 Virginia Carla de Almeida Falcão1
Virginia Carla de Almeida Falcão1 Alexia de Matos Czeczot2,3
Alexia de Matos Czeczot2,3 Mohammad Maqusood Alam4†
Mohammad Maqusood Alam4† Kyu-Ho Paul Park5
Kyu-Ho Paul Park5 Jinyeong Heo5Minjeong Woo1
Jinyeong Heo5Minjeong Woo1 Ana Micaela Camini6
Ana Micaela Camini6 Luis Fernando Saraiva Macedo Timmers2,6,7David Shum5Marcia Alberton Perelló2
Luis Fernando Saraiva Macedo Timmers2,6,7David Shum5Marcia Alberton Perelló2 Luiz Augusto Basso2,3,8
Luiz Augusto Basso2,3,8 Pablo Machado2,3,8
Pablo Machado2,3,8 Cristiano Valim Bizarro2,8
Cristiano Valim Bizarro2,8 Vincent Delorme1*†
Vincent Delorme1*†- 1Tuberculosis Research Laboratory, Institut Pasteur Korea, Seongnam, Gyeonggi, Republic of Korea
- 2Instituto Nacional de Ciência e Tecnologia em Tuberculose, Centro de Pesquisas em Biologia Molecular e Funcional, Pontifícia Universidade Católica do Rio Grande do Sul, Porto Alegre, Brazil
- 3Programa de Pós-Graduação em Medicina e Ciências da Saúde, Pontifícia Universidade Católica do Rio Grande do Sul, Porto Alegre, Brazil
- 4Chemistry Platform, Institut Pasteur Korea, Seongnam, Gyeonggi, Republic of Korea
- 5Screening Discovery Platform, Institut Pasteur Korea, Seongnam, Gyeonggi, Republic of Korea
- 6Programa de Pós-Graduação em Biotecnologia e Universidade do Vale do Taquari, Lajeado, Brazil
- 7Programa de Pós-Graduação em Ciências Médicas e Universidade do Vale do Taquari, Lajeado, Brazil
- 8Programa de Pós-Graduação em Biologia Celular e Molecular, Pontifícia Universidade Católica do Rio Grande do Sul, Porto Alegre, Brazil
Introduction: Antifolates were among the first broad-spectrum compounds used as antimycobacterial agents and can still be of use when no other therapeutic options are available. The discovery of compounds targeting the essential folate synthesis pathway could lead to new therapeutic agents to treat tuberculosis (TB). In particular, the enzyme required for the conversion of 7,8-dihydroneopterin (DHNP) to 6-hydroxymethyl-7,8-dihydropterin (HP) and glycolaldehyde (GA) in the folate pathway (MtbFolB, a dihydroneopterin aldolase - DHNA, EC 4.1.2.25), has received little attention as a potential drug target so far, as it is acting upstream of the clinically validated targets dihydropteroate synthase (DHPS; EC 2.5.1.15) and dihydrofolate reductase (DHFR; EC 1.5.1.3).
Methods: We conducted a small-scale diversity screening to identify MtbFolB inhibitors using a microplate-based enzyme inhibition assay. A total of 6,074 compounds were assembled, tested and confirmed in dose-response studies. A preliminary structure activity analysis was performed for the validated hit compounds, along with kinetic inhibition, time-dependent inhibition, as well as docking studies.
Results: The screening resulted in the selection of 19 hits spanning 5 independent clusters. Dose-response studies of re-synthesized hits and newly synthesized derivatives displayed compounds with IC50 values ranging from 2.6 to 47 µM. The structure activity analysis revealed that bi-sulfonamide compounds could be explored for further optimizations. Docking studies highlighted two modes of binding for pyrazol-3-one compounds and, for the sulfonamide series, indicated several interactions with the catalytic Tyrosine-54 (Tyr54D) and Lysine-99 (Lys99A) residues of MtbFolB.
Discussion: Through this work, we established that the MtbFolB assay was able to select small molecules with inhibitory activities, opening prospects for larger scale screening. The sulfonamide compound 13 was also identified as the first compound directed against MtbFolB with an antimycobacterial activity.
Introduction
Folates are essential cofactors participating in key biosynthetic processes in both prokaryotic and eukaryotic cells, notably the synthesis of methionine, thymine and purine bases. While a folate transport system based on membrane-associated proteins exist in mammals for the salvage of folates (1), most micro-organisms, including Mycobacterium tuberculosis (Mtb), must synthesize them de novo. This requirement led to the discovery of small molecules inhibiting the folate biosynthesis pathway (Figure 1), in particular the sulfonamides (2) and diaminopyrimidines (3), leading to an arrest of cellular growth. Such molecules, termed antifolates, were among the first broad-spectrum, synthetic antibiotics and have been widely used for the treatment of urinary tract infections (4), as well as leprosy, caused by Mycobacterium leprae (5). The treatment of tuberculosis (TB) with synthetic drugs also started as early as 1946 with para-aminosalicylic acid (PAS) (6), a pro-drug incorporated in the folate pathway and leading to its downstream inhibition (7–10). Despite these successes, antifolates are now largely absent from the modern treatment of TB, as more potent, bactericidal drugs are available. The emergence of drug-resistant infections however warrants their usage when no other therapeutic options are available (11).
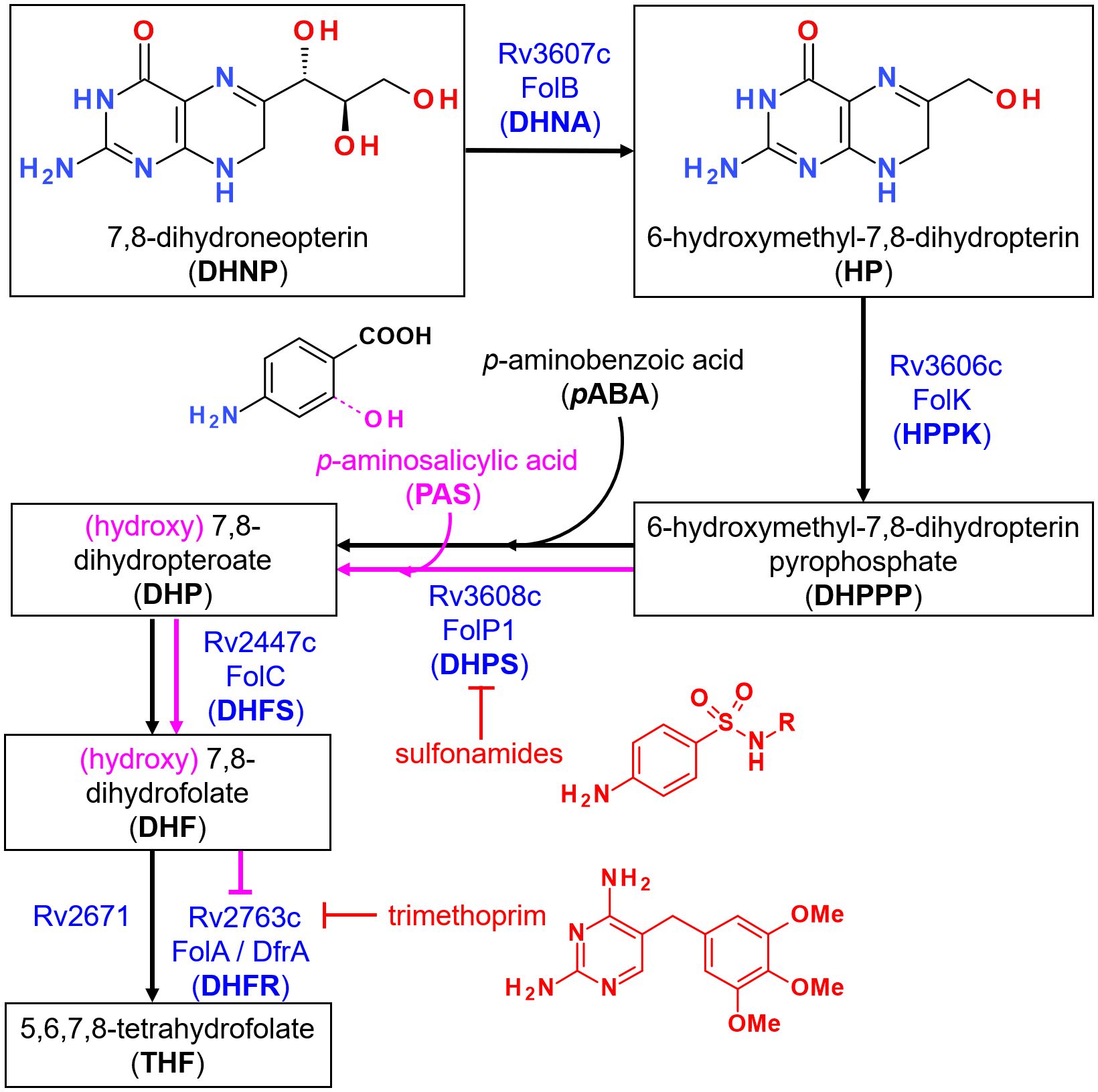
Figure 1 Overview of the folate synthesis pathway in M. tuberculosis. Steps leading to the formation of 7,8-dihydroneopterin (DHNP) are omitted for clarity. The name of the enzyme(s) proposed to catalyze each reaction is indicated in blue, along with known inhibitors in red. Incorporation of p-aminosalicylic acid (PAS) instead of p-aminobenzoic acid (PABA) at the third depicted step leads to the formation of a hydroxylated intermediate that is propagated downstream within the pathway, as indicated in pink. The metabolite ultimately inhibits FolA but not Rv2371, which has been recently identified as having a dihydrofolate reductase (DHFR) activity. Trimethoprim however is able to inhibit both enzymes at this step.
Antifolates as antibacterials have so far mostly been designed to target dihydropteroate synthase (DHPS; EC 2.5.1.15) and dihydrofolate reductase (DHFR; EC 1.5.1.3) due to their attractive active site features well conserved across bacterial species and, for DHPS, the absence of mammalian counterparts (12). The availability of crystal structures for the two orthologues in Mtb [MtbDHPS, Rv3608c (13); MtbDHFR, Rv2763c (14)] also greatly promoted new drug design and discovery of inhibitors with whole-cell activities against this bacteria, particularly for MtbDHFR (15, 16). The fact that Rv2671 (wrongly annotated as a riboflavin biosynthesis protein, RibD) also displays DHFR activity, although to a lower extent than MtbDHFR, may however highlight additional challenges to overcome for the success of such inhibitors (17, 18). In contrast, the catalytic reactions upstream MtbDHPS assigned to Rv3606c (HPPK or FolK; EC 2.7.6.3) and Rv3607c (DHNA or FolB; EC 4.1.2.25) (Figure 1), have received little attention so far. In a preliminary work conducted to identify inhibitors of FolB in Staphylococcus aureus, guanine analogues (triazolo[4,5-d]pyrimidin-7-one) were found to display submicromolar inhibition in vitro despite absence of antibacterial activity (19). Based on the results of this work and others, 8-mercaptoguanine analogues were also recently designed and tested against MtbFolB, revealing also submicromolar in vitro activities but absence of whole cell activity (20).
Previous work highlighted distinctive features of MtbFolB compared to other bacterial orthologues, in that it can catalyze an oxygenase reaction leading to the formation of 7,8-dihydroxanthopterin (DHXP) in addition to the conventional aldolase and epimerase reactions, without the need of metals or cofactor (21). The recent confirmation that MtbFolB aldolase/epimerase activity is essential for bacterial growth (22), together with this body of preliminary work, prompted us to conduct additional screening to identify inhibitors of MtbFolB with greater structural diversity and less resemblance to purines. Here, we report the results of a small-scale diversity screening conducted on MtbFolB and describe the initial structure-activity relationship results obtained for selected hit scaffolds.
Materials and methods
Reagents and chemicals
A library of 5,674 compounds from ChemDiv (diversity set) and 400 compounds from Prestwick (F2L library) were assembled for the screening, arrayed in 96-well plates at concentration of 20 mM in DMSO and stored at -20°C. When available, the hits selected for dose-response validation were repurchased from the same suppliers. The 7,8-dihydro-D-erythro-neopterin (DHNP) substrate for MtbFolB was purchased from Sigma-Aldrich. The 6-hydroxymethyl-7,8-dihydropterin hydrochloride (HP) substrate and the 7,8-dihydroxanthopterin (DHXP) reaction by-product were both purchased from Schircks Laboratories.
Cloning, expression and purification of MtbFolB
The coding region of MtbFolB (Rv3607c; NCBI RefSeq GeneID: 885345) was amplified by PCR using genomic DNA from Mtb H37Rv as a template, using primers 5’-TTTTCCATGGCTGACCGAATCGAA-3’ (Forward) and 5’-TTTTAAGCTTCATACCGCGCCGC-3’ (Reverse), and cloned in pET28a(+) vector using the NcoI/HindIII restriction sites (underlined in the primer sequences). The resulting plasmid pET28::folB, allowing over-expression of the native MtbFolB without tags, was transformed into competent E. coli BL21 Star (DE3) cells (Invitrogen). Transformants were selected on LB agar plates supplemented with 50 μg/mL kanamycin. Single colonies were grown overnight at 37°C in LB medium (50 mL) supplemented with 50 μg/mL kanamycin. Part of the culture (8.5 mL) was inoculated into 500 mL of LB medium supplemented with 50 μg/mL kanamycin and grown at 37°C in shaker at 180 rpm until reaching an optical density (OD600 nm) of 0.4-0.6. The protein expression was induced by addition of 0.1 mM isopropyl-β-D-thiogalactoside (IPTG, Sigma-Aldrich) for 6 hours at 37°C. The cells were then harvested by centrifugation at 11,800 × g for 30 min at 4°C. Approximately 2 g of cell paste was resuspended in 10 mL of buffer A (25 mM Tris-HCl pH 8.0, 50 mM NaCl, 5% glycerol) containing a protease inhibitor cocktail tablet (Complete EDTA-free, Roche Diagnostics) and gently stirred for 30 min in the presence of 0.2 mg/mL lysozyme (Sigma Aldrich). Cells were subsequently disrupted by sonication with a 500W sonicator equipped with a 1/2” diameter probe (Qsonica), using ten pulses of 10s, with cooling intervals of 1 min, at 60% amplitude and the lysate was centrifuged at 38,900 × g for 30 min at 4°C. The supernatant was treated with 1% (w/v) streptomycin sulphate (final concentration) for 30 min to precipitate nucleic acids and ribonuclear proteins (23) under slow agitation and centrifuged at 38,900 × g for 30 min at 4°C. After centrifugation, the supernatant was dialyzed against buffer A, and loaded onto a DEAE Sepharose CL6B anionic exchange column (GE Healthcare) equilibrated with buffer A. The column was washed with 3 column volumes (CV) of buffer A and adsorbed proteins were eluted by a linear gradient (0-1M NaCl) with 14.3 CV in buffer B (25 mM Tris-HCl pH 8.0, 1M NaCl, 5% glycerol) at a flow rate of 1 mL/min. The fractions eluted from anionic exchange column were incubated with ammonium sulfate to a final concentration of 1M and loaded on a Butyl Sepharose High Performance HiLoad 16/10 (GE Healthcare) equilibrated with buffer C (25 mM Tris-HCl pH 8.0, 1M ammonium sulfate, 5% glycerol). The column was washed with 7 CV of buffer C and the adsorbed proteins were eluted by a linear gradient (1 to 0M ammonium sulfate) with 20 CV in buffer C at a flow rate of 1 mL/min. The fractions containing the enzyme, first identified by the absorbance (OD280 nm) and confirmed by SDS-Page, were pooled, concentrated using an Amicon Ultra centrifugal filter with a 10 kDa cut-off (Millipore) and loaded on a HiLoad Superdex 200 26/60 size exclusion column (GE Healthcare) previously equilibrated with buffer A. Protein was isocratically eluted with 1 CV of buffer A at a flow rate of 0.3 mL/min. Fractions containing pure FolB were pooled and concentrated to 0.4-0.6 mg/mL, aliquoted and flash-frozen for conservation at -80°C.
Microplate-based enzyme inhibition assay and screening
All enzymatic reactions were conducted in 384-well plates (Greiner, Cat #781900) using buffer A (25 mM Tris-HCl pH 8.0, 50 mM NaCl, 5% glycerol), in presence of 5 μM 7,8-dihydroneopterin (DHNP, Sigma-Aldrich) and 10 ng of recombinant MtbFolB (14 nM) in a total volume of 50 μL. Prior optimization experiments indicated that these concentrations of enzyme/substrate yielded reproducible end-points, while maintaining substrate saturation and minimizing the amount of enzyme needed per well. Incubations were done at room temperature (24°C) for 4h and the end-point fluorescence reading obtained with a multimode microplate reader (Envision or EnSight, Perkin Elmer) equipped with excitation 405 nm and emission 535 nm filters. Each assay plate included 4 full columns of controls: 2 columns for the positive control (1% DMSO) and 2 columns for the negative control (heat-inactivated MtbFolB), to assess the assay performance. For the negative control, an aliquot of the pure MtbFolB preparation used for the assay was taken and inactivated at 95°C for 10 min the same day.
For the screening, a fresh powder aliquot of substrate (DHNP) was added to buffer A to reach a concentration of 5 mM and solubilized by water-bath sonication for 1h in the dark. This stock was further diluted in buffer A to prepare a working solution at 12.5 μM. Stock solution of compounds at 20 mM in 100% DMSO were extemporaneously diluted at 2 mM in 100% DMSO and 0.5 µL of this intermediate dilution was added to the assay plate, which was pre-filled with 10 μL of buffer A. Using an automated dispenser, the substrate (20 μL at 12.5 μM; 5 μM final concentration) and the enzyme (10 ng in 20 μL; 14 nM final concentration) solutions were immediately added, for a total of 50 μL per well. The compounds were thus at a final concentration of 20 μM and a residual DMSO concentration of 1%. The fluorescence values obtained for each compound were normalized with that of the controls for the corresponding plates, using the average value for the positive control (1% DMSO) as reference for 0% inhibition and the average value of the negative control (heat-inactivated MtbFolB) as reference for 100% inhibition.
Dose-response assay (end-point assay)
For determination of compound activity by dose-response, a similar procedure was followed using compound concentration starting at 100 μM and a total of 10 doses with 2-fold dilution between each dose, with 1% DMSO final concentration in each well. Each dose-response was performed as a technical duplicate and both data pooled and used together for calculations. Raw fluorescence values were plotted as a function of the compound concentration, expressed in log scale, and the data fitted against a sigmoidal dose-response model (4 parameters) using the software Prism v6 (GraphPad). The concentration required to inhibit the enzyme activity by 50% (IC50) was obtained as the best fit value for the IC50 parameter when the fitting converged and the 95% CI are reported. When necessary (absence of low plateau), constraints were added to the bottom value of the dose-response, constraining the value of this parameter above or equal to that of the negative control (inactivated enzyme). In such cases (no plateau), the 95% CI values were omitted (interval too large).
Kinetic assay
Enzyme inhibition studies were performed using a RF-5301 spectrofluorometer (Shimadzu), monitoring an increase in fluorescence, corresponding to the formation of HP, with excitation wavelength at 365 nm and fluorescence emission at 525 nm. The slits were 10 nm for excitation and 15 nm for emission. Reactions were conducted as previously described (20), during 6 min at 25°C using buffer A (25 mM Tris-HCl pH 8.0, 50 mM NaCl, 5% glycerol), in presence of 1.5 μM DHNP (non-saturating concentration) and 300 nM of recombinant MtbFolB in a total volume of 500 μL (quartz cuvette). The DMSO concentration was fixed at 10% and the maximal rate of enzymatic reaction (100% of MtbFolB activity) was determined in these conditions, in absence of inhibitor. As controls, buffer, substrate, inhibitor and enzyme spectra were performed under the same conditions to subtract fluorescence intensities that were not coming from the reaction product. Reaction velocities (μM.min-1) were determined from the fluorescence kinetic measurement by fitting the data points (20) to a straight line by linear regression, using compound concentrations ranging from 50 μM to 1 mM. For each compound, velocities were then plotted as a function of concentration and the plot fitted by non-linear regression using the equation below:
in which Vi and V0 are, respectively, the reaction velocity in the presence and in the absence of inhibitor (I).
Time-dependent enzyme inhibition
Prior to activity determination, MtbFolB (300 nM) was incubated at room temperature in buffer A together with the compound (3, 9 or 20). At the corresponding time-point (0, 30 and 60 min), the substrate was added, and the enzyme velocity determined as described above (kinetic assay). Different incubation and test conditions were used while maintaining the concentration of MtbFolB fixed at 300 nM. i) Normal condition: MtbFolB was incubated with the compound at 300 μM and 2% DMSO, then the velocity measured in presence of the compound (300 μM), 1.5 μM of substrate and 2% DMSO. ii) Excess of substrate: MtbFolB was incubated with the compound at 300 μM and 2% DMSO, then the velocity measured in presence of the compound (300 μM), 3 μM (2×) of substrate and 2% DMSO. iii) Excess of inhibitor: MtbFolB was incubated with the compound at 3 mM (10×) and 15% DMSO, then the velocity measured in presence of the compound (300 μM) and 1.5 μM of substrate with DMSO at a final concentration of 2%.
Chemistry
All procedures for the synthesis of the hit of interests and their derivatives, along with NMR and mass spectrometry characterization data, are available in the Supplementary Methods of this article.
Docking studies
All docking procedures were performed using AutoDock4.2 program. To ensure that all compounds were properly docked, a 3D-grid with dimensions 50 × 50 × 50 with spacing of 0.375 Å was used to define the active site. The grid was defined in the 6-hydroxymethyl-7,8-dihydropterin (HP) binding region, which is located at the interface of two subunits, based on the octameric structure of MtbFolB obtained by crystallography (PDB: 1NBU) (24). A re-docking procedure was performed to evaluate whether the program could reproduce the ligand location found in the structure. The Lamarckian Genetic search Algorithm was used with 60 runs, and the other parameters were set to their default values, except for the number of evaluations and the number of individuals in population, which were set to 4,000,000 and 450, respectively. Docking results were prepared using PyMOL v2.5.3 (DeLano Scientific).
Mycobacterium tuberculosis inhibition assay
The activity of the compounds 3, 9, 13, 16, 20, 21, 22, and 23 was evaluated against M. tuberculosis H37Rv reference strain (ATCC 27294) using a colorimetric assay based on resazurin to determine their minimum inhibitory concentration (MIC), as previously described (25). Briefly, bacteria were seeded in 96-well plates at an OD600 nm of 0.006 and incubated with the compounds at 37°C for 7 days (200 µL per well total). The resazurin solution (60 µL of 0.01%) was then added and the plates further incubated at 37°C for 2 days. A change in color from blue to pink indicated bacterial growth, and the MIC was visually defined as the lowest drug concentration that prevented the color change. Isoniazid (MIC = 2.3 µM) and rifampicin (MIC = 0.2 µM) were used as controls in this experiment. The MIC determination procedures were carried out in Difco™ Middlebrook 7H9 broth (Becton Dickinson – BD) supplemented with 10% (v/v) BBL™ Middlebrook ADC enrichment (albumin, dextrose and catalase – BD) at a final concentration of DMSO equal to 2.5%. A higher concentration of DMSO was chosen to favor compound solubilization at the highest concentrations. The compounds were evaluated using 8-point, 2-fold serial dilutions, with the maximum concentration being 200 µM for the compounds 3, 9, 16, 20, 21, 22, 23 and 20 µg/mL (equivalent to 55.4 µM) for the compound 13. Three independent experiments were conducted for each compound (each experiment included a single dose-response), and the MIC was considered as the most frequent value among the three obtained.
Results and discussion
Assay development, screening and hit confirmation
Based on previous work performed using FolB from S. aureus (19) and Mtb (20, 21), we established an in vitro enzymatic assay for the evaluation of compound libraries arrayed in 384-well plates. The assay is based on the fluorescence of 6-hydroxymethyl-7,8-dihydropterin (HP), the kinetic product formed by action of MtbFolB on the substrate 7,8-dihydroneopterin (DHNP). The fluorescence of the mixture containing the substrate alone was found to be stable, even after 20h incubation (Supplementary Figure S1A). The thermodynamic product, 7,8-dihydroxanthopterin (DHXP), has a weaker fluorescence intensity at the wavelength used (Supplementary Figure S1B) and is formed at a smaller rate (21), allowing for a stable end-point measurement in a time-window from 3h and up to 20h (Supplementary Figure S1C). The 4h time-point was selected for the screening to ensure maximum conversion of the substrate while minimizing formation of DHXP.
The assay developed was then used to screen a small library of diverse molecules (total 6,074 compounds) and the fluorescence results normalized to that of the controls on a plate-by-plate basis (Figure 2). Hits were selected based on a 30% inhibition threshold, yielding 19 compounds that were selected for dose-response studies (Supplementary Figure S2). These molecules could be grouped into 5 independent clusters (numbered I-V), except for 4 compounds that were singletons. Hits were re-purchased or re-synthesized and tested at 10 different doses in duplicate using the same assay, with concentrations starting at 100 μM. Results are summarized in Table 1. Based on these confirmation data, clusters I, IV and V were eliminated due to lack of activity. The activity of the singletons 2, 14 and 18 were confirmed, but we could not re-test compound 17 (not available to re-purchase). Clusters II and III were also found to be active and we decided to prioritize our efforts on the characterization of these two clusters.
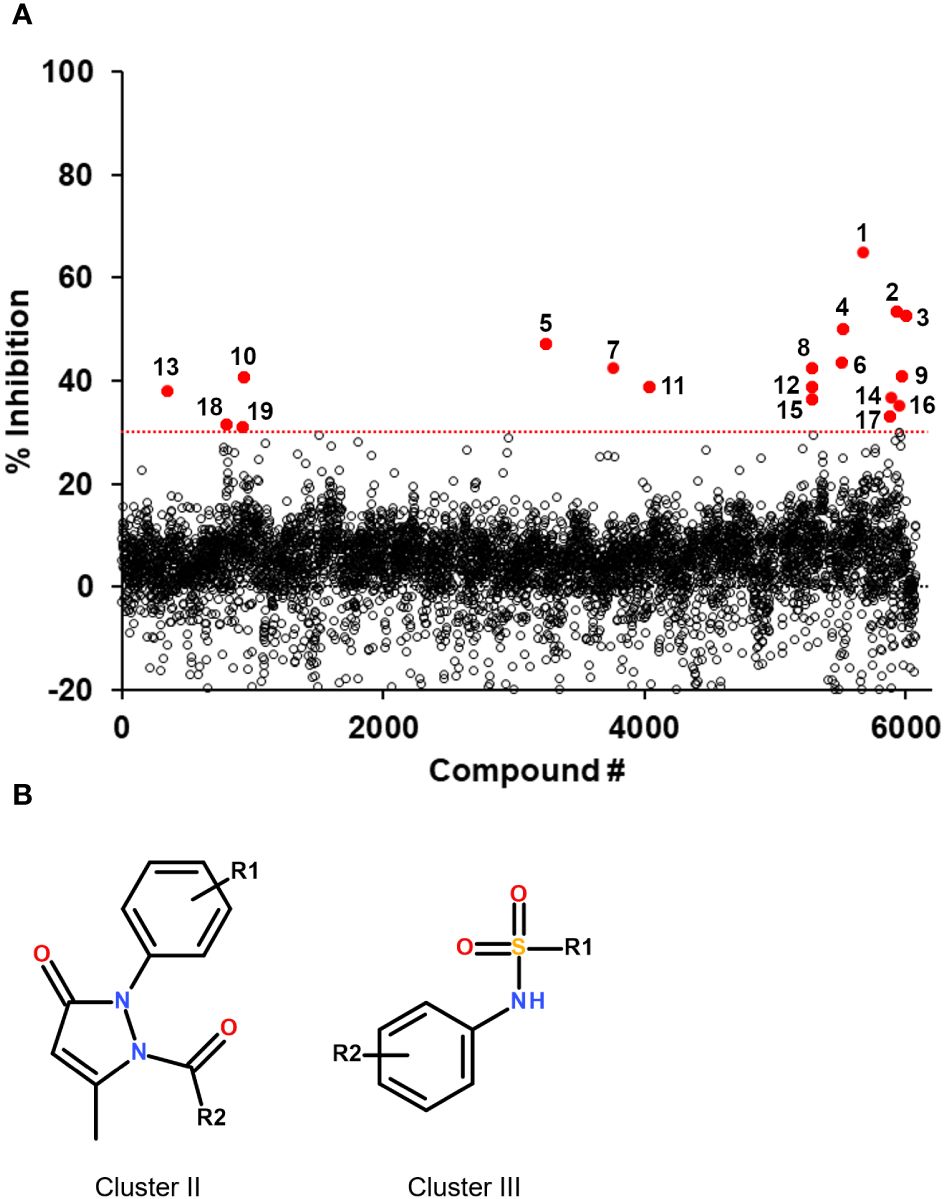
Figure 2 Screening results of the 6,074 compounds and selected hit scaffolds. (A) Summary of the percent inhibition obtained during the primary screening, for each compound tested. The threshold for hit selection was fixed at 30% in order to maximize the number of hits while minimizing the selection of potential false positives. The threshold is indicated by a red dotted line. Compounds above the threshold are shown in red and their compound number is indicated (numbering corresponds to that of Table 1). (B) Core structure of the two hit scaffolds that were selected following validation by dose-response.
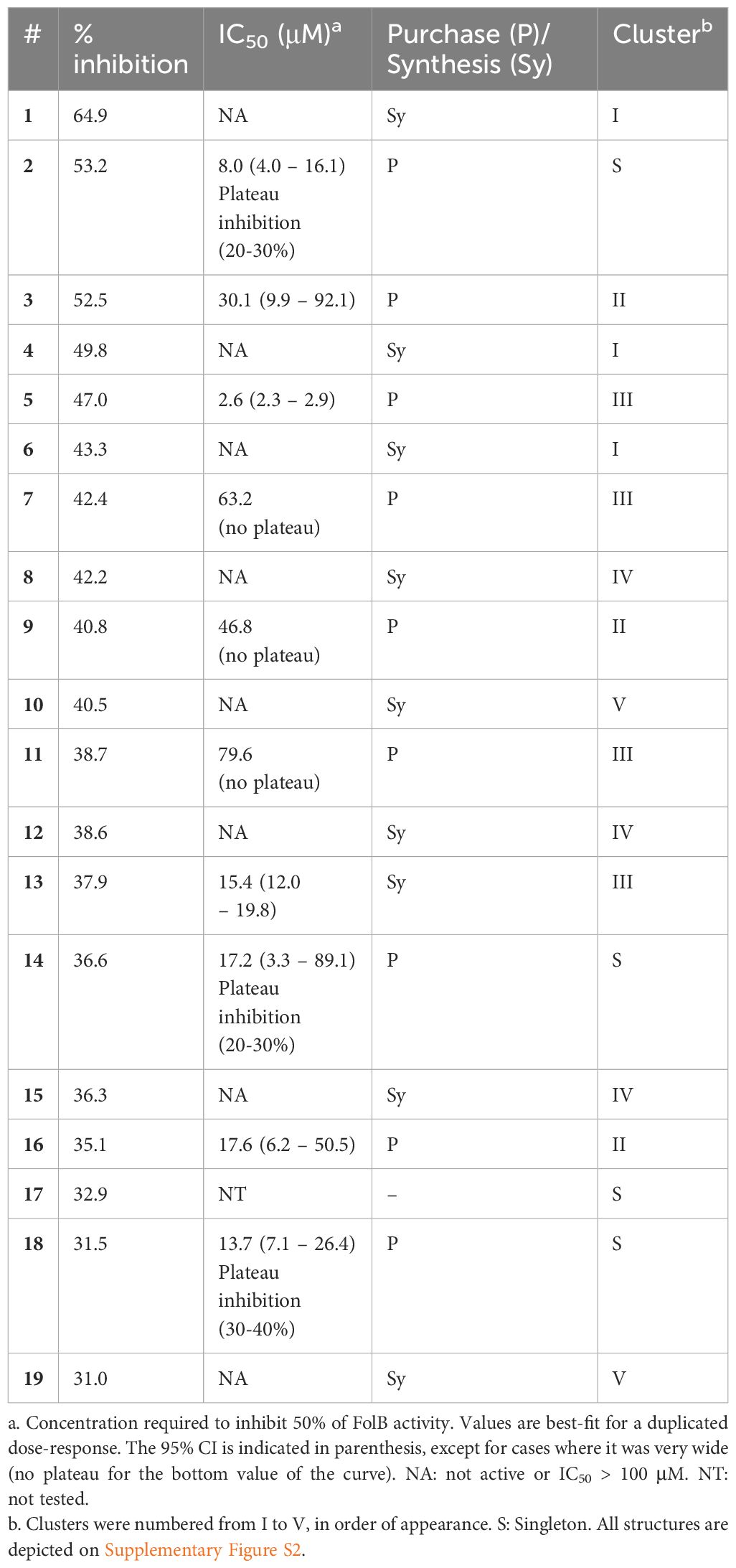
Table 1 Summary of the hits selected at 30% inhibition threshold and their validation by dose-response.
Pyrazol-3-one series (cluster II)
The 3 active hits for this series (3, 9 and 16) had IC50 values ranging from 18 to 47 μM in the dose-response assay. Interestingly, 4 other compounds from the initial screening library displayed similar structures and, although not selected from the primary screening, had percentages of inhibition close to the 30% threshold. These 4 compounds were re-purchased and tested in dose-response, revealing that all were active with IC50 values within the same range as the 3 selected hits (Table 2). Kinetic inhibition studies also indicated a dose-dependent inhibition of the reaction rate (Supplementary Figure S3), but higher doses of compounds were required to see a reduction in velocity during the first minutes of the reaction, as shown by the higher IC50 values obtained (Table 2). Time-dependent inhibition studies conducted with compounds 3, 9 and 20 indicated that the inhibition of the reaction rate was stable in time for at least 1h, reaching 40 to 60% inhibition at 300 μM (Supplementary Figure S4). Pre-incubating the enzyme with higher doses of compounds (up to 3 mM) did not affect the inhibition of the reaction, nor did higher substrate concentrations (Supplementary Figure S4).
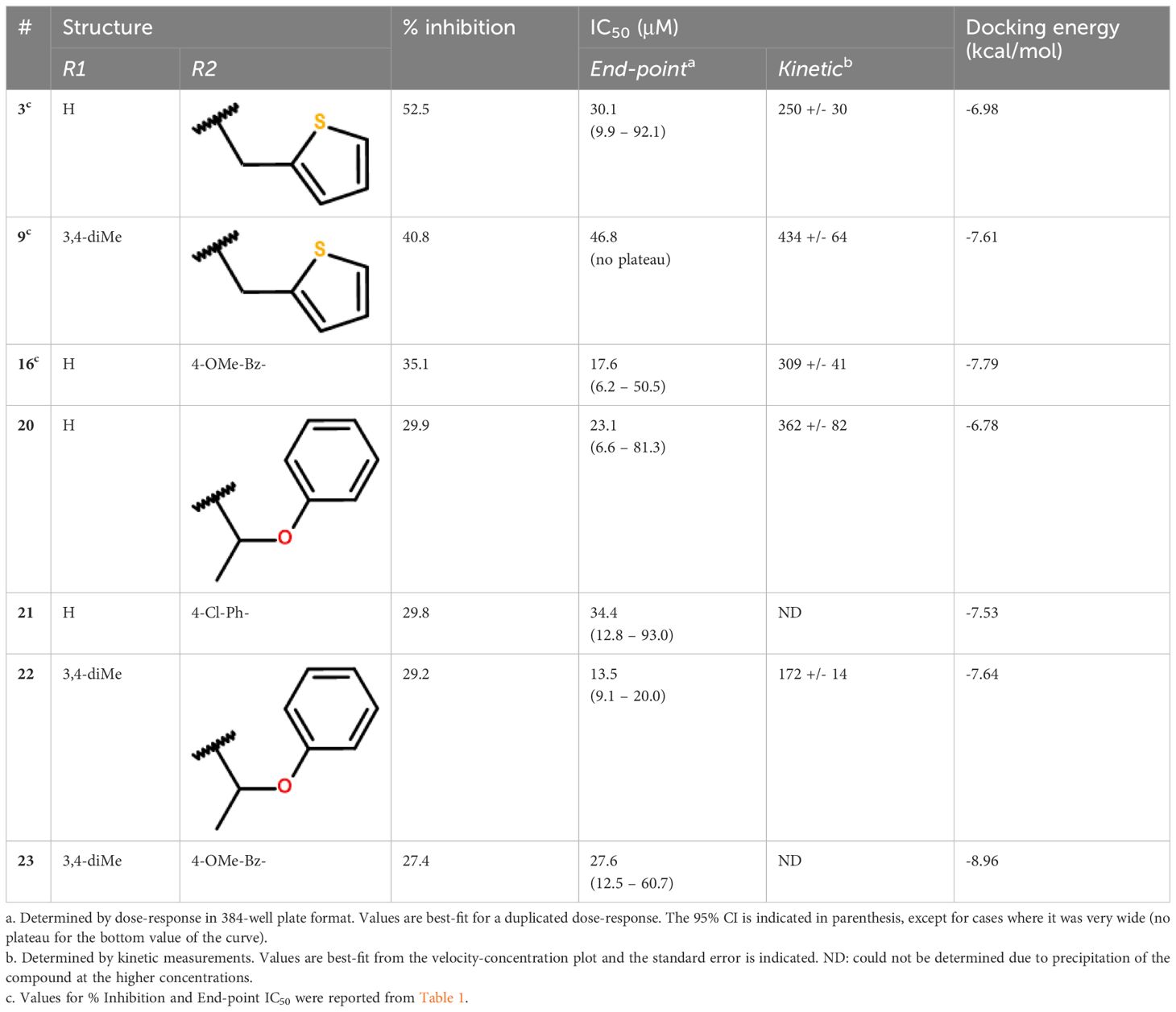
Table 2 Activity profile of the compounds from cluster II.
Because MtbFolB is able to transform the initial substrate into 3 different species (including 2 reversibly) (21), complex experiments would be needed to conclude on the inhibition mechanism. Here, we assumed that the compounds could bind within the active site (formed at the junction between 2 monomers of MtbFolB) and conducted docking studies to explore their mode of binding. The estimated free energies of the top docking poses are indicated in Table 2. Interestingly, not all 7 derivatives adopted a similar pose and two distinct modes of binding were observed. In the first pose (compounds 9, 16, 22 and 23), the carbonyl of the pyrazol-3-one core is engaged in hydrogen-bonding with the tyrosine and lysine of the active site (Tyr54D, Lys99A; A, D refer to the MtbFolB subunit: two are necessary to form an active site), as well as π-π stacking interactions with Tyr54 (Figure 3A; Supplementary Figure S5). The N-carbonyl side-chains were found to interact either through hydrophobic contacts (9, 22) or hydrogen-bonding (16, 23) with nearby residues. In the second pose however (compounds 3, 20 and 21), only the N-carbonyl side-chains were found to interact with the Tyr54D of the active site, through π-π stacking interactions (Figure 3B and Supplementary Figure S5). Although the first pose is likely to induce inhibition of the catalytic activity, the second pose appears to have less affinity for the catalytic residues, which may lead to an easy displacement of the compound by the natural substrate. The existence of this second pose, less productive, may explain the low inhibition potential of the compounds from this cluster. Indeed, no compound in this series showed inhibitory activity against M. tuberculosis (data not shown).
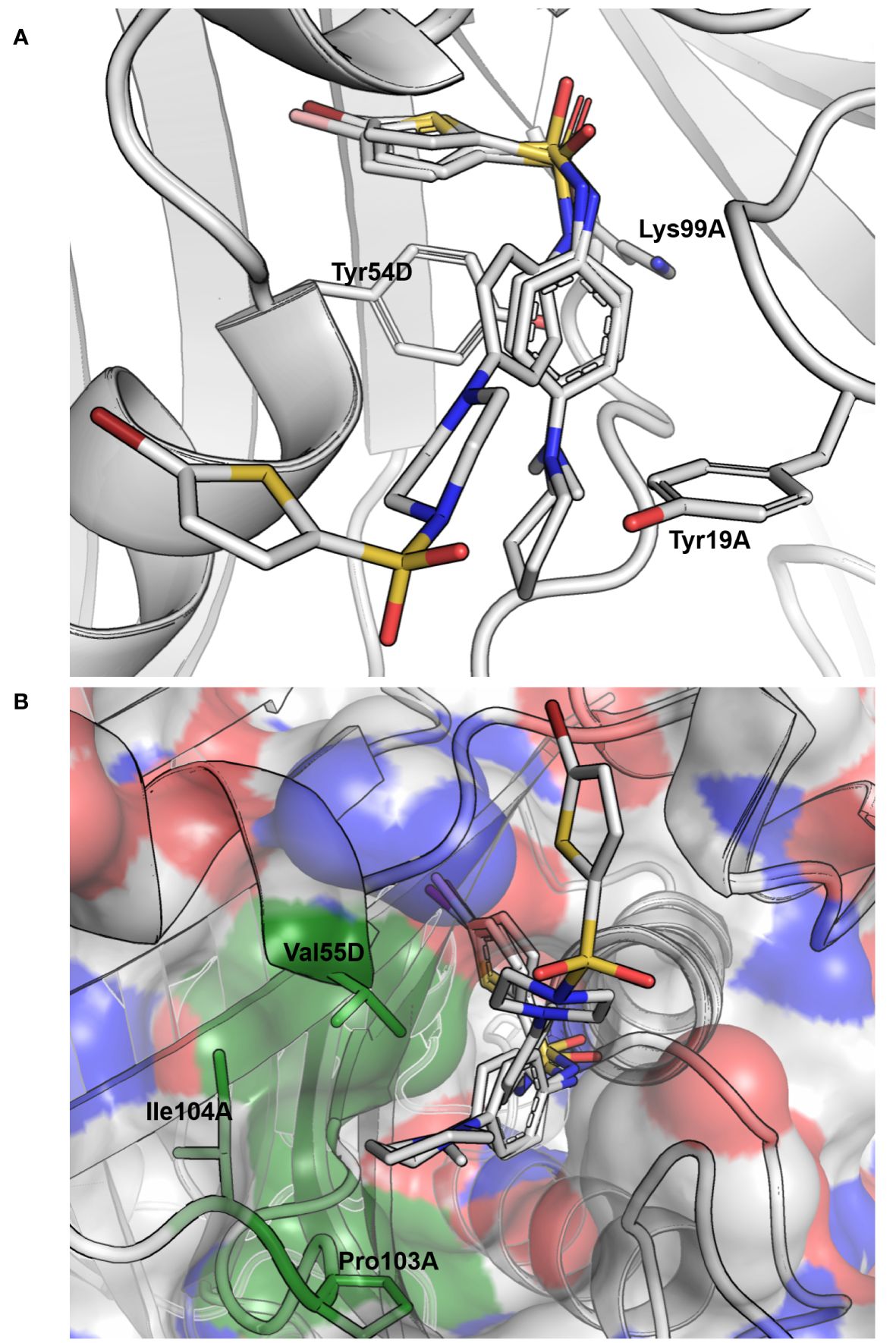
Figure 3 Docking results for compounds 13, 32 and 39 of cluster III. (A) Docking pose obtained for the three compounds, where they are seen interacting with the tyrosine of the active site (Tyr54D; A, D refer to the MtbFolB subunit: two are necessary to form an active site) through π-π stacking interactions with both the benzyl and thiophene rings on each side of the sulfonamide. Hydrogen-bonding is also possible between the lysine of the active site (Lys99A) and the oxygen of the sulfonamides (distance ~3.0 Å). Compound backbones are indicated in gray, with nitrogen in blue, sulfur in light orange and oxygen in red. PDB: 1NBU. (B) Surface view of the MtbFolB dimer, showing that in this docking pose the S-moiety of these sulfonamides lye within the catalytic pocket, while the N-moieties rest at the entrance of the cavity, lined by residues Val55D, Ile104A and Pro103A. Surface transparency is 20% and colors are gray for carbon, blue for nitrogen and red for oxygen. The three residues lining the cavity entrance are labeled and colored in green. PDB: 1NBU.
Sulfonamide series (cluster III)
The second series included 2 promising derivatives, namely 5 and 13, displaying IC50 values of 2.6 and 15.4 μM, respectively. Considering the relative ease of access to these compounds and other derivatives (1 step reaction with the appropriate reagents), we decided to draw a preliminary structure activity profile of this series of compounds. In a first step, we explored the effect of varying the substituent connected to the sulfur atom of the sulfonamide (S-series, Table 3). Our results indicated that the activity could be retained for a variety of substituents. In particular, the pyrazole 25 and the isoxazole 27 both showed good activities (IC50 values of 6.8 μM and 9.2 μM, respectively) and the longer, flexible diphenyl ether chain of 30 could also be tolerated, although the resulting inhibition was more modest (IC50 value of 31.7 μM). In a second step, we explored the substituents on the amide side (N-series) and noted that variations at this position might be more constrained (Table 4). While the piperidine 32 had slightly better activity compared to the reference secondary amine (IC50 value of 11.3 μM against 15.4 μM), the primary amine 31 was less active (IC50 value of 37.8 μM) and the imidazole 33 together with the amide 34 were both inactive. Substituting the piperidine led to either decrease (35, 36) or loss (38) of the activity. Replacing the secondary amine with an isopropyl also led to the loss of activity, as seen in compound 37. Interestingly however, the phenyl piperazine-linked bi-sulfonamide 39 maintained a good activity (IC50 value of 10.6 μM), indicating that this type of bi-sulfonamide could present a viable strategy for further optimizations.

Table 3 Activity profile of the derivatives from cluster III (S-series).
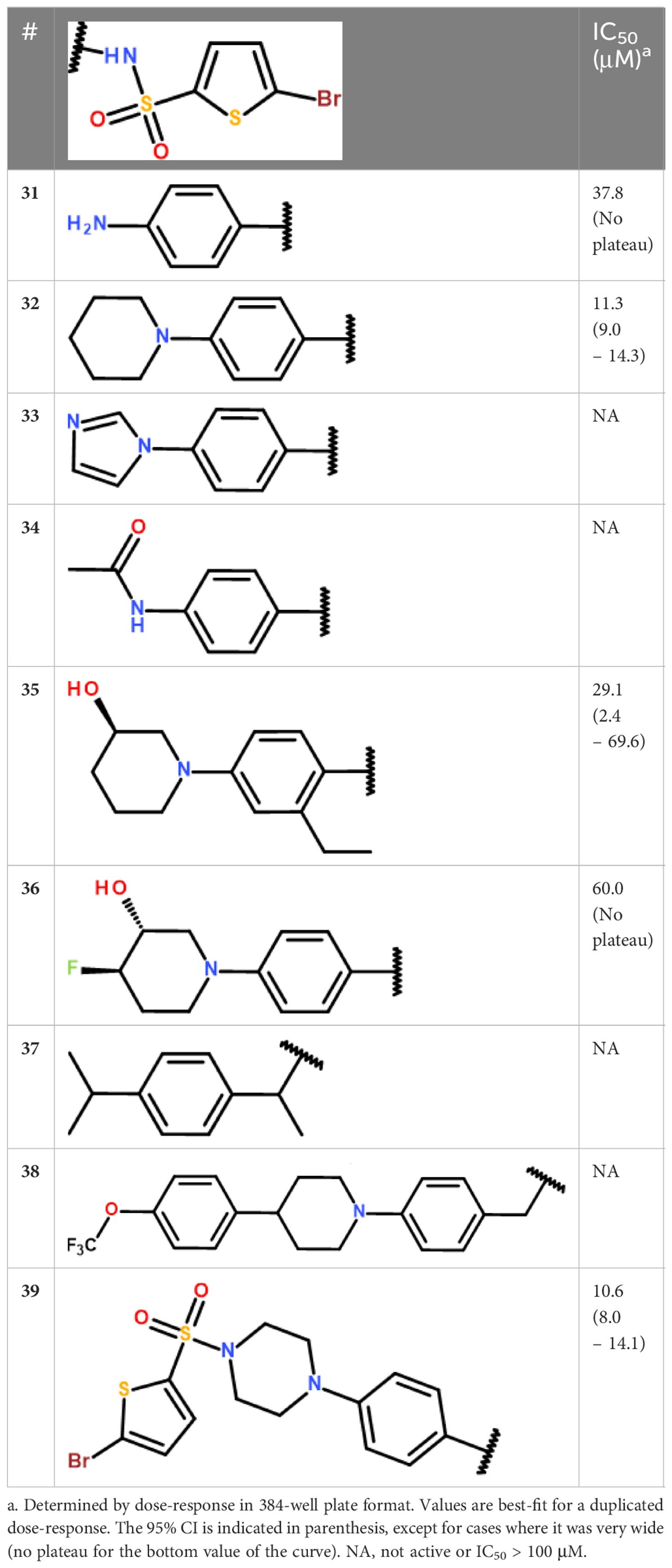
Table 4 Activity profile of the derivatives from cluster III (N-series).
Similarly to the compounds of the pyrazol-3-one series (Cluster II), kinetic and time-dependent inhibition studies were initiated for the sulfonamide series. Despite reasonable computed logP and polar surface area (TPSA, Å2) values (< 3 and< 120, respectively; Supplementary Table S1), solubility issues appeared for most compounds of this family preventing studies at concentrations exceeding 20 to 40 µM, which was insufficient to complete the required experiments. We thus decided to focus instead on docking studies in order to gain more insight into the potential mode of binding of these compounds in the active site of MtbFolB. We chose to include compounds 5, 13, 27, 32 and 39 as they were among the most active derivatives identified for this series. Compounds 24 and 34, which were found to be inactive, were also included for comparison. As shown in Figure 4A, compounds 13, 32 and 39 aligned on the same pose within the active site and displayed several interactions with the tyrosine and lysine of the active site (Tyr54D, Lys99A), through π-π stacking interactions with Tyr54D and both the benzyl and thiophene rings on each side of the sulfonamide, as well as hydrogen-bonding between the Lys99A and the oxygen of the sulfonamide (distance ~3.0 Å). While the S-moiety of the sulfonamide lied within the catalytic pocket, the N-moiety rested at the entrance of the cavity, lined by alkyl residues Val55D and Ile104A (Figure 4B). Compound 27 had a similar orientation but displayed a different pose in the active site, so that the Lys99A was apparently not engaged in the interaction (Supplementary Figures S6A, S7). Compound 5 in contrast showed an opposite orientation, with the S-moiety pointing towards the outside, similarly to the two inactive compounds 24 and 34 (Supplementary Figure S6). The presence of two hydrophobic and bulky amino-acids at the entrance of the active site (Val55D and Ile104A) may explain the lower tolerance for variation on this side of the molecule, in particular the loss of activity seen for the quite rigid and polar imidazole 33 and the rigid and bulky 4-isopropylbenzyl 37. It is more complicated however to understand why the amine 31 and amide 34 are inactive solely based on these docking studies. Molecular dynamic experiments would be required to better appreciate the interactions within the active site. Other parameters might also have to be considered to explain this lack of activity, like non-specific binding elsewhere in the MtbFolB tetramer prior to the formation of the octamer, which is expected to occur upon substrate binding (24).
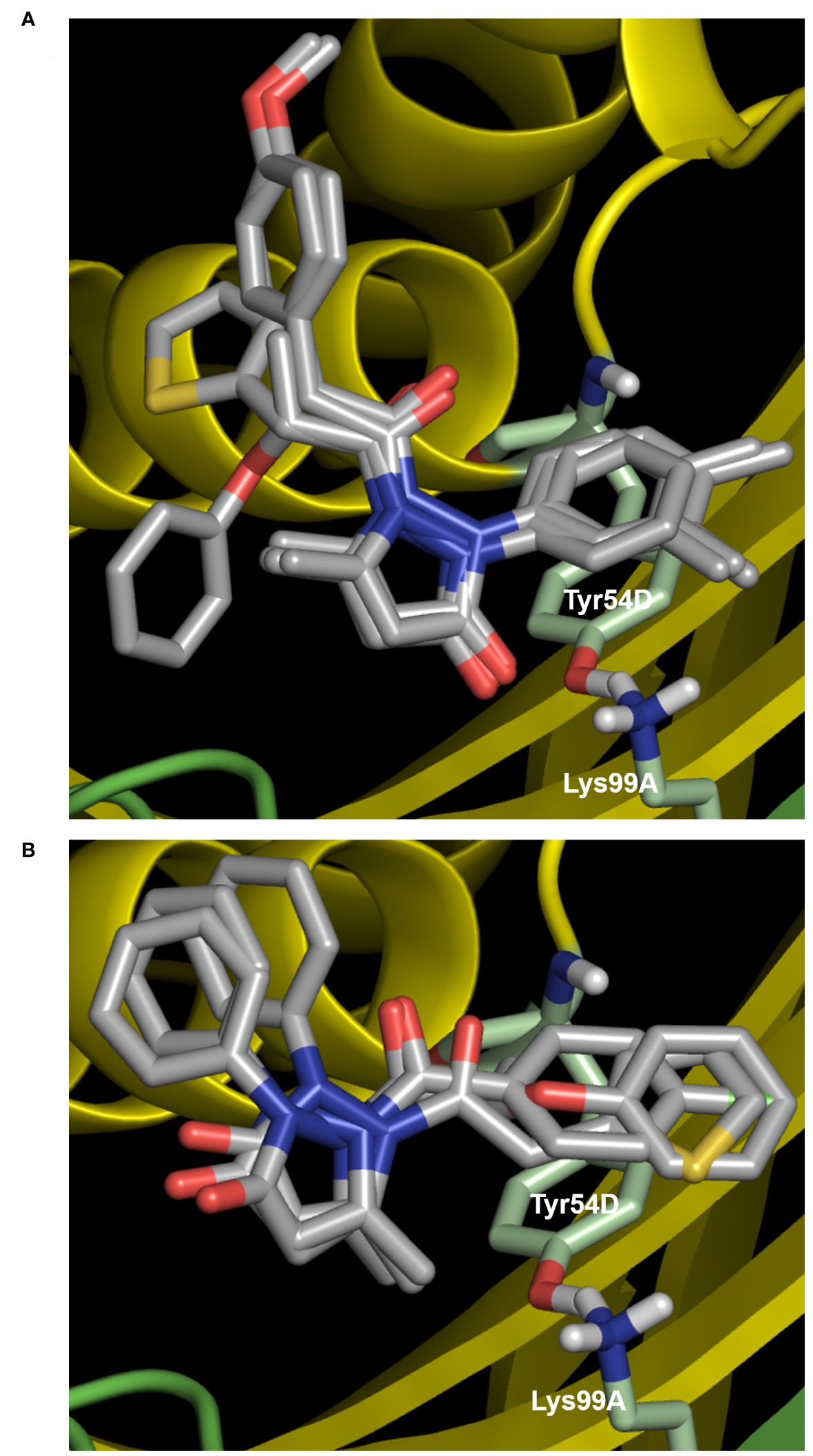
Figure 4 Docking results for compounds of cluster II. (A) First docking pose, obtained with compounds 9, 16, 22 and 23. The carbonyl of the pyrazol-3-one core is engaged in hydrogen-bonding with the tyrosine and lysine of the active site (Tyr54D, Lys99A; A, D refer to the MtbFolB subunit: two are necessary to form an active site), as well as p-p stacking interactions with Tyr54D. (B) Second docking pose (compounds 3, 20 and 21), where only the N-carbonyl side-chains interact with the Tyr54D of the active site through p-p stacking interactions. For both panels, compound backbones are indicated in gray, with nitrogen in blue, sulfur in light orange and oxygen in red. Residues of the active site are indicated in light green and labeled in white, with nitrogen in blue, hydrogen in light gray and oxygen in red. The two MtbFolB monomers required to compose the active site are shown as yellow or green ribbon. PDB: 1NBU.
The main challenge with target-based screening is to identify compounds that remain active against the bacteria, especially in the case of Mtb. The results of the MIC study indicated that compound 13 showed a modest inhibitory activity against the bacillus, with a MIC of 55.4 µM. This was quite interesting as, to our knowledge, this is the first compound directed against MtbFolB that is also found to display an antimycobacterial activity. Although more experiments will be needed to more precisely evaluate the effect this compound can have on the bacteria, notably with the addition of additional clinical strains of Mtb, this finding nonetheless paves the way for further medicinal chemistry efforts to improve the antimycobacterial potency of this lead compound 13.
In summary, we have developed a high-throughput screening assay directed against MtbFolB, in an effort to identify new scaffolds that could prove effective at inhibiting this essential enzyme. We present here the results of a small 6,074-compound screening, together with the validation campaign performed on the selected hits and their newly synthesized derivatives. Through this work, we established that the assay was functional and able to select small molecules with inhibitory activities, opening prospects for larger scale screening targeting MtbFolB. We also highlighted compound 13 as a potential starting point for further studies, as it was also found to be active against Mtb in contrast to all the other hits reported in this work and elsewhere in the literature for MtbFolB inhibitors.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
Author contributions
VF: Data curation, Investigation, Methodology, Writing – review & editing. AC: Data curation, Investigation, Methodology, Writing – review & editing. MA: Data curation, Investigation, Methodology, Writing – review & editing. KP: Data curation, Formal analysis, Investigation, Methodology, Supervision, Writing – review & editing. JH: Data curation, Investigation, Writing – review & editing. MW: Data curation, Investigation, Writing – review & editing. AC: Data curation, Investigation, Writing – review & editing. LT: Data curation, Investigation, Methodology, Visualization, Writing – review & editing. DS: Data curation, Project administration, Resources, Supervision, Writing – review & editing. MP: Data curation, Investigation, Writing – review & editing. LB: Data curation, Investigation, Writing – review & editing. PM: Data curation, Investigation, Writing – review & editing. CB: Data curation, Investigation, Methodology, Project administration, Resources, Supervision, Writing – original draft, Writing – review & editing. VD: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Supervision, Validation, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the national research foundation of Korea (NRF), funded by the Korean ministry of science, ICT and technology (MSIT, project numbers NRF-2017M3A9G6068246, NRF-2019R1A2C2084652 and RS-2024-00398073) as well as the Gyeonggi province, Korea. VD was supported by the French ministry of foreign affairs. This work was also supported by the National Institute of Science and Technology on Tuberculosis (CNPq-FAPERGS-CAPES) [grant number 421703–2017-2 and 17–1265-8]. C. V. Bizarro, L. A. Basso, and P. Machado are Research Career Awardees of the National Research Council of Brazil (CNPq). This study was financed in part by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior, Brazil (CAPES), Finance Code 001.
Acknowledgments
Institut Pasteur Korea is a member of the Institut Pasteur International Network (https://pasteur-network.org/).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fitd.2024.1402321/full#supplementary-material
References
1. Henderson G, Huennekens F. Membrane-Associated folate transport proteins. Methods Enzymol. (1986), 260–9. doi: 10.1016/0076-6879(86)22180-1
2. Woods DD. The relation of p-aminobenzoic acid to the mechanism of the action of sulphanilamide. Br J Exp Pathol. (1940) 122 21:74.
3. Hitchings GH, Elion GB, Vander Werff H, Falco EA. Pyrimidine derivatives as antagonists of pteroylglutamic acid. J Biol Chem. (1948) 174:765–6. doi: 10.1016/S0021-9258(18)57361-0
4. Keys T. Antimicrobials commonly used for urinary tract infection: sulfonamides, trimethoprim-sulfamethoxazole, nitrofurantoin, nalidixic acid. Mayo Clinic Proc. (1977) 52(11):p680–682.
5. Doull J. Sulfone therapy of leprosy. Background, early history and present status. Int J Leprosy. (1963) 31:143–60. doi: 10.5555/19642901334
6. Lehmann J. The treatment of tuberculosis in Sweden with para-aminosalicylic acid (PAS): A review. Dis Chest. (1949) 16:684–703. doi: 10.1378/chest.16.6.684
7. Chakraborty S, Gruber T, Barry CE, Boshoff HI, Rhee KY. Para-aminosalicylic acid acts as an alternative substrate of folate metabolism in Mycobacterium tuberculosis. Science. (2013) 339:88–91. doi: 10.1126/science.1228980
8. Minato Y, Thiede JM, Kordus SL, McKlveen EJ, Turman BJ, Baughn AD. Mycobacterium tuberculosis folate metabolism and the mechanistic basis for para-aminosalicylic acid susceptibility and resistance. Antimicrob Agents Chemother. (2015) 59:5097–106. doi: 10.1128/AAC.00647-15
9. Zhao F, Wang X-D, Erber LN, Luo M, Guo A-z, Yang S-s, et al. Binding pocket alterations in dihydrofolate synthase confer resistance to para-aminosalicylic acid in clinical isolates of Mycobacterium tuberculosis. Antimicrob Agents Chemother. (2014) 58:1479–87. doi: 10.1128/AAC.01775-13
10. Zheng J, Rubin EJ, Bifani P, Mathys V, Lim V, Au M, et al. para-Aminosalicylic acid is a prodrug targeting dihydrofolate reductase in Mycobacterium tuberculosis. J Biol Chem. (2013) 288:23447–56. doi: 10.1074/jbc.M113.475798
11. WHO. WHO consolidated guidelines on tuberculosis. Module 4: treatment - drug-resistant tuberculosis treatment. Geneva: World Health Organization (2020).
12. Bermingham A, Derrick JP. The folic acid biosynthesis pathway in bacteria: evaluation of potential for antibacterial drug discovery. BioEssays. (2002) 24:637–48. doi: 10.1002/bies.10114
13. Baca AM, Sirawaraporn R, Turley S, Sirawaraporn W, Hol WG. Crystal structure of Mycobacterium tuberculosis 6-hydroxymethyl-7, 8-dihydropteroate synthase in complex with pterin monophosphate: new insight into the enzymatic mechanism and sulfa-drug action. J Mol Biol. (2000) 302:1193–212. doi: 10.1006/jmbi.2000.4094
14. Li R, Sirawaraporn R, Chitnumsub P, Sirawaraporn W, Wooden J, Athappilly F, et al. Three-dimensional structure of M. tuberculosis dihydrofolate reductase reveals opportunities for the design of novel tuberculosis drugs. J Mol Biol. (2000) 295:307–23. doi: 10.1006/jmbi.1999.3328
15. Hajian B, Scocchera E, Keshipeddy S, G-Dayanandan N, Shoen C, Krucinska J, et al. Propargyl-linked antifolates are potent inhibitors of drug-sensitive and drug-resistant Mycobacterium tuberculosis. PloS One. (2016) 11:e0161740. doi: 10.1371/journal.pone.0161740
16. Ribeiro JA, Hammer A, Libreros-Zuniga GA, Chavez-Pacheco SM, Tyrakis P, de Oliveira GS, et al. Using a fragment-based approach to identify alternative chemical scaffolds targeting dihydrofolate reductase from Mycobacterium tuberculosis. ACS Infect Dis. (2020) 6:2192–201. doi: 10.1021/acsinfecdis.0c00263
17. Cheng Y-S, Sacchettini JC. Structural insights into Mycobacterium tuberculosis Rv2671 protein as a dihydrofolate reductase functional analogue contributing to para-aminosalicylic acid resistance. Biochemistry. (2016) 55:1107–19. doi: 10.1021/acs.biochem.5b00993
18. Hajian B, Scocchera E, Shoen C, Krucinska J, Viswanathan K, Narendran G, et al. Drugging the folate pathway in Mycobacterium tuberculosis: the role of multi-targeting agents. Cell Chem Biol. (2019) 26:781–791. e6. doi: 10.1016/j.chembiol.2019.02.013
19. Sanders WJ, Nienaber VL, Lerner CG, McCall JO, Merrick SM, Swanson SJ, et al. Discovery of potent inhibitors of dihydroneopterin aldolase using CrystaLEAD high-throughput X-ray crystallographic screening and structure-directed lead optimization. J Medicinal Chem. (2004) 47:1709–18. doi: 10.1021/jm030497y
20. Czeczot A, Roth CD, Ducati RG, Pissinate K, Rambo RS, Timmers LFSM, et al. 8-Mercaptoguanine-based inhibitors of Mycobacterium tuberculosis dihydroneopterin aldolase: synthesis, in vitro inhibition and docking studies. J Enzyme Inhibition Medicinal Chem. (2021) 36:847–55. doi: 10.1080/14756366.2021.1900157
21. Czekster CM, Blanchard JS. One substrate, five products: reactions catalyzed by the dihydroneopterin aldolase from Mycobacterium tuberculosis. J Am Chem Soc. (2012) 134:19758–71. doi: 10.1021/ja308350f
22. Falcão V, Villela A, Rodrigues-Junior V, Pissinate K, Eichler P, Pinto A, et al. Validation of Mycobacterium tuberculosis dihydroneopterin aldolase as a molecular target for anti-tuberculosis drug development. Biochem Biophys Res Commun. (2017) 485:814–9. doi: 10.1016/j.bbrc.2017.02.137
23. Cohen SS. Streptomycin and desoxyribonuclease in the study of variations in the properties of a bacterial virus. J Biol Chem. (1947) 168:511–26. doi: 10.1016/S0021-9258(17)30911-0
24. Goulding CW, Apostol MI, Sawaya MR, Phillips M, Parseghian A, Eisenberg D. Regulation by oligomerization in a mycobacterial folate biosynthetic enzyme. J Mol Biol. (2005) 349:61–72. doi: 10.1016/j.jmb.2005.03.023
Keywords: Mycobacterium tuberculosis, dihydroneopterin aldolase (DHNA), Rv3607c (FolB), folate precursor synthesis, drug screening
Citation: Falcão VCdA, Czeczot AdM, Alam MM, Park K-HP, Heo J, Woo M, Camini AM, Timmers LFSM, Shum D, Perelló MA, Basso LA, Machado P, Bizarro CV and Delorme V (2024) Identification and characterization of new structural scaffolds modulating the activity of Mycobacterium tuberculosis dihydroneopterin aldolase (FolB) in vitro. Front. Trop. Dis 5:1402321. doi: 10.3389/fitd.2024.1402321
Received: 17 March 2024; Accepted: 15 May 2024;
Published: 13 June 2024.
Edited by:
Radha Gopalaswamy, National Institute of Research in Tuberculosis (ICMR), IndiaReviewed by:
Selvakumar Subbian, The State University of New Jersey, United StatesMoti Chapagain, Praedicare Laboratories, United States
Copyright © 2024 Falcão, Czeczot, Alam, Park, Heo, Woo, Camini, Timmers, Shum, Perelló, Basso, Machado, Bizarro and Delorme. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vincent Delorme, dmkuZGVsb3JtZUBnbWFpbC5jb20=
†Present addresses: Mohammad Maqusood Alam, Neuroscience Research Institute, Gachon University, Incheon, Republic of Korea
Vincent Delorme, R&D Center, Insol Co., Ltd., Hanam, Gyeonggi, Republic of Korea