus vector carrying brain-derived neurotrophic factor gene prevents loss of mouse (twy/twy) anterior horn neurons in vivo sustaining mechanical compression. Spine. 31, 1867–1874. doi: 10.1097/01.brs.0000228772.53598.cc
Yamaguchi, Y. (1980). Electron microscopic studies on the experimental cervical myelopathy. Nihon Geka Hokan 49, 713–738.
Yamamoto, S., Kurokawa, R., and Kim, P. (2014). Cilostazol, a selective Type III phosphodiesterase inhibitor: prevention of cervical myelopathy in a rat chronic compression model. J. Neurosurg. Spine 20, 93–101. doi: 10.3171/2013.9.SPINE121136
Yamaura, I., Yone, K., Nakahara, S., et al. (2002). Mechanism of destructive pathologic changes in the spinal cord under chronic mechanical compression. Spine 27, 21–26. doi: 10.1097/00007632-200201010-00008
Yato, Y., Fujimura, Y., Nakamura, M., Watanabe, M., and Yabe, Y. (1997). Decreased choline acetyltransferase activity in the murine spinal cord motoneurons under chronic mechanical compression. Spinal Cord 35, 729–734. doi: 10.1038/sj.sc.3100529
Yoshizumi, T., Murata, H., Yamamoto, S., Kurokawa, R., Kim, P., and Kawahara, N. (2016). Granulocyte colony-stimulating factor improves motor function in rats developing compression myelopathy. Spine 41, 1380–1387 doi: 10.1097/BRS.0000000000001659
Yu, W., Liu, R. T., Kiehl, T. R., and Fehlings, M. G. (2011). Human neuropathological and animal model evidence supporting a role for Fas-mediated apoptosis and inflammation in cervical spondylotic myelopathy. Brain 134, 1277–1292. doi: 10.1093/brain/awr054
Yu, W. R., Baptiste, D. C., Liu, T., Odrobina, E., Stanisz, G. J., and Fehlings, M. G. (2009). Molecular mechanisms of spinal cord dysfunction and cell death in the spinal hyperostotic mouse: implications for the pathophysiology of human cervical spondylotic myelopathy. Neurobiol. Dis. 33, 149–163. doi: 10.1016/j.nbd.2008.09.024
Keywords: neuronal loss, apoptosis, pathogenesis, spine, degeneration
Citation: Akter F, Yu X, Qin X, Yao S, Nikrouz P, Syed YA and Kotter M (2020) The Pathophysiology of Degenerative Cervical Myelopathy and the Physiology of Recovery Following Decompression. Front. Neurosci. 14:138. doi: 10.3389/fnins.2020.00138
Received: 27 October 2019; Accepted: 03 February 2020;
Published: 30 April 2020.
Edited by:
Cristina Moglia, University of Turin, ItalyReviewed by:
Sukhvinder Kalsi-Ryan, University Health Network (UHN), CanadaSpyridon Karadimas, University of Toronto, Canada
Copyright © 2020 Akter, Yu, Qin, Yao, Nikrouz, Syed and Kotter. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Farhana Akter, RmEzNDdAYWx1bW5pLmNhbS5hYy51aw==
†These authors share first authorship