 Ji-Heui Sohn1,2†
Ji-Heui Sohn1,2† Dong-Yeop Lee3†
Dong-Yeop Lee3† Tae-Hyeon Kim4Hyun-Jeong Sung2
Tae-Hyeon Kim4Hyun-Jeong Sung2 Hyomi Jang2
Hyomi Jang2 Jung-Hyun Kim1
Jung-Hyun Kim1 Dong-Hun Lee3*
Dong-Hun Lee3*- 1Department of Veterinary Internal Medicine, College of Veterinary Medicine, Konkuk University, Seoul, Republic of Korea
- 2VIP Animal Medical Center, Seoul, Republic of Korea
- 3Wildlife Health Laboratory, College of Veterinary Medicine, Konkuk University, Seoul, Republic of Korea
- 4Avian Disease Laboratory, College of Veterinary Medicine, Konkuk University, Seoul, Republic of Korea
1 Introduction
Feline calicivirus (FCV) is a prominent infectious pathogen of cats (1–4). Classical FCV causes mild symptoms, such as respiratory disease and oral inflammation, which tend to be self-limiting and are referred to as oral respiratory diseases (ORD) (4). However, virulent systemic feline calicivirus (VS-FCV) causes virulent systemic disease (VSD) and high mortality. This highly virulent form has spread to Europe, the United States of America, and, more recently, China and South Korea (5–11). Although sporadic clinical cases have been reported in South Korea, there is a lack of genetic analysis based on whole-genome sequencing (WGS) of VS-FCV isolated from these cases (10, 12).
FCV is a single-stranded (1), positive-sense, non-enveloped RNA virus of the genus Vesivirus, family Caliciviridae (2, 3). The FCV genome encodes three open reading frames (ORFs): ORF1 for nonstructural proteins, ORF2 for the major capsid protein VP1, and ORF3 for the minor capsid protein VP2 (13). FCV exhibit considerable genetic and antigenic diversity, with region E, comprising amino acids (aa) 426–521 in the protruding domain of VP1, being the primary contributor to this variability. FCV vaccines have been available for over four decades; however, they do not provide complete immunity, leading to the widespread distribution of diverse FCV strains (14).
In this study, oropharyngeal swab samples from FCV infected cats were collected from an animal hospital in South Korea and analyzed for their whole genome sequences using multiplex tiling reverse transcription polymerase chain reaction and Illumina next-generation sequencing.
2 Materials and methods
2.1 Primer design and tiling amplicon PCR for whole genome sequencing
A multiplex tiling RT-PCR approach was developed to assess the full viral genome coverage of clinical samples. Initially, all available FCV genomes were downloaded from the National Center for Biotechnology Information database and aligned to generate a consensus sequence. Based on this, three pairs of primers were designed using PrimalScheme software, amplifying the entire genome of 7,800 bp with ~100–200 bp overlaps (15) (Table 1). Oropharyngeal swabs from six cats were subjected to RT-PCR using the OneStep RT-PCR Kit (Qiagen, Hilden, Germany) following the manufacturer's instructions. The PCR mixture was prepared by mixing 10 ul 5x QIAGEN One-Step RT-PCR buffer, 10 ul dNTP mix, 0.6 uM of each primer, 2 ul of template RNA, and 31 ul nuclease-free water. PCR amplification conditions were: 45°C for 30 min, 95°C for 15 min, followed by 40 cycles of 94°C for 30 s, 59 (primer set 1)/65 (primer set 2, 3)°C for 1 min, and 68°C for 3 min. The PCR products were then visualized via electrophoresis on 1% agarose gels, showing ~1.3–2.9 kbp amplicons. PCR products were pooled, and ~100,000 next-generation sequencing (NGS) reads of 150 bp per sample were produced using Illumina DNA prep kit and Nextseq 500 NGS system (Illumina, USA) according to the manufacturer‘s instructions to achieve >1,000 × genome coverage.
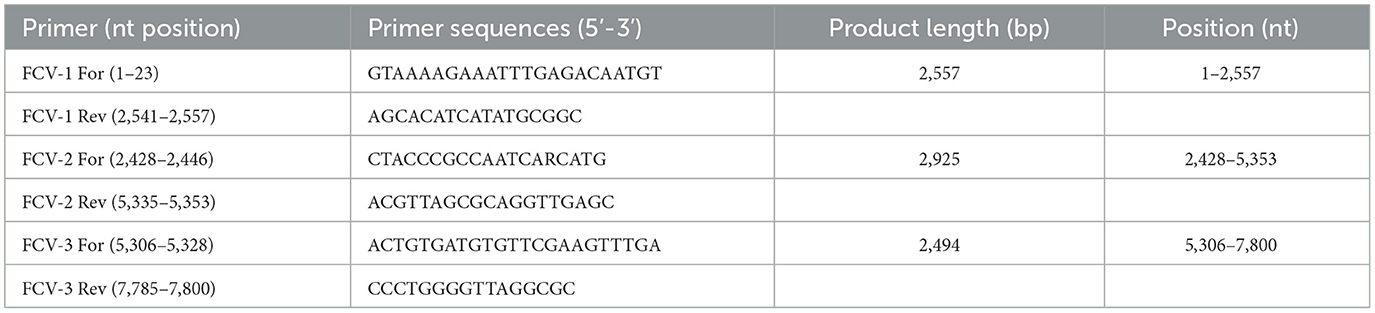
Table 1. Tiling amplicon PCR primer sets used for amplification of feline calicivirus genome.
2.2 Genome assembly and phylogenetic analysis
The raw reads obtained from NGS were subjected to trimming and filtering to remove adapters and low-quality bases using BBDuk (v38.84) with a minimum quality threshold set at 30 and a minimum length of 50 bp. Subsequently, de novo and reference-based assemblies of the genome sequences were conducted using Geneious Prime version 2024.0.4 (Biomatters Ltd., Auckland, New Zealand). For reference-based assembly, the trimmed reads were mapped to the KP361 viral genome (GenBank accession number: MZ542330) using Geneious Mapper with default options.
To perform genetic analysis, 27 FCV sequences were downloaded from the NCBI GenBank database and aligned using MAFFT Multiple Sequence Alignment software (v7.490) (16). Maximum likelihood (ML) phylogenetic analysis was performed using RaxML (v8.2.13) with a GTR GAMMA model and a rapid bootstrapping option set to 1,000 replicates (17).
To further investigate the possible genetic relationship between our strains and VS-FCVs, the inferred amino acid (aa) sequences of the hypervariable region E of the viruses were mapped to identify seven aa residue positions (438, 440, 448, 452, 455, 465, and 492), whose physical and chemical properties were previously shown to be statistically significant for differentiation between the upper respiratory tract and VS-FCV pathotypes (Supplementary Tables S1, S2) (18).
A recent study identified two linear epitopes within the P2 sub-domain of VP1, specifically in the E5 antigenic hypervariable region. The first epitope, spanning aa 431–435 (PAGDY), is highly conserved and induces a non-neutralizing immune response. The second epitope, located at aa 445–451 (ITTANQY), is highly variable and elicits a neutralizing immune response (19). We comparatively analyzed the linear epitope sequences of the viruses from this study and other previously reported viruses.
3 Descriptive results
In 2023, a total of six cats presented to the clinic with clinical symptoms and tested positive for calicivirus via real-time RT-PCR. Each cat exhibited systemic symptoms, including tongue and skin ulcers, respiratory distress, anorexia, fever, anemia, and pleural effusion. Of the six cats, three survived, whereas the other three died. All cats completed their primary vaccination series early in life and received at least one booster vaccination. Based on molecular FCV detection and clinical findings, we presumptively diagnosed VS-FCV infection. However, systemic infection was not definitively confirmed in these cases, which represents a limitation, as it does not fully meet the established criteria for VS-FCV.
The tiling amplicon PCR method has demonstrated remarkable efficiency and productivity in generating complete viral genome sequences directly from clinical samples (15). In this study, we developed multiplex tiling RT-PCR primer panels and successfully obtained the complete genomes of VS-FCVs directly from the clinical samples of five cats using the newly designed primer set. The sample obtained from cat 6 contained insufficient sequencing material. The samples obtained from cat 1–5 shared a high nucleotide sequence identity of >99.8%. Based on the phylogenetic analysis of the VP1 gene, FCV has been categorized into two genotypes: GI and GII (20). The GI genotype is globally dominant and widespread, whereas all identified GII strains have been isolated in Asia. The ML phylogenetic analysis showed that the FCVs sequenced in this study formed a monophyletic clade and belonged to the genogroup I (Figure 1). They showed the closest genetic relationship with an FCV strain identified in South Korea in 2014 (GenBank Accession no. MT123328; nucleotide sequence identity: 85.05–85.22%). However, they formed a long branch in the ML phylogeny, due to the scarcity of FCV genome sequences.
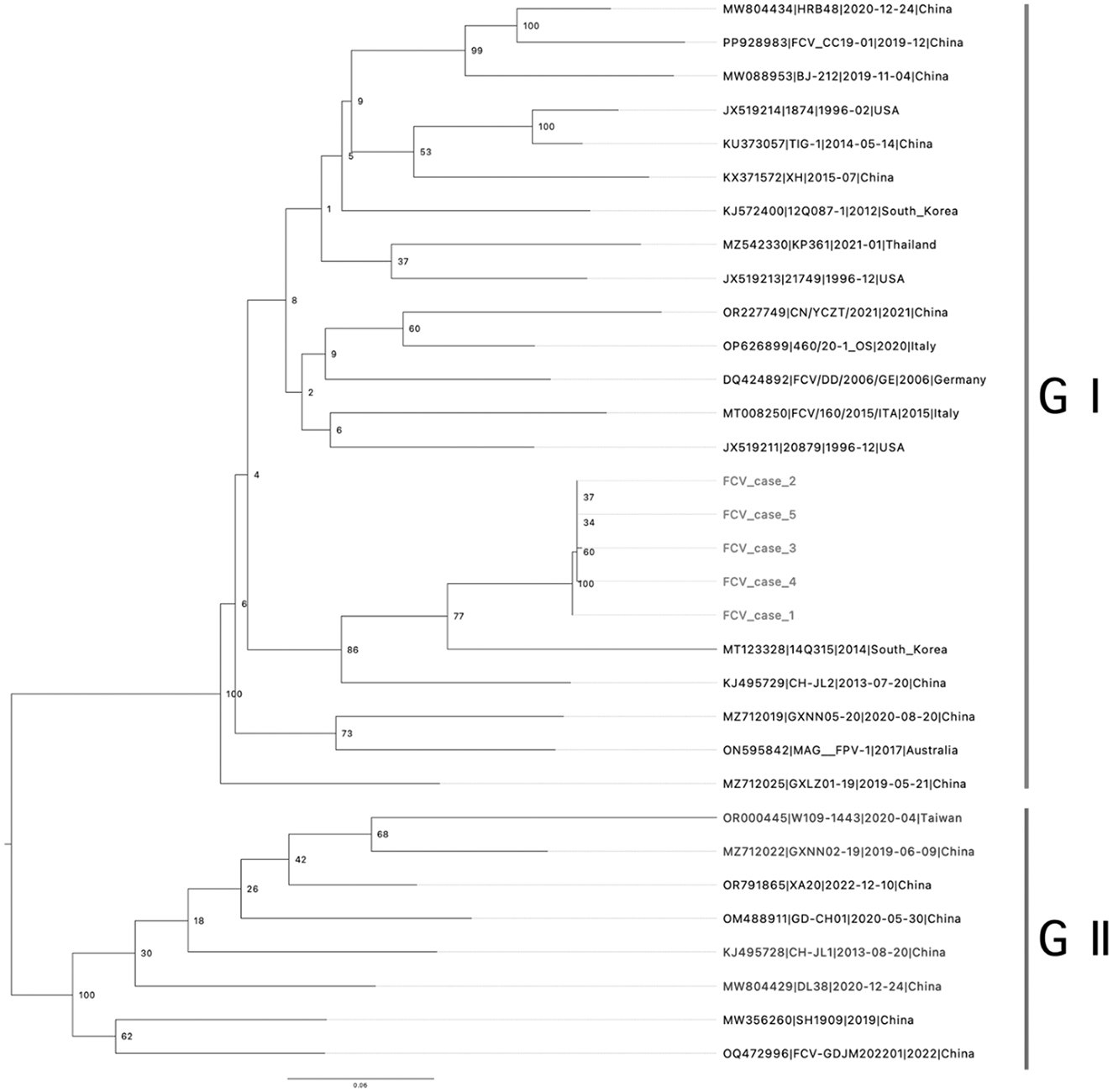
Figure 1. Phylogenic tree of viral protein 1 (VP1) capsid protein: phylogenetic analysis based on the amino acid sequence of the full-length VP1 capsid protein was generated using the maximum likelihood, supplying statistical support with bootstrapping of 1,000 replicates. Rooted to midpoint. The scale bars show the number of substitutions per site. The numerical values represent 1,000 bootstrap replicate values expressed as a percentage. Genotypes are indicated by vertical bars.
The high degree of genomic plasticity in FCV led to the emergence of various variants, some of which are associated with severe clinical diseases. To further investigate the possible genetic relationship between our FCVs and other VS-FCVs, the deduced aa sequences of the hypervariable region E were mapped to compare seven aa residue positions (438, 440, 448, 452, 455, 465, and 492), whose physical and chemical properties were previously shown to be statistically significant for differentiation between the ORD and VSD-FCV pathotypes (18). In our analysis, the predicted properties for virulent pathotypes were observed in four of the seven residues (positions 448, 452, 455, and 492) in the hypervariable region E (Supplementary Table S1). We assume that these VSD markers may not reliably distinguish between current VSD and less pathogenic viruses in South Korea. Further research is required to establish a clear, stringent differentiation between VSD and ORD FCV strains clinically and at the molecular level (2).
We conducted a comparative analysis of the linear epitope sequences in VP1 and found significant sequence differences in the neutralizing epitope (aa 445–451) among the FCV strains. The pairwise identity of the neutralizing epitope was relatively lower (52.0%) than 87.5% of the non-neutralizing epitope and 86.7% for the entire VP1 (Supplementary Table S2). We assume that these genetic variations may result in vaccine-generated antibodies being less effective at neutralizing the virus during actual infections. Some endemic calicivirus strains occurring sporadically have shown genetic diversity compared with vaccine strains (21, 22), and reports have revealed the emergence of vaccine-resistant viruses (23, 24).
Given the recent reports of VS-FCV infections and outbreaks resulting in high virulence in cats within veterinary hospitals, complete genome sequencing of suspected cases would be helpful in monitoring the evolution and transmission of FCVs. The multiplex tiling RT-PCR and NGS approach used in this study demonstrates significant potential for integration into diagnostic workflows, providing a rapid and reliable method for complete genome sequencing and molecular epidemiological investigation of FCV from clinical samples. This study highlights the need to develop more effective vaccines based on genomic surveillance data to address the diverse and rapidly evolving strains of FCV.
Data availability statement
The genome sequences generated in this study can be found in the GenBank under accession numbers PV054606-PV054611.
Ethics statement
The animal studies were approved by VIP Animal Clinic Committee, Seoul, South Korea. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent was obtained from the owners for the participation of their animals in this study.
Author contributions
J-HS: Writing – original draft. D-YL: Writing – original draft. T-HK: Writing – original draft. H-JS: Writing – original draft. HJ: Writing – original draft. J-HK: Writing – review & editing. D-HL: Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This research was financially supported by Konkuk University in 2024.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Gen AI was used in the creation of this manuscript.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fvets.2025.1570761/full#supplementary-material
References
1. Radford AD, Coyne KP, Dawson S, Porter CJ, Gaskell RM. Feline calicivirus. Vet Res. (2007) 38:319–35. doi: 10.1051/vetres:2006056
2. Bordicchia M, Fumian TM, Van Brussel K, Russo AG, Carrai M, Le SJ, et al. Feline calicivirus virulent systemic disease: clinical epidemiology, analysis of viral isolates and in vitro efficacy of novel antivirals in australian outbreaks. Viruses. (2021) 13:2040. doi: 10.3390/v13102040
3. Seal BS, Ridpath JF, Mengeling WL. Analysis of feline calicivirus capsid protein genes: identification of variable antigenic determinant regions of the protein. J Gen Virol. (1993) 74:2519–24. doi: 10.1099/0022-1317-74-11-2519
4. Berger A, Willi B, Meli ML, Boretti FS, Hartnack S, Dreyfus A, et al. Feline calicivirus and other respiratory pathogens in cats with feline calicivirus-related symptoms and in clinically healthy cats in Switzerland. BMC Vet Res. (2015) 11:282. doi: 10.1186/s12917-015-0595-2
5. Pedersen NC, Elliott JB, Glasgow A, Poland A, Keel K. An isolated epizootic of hemorrhagic-like fever in cats caused by a novel and highly virulent strain of feline calicivirus. Vet Microbiol. (2000) 73:281–300. doi: 10.1016/S0378-1135(00)00183-8
6. Hurley KE, Pesavento PA, Pedersen NC, Poland AM, Wilson E, Foley JE. An outbreak of virulent systemic feline calicivirus disease. J Am Vet Med Assoc. (2004) 224:241–9. doi: 10.2460/javma.2004.224.241
7. Pesavento PA, MacLachlan NJ, Dillard-Telm L, Grant CK, Hurley KF. Pathologic, immunohistochemical, and electron microscopic findings in naturally occurring virulent systemic feline calicivirus infection in cats. Vet Pathol. (2004) 41:257–63. doi: 10.1354/vp.41-3-257
8. Guo H, Miao Q, Zhu J, Yang Z, Liu G. Isolation and molecular characterization of a virulent systemic feline calicivirus isolated in China. Infect Genet Evol. (2018) 65:425–9. doi: 10.1016/j.meegid.2018.08.029
9. Radford AD, Addie D, Belak S, Boucraut-Baralon C, Egberink H, Frymus T, et al. Feline calicivirus infection. ABCD guidelines on prevention and management. J Feline Med Surg. (2009) 11:556–64. doi: 10.1016/j.jfms.2009.05.004
10. Park J-I, Suh S-I, Hyun C. Virulent systemic feline calicivirus infection in a kitten. J Vet Clin. (2015) 32:445–8. doi: 10.17555/jvc.2015.10.32.5.445
11. Wang Z, Xin T, Wei J, Jiang Y, Liu X, Song W, et al. Isolation and phylogenetic analysis of strains of feline calicivirus in Beijing, China. Arch Virol. (2021) 166:2521–7. doi: 10.1007/s00705-021-05163-2
12. Park J, Lee D, Hong YJ, Hwang CY, Hyun JE. Outbreaks of nosocomial feline calicivirus-associated virulent systemic disease in Korea. J Vet Sci. (2024) 25:e51. doi: 10.4142/jvs.24030
13. Bhella D, Goodfellow IG. The cryo-electron microscopy structure of feline calicivirus bound to junctional adhesion molecule a at 9-angstrom resolution reveals receptor-induced flexibility and two distinct conformational changes in the capsid protein VP1. J Virol. (2011) 85:11381–90. doi: 10.1128/JVI.05621-11
14. Radford AD, Dawson S, Ryvar R, Coyne K, Johnson DR, Cox MB, et al. High genetic diversity of the immunodominant region of the feline calicivirus capsid gene in endemically infected cat colonies. Virus Genes. (2003) 27:145–55. doi: 10.1023/a:1025772409539
15. Quick J, Grubaugh ND, Pullan ST, Claro IM, Smith AD, Gangavarapu K, et al. Multiplex PCR method for minion and illumina sequencing of zika and other virus genomes directly from clinical samples. Nat Protoc. (2017) 12:1261–76. doi: 10.1038/nprot.2017.066
16. Katoh K, Standley DM. Mafft multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. (2013) 30:772–80. doi: 10.1093/molbev/mst010
17. Stamatakis A. Raxml version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. (2014) 30:1312–3. doi: 10.1093/bioinformatics/btu033
18. Brunet S, Sigoillot-Claude C, Pialot D, Poulet H. Multiple correspondence analysis on amino acid properties within the variable region of the capsid protein shows differences between classical and virulent systemic feline calicivirus strains. Viruses. (2019) 11:1090. doi: 10.3390/v11121090
19. Cubillos-Zapata C, Angulo I, Almanza H, Borrego B, Zamora-Ceballos M, Caston JR, et al. Precise location of linear epitopes on the capsid surface of feline calicivirus recognized by neutralizing and non-neutralizing monoclonal antibodies. Vet Res. (2020) 51:59. doi: 10.1186/s13567-020-00785-x
20. Yang Y, Liu Z, Chen M, Feng K, Qi R, Zheng Y, et al. Classification of genotypes based on the VP1 gene of feline calicivirus and study of cross-protection between different genotypes. Front Microbiol. (2023) 14:1226877. doi: 10.3389/fmicb.2023.1226877
21. Coyne KP, Christley RM, Pybus OG, Dawson S, Gaskell RM, Radford AD. Large-scale spatial and temporal genetic diversity of feline calicivirus. J Virol. (2012) 86:11356–67. doi: 10.1128/JVI.00701-12
22. Spiri AM, Theze J, Meli ML, Cattori V, Berger A, Steinrigl A, et al. Genetic diversity and phenotypic associations of feline caliciviruses from cats in Switzerland. J Gen Virol. (2016) 97:3253–66. doi: 10.1099/jgv.0.000622
23. Smith SL, Afonso MM, Pinchbeck GL, Gaskell RM, Dawson S, Radford AD. Temporally separated feline calicivirus isolates do not cluster phylogenetically and are similarly neutralised by high-titre vaccine strain Fcv-F9 antisera in vitro. J Feline Med Surg. (2020) 22:602–7. doi: 10.1177/1098612X19866521
Keywords: feline calicivirus, phylogenetic analysis, reverse transcription polymerase chain reaction, virulent systemic disease, whole-genome sequencing
Citation: Sohn J-H, Lee D-Y, Kim T-H, Sung H-J, Jang H, Kim J-H and Lee D-H (2025) Whole-genome sequencing of feline calicivirus in domestic cats, South Korea, 2023. Front. Vet. Sci. 12:1570761. doi: 10.3389/fvets.2025.1570761
Received: 04 February 2025; Accepted: 09 April 2025;
Published: 28 April 2025.
Edited by:
Iryna Goraichuk, Agricultural Research Service (USDA), United StatesReviewed by:
Federica Di Profio, Università degli Studi di Teramo, ItalyRebecca P. Wilkes, Purdue University, United States
Copyright © 2025 Sohn, Lee, Kim, Sung, Jang, Kim and Lee. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dong-Hun Lee, ZG9uZ2h1bmxlZUBrb25rdWsuYWMua3I=
†These authors have contributed equally to this work