 Neelam Younas1,2*
Neelam Younas1,2* Inga Zerr1,2
Inga Zerr1,2- 1Department of Neurology, National Reference Center for Surveillance of TSE, University Medical Center Göttingen, Göttingen, Germany
- 2German Center for Neurodegenerative Diseases (DZNE), University of Göttingen, Göttingen, Germany
Intracellular aggregation of proteins such as Tau, TDP43, FUS, prion protein, and α-synuclein is a major hallmark of many major neurodegenerative diseases. Aberrant stress granules (SGs) are emerging as key contributors to the nucleation of toxic protein aggregates in these disorders. SGs are dynamic, membrane less cytoplasmic assemblies that form transiently through liquid–liquid phase separation (LLPS) of RNA binding proteins (RBPs) containing low complexity domains, together with stalled mRNAs, to help cells cope with stress. While physiological SGs facilitate cellular resilience to acute stress and undergo rapid disassembly, chronic or excessive stress leads to persistent SGs, driving pathological protein aggregation characteristic of age related neurodegeneration. The inherent reversible aggregation of RBPs crucial for cellular function paradoxically exposes them to misfolding disorders. Notably, recent findings expand this paradigm by demonstrating that Tau itself participates in SG formation, with Tau–SG interactions potentiating Tau aggregation and disease progression in tauopathies. Despite these insights, the precise cellular stressors and posttranslational modifications (PTMs) governing the shift from physiological granules to pathological aggregates remain poorly defined. Emerging evidence highlights oxidative stress as a central upstream mediator of this transition. In this perspective, we synthesize current understanding of how SG dynamics intersect with oxidative stress to potentiate protein aggregation, proposing molecular mechanisms that bridge SG biology and neurodegenerative disease. Elucidating these pathways is essential for the development of targeted therapeutic interventions for disorders such as Alzheimer’s disease and related tauopathies.
1 Introduction
A key hallmark of major neurodegenerative disorders—including Alzheimer’s disease (AD), amyotrophic lateral sclerosis (ALS), frontotemporal dementia (FTD), synucleinopathies and prion diseases—is the intracellular accumulation of aggregated proteins such as MAPT, TDP43, FUS, PRNP and SNCA (Wilson et al., 2023).
Although significant progress has been made in delineating the aggregation process, the exact cellular mechanisms that precipitate pathological aggregation remain inadequately resolved.
Newly identified elements of these aggregates are stress granules (SGs)–membrane less cytoplasmic assemblies of RNA-binding proteins and stalled RNA translational machinery–as crucibles of aggregation (Anderson and Kedersha, 2008; Fomicheva and Ross, 2021). Stress granule dynamics are conserved throughout eukaryotes and represent a universal cytoprotective mechanism against diverse environmental stresses (Kedersha et al., 2013) including oxidative stress, heat shock, osmotic stress, UV irradiation, and nutrient deprivation (Cabral et al., 2022; Glauninger et al., 2022; Kedersha et al., 1999). Under physiological conditions, SGs are transient, rapidly dissolving upon stress resolution. However, chronic or persistent SGs—often observed with aging or continuous stress exposure—may act as nucleation centers for disease-associated protein aggregation, thereby contributing to neurodegenerative pathology (Figure 1).
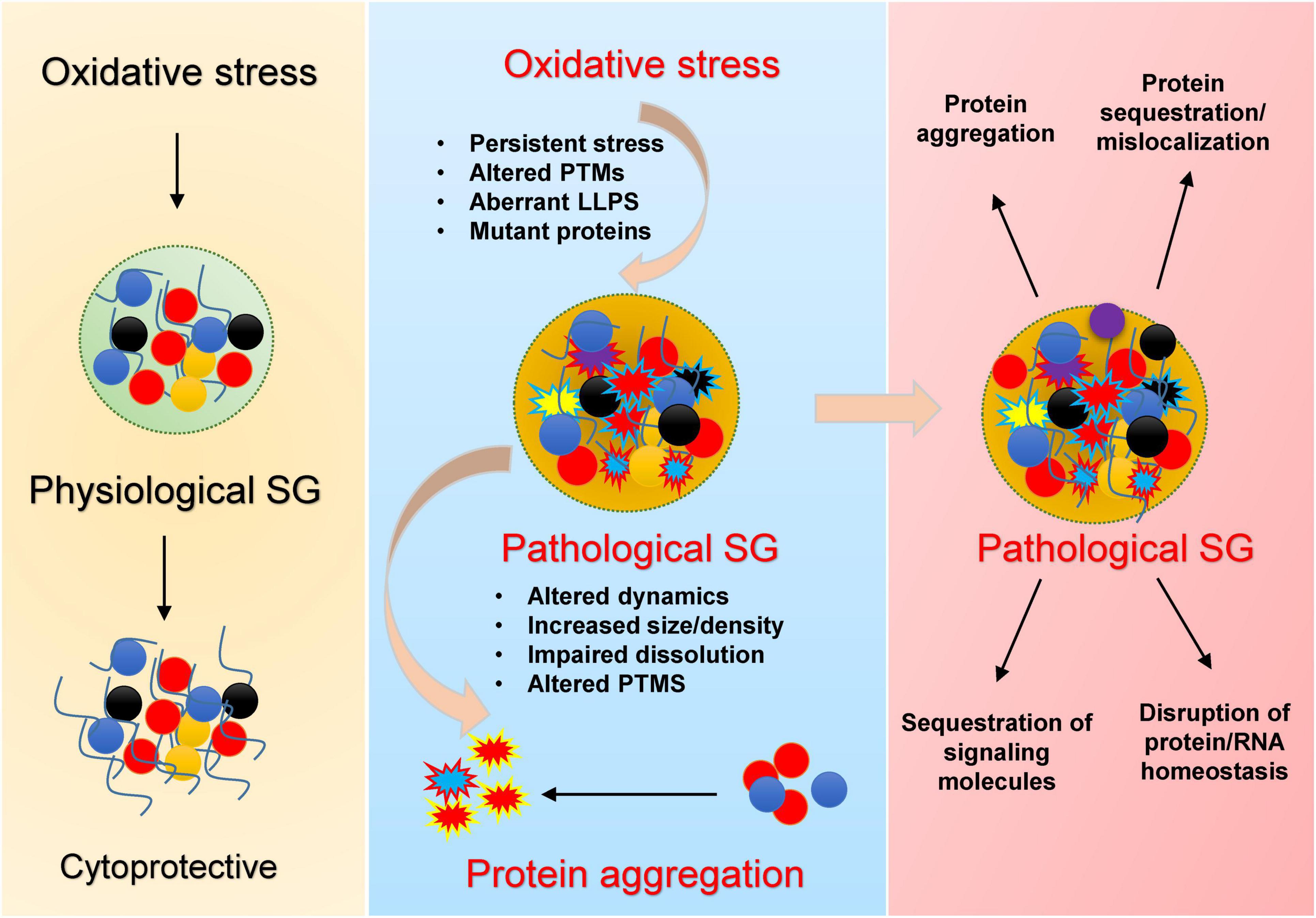
Figure 1. Mechanistic overview by which oxidative stress promotes pathological stress granule (SG) formation and protein aggregation. Under acute stress, SGs transiently assemble, sequester RNAs and proteins, and regulate translation, supporting cell survival during stress (left). Chronic/recurrent oxidative stress promotes persistent SGs, oxidation of key residues, abnormal PTMs, and LLPS transitions, especially in the presence of disease mutations (middle), leading to conversion of dynamic liquid-like SGs into dense, pathological condensates. These aberrant SGs display impaired dissolution, altered composition, and serve as nucleation sites for neurodegeneration-associated protein aggregates. Their persistence (right) drives protein misfolding and mislocalization, sequestration of signaling factors, disruption of protein/RNA homeostasis, and ultimately neuronal dysfunction.
Multiple lines of evidence provide a strong evidence that SGs in cells exposed to environmental stress play the role of seeds for pathogenic protein aggregation in neurodegenerative disorders. First, several proteins [MAPT (Maziuk et al., 2018; Vanderweyde et al., 2016; Younas et al., 2020), TDP43 (Yan et al., 2025), FUS, SNCA (Younas et al., 2023b) and PRNP (Younas et al., 2023b)] that mount up in cytoplasmic aggregates are also components of these SGs (Ash et al., 2021; LeBlang et al., 2020; Yan et al., 2025; Younas et al., 2023b). Second, long lifespan and high metabolism of neurons make them specifically vulnerable to successive stress episodes and to cycles of SG assembly-disassembly (Bishop et al., 2010). Third, mutations in the disease-linked proteins can disrupt normal dynamics of SGs (Lenzi et al., 2015; Mateju et al., 2017; Murakami et al., 2015). Additionally, mutations in other components of SGs e.g., hnRNPA1 (Clarke et al., 2021), TIA1 (Apicco et al., 2018; Baradaran-Heravi et al., 2020; Jiang et al., 2022), ATXN2 (Guevara-García et al., 2012; Kato et al., 2019), UBQLN2 (Peng et al., 2022; Yan et al., 2025) can also disrupt SG dynamics (Table 1). Fourth, SG dense cores act as hotspots for protein aggregation (Maziuk et al., 2018; Vanderweyde et al., 2016; Yan et al., 2025). Stress granules (SGs) have emerged as a critical missing link that integrates genetic, cellular, and pathological evidence, completing the puzzle of aberrant protein aggregation cascade in neurodegenerative diseases (Cui et al., 2024; Dudman and Qi, 2020; Gu et al., 2024; Yuan et al., 2025). A summary of key proteins, their associated stressors, reported mutations, models used for study, post-translational modifications (PTM), and supporting references are described in Table 1.
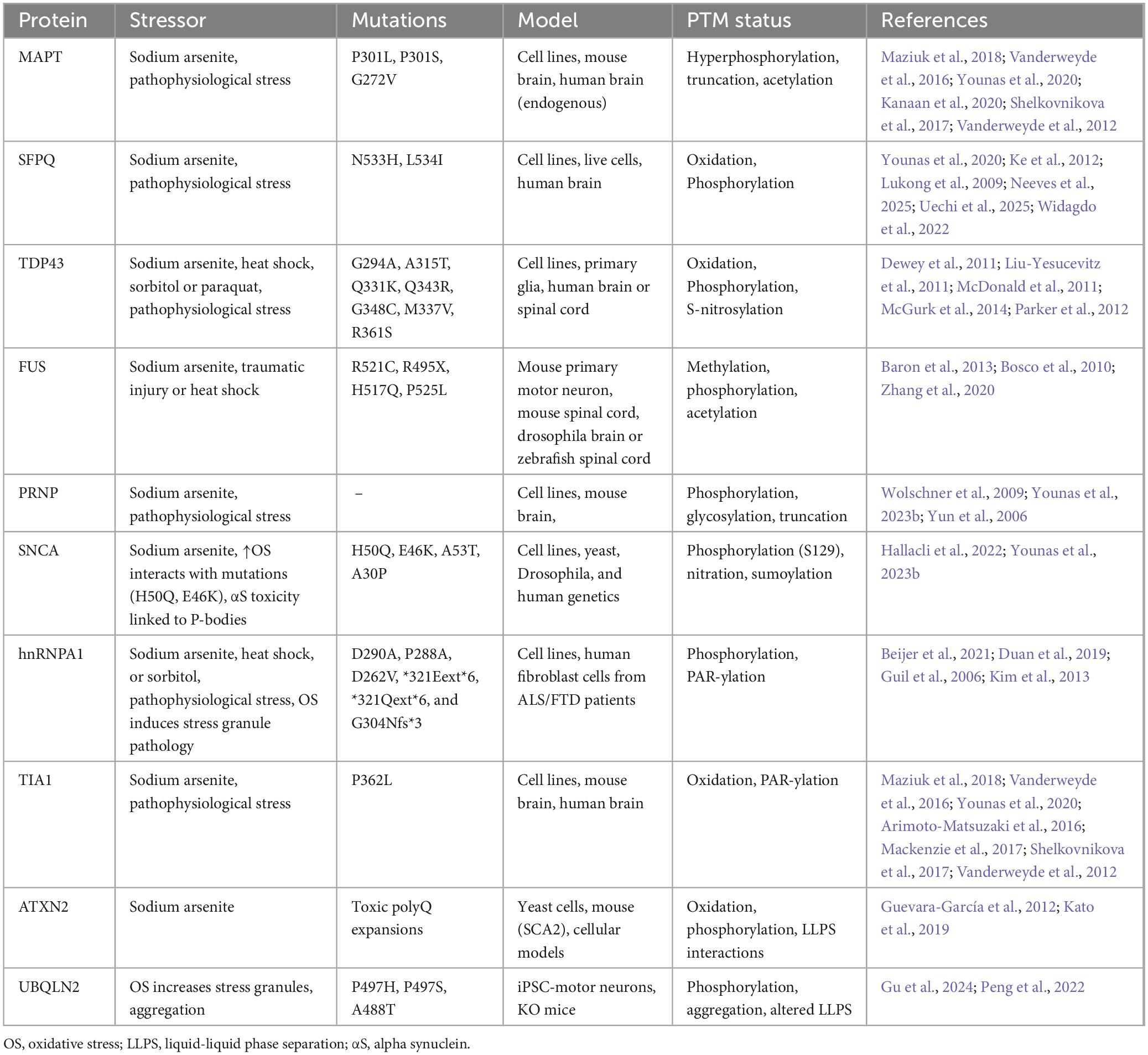
Table 1. Summary of different RBPs or non-RBPs linked with specifically oxidative stress-induced stress granule pathology in neurodegenerative diseases.
2 Oxidative stress-induced stress granules as key drivers of pathological aggregation
A central unresolved question in the field is whether specific stressors preferentially drive the conversion of physiological protein condensates into pathogenic aggregates in vivo, especially under prolonged or recurrent stress. A growing body of research points toward oxidative stress as a principal upstream trigger in this pathogenic cascade (Gu et al., 2024; Ratti et al., 2020). Recent work by Yan et al. (2025) demonstrates that the aggregation of TDP-43 within SGs not only requires high protein concentrations but also strictly depends on oxidative insults. Oxidative agents such as arsenite and paraquat oxidize cysteine residues, which is crucial for induction of TDP-43 demixing within condensates, finally converting it into pathological aggregates. By contrast, stress granules induced by non-oxidative agents (e.g., puromycin) fail to trigger aggregation—even at high TDP-43 levels and with impaired proteasomal or chaperone systems—unless oxidative stress is also present (Yan et al., 2025).
Similar mechanisms appear operative with Tau protein, the signature protein in AD and related tauopathies (Younas et al., 2020). Oxidative stress (induced by sodium arsenite) lead to the upregulation of TIA-1 (a classical marker of SGs) and phosphorylation of Tau in cellular models in both neuronal and non-neuronal cell lines, and human brain of AD cases (Alavi Naini and Soussi-Yanicostas, 2015; Younas et al., 2020). Tau phosphorylation–a hallmark of AD–promotes its phase separation and aggregation in vitro (Ambadipudi et al., 2017). Chronic or repeated oxidative stress, coupled with elevated levels of TIA-1 and phosphorylated Tau, may create a microenvironment within SGs that nucleates pathological aggregation. Thus, stress granules may represent the missing mechanistic link bridging oxidative damage, protein modification, and aggregate formation in neurodegenerative diseases (Figure 1).
Outstanding mechanistic questions remain, especially regarding which molecular species of Tau participate in SG formation. Are they distinct from the Tau species involved in the stabilization of microtubules? Previously it has been reported that a shift in localization of Tau toward somatodendritic compartments occurs to facilitate formation of SGs (Vanderweyde et al., 2016). However, it is unlikely that the same Tau species involved in microtubule stabilization are also responsible for stress granule formation, as this would compromise microtubules integrity during stress. It is more plausible that nuclear Tau species (Younas et al., 2023a) might be involved in the formation of SGs. Tau has six isoforms and many posttranslational modifications. Post-translational modifications also affect stress granules dynamics (Wang et al., 2023). Certain pathological Tau species—especially those that are hyperphosphorylated—show enhanced association with SGs, impacting SG dynamics and contributing to neurodegenerative mechanisms. This mechanistic link underscores the importance of isoform diversity and PTMs in how Tau modulates SG formation and pathological aggregation. Further research is needed to clarify this important issue. By focusing future research on the molecular interactions between oxidative stress, SG dynamics, and protein aggregation, this emerging concept has the potential to transform our strategies for early diagnosis, disease monitoring, and the development of novel treatments for Alzheimer’s disease and related tauopathies.
Chronic and recurrent oxidative stress in the context of mutant proteins—whether RNA binding or not—can drive the formation of pathological SGs (Table 1). Notably, mutations in SG-associated genes—including MAPT, TARDBP (TDP-43), FUS, ATXN2, TIA1, and HNRNPA1—have been shown to disrupt SG dynamics, impair disassembly, and enhance aggregation propensity, thereby contributing to neurodegenerative disease pathogenesis (Table 1).
This is also important to note that the vast majority of knowledge about oxidative stress and stress granule dynamics is derived from experimental exposure to artificial oxidants (e.g., sodium arsenite, H2O2) in cellular models. There is limited direct evidence demonstrating that physiologically relevant endogenous reactive oxygen species levels induce the same oxidative modifications and phase behaviors under disease conditions in vivo (Table 1). The conversion of protective, metastable condensates into pathogenic aggregates within stress granules is determined by a complex interplay of the protein involved, nature of oxidative insult, and the posttranslational landscape.
Taken together, these findings suggest that stress granules could represent a missing mechanistic link connecting oxidative damage, protein modification, and aggregate formation in neurodegenerative diseases (Figure 1). This mechanistic specificity underscores the potential for developing targeted, redox-oriented neuroprotective therapies aimed at modulating SG composition and stability under oxidative conditions. Advancement in this area will require deeper investigations into physiologically relevant stress profiles, diversity of isoforms and posttranslational modifications, and validation in robust in vivo models. These efforts hold promise for translating mechanistic insights into tangible clinical interventions that halt or reverse the aggregation process.
3 Conclusion
In conclusion, converging evidence positions stress granules as a mechanistic nexus where oxidative stress and altered protein homeostasis intersect to drive pathological aggregation in neurodegenerative disorders. The conversion of metastable, protective condensates into pathogenic aggregates appears to be highly contingent on the nature and duration of cellular insults—particularly oxidative stress—rather than on SG formation per se. This duality underscores the therapeutic potential of targeting stress granule dynamics and redox homeostasis, but also highlights the need for more granular understanding of the molecular players and contextual triggers involved. As our mechanistic insights deepen, SGs can offer a promising frontier for both biomarker discovery and intervention in proteinopathic neurodegeneration.
Data availability statement
The original contributions presented in this study are included in this article, further inquiries can be directed to the corresponding author.
Author contributions
NY: Conceptualization, Writing – original draft, Writing – review & editing. IZ: Conceptualization, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. We acknowledge support by the Open Access Publication Funds of the Göttingen University.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The authors declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Alavi Naini, S. M., and Soussi-Yanicostas, N. (2015). Tau hyperphosphorylation and oxidative stress, a critical vicious circle in neurodegenerative tauopathies? Oxid. Med. Cell Longev. 2015:151979. doi: 10.1155/2015/151979
Ambadipudi, S., Biernat, J., Riedel, D., Mandelkow, E., and Zweckstetter, M. (2017). Liquid-liquid phase separation of the microtubule-binding repeats of the Alzheimer-related protein Tau. Nat. Commun. 8:275. doi: 10.1038/s41467-017-00480-0
Anderson, P., and Kedersha, N. (2008). Stress granules: The Tao of RNA triage. Trends Biochem. Sci. 33, 141–150. doi: 10.1016/j.tibs.2007.12.003
Apicco, D. J., Ash, P. E. A., Maziuk, B., LeBlang, C., Medalla, M., Al Abdullatif, A., et al. (2018). Reducing the RNA binding protein TIA1 protects against tau-mediated neurodegeneration in vivo. Nat. Neurosci. 21, 72–80. doi: 10.1038/s41593-017-0022-z
Arimoto-Matsuzaki, K., Saito, H., and Takekawa, M. (2016). TIA1 oxidation inhibits stress granule assembly and sensitizes cells to stress-induced apoptosis. Nat. Commun. 7:10252. doi: 10.1038/ncomms10252
Ash, P. E. A., Lei, S., Shattuck, J., Boudeau, S., Carlomagno, Y., Medalla, M., et al. (2021). TIA1 potentiates tau phase separation and promotes generation of toxic oligomeric tau. Proc. Natl. Acad. Sci. U. S. A. 118:e2014188118. doi: 10.1073/pnas.2014188118
Baradaran-Heravi, Y., Van Broeckhoven, C., and van der Zee, J. (2020). Stress granule mediated protein aggregation and underlying gene defects in the FTD-ALS spectrum. Neurobiol. Dis. 134:104639. doi: 10.1016/j.nbd.2019.104639
Baron, D. M., Kaushansky, L. J., Ward, C. L., Sama, R. R., Chian, R. J., Boggio, K. J., et al. (2013). Amyotrophic lateral sclerosis-linked FUS/TLS alters stress granule assembly and dynamics. Mol. Neurodegener. 8:30. doi: 10.1186/1750-1326-8-30
Beijer, D., Kim, H. J., Guo, L., O’Donovan, K., Mademan, I., Deconinck, T., et al. (2021). Characterization of HNRNPA1 mutations defines diversity in pathogenic mechanisms and clinical presentation. JCI Insight 6:e148363. doi: 10.1172/jci.insight.148363
Bishop, N. A., Lu, T., and Yankner, B. A. (2010). Neural mechanisms of ageing and cognitive decline. Nature 464, 529–535. doi: 10.1038/nature08983
Bosco, D. A., Lemay, N., Ko, H. K., Zhou, H., Burke, C., Kwiatkowski, T. J., et al. (2010). Mutant FUS proteins that cause amyotrophic lateral sclerosis incorporate into stress granules. Hum. Mol. Genet. 19, 4160–4175. doi: 10.1093/hmg/ddq335
Cabral, A. J., Costello, D. C., and Farny, N. G. (2022). The enigma of ultraviolet radiation stress granules: Research challenges and new perspectives. Front. Mol. Biosci. 9:1066650. doi: 10.3389/fmolb.2022.1066650
Clarke, J. W. E., Thibault, P. A., Salapa, H. E., Kim, D. E., Hutchinson, C., and Levin, M. C. (2021). Multiple sclerosis-associated hnRNPA1 mutations alter hnRNPA1 dynamics and influence stress granule formation. Int. J. Mol. Sci. 22:2909. doi: 10.3390/ijms22062909
Cui, Q., Liu, Z., and Bai, G. (2024). Friend or foe: The role of stress granule in neurodegenerative disease. Neuron 112, 2464–2485. doi: 10.1016/j.neuron.2024.04.025
Dewey, C. M., Cenik, B., Sephton, C. F., Dries, D. R., Mayer, P., Good, S. K., et al. (2011). TDP-43 is directed to stress granules by sorbitol, a novel physiological osmotic and oxidative stressor. Mol. Cell Biol. 31, 1098–1108. doi: 10.1128/MCB.01279-10
Duan, Y., Du, A., Gu, J., Duan, G., Wang, C., Gui, X., et al. (2019). PARylation regulates stress granule dynamics, phase separation, and neurotoxicity of disease-related RNA-binding proteins. Cell Res. 29, 233–247. doi: 10.1038/s41422-019-0141-z
Dudman, J., and Qi, X. (2020). Stress granule dysregulation in amyotrophic lateral sclerosis. Front. Cell Neurosci. 14:598517. doi: 10.3389/fncel.2020.598517
Fomicheva, A., and Ross, E. D. (2021). From prions to stress granules: Defining the compositional features of prion-like domains that promote different types of assemblies. Int. J. Mol. Sci. 22:1251. doi: 10.3390/ijms22031251
Glauninger, H., Wong Hickernell, C. J., Bard, J. A. M., and Drummond, D. A. (2022). Stressful steps: Progress and challenges in understanding stress-induced mRNA condensation and accumulation in stress granules. Mol. Cell 82, 2544–2556. doi: 10.1016/j.molcel.2022.05.014
Gu, A., Zhang, Y., He, J., Zhao, M., Ding, L., Liu, W., et al. (2024). Chronic oxidative stress and stress granule formation in UBQLN2 ALS neurons: Insights into neuronal degeneration and potential therapeutic targets. Int. J. Mol. Sci. 25:13448. doi: 10.3390/ijms252413448
Guevara-García, M., Gil-del Valle, L., Velásquez-Pérez, L., and García-Rodríguez, J. C. (2012). Oxidative stress as a cofactor in spinocerebellar ataxia type 2. Redox Rep. 17, 84–89. doi: 10.1179/1351000212Y.0000000005
Guil, S., Long, J. C., and Cáceres, J. F. (2006). hnRNP A1 relocalization to the stress granules reflects a role in the stress response. Mol. Cell Biol. 26, 5744–5758. doi: 10.1128/MCB.00224-06
Hallacli, E., Kayatekin, C., Nazeen, S., Wang, X. H., Sheinkopf, Z., Sathyakumar, S., et al. (2022). The Parkinson’s disease protein alpha-synuclein is a modulator of processing bodies and mRNA stability. Cell 185, 2035–2056.e33. doi: 10.1016/j.cell.2022.05.008
Jiang, L. L., Guan, W. L., Wang, J. Y., Zhang, S. X., and Hu, H. Y. (2022). RNA-assisted sequestration of RNA-binding proteins by cytoplasmic inclusions of the C-terminal 35-kDa fragment of TDP-43. J. Cell Sci. 135:jcs259380. doi: 10.1242/jcs.259380
Kanaan, N. M., Hamel, C., Grabinski, T., and Combs, B. (2020). Liquid-liquid phase separation induces pathogenic tau conformations in vitro. Nat. Commun. 11:2809. doi: 10.1038/s41467-020-16580-3
Kato, M., Yang, Y. S., Sutter, B. M., Wang, Y., McKnight, S. L., and Tu, B. P. (2019). Redox state controls phase separation of the yeast ataxin-2 protein via reversible oxidation of its methionine-rich low-complexity domain. Cell 177, 711–721.e8. doi: 10.1016/j.cell.2019.02.044
Ke, Y. D., Dramiga, J., Schütz, U., Kril, J. J., Ittner, L. M., Schröder, H., et al. (2012). Tau-mediated nuclear depletion and cytoplasmic accumulation of SFPQ in Alzheimer’s and Pick’s disease. PLoS One 7:e35678. doi: 10.1371/journal.pone.0035678
Kedersha, N. L., Gupta, M., Li, W., Miller, I., and Anderson, P. (1999). RNA-binding proteins TIA-1 and TIAR link the phosphorylation of eIF-2 alpha to the assembly of mammalian stress granules. J. Cell Biol. 147, 1431–1442. doi: 10.1083/jcb.147.7.1431
Kedersha, N., Ivanov, P., and Anderson, P. (2013). Stress granules and cell signaling: More than just a passing phase? Trends Biochem. Sci. 38, 494–506. doi: 10.1016/j.tibs.2013.07.004
Kim, H. J., Kim, N. C., Wang, Y. D., Scarborough, E. A., Moore, J., Diaz, Z., et al. (2013). Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature 495, 467–473. doi: 10.1038/nature11922
LeBlang, C. J., Medalla, M., Nicoletti, N. W., Hays, E. C., Zhao, J., Shattuck, J., et al. (2020). Reduction of the RNA binding protein TIA1 exacerbates neuroinflammation in tauopathy. Front. Neurosci. 14:285. doi: 10.3389/fnins.2020.00285
Lenzi, J., De Santis, R., de Turris, V., Morlando, M., Laneve, P., Calvo, A., et al. (2015). ALS mutant FUS proteins are recruited into stress granules in induced pluripotent stem cell-derived motoneurons. Dis. Model Mech. 8, 755–766. doi: 10.1242/dmm.020099
Liu-Yesucevitz, L., Bilgutay, A., Zhang, Y. J., Vanderweyde, T., Citro, A., Mehta, T., et al. (2011). Tar DNA binding protein-43 (TDP-43) associates with stress granules: Analysis of cultured cells and pathological brain tissue. PLoS One 5:e13250. doi: 10.1371/journal.pone.0013250
Lukong, K. E., Huot, M. E., and Richard, S. B. (2009). BRK phosphorylates PSF promoting its cytoplasmic localization and cell cycle arrest. Cell Signal. 21, 1415–1422. doi: 10.1016/j.cellsig.2009.04.008
Mackenzie, I. R., Nicholson, A. M., Sarkar, M., Messing, J., Purice, M. D., Pottier, C., et al. (2017). TIA1 mutations in amyotrophic lateral sclerosis and frontotemporal dementia promote phase separation and alter stress granule dynamics. Neuron 95, 808–816.e9. doi: 10.1016/j.neuron.2017.07.025
Mateju, D., Franzmann, T. M., Patel, A., Kopach, A., Boczek, E. E., Maharana, S., et al. (2017). An aberrant phase transition of stress granules triggered by misfolded protein and prevented by chaperone function. EMBO J. 36, 1669–1687. doi: 10.15252/embj.201695957
Maziuk, B. F., Apicco, D. J., Cruz, A. L., Jiang, L., Ash, P. E. A., da Rocha, E. L., et al. (2018). RNA binding proteins co-localize with small tau inclusions in tauopathy. Acta Neuropathol. Commun. 6:71. doi: 10.1186/s40478-018-0574-5
McDonald, K. K., Aulas, A., Destroismaisons, L., Pickles, S., Beleac, E., Camu, W., et al. (2011). TAR DNA-binding protein 43 (TDP-43) regulates stress granule dynamics via differential regulation of G3BP and TIA-1. Hum. Mol. Genet. 20, 1400–1410. doi: 10.1093/hmg/ddr021
McGurk, L., Lee, V. M., Trojanowksi, J. Q., Van Deerlin, V. M., Lee, E. B., and Bonini, N. M. (2014). Poly-A binding protein-1 localization to a subset of TDP-43 inclusions in amyotrophic lateral sclerosis occurs more frequently in patients harboring an expansion in C9orf72. J. Neuropathol. Exp. Neurol. 73, 837–845. doi: 10.1097/NEN.0000000000000102
Murakami, T., Qamar, S., Lin, J. Q., Schierle, G. S., Rees, E., Miyashita, A., et al. (2015). ALS/FTD mutation-induced phase transition of FUS liquid droplets and reversible hydrogels into irreversible hydrogels impairs RNP granule function. Neuron 88, 678–690. doi: 10.1016/j.neuron.2015.10.030
Neeves, J., Petrić Howe, M., Ziff, O. J., Callaghan, B., Jutzi, D., Pal, K., et al. (2025). An alternative cytoplasmic SFPQ isoform with reduced phase separation potential is up-regulated in ALS. Sci. Adv. 11:eadt4814. doi: 10.1126/sciadv.adt4814
Parker, S. J., Meyerowitz, J., James, J. L., Liddell, J. R., Crouch, P. J., Kanninen, K. M., et al. (2012). Endogenous TDP-43 localized to stress granules can subsequently form protein aggregates. Neurochem. Int. 60, 415–424. doi: 10.1016/j.neuint.2012.01.019
Peng, G., Gu, A., Niu, H., Chen, L., Chen, Y., Zhou, M., et al. (2022). Amyotrophic lateral sclerosis (ALS) linked mutation in Ubiquilin 2 affects stress granule assembly via TIA-1. CNS Neurosci. Ther. 28, 105–115. doi: 10.1111/cns.13757
Ratti, A., Gumina, V., Lenzi, P., Bossolasco, P., Fulceri, F., Volpe, C., et al. (2020). Chronic stress induces formation of stress granules and pathological TDP-43 aggregates in human ALS fibroblasts and iPSC-motoneurons. Neurobiol. Dis. 145:105051. doi: 10.1016/j.nbd.2020.105051
Shelkovnikova, T. A., Dimasi, P., Kukharsky, M. S., An, H., Quintiero, A., Schirmer, C., et al. (2017). Chronically stressed or stress-preconditioned neurons fail to maintain stress granule assembly. Cell Death Dis. 8:e2788. doi: 10.1038/cddis.2017.199
Uechi, H., Sridharan, S., Nijssen, J., Bilstein, J., Iglesias-Artola, J. M., Kishigami, S., et al. (2025). Small-molecule dissolution of stress granules by redox modulation benefits ALS models. Nat. Chem. Biol. 21, 1577–1588. doi: 10.1038/s41589-025-01893-5
Vanderweyde, T., Apicco, D. J., Youmans-Kidder, K., Ash, P. E. A., Cook, C., Lummertz da Rocha, E., et al. (2016). Interaction of tau with the RNA-Binding protein TIA1 regulates tau pathophysiology and toxicity. Cell Rep. 15, 1455–1466. doi: 10.1016/j.celrep.2016.04.045
Vanderweyde, T., Yu, H., Varnum, M., Liu-Yesucevitz, L., Citro, A., Ikezu, T., et al. (2012). Contrasting pathology of the stress granule proteins TIA-1 and G3BP in tauopathies. J. Neurosci. 32, 8270–8283. doi: 10.1523/JNEUROSCI.1592-12.2012
Wang, Z., Zhang, C., Fan, C., and Liu, Y. (2023). Post-translational modifications in stress granule and their implications in neurodegenerative diseases. Biochim. Biophys. Acta Gene Regul. Mech. 1866:194989. doi: 10.1016/j.bbagrm.2023.194989
Widagdo, J., Udagedara, S., Bhembre, N., Tan, J. Z. A., Neureiter, L., Huang, J., et al. (2022). Familial ALS-associated SFPQ variants promote the formation of SFPQ cytoplasmic aggregates in primary neurons. Open Biol. 12:220187. doi: 10.1098/rsob.220187
Wilson, D. M., Cookson, M. R., Van Den Bosch, L., Zetterberg, H., Holtzman, D. M., and Dewachter, I. (2023). Hallmarks of neurodegenerative diseases. Cell 186, 693–714. doi: 10.1016/j.cell.2022.12.032
Wolschner, C., Giese, A., Kretzschmar, H. A., Huber, R., Moroder, L., and Budisa, N. (2009). Design of anti- and pro-aggregation variants to assess the effects of methionine oxidation in human prion protein. Proc. Natl. Acad. Sci. U. S. A. 106, 7756–7761. doi: 10.1073/pnas.0902688106
Yan, X., Kuster, D., Mohanty, P., Nijssen, J., Pombo-García, K., Garcia Morato, J., et al. (2025). Intra-condensate demixing of TDP-43 inside stress granules generates pathological aggregates. Cell 188, 4123–4140.e18. doi: 10.1016/j.cell.2025.04.039
Younas, N., Saleem, T., Younas, A., and Zerr, I. (2023a). Nuclear face of Tau: An inside player in neurodegeneration. Acta Neuropathol. Commun. 11:196. doi: 10.1186/s40478-023-01702-x
Younas, N., Zafar, S., Saleem, T., Fernandez Flores, L. C., Younas, A., Schmitz, M., et al. (2023b). Differential interactome mapping of aggregation prone/prion-like proteins under stress: Novel links to stress granule biology. Cell Biosci. 13:221. doi: 10.1186/s13578-023-01164-7
Younas, N., Zafar, S., Shafiq, M., Noor, A., Siegert, A., Arora, A. S., et al. (2020). SFPQ and Tau: Critical factors contributing to rapid progression of Alzheimer’s disease. Acta Neuropathol. 140, 317–339. doi: 10.1007/s00401-020-02178-y
Yuan, L., Mao, L. H., Huang, Y. Y., Outeiro, T. F., Li, W., Vieira, T. C. R. G., et al. (2025). Stress granules: Emerging players in neurodegenerative diseases. Transl. Neurodegener. 14:22. doi: 10.1186/s40035-025-00482-9
Yun, S. W., Gerlach, M., Riederer, P., and Klein, M. A. (2006). Oxidative stress in the brain at early preclinical stages of mouse scrapie. Exp. Neurol. 201, 90–98. doi: 10.1016/j.expneurol.2006.03.025
Keywords: oxidative stress, stress granules, pathological aggregation, persistent SGs, tauopathies
Citation: Younas N and Zerr I (2025) Oxidative stress-induced stress granules: a central link to protein aggregation in neurodegenerative diseases. Front. Neurosci. 19:1686571. doi: 10.3389/fnins.2025.1686571
Received: 15 August 2025; Accepted: 23 October 2025;
Published: 07 November 2025.
Edited by:
Willayat Yousuf Wani, Northwestern University, United StatesReviewed by:
Nandkishore Belur, Northwestern University, United StatesCopyright © 2025 Younas and Zerr. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Neelam Younas, bmVlbGFtLnlvdW5hc0BtZWQudW5pLWdvZXR0aW5nZW4uZGU=