Abstract
Neurotoxicity is a common toxic reaction associated with the use of the anesthetic drug propofol. With the widespread use of propofol, the issue of neurotoxicity has garnered significant attention. Mitochondria are the energy metabolism centers of cells and play a crucial role in biological processes such as cell growth and development, invasion and metastasis, division and differentiation, and apoptosis. Dynamin-related protein 1 (Drp1) is a key regulator of mitochondrial fission that can modulate the dynamic balance of mitochondria and plays an important role in maintaining mitochondrial morphology and function. The abnormal expression of Drp1 is closely related to the occurrence and development of various pathological conditions. Through a systematic review of multi-species animal and cellular studies, we elucidated the correlation between Drp1 and propofol-induced neurotoxicity. By analyzing Drp1-mediated mitochondrial fragmentation across different organ systems, this work provides crucial theoretical foundations for developing Drp1-targeted strategies in propofol neurotoxicity detection, prevention, and pharmacological intervention.
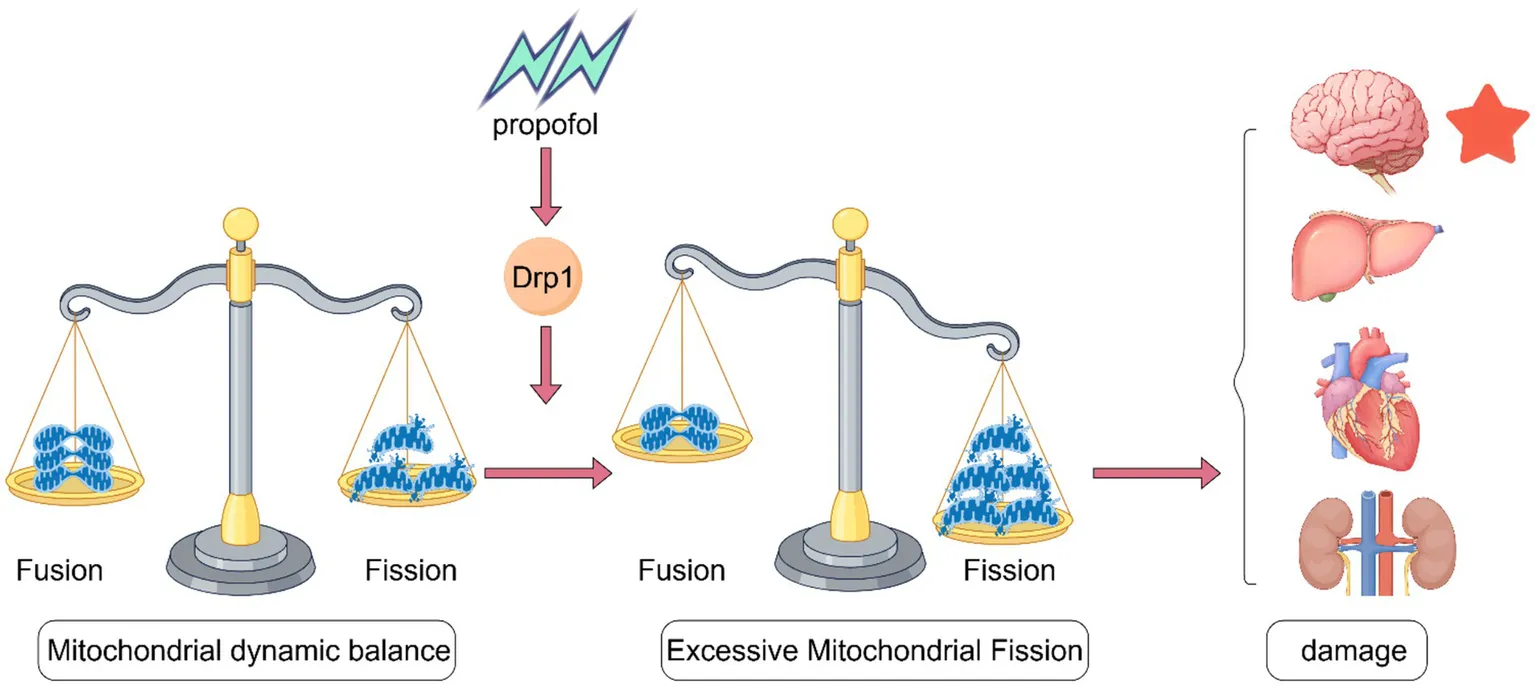
Created with Figdraw.
1 Introduction
Neurotoxicity refers to damage to the nervous system, which can lead to various clinical manifestations such as altered consciousness, motor function impairment, and cognitive decline. Neurotoxicity can be caused by a variety of factors, including drugs, toxins, and infections (Della Giovampaola et al., 2022). The neurotoxic mechanism of propofol mainly induces apoptosis, but the specific mechanism of its action is currently unclear and requires further research. The protein Drp1 plays a central role in regulating mitochondrial morphological changes, especially when cells respond to various physiological and pathological stimuli. Studies have shown that excessive activation of the Drp1 can lead to excessive mitochondrial fission, resulting in dysfunctional mitochondria, which in turn triggers apoptosis and tissue damage (Wang and Qiu, 2023; Zhang B.-F., et al., 2024). The dynamic balance of mitochondria relies on the coordination of fission and fusion processes. Drp1, by promoting mitochondrial fission, can help cells adapt to changes in energy demand and respond to challenges such as oxidative stress (Rosdah et al., 2020; Yang et al., 2022). The balance between mitochondrial fusion and fission is disrupted in hippocampal neuron injuries, and the main protein involved, Drp1, is linked to neurodevelopment, neural plasticity, and neurological issues (Gao et al., 2022). Drp1’s role in mitochondrial fission is essential for repairing injuries in hippocampal neurons. This article reviews the structure and function of Drp1, discusses its role in propofol neurotoxicity based on animal studies, examines the mechanistic link between Drp1 and neurotoxicity, analyzes Drp1’s involvement in mitochondrial dysfunction across different organs, and suggests targeting Drp1 as a strategy to prevent neurotoxicity. In summary, the discussion in this article provides important insights for the detection, prevention, and drug development of propofol-induced neurotoxicity from the perspective of Drp1.
2 Structure and function of Drp1 protein
Drp1 is an important GTPase that primarily participates in the division and dynamic regulation of mitochondria. Drp1 has multiple protein isoforms, which arise from the alternative splicing of pre-mRNA transcripts encoded by a single gene. The structure of Drp1 mainly consists of a GTPase domain, a GTP-binding domain, and a C-terminal helical domain. At its N-terminus, Drp1 also has a proline-rich region. The GTPase activity of Drp1 enables it to bind GTP and drive the mitochondrial division process through GTP hydrolysis. The domains of Drp1 play a crucial role not only in its self-assembly and activity regulation but also in regulating mitochondrial morphology and function through interactions with other proteins. Drp1 interacts with mitochondrial fission proteins such as Fis1, Mff, MiD49, and MiD51 (Nolden et al., 2023). For example, Drp1 promotes the process of mitochondrial fission by binding to adaptor proteins (such as Mff and Fis1) on the outer mitochondrial membrane through its C-terminal helical domain (Nolden et al., 2023; Torcasio et al., 2024). Research shows that the direct interaction between Fis1 and Drp1 can maintain mitochondrial morphology, with Fis1 influencing the balance of mitochondrial fission and fusion by regulating the assembly state of Drp1 (Choi et al., 2020; Egner et al., 2022). Drp1 is involved in the process of mitochondrial fission, including recruitment from the cytosol to the outer mitochondrial membrane (OMM), high-level self-assembly, GTP hydrolysis, and eventual fragmentation (Feng et al., 2020).
In recent years, it has been reported that the function of Drp1 is not limited to mitochondrial fission but also involves various biological processes in cells, including cellular stress responses, metabolic regulation, and apoptosis. There is evidence that mitochondria in the spinal dorsal horn (SDH) are sensitive to neuropathic pain (NP), and targeting mitochondrial Drp1 overexpression alleviates pain hypersensitivity, providing a new therapeutic target for pain treatment (Zhang K.-L., et al., 2022).
Abnormal levels or dysfunction of Drp1 are closely linked to the development of several diseases, including neurodegenerative disorders, cardiovascular diseases, and cancers (Zeng et al., 2020). Because the nervous system requires a large amount of energy, mitochondrial damage or dysfunction greatly affects neuronal activity and can even lead to their death.
3 Propofol and neurotoxicity
3.1 The mechanism of apoptosis-induced neurotoxicity
Propofol enhances the effects of the inhibitory neurotransmitter gamma-aminobutyric acid (GABA) by binding to the β+/α-subunits of the GABAA receptor, leading to hyperpolarization of the neuronal membrane and reduced neuronal excitability. This allows propofol to rapidly induce loss of consciousness and maintain anesthesia. Propofol may also regulate the balance of excitation and inhibition in the brain by affecting other neurotransmitter systems, such as the glutamatergic system (Li et al., 2020). The apoptotic pathway induced by propofol is an important aspect of studying its neurotoxic mechanisms. Propofol can lead to the apoptosis of nerve cells and negatively affect their survival. Apoptosis is a programmed pattern of cell death that typically occurs under stress conditions. At this time, the permeability of the mitochondrial membrane increases, and the membrane potential decreases, leading to the release of cytochrome C into the cytoplasm (Gottlieb et al., 2003), which in turn activates downstream caspases. This process is usually triggered by the opening of the mitochondrial permeability transition pore (mPTP). The opening of mPTP is associated with various factors, including calcium ion concentration, oxidative stress, and changes in the mitochondrial internal environment (Li et al., 2024). The respiratory chain consists of four multi-protein complexes (I, II, III, IV), two mobile electron carriers, ubiquinone (coenzyme Q), and cytochrome C. Propofol inhibits mitochondrial complexes II and III in the hippocampus. This inhibition can impair electron transfer and reduce the mitochondrial membrane potential (Finsterer and Frank, 2016). More and more studies have shown that propofol promotes changes in the expression of apoptosis-related proteins such as Bcl-2 and Bax, thereby affecting cell survival (Xu et al., 2022). For instance, Maternal propofol exposure during mid-gestation significantly increased Cleaved-Caspase3 levels and pro-apoptotic Bax expression while decreasing anti-apoptotic Bcl-2 in fetal rats, ultimately resulting in cognitive deficits (particularly in learning and memory functions) in male offspring (Mei et al., 2022a). A study found that low concentrations of propofol can promote the survival of nerve cells, while high concentrations of propofol significantly increase the rate of cell apoptosis, demonstrating a concentration-dependent effect (Xu et al., 2022) (Figure 1).
Figure 1

The basic process of cell apoptosis. Created with Figdraw.
3.1.1 MAPK and cell apoptosis
Propofol promotes neuronal apoptosis by inhibiting the MAPK/ERK signaling pathway, disrupting the dynamic balance between pro-apoptotic and anti-apoptotic signals. Studies have found that in human neuronal SH-SY5Y neuroblastoma cells and rat hippocampal tissue cells, the levels of ERK1/2 and its phosphorylated forms (pERK1/2) significantly decrease after propofol treatment, while the phosphorylation level of NF-κB p65 also declines. Propofol treatment also leads to the upregulation of microRNA-34a, which can directly target and inhibit the expression of key molecules in the MAPK/ERK pathway (such as ERK1/2), forming a positive feedback mechanism that further exacerbates ERK signal inhibition (Li et al., 2018).
Phosphorylated CREB is an important survival signaling molecule downstream of the ERK signaling pathway and plays a role in the apoptosis of nerve cells induced by propofol. In early animal experiments, we observed that propofol anesthesia applied to rats during mid-pregnancy resulted in cognitive impairments in their offspring. A key mechanism behind this phenomenon is the activation of histone deacetylase 2 (HDAC2), which in turn inhibits the CREB and NMDA receptor subtype (NR2B) signaling pathway, leading to impaired synaptic plasticity in the hippocampus. Over-phosphorylation of HDAC2 can reduce the phosphorylation level of CREB, thereby decreasing the expression of CREB protein (Mei et al., 2022c).
p38MAPK, as a member of the MAPK family, can be activated by stress factors such as pro-inflammatory cytokines and toxins, leading to inhibition of cell growth and increased apoptosis. The action of propofol can activate the p38MAPK signaling pathway, thereby reducing the vitality of nerve cells and increasing the occurrence of apoptosis (Xu et al., 2020).
3.1.2 Mitochondria and cell apoptosis
Propofol affects a series of mitochondrial-related proteins, including Cyclin-dependent kinase 1 (CDK1), phosphorylated dynamin-related protein 1 (p-DRP1), Parkin1, and DJ-1, by inhibiting Pink1 in developing neurons. This inhibitory effect leads to an increase in the expression levels of these proteins under propofol exposure, resulting in mitochondrial deformation, vacuolization, and swelling, ultimately causing dysfunction, which affects the proliferation, differentiation, and apoptosis of nerve cells, leading to neurotoxicity (Liang et al., 2021). In the CNS, glucose enters neurons through glucose transporter 3 (GLUT3), and then within the cell, mitochondria convert glucose into ATP through oxidative phosphorylation. However, exposure to propofol leads to a limitation in the energy supply of hippocampal neurons, primarily mediated by the downregulation of GLUT3 protein expression, which affects glucose uptake and transport (Xiao et al., 2020). This not only increases the levels of oxidative stress but also promotes the generation of reactive oxygen species (ROS), further exacerbating the damage to nerve cells (Zhang W. et al., 2022).
3.1.3 Ferroptosis and apoptosis
Ferroptosis is a programmed cell death mechanism distinct from apoptosis. Research indicates that propofol may cause an imbalance in the production and degradation of lipid reactive oxygen species within cells. This imbalance can activate ferroptosis, leading to neuronal apoptosis and resulting in neurotoxicity (Chen J. et al., 2023). In the early stages of iron overload, ferroptosis is triggered through the p53 pathway, while in the later stages, it leads to apoptosis accompanied by mitochondrial dysfunction (Zhang, 2020). Thus, ferroptosis and apoptosis may occur sequentially or synergistic regulatory relationship.
3.1.4 Long non-coding RNA and cell apoptosis
Long non-coding RNAs (lncRNAs) play pivotal roles in transcriptional regulation and chromatin remodeling complex formation, and are also capable of modulating mRNA and protein expression levels. Exposure to propofol leads to changes in lncRNA, mRNA, and their downstream synergistic signaling networks. These lncRNAs co-express with calcium signaling pathways (such as CaMKII) and genes related to synaptogenesis, which may trigger neurotoxic responses in mice (Logan et al., 2018). However, the specific mechanisms remain to be further studied. In summary, apoptotic mechanisms play an indispensable role in the pathogenesis of neurotoxicity. The pathological process of apoptosis is intricate, but understanding it offers valuable insights into investigating the neuroprotective effects.
3.2 Neurotoxicity of propofol in different developmental stages of the population
The timing of synaptogenesis in humans ranges from late pregnancy to the third year after birth. During this developmental stage, propofol can easily cross the blood–brain barrier, leading to neurotoxic effects on the developing brain. This toxicity primarily manifests as damage to nerve cells, which may negatively impact learning and memory abilities (Zhang W., et al., 2024; Twaroski et al., 2015).
The mid-pregnancy period is a critical time for neurogenesis and neuronal migration, during which the fetus’s sensitivity to external environmental factors significantly increases. Based on previous animal experiments conducted by our research group, we found that maternal rats exposed to propofol during mid-gestation gave birth to offspring showing significantly prolonged escape latency and reduced platform crossings in behavioral tests. Additionally, the number of neurons in the hippocampal region was significantly decreased, while the expression levels of Bax, Cleaved-Caspase3, and CaMKII were increased, and the expression of Bcl-2 in the hippocampus was suppressed (Mei et al., 2022a).
In the pediatric stage, there is a period known as the “danger window,” which refers to a specific age group where exposure to multiple general anesthetics (mGA) may lead to long-term neurocognitive disorders. It is generally believed that the most sensitive period is before the age of 3 or 4. During this developmental stage, the brain undergoes significant remodeling processes, and anesthetic agents may interfere with the mechanisms of synaptogenesis, neurogenesis, and cell survival in neurons (Colletti et al., 2023), thereby affecting long-term learning and memory capabilities.
For patients aged 60 and above, the incidence of postoperative cognitive dysfunction is more than twice that of patients under 60. This may be related to the fact that elderly patients typically face more risk factors for neurovascular diseases, increased white matter damage, and reduced cognitive reserve (Berger et al., 2015).
In patients with pre-existing neurological disorders, such as Parkinson’s disease and Alzheimer’s disease, the use of propofol may further exacerbate their neurological impairment or accelerate disease progression. In Alzheimer’s patients, the interaction between intrabrain Aβ plaques and hyperphosphorylated Tau protein synergizes with the effects of propofol: propofol promotes Tau phosphorylation by activating GSK3β, while abnormal Tau further inhibits mitochondrial transport, creating a vicious cycle (Eryilmaz et al., 2024). In patients in the preclinical stage of Parkinson’s disease, propofol may worsen the damage to mitochondria caused by α-synuclein oligomers. Overall, the neurotoxicity of propofol varies across different developmental stages. Understanding these differences.
3.3 Neurotoxicity of propofol in different types of nerve cells
The effects of propofol on inhibitory synaptic currents vary across different brain nuclei, indicating that it may have specific effects in different types of neurons (Liu et al., 2021). Astrocytes are the most common glial cells in the central nervous system and play important roles in overall brain metabolism, synaptic transmission, and neuronal protection (Gollihue and Norris, 2020). Astrocytes are responsible for providing energy and metabolic support to neurons by releasing extracellular vesicles. Studies have found that aging, injury, and disease can lead to mitochondrial dysfunction in astrocytes, which can have harmful effects on neurons. Co-culturing neurons with propofol and astrocytes with propofol revealed that propofol reduces neuronal viability and ATP levels, increases neuronal death rates, decreases mitochondrial membrane potential, and increases ROS levels and apoptosis rates, indicating that propofol-induced mitochondrial damage in neurons is more pronounced compared to astrocytes (Zhou et al., 2024), and that the two types of cells respond differently to the neurotoxicity of propofol.
4 The systemic toxicity of propofol and the role of Drp1 in mitochondrial dysfunction in different organs
Propofol’s toxic reactions extend beyond the nervous system, primarily causing dose-dependent suppression of the cardiovascular and respiratory systems, with symptoms such as hypotension, bradycardia, and apnea. An overdose can result in significant hypotension, arrhythmias, or even cardiac arrest. Moreover, prolonged high-dose use of propofol (exceeding 4 mg/kg/h) for over 48 h may trigger propofol infusion syndrome (PRIS), characterized by severe conditions such as metabolic acidosis, rhabdomyolysis, hyperkalemia, and multiple organ failure (Singh and Anjankar, 2022). Other possible adverse reactions include injection pain, allergic reactions, and, although rare, lipid metabolism disorders that can be fatal. The risk of toxicity significantly increases in children, critically ill patients, and with prolonged use, necessitating strict monitoring of vital signs and medication dosage. The corresponding toxic reactions of propofol on different organs are as follows.
Propofol is mainly metabolized in the liver, relying on hepatic blood flow, though extrahepatic metabolism also occurs (Kazi et al., 2025). In adults, UGT1A9 primarily mediates hepatic metabolism, whereas in neonates, cytochrome P450 plays a key role (Sandra et al., 2021). The use of propofol can produce ROS that overwhelm the body’s antioxidant defenses, leading to oxidative stress and potential liver damage. Research indicates that propofol doses exceeding clinical levels (e.g., an IC₅₀ of 254.904 μg/mL) can be toxic to hepatic fibroblasts (AML12) due to elevated intracellular ROS, which triggers mitochondrial pathways that lead to apoptosis (Pehlivan et al., 2023). Mitochondrial fission plays a crucial role in maintaining hepatocyte homeostasis and preventing liver tumorigenesis (Ma et al., 2025).
When activated, Drp1 moves to the mitochondria in cardiac cells, leading to significant mitochondrial division. The excessive fragmentation of mitochondria disrupts the structure of the heart and affects blood flow. This disruption can lead to multi-organ dysfunction and higher mortality rates (Liu et al., 2024). The main pathophysiological mechanism behind PRIS is the disruption of the mitochondrial respiratory chain, which hampers ATP synthesis and leads to cellular hypoxia. For example, propofol causes coenzyme Q-sensitive leakage in the mitochondria of cardiac myocytes. This leakage uncouples immature cardiomyocytes and disrupts coenzyme Q’s role in the electron transport chain (Barajas et al., 2022).
Furthermore, human embryonic kidney cells (HEK-293) serve as an excellent in vitro model for examining renal cell physiology. Studies indicate that with an increase in propofol dosage, there is a corresponding decline in cell viability alongside an increase in ROS, culminating in cell apoptosis and ultimately cell death (Pehlivan et al., 2024). Similar trends have been observed in other cell types, such as human promyelocytic leukemia cells, where elevated doses of isoflurane also hinder oxygen utilization and initiate apoptosis. The extent to which propofol exhibits either complete or primarily mitochondrial toxicity remains uncertain, particularly in individuals with mitochondrial defects. However, it can be inferred that in patients with pre-existing mitochondrial disorders, propofol may further aggravate mitochondrial dysfunction (Finsterer and Frank, 2016).
In summary, propofol causes varying degrees of damage to the nervous system, liver, heart, and kidneys. A deeper exploration of the effects of propofol on different systems and organs provides important clues for our comprehensive understanding of the toxic reactions of propofol.
5 The mechanism by which Drp1 affects the neurotoxicity of propofol
The mitochondrial quality control system, which includes mitochondrial fusion and fission, mitophagy, and mitochondrial biogenesis, is crucial for maintaining mitochondrial homeostasis and function (Eldeeb et al., 2022). Mitochondrial fission is regulated by a series of proteins, with Drp1 being the dominant one. Overexpression of Drp1 leads to excessive mitochondrial fission, resulting in dysfunctional mitochondria. In neurons, abnormal mitochondrial fission is closely related to the occurrence of various neurodegenerative diseases such as Alzheimer’s disease (Joshi et al., 2018), Huntington’s disease (Roe and Qi, 2018), and Parkinson’s disease (Zhang et al., 2020). Therefore, inhibiting the overactivation of Drp1 or regulating mitochondrial dynamics is a method to promote neuroprotection (He et al., 2024).
5.1 Phosphorylation state of Drp1 and neurotoxicity
The activity and function of Drp1 are regulated by various post-translational modifications (PTMs), including phosphorylation, SUMOylation, ubiquitination, palmitoylation, S-nitrosylation, and O-GlcNAcylation (He et al., 2024). Among numerous studies, the phosphorylation modification of Drp1 has attracted particular attention. Specifically, serine 616 (S616) and serine 637 (S637) in Drp1 have been identified as the main phosphorylation sites. S616 can promote mitochondrial fission (Cho et al., 2013), while S637 promotes mitochondrial fusion. Cyclins and cyclin-dependent kinases (CDKs) regulate the phosphorylation of Drp1 Ser616, which then activates Drp1, leading to oligomerization at the fission sites of the mitochondrial outer membrane and initiating mitochondrial constriction and division, facilitating the transition of the cell from G1 to S phase (Dasgupta et al., 2020). A recent study showed that after co-culturing with propofol, propofol treatment significantly increased the phosphorylation level of Drp1 at the Ser616 site in neurons, resulting in enhanced mitochondrial fission and the formation of a fragmented mitochondrial network, ultimately triggering neurotoxicity (Zhou et al., 2024). PINK1 can mediate Drp1 S616 phosphorylation to promote mitochondrial fission, thereby regulating the development and maturation of excitatory circuits and synaptic plasticity in the mouse hippocampus and cortex (Gao et al., 2022). Additionally, PINK1 also influences mitophagy.
There is a significant association between the post-translational modifications of Drp1 and neurodegenerative diseases. For example, blocking Drp1 Ser579 phosphorylation inhibits Aβ1-42 induced neurotoxicity (Xu et al., 2021); in the brains of AD patients, S-nitrosylation of Drp1 promotes the formation of dimers and enhances GTPase activity. This change accelerates mitochondrial fission, ultimately leading to damage to neuronal synapses or cell death (Cho et al., 2009). Currently, there is a lack of research and relevant information on other post-translational modifications of Drp1, apart from dephosphorylation, in regulating the neurotoxic mechanisms in the propofol exposure model. Therefore, further research should be conducted on the post-translational modifications of Drp1, comparing their similarities and differences in neurodegenerative lesions and the propofol exposure model.
5.2 Drp1-mediated signaling pathway and neurotoxicity
In addition, the activity and function of Drp1 are regulated by various intracellular signaling pathways, including AMPK and ERK signaling pathways, which play a key role in cellular metabolic adaptation and survival (Timmins et al., 2024). The translocation of Drp1 to the outer mitochondrial membrane is a necessary step for CaMKII to exert its function. Drp1 can also regulate mitochondrial dynamics through interactions with signaling molecules such as TBK1, thereby affecting the immune response and metabolic state of the cell (Chen et al., 2020). Data indicate that exposure to propofol in human embryonic stem cell (hESC)-derived neurons leads to Drp1 activation via CDK1, increased mitochondrial fission, reduced mitochondrial membrane potential, and opening of the mPTP, all of which collectively induce cell death (Twaroski et al., 2015), but do not significantly alter the expression of mitochondrial fusion-related proteins.
5.3 ROS produced by Drp1 and neurotoxicity
By regulating mitochondrial fission, Drp1 can affect intracellular energy metabolism and oxidative stress, leading to apoptosis. For example, overexpression of Drp1 has been found to induce the production of intracellular ROS, activating apoptotic signaling pathways and promoting cell death (You et al., 2020). The activity of Drp1 is also influenced by changes in calcium ion concentration, with calcium overload activating Drp1 (Feng et al., 2020). The activation of Drp1 and the production of reactive oxygen species (ROS) create a vicious cycle: ROS promote the activation of Drp1, while excessive activation of Drp1 leads to the production of more ROS. Increased ROS levels can lead to the rise of inflammatory factors IL-6, TNF-α, IL-18, and IL-1β, causing damage to neurons (Chen P. et al., 2023). Propofol inhibits PINK1 levels, increasing ROS production.
5.4 The effect of Drp1 and mitophagy on neurotoxicity
Mitophagy refers to the selective recognition and clearance of damaged, dysfunctional, or superfluous mitochondria by cells through the autophagy mechanism. Drp1 interacts with mitochondrial autophagy-related proteins such as PINK1 and Parkin to regulate mitochondrial quality control. In the context of cellular stress and apoptosis, Drp1 promotes mitochondrial fission, increases the occurrence of mitochondrial autophagy, and helps cells eliminate damaged mitochondria, thereby maintaining cell survival (Hasani et al., 2023). Animal studies have found that propofol induces apoptosis in nerve cells but inhibits the mTOR signaling pathway, leading to an increase in autophagy markers LC3-II and Beclin-1, enhancing the level of cellular autophagy and initiating adaptive protective responses in the body (Mei et al., 2022b).
In summary, the mechanisms by which Drp1 influences propofol-induced neurotoxicity are not independent; rather, they are interconnected and intricately intertwined (Figure 2).
Figure 2

The mechanism of propofol in promoting apoptosis via Drp1 regulation. Created with Figdraw.
6 Targeting Drp1 as a therapeutic target to prevent propofol neurotoxicity
It is known that astrocytes can release healthy mitochondria to migrate to damaged neurons to alleviate propofol-induced neurotoxicity. Inhibitors of Drp1, such as Mdivi-1, have been shown to weaken the effects of propofol on NSC proliferation, differentiation, apoptosis, and ROS production, as well as mitigate propofol-induced mitochondrial ultrastructural changes and MMP inhibition, effectively reducing pathological changes in neurodegenerative and metabolic diseases (Liang et al., 2021; Furuya et al., 2023; Sun et al., 2024). However, it is noteworthy that pretreatment with Mdivi-1 only partially attenuates, rather than completely reverses, propofol-induced cell death, which may be associated with the potential role of uninvestigated components. In contrast, pretreatment with the CDK1 inhibitor Roscovitine rescues propofol-induced cell death, Drp1 activation, and increased mitochondrial fission (Hogarth et al., 2023). Hypoxic preconditioning can increase the expression of pDrp1 protein and glucose transporters (GLUT1 and GLUT3), providing neuroprotection and promoting brain recovery (Xiao et al., 2020). Pharmacological activation of AMPK using agents such as AICAR effectively suppresses propofol-induced p53 expression and subsequent apoptotic signaling activation, thereby attenuating propofol-mediated neurotoxicity (Xiao et al., 2022). Experimental evidence demonstrates that Liuwei Dihuang Wan enhances cognitive function and confers neuroprotective effects against brain aging by modulating mitochondrial dynamics. Specifically, LDW upregulates mitochondrial fusion proteins while downregulating Drp1 expression, thereby attenuating mitochondrial impairment and restoring the balance between mitochondrial fusion and fission (Wang et al., 2023).
7 Future research directions and challenges
To fully understand the regulatory mechanisms of Drp1, future research needs to employ various technical approaches, including gene editing, protein interaction analysis, and metabolomics, to reveal the functions of Drp1 under different physiological and pathological conditions. The widespread application of CRISPR/Cas9 gene editing technology allows researchers to quickly construct cell models to explore the roles of specific genes in the cell cycle and apoptosis. Additionally, the emergence of single-cell RNA sequencing technology enables researchers to analyze the dynamic changes in gene expression during the cell cycle and apoptosis at the single-cell level. Furthermore, exploring the combined application of Drp1 with other neurotoxic markers (such as Pink1 and Mfn2) will help improve the predictive and detection capabilities of propofol neurotoxicity and provide references for clinical translational research (Froehlich et al., 2024). The ways in which Drp1 influences Bax and Bcl-2 to initiate the caspase cascade reaction, thereby triggering neuronal apoptosis, also need to be explored in depth in the future. A critical question warranting further investigation is whether the combined use of anesthetic agents in clinical practice elicits differential Drp1-mediated effects on neuronal cells compared to propofol monotherapy.
8 Conclusion
Drp1 reduces the neurotoxicity of propofol by decreasing the production of ROS, adjusting the phosphorylation state of Drp1 to affect the cell cycle, regulating signaling pathways, and activating autophagy. However, the mechanisms and intrinsic relationships among these factors still require further investigation. There is considerable evidence from animal experiments and in vitro studies, but a lack of data at the human level. Although propofol exposure causes damage to developing hippocampal neurons, the mechanisms are not yet clear, but current research evidence points to changes in the expression of apoptosis-related proteins such as Bcl-2 and Bax, thereby affecting neuronal survival. However, this paper has limited clinical relevance and human data references, as it mainly relies on animal studies. As the research advances, we will better understand propofol’s neurotoxicity during human development and how Drp1 regulates this neurotoxicity.
Statements
Author contributions
PG: Writing – original draft. JM: Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was funded by the Natural Science Foundation of Xinjiang Uygur Autonomous Region, China (No. 2023D01C79).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The authors declare that no Gen AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1
Barajas M. B. Brunner S. D. Wang A. Griffiths K. K. Levy R. J. (2022). Propofol toxicity in the developing mouse heart mitochondria. Pediatr. Res.92, 1341–1349. doi: 10.1038/s41390-022-01985-1
2
Berger M. Nadler J. W. Browndyke J. Terrando N. Ponnusamy V. Cohen H. J. et al . (2015). Postoperative cognitive dysfunction: minding the gaps in our knowledge of a common postoperative complication in the elderly[J]. Anesthesiology Clinics, 33, 517–550.
3
Chen P. Chen F. Lei J. Zhou B. (2023). Pomegranate polyphenol Punicalagin improves learning memory deficits, redox homeostasis, and neuroinflammation in aging mice. Phytother. Res.37, 3655–3674. doi: 10.1002/ptr.7848
4
Chen S. Liu S. Wang J. Wu Q. Wang A. Guan H. et al . (2020). TBK1-mediated DRP1 targeting confers nucleic acid sensing to reprogram mitochondrial dynamics and physiology. Mol. Cell80, 810–827.e7. doi: 10.1016/j.molcel.2020.10.018
5
Chen J. Xiao F. Chen L. Zhou Z. Wei Y. Zhong Y. et al . (2023). Role of ferroptosis in hypoxic preconditioning to reduce propofol neurotoxicity. Front. Pharmacol.14:1121280. doi: 10.3389/fphar.2023.1121280
6
Cho B. Choi S. Y. Cho H. M. Kim H. J. Sun W. (2013). Physiological and pathological significance of dynamin-related protein 1 (Drp1)-dependent mitochondrial fission in the nervous system. Exp. Neurobiol.22, 149–157. doi: 10.5607/en.2013.22.3.149
7
Cho D.-H. Nakamura T. Fang J. Cieplak P. Godzik A. Zezong G. et al . (2009). S-Nitrosylation of Drp1 mediates β-amyloid-related mitochondrial fission and neuronal injury. Science324, 102–105. doi: 10.1126/science.1171091
8
Choi S. Y. Lee J.-H. Chung A.-Y. Jo Y. Shin J.-H. Park H.-C. et al . (2020). Prevention of mitochondrial impairment by inhibition of protein phosphatase 1 activity in amyotrophic lateral sclerosis. Cell Death Dis.11:888. doi: 10.1038/s41419-020-03102-8
9
Colletti G. Di Bartolomeo M. Negrello S. Geronemus R. G. Cohen B. Chiarini L. et al . (2023). Multiple general anesthesia in children: a systematic review of its effect on neurodevelopment. J. Pers. Med.13:867. doi: 10.3390/jpm13050867
10
Dasgupta A. Chen K.-H. Wu D. Hoskin V. Mewburn J. Lima P. D. A. et al . (2020). An epigenetic increase in mitochondrial fission by MiD49 and MiD51 regulates the cell cycle in cancer: diagnostic and therapeutic implications. FASEB J.34, 5106–5127. doi: 10.1096/fj.201903117R
11
Della Giovampaola M. Cavalli I. Mascia L. (2022). Neuropsychological outcome of critically ill patients with severe infection. Biomedicines10:526. doi: 10.3390/biomedicines10030526
12
Egner J. M. Nolden K. A. Harwig M. C. Bonate R. P. De Anda J. Tessmer M. H. et al . (2022). Structural studies of human fission protein FIS1 reveal a dynamic region important for GTPase DRP1 recruitment and mitochondrial fission. J. Biol. Chem.298:102620. doi: 10.1016/j.jbc.2022.102620
13
Eldeeb M. A. Thomas R. A. Ragheb M. A. Fallahi A. Fon E. A. (2022). Mitochondrial quality control in health and in Parkinson’s disease. Physiol. Rev.102, 1721–1755. doi: 10.1152/physrev.00041.2021
14
Eryilmaz N. C. Arslan M. Kucuk A. Tuna A. T. Guney S. Kaplanoglu G. T. et al . (2024). Evaluation of the effects of repetitive anaesthesia administration on the brain tissues and cognitive functions of rats with experimental Alzheimer’s disease. Medicina60:1266. doi: 10.3390/medicina60081266
15
Feng S.-T. Wang Z.-Z. Yuan Y.-H. Wang X.-L. Sun H.-M. Chen N.-H. et al . (2020). Dynamin-related protein 1: a protein critical for mitochondrial fission, mitophagy, and neuronal death in Parkinson’s disease. Pharmacol. Res.151:104553. doi: 10.1016/j.phrs.2019.104553
16
Finsterer J. Frank M. (2016). Propofol is mitochondrion-toxic and may unmask a mitochondrial disorder. J. Child Neurol.31, 1489–1494. doi: 10.1177/0883073816661458
17
Froehlich T. Jenner A. Cavarischia-Rega C. Fagbadebo F. O. Lurz Y. Frecot D. I. et al . (2024). Nanobodies as novel tools to monitor the mitochondrial fission factor Drp1. Life Sci. Alliance7:e202402608. doi: 10.26508/lsa.202402608
18
Furuya T. Lin J. Afanaseva A. Molz L. Lagu B. Ma B. (2023). Discovery of potent allosteric DRP1 inhibitors by disrupting protein-protein interaction with MiD49. ACS Med. Chem. Lett.14, 1095–1099. doi: 10.1021/acsmedchemlett.3c00223
19
Gao Q. Tian R. Han H. Slone J. Wang C. Ke X. et al . (2022). PINK1-mediated Drp1S616 phosphorylation modulates synaptic development and plasticity via promoting mitochondrial fission. Signal Transduct. Target. Ther.7:103. doi: 10.1038/s41392-022-00933-z
20
Gollihue J. L. Norris C. M. (2020). Astrocyte mitochondria: central players and potential therapeutic targets for neurodegenerative diseases and injury. Ageing Res. Rev.59:101039. doi: 10.1016/j.arr.2020.101039
21
Gottlieb E. Armour S. M. Harris M. H. Thompson C. B. (2003). Mitochondrial membrane potential regulates matrix configuration and cytochrome c release during apoptosis. Cell Death Differ.10, 709–717. doi: 10.1038/sj.cdd.4401231
22
Hasani S. Young L. E. A. Van Nort W. Banerjee M. Rivas D. R. Kim J. et al . (2023). Inhibition of mitochondrial fission activates glycogen synthesis to support cell survival in colon cancer. Cell Death Dis.14. doi: 10.1038/s41419-023-06202-3
23
He X. Wang L. Tsang H. Y. Liu X. Yang X. Pu S. et al . (2024). GTPBP8 modulates mitochondrial fission through a Drp1-dependent process. J. Cell Sci.137:jcs261612. doi: 10.1242/jcs.261612
24
Hogarth K. Tarazi D. Maynes J. T. (2023). The effects of general anesthetics on mitochondrial structure and function in the developing brain. Front. Neurol.14:1179823. doi: 10.3389/fneur.2023.1179823
25
Joshi A. U. Saw N. L. Shamloo M. Mochly-Rosen D. (2018). Drp1/Fis1 interaction mediates mitochondrial dysfunction, bioenergetic failure and cognitive decline in Alzheimer’s disease. Oncotarget9, 6128–6143. doi: 10.18632/oncotarget.23640
26
Kazi M. Gaskari A. Shahba A. A. Ahmad S. Aldughaim M. S. Hussain M. D. (2025). Propofol: current updates, challenges, and strategies for improved self-nanoemulsifying formulation. ACS Pharmacol. Transl. Sci.8, 1013–1027. doi: 10.1021/acsptsci.4c00745
27
Li G. F. Zhi B. L. Su J. Z. Guang C. Li. (2018). Inhibition of microRNA-34a protects against propofol anesthesia-induced neurotoxicity and cognitive dysfunction via the MAPK/ERK signaling pathway. Neurosci. Lett.675, 152–159. doi: 10.1016/j.neulet.2018.03.052
28
Li Y. Chen L. Zhu D. Chen Y. Qin W. Cui J. (2020). Propofol downregulates the activity of glutamatergic neurons in the basal forebrain via affecting intrinsic membrane properties and postsynaptic GABAARs. Neuroreport31, 1242–1248. doi: 10.1097/WNR.0000000000001540
29
Li J.-y. Sun X.-a. Wang X. Yang N.-h. Xie H.-y. Guo H.-j. et al . (2024). PGAM5 exacerbates acute renal injury by initiating mitochondria-dependent apoptosis by facilitating mitochondrial cytochrome c release. Acta Pharmacol. Sin.45, 125–136. doi: 10.1038/s41401-023-01151-1
30
Liang C. Sun M. Zhong J. Miao C. Han X. (2021). The role of Pink1-mediated mitochondrial pathway in Propofol-induced developmental neurotoxicity. Neurochem. Res.46, 2226–2237. doi: 10.1007/s11064-021-03359-1
31
Liu P.-F. Wang Y. Zhang R. Xu L. Li J.-B. Mu D. (2021). Propofol modulates inhibitory inputs in paraventricular thalamic nucleus of mice. Neurosci. Lett.756:135950. doi: 10.1016/j.neulet.2021.135950
32
Liu Y. Wu Y. Zhu Y. Li Q. Peng X. Zhang Z. et al . (2024). Role of excessive mitochondrial fission in seawater immersion aggravated hemorrhagic shock-induced cardiac dysfunction and the protective effect of mitochondrial division inhibitor-1. Antioxid. Redox Signal.41, 462–478. doi: 10.1089/ars.2022.0167
33
Logan S. Jiang C. Yan Y. Inagaki Y. Arzua T. Bai X. (2018). Propofol alters long non-coding RNA profiles in the neonatal mouse hippocampus: implication of novel mechanisms in anesthetic-induced developmental neurotoxicity. Cell. Physiol. Biochem.49, 2496–2510. doi: 10.1159/000493875
34
Ma X. Wei X. Niu M. Zhang C. Peng Z. Liu W. et al . (2025). Disruption of mitochondrial dynamics and stasis leads to liver injury and tumorigenesis. Cell Biol. doi: 10.1101/2025.02.11.637688
35
Mei J. La H. Xu G. P. (2022a). Effects of propofol anesthesia at different concentrations on learning and memory function of male offspring in mid-pregnant rats and its mechanism. Guangxi Med. J.9, 984–991. doi: 10.11675/j.issn.0253-4304.2022.09.13
36
Mei J. La H. Xu G. P. (2022b). Role of autophagy activation in inhibiting propofol-induced neuronal apoptosis. J. Anhui Med. Univ.57, 1552–1558. doi: 10.19405/j.cnki.issn1000-1492.2022.10.008
37
Mei J. Lin M. Guiping X. (2022c). Effects of propofol anesthesia on learning and memory function and HDAC2-CREB-NR2B signaling pathway in offspring of mid-pregnant rats. Chin. J. Pharm.3, 53–59. doi: 10.3969/j.issn.1006-4931.2022.03.014
38
Nolden K. A. Harwig M. C. Hill R. B. (2023). Human Fis1 directly interacts with Drp1 in an evolutionarily conserved manner to promote mitochondrial fission. J. Biol. Chem.299:105380. doi: 10.1016/j.jbc.2023.105380
39
Pehlivan V. F. Pehlivan B. Duran E. Koyuncu İ. (2024). Comparing the effects of propofol and thiopental on human renal HEK-293 cells with a focus on reactive oxygen species (ROS) production, cytotoxicity, and apoptosis: insights into dose-dependent toxicity. Cureus. doi: 10.7759/cureus.74120
40
Pehlivan B. Pehlivan V. F. Koyuncu I. Duran E. Erdogdu H. (2023). Comparison of cytotoxic, reactive oxygen species (ROS) and apoptotic effects of propofol, thiopental and dexmedetomidine on liver cells at accumulative doses (AML12). Eur. Rev. Med. Pharmacol. Sci.27, 1336–1345. doi: 10.26355/eurrev_202302_31367
41
Roe A. J. Qi X. (2018). Drp1 phosphorylation by MAPK1 causes mitochondrial dysfunction in cell culture model of Huntington’s disease. Biochem. Biophys. Res. Commun.496, 706–711. doi: 10.1016/j.bbrc.2018.01.114
42
Rosdah A. A. Smiles W. J. Oakhill J. S. Scott J. W. Langendorf C. G. Delbridge L. M. D. et al . (2020). New perspectives on the role of Drp1 isoforms in regulating mitochondrial pathophysiology. Pharmacol. Ther.213:107594. doi: 10.1016/j.pharmthera.2020.107594
43
Sandra L. Smits A. Allegaert K. Nicolaï J. Annaert P. Bouillon T. (2021). Population pharmacokinetics of Propofol in neonates and infants: gestational and postnatal age to determine clearance maturation. Br. J. Clin. Pharmacol.87, 2089–2097. doi: 10.1111/bcp.14620
44
Singh A. Anjankar A. P. (2022). Propofol-related infusion syndrome: a clinical review. Cureus14:e30383. doi: 10.7759/cureus.30383
45
Sun F. Fang M. Zhang H. Song Q. Li S. Li Y. et al . (2024). Drp1: focus on diseases triggered by the mitochondrial pathway. Cell Biochem. Biophys.82, 435–455. doi: 10.1007/s12013-024-01245-5
46
Timmins L. R. Ortiz-Silva M. Joshi B. Li Y. L. Dickson F. H. Wong T. H. et al . (2024). Caveolin-1 promotes mitochondrial health and limits mitochondrial ROS through ROCK/AMPK regulation of basal mitophagic flux. FASEB J.38:e23343. doi: 10.1096/fj.202201872RR
47
Torcasio R. Gallo Cantafio M. E. Veneziano C. De Marco C. Ganino L. Valentino I. et al . (2024). Targeting of mitochondrial fission through natural flavanones elicits anti-myeloma activity. J. Transl. Med.22:208. doi: 10.1186/s12967-024-05013-0
48
Twaroski D. M. Yan Y. Zaja I. Clark E. Bosnjak Z. J. Bai X. (2015). Altered mitochondrial dynamics contributes to propofol-induced cell death in human stem cell–derived neurons. Anesthesiology123:1067. doi: 10.1097/ALN.0000000000000857
49
Wang Q. Qiu H. (2023). Deubiquitinase USP16 induces gouty arthritis via Drp1-dependent mitochondrial fission and NLRP3 inflammasome activation. Arthritis Res. Ther.25:126. doi: 10.1186/s13075-023-03095-7
50
Wang S. J. Yang J. Ma H. (2023). Neuroprotective effect of liuwei dihuangwan on aged mice based on the mitochondrial quality control system. Chin J Comp Med, 33, 24–34. doi: 10.3969/j.issn.1671-7856.2023.06.004
51
Xiao F. Lv J. Liang Y. B. Chen Y. H. Tu Y. B. Guan R. C. et al . (2020). The expression of glucose transporters and mitochondrial division and fusion proteins in rats exposed to hypoxic preconditioning to attenuate propofol neurotoxicity. Int. J. Neurosci.130, 161–169. doi: 10.1080/00207454.2019.1667784
52
Xiao F. Qin Y. Chen J. Li C. Qin Y. Wei Y. et al . (2022). The propofol-induced mitochondrial damage in fetal rat hippocampal neurons via the AMPK/P53 signaling pathway. Ann. Transl. Med.10:1106. doi: 10.21037/atm-22-4374
53
Xu Y.-H. Luo Y. Cao J.-B. Liu Y.-H. Song Y.-X. Zhang X.-Y. et al . (2022). LncRNA BDNF-AS attenuates propofol-induced apoptosis in HT22 cells by modulating the BDNF/TrkB pathway. Mol. Neurobiol.59, 3504–3511. doi: 10.1007/s12035-022-02757-y
54
Xu X. Wu G. Liu Y. Zhang L. (2020). Effects of propofol on hippocampal neuron viability. Childs Nerv. Syst.36, 1995–2002. doi: 10.1007/s00381-020-04548-z
55
Xu D. Yang P. Yang Z.-J. Li Q.-G. Ouyang Y.-T. Yu T. et al . (2021). Blockage of Drp1 phosphorylation at Ser579 protects neurons against Aβ1-42-induced degeneration. Mol. Med. Rep.24:657. doi: 10.3892/mmr.2021.12296
56
Yang H. Sibilla C. Liu R. Yun J. Hay B. A. Blackstone C. et al . (2022). Clueless/CLUH regulates mitochondrial fission by promoting recruitment of Drp1 to mitochondria. Nat. Commun.13:1582. doi: 10.1038/s41467-022-29071-4
57
You Y. He Q. Lu H. Zhou X. Chen L. Liu H. et al . (2020). Silibinin induces G2/M cell cycle arrest by activating Drp1-dependent mitochondrial fission in cervical cancer. Front. Pharmacol.11:271. doi: 10.3389/fphar.2020.00271
58
Zeng M. He Y. Du H. Yang J. Wan H. (2020). Output regulation and function optimization of mitochondria in eukaryotes. Front. Cell Dev. Biol.8:598112. doi: 10.3389/fcell.2020.598112
59
Zhang P. (2020). Ferroptosis was more initial in cell death caused by iron overload and its underlying mechanism in Parkinson’s disease. Free Radic. Biol. Med.152, 227–234. doi: 10.1016/j.freeradbiomed.2020.03.015
60
Zhang X. Huang W. Shao Q. Yang Y. Xu Z. Chen J. et al . (2020). Drp1, a potential therapeutic target for Parkinson’s disease, is involved in olfactory bulb pathological alteration in the rotenone-induced rat model. Toxicol. Lett.325, 1–13. doi: 10.1016/j.toxlet.2020.02.009
61
Zhang K.-L. Li S.-J. Pu X.-Y. Wu F.-F. Liu H. Wang R.-Q. et al . (2022). Targeted up-regulation of Drp1 in dorsal horn attenuates neuropathic pain hypersensitivity by increasing mitochondrial fission. Redox Biol.49:102216. doi: 10.1016/j.redox.2021.102216
62
Zhang W. Liu Q. Wang J. Liu L. (2024). Anaesthesia and brain development: a review of Propofol-induced neurotoxicity in pediatric populations. J. Dev. Orig. Health Dis.15:e2. doi: 10.1017/S2040174424000059
63
Zhang W. Liu Q. Zhu H. Ma C. Luo Q. Ji M. et al . (2022). Propofol induces the apoptosis of neural stem cells via microRNA-9-5p/chemokine CXC receptor 4 signaling pathway. Bioengineered13, 1062–1072. doi: 10.1080/21655979.2021.2017590
64
Zhang B.-F. Wu Z.-H. Chen K. Jin H.-J. Wu J. Huang Z.-Y. et al . (2024). Dynamin-related protein 1 mediates the therapeutic effect of isoliquiritigenin in diabetic intimal hyperplasia via regulation of mitochondrial fission. Hypertens. Res.47, 1908–1924. doi: 10.1038/s41440-024-01681-z
65
Zhou Z. Dai W. Liu T. Shi M. Wei Y. Chen L. et al . (2024). Transfer of massive mitochondria from astrocytes reduce Propofol neurotoxicity. Neurosci. Lett.818:137542. doi: 10.1016/j.neulet.2023.137542
Summary
Keywords
Drp1, propofol, neurotoxicity, mitochondria, neuroprotection
Citation
Guo P and Mei J (2025) Exploring the correlation between Drp1 protein and the neurotoxicity of propofol. Front. Neurosci. 19:1614362. doi: 10.3389/fnins.2025.1614362
Received
22 April 2025
Accepted
07 July 2025
Published
21 July 2025
Volume
19 - 2025
Edited by
Petra Scholze, Medical University of Vienna, Austria
Reviewed by
Rohan Gupta, University of South Carolina, United States
Veli Pehlivan, Harran University, Türkiye
Updates
Copyright
© 2025 Guo and Mei.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jing Mei, 2339672366@qq.com
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.