 Nao Nakagawa-Tamagawa
Nao Nakagawa-Tamagawa Emi Kirino1
Emi Kirino1 Kohtaroh Sugao
Kohtaroh Sugao Hidetaka Nagata
Hidetaka Nagata Yoshiaki Tagawa
Yoshiaki Tagawa- 1Department of Physiology, Graduate School of Medical and Dental Sciences, Kagoshima University, Kagoshima, Japan
- 2Laboratory for Molecular Analysis of Higher Brain Function, RIKEN Center for Brain Science, Saitama, Japan
- 3Platform Technology Research Unit, Sumitomo Dainippon Pharma Co., Ltd., Osaka, Japan
Introduction: Gain-of-function mutations in the L-type Ca2+ channel Cav1.2 cause Timothy syndrome (TS), a multisystem disorder associated with neurologic symptoms, including autism spectrum disorder (ASD), seizures, and intellectual disability. Cav1.2 plays key roles in neural development, and its mutation can affect brain development and connectivity through Ca2+-dependent and -independent mechanisms. Recently, a gain-of-function mutation, I1166T, in Cav1.2 was identified in patients with TS-like disorder. Its channel properties have been analyzed in vitro but in vivo effects of this mutation on brain development remain unexplored.
Methods: In utero electroporation was performed on ICR mice at embryonic day 15 to express GFP, wild-type, and mutant Cav1.2 channels into cortical layer 2/3 excitatory neurons in the primary somatosensory area. The brain was fixed at postnatal days 14–16, sliced, and scanned using confocal microscopy. Neuronal migration of electroporated neurons was examined in the cortex of the electroporated hemisphere, and callosal projection was examined in the white matter and contralateral hemisphere.
Results: Expression of the I1166T mutant in layer 2/3 neurons caused migration deficits in approximately 20% of electroporated neurons and almost completely diminished axonal arborization in the contralateral hemisphere. Axonal projection in the white matter was not affected. We introduced second mutations onto Cav1.2 I1166T; L745P mutation blocks Ca2+ influx through Cav1.2 channels and inhibits the Ca2+-dependent pathway, and the W440A mutation blocks the interaction of the Cav1.2 α1 subunit to the β subunit. Both second mutations recovered migration and projection.
Conclusion: This study demonstrated that the Cav1.2 I1166T mutation could affect two critical steps during cerebrocortical development, migration and axonal projection, in the mouse brain. This is mediated through Ca2+-dependent pathway downstream of Cav1.2 and β subunit-interaction.
Introduction
Autism spectrum disorder (ASD) is a neurodevelopmental disease. Its etiology has been extensively studied but remains largely unknown, primarily because the developing brain undergoes complex processes influenced by many genetic and environmental factors (Abrahams and Geschwind, 2008; Courchesne et al., 2020). However, there are some cases where a single mutation in a certain gene is linked to ASD. One such case is a gain-of-function mutation, G406R, in the L-type Ca2+ channel Cav1.2 that causes Timothy syndrome (TS). TS is a multisystem disorder associated with long QT syndrome (LQTS type 8) in the heart, syndactyly, and neurologic symptoms, including ASD, seizures, and intellectual disability (Splawski et al., 2004). G406R mutation alters ion channel kinetics, leading to an abnormal Ca2+ overload and Ca2+-dependent and -independent gene expression in neuronal cells, which are thought to be the basis for the disease (Paşca et al., 2011; Marcantoni et al., 2020).
Recently, a mutation, I1166T, in Cav1.2 was identified in patients with TS-like disorder (Boczek et al., 2015; Wemhöner et al., 2015). Among the mutations reported in those studies, the patient with I1166T mutation had brain symptoms. The G406R (the original mutation) and I1166T mutations are considered to be gain-of-function; both elicit excess Ca2+ influx. Because G406R was the original mutation identified in TS patients, many in vitro and in vivo studies have been conducted on the G406R mutant channel. As for the I1166T mutant, channel properties have been thoroughly analyzed in vitro (Boczek et al., 2015; Wemhöner et al., 2015), but in vivo effects of this mutation on brain development have not been reported.
The Cav1.2 Ca2+ channel plays important roles in neural development (Krey et al., 2013; Kabir et al., 2017; Horigane et al., 2019). Ca2+ influx through the channel and the subsequent intracellular Ca2+ signaling are essential for neurodevelopmental processes, such as neurogenesis, migration, and neurite morphogenesis in the neocortex (Kamijo et al., 2018; Panagiotakos et al., 2019). One of the key molecules downstream of Cav1.2 is calmodulin (CaM). Ca2+-CaM binds to the C terminus of the Cav1.2 α1 subunit and modulates channel activity. Ca2+-CaM also activates kinases (such as CaMK) and phosphatases (such as calcineurin; CaN) in the cytoplasm and eventually regulates Ca2+-dependent gene expression in the nucleus (Deisseroth et al., 2003; Yap and Greenberg, 2018). However, recent studies suggested another downstream signaling pathway—the Cav1.2 β subunit-dependent pathway (Servili et al., 2018)—which depends on extracellular Ca2+ and Ca2+ occupancy of the open channel but is Ca2+-influx independent. Thus, there are at least two important signaling pathways downstream of Cav1.2 (Krey et al., 2013; Servili et al., 2018).
In this study, we asked two questions. First, what is the effect of the I1166T mutant channel expression on neocortical development? We expressed the I1166T mutant channel in in vivo mouse cortical neurons by in utero electroporation, and found that neuronal migration was impaired and callosal axon projections were disturbed. Second, which signaling pathway is involved in the defects of migration and callosal projections caused by the I1166T mutant? We attempted to assess the contribution of Ca2+ influx-dependent and β subunit-dependent pathways downstream of the Cav1.2 I1166T mutant channel to the circuit formation deficits. We found that the Ca2+ influx-dependent pathway surely contributed to the phenotypes in brain development and that the interaction of the Cav1.2 α1 subunit to the β subunit was also involved in the deficits caused by the I1166T mutation. Thus, our findings suggest that the Cav1.2 I1166T mutation affects brain development and connectivity through Ca2+-dependent pathway and β subunit-interaction.
Materials and Methods
Mice
All experiments were approved by the Animal Experiments Committee and the Genetic Recombinant Experiment Safety Committee of Kagoshima University and performed according to the guidelines of the university. Pregnant ICR mice were purchased from Japan SLC and used in all experiments.
Plasmid Construction
All plasmid vectors for the expression of the human Cav1.2 α1 subunit of the wild-type (WT) and all mutants, α2δ1 subunit, and β3 subunit were synthesized by Genscript. The Cav1.2 channel subunit sequences were under the chicken-actin-globin (CAG) promoter and followed by the WPRE and BGH polyA signal. The enhanced green fluorescent protein (EGFP) was expressed by pCAsalEGFP vector (Bando et al., 2014). The signal pathways downstream of Cav1.2 that regulate gene expression, and the I1166T and four additional mutations used in this study are shown in Figure 1.
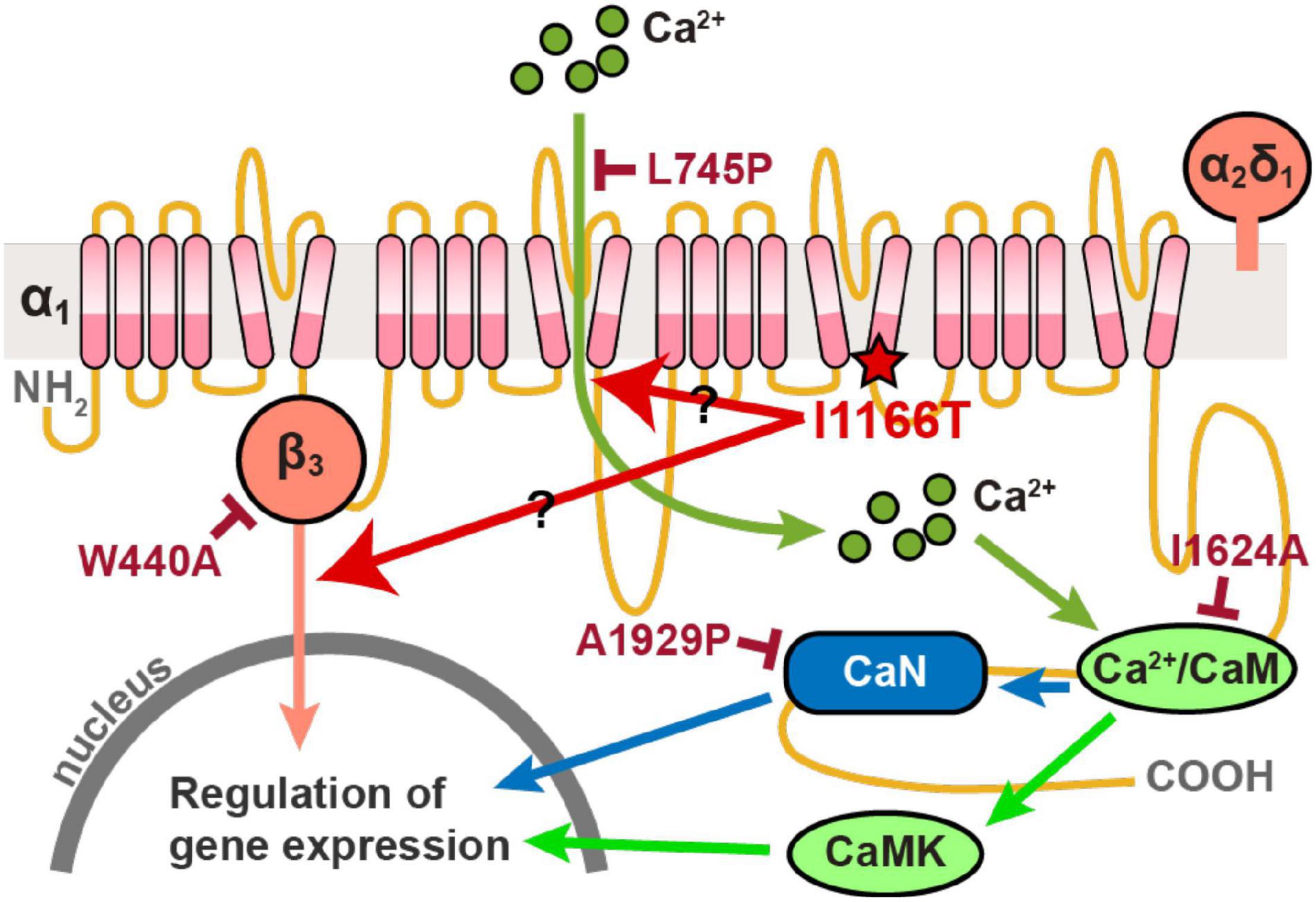
Figure 1. Diagram of signal pathways downstream of Cav1.2 that regulate gene expression. The I1166T and four additional mutations used in this study are also shown.
In utero Electroporation
In utero electroporation was performed as reported previously (Saito and Nakatsuji, 2001; Tabata and Nakajima, 2001). In brief, all the plasmid vectors were prepared at 1 μg/mL with 0.2 μg/mL Fast Green FCF (Nacalai Tesque) in 1/10 Tris-EDTA Buffer. To express the α1 subunit of the WT and all mutant Cav1.2 channels, α2δ1 and β3 subunits, and EGFP were expressed together otherwise noted. For the GFP-alone control experiments, EGFP was expressed alone. ICR mice at embryonic day 15 (E15) were anesthetized with an intraperitoneal injection of pentobarbital sodium salt (64.8 mg/mL, Tokyo Chemical Industry) in saline and maintained with isoflurane (0.7–1.2%, Forane). The mouse uteruses were exposed, and approximately 0.5–1 μL of DNA solution was injected into the lateral ventricle. The embryo heads were pinched with a tweezer CUY650P5 (Nepa Gene), and five electric pulses of 50 V for 50 ms at 1 Hz were applied using an electroporator NEPA21 (Nepa Gene) or an electroporator CUY21 (BEX). We introduced plasmid vectors into neurons in the middle part of the anterior-posterior axis in the neocortex, centered by the primary somatosensory area (S1). One section per mouse was used for all the analyses. The number of in utero electroporation performed for each test group was shown in Supplementary Figure 1.
Fixation and Sectioning
The pups at postnatal days 14–16 were perfused transcardially with ice-cold 0.1 M phosphate-buffered saline (PBS), followed by fixation with 4% paraformaldehyde (PFA) in 0.1 M phosphate buffer. After the brains were dissected, they were further fixed with PFA overnight at 4°C, and then incubated in 20% sucrose in 0.1 M PBS overnight at 4°C. Coronal sections of 80 μm were cut using a microtome REM-710 (Yamato Kohki) and were mounted on glass slides and embedded in Vectashield with DAPI (Vector).
Microscopy
All the fluorescent images were acquired using a confocal laser-scanning microscope TCS SP8 (Leica) with a 10 × dry objective. Z-stack images of the slices were acquired in a 2.5 μm-interval.
Migration Analysis
GFP-positive cells were imaged in the S1 barrel field of the electroporated hemisphere. The three-dimensional positions of cells were determined manually using a custom-made program in MATLAB (MathWorks). A “radial index,” defined as the radial position of the lower edge of white matter, was set to zero and that of pia was set to 1, and a radial index of each neuron was determined. A histogram of cell density along the radial axis was obtained by dividing the cortex into 20 bins and calculating the fraction of GFP-positive cells in each bin. The cells were also categorized into layer 2/3 (“L2/3”), white matter (“WM”), and in between (“Middle”) as follows: the majority of cells were clustered in the upper layer in all samples and were categorized as L2/3, cells located in the white matter were categorized as WM, and cells located between the upper cluster and the white matter were categorized as Middle. WM had a radial index of 0–0.1. The cell numbers analyzed per animal were; GFP-alone, 220.0 ± 71.2; Cav1.2WT, 160.0 ± 55.2; Cav1.2I1166T, 232.9 ± 108.7; Cav1.2I1166T/L745P, 211.9 ± 118.2; Cav1.2I1166T/W440A, 181.0 ± 51.3; Cav1.2I1166T/L745P/E363A/E1115A, 161.0 ± 74.6; Cav1.2I1166T/I1624A, 128.7 ± 57.2; Cav1.2I1166T/A1929P, 162.3 ± 46.8; Cav1.2WT α1-only, 308.1 ± 94.5; Cav1.2I1166T α1-only, 275.9 ± 53.0 (Mean ± SD).
Callosal Projection Analysis
The GFP signals of callosal axons were imaged at the border between primary and secondary somatosensory area (S1/S2 border), where callosal axons arbor densely, contralateral to the electroporated hemisphere. The intensity distribution of background signal was fitted by a Gaussian function and median + 3 × standard deviation of the Gaussian curve was defined as a threshold, and each pixel was determined to have axonal signal (1) or not (0) by whether it had signal intensity higher than the threshold or not. The binarized images of 500 μm width were averaged tangentially to obtain the proportion of pixels with an axonal signal in each radial position. A strong GFP signal was seen with a radial index of 0.4–0.6 and 0.8–1.0, which may correspond to layer 5 and upper layers where callosal axons are arborized densely (Mizuno et al., 2007); therefore, they were assigned “L5” and “L1–3,” respectively. Because the proportion in the white matter, which represents the amount of GFP-positive axons that reached the S1/S2 border, varied between test groups and between samples, the proportion was normalized by the maximum proportion in the white matter and compared between test groups. Several samples had a proportion of WM that was too small (<0.2) to analyze the change along the radial axis; these were excluded from the analysis.
White Matter Projection Analysis
Axonal GFP signals in the white matter were imaged from the ipsilateral to the contralateral hemisphere under the same scanning conditions. To analyze the axonal GFP signal in the white matter, images of 200 μm × 50 μm (radial axis × tangential axis) centered with the white matter were taken at the ipsilateral area (“Ipsi”), midline (“Midline”), and two contralateral areas (“Contra” and “Contra2;” Contra2 is twice the distance from Midline than Contra). Ipsi and Contra positions were determined where white matter was horizontal. GFP signals were extracted by subtracting the background signal intensity (median + 2 × standard deviation of the Gaussian curve fitted the intensity distribution of background signal) from the images. GFP intensities in each image were normalized with that in the Ipsi area and compared between test groups. To compare the broadness of axon bundles in the white matter, GFP images were averaged in the tangential axis, and Gaussian fit was conducted, and the standard deviations of Gaussian curves were compared between test groups. The few samples in which the Gaussian fit was not appropriately conducted in either Ipsi, Midline, or Contra were excluded from the analysis of bundle broadness. All the analyses of migration, callosal projection, and white matter projection were conducted using MATLAB.
Statistical Analysis
Error bars represent the standard error of the mean (SEM). Mann-Whitney-Wilcoxon tests with Holm-Bonferroni correction were used to compare test groups. The statistical analyses were conducted using MATLAB.
Results
The I1166T Mutation Elicits a Migration Deficit
An in utero electroporation-mediated gene transfer was performed at E15, an age when efficient and specific gene transfer into cortical L2/3 neurons has been reported (Saito and Nakatsuji, 2001; Tabata and Nakajima, 2001). At P14–16, almost all neurons expressing GFP-alone migrated to the upper layer and distributed in a band (98.8 ± 0.2%, N = 16, Figure 2A). For neurons with forced expression of Cav1.2WT, though a small proportion of neurons failed to migrate (3.0 ± 0.6%, N = 17; vs. GFP-alone, P = 0.072), neurons migrated primarily to the upper layer (Figures 2B,D,E). On the other hand, a considerable number of neurons with a forced expression of Cav1.2I1166T failed to migrate to the upper layer (20.6 ± 2.7%, N = 17) and were scattered across the full depth of the cortex (Figure 2C). These neurons were located relatively densely in the white matter and at the middle of the cortical depth (radial index = 0.5, Figures 2C–E). The ratio of migration-failed neurons in the Cav1.2I1166T group was larger than that of the Cav1.2WT group (P = 1.2e−5; Figures 2D,E). These results indicate that the I1166T mutation causes migration failure.
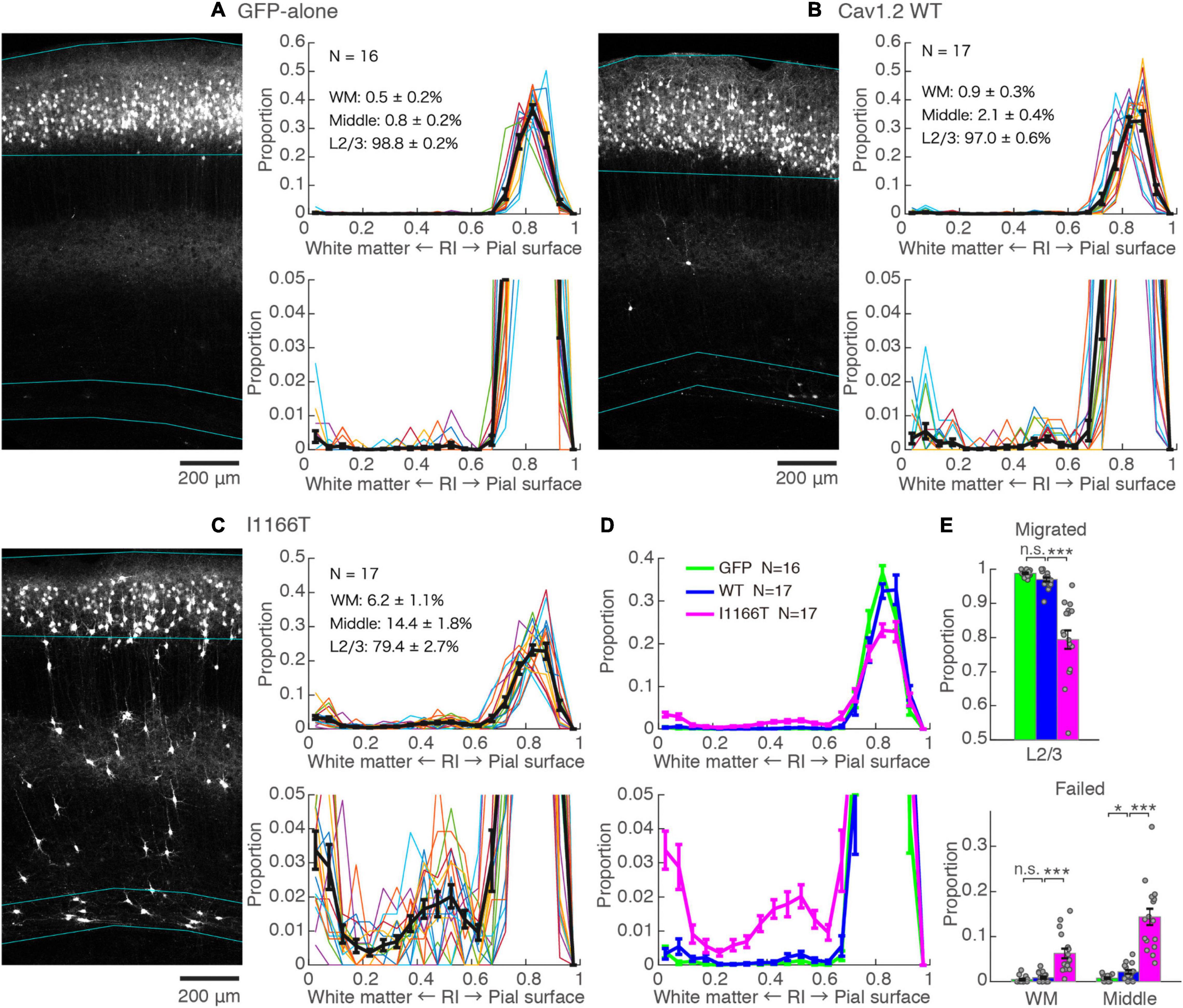
Figure 2. Expression of the Cav1.2 I1166T mutant caused neuronal migration deficit. (A) Left, GFP images of electroporated cortex in the GFP-alone group. Cyan lines represent the top; pial surface, the bottom edge of the GFP-positive neuron cluster, and the top and bottom edge of white matter (see section “Materials and Methods”). Right, histogram of radial index (RI) of the GFP-positive neurons. Colors, each sample. Black, mean ± SEM. Y-axis is enlarged and shown at the bottom. (B,C) Images and histograms of the Cav1.2WT group (B) and Cav1.2I1166T group (C), as shown in (A). (D) Histogram of the mean ± SEM are merged for GFP-alone, Cav1.2WT, and Cav1.2I1166T groups. (E) Proportion of correctly migrated neurons (top) and migration-failed neurons in WM and Middle (bottom). Independent values and mean ± SEM are shown. ***P < 0.001; *P < 0.05; n.s., P ≥ 0.05.
Calcium-Dependent Pathway and β Subunit-Interaction Are Responsible for the Migration Deficit Caused by Cav1.2I1166T Expression
The I1166T mutation of Cav1.2 elicits excess Ca2+ influx, which has been thought to be the cause of the brain phenotypes in the patients (Boczek et al., 2015; Wemhöner et al., 2015). Therefore, we introduced the second mutation, L745P, that inhibits Ca2+ influx (Servili et al., 2018) onto Cav1.2I1166T and examined migration. Neurons expressing Cav1.2I1166T/L745P showed almost normal migration (96.3 ± 0.6%, N = 14; vs. I1166T, P = 6.0e−5; Figures 3A,C,D), which was equivalent to neurons expressing Cav1.2WT (P = 0.55, Figure 3D). This result suggests that Ca2+-dependent pathway is responsible for the migration deficit caused by the I1166T mutation. In addition, another TS mutation, G406R, has been reported to enhance gene expression under a calcium-independent, β subunit-dependent pathway (Servili et al., 2020). Therefore, we introduced another mutation, W440A, that inhibits the interaction of Cav1.2 α1 subunit to the β subunit. We expected that W440A mutation would inhibit a β subunit-dependent signaling pathway, as suggested in other studies (Berrou et al., 2002; Servili et al., 2018; Yang et al., 2019) (but for a possible effect of this mutation on channel trafficking, please see section “Discussion”). Though the fraction of Cav1.2I1166T/W440A-expressing neurons in the upper layer (93.0 ± 0.8%, N = 14) was smaller than that of Cav1.2WT-expressing neurons (P = 0.0041, Figure 3D), migration proceeded normally compared with that of Cav1.2I1166T-expressing neurons (P = 4.5e−4, Figure 3C). Neurons that failed migration were mostly located in the WM (Figures 3B–D). This result suggests that the β subunit-interaction contributes to the migration deficit caused by the I1166T mutation. The introduction of the double pore mutation L745P/E363A/E1115A, which virtually eliminates Ca2+-dependent gene expression (Servili et al., 2018), onto Cav1.2I1166T elicited migration deficits with additional phenotypes (Supplementary Figure 2), suggesting that the normal level of Ca2+ influx is necessary for normal development.
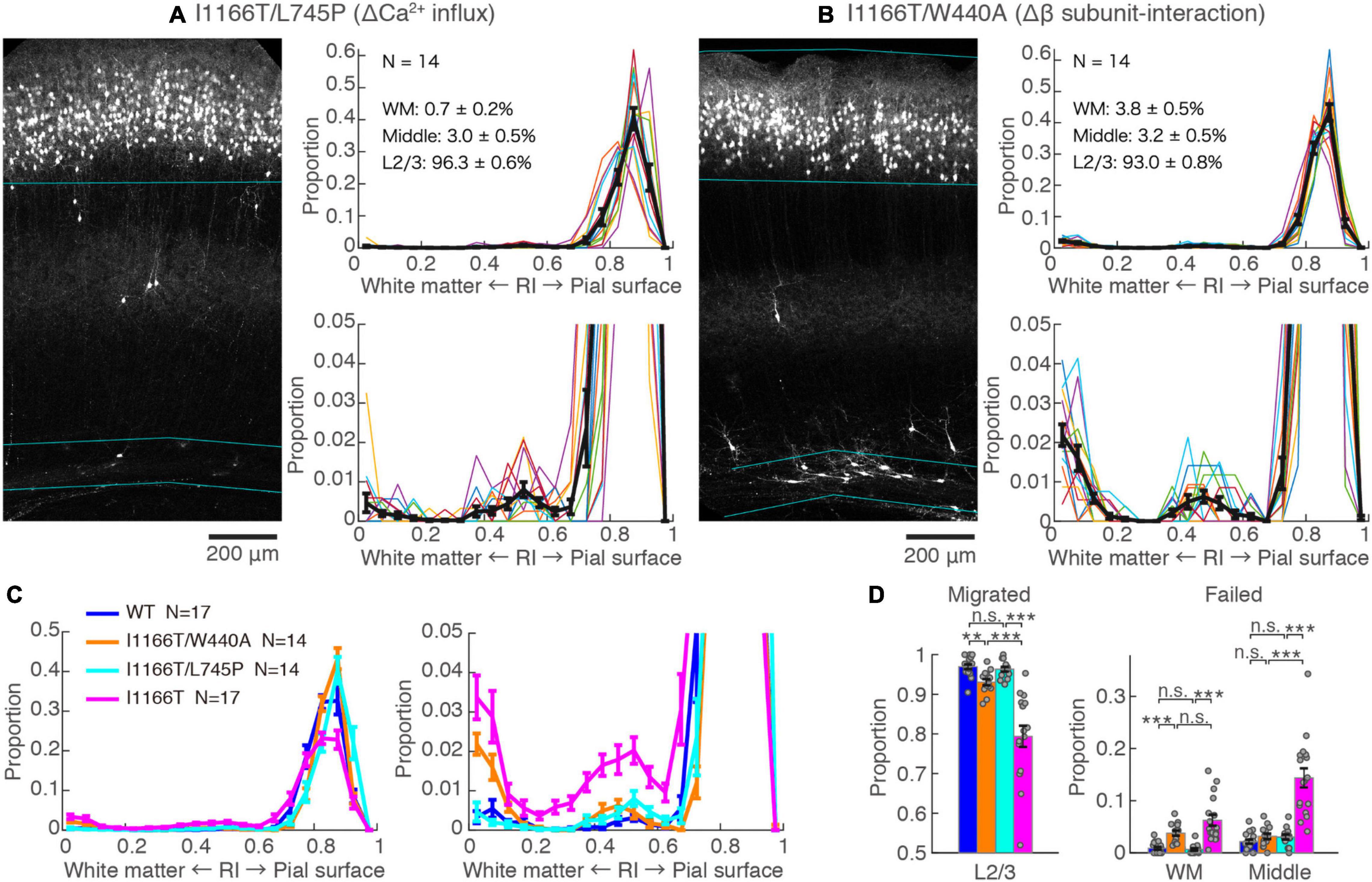
Figure 3. Blockade of Ca2+ influx and β subunit-interaction improves migration deficit caused by the I1166T mutation. (A,B) GFP images and histograms of the Cav1.2I1166T/L745P (A) and Cav1.2I1166T/W440A group (B), as shown in Figure 2A. (C) Histogram of the mean ± SEM are merged for Cav1.2WT, Cav1.2I1166T/W440A, Cav1.2I1166T/L745P, and Cav1.2I1166T groups. (D) Proportion of correctly migrated neurons (left) and that of migration-failed neurons located in WM and Middle (right). Independent values and mean ± SEM are shown. ***P < 0.001; **P < 0.01; n.s., P ≥ 0.05.
The Calmodulin Pathway but Not the Calcineurin Pathway May Be Responsible for the Migration Deficit by Cav1.2I1166T Expression
Next, we examined the possible involvement of two major factors downstream of the Ca2+-influx, CaM and CaN, in Cav1.2I1166T-mediated migration defects. Both CaM and CaN bind to the C-terminal region of the Cav1.2 α1 subunit and modulate channel activity through the so-called Ca2+/CaM- and voltage-dependent inactivation (CDI and VDI, respectively) (Barrett and Tsien, 2008; Cohen-Kutner et al., 2012). CaM and CaN are also key players involved in Ca2+-dependent gene expression. First, we used the I1624A mutation, which is located at the C-terminal IQ motif of the Cav1.2 α1 subunit and impairs CaM binding (Zühlke et al., 1999). The vast majority of neurons expressing Cav1.2I1166T/I1624A migrated normally (91.5 ± 2.0%, N = 7, Figures 4A,C,D). Although the fraction was slightly smaller than neurons expressing Cav1.2I1166T/L745P (P = 0.12, Figure 4D), it was significantly larger than Cav1.2I1166T expressing neurons (P = 0.026, Figure 4D). This result suggests that the migration deficit caused by the I1166T mutation was “rescued” by the blockade of the CaM interaction. In some samples, neuron numbers seemed to be lower than others (e.g., in Figure 4A). Therefore, we compared neuron densities between test groups (Supplementary Figure 3). Cav1.2I1166T/I1624A group had slightly smaller density than I1166T group (P = 0.043). This result could be due to the disappearance of migration-failed neurons in Cav1.2I1166T/I1624A group, or be due to the fact that the number of electroporated neurons by in utero electroporation generally varies widely between pups. Next, we used an A1929P mutation, which is located at the CaN-binding site of the C-terminal tail of the human Cav1.2 α1 subunit, which was shown to abrogate binding of CaN to the channel (Tandan et al., 2009; Xu et al., 2010). A considerable number of Cav1.2I1166T/A1929P-expressing neurons failed to migrate to the upper layer (17.0 ± 3.8%, N = 6, Figures 4B–D). The fraction of correctly migrated neurons was similar to that of Cav1.2I1166T-expressing neurons (P = 0.55, Figure 4D) and was significantly smaller than that of Cav1.2I1166T/L745P-expressing neurons (P = 0.012, Figure 4D). Thus, the migration deficit by I1166T was not “rescued” by the blockade of the CaN interaction.
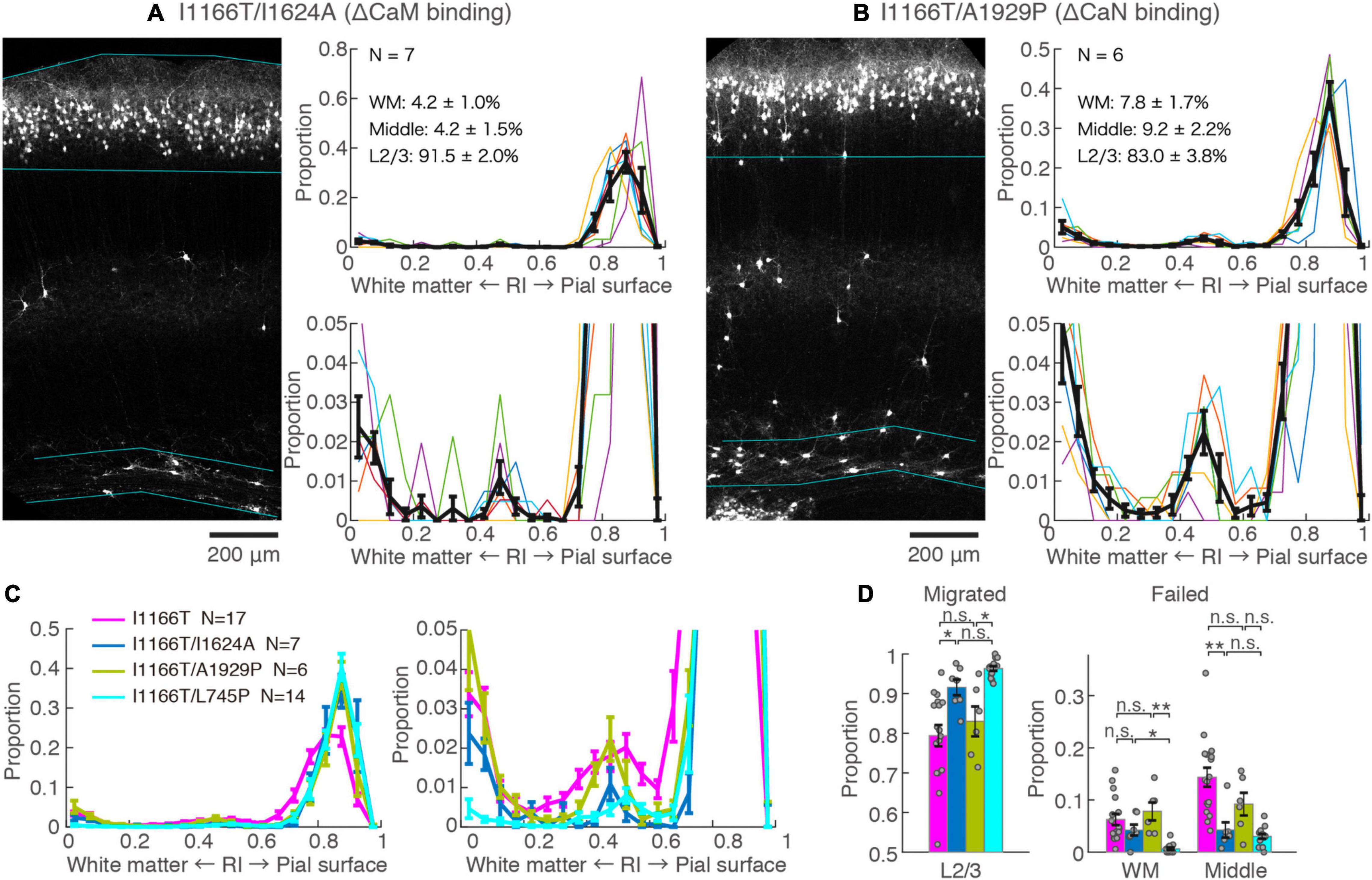
Figure 4. Blockade of CaM-binding but not CaN-binding rescues the migration deficit. (A,B) GFP images and histograms of the Cav1.2I1166T/I1624A (A) and Cav1.2I1166T/A1929P group (B), as shown in Figure 2A. (C) Histogram of the mean ± SEM are merged for Cav1.2WT, Cav1.2I1166T/I1624A, Cav1.2I1166T/A1929P, and Cav1.2I1166T/L745P groups. (D) Proportion of correctly migrated neurons (left) and that of migration-failed neurons located in WM and Middle (right). Independent values and mean ± SEM are shown. **P < 0.01; *P < 0.05; n.s., P ≥ 0.05.
Cav1.2 I1166T Mutation Interrupts Callosal Axon Arborization in the Contralateral Cortex Through Calcium-Dependent Pathway and β Subunit-Interaction
L2/3 neurons provide most axons projecting to the contralateral hemisphere (Fame et al., 2011). Such callosal axons arbor densely at the S1/S2 border. The arborization was dense in the upper layers and L5 (Mizuno et al., 2007). These projection patterns were seen in samples with neurons expressing GFP-alone (Figure 5A). Axons clustered at the S1/S2 border and densely arborized in the upper layers and L5. Callosal axons of the Cav1.2WT group were less arborized in the contralateral cortex and showed a moderate clustering in the S1/S2 border and in the upper layers and L5 (Figure 5B). On the other hand, callosal axons were largely diminished in the Cav1.2I1166T group, and clustering was unclear (Figure 5C). The axonal GFP signal strength was significantly lower in the Cav1.2WT group than in the GFP-alone group (L5, P = 8.2e−4; upper layer, P = 0.025), and in the Cav1.2I1166T group than in the Cav1.2WT group (L5, P = 0.70; upper layer, P = 0.023; Figures 5D,F). To further demonstrate that the I1166T mutation decreases callosal projection, we electroporated WT or I1166T α1 subunit-only (α2δ1 and β3 subunits were omitted). In this condition, exogenous α1 subunits should make a complex with endogenous α2δ1 and β3 subunits, thus the channel number increase would be alleviated. As we expected, both Cav1.2WT α1-only and Cav1.2I1166T α1-only groups showed increased callosal axon arborization compared with those expressing α2δ1 and β3 subunits together (P < 0.05 for all; Supplementary Figures 4A–E). The arborization of Cav1.2I1166T α1-only group was highly less than that of Cav1.2WT α1-only group (P = 0.0022 for both L5 and upper layer; Supplementary Figures 4D,E). Migration deficit was also worse in Cav1.2I1166T α1-only group than Cav1.2WT α1-only group (Supplementary Figures 4F–I). These results suggest that not only the Cav1.2 channel number but also the I1166T mutation decreases axon arborization in the contralateral cortex.
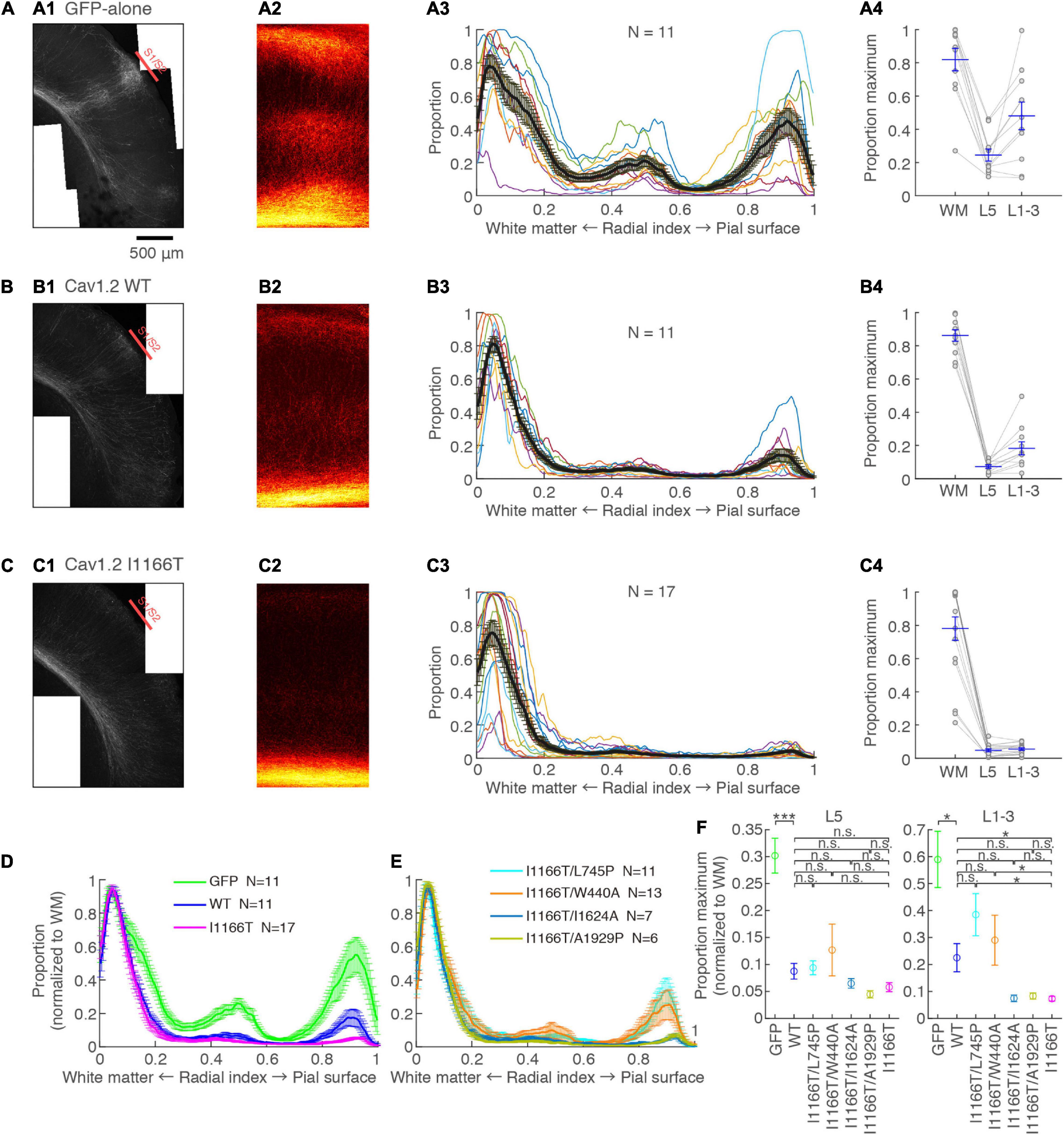
Figure 5. Axonal arborization in the contralateral cortex is affected by I1166T mutation, which is rescued by the blockade of calcium overload and β subunit-interaction. (A) Axonal arborization of the GFP-alone group. (A1) An example image of axonal arborization in the contralateral somatosensory area. (A2) Binarized images around the S1/S2 border of multiple samples were averaged and are shown. (A3) Proportion of pixels that have axonal signals are shown against the radial position. Colors for each sample: black, the mean ± SEM. (A4) Maximum proportion in the white matter (WM; Radial index = 0–0.1), layer 5 (L5; Radial index = 0.4–0.6), and upper layers (L1–3; Radial index = 0.8–1) of each sample; the mean ± SEM are shown. (B,C) Axonal arborization of Cav1.2WT- (B) and Cav1.2I1166T-expressing neurons (C) are shown as in (A). (D) Proportions that were normalized with the maximum proportion in the WM are shown for the GFP-alone, Cav1.2WT, and Cav1.2I1166T groups by the mean ± SEM. (E) Proportions for Cav1.2I1166T/L745P, Cav1.2I1166T/W440A, Cav1.2I1166T/I1624A, and Cav1.2I1166T/A1929P groups are shown as in (D). (F) Proportion in L5 (left) and L1–3 (right) for GFP, WT, and all mutation groups used in this study are shown by the mean ± SEM. ***P < 0.001; *P < 0.05; n.s., P ≥ 0.05.
We analyzed contralateral projection for all mutant groups used for migration analyses. Whereas projection significantly decreased in all mutant groups compared with the GFP-alone group, the Cav1.2I1166T/L745P and Cav1.2I1166T/W440A group had a greater projection than the Cav1.2I1166T group and was comparable to the Cav1.2WT group (Figures 5E,F and Supplementary Figure 5). However, the Cav1.2I1166T/I1624A group and the Cav1.2I1166T/A1929P group failed to improve projection (Figures 5E,F and Supplementary Figure 5). These results suggest that Ca2+ influx and β subunit-interaction are responsible for the callosal projection deficit in Cav1.2I1166T-expressing neurons.
Cav1.2 I1166T Mutation Does Not Affect Local Axon Arborizing in Layer 5 and Axonal Projection in the White Matter
L2/3 neurons make axonal branches densely in layer 5 in the same ipsilateral cortical area (Mizuno et al., 2007). A strong axonal GFP signal was seen in the middle of the cortical depth in the GFP-alone group (Figure 6A; Radial index = 0.4–0.6), which may correspond to layer 5. Axonal GFP signal intensities in layer 5 were similar among the GFP-alone, Cav1.2WT, and Cav1.2I1166T groups (Figure 6). Interestingly, migration-failed neurons in all Cav1.2 mutant-expressing groups tended to locate at the same depth (Supplementary Figures 6E,F). Layer 5 could attract not only L2/3 axons but also migrating L2/3 neurons in the developmental stage.
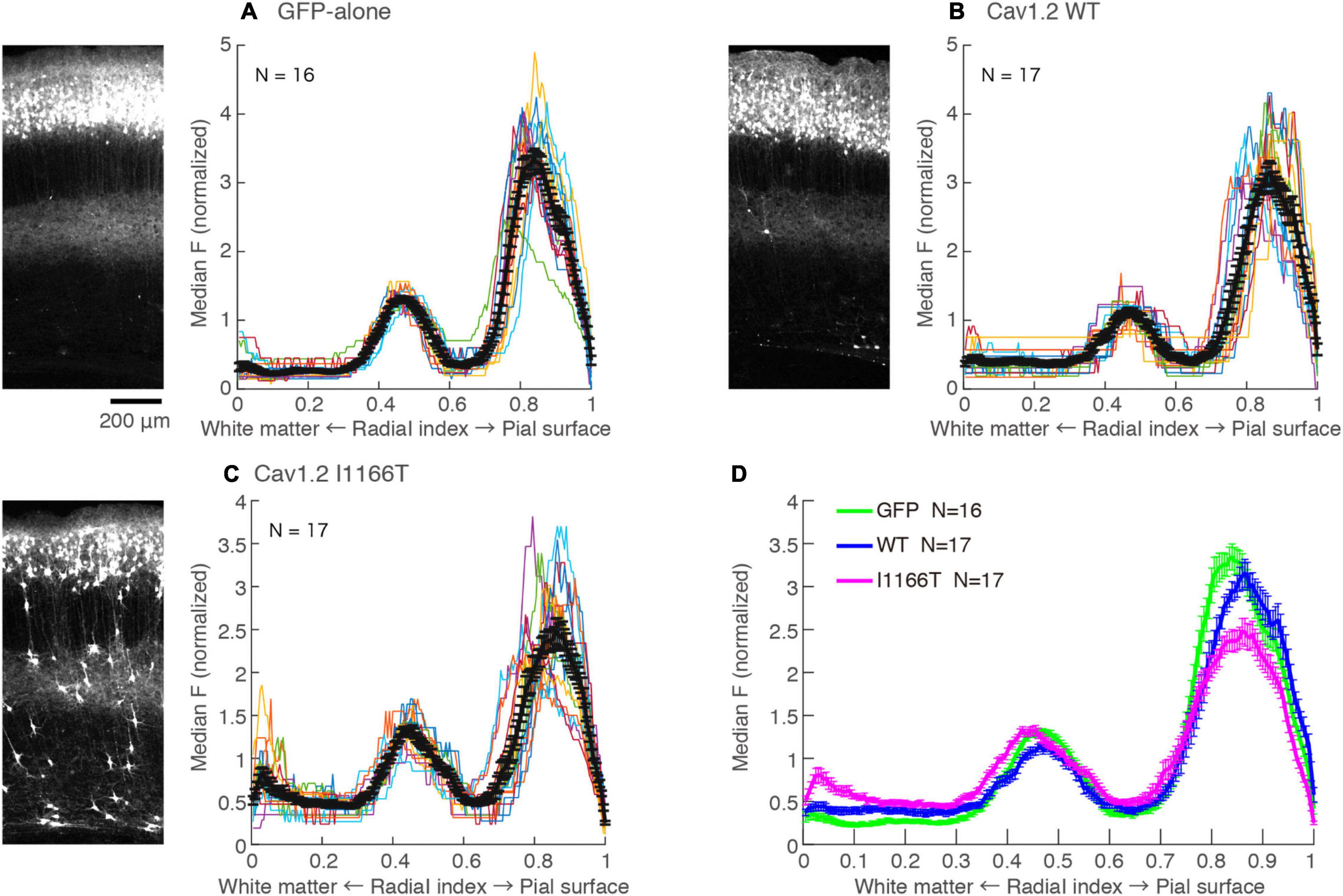
Figure 6. Local axonal arborization in the ipsilateral cortex is not affected by the expression of Cav1.2 mutants. (A) Left, An example GFP image in the electroporated hemisphere of GFP-alone group. A strong GFP signal was seen in the upper layer, representing cell bodies, dendrites, and axons of GFP-expressing neurons. A strong signal was also seen in the middle of the cortex, representing axons of neurons migrated to the L2/3. Right, the medians of GFP intensities are calculated along the tangential axis of the images. Colors, each sample. Black, the mean ± SEM. (B,C) GFP images and median intensities of Cav1.2WT group (B) and Cav1.2I1166T group (C), as shown in (A). Because migration-failed neurons were sparse in the middle of the cortex, their strong signals could be excluded and axonal signals could be obtained by calculating the medians. (D) The values of mean ± SEM of three groups are merged.
Next, we examined whether the mass of axons projecting in the white matter to the contralateral area differed. GFP intensities in the white matter were tested at the ipsilateral area, midline, and contralateral areas and were normalized with the intensity at the ipsilateral area. Similar signal strengths were seen in all locations among the GFP-alone, Cav1.2WT, and Cav1.2I1166T groups (Figures 7A–D). Because the axonal position in the white matter is one of the factors regulating the callosal projection (Zhou et al., 2013), we also analyzed whether axon bundles broadened in the white matter in the Cav1.2I1166T group. The broadness was also similar among the three groups (Figures 7A–C,E). Axonal projections in ipsilateral layer 5 and the white matter were similar for all mutant groups tested (Supplementary Figures 6A–E,G,H). These results suggest that signal cascades starting from Cav1.2 are less responsible for local axonal arborization and axonal elongation in the white matter but are crucial for axon invasion and arborization in the contralateral cortex.
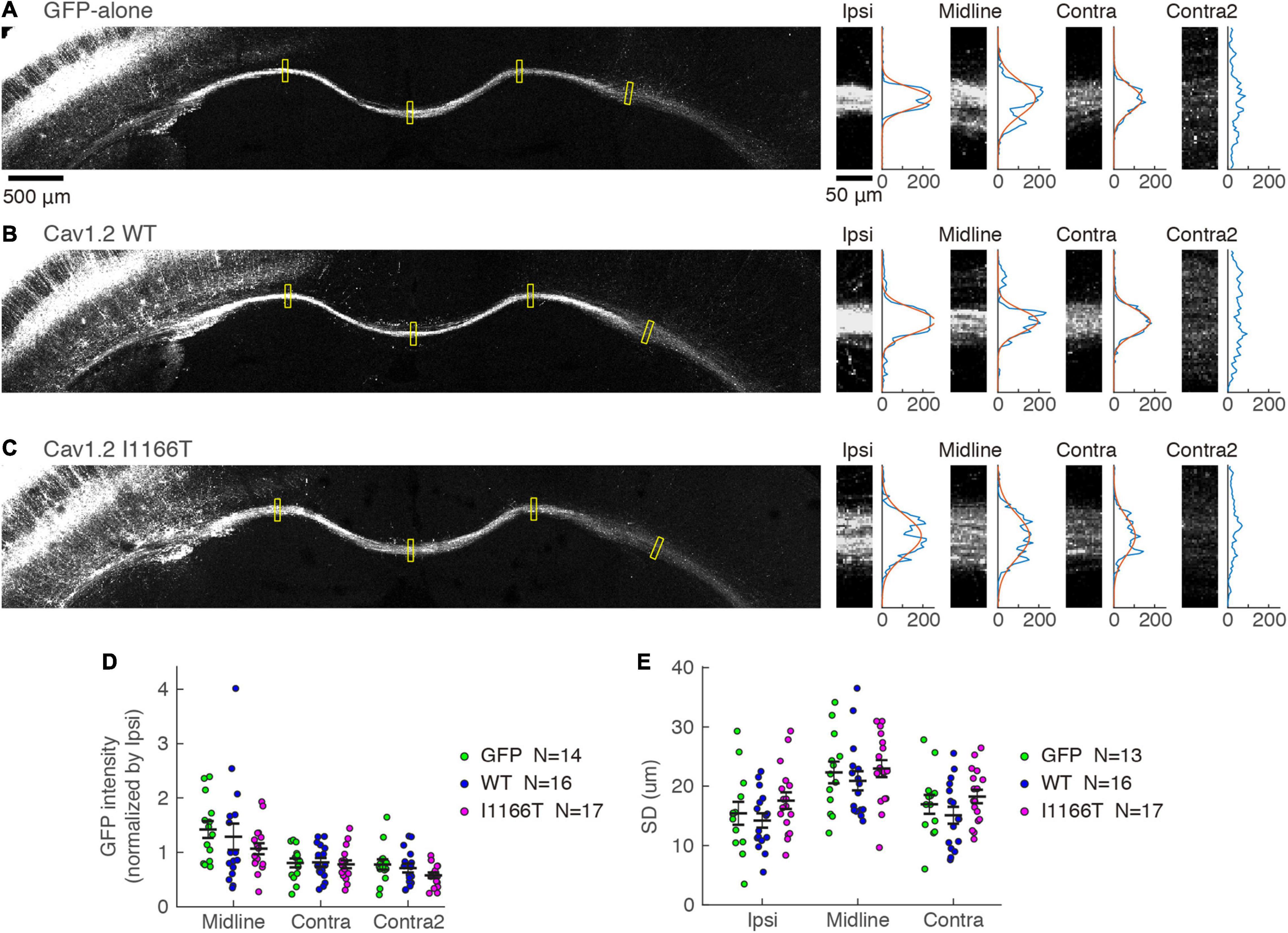
Figure 7. Axonal projection in the white matter is not affected by the Cav1.2 I1166T mutation. (A) Left, GFP images of white matter in the GFP-alone group. Yellow squares represent the four areas shown on the right. Right, GFP images of Ipsi, Midline, Contra, and Contra2 areas. Intensities of each area were averaged in the horizontal axis and shown on the right. Gaussian fit to the intensity distribution is shown in red. (B,C) Axonal projections of the Cav1.2WT (B) and Cav1.2I1166T (C) groups are shown as in (A). (D) GFP intensities normalized by that of Ipsi are shown for each sample and the mean ± SEM for the GFP-alone, Cav1.2WT, and Cav1.2I1166T groups. All the combination of groups had insignificant differences (P > 0.09). (E) Broadness of axon bundles in white matter, represented by the standard deviation of Gaussian curves, are shown for each sample and the mean ± SEM for the GFP-alone, Cav1.2WT, and Cav1.2I1166T groups. All the combination of groups had insignificant differences (P > 0.1).
Discussion
In this study, we used mouse layer 2/3 cortical neurons as a model to demonstrate that expressing a disease-causing Cav1.2 mutant, I1166T, affected migration and axonal projections. Neuronal migration is crucial for the formation of the organized structure of the cerebral cortex, and migration deficits can be associated with ASD, seizures, and intellectual disability (Pan et al., 2019). Callosal axons connect the two cortical hemispheres; thus, they are important for higher cognitive functions. Alterations in the corpus callosum have been noted in patients with ASD and other psychiatric and developmental disorders (Egaas et al., 1995; Paul, 2011). Our findings suggest that brain development and neural circuit connectivity can be affected by the Cav1.2 I1166T mutation. Because patients with the I1166T mutation are rare and the possible abnormalities in the brain have been rarely studied, our study raises the possibility that patients with the I1166T mutation have defects in neuronal migration and/or callosal projections.
Regarding neuronal migration, the I1166T mutation altered migration, and the addition of the L745P mutation that decreases calcium influx improved migration. The addition of the I1624A mutation inhibiting CaM-binding to the α1 subunit partially recovered migration, whereas the A1929P mutation that inhibits CaN-binding to the α1 subunit did not improve migration. The most plausible explanation for these observations may be that the increase in calcium influx to neurons with the I1166T mutation altered Ca2+-dependent gene expression via Ca2+-CaM binding to the channel and impaired migration.
What is the mechanism underlying the migration recovery in Cav1.2I1166T/I1624A in the current study? I1624 is responsible for binding to CaM; Ca2+-CaM binds to the C terminus of the Cav1.2 α1 subunit and modulates channel activity through the so-called Ca2+-dependent inactivation (CDI) and voltage-dependent inactivation (VDI) (Zühlke et al., 1999; Barrett and Tsien, 2008). Ca2+-CaM also regulates Ca2+-dependent gene expression (Deisseroth et al., 2003; Yap and Greenberg, 2018). The additional I1624A mutation may “cancel” the increased calcium influx caused by the I1166T mutation and/or normalize gene expression, by which the recovery of migration was observed in Cav1.2I1166T/I1624A. Consistent with this, it was shown that the I1624A mutation accelerated VDI in WT Cav1.2 (but not Cav1.2G406R; Barrett and Tsien, 2008). However, the inhibition of CaN-binding by the A1929P mutation has been shown to accelerate VDI of both the WT and G406R (Cohen-Kutner et al., 2012), but the Cav1.2I1166T/A1929P mutation failed to improve migration. The mutation could also impair CaN-dependent gene expression. The effects of the A1929P mutation on channel modulation and possible gene expression regulated by CaN might not have been strong enough to rescue the phenotype caused by the I1166T mutation.
A previous study showed that expression of Cav1.2 G406R, which is the original mutation identified in patients with TS, caused migration defects in the mouse cerebral cortex (Kamijo et al., 2018). Callosal axon projections were not assessed in that study. Both the G406R and I1166T mutations are considered to be gain-of-function, though the channel property of Cav1.2 G406R and I1166T appears somewhat different (Boczek et al., 2015; Wemhöner et al., 2015). Whether the G406R mutant channel also impairs callosal axon projections is an intriguing question. Additionally, many other “gain-of-function” mutations associated with patients are reported in Cav1.2 (Ozawa et al., 2018; Zhang et al., 2018; Marcantoni et al., 2020). All these patients show LQTS type 8 in their hearts, but they exhibit various neurologic symptoms. It is important to test whether these gain-of-function mutant channels affect brain development and connectivity in the mouse model.
Critical roles of Ca2+ signaling in cortical neuron migration and axonal projections have been recognized (Ageta-Ishihara et al., 2009; Hutchins et al., 2011; Sutherland et al., 2014; Horigane et al., 2019). In this study, approximately 80% of neurons migrated to the correct layer (i.e., layer 2/3), although they were transfected with Cav1.2 I1166T. Nonetheless, the callosal projections were almost completely perturbed by the Cav1.2 I1166T expression. The expression of Cav1.2WT only slightly affected migration but significantly affected callosal projections. These results suggest that axonal projection is more sensitive to Ca2+ overload than migration. The precise intracellular signaling mechanism by which the Cav1.2I1166T channel leads to the deficits remains elusive, and further studies are required.
Interestingly, the overexpression of the Cav1.2 WT or all the mutants tested altered axonal growth in the contralateral cortex but not in the white matter (Figures 5, 7). Thus, axonal elongation and midline crossing were less perturbed, and defects were evident after callosal axons entered the gray matter. It has been shown that various players that regulate the intracellular calcium signaling (i.e., Cav1.2, Wnt, and TRP channels) are critically involved in callosal axon elongation, midline crossing, and axonal branching (Ageta-Ishihara et al., 2009; Hutchins et al., 2011). Among them, Cav1.2-mediated calcium signaling may play a part in the gray matter but not in the white matter.
Our results from experiments using Cav1.2I1166T/W440A can be explained by two possible mechanisms. One is a crucial role of the interaction of Cav1.2 α1 subunit to the β subunit for membrane trafficking of the channels. In the literature, there are reports showing that W440A mutation affects channel trafficking and membrane expression of the WT channel (Obermair et al., 2010). If channel trafficking of the I1166T mutant is also dependent on the interaction to the β subunit through W440, then Cav1.2I1166T/W440A would not be transported to the membrane and therefore show the “rescued” phenotypes. The other possible mechanism is the involvement of the β subunit-dependent signaling pathway (Servili et al., 2018). There are reports showing that W440A does not affect channel trafficking but influence downstream signaling (Berrou et al., 2002; Servili et al., 2018; Yang et al., 2019). Previous in vitro expression studies using HEK cells showed the relevance of the Ca2+-independent, β subunit-dependent pathway for the regulation of gene expression downstream of the Cav1.2 WT and G406R channels (Servili et al., 2018, 2020). This pathway recruits Ras/ERK/CREB to trigger c-Fos and MeCP2 activation. In the current study (Figures 3, 5), the W440A mutation might not have had major effect on channel trafficking in our experimental conditions. For example, if Cav1.2I1166T/W440A was not transported to the membrane, we would have seen the migration and axonal projections of the Cav1.2I1166T/W440A group to the level equivalent to the “GFP only” group. In fact, this was not the case; the result suggests that Cav1.2I1166T/W440A would have been (at least partially) transported to the membrane and therefore have affected the migration and axonal projections. However, (1) to what extent the interaction with β subunit is essential for the membrane trafficking of the α1 subunit and (2) to what extent the specific amino acid residue W440 influences the channel trafficking and the downstream signaling remain to be elucidated in this study, and further studies are required.
There are some caveats in this study. First, this study used an overexpression strategy to reveal the effects of mutant channel expression on neuronal development. The strategy introduces mutant channels additional to the endogenously expressed channels; thus, changing the number of channels. Indeed, projections in the contralateral cortex were highly impaired in the WT group. Second, in utero electroporation, a gene transfection technique used in this study can introduce genes in a small number of cells in the brain. This is suitable for assessing the effect of expression at the cellular level, but linking the cellular phenotypes to behavior is difficult. A brain-wide knock-in strategy would be desirable for a more accurate study of the disease.
Ion channels play key roles in the formation and function of the nervous system (Medvedeva and Pierani, 2020; Smith and Walsh, 2020). Cav1.2 is critically involved in neural development. It also plays crucial roles in the heart, and patients with its mutations are relatively easily detectable by cardiac symptoms or testing. Indeed, many disease-causing, gain-of-function mutations are reported in Cav1.2, and their channel properties have been characterized in in vitro expression studies. Combining those studies with in vivo expression studies would facilitate our understanding of how each mutant channel alters the development, connectivity, and functions of the brain, which will eventually lead to a better understanding of the etiology of the neurological manifestations associated with the disease.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
Ethics Statement
The animal study was reviewed and approved by the Animal Experiments Committee and the Genetic Recombinant Experiment Safety Committee of Kagoshima University.
Author Contributions
HN and YT designed the study. KS designed the plasmids expressing Cav1.2. NN-T and EK conducted the experiments and analyzed the data. NN-T and YT wrote the manuscript. All authors checked and approved the manuscript.
Funding
This work was supported by the collaboration research grant with Sumitomo Dainippon Pharma Co., Ltd.; Japan Society for Promotions of Sciences (JSPS) KAKENHI (Grant Nos. 16K06992 and 21K06374 to YT; 16K14565, 19K06887, and 20K06876 to NN-T), “Dynamic Regulation of Brain Function by Scrap and Build System” (17H05745 and 19H04756 to YT), Astellas Foundation for Research on Metabolic Disorders (to YT), the Kodama Memorial Fund for Medical Research (to YT and NN-T), the Novartis Foundation (Japan) for the Promotion of Science (to YT), the Uehara Memorial Foundation (to YT), the Core Research Program for “Neuroscience Core Unit” by Kagoshima University, the Kato Memorial Bioscience Foundation (to NN-T), and the Sasakawa Scientific Research Grant (to NN-T).
Conflict of Interest
KS and HN were employees of Sumitomo Dainippon Pharma Co., Ltd. This study received funding from Sumitomo Dainippon Pharma Co., Ltd. The funder had the following involvement with the study: study design. The funder was not involved in data collection, analysis, interpretation, the writing of this article or the decision to submit it for publication.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank C. Kawagoe, K. Tashiro, K. Fujisawa, K. Omori (Kagoshima University) for the help in conducting experiments; the Joint Research Center for Human Retrovirus Infection in Kagoshima University for the use of confocal laser-scanning microscope; all the members of Tagawa and Okuno laboratories and E. Kuramoto (Kagoshima University) for useful discussions and support.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnins.2021.747951/full#supplementary-material
References
Abrahams, B. S., and Geschwind, D. H. (2008). Advances in autism genetics: on the threshold of a new neurobiology. Nat. Rev. Genet. 9, 341–355. doi: 10.1038/nrg2346
Ageta-Ishihara, N., Takemoto-Kimura, S., Nonaka, M., Adachi-Morishima, A., Suzuki, K., Kamijo, S., et al. (2009). Control of cortical axon elongation by a GABA-driven Ca2+/ calmodulin-dependent protein kinase cascade. J. Neurosci. 29, 13720–13729. doi: 10.1523/JNEUROSCI.3018-09.2009
Bando, Y., Hirano, T., and Tagawa, Y. (2014). Dysfunction of KCNK potassium channels impairs neuronal migration in the developing mouse cerebral cortex. Cereb. Cortex 24, 1017–1029. doi: 10.1093/cercor/bhs387
Barrett, C. F., and Tsien, R. W. (2008). The Timothy syndrome mutation differentially affects voltage- and calcium-dependent inactivation of CaV1.2 L-type calcium channels. Proc. Natl. Acad. Sci. U. S. A. 105, 2157–2162. doi: 10.1073/pnas.0710501105
Berrou, L., Klein, H., Bernatchez, G., and Parent, L. (2002). A specific tryptophan in the I-II linker is a key determinant of beta-subunit binding and modulation in Ca(V)2.3 calcium channles. Biophys. J. 83, 1429–1442. doi: 10.1016/S0006-3495(02)73914-3
Boczek, N. J., Miller, E. M., Ye, D., Nesterenko, V. V., Tester, D. J., Antzelevitch, C., et al. (2015). Novel Timothy syndrome mutation leading to increase in CACNA1C window current. Heart Rhythm 12, 211–219. doi: 10.1016/j.hrthm.2014.09.051
Cohen-Kutner, M., Yahalom, Y., Trus, M., and Atlas, D. (2012). Calcineurin controls Voltage-Dependent-Inactivation (VDI) of the normal and timothy cardiac channels. Sci. Rep. 2:366. doi: 10.1038/srep00366
Courchesne, E., Gazestani, V. H., and Lewis, N. E. (2020). Prenatal origins of ASD: the when, what, and how of ASD development. Trends Neurosci. 43, 326–342. doi: 10.1016/j.tins.2020.03.005
Deisseroth, K., Mermelstein, P. G., Xia, H., and Tsien, R. W. (2003). Signaling from synapse to nucleus: the logic behind the mechanisms. Curr. Opin. Neurobiol. 13, 354–365. doi: 10.1016/S0959-4388(03)00076-X
Egaas, B., Courchesne, E., and Saitoh, O. (1995). Reduced Size of Corpus Callosum in Autism. Arch. Neurol. 52, 794–801. doi: 10.1001/archneur.1995.00540320070014
Fame, R. M., MacDonald, J. L., and Macklis, J. D. (2011). Development, specification, and diversity of callosal projection neurons. Trends Neurosci. 34, 41–50. doi: 10.1016/j.tins.2010.10.002
Horigane, S.-I., Ozawa, Y., Yamada, H., and Takemoto-Kimura, S. (2019). Calcium signalling: a key regulator of neuronal migration. J. Biochem. 165, 401–409. doi: 10.1093/jb/mvz012
Hutchins, B. I., Li, L., and Kalil, K. (2011). Wnt/calcium signaling mediates axon growth and guidance in the developing corpus callosum. Dev. Neurobiol. 71, 269–283. doi: 10.1002/dneu.20846
Kabir, Z. D., Martínez-Rivera, A., and Rajadhyaksha, A. M. (2017). From gene to behavior: L-Type calcium channel mechanisms underlying neuropsychiatric symptoms. Neurotherapeutics 14, 588–613. doi: 10.1007/s13311-017-0532-0
Kamijo, S., Ishii, Y., Horigane, S.-I., Suzuki, K., Ohkura, M., Nakai, J., et al. (2018). A Critical neurodevelopmental role for L-Type Voltage-Gated calcium channels in neurite extension and radial migration. J. Neurosci. 38, 5551–5566. doi: 10.1523/JNEUROSCI.2357-17.2018
Krey, J. F., Paşca, S. P., Shcheglovitov, A., Yazawa, M., Schwemberger, R., Rasmusson, R., et al. (2013). Timothy syndrome is associated with activity-dependent dendritic retraction in rodent and human neurons. Nat. Neurosci. 16, 201–209. doi: 10.1038/nn.3307
Marcantoni, A., Calorio, C., Hidisoglu, E., Chiantia, G., and Carbone, E. (2020). Cav1.2 channelopathies causing autism: new hallmarks on Timothy syndrome. Pflugers Arch. 472, 775–789. doi: 10.1007/s00424-020-02430-0
Medvedeva, V. P., and Pierani, A. (2020). How do electric fields coordinate neuronal migration and maturation in the developing cortex? Front. Cell Dev. Biol. 8:580657. doi: 10.3389/fcell.2020.580657
Mizuno, H., Hirano, T., and Tagawa, Y. (2007). Evidence for activity-dependent cortical wiring: formation of interhemispheric connections in neonatal mouse visual cortex requires projection neuron activity. J. Neurosci. 27, 6760–6770. doi: 10.1523/JNEUROSCI.1215-07.2007
Obermair, G. J., Schlick, B., Biase, V. D., Subramanyam, P., Gebhart, M., Baumgartner, S., et al. (2010). Reciprocal interactions regulate targeting of calcium channel beta subunits and membrane expression of alpha1 subunits in cultured hippocampal neurons. J. Biol. Chem. 285, 5776–5791. doi: 10.1074/jbc.M109.044271
Ozawa, J., Ohno, S., Saito, H., Saitoh, A., Matsuura, H., and Horie, M. (2018). A novel CACNA1C mutation identified in a patient with Timothy syndrome without syndactyly exerts both marked loss- and gain-of-function effects. HeartRhythm Case Rep. 4, 273–277. doi: 10.1016/j.hrcr.2018.03.003
Pan, Y.-H., Wu, N., and Yuan, X.-B. (2019). Toward a better understanding of neuronal migration deficits in autism spectrum disorders. Front. Cell Dev. Biol. 7:205. doi: 10.3389/fcell.2019.00205
Panagiotakos, G., Haveles, C., Arjun, A., Petrova, R., Rana, A., Portmann, T., et al. (2019). Aberrant calcium channel splicing drives defects in cortical differentiation in timothy syndrome. Elife 8:e51037. doi: 10.7554/eLife.51037
Paşca, S. P., Portmann, T., Voineagu, I., Yazawa, M., Shcheglovitov, A., Paşca, A. M., et al. (2011). Using iPSC-derived neurons to uncover cellular phenotypes associated with Timothy syndrome. Nat. Med. 17, 1657–1662. doi: 10.1038/nm.2576
Paul, L. K. (2011). Developmental malformation of the corpus callosum: a review of typical callosal development and examples of developmental disorders with callosal involvement. J. Neurodev. Disord. 3, 3–27. doi: 10.1007/s11689-010-9059-y
Saito, T., and Nakatsuji, N. (2001). Efficient gene transfer into the embryonic mouse brain using in vivo electroporation. Dev. Biol. 240, 237–246. doi: 10.1006/dbio.2001.0439
Servili, E., Trus, M., Maayan, D., and Atlas, D. (2018). β-Subunit of the voltage-gated Ca2+ channel Cav1.2 drives signaling to the nucleus via H-Ras. Proc. Natl. Acad. Sci. U. S. A. 115, E8624–E8633. doi: 10.1073/pnas.1805380115
Servili, E., Trus, M., Sajman, J., Sherman, E., and Atlas, D. (2020). Elevated basal transcription can underlie timothy channel association with autism related disorders. Progr. Neurobiol. 191:101820. doi: 10.1016/j.pneurobio.2020.101820
Smith, R. S., and Walsh, C. A. (2020). Ion channel functions in early brain development. Trends Neurosci. 43, 103–114. doi: 10.1016/j.tins.2019.12.004
Splawski, I., Timothy, K. W., Sharpe, L. M., Decher, N., Kumar, P., Bloise, R., et al. (2004). Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell 119, 19–31. doi: 10.1016/j.cell.2004.09.011
Sutherland, D. J., Pujic, Z., and Goodhill, G. J. (2014). Calcium signaling in axon guidance. Trends Neurosci. 37, 424–432. doi: 10.1016/j.tins.2014.05.008
Tabata, H., and Nakajima, K. (2001). Efficient in utero gene transfer system to the developing mouse brain using electroporation: visualization of neuronal migration in the developing cortex. Neuroscience 103, 865–872. doi: 10.1016/s0306-4522(01)00016-1
Tandan, S., Wang, Y., Wang, T. T., Jiang, N., Hall, D. D., Hell, J. W., et al. (2009). Physical and functional interaction between calcineurin and the cardiac L-type Ca2+ channel. Circ. Res. 105, 51–60. doi: 10.1161/CIRCRESAHA.109.199828
Wemhöner, K., Friedrich, C., Stallmeyer, B., Coffey, A. J., Grace, A., Zumhagen, S., et al. (2015). Gain-of-function mutations in the calcium channel CACNA1C (Cav1.2) cause non-syndromic long-QT but not Timothy syndrome. J. Mol. Cell. Cardiol. 80, 186–195. doi: 10.1016/j.yjmcc.2015.01.002
Xu, H., Ginsburg, K. S., Hall, D. D., Zimmermann, M., Stein, I. S., Zhang, M., et al. (2010). Targeting of protein phosphatases PP2A and PP2B to the C-terminus of the L-type calcium channel Ca v1.2. Biochemistry 49, 10298–10307. doi: 10.1021/bi101018c
Yang, L., Katchman, A., Kushner, J., Kushnir, A., Zakharov, S. I., Chen, B. X., et al. (2019). Cardiac CaV1.2 channels require b subunits for b-adrenergic-mediated modulation but not trafficking. J. Clin. Investig. 129, 647–658. doi: 10.1172/JCI123878
Yap, E.-L., and Greenberg, M. E. (2018). Activity-regulated transcription: bridging the gap between neural activity and behavior. Neuron 100, 330–348. doi: 10.1016/j.neuron.2018.10.013
Zhang, Q., Chen, J., Qin, Y., Wang, J., and Zhou, L. (2018). Mutations in voltage-gated L-type calcium channel: implications in cardiac arrhythmia. Channels 12, 201–218. doi: 10.1080/19336950.2018.1499368
Zhou, J., Wen, Y., She, L., Sui, Y. N., Liu, L., Richards, L. J., et al. (2013). Axon position within the corpus callosum determines contralateral cortical projection. Proc. Natl. Acad. Sci. U. S. A. 110, E2714–E2723. doi: 10.1073/pnas.1310233110
Keywords: Timothy syndrome, calcium channel, radial migration, callosal projection, neocortex
Citation: Nakagawa-Tamagawa N, Kirino E, Sugao K, Nagata H and Tagawa Y (2021) Involvement of Calcium-Dependent Pathway and β Subunit-Interaction in Neuronal Migration and Callosal Projection Deficits Caused by the Cav1.2 I1166T Mutation in Developing Mouse Neocortex. Front. Neurosci. 15:747951. doi: 10.3389/fnins.2021.747951
Received: 27 July 2021; Accepted: 11 November 2021;
Published: 08 December 2021.
Edited by:
Hidenobu Mizuno, Kumamoto University, JapanReviewed by:
Sayaka Takemoto-Kimura, Nagoya University, JapanLorenzo A. Cingolani, Italian Institute of Technology (IIT), Italy
Copyright © 2021 Nakagawa-Tamagawa, Kirino, Sugao, Nagata and Tagawa. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yoshiaki Tagawa, dGFnYXdhQG0ua3VmbS5rYWdvc2hpbWEtdS5hYy5qcA==