 Luca Goffin
Luca Goffin Damien Lemoine
Damien Lemoine Frédéric Clotman
Frédéric Clotman- Université catholique de Louvain, Louvain Institute of Biomolecular Science and Technology, Animal Molecular and Cellular Biology, Louvain-la-Neuve, Belgium
Amyotrophic lateral sclerosis (ALS) consists of a group of adult-onset fatal and incurable neurodegenerative disorders characterized by the progressive death of motor neurons (MNs) throughout the central nervous system (CNS). At first, ALS was considered to be an MN disease, caused by cell-autonomous mechanisms acting specifically in MNs. Accordingly, data from ALS patients and ALS animal models revealed alterations in excitability in multiple neuronal populations, including MNs, which were associated with a variety of cellular perturbations such as protein aggregation, ribonucleic acid (RNA) metabolism defects, calcium dyshomeostasis, modified electrophysiological properties, and autophagy malfunctions. However, experimental evidence rapidly demonstrated the involvement of other types of cells, including glial cells, in the etiopathogenesis of ALS through non-cell autonomous mechanisms. Surprisingly, the contribution of pre-motor interneurons (INs), which regulate MN activity and could therefore critically modulate their excitability at the onset or during the progression of the disease, has to date been severely underestimated. In this article, we review in detail how spinal pre-motor INs are affected in ALS and their possible involvement in the etiopathogenesis of the disease.
Introduction
Amyotrophic lateral sclerosis (ALS), also known as Motor Neurone Disease (MND) or Lou Gehrig's disease in the US, consists of a group of adult-onset fatal and incurable neurodegenerative disorders characterized by the progressive death of both cortical upper motor neurons (UMNs) and of brainstem or spinal lower motor neurons (LMNs), leading to fasciculation, spasticity, muscular atrophy and weakness, and eventually paralysis and respiratory failure (Ravits et al., 2007). In Europe, about 50.000 individuals of middle age are affected by ALS, and about 10.000 people die from ALS each year. Worldwide 450.000 people suffer from this pathology, and 120,000 people are diagnosed with ALS annually (Masrori and Van Damme, 2020). The clinical onset of ALS typically occurs between the ages of 40 and 70, and once diagnosed, the disease progresses rapidly, usually leading to the death of the patient from respiratory failure within 3–5 years (Dhasmana et al., 2022). Approximately 90% of ALS cases, known as sporadic ALS (sALS), occur without a familial history. The etiology of sALS remains largely unknown, but some sALS patients have de novo mutations found in familial forms. Indeed, the remaining cases, known as familial ALS (fALS), are linked to heritable mutations. Among the most common mutated genes are chromosome 9 open reading frame 72 (c9orf72, ≈ 40% of fALS), superoxide dismutase 1 (SOD1, ≈ 20%), fused in sarcoma (FUS, ≈ 4%), optineurin (OPTN2, ≈ 4%), and TARDBP (TDP-43, ≈ 5%) (Taylor et al., 2016). Interestingly, cytoplasmic inclusions of TDP-43 are found in a large majority (≈95%) of all ALS cases, regardless of familial or sporadic status (Kwong et al., 2007). Although ALS affects people of all ethnicities, the prevailing rates of these mutations and clinical symptoms of the disease vary across ethnic groups (Mesaros et al., 2021).
ALS symptoms can be classified into two categories based on the central nervous system (CNS) region where MN degeneration begins. Bulbar symptoms, such as dysarthria and dysphagia, are caused by the loss of brachial/cervical MNs. Spinal symptoms, such as skeletal muscular atrophy and loss of strength in the limbs, result from spinal MN degeneration. However, in both cases, the disease extends over time, eventually affecting all regions of the CNS. The primary cause of death in ALS is respiratory failure due to alterations in MNs controlling the muscles involved in ventilation (see below). Visceral MNs, which innervate cardiac muscle fibers, smooth muscles of visceral organs, and glands, are rarely affected by the disease. In contrast, the most vulnerable MN types in ALS are somatic MNs that innervate fast-twitch fatigable (FF) muscle fibers (Pun et al., 2002; Wootz et al., 2013; Sharma et al., 2016; Spiller et al., 2016). The differential vulnerability of MN subtypes to ALS can be linked to several intrinsic or extrinsic factors such as soma or dendritic tree size, intracellular calcium dynamics, and excitatory/inhibitory synaptic inputs (Nijssen et al., 2017; Ovsepian et al., 2024).
ALS was initially considered an MN disease, caused by autonomous mechanisms affecting specifically these cells. Thus, the research efforts were initially focused on MN and enabled the uncovering of different intrinsic pathological mechanisms. ALS appears to be caused by changes in excitability in multiple neuronal populations, including MNs (Benedetti et al., 2016; Chenji et al., 2016; Kim et al., 2017; Scekic-Zahirovic et al., 2024), which are caused by or associated with a variety of perturbations such as protein aggregation, ribonucleic acid (RNA) metabolism defects, calcium dyshomeostasis, modified electrophysiological properties, and autophagy malfunctions, among others (Srinivasan and Rajasekaran, 2020). However, although the neuronal expression of mutated genes associated with fALS forms is sufficient to induce symptoms related to ALS (Jaarsma et al., 2008; Wang et al., 2009; Huang et al., 2012; Sharma et al., 2016), experimental evidence rapidly demonstrated the involvement of other cell types through non-cell autonomous mechanisms (reviewed in Crabe et al., 2020; Srinivasan and Rajasekaran, 2020; Van Harten et al., 2021). Accordingly, genetic manipulations of ALS genes in astrocytes (Yamanaka et al., 2008b), oligodendrocytes (Kim et al., 2019), and microglial cells (Boillee et al., 2006; Wang et al., 2009) resulted in changes in the progression of the disease in animal models (Yamanaka et al., 2008a). Cell transplantation and co-culture or chimera studies have confirmed the deleterious contribution of astrocytes (Nagai et al., 2007; Qian et al., 2017; Arredondo et al., 2022) and other glial cells (Clement et al., 2003). Surprisingly, the contribution of brainstem or spinal cord pre-motor interneurons (INs), which regulate the activity of LMNs and could therefore critically modulate their excitability at the beginning or during the progression of the disease, has to date been severely underestimated (Gunes et al., 2020; Van Harten et al., 2021).
Motor circuitry of the spinal cord and ALS
Spinal somatic motor circuits are composed of MNs, which are the effectors of the system and directly control the contraction of skeletal muscles, as well as pre-motor INs, which are connected to each other or directly to MNs and eventually regulate MN activity. Despite numerous limitations in properly defining the diversity of INs in the adult spinal cord (Dougherty, 2023; Roome and Levine, 2023; Dominguez-Bajo and Clotman, 2024), these cells are usually classified according to their embryonic origin and targeted for genetic modifications or lineage-tracing using embryonic-specific markers (Lu et al., 2015). The ventral neural tube contains five IN progenitor domains (pd6, p0, p1, p2, and p3) that produce different pre-motor IN populations (dI6, V0, V1, V2, and V3, respectively) (Figure 1). These cardinal classes of INs have been divided into main populations characterized by unique markers, such as Chx10 for V2a, Gata3 for V2b, Sox1 for V2c, and Shox2 for V2d INs (Wilson and Sweeney, 2023), which can be further subdivided based on the expression of other genes, such as Sp8, FoxP2, Pou6f2, or MafA for V1 INs or Onecut factors Prdm8, Bhlhb5, MafA, or cMaf for V2 INs (Francius et al., 2013; Bikoff et al., 2016; Gosgnach et al., 2017; Sweeney et al., 2018; Harris et al., 2019). Consequently, subpopulations of INs within the same cardinal class can vary in their location, synaptic targets, activity, and function (Borowska et al., 2013, 2015; Bikoff et al., 2016; Sweeney et al., 2018; Deska-Gauthier et al., 2024). Pre-motor INs regulate MN activity through excitatory, inhibitory, or modulatory synapses. V0v, V0g, V2a, V3, and Hb9-INs are excitatory glutamatergic neurons; V0d, V1, and V2b INs are inhibitory gamma-aminobutyric acid (GABA)ergic/glycinergic neurons; V0c are modulatory cholinergic INs projecting directly on MNs and on V1-derived Ia INs (IaINs) (see below); neurotransmitters used by the other IN subsets have not been identified yet (Figure 1; reviewed among others in Ziskind-Conhaim and Hochman, 2017). At the brachial and lumbar levels, most of these IN populations collectively constitute central pattern generators (CPGs), i.e., the neuronal networks responsible for the general coordination of locomotion (Ramirez-Jarquin and Tapia, 2018). Together, INs and MNs form motor circuitry units within the spinal cord, allowing an accurate activation pattern of spinal MNs for specific motor tasks such as object grasping, body orientation, or locomotion.
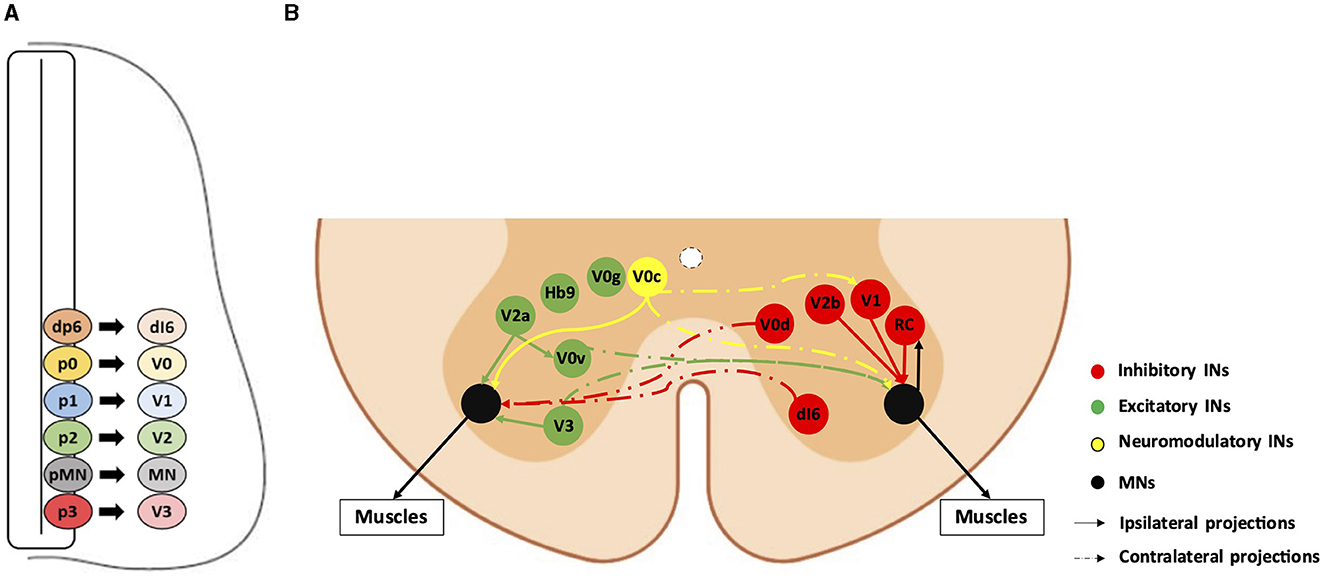
Figure 1. Diversity and connectivity of spinal ventral neuronal populations. A schematic representation of an embryonic transverse hemisection (A) or the ventral half of an adult transverse section (B) in the spinal cord is shown here. Ventral is to the bottom, dorsal is to the top. (A) The six most ventral progenitor domains (dp6, p0, p1, p2, pMN, and p3) and each cardinal neuronal population (dI6, V0, V1, V2, and V3 interneurons [INs], and motor neurons [MN]) arising from their respective progenitor domains. The other half of the spinal cord, as well as the dorsal progenitor domains and neuronal populations, are not represented. (B) The MNs and the main pre-motor IN populations involved in the spinal motor circuitry and their known connectivity are represented by colored bubbles (cell body localization) and arrows (projections). For clarity, excitatory or neuromodulatory populations are represented on the left hemicord, and inhibitory populations are represented on the right hemicord, although all these populations are evidently present on both sides. Hb9-IN (the origin of which remains elusive) and V0g connectivity are not precisely known. RC: Renshaw cells (V1 IN subset).
A tight balance between the excitation and inhibition levels in motor circuits is crucial to maintaining proper neuronal activity. MN excitotoxicity is one of the mechanisms that contribute to ALS, although the extent of this contribution remains debated (Leroy et al., 2014; Leroy and Zytnicki, 2015). Excitotoxicity, defined as a detrimental response to excitatory stimulations, notably due to poor handling of excessive Ca2+, can result from intrinsic hyperresponsiveness of the postsynaptic neuron to physiological glutamate levels or from extrinsic factors including increased extracellular synaptic glutamate concentrations or presynaptic excitation/inhibition imbalance (Van Den Bosch et al., 2006; King et al., 2016). In ALS mouse or zebrafish models, prenatal or postnatal MNs display hyperexcitability in synaptic and firing activities (van Zundert et al., 2008; Martin et al., 2013; Leroy et al., 2014; Benedetti et al., 2016; Jiang et al., 2017) and are intrinsically more responsive to stimulation (Jensen et al., 2020), although hypoexcitability of an MN subset has also been reported (Filipchuk et al., 2021). These defects could be amplified by the alteration of synaptic glutamate recapture by astrocytes via excitatory amino acid transporter 2 (EAAT2, also named glutamate transporter 1 or GLT1) dysfunction (Rosenblum and Trotti, 2017), suggesting a significant contribution of astrocytes to excitotoxicity. However, despite evidence that this mechanism participates in disease progression, inhibition of glutamate uptake alone is not sufficient to recapitulate spinal MN degeneration (Tovar-Y-Romo et al., 2009; Yin and Weiss, 2012). Furthermore, restoration of the glutamate retake function using GLT1 overexpression does not protect mice expressing human sod1 carrying the G93A mutation found in fALS forms from motor impairment and premature death or delay disease progression (Li et al., 2015). Together, these findings suggest that glutamate transport alterations worsen disease progression in already weakened MNs but fail to explain disease onset.
In parallel to cell-autonomous changes that may modify the excitability of spinal MNs and to the extrinsic contribution of glial cells to these modifications, alterations of pre-motor INs could modulate MN excitability in a non-cell autonomous manner and thereby contribute to the onset or progression of ALS (Morrison et al., 1998; Stephens et al., 2006; Jiang et al., 2009; Hossaini et al., 2011; Benedetti et al., 2016; Qian et al., 2017; Quinlan et al., 2017; Brownstone and Lancelin, 2018). Consistently, in different animal models, INs have been reported to show alterations before MN degeneration in the course of the disease (Niessen et al., 2007; Chang and Martin, 2009; Martin and Chang, 2012; Casas et al., 2013; McGown et al., 2013; Gallart-Palau et al., 2014; Milan et al., 2015; Medelin et al., 2016; Cavarsan et al., 2023). Furthermore, a genome-wide association study (GWAS) in patients provides evidence for cell-autonomous pathology initiation in glutamatergic neurons (van Rheenen et al., 2021), potentially including excitatory spinal INs and resulting in subsequent alterations of spinal MN activity. Consistently, transplanting spinal neural progenitors derived from human sALS-induced pluripotent stem cells in the spinal cord of severe combined immunodeficient (SCID) mice results in endogenous IN alterations that precede MN loss (Qian et al., 2017), suggesting that spinal INs may be more vulnerable than MNs in ALS. Alternatively, the study of a rat model with TARDBP overexpression indicated that some IN populations may exert a protective action against MN degeneration (Huang et al., 2012). In this article, we will review in detail how spinal pre-motor INs are affected in ALS and their possible involvement in the etiopathogenesis of the disease.
Alterations of spinal inhibitory pre-motor INs
Spinal MN activity is tightly dampened by multiple pre-motor GABAergic and/or glycinergic inhibitory IN populations (Figure 1). Numerous studies have reported modifications in GABAergic/glycinergic signaling in the context of ALS. In transgenic SOD1G93A mice, the inhibitory V1 INs (derived from embryonic Engrailed1-positive cells) are reduced in number from the early presymptomatic stages of the disease (Salamatina et al., 2020; Allodi et al., 2021; Montanana-Rosell et al., 2024). The V1 cardinal population contains IaINs involved in the alternation of flexor- or extensor-innervating MN activity during movement (Zhang et al., 2014; Britz et al., 2015), Renshaw cells, which ensure recurrent inhibition on MNs (Alvarez et al., 2005), and other yet unidentified populations. Each V1 IN subset is not equally affected in ALS, suggesting that V1 inhibitory neurons fall into ALS-susceptible or ALS-resistant subpopulations (Salamatina et al., 2020; Montanana-Rosell et al., 2024). This finding is in line with previous results obtained with the same ALS mouse model showing that the number of Renshaw cells is significantly reduced only at the late stages of the disease (Chang and Martin, 2009; Wootz et al., 2013). However, the later degeneration of Renshaw cells may be secondary to the loss of cholinergic input from MNs that parallels MN degeneration (Casas et al., 2013; Wootz et al., 2013). Reduction in neuronal activity of the V1 INs results in a slowing of the motor speed similar to that observed in the early stages of ALS progression in SOD1G93A mice (Gosgnach et al., 2006; Salamatina et al., 2020), suggesting that V1 alterations could be causal to the onset of the disease (Figure 2). Consistently, the presynaptic organizer Extended Synaptotagmin 1 (Esyt1) is downregulated early in V1 INs, and its viral delivery in the spinal cord or overexpression in V1 INs restored V1-MN connectivity, reduced the onset of motor impairment, increased MN survival, and ameliorated motor phenotype (Mora et al., 2024). In contrast, dampening of the V1 activity after the onset of symptoms does not change the SOD1G93A locomotor phenotype (Allodi et al., 2021), indicating that subsequent V1 alterations may not further contribute to the progression of the pathology.

Figure 2. Spinal neuronal inhibitory circuitry and its fate in ALS. Inhibitory interneurons (INs) are represented in red, and motor neurons (MNs) in gray. Altered INs and MNs are represented in light red and light gray, respectively. (A) Inhibitory circuitry in a non-pathological situation. (B) Early alteration of the inhibitory circuitry before the apparition of the first symptoms. The decrease in glycinergic signaling and in the recurrent inhibition loop ensured by Renshaw cells might act as compensatory mechanisms that preserve motor activity despite low-noise early MN alterations before deleterious breakdown of the circuitry. This possibly results in MN hyperexcitability, represented by spikes on the cell body. This hyperexcitability may contribute to MN excitotoxicity. Panel B summarizes in a single scheme the different alterations involving spinal inhibitory INs, including Renshaw cells, whatever their chronology (for more details, see the proposed working hypothesis in the last section of this article). (C) Alterations of the inhibitory circuitry during the symptomatic phase of the disease. Degenerative cells are represented by dotted lines on the cell body and projections. It is still unclear whether degeneration of MNs occurs before or after degeneration of inhibitory INs and if one is causative of the other. RC, Renshaw cells.
V1 INs establish GABAergic and glycinergic synapses on MNs. Glycinergic synapses are more abundant on MNs innervating fast muscle fibers (fast MNs) than on slow MNs, and V1 has stronger inputs on fast MNs than on slow MNs (Allodi et al., 2021). Interestingly, fast MNs are more affected than slow MNs during ALS progression (see above). Furthermore, SOD1G93A mice or zebrafish display a selective loss of glycinergic inputs on fast MNs before MN degeneration (Hayashi et al., 1981; Pun et al., 2006; Chang and Martin, 2011a,b; Hossaini et al., 2011; McGown et al., 2013; Medelin et al., 2016; Spiller et al., 2016), and global V1 IN input on fast MNs is selectively reduced before symptoms (Allodi et al., 2021). Since other inhibitory IN populations, including V0d and V2b (Figure 1), are not reported to be affected at these early stages of the disease, these observations strongly suggest that this early loss of glycinergic inputs on fast MNs originates at least partly from V1 INs (Figure 2). This global reduction in glycinergic inputs seems greater than the loss of V1 INs (Allodi et al., 2021), which can be explained either by glycinergic synapse disruptions occurring partially without V1 cell death and/or by the possible implication of other glycinergic INs that are yet to be identified. Interestingly, in the early phase of the disease, the number of V1 synapses on stressed or dying MNs is transiently increased, suggesting a possible compensatory mechanism that may be established as an attempt to counteract the hyperactivity of the affected MNs (Wootz et al., 2013; Salamatina et al., 2020). Consistently, a prolongation of inhibitory synaptic events has been observed in fetal SOD1G93A MNs (Branchereau et al., 2019).
In contrast to glycinergic inputs, spinal alterations of GABAergic synapses have not been reported in ALS animal models (Chang and Martin, 2009; Martin and Chang, 2012; Ramirez-Jarquin and Tapia, 2018), although in situ hybridization or high-resolution magnetic resonance spectroscopy studies suggested a steady decrease in GABA in the spinal cord from presymptomatic to late disease stages (Niessen et al., 2007; Hossaini et al., 2011), and cortical GABAergic inhibition is reduced in different animal models (Nieto-Gonzalez et al., 2011; Clark et al., 2018) and in human patients (Vucic et al., 2009). Of note, in SOD1G93A mice, rescuing this altered cortical inhibition by activating inhibitory INs delays the onset of the disease (Khademullah et al., 2020). However, functionality and expression of GABAA receptors are altered in SOD1G93A spinal MNs, inducing a higher Cl− influx into the cell with a possible, although counterintuitive, consequent acceleration of neuronal excitotoxicity (Carunchio et al., 2008). Cl− homeostasis alterations were even observed in SOD1G93A prenatal spinal MNs, along with reduced GABAergic/glycinergic activity, suggesting very early inhibitory impairments (Branchereau et al., 2019). Furthermore, organotypic cultures from SOD1G93A spinal cords evidenced that GABA co-release may speed the decay of glycine responses, supported by the detection of a longer persistence of GABA in glycinergic terminals (Medelin et al., 2016). Thus, possible alterations of GABA-dependent inhibition or interactions between GABAergic and glycinergic signaling could contribute to perturbed MN excitability in the onset or progression of ALS (Figure 2), which remains to be investigated in animal models. Furthermore, a more complete characterization of the V1 IN subsets is necessary to provide a more comprehensive understanding of their possible contribution to the pathology. Finally, recent observations reported above highlight the importance of distinguishing different stages of the disease-affected or non-affected neuronal populations in relation to the disease stage and the specificity of each neuronal subtype regarding activity, connectivity, and function.
Recently, several studies investigated the fitness of spinal inhibitory circuitry in human ALS patients using electrostimulation and motor response measuring approaches, notably by assessing Hoffman's reflex (H-reflex) changes. In accordance with the results obtained in animal models, spinal inhibition is reduced in human patients as the disease progresses. Observations suggested that recurrent inhibition involving Renshaw cells (see above) is decreased in patients showing spinal symptoms (Howells et al., 2020; Ozyurt et al., 2020; Sangari et al., 2022; Castro et al., 2024). Interestingly, an increase in recurrent inhibition was observed in the early phase of the disease in one of these studies, supporting the hypothesis of an initial compensatory mechanism preventing MN decline (Sangari et al., 2022). Although conclusions from these studies regarding precise cellular mechanisms, involved IN populations, and timing of occurrence of the different pathological events remain limited, these results are in line with observations in animal models, highlighting their relevance and strongly supporting the hypothesis that spinal interneuronopathy is an important feature of ALS.
Taken together, these data suggest that early changes in inhibitory signaling contribute to the onset of perturbations in spinal MN activity in ALS, possibly resulting in the establishment of transient compensatory mechanisms that may temporarily slow down disease progression before a deleterious breakdown of the motor circuitry (Figure 2).
Alterations of spinal excitatory pre-motor INs
In addition to a reduction in inhibitory inputs, hyperactivation of MNs that can lead to excitotoxicity may result from increased activity of pre-motor excitatory INs. Using an in vitro preparation of the sacral cord from SOD1G93A mice, electrophysiology measurements demonstrated that spinal IN activity increases after symptom onset, which may contribute to excitotoxicity, an alteration that involves N-methyl-D-aspartate (NMDA) receptors located either on INs or on MNs (Jiang et al., 2009). Consistently, in zebrafish carrying a human TARDBP gene (encoding the TDP-43 protein) with a mutation found in fALS forms, optogenetic activation of reticulospinal neurons and of spinal V2a INs was sufficient to elicit ALS-like symptoms characteristic of this animal model, indicating a functional alteration of glutamatergic synapses in the spinal cord (Petel Legare et al., 2019). However, as suggested for inhibitory inputs on MNs (see above), excitatory INs may increase their activity to compensate for the onset of alterations in MN activity and survival (Figure 3). These compensatory changes might also partly explain the maintenance of motor functions despite affected MNs in the earlier stages of the disease. As an example, accessory respiratory muscle (ARM) activity contributes to preserving ventilation despite diaphragm dysfunctions due to respiratory MN loss in different neurodegenerative disorders, including ALS (Johnson and Mitchell, 2013), and recruitment of the ARM is increased in SOD1G93A mice (Romer et al., 2017). Interestingly, V2a INs innervate ARM MNs and are sufficient to drive increased ARM activity at rest in healthy mice (Romer et al., 2017). Therefore, increased V2a activity may contribute to compensating for the reduced activity of the phrenic MNs in the early stages of ALS. However, V2a INs themselves degenerate in the course of the disease, in parallel with MN degeneration (Romer et al., 2017; Salamatina et al., 2020; Montanana-Rosell et al., 2024). Thus, loss of V2a INs during the progression of the disease will progressively erode the initially activated compensatory mechanism and eventually lead to respiratory failure, the cause of death in most of the ALS cases (Figure 3).
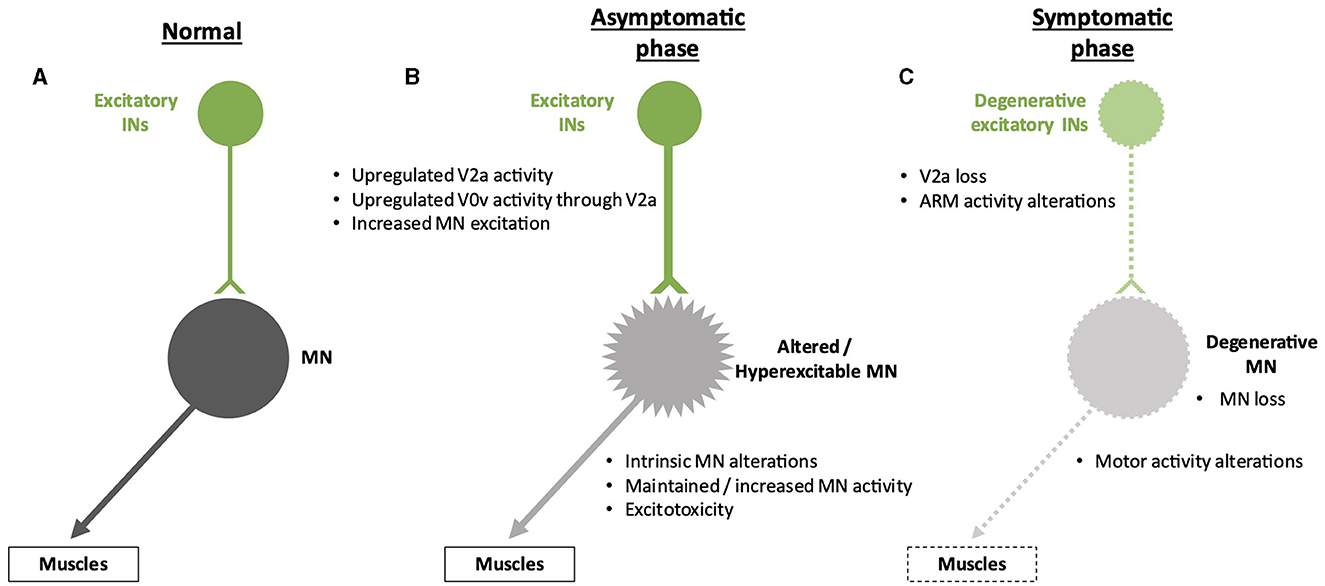
Figure 3. Spinal neuronal excitatory circuitry and its fate in ALS. Excitatory interneurons (INs) are represented in green, and motor neurons (MNs) are represented in gray. Altered INs and MNs are represented in light green and light gray, respectively. Degenerative cells are represented by dotted lines on the cell body and projections. (A) Excitatory circuitry in a non-pathological situation. (B) Early increase in activity of the excitatory circuitry before the apparition of the first symptoms. This change might preserve motor activity despite low-noise early MN alterations but may result in excitotoxicity for the MNs. The hyperexcitability of the MNs is represented by spikes on the cell body. (C) Alterations of the excitatory circuitry during the symptomatic phase of the disease. It is still unclear whether degeneration of MNs occurs before or after degeneration of excitatory INs and if one is causative of the other.
Surprisingly, the contribution of other spinal excitatory INs to the onset or progression of ALS has currently not been addressed. For example, glutamatergic commissural V0v INs are part of an excitatory V2a-V0v circuitry involved in left-right alternation during locomotion (Figure 1) (Lanuza et al., 2004; Crone et al., 2008; Talpalar et al., 2013; Shevtsova, 2015; Ramirez-Jarquin and Tapia, 2018), but the possible contribution of V0v to V2a or MN alterations in ALS has not been reported yet. Similarly, spinal V3 INs contribute to left-right coordination and the robustness of locomotor circuit activity (Figure 1) (Zhang et al., 2008, 2022; Danner et al., 2019), yet their involvement and fate in ALS remain unknown.
Alterations of spinal cholinergic pre-motor INs
In the brainstem and spinal cord, in addition to inhibitory or excitatory synapses, MNs also receive neuromodulatory inputs. Among these, large cholinergic synapses called C-boutons modulate MN activity by increasing their excitability through m2-muscarinic receptor activation (Miles et al., 2007). Interestingly, C-boutons are found predominantly on fast MNs, the most affected in ALS compared to slow MNs (Hellstrom et al., 2003). A subset of spinal V0 INs, characterized by the expression of the Pitx2 transcription factor and called cholinergic V0 or V0c (Figure 1), has been identified as the sole source of C-boutons onto MNs (Zagoraiou et al., 2009; Siembab et al., 2010). Of note, V0c also innervate inhibitory V1 IaINs and could therefore indirectly modulate inhibitory inputs on MNs (Siembab et al., 2010).
Changes in C-boutons in ALS have been observed both in humans (Nagao et al., 1998) and in mouse models (Chang and Martin, 2009; Pullen and Athanasiou, 2009; Herron and Miles, 2012; Gallart-Palau et al., 2014; Milan et al., 2015). However, it remains unclear whether these changes are compensatory to early MN alterations or intrinsic to V0c INs (Brownstone and Lancelin, 2018). In SOD1 mouse models, the density of C-boutons around MN soma was transiently increased at early presymptomatic stages (Herron and Miles, 2012; Gallart-Palau et al., 2014; Milan et al., 2015), possibly only on slow MNs (Bak et al., 2023). Furthermore, the size of the C-boutons was increased as early as postnatal day 8, long before symptom onset and MN degeneration (Pullen and Athanasiou, 2009; Herron and Miles, 2012; Milan et al., 2015). Interestingly, the persistence of this early change until adult stages was only observed in male SOD1 mice, suggesting a gender effect on cholinergic alterations in ALS (Herron and Miles, 2012). A similar gender bias has been reported in a mutated TARDBP-based ALS mouse model regarding changes in C-bouton numbers on fast or slow MNs (Bak et al., 2023). These observations could relate to the higher prevalence of ALS in men than in women (Zamani et al., 2024). Given that C-boutons increase MN excitability (Zagoraiou et al., 2009; Witts et al., 2014; Nascimento et al., 2020; Wells et al., 2021), an early increase in density and enlargement could contribute to MN hyperexcitability and participate in excitotoxic mechanisms leading to MN degeneration (Figure 4). However, in the same model, ChAT levels are shown to be reduced in MNs, V0c INs, and C-boutons in the presymptomatic phase (Casas et al., 2013). This suggests that enlargement of C-boutons could compensate for early cholinergic alterations in MNs or at C-boutons rather than being causative of MN hyperexcitability. Consistently, C-boutons activation was increased in low-intensity tasks like walking in SOD1G93A mice as compared to wild-type mice (Wells et al., 2021). This rescue hypothesis was tested by genetically inactivating C-boutons in the SOD1 mouse model, but studies reported opposite results. In the earlier study, C-bouton inactivation resulted in an acceleration in the apparition of the disease phenotype (Landoni et al., 2019), suggesting that these cholinergic synapses may protect MN against early alterations (Wells et al., 2021), possibly through C-bouton enlargement. This compensation of motor impairments by V0c would decrease as the C-boutons are lost at later stages of the disease (Nagao et al., 1998; Pullen and Athanasiou, 2009; Casas et al., 2013; Bak et al., 2023). In later studies, mice performed slightly better in ALS-related motor tasks, and innervation of fast-twitch muscles was longer preserved after C-bouton inactivation, although without any change in the “humane endpoint,” suggesting a deleterious effect of cholinergic inputs early during disease progression (Konsolaki et al., 2020; Wells et al., 2021). In line with this finding, deletion of TMEM16F, a Ca2+-activated Cl− channel present at the postsynaptic face of C-boutons and involved in neuromodulation of MN activity, delays disease onset in SOD1G93A mice, again only in male mice (Soulard et al., 2020). Thus, modifications in C-bouton numbers and size may initially compensate for early alterations in MN fitness but may later become detrimental for MN innervation and survival, resulting in defective motor activity (Figure 4).

Figure 4. Spinal neuronal cholinergic circuitry and its fate in ALS. Cholinergic interneurons (INs) are represented in yellow, and motor neurons (MNs) in gray. Altered INs and MNs are represented in light yellow and light gray, respectively. Degenerative cells are represented by dotted lines on the cell body and projections. (A) Cholinergic circuitry in a non-pathological situation. (B) Early increase in activity of the cholinergic circuitry before the apparition of the first symptoms. This modified activity might preserve motor activity despite low-noise early MN alterations but may result in excitotoxicity for the MNs. The hyperexcitability of the MNs is represented by spikes on the cell body. This hyperexcitability may contribute to MN excitotoxicity. (C) Alterations of the cholinergic circuitry during the symptomatic phase of the disease. It is still unclear whether degeneration of MN occurs before or after degeneration of cholinergic INs and if one is causative of the other.
In any case, despite contradictory observations on C-bouton fate and contribution to MN degeneration in ALS, all these studies highlighted early presymptomatic alterations of cholinergic inputs on MNs before MN degeneration occurs. Although their number is transiently increased at these early stages (Herron and Miles, 2012; Milan et al., 2015; Bak et al., 2023), a later reduction in C-boutons and degeneration of the V0c INs is observed in symptomatic SOD1 or TDP43 mouse models of ALS (Chang and Martin, 2009; Casas et al., 2013; Milan et al., 2015; Wells et al., 2021; Bak et al., 2023). The reduction in C-boutons in the symptomatic phase of the disease may result from MN degeneration losing its afferent cholinergic synapses (Wells et al., 2021). In line with this hypothesis, Neuregulin-1 expression, a factor present at the postsynaptic face of C-boutons, is diminished in MNs of an SOD1 mouse model and of ALS patients (Gallart-Palau et al., 2014; Lasiene et al., 2016), and its administration restores C-bouton numbers in mice (Lasiene et al., 2016). Whether the subsequent loss of V0c INs is a consequence of MN degeneration or results from intrinsic alterations that parallel MN defects remains to be investigated.
Taken together, although alterations of C-boutons and V0c INs in ALS have been demonstrated, a more systematic investigation of each alteration at each stage of disease progression in each available model should be carefully conducted to identify the involved cellular and molecular mechanisms, possible gender bias, whether these changes are beneficial or detrimental for ALS onset or progression, and how much these could constitute potential therapeutic targets to slow down the progression of the pathology. This said, it is plausible that the contribution of C-boutons is not strictly protective or deleterious but may differ depending on the stage of the disease. An increase in V0c cholinergic inputs on MNs in the presymptomatic phase discussed above might initially occur as an early effort to preserve motor unit activity by increasing MN excitability. However, this increased MN excitability, along with weakened MN and altered excitation/inhibition balance (see above), could eventually be detrimental to MN integrity through excitotoxic mechanisms and thus participate in their degeneration (Figure 4).
Conclusion and perspectives
ALS is a complex disease that encompasses several intrinsic and extrinsic mechanisms leading to MN death, inevitably affecting the whole motor control circuitry. In such a context, the proof of a single pathological trigger is still missing, and its very existence might be questioned. A more plausible hypothesis would favor a combination of multiple pathological events resulting in MN degeneration.
An accurate illustration of this conclusion resides in the involvement of spinal INs in the disease course. In the spinal cord motor circuitry, excitatory, inhibitory, or cholinergic INs are tightly regulating MN activity and excitability by establishing a versatile but well-controlled excitation/inhibition balance. Alterations in IN populations observed in ALS are deregulating this balance and thus most likely contribute to MN degeneration, notably through excitotoxic mechanisms. Based on the publications reviewed above, a working hypothesis can be proposed. Each type of spinal IN, i.e., inhibitory, excitatory, or cholinergic INs, shows changes in the number or activity of the synapses they form on MNs before the onset of MN degeneration or symptom apparition, either as a result of intrinsic alterations or as an attempt to compensate for early low-noise perturbations in MN activity. These early changes, although transiently preserving the fitness of the motor circuitry, may progressively result in the hyperexcitability of MNs, which are weakened by intrinsic alterations or other non-cell autonomous mechanisms. Even though this hyperexcitability has also been described as a possible protective mechanism (Leroy and Zytnicki, 2015; Huh et al., 2021), it would eventually lead to excitotoxicity and MN degeneration, possibly causing parallel degeneration of the pre-motor IN populations (Figures 2–4). The currently available therapy for ALS, a glutamate antagonist that reduces excitotoxicity, may transiently slow down MN degeneration caused by a combination of these mechanisms but becomes rapidly inefficient to counteract the general collapse of all the components of the spinal motor circuitry.
The identification of novel therapeutic options will require a systematic characterization of the contribution of each spinal IN population, alone or in combination with MNs, other INs, or glial cells, to the onset or progression of ALS. Furthermore, forthcoming studies should consider any gender bias, carefully report on the precise disease stage of each analysis, and systematically discriminate the type of motor units that are investigated. However, most of the reported observations have been obtained in SOD1-based animal models of the disease. Importantly, Sod1 mutations only affect a small fraction of ALS cases in humans, raising questions about the validity of these models to identify general mechanisms of the pathology. In contrast, TDP43-based models recapitulate a feature found in more than 95% of all ALS cases, i.e., cytoplasmic TDP43 aggregation (Suk and Rousseaux, 2020), and might therefore constitute more promising approaches to studying the disease (Spiller et al., 2016).
Author contributions
LG: Writing – original draft, Writing – review & editing. DL: Writing – review & editing. FC: Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study is supported by grants from the ≪ Fonds spéciaux de recherche ≫ (FSR) of the Université Catholique de Louvain, by a Projet de Recherche (PDR) funding (No. T.0039.21) of the Fonds de la Recherche Scientifique (F.R.S.-FNRS, Belgium) and by the Association Belge contre les Maladies neuro-Musculaires (ABMM). LG holds a PhD grant from the FRIA (F.R.S.-FNRS) and FC is a Research Director of the F.R.S.-FNRS.
Acknowledgments
Authors thank FSR, F.R.S.-FNRS, Belgium, and ABMM for their support.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Allodi, I., Montanana-Rosell, R., Selvan, R., Low, P., and Kiehn, O. (2021). Locomotor deficits in a mouse model of ALS are paralleled by loss of V1-interneuron connections onto fast motor neurons. Nat. Commun. 12:3251. doi: 10.1038/s41467-021-23224-7
Alvarez, F. J., Jonas, P. C., Sapir, T., Hartley, R., Berrocal, M. C., Geiman, E. J., et al. (2005). Postnatal phenotype and localization of spinal cord V1 derived interneurons. J. Comp. Neurol. 493, 177–192. doi: 10.1002/cne.20711
Arredondo, C., Cefaliello, C., Dyrda, A., Jury, N., Martinez, P., Diaz, I., et al. (2022). Excessive release of inorganic polyphosphate by ALS/FTD astrocytes causes non-cell-autonomous toxicity to motoneurons. Neuron 110, 1656–1670.e1612. doi: 10.1016/j.neuron.2022.02.010
Bak, A. N., Djukic, S., Kadlecova, M., Braunstein, T. H., Jensen, D. B., and Meehan, C. F. (2023). Cytoplasmic TDP-43 accumulation drives changes in C-bouton number and size in a mouse model of sporadic Amyotrophic Lateral Sclerosis. Mol. Cell. Neurosci. 125:103840. doi: 10.1016/j.mcn.2023.103840
Benedetti, L., Ghilardi, A., Rottoli, E., De Maglie, M., Prosperi, L., Perego, C., et al. (2016). INaP selective inhibition reverts precocious inter- and motorneurons hyperexcitability in the Sod1-G93R zebrafish ALS model. Sci. Rep. 6:24515. doi: 10.1038/srep24515
Bikoff, J. B., Gabitto, M. I., Rivard, A. F., Drobac, E., Machado, T. A., Miri, A., et al. (2016). Spinal inhibitory interneuron diversity delineates variant motor microcircuits. Cell 165, 207–219. doi: 10.1016/j.cell.2016.01.027
Boillee, S., Yamanaka, K., Lobsiger, C. S., Copeland, N. G., Jenkins, N. A., Kassiotis, G., et al. (2006). Onset and progression in inherited ALS determined by motor neurons and microglia. Science 312, 1389–1392. doi: 10.1126/science.1123511
Borowska, J., Jones, C. T., Deska-Gauthier, D., and Zhang, Y. (2015). V3 interneuron subpopulations in the mouse spinal cord undergo distinctive postnatal maturation processes. Neuroscience 295, 221–228. doi: 10.1016/j.neuroscience.2015.03.024
Borowska, J., Jones, C. T., Zhang, H., Blacklaws, J., Goulding, M., and Zhang, Y. (2013). Functional subpopulations of V3 interneurons in the mature mouse spinal cord. J. Neurosci. 33, 18553–18565. doi: 10.1523/JNEUROSCI.2005-13.2013
Branchereau, P., Martin, E., Allain, A. E., Cazenave, W., Supiot, L., Hodeib, F., et al. (2019). Relaxation of synaptic inhibitory events as a compensatory mechanism in fetal SOD spinal motor networks. Elife 8:e51402. doi: 10.7554/eLife.51402
Britz, O., Zhang, J., Grossmann, K. S., Dyck, J., Kim, J. C., Dymecki, S., et al. (2015). A genetically defined asymmetry underlies the inhibitory control of flexor-extensor locomotor movements. Elife 4:e04718. doi: 10.7554/eLife.04718
Brownstone, R. M., and Lancelin, C. (2018). Escape from homeostasis: spinal microcircuits and progression of amyotrophic lateral sclerosis. J. Neurophysiol. 119, 1782–1794. doi: 10.1152/jn.00331.2017
Carunchio, I., Mollinari, C., Pieri, M., Merlo, D., and Zona, C. (2008). GAB(A) receptors present higher affinity and modified subunit composition in spinal motor neurons from a genetic model of amyotrophic lateral sclerosis. Eur. J. Neurosci. 28, 1275–1285. doi: 10.1111/j.1460-9568.2008.06436.x
Casas, C., Herrando-Grabulosa, M., Manzano, R., Mancuso, R., Osta, R., and Navarro, X. (2013). Early presymptomatic cholinergic dysfunction in a murine model of amyotrophic lateral sclerosis. Brain Behav. 3, 145–158. doi: 10.1002/brb3.104
Castro, J., Oliveira Santos, M., Swash, M., and de Carvalho, M. (2024). Segmental motor neuron dysfunction in amyotrophic lateral sclerosis: Insights from H reflex paradigms. Muscle Nerve 69, 303–312. doi: 10.1002/mus.28035
Cavarsan, C. F., Steele, P. R., Genry, L. T., Reedich, E. J., McCane, L. M., LaPre, K. J., et al. (2023). Inhibitory interneurons show early dysfunction in a SOD1 mouse model of amyotrophic lateral sclerosis. J. Physiol. 601, 647–667. doi: 10.1113/JP284192
Chang, Q., and Martin, L. J. (2009). Glycinergic innervation of motoneurons is deficient in amyotrophic lateral sclerosis mice: a quantitative confocal analysis. Am. J. Pathol. 174, 574–585. doi: 10.2353/ajpath.2009.080557
Chang, Q., and Martin, L. J. (2011a). Glycine receptor channels in spinal motoneurons are abnormal in a transgenic mouse model of amyotrophic lateral sclerosis. J. Neurosci. 31, 2815–2827. doi: 10.1523/JNEUROSCI.2475-10.2011
Chang, Q., and Martin, L. J. (2011b). Motoneuron subtypes show specificity in glycine receptor channel abnormalities in a transgenic mouse model of amyotrophic lateral sclerosis. Channels 5, 299–303. doi: 10.4161/chan.5.4.16206
Chenji, S., Jha, S., Lee, D., Brown, M., Seres, P., Mah, D., et al. (2016). Investigating default mode and sensorimotor network connectivity in Amyotrophic Lateral Sclerosis. PLoS ONE 11:e0157443. doi: 10.1371/journal.pone.0157443
Clark, R. M., Brizuela, M., Blizzard, C. A., and Dickson, T. C. (2018). Reduced excitability and increased neurite complexity of cortical interneurons in a familial mouse model of amyotrophic lateral sclerosis. Front. Cell. Neurosci. 12:328. doi: 10.3389/fncel.2018.00328
Clement, A. M., Nguyen, M. D., Roberts, E. A., Garcia, M. L., Boillee, S., Rule, M., et al. (2003). Wild-type nonneuronal cells extend survival of SOD1 mutant motor neurons in ALS mice. Science 302, 113–117. doi: 10.1126/science.1086071
Crabe, R., Aimond, F., Gosset, P., Scamps, F., and Raoul, C. (2020). How degeneration of cells surrounding motoneurons contributes to amyotrophic lateral sclerosis. Cells 9:2550. doi: 10.3390/cells9122550
Crone, S. A., Quinlan, K. A., Zagoraiou, L., Droho, S., Restrepo, C. E., Lundfald, L., et al. (2008). Genetic ablation of V2a ipsilateral interneurons disrupts left-right locomotor coordination in mammalian spinal cord. Neuron 60, 70–83. doi: 10.1016/j.neuron.2008.08.009
Danner, S. M., Zhang, H., Shevtsova, N. A., Borowska-Fielding, J., Deska-Gauthier, D., Rybak, I. A., et al. (2019). Spinal V3 interneurons and left-right coordination in mammalian locomotion. Front. Cell. Neurosci. 13:516. doi: 10.3389/fncel.2019.00516
Deska-Gauthier, D., Borowska-Fielding, J., Jones, C., Zhang, H., MacKay, C. S., Michail, R., et al. (2024). Embryonic temporal-spatial delineation of excitatory spinal V3 interneuron diversity. Cell Rep. 43:113635. doi: 10.1016/j.celrep.2023.113635
Dhasmana, S., Dhasmana, A., Narula, A. S., Jaggi, M., Yallapu, M. M., and Chauhan, S. C. (2022). The panoramic view of amyotrophic lateral sclerosis: a fatal intricate neurological disorder. Life Sci. 288:120156. doi: 10.1016/j.lfs.2021.120156
Dominguez-Bajo, A., and Clotman, F. (2024). Potential roles of specific subclasses of premotor interneurons in spinal cord function recovery after traumatic spinal cord injury in adults. Cells 13:652. doi: 10.3390/cells13080652
Dougherty, K. J. (2023). Distinguishing subtypes of spinal locomotor neurons to inform circuit function and dysfunction. Curr. Opin. Neurobiol. 82:102763. doi: 10.1016/j.conb.2023.102763
Filipchuk, A., Pambo-Pambo, A., Gaudel, F., Liabeuf, S., Brocard, C., Gueritaud, J. P., et al. (2021). Early hypoexcitability in a subgroup of spinal motoneurons in superoxide dismutase 1 transgenic mice, a model of amyotrophic lateral sclerosis. Neuroscience 463, 337–353. doi: 10.1016/j.neuroscience.2021.01.039
Francius, C., Harris, A., Rucchin, V., Hendricks, T. J., Stam, F. J., Barber, M., et al. (2013). Identification of multiple subsets of ventral interneurons and differential distribution along the rostrocaudal axis of the developing spinal cord. PLoS ONE 8:e70325. doi: 10.1371/journal.pone.0070325
Gallart-Palau, X., Tarabal, O., Casanovas, A., Sabado, J., Correa, F. J., Hereu, M., et al. (2014). Neuregulin-1 is concentrated in the postsynaptic subsurface cistern of C-bouton inputs to alpha-motoneurons and altered during motoneuron diseases. FASEB J. 28, 3618–3632. doi: 10.1096/fj.13-248583
Gosgnach, S., Bikoff, J. B., Dougherty, K. J., El Manira, A., Lanuza, G. M., and Zhang, Y. (2017). Delineating the diversity of spinal interneurons in locomotor circuits. J. Neurosci. 37, 10835–10841. doi: 10.1523/JNEUROSCI.1829-17.2017
Gosgnach, S., Lanuza, G. M., Butt, S. J., Saueressig, H., Zhang, Y., Velasquez, T., et al. (2006). V1 spinal neurons regulate the speed of vertebrate locomotor outputs. Nature 440, 215–219. doi: 10.1038/nature04545
Gunes, Z. I., Kan, V. W. Y., Ye, X., and Liebscher, S. (2020). Exciting complexity: the role of motor circuit elements in ALS pathophysiology. Front. Neurosci. 14:573. doi: 10.3389/fnins.2020.00573
Harris, A., Masgutova, G., Collin, A., Toch, M., Hidalgo-Figueroa, M., Jacob, B., et al. (2019). Onecut factors and Pou2f2 regulate the distribution of V2 interneurons in the mouse developing spinal cord. Front. Cell. Neurosci. 13:184. doi: 10.3389/fncel.2019.00184
Hayashi, H., Suga, M., Satake, M., and Tsubaki, T. (1981). Reduced glycine receptor in the spinal cord in amyotrophic lateral sclerosis. Ann. Neurol. 9, 292–294. doi: 10.1002/ana.410090313
Hellstrom, J., Oliveira, A. L., Meister, B., and Cullheim, S. (2003). Large cholinergic nerve terminals on subsets of motoneurons and their relation to muscarinic receptor type 2. J. Comp. Neurol. 460, 476–486. doi: 10.1002/cne.10648
Herron, L. R., and Miles, G. B. (2012). Gender-specific perturbations in modulatory inputs to motoneurons in a mouse model of amyotrophic lateral sclerosis. Neuroscience 226, 313–323. doi: 10.1016/j.neuroscience.2012.09.031
Hossaini, M., Cardona Cano, S., van Dis, V., Haasdijk, E. D., Hoogenraad, C. C., Holstege, J. C., et al. (2011). Spinal inhibitory interneuron pathology follows motor neuron degeneration independent of glial mutant superoxide dismutase 1 expression in SOD1-ALS mice. J. Neuropathol. Exp. Neurol. 70, 662–677. doi: 10.1097/NEN.0b013e31822581ac
Howells, J., Sangari, S., Matamala, J. M., Kiernan, M. C., Marchand-Pauvert, V., and Burke, D. (2020). Interrogating interneurone function using threshold tracking of the H reflex in healthy subjects and patients with motor neurone disease. Clin. Neurophysiol. 131, 1986–1996. doi: 10.1016/j.clinph.2020.03.028
Huang, C., Tong, J., Bi, F., Zhou, H., and Xia, X. G. (2012). Mutant TDP-43 in motor neurons promotes the onset and progression of ALS in rats. J. Clin. Invest. 122, 107–118. doi: 10.1172/JCI59130
Huh, S., Heckman, C. J., and Manuel, M. (2021). Time course of alterations in adult spinal motoneuron properties in the SOD1(G93A) mouse model of ALS. eNeuro 8:e378. doi: 10.1101/2020.05.19.105007
Jaarsma, D., Teuling, E., Haasdijk, E. D., De Zeeuw, C. I., and Hoogenraad, C. C. (2008). Neuron-specific expression of mutant superoxide dismutase is sufficient to induce amyotrophic lateral sclerosis in transgenic mice. J. Neurosci. 28, 2075–2088. doi: 10.1523/JNEUROSCI.5258-07.2008
Jensen, D. B., Kadlecova, M., Allodi, I., and Meehan, C. F. (2020). Spinal motoneurones are intrinsically more responsive in the adult G93A SOD1 mouse model of amyotrophic lateral sclerosis. J. Physiol. 598, 4385–4403. doi: 10.1113/JP280097
Jiang, M., Schuster, J. E., Fu, R., Siddique, T., and Heckman, C. J. (2009). Progressive changes in synaptic inputs to motoneurons in adult sacral spinal cord of a mouse model of amyotrophic lateral sclerosis. J. Neurosci. 29, 15031–15038. doi: 10.1523/JNEUROSCI.0574-09.2009
Jiang, M. C., Adimula, A., Birch, D., and Heckman, C. J. (2017). Hyperexcitability in synaptic and firing activities of spinal motoneurons in an adult mouse model of amyotrophic lateral sclerosis. Neuroscience 362, 33–46. doi: 10.1016/j.neuroscience.2017.08.041
Johnson, R. A., and Mitchell, G. S. (2013). Common mechanisms of compensatory respiratory plasticity in spinal neurological disorders. Respir. Physiol. Neurobiol. 189, 419–428. doi: 10.1016/j.resp.2013.05.025
Khademullah, C. S., Aqrabawi, A. J., Place, K. M., Dargaei, Z., Liang, X., Pressey, J. C., et al. (2020). Cortical interneuron-mediated inhibition delays the onset of amyotrophic lateral sclerosis. Brain 143, 800–810. doi: 10.1093/brain/awaa034
Kim, J., Hughes, E. G., Shetty, A. S., Arlotta, P., Goff, L. A., Bergles, D. E., et al. (2017). Changes in the excitability of neocortical neurons in a mouse model of amyotrophic lateral sclerosis are not specific to corticospinal neurons and are modulated by advancing disease. J. Neurosci. 37, 9037–9053. doi: 10.1523/JNEUROSCI.0811-17.2017
Kim, S., Chung, A. Y., Na, J. E., Lee, S. J., Jeong, S. H., Kim, E., et al. (2019). Myelin degeneration induced by mutant superoxide dismutase 1 accumulation promotes amyotrophic lateral sclerosis. Glia 67, 1910–1921. doi: 10.1002/glia.23669
King, A. E., Woodhouse, A., Kirkcaldie, M. T., and Vickers, J. C. (2016). Excitotoxicity in ALS: overstimulation, or overreaction? Exp. Neurol. 275, 162–171. doi: 10.1016/j.expneurol.2015.09.019
Konsolaki, E., Koropouli, E., Tsape, E., Pothakos, K., and Zagoraiou, L. (2020). Genetic inactivation of cholinergic c bouton output improves motor performance but not survival in a mouse model of amyotrophic lateral sclerosis. Neuroscience 450, 71–80. doi: 10.1016/j.neuroscience.2020.07.047
Kwong, L. K., Neumann, M., Sampathu, D. M., Lee, V. M., and Trojanowski, J. Q. (2007). TDP-43 proteinopathy: the neuropathology underlying major forms of sporadic and familial frontotemporal lobar degeneration and motor neuron disease. Acta Neuropathol. 114, 63–70. doi: 10.1007/s00401-007-0226-5
Landoni, L. M., Myles, J. R., Wells, T. L., Mayer, W. P., and Akay, T. (2019). Cholinergic modulation of motor neurons through the C-boutons are necessary for the locomotor compensation for severe motor neuron loss during amyotrophic lateral sclerosis disease progression. Behav. Brain Res. 369:111914. doi: 10.1016/j.bbr.2019.111914
Lanuza, G. M., Gosgnach, S., Pierani, A., Jessell, T. M., and Goulding, M. (2004). Genetic identification of spinal interneurons that coordinate left-right locomotor activity necessary for walking movements. Neuron 42, 375–386. doi: 10.1016/S0896-6273(04)00249-1
Lasiene, J., Komine, O., Fujimori-Tonou, N., Powers, B., Endo, F., Watanabe, S., et al. (2016). Neuregulin 1 confers neuroprotection in SOD1-linked amyotrophic lateral sclerosis mice via restoration of C-boutons of spinal motor neurons. Acta Neuropathol. Commun. 4:15. doi: 10.1186/s40478-016-0286-7
Leroy, F., Lamotte d'Incamps, B., Imhoff-Manuel, R. D., and Zytnicki, D. (2014). Early intrinsic hyperexcitability does not contribute to motoneuron degeneration in amyotrophic lateral sclerosis. Elife 3:e04046. doi: 10.7554/eLife.04046
Leroy, F., and Zytnicki, D. (2015). Is hyperexcitability really guilty in amyotrophic lateral sclerosis? Neural Regen. Res. 10, 1413–1415. doi: 10.4103/1673-5374.165308
Li, K., Hala, T. J., Seetharam, S., Poulsen, D. J., Wright, M. C., and Lepore, A. C. (2015). GLT1 overexpression in SOD1(G93A) mouse cervical spinal cord does not preserve diaphragm function or extend disease. Neurobiol. Dis. 78, 12–23. doi: 10.1016/j.nbd.2015.03.010
Lu, D. C., Niu, T., and Alaynick, W. A. (2015). Molecular and cellular development of spinal cord locomotor circuitry. Front. Mol. Neurosci. 8:25. doi: 10.3389/fnmol.2015.00025
Martin, E., Cazenave, W., Cattaert, D., and Branchereau, P. (2013). Embryonic alteration of motoneuronal morphology induces hyperexcitability in the mouse model of amyotrophic lateral sclerosis. Neurobiol. Dis. 54, 116–126. doi: 10.1016/j.nbd.2013.02.011
Martin, L. J., and Chang, Q. (2012). Inhibitory synaptic regulation of motoneurons: a new target of disease mechanisms in amyotrophic lateral sclerosis. Mol. Neurobiol. 45, 30–42. doi: 10.1007/s12035-011-8217-x
Masrori, P., and Van Damme, P. (2020). Amyotrophic lateral sclerosis: a clinical review. Eur. J. Neurol. 27, 1918–1929. doi: 10.1111/ene.14393
McGown, A., McDearmid, J. R., Panagiotaki, N., Tong, H., Al Mashhadi, S., Redhead, N., et al. (2013). Early interneuron dysfunction in ALS: insights from a mutant sod1 zebrafish model. Ann. Neurol. 73, 246–258. doi: 10.1002/ana.23780
Medelin, M., Rancic, V., Cellot, G., Laishram, J., Veeraraghavan, P., Rossi, C., et al. (2016). Altered development in GABA co-release shapes glycinergic synaptic currents in cultured spinal slices of the SOD1(G93A) mouse model of amyotrophic lateral sclerosis. J. Physiol. 594, 3827–3840. doi: 10.1113/JP272382
Mesaros, M., Lenz, S., Lim, W., Brown, J., Drury, L., and Roggenbuck, J. (2021). Investigating the genetic profile of the Amyotrophic Lateral Sclerosis/Frontotemporal Dementia (ALS-FTD) continuum in patients of diverse race, ethnicity and ancestry. Genes (Basel) 13:76. doi: 10.3390/genes13010076
Milan, L., Courtand, G., Cardoit, L., Masmejean, F., Barriere, G., Cazalets, J. R., et al. (2015). Age-related changes in pre- and postsynaptic partners of the cholinergic C-boutons in wild-type and SOD1G93A lumbar motoneurons. PLoS ONE 10:e0135525. doi: 10.1371/journal.pone.0135525
Miles, G. B., Hartley, R., Todd, A. J., and Brownstone, R. M. (2007). Spinal cholinergic interneurons regulate the excitability of motoneurons during locomotion. Proc. Natl. Acad. Sci. USA. 104, 2448–2453. doi: 10.1073/pnas.0611134104
Montanana-Rosell, R., Selvan, R., Hernandez-Varas, P., Kaminski, J. M., Sidhu, S. K., Ahlmark, D. B., et al. (2024). Spinal inhibitory neurons degenerate before motor neurons and excitatory neurons in a mouse model of ALS. Sci. Adv. 10:eadk3229. doi: 10.1126/sciadv.adk3229
Mora, S., Stuckert, A., von Huth Friis, R., Pietersz, K., Noes-Holt, G., Montanana-Rosell, R., et al. (2024). Stabilization of V1 interneuron-motor neuron connectivity ameliorates motor phenotype in a mouse model of ALS. Nat. Commun. 15:4867. doi: 10.1038/s41467-024-48925-7
Morrison, B. M., Janssen, W. G., Gordon, J. W., and Morrison, J. H. (1998). Time course of neuropathology in the spinal cord of G86R superoxide dismutase transgenic mice. J. Comp. Neurol. 391, 64–77. doi: 10.1002/(SICI)1096-9861(19980202)391:1<64::AID-CNE6>3.0.CO;2-P
Nagai, M., Re, D. B., Nagata, T., Chalazonitis, A., Jessell, T. M., Wichterle, H., et al. (2007). Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons. Nat. Neurosci. 10, 615–622. doi: 10.1038/nn1876
Nagao, M., Misawa, H., Kato, S., and Hirai, S. (1998). Loss of cholinergic synapses on the spinal motor neurons of amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 57, 329–333. doi: 10.1097/00005072-199804000-00004
Nascimento, F., Broadhead, M. J., Tetringa, E., Tsape, E., Zagoraiou, L., and Miles, G. B. (2020). Synaptic mechanisms underlying modulation of locomotor-related motoneuron output by premotor cholinergic interneurons. Elife 9:e54170. doi: 10.7554/eLife.54170
Niessen, H. G., Debska-Vielhaber, G., Sander, K., Angenstein, F., Ludolph, A. C., Hilfert, L., et al. (2007). Metabolic progression markers of neurodegeneration in the transgenic G93A-SOD1 mouse model of amyotrophic lateral sclerosis. Eur. J. Neurosci. 25, 1669–1677. doi: 10.1111/j.1460-9568.2007.05415.x
Nieto-Gonzalez, J. L., Moser, J., Lauritzen, M., Schmitt-John, T., and Jensen, K. (2011). Reduced GABAergic inhibition explains cortical hyperexcitability in the wobbler mouse model of ALS. Cereb. Cortex 21, 625–635. doi: 10.1093/cercor/bhq134
Nijssen, J., Comley, L. H., and Hedlund, E. (2017). Motor neuron vulnerability and resistance in amyotrophic lateral sclerosis. Acta Neuropathol. 133, 863–885. doi: 10.1007/s00401-017-1708-8
Ovsepian, S. V., O'Leary, V. B., and Martinez, S. (2024). Selective vulnerability of motor neuron types and functional groups to degeneration in amyotrophic lateral sclerosis: review of the neurobiological mechanisms and functional correlates. Brain Struct. Funct. 229, 1–14. doi: 10.1007/s00429-023-02728-6
Ozyurt, M. G., Topkara, B., Isak, B., and Turker, K. S. (2020). Amyotrophic lateral sclerosis weakens spinal recurrent inhibition and post-activation depression. Clin. Neurophysiol. 131, 2875–2886. doi: 10.1016/j.clinph.2020.09.021
Petel Legare, V., Harji, Z. A., Rampal, C. J., Allard-Chamard, X., Rodriguez, E. C., and Armstrong, G. A. B. (2019). Augmentation of spinal cord glutamatergic synaptic currents in zebrafish primary motoneurons expressing mutant human TARDBP (TDP-43). Sci. Rep. 9:9122. doi: 10.1038/s41598-019-45530-3
Pullen, A. H., and Athanasiou, D. (2009). Increase in presynaptic territory of C-terminals on lumbar motoneurons of G93A SOD1 mice during disease progression. Eur. J. Neurosci. 29, 551–561. doi: 10.1111/j.1460-9568.2008.06602.x
Pun, S., Santos, A. F., Saxena, S., Xu, L., and Caroni, P. (2006). Selective vulnerability and pruning of phasic motoneuron axons in motoneuron disease alleviated by CNTF. Nat. Neurosci. 9, 408–419. doi: 10.1038/nn1653
Pun, S., Sigrist, M., Santos, A. F., Ruegg, M. A., Sanes, J. R., Jessell, T. M., et al. (2002). An intrinsic distinction in neuromuscular junction assembly and maintenance in different skeletal muscles. Neuron 34, 357–370. doi: 10.1016/S0896-6273(02)00670-0
Qian, K., Huang, H., Peterson, A., Hu, B., Maragakis, N. J., Ming, G. L., et al. (2017). Sporadic ALS astrocytes induce neuronal degeneration in vivo. Stem Cell Reports 8, 843–855. doi: 10.1016/j.stemcr.2017.03.003
Quinlan, K. A., Kajtaz, E., Ciolino, J. D., Imhoff-Manuel, R. D., Tresch, M. C., Heckman, C. J., et al. (2017). Chronic electromyograms in treadmill running SOD1 mice reveal early changes in muscle activation. J. Physiol. 595, 5387–5400. doi: 10.1113/JP274170
Ramirez-Jarquin, U. N., and Tapia, R. (2018). Excitatory and inhibitory neuronal circuits in the spinal cord and their role in the control of motor neuron function and degeneration. ACS Chem. Neurosci. 9, 211–216. doi: 10.1021/acschemneuro.7b00503
Ravits, J., Paul, P., and Jorg, C. (2007). Focality of upper and lower motor neuron degeneration at the clinical onset of ALS. Neurology 68, 1571–1575. doi: 10.1212/01.wnl.0000260965.20021.47
Romer, S. H., Seedle, K., Turner, S. M., Li, J., Baccei, M. L., and Crone, S. A. (2017). Accessory respiratory muscles enhance ventilation in ALS model mice and are activated by excitatory V2a neurons. Exp. Neurol. 287, 192–204. doi: 10.1016/j.expneurol.2016.05.033
Roome, R. B., and Levine, A. J. (2023). The organization of spinal neurons: Insights from single cell sequencing. Curr. Opin. Neurobiol. 82:102762. doi: 10.1016/j.conb.2023.102762
Rosenblum, L. T., and Trotti, D. (2017). EAAT2 and the molecular signature of amyotrophic lateral sclerosis. Adv. Neurobiol. 16, 117–136. doi: 10.1007/978-3-319-55769-4_6
Salamatina, A., Yang, J. H., Brenner-Morton, S., Bikoff, J. B., Fang, L., Kintner, C. R., et al. (2020). Differential loss of spinal interneurons in a mouse model of ALS. Neuroscience 450, 81–95. doi: 10.1016/j.neuroscience.2020.08.011
Sangari, S., Peyre, I., Lackmy-Vallee, A., Bayen, E., Pradat, P. F., and Marchand-Pauvert, V. (2022). Transient increase in recurrent inhibition in amyotrophic lateral sclerosis as a putative protection from neurodegeneration. Acta Physiol. 234:e13758. doi: 10.1111/apha.13758
Scekic-Zahirovic, J., Benetton, C., Brunet, A., Ye, X., Logunov, E., Douchamps, V., et al. (2024). Cortical hyperexcitability in mouse models and patients with amyotrophic lateral sclerosis is linked to noradrenaline deficiency. Sci. Transl. Med. 16:eadg3665. doi: 10.1126/scitranslmed.adg3665
Sharma, A., Lyashchenko, A. K., Lu, L., Nasrabady, S. E., Elmaleh, M., Mendelsohn, M., et al. (2016). ALS-associated mutant FUS induces selective motor neuron degeneration through toxic gain of function. Nat. Commun. 7:10465. doi: 10.1038/ncomms10465
Shevtsova, N. A. (2015). Organization of left-right coordination of neuronal activity in the mammalian spinal cord: Insights from computational modelling. J. Physiol. 593, 2403–2426. doi: 10.1113/JP270121
Siembab, V. C., Smith, C. A., Zagoraiou, L., Berrocal, M. C., Mentis, G. Z., and Alvarez, F. J. (2010). Target selection of proprioceptive and motor axon synapses on neonatal V1-derived Ia inhibitory interneurons and Renshaw cells. J. Comp. Neurol. 518, 4675–4701. doi: 10.1002/cne.22441
Soulard, C., Salsac, C., Mouzat, K., Hilaire, C., Roussel, J., Mezghrani, A., et al. (2020). Spinal motoneuron TMEM16F acts at C-boutons to modulate motor resistance and contributes to ALS pathogenesis. Cell Rep. 30, 2581–2593.e2587. doi: 10.1016/j.celrep.2020.02.001
Spiller, K. J., Cheung, C. J., Restrepo, C. R., Kwong, L. K., Stieber, A. M., Trojanowski, J. Q., et al. (2016). Selective motor neuron resistance and recovery in a new inducible mouse model of TDP-43 proteinopathy. J. Neurosci. 36, 7707–7717. doi: 10.1523/JNEUROSCI.1457-16.2016
Srinivasan, E., and Rajasekaran, R. (2020). A systematic and comprehensive review on disease-causing genes in Amyotrophic Lateral Sclerosis. J. Mol. Neurosci. 70, 1742–1770. doi: 10.1007/s12031-020-01569-w
Stephens, B., Guiloff, R. J., Navarrete, R., Newman, P., Nikhar, N., and Lewis, P. (2006). Widespread loss of neuronal populations in the spinal ventral horn in sporadic motor neuron disease. A morphometric study. J. Neurol. Sci. 244, 41–58. doi: 10.1016/j.jns.2005.12.003
Suk, T. R., and Rousseaux, M. W. C. (2020). The role of TDP-43 mislocalization in amyotrophic lateral sclerosis. Mol. Neurodegener. 15:45. doi: 10.1186/s13024-020-00397-1
Sweeney, L. B., Bikoff, J. B., Gabitto, M. I., Brenner-Morton, S., Baek, M., Yang, J. H., et al. (2018). Origin and segmental diversity of spinal inhibitory interneurons. Neuron 97, 341–355.e343. doi: 10.1016/j.neuron.2017.12.029
Talpalar, A. E., Bouvier, J., Borgius, L., Fortin, G., Pierani, A., and Kiehn, O. (2013). Dual-mode operation of neuronal networks involved in left-right alternation. Nature 500, 85–88. doi: 10.1038/nature12286
Taylor, J. P., Brown, R. H. Jr., and Cleveland, D. W. (2016). Decoding ALS: from genes to mechanism. Nature 539, 197–206. doi: 10.1038/nature20413
Tovar-Y-Romo, L. B., Santa-Cruz, L. D., Zepeda, A., and Tapia, R. (2009). Chronic elevation of extracellular glutamate due to transport blockade is innocuous for spinal motoneurons in vivo. Neurochem. Int. 54, 186–191. doi: 10.1016/j.neuint.2008.09.015
Van Den Bosch, L., Van Damme, P., Bogaert, E., and Robberecht, W. (2006). The role of excitotoxicity in the pathogenesis of amyotrophic lateral sclerosis. Biochim. Biophys. Acta 1762, 1068–1082. doi: 10.1016/j.bbadis.2006.05.002
Van Harten, A. C. M., Phatnani, H., and Przedborski, S. (2021). Non-cell-autonomous pathogenic mechanisms in amyotrophic lateral sclerosis. Trends Neurosci. 44, 658–668. doi: 10.1016/j.tins.2021.04.008
van Rheenen, W., van der Spek, R. A. A., Bakker, M. K., van Vugt, J., Hop, P. J., Zwamborn, R. A. J., et al. (2021). Common and rare variant association analyses in amyotrophic lateral sclerosis identify 15 risk loci with distinct genetic architectures and neuron-specific biology. Nat. Genet. 53, 1636–1648. doi: 10.1038/s41588-021-00973-1
van Zundert, B., Peuscher, M. H., Hynynen, M., Chen, A., Neve, R. L., Brown, R. H. Jr., et al. (2008). Neonatal neuronal circuitry shows hyperexcitable disturbance in a mouse model of the adult-onset neurodegenerative disease amyotrophic lateral sclerosis. J. Neurosci. 28, 10864–10874. doi: 10.1523/JNEUROSCI.1340-08.2008
Vucic, S., Cheah, B. C., and Kiernan, M. C. (2009). Defining the mechanisms that underlie cortical hyperexcitability in amyotrophic lateral sclerosis. Exp. Neurol. 220, 177–182. doi: 10.1016/j.expneurol.2009.08.017
Wang, L., Sharma, K., Grisotti, G., and Roos, R. P. (2009). The effect of mutant SOD1 dismutase activity on non-cell autonomous degeneration in familial amyotrophic lateral sclerosis. Neurobiol. Dis. 35, 234–240. doi: 10.1016/j.nbd.2009.05.002
Wells, T. L., Myles, J. R., and Akay, T. (2021). C-boutons and their influence on amyotrophic lateral sclerosis disease progression. J. Neurosci. 41, 8088–8101. doi: 10.1523/JNEUROSCI.0660-21.2021
Wilson, A. C., and Sweeney, L. B. (2023). Spinal cords: symphonies of interneurons across species. Front. Neural Circuits 17:1146449. doi: 10.3389/fncir.2023.1146449
Witts, E. C., Zagoraiou, L., and Miles, G. B. (2014). Anatomy and function of cholinergic C bouton inputs to motor neurons. J. Anat. 224, 52–60. doi: 10.1111/joa.12063
Wootz, H., Fitzsimons-Kantamneni, E., Larhammar, M., Rotterman, T. M., Enjin, A., Patra, K., et al. (2013). Alterations in the motor neuron-renshaw cell circuit in the Sod1(G93A) mouse model. J. Comp. Neurol. 521, 1449–1469. doi: 10.1002/cne.23266
Yamanaka, K., Boillee, S., Roberts, E. A., Garcia, M. L., McAlonis-Downes, M., Mikse, O. R., et al. (2008a). Mutant SOD1 in cell types other than motor neurons and oligodendrocytes accelerates onset of disease in ALS mice. Proc. Natl. Acad. Sci. USA. 105, 7594–7599. doi: 10.1073/pnas.0802556105
Yamanaka, K., Chun, S. J., Boillee, S., Fujimori-Tonou, N., Yamashita, H., Gutmann, D. H., et al. (2008b). Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat. Neurosci. 11, 251–253. doi: 10.1038/nn2047
Yin, H. Z., and Weiss, J. H. (2012). Marked synergism between mutant SOD1 and glutamate transport inhibition in the induction of motor neuronal degeneration in spinal cord slice cultures. Brain Res. 1448, 153–162. doi: 10.1016/j.brainres.2012.02.005
Zagoraiou, L., Akay, T., Martin, J. F., Brownstone, R. M., Jessell, T. M., and Miles, G. B. (2009). A cluster of cholinergic premotor interneurons modulates mouse locomotor activity. Neuron 64, 645–662. doi: 10.1016/j.neuron.2009.10.017
Zamani, A., Thomas, E., and Wright, D. K. (2024). Sex biology in amyotrophic lateral sclerosis. Ageing Res. Rev. 95:102228. doi: 10.1016/j.arr.2024.102228
Zhang, H., Shevtsova, N. A., Deska-Gauthier, D., Mackay, C., Dougherty, K. J., Danner, S. M., et al. (2022). The role of V3 neurons in speed-dependent interlimb coordination during locomotion in mice. Elife 11:e73424. doi: 10.7554/eLife.73424
Zhang, J., Lanuza, G. M., Britz, O., Wang, Z., Siembab, V. C., Zhang, Y., et al. (2014). V1 and v2b interneurons secure the alternating flexor-extensor motor activity mice require for limbed locomotion. Neuron 82, 138–150. doi: 10.1016/j.neuron.2014.02.013
Zhang, Y., Narayan, S., Geiman, E., Lanuza, G. M., Velasquez, T., Shanks, B., et al. (2008). V3 spinal neurons establish a robust and balanced locomotor rhythm during walking. Neuron 60, 84–96. doi: 10.1016/j.neuron.2008.09.027
Keywords: amyotrophic lateral sclerosis, motor neurons, pre-motor interneurons, spinal cord, non-cell autonomous mechanisms, neurodegenerative disease
Citation: Goffin L, Lemoine D and Clotman F (2024) Potential contribution of spinal interneurons to the etiopathogenesis of amyotrophic lateral sclerosis. Front. Neurosci. 18:1434404. doi: 10.3389/fnins.2024.1434404
Received: 17 May 2024; Accepted: 21 June 2024;
Published: 18 July 2024.
Edited by:
Thibaut Burg, VIB & KU Leuven Center for Brain & Disease Research, BelgiumReviewed by:
José Castro, Universidade de Lisboa, PortugalPascal Branchereau, Université de Bordeaux, France
Mamede De Carvalho, University of Lisbon, Portugal
Copyright © 2024 Goffin, Lemoine and Clotman. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Frédéric Clotman, ZnJlZGVyaWMuY2xvdG1hbkB1Y2xvdXZhaW4uYmU=
†Present address: Damien Lemoine, Lille Neuroscience and Cognition, Lille, France