 Gerard Badia-Bringué
Gerard Badia-Bringué Victoria Asselstine
Victoria Asselstine Ángela Cánovas
Ángela Cánovas Marta Alonso-Hearn
Marta Alonso-Hearn- 1Department of Animal Health, NEIKER-Basque Institute of Agricultural Research and Development, Basque Research and Technology Alliance (BRTA), Derio, Spain
- 2Department of Animal Biosciences, Center for Genetic Improvement of Livestock, University of Guelph, Guelph, ON, Canada
Bovine paratuberculosis (PTB) is a chronic enteritis caused by Mycobacterium avium subsp. paratuberculosis (MAP), which results in significant economic losses to the dairy industry worldwide. Long non-coding RNAs (lncRNAs) play a crucial role in regulating the host immune response due to their interaction with transcripts in proximity. However, their annotation in cattle remains limited, and their role in cattle naturally infected with MAP has not been fully explored. In this study, lncRNAs were identified in the transcriptome of ileocecal valve samples from control cows without lesions (N = 4) and with PTB-associated focal (N = 5) and diffuse (N = 5) lesions in intestinal tissues using RNA sequencing. The raw reads were uploaded into the CLC Bio Genomics Workbench, and the trimmed reads were mapped to the Bos taurus ARS_UCD1.2.109 reference genome using the Large Gap Read Mapping tool. The resulting annotation allowed the identification of 1,434 LncRNAs, 899 of which were novel, using the FlExible Extraction of LncRNA pipeline. LncRNA differential expression (DE) analysis performed with DESeq2 allowed the identification of 1, 6, and 2 DE lncRNAs in the comparisons of cows with focal lesions versus (vs) controls, diffuse lesions vs. controls, and diffuse vs. focal lesions, respectively. Best lncRNA partner analysis identified expression correlations between the lncRNA1086.1, lncRNA ENSBTAG00000050406, and lncRNA_2340.1, and the Inactive Phosphatidylinositol 3-Phosphatase 9 (MTMR9), GM Domain Family member B (RGMB), and the homeobox A6 (HOXA6), respectively. The MTMR9 negatively regulates apoptosis, the RGMB positively regulates IL-6 expression, and the HOXA6 regulates cell differentiation and inflammation. The results of the quantitative trait locus (QTL) enrichment analysis showed that the DE lncRNAs were located in genomic regions previously associated with clinical mastitis, HDL cholesterol, bovine tuberculosis, paratuberculosis, and bovine leukosis susceptibility. The identified DE lncRNAs could allow the development of novel PTB diagnostic tools and have potential applications in breeding strategies for PTB-resistant cattle.
1 Introduction
Bovine paratuberculosis (PTB), also known as Johne’s disease, is a chronic granulomatous enteritis that affects ruminants worldwide. The disease is caused by Mycobacterium avium subsp. paratuberculosis (MAP) and is primarily characterized by reduced milk production and weight loss. Global economic losses due to PTB are estimated to exceed $1.5 billion annually, with $198.42 million in the United States and $364.31 million in Europe (1, 2). These losses are mainly due to decreased milk production, decreased pregnancy rates, increased management costs, and premature culling of infected animals. Indeed, bovine PTB is considered endemic in the United States and Europe, with more than 50% of herds testing positive for anti-MAP antibodies by ELISA (3). Furthermore, scientific evidence links MAP to human inflammatory bowel disease (IBD), Crohn’s disease, autoimmune diseases, colorectal cancer, and Alzheimer’s disease (4–6). This potential threat to human health has increased interest in PTB and the development of more sensitive diagnostic and control methods.
Transmission of MAP typically occurs early in an animal’s life through the ingestion of MAP-contaminated feces, water, or milk. MAP shows an evident tropism for the gastrointestinal tract, crosses the intestinal barrier by interacting with M cells and epithelial cells, and can survive within subepithelial macrophages by inhibiting apoptosis, phagosome acidification, and antigen presentation to the immune system (7–9). MAP-infected animals can progress to the subclinical phase that is characterized by the inhibition of the Th1 pro-inflammatory immune response and MAP persistence within macrophages (10, 11). As the infection progresses, granulomas form, and a host Th1 response is induced, causing intestinal mucosa damage (12, 13). In advanced clinical stages of the disease, a Th2 humoral response emerges, indicating that MAP bacilli escaped granulomas and spread to other tissues and organs (14). MAP control strategies primarily involve identifying and culling infected animals. Currently used diagnostic tests include ELISA for detecting anti-MAP antibodies and real-time qPCR for detecting MAP DNA in fecal samples. While serum ELISA is a simple, fast, and cost-effective diagnostic method, it has low sensitivity in detecting subclinical animals (15). Detecting subclinical infections remains a challenge, and novel diagnostic tools are needed to identify MAP-infected animals in the early and subclinical stages of the infection.
LncRNAs are a type of non-coding RNA (ncRNA) with a length >200 nt that can be coded almost anywhere in the genome, within intergenic regions, within protein-coding genes but on the opposite strand, and within introns and pseudogenes (16). LncRNAs share many characteristics with mRNAs, are transcribed by RNA polymerase II, can be alternatively spliced, are either single-exonic or multi-exonic, are differentially expressed (DE), and are usually polyadenylated at their 3′ ends (17). LncRNAs are relatively shorter than protein-coding genes, exhibit lower expression levels, have fewer but longer exons, have a higher degree of tissue specificity, and have lower levels of homology across species (18, 19). However, lncRNAs from related species may have well-conserved secondary or tertiary structures in structural motifs. Consequently, lncRNA-related gene regulatory roles may be preserved despite a lack of primary sequence conservation (20). The cellular expression levels of lncRNAs are highly correlated with the expression of adjacent protein-coding genes (21). LncRNAs participate in many important regulatory processes such as X chromosome silencing, genome imprinting, chromatic modification, DNA methylation, histone modification, transcription activation, and inhibition, and are useful biomarkers for diagnosis and treatment in various diseases (22–24).
Identification and annotation of novel lncRNAs in the transcriptome of bovine tissues including lung, liver, kidney, white blood cells, mammary gland tissue, and milk somatic cells were previously performed (23, 25–29). However, lncRNAs remain poorly identified and annotated in the bovine genome in comparison to other species such as humans and mice (30). In addition, little is known about the diverse functions of bovine lncRNAs and how they can ultimately impact complex phenotypes including disease outcomes. In MAP-infected macrophages, previous studies have identified novel lncRNAs potentially associated with the regulation of several immune responses including neutrophil degranulation and activation, RNA polymerase function, NF-ƙß signaling pathway, chemokine signaling pathway, cytokine–cytokine receptor interaction, and NOD-like and toll-like receptor signaling pathways (31–33). However, the identification of lncRNAs in the ileocecal valve (ICV) from cows in different stages of the disease has not been addressed before. The objectives of this study were to (1) identify previously annotated and novel lncRNAs present in the bovine ICV transcriptome of 14 Holstein cattle with distinct histopathological lesions in gut tissues and without lesions (controls) using RNA-Seq; (2) identify lncRNAs that are DE between cows with distinct PTB-associated lesions versus (vs) controls; (3) predict the target genes of the identified lncRNAs using mRNA expression data from the same animals; and (4) perform quantitative trait locus (QTL) annotation and QTL enrichment analysis using the genomic regions where the DE lncRNAs were located to find additional evidence of their involvement in the regulation of the host immune response against MAP infection. The goal was to provide novel insights into lncRNA regulatory functions in gut tissues of MAP-infected cattle, investigate whether differences in lncRNA profiles reflected in the mRNA data, and explore how they might explain the different disease outcomes.
2 Materials and methods
2.1 Ethical statement
All methods were conducted in accordance with relevant guidelines and regulations. The study is reported according to the ARRIVE guidelines. The Animal Ethics Committee of the Servicio Regional de Investigación y Desarrollo Agroalimentario (SERIDA) approved the procedures for the animals included in this study. All procedures were authorized by the Regional Consejería de Agroganadería y Recursos Autóctonos of the Principality of Asturias (approval codes PROAE 29/2015 and PROAE 66/2019) and were carried out in compliance with the European Guidelines for the Care and Use of Animals for Research Purposes (2012/63/EU). The cows from which the samples were taken were euthanized for a reason other than collecting samples. The samples were collected by trained personnel following good veterinary practices.
2.2 RNA extraction, RNA-Seq library preparation, sequencing, and differential expression analysis
RNA-Seq data used in this study (Gene Expression Omnibus public repository, GEO Accession: GSE137395) were obtained from 14 Holstein cows from a single commercial dairy farm in Asturias, Spain. The cows were classified into three groups based on histopathological analysis: 4 animals without lesions (controls), 5 with focal lesions, and 5 with diffuse lesions, as previously described (34, 35). Total RNA isolation, RNA-Seq library preparation, and sequencing were previously performed (34). In brief, ICV samples were collected at the time of slaughter, stored in RNAlater (Sigma, St Louis, MP), and processed using the RNeasy Mini Kit, according to the manufacturer’s instructions (Qiagen, Hilden, Germany). RNA-Seq libraries were generated using the Illumina NEBNext® Ultra Directional RNA Library preparation kit (Illumina Inc., CA, USA) and were single-end sequenced (1 × 75) using an Illumina NextSeq500 sequencer.
2.3 RNA-Seq read trimming, quality control, and transcriptome assembly
To distinguish lncRNA transcripts from unannotated genes and protein-coding genes, a computational pipeline adapted from a previous study was implemented (25). The pipeline is shown in Figure 1. In brief, raw reads were uploaded to CLC Genomics Workbench (CLC Bio, Aarhus, Denmark) and were trimmed using the automatic trimmer function with a quality trimming score of 0.05. After the reads were trimmed, quality control was performed using the NGS quality control tool of CLC Genomics Workbench, as described by Cánovas et al. (36). The trimmed reads were mapped to the bovine reference genome ARS_UCD1.2.109 using the Large Gap Read Mapping (LGRM) tool in CLC Genomics Workbench, with a length fraction and similarity of 0.7 to exclude paralogous sequence variants and with a minimum contig length of 200 bp, with the following settings: match score: 1, mismatch cost: 2, deletion cost: 3, and insertion cost: 3. The LGRM tool maps reads that span introns without requiring prior transcript annotation, allowing large gaps in-between (37). First, the tool maps the first region of a read with the best match. Second, if 15 bp or more are still unmapped, it returns to the first step and tries to map the remaining sequence. This process continues until the sequence is too short or until the software cannot map it.
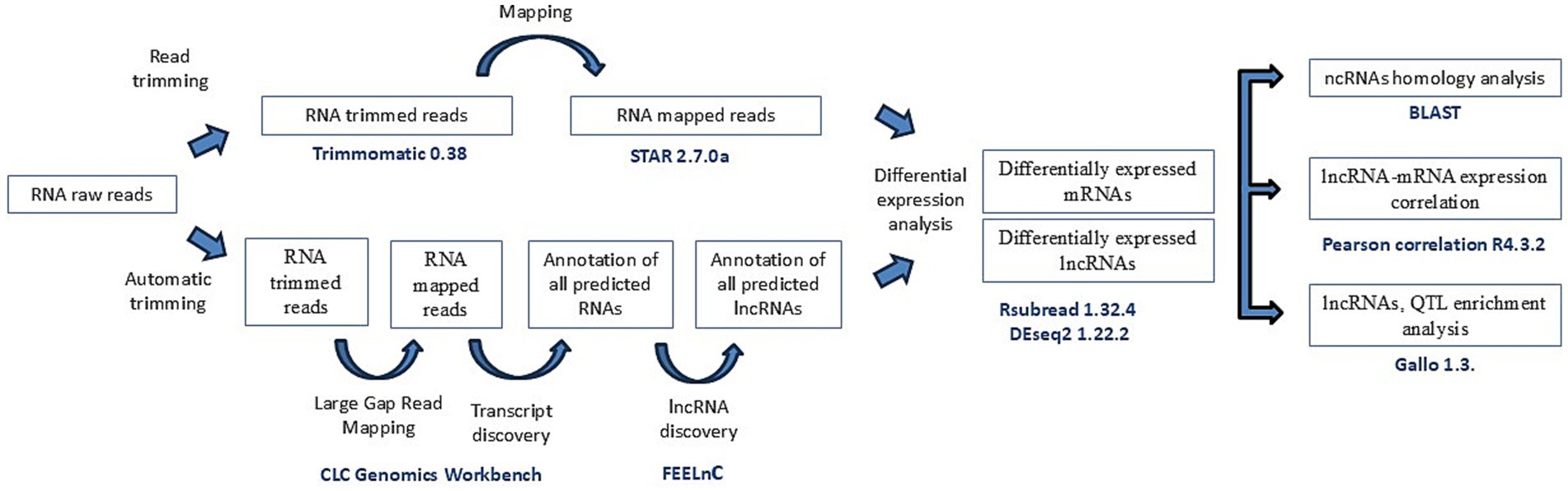
Figure 1. Workflow of the filtering pipeline used in this study. Study workflow from raw fastq reads to lnRNAs identification and differential expression of lncRNAs and nearest genes.
2.4 Novel and annotated lncRNA identification
The LGRM assembly generated by CLC Genomics Workbench and the bovine reference genome ARS_UCD1.2.109 were used to perform transcript discovery with the following settings: gene merging distance = 50; minimum reads in gene = 10; and minimum predicted gene length = 200 bp. The resulting gtf file containing all the predicted RNAs was used as input of the FlExible Extraction of LncRNA (FEELnc) pipeline to identify lncRNAs. The pipeline generated a new gtf file containing all the identified lncRNAs, which was combined with the gtf of the annotated reference genome.
2.5 RNA-Seq alignment and read count
Quality control of the merged file from FeelNC and the annotated gtf reference file was performed using FastQC 0.12 to identify sequencing read artifacts including sites with low-quality reads (Phred score < 30), duplicated reads, uncalled bases, and potentially contaminated reads (36, 37). Next, the reads were trimmed to remove Illumina adapters and low-quality bases at the start and end of each read using Trimmomatic 0.38 (38). In addition, reads with an average quality score below 20, within a sliding window of 5 nucleotides and with a length <75 nucleotides, were removed. Trimmed reads were mapped to the ARS-UCD1.2.109 reference genome using the Spliced Transcripts Alignment to a Reference aligner STAR 2.7.0a (39) with default settings. Read counts were extracted from the alignment files using the function “feature counts” of the R package Rsubread 1.32.4 (40) with the gtf file obtained in the FEELnc pipeline.
2.6 Differential expression analysis
Differential expression analysis of all transcripts was performed between all groups (cows with focal lesions vs. controls, with diffuse lesions vs. controls, and with diffuse vs. focal lesions) using the R package DESeq2 1.22.2 (41). Transcripts were considered as DE between groups when the false discovery rate (FDR) was lower than 0.05. Only those transcripts identified as lncRNAs by the FEELnc pipeline were considered for further analysis. The sequence of each DE lncRNA was extracted from the ARS-UCD1.2.109 reference genome using the coordinates specified in the gtf file. A BLAST analysis was performed to assess if the predicted lncRNAs had homologous sequences with ncRNAs of other species.
2.7 Expression of lncRNA-mRNA pairs
LncRNAs tend to exhibit similar expression level patterns to the protein-coding genes that are located nearby (22). To assess if the identified lncRNAs modified the expression of the nearby coding transcript, Pearson’s correlation tests were performed in R using the counts of each lncRNA–mRNA predicted pair, considering a test p < 0.05 as significant.
2.8 QTL annotation and enrichment analysis
QTL annotation was performed 500 Kbp upstream of the start and 500 Kbp downstream of the end of each DE lncRNA using the R package Genomic functional Annotation in Livestock for positional candidate LOci (GALLO) (42). In addition, to evaluate which identified QTL was significantly overrepresented in the database, a QTL enrichment analysis using the qtl_enrich() function from GALLO was performed. Enriched QTL had an adjusted p < 0.05. The genome coordinates of the DE lncRNAs were used, and the QTL gff annotation file was retrieved from release 52 of the Animal QTLdb (43).
3 Results
3.1 RNA-Seq data and lncRNA identification in ICV samples
High-throughput RNA-Seq data derived from ICV samples of 14 animals (4 without lesions, 5 with focal lesions, and 5 with diffuse lesions) were analyzed using the CLC Genomics Workbench (CLC Bio, Aarhus, Denmark), obtaining a mean of 21,579,876 reads per sample. Using LGRM, we observed that a mean of 19,435,468 (90.05%) of these reads mapped to a single location of the bovine reference genome (Supplementary Table 1). A computational pipeline adapted from a previous study (25) was implemented to identify lncRNAs (Figure 1). The FEELnc pipeline identified 1,434 lncRNAs with specificity and sensitivity of 0.962 (Supplementary Figure 1). Figure 2 shows the distribution of the identified lncRNAs across the genome of the cow. Chromosomes BTA19 and BTA18 contained more lncRNAs in comparison to the rest of chromosomes, with each containing 93 and 90 lncRNAs, respectively. Among the 1,434 lncRNAs, 899 (62%) were not annotated in the ARS-UCD1.2.109 bovine reference genome and were considered novel. Interestingly, chromosomes BTA19 and BTA25 account for the highest number of novel lncRNAs; 69 and 68, respectively. Among the 1,434 identified lncRNAs, 1,315 had a predicted gene target. From these 1,315 lncRNAs with a target gene, 600 (46%) were located upstream, 445 (34%) downstream, 195 (15%) in intronic regions, and 75 (5%) in exons.
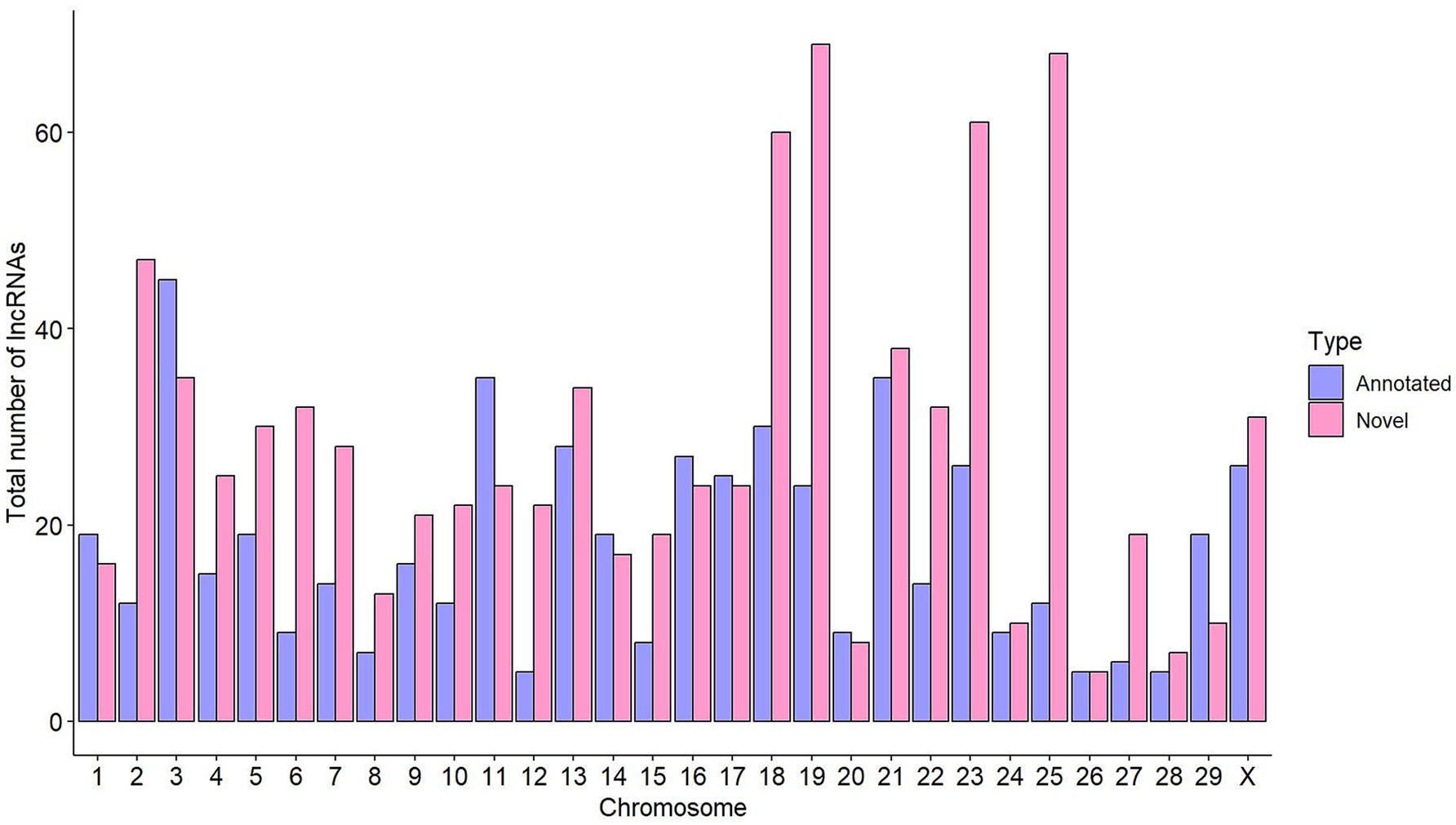
Figure 2. Distribution of lncRNAs across the cow genome. The total number of annotated lncRNAs per chromosome is shown in purple, while the number of novel lncRNAs is indicated in pink.
3.2 Differential expression of lncRNAs in ICV from cows with distinct PTB-associated lesions
In total, 1, 6, and 2 lncRNAs were DE in cows with focal lesions vs. controls, with diffuse lesions vs. controls, and with diffuse vs. focal lesions, respectively (Table 1). The DE lncRNAs were located in BTA2, 4, 6, 7, and 12 and mainly upstream of their target mRNA. The longest DE lncRNA (lnRNA_23401.1) was located on chromosome BTA4, while the shortest DE lncRNA (lncRNA_18413.2) was located on chromosome BTA12. The most highly DE lncRNA, the lncRNA_18413.2, was downregulated (log2 fold = −5.580) in cows with diffuse lesions compared to control cows.
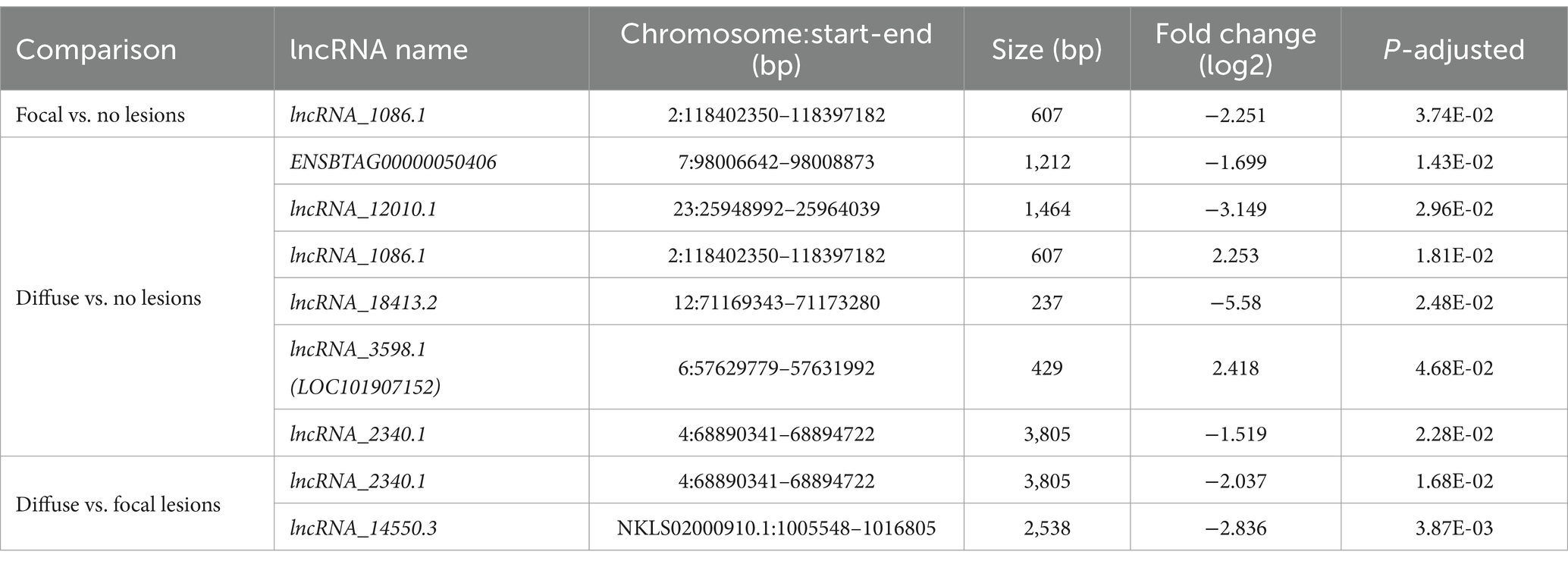
Table 1. Differentially expressed lncRNAs in ICV from Holstein cattle with PTB-associated lesions vs. animals without lesions in gut tissues.
The lncRNA_1086.1 was identified in two comparisons: in cows with focal lesions vs. controls and with diffuse lesions vs. controls. In addition, the lncRNA_2340.1 was identified in cows with diffuse lesions vs. controls and with diffuse vs. focal lesions. Only one DE lncRNA (ENSBTAG00000050406) was annotated in the ARS-UCD1.2.109 bovine reference genome. BLAST analysis revealed that the lncRNA_3,598 has been recently annotated in the new version of the bovine reference genome (ARS-UCD1.3) as LOC101907152. BLAST alignment analysis revealed that all the DE lncRNAs shared homology with at least one annotated mammal ncRNA except the lncRNA_18413.2. For instance, the lncRNA_1086.1, lncRNA_2340.1, and lncRNA_14550.3 shared homology with the human prostate cancer-associated lncRNA (PRNCR1), HOXA-AS3 lncRNA, and the predicted lncRNA TCONS-00241516, respectively.
3.3 Expression of lncRNA–mRNA pairs
Pearson’s correlation tests between lncRNAs and their best-predicted interaction partner revealed that most lncRNAs had significant positive or negative Pearson’s correlation coefficient (ρ) with their predicted partners (Table 2). The lncRNA_2340.1, lncRNA ENSBTAG00000050406, and lncRNA_12010.1 had a ρ of 0.801, 0.905, and 0.686 with their best interaction partners: the Homeobox A6 (HOXA6), the repulsive Guidance Molecule BMP Co-Receptor B (RGMB), and the transcript ENSBTAT00000037068, respectively. The lncRNA1086.1 had a significant ρ of −0.575 with its best interaction partner, the myotubularin-related protein 9 (MTMR9). The lncRNA_18413.2 and lncRNA_14550.3 had no significant correlation with their best-predicted interaction partners, and lncRNA_3598.1 had no predicted interaction partner.

Table 2. Pearson’s correlation (ρ) tests between lncRNAs and their best-predicted partner.
3.4 QTL annotation and enrichment analysis
QTL annotation 500 Kbp upstream and downstream of the start and end of each DE lncRNA and QTL enrichment analysis were performed using GALLO. A total of 37, 123, and 11 QTLs were identified in cows with focal lesions vs. controls, with diffuse lesions vs. controls, and with diffuse vs. focal lesions, respectively (Supplementary Table 2). QTL enrichment analysis was performed to correct the large number of QTL in the database associated with the more studied traits (Figure 3). In cows with focal lesions vs. controls, significant enrichment of QTLs previously associated with clinical mastitis was observed. In the comparison of cows with diffuse lesions vs. controls, significantly enriched QTLs were associated with HDL cholesterol, bovine leukemia, bovine tuberculosis, and paratuberculosis susceptibility.
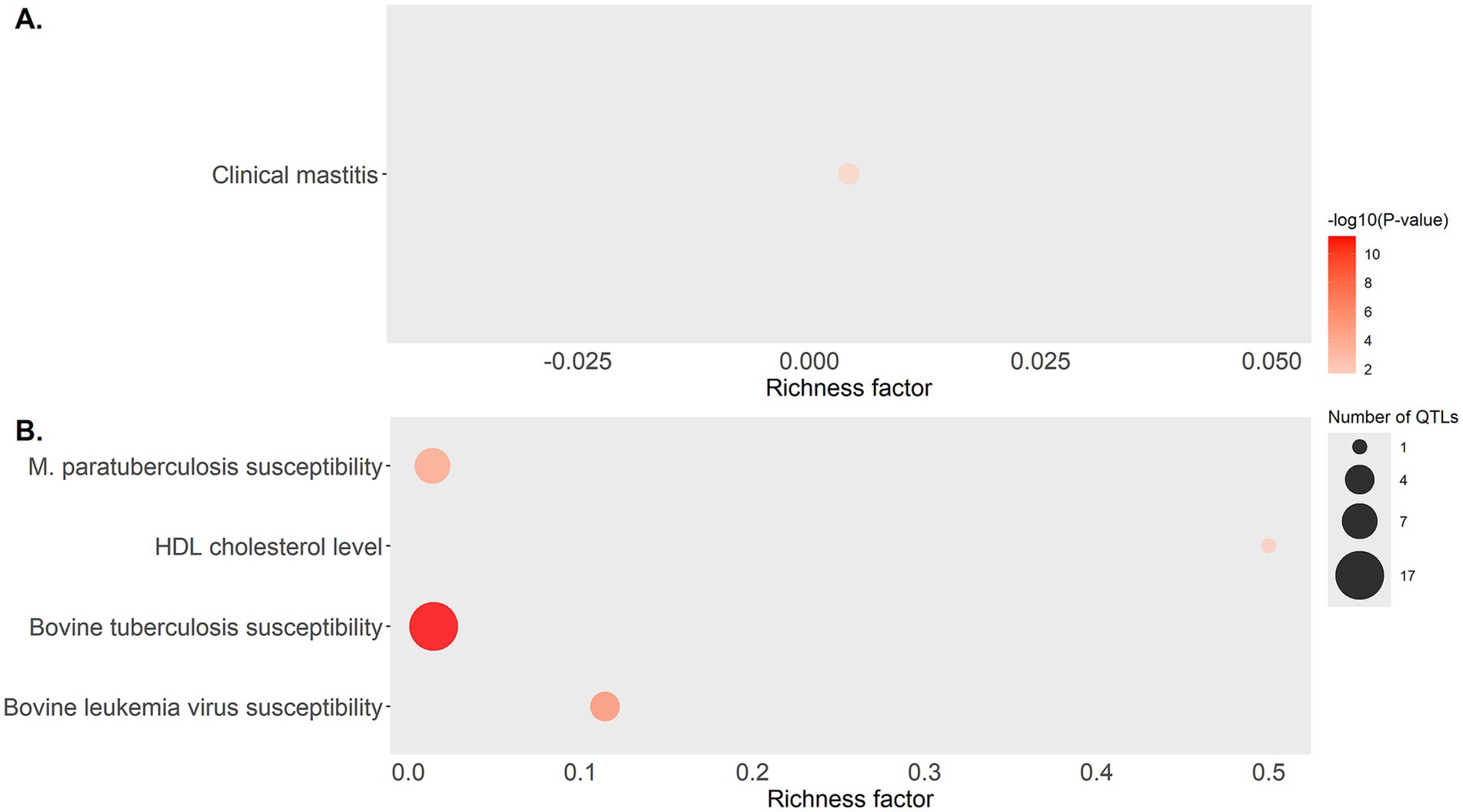
Figure 3. Bubble plot displaying the QTL associated with health traits that were found enriched in the QTL enrichment analysis. QTL enrichment analysis was performed using annotated QTLs located within a 500 Kbp interval of the DE lncRNAs identified in (A) cows with focal lesions compared to controls, and (B) cows with diffuse lesions compared to controls. The area of the bubbles represents the number of QTL for that QTL class, and the color represents the p-value scale (darker color = smaller p-value). The richness factor for each QTL represents the ratio of the number of QTL and the expected number of QTL.
4 Discussion
LncRNAs participate in host–pathogen interactions that alter the resulting immune response (44), but their functional annotation in cattle is very limited, and their role in the regulation of the host immune response against mycobacterial infections remains unknown. Furthermore, the identification of lncRNAs is challenging due to their lower expression levels than mRNAs. Recent studies have shown that the low expression of lncRNAs can be essential for their functional role by ensuring specificity to their regulated targets (45). A common approach to discover and annotate putative lncRNAs is to consider transcripts that have nucleotide lengths >200 nt and that show little to no evidence of protein-coding potential (26, 30). In this study, RNA-Seq data from ICV samples of 14 Holstein cows without lesions and with two different PTB-associated lesions in gut tissues (focal and diffuse) allowed the identification of 1,434 lncRNAs, 899 of which were novel. The genomic features of the identified lncRNAs were in concordance with earlier observations in cattle and in other animal species. For instance, the identified lncRNAs had lower expression levels than their target mRNAs, and their length was shorter due to the lower number of exons in the transcripts. In our study, the predominant distribution of the identified lncRNAs in chromosomes BTA18, BTA19, and BTA25 was also consistent with the predominant location of the lncRNAs previously identified in MAP-infected macrophages (32, 33). To the best of our knowledge, this is the first study that provides novel insights into the role of lncRNAs in the bovine ICV, the primary site of MAP infection, and their interplay with mRNAs during MAP infection in cattle. Whether the identified lncRNAs are specific to MAP infection and may serve as potential biomarkers to detect infected animals needs to be further investigated.
Eight lncRNAs were DE between cows with different PTB-associated lesions vs. controls, five of which were unannotated and could be considered novel (lncRNA_1086.1, lncRNA_12010.1, lncRNA_18413.2, lncRNA_2340.1, and lncRNA_14550.3). Four of the five novel DE lncRNAs (lncRNA_1086.1, lncRNA_12010.1, lncRNA_2340.1, and lncRNA_14550.3) had partial homology with at least one annotated mammal ncRNA. We observed no overlap between the DE lncRNAs and previously published bovine lncRNAs identified in MAP-infected macrophages (31, 33, 46) and jejunum samples from MAP-infected cattle (47). This might be because the bovine lncRNA library is not fully annotated and some lncRNAs show tissue-specific expression patterns (48). The lack of overlap between the DE lncRNAs identified in this study and those reported in previous studies may be due to other factors such as methodological differences, variations in lncRNA discovery algorithms, or differences in study samples. None of the genes close to DE lncRNAs were previously associated with susceptibility to PTB in the literature.
Inferring lncRNA biological roles is challenging, particularly for dairy cattle, where the functional annotation of lncRNAs is limited. We inferred the putative biological roles of the identified lncRNAs by assessing the function of neighboring protein-coding genes and the lncRNAs-mRNA co-expression patterns. In cows with focal lesions vs. controls, the lncRNA1086.1 was downregulated (log2 fold = −2.251). In contrast, in cows with diffuse lesions vs. controls, the lncRNA1086.1 was upregulated (log2 fold = 2.253). In both comparisons, the lncRNA1086.1 was predicted to interact with the MTMR9 gene, and the expression levels of the lncRNA1086.1 and the MTMR9 were negatively correlated (ρ = −0.575). The MTMR9 interacts with the MTMR6, and this interaction is critical for lipid binding, catalytic activity, and protein stability of MTMR6, and the depletion of both proteins promotes apoptosis (49). Therefore, we hypothesize that the decrease in the lncRNA1086.1 expression observed in the cows with focal lesions vs. controls (log2 fold = −2.251) would cause an increase in MTMR9 expression and an inhibition of apoptosis during the subclinical stage of MAP infection, which leads to MAP intracellular survival and persistence without infected macrophages. In contrast, the increase in lncRNA1086.1 expression (log2 fold = 2.253) in cows with diffuse lesions vs. controls would have the opposite effect. Therefore, our results suggest that the lncRNA1086.1 could be modulating apoptosis in distinct stages of MAP infection. Interestingly, the sequence of the lncRNA_1086.1 shared 91% homology with the PRNCR1. This lncRNA has been reported to be an oncogenic transcript participating in the pathogenesis of several kinds of cancers, and some single-nucleotide polymorphisms within this lncRNA affect cancer risk (50, 51). PRNCR1 levels in tumors were shown to have diagnostic and prognostic importance, and comparatively higher expression levels of this lncRNA were also found in blood exosomes of patients with pancreatic carcinoma, hepatocellular carcinoma, and colorectal cancer than that of normal controls.
In cows with diffuse lesions vs. controls, downregulation of the ENSBTAG00000050406 lncRNA was observed (log2 fold = −1.699). The ENSBTAG00000050406 was predicted to interact with the RGMB gene, resulting in a downregulation of the RGMB expression (ρ = 0.905). The dysregulation of RGMB has been implicated in the development and progression of various malignancies in humans, including breast, prostate, lung, and colorectal cancers; melanomas, and osteosarcomas (52, 53). Interestingly, the inhibition of RGBM expression in RAW264.7 or J774 macrophages increased interleukin 6 (IL-6) expression, while RGMB overexpression inhibited IL-6 expression in a ligand-dependent manner (54). In our study, therefore, the observed decrease in the expression of the lncRNA ENSBTAG00000050406 (log2 fold = −1.699) could cause downregulation of the RGMB (log2 fold = −0.611) and upregulation of IL-6, thus enhancing the pro-inflammatory immune response. Rapid production of IL-6 contributes to host defense during infection and tissue injury, but excessive IL-6 synthesis is involved in PTB pathology. Other lncRNAs with differing expression levels in the comparisons of cows with diffuse lesions vs. controls were the lncRNA 12010.1 (log2 fold = −3.149), lncRNA 18413.2 (log2 fold = −5.580), and lncRNA 3598.1 (log2 fold = 2.418).
In the cows with diffuse lesions, downregulation of the lncRNA_2340.1 was observed. The lncRNA_2340.1 was predicted to interact and downregulate HOXA6 gene expression (ρ = 0.801). The homeobox genes (HOX) are a group of important transcriptional regulatory factors that act on target genes and are highly conserved in evolution (55–57). They can participate in embryonic development, cell identification, cell differentiation, cell metabolism, apoptosis, autophagy, and other processes. In gastric cancer and colorectal cancer, HOXA6 can inhibit the apoptosis of tumor cells by binding to other genes or acting on other pathways to promote the occurrence and progression of tumors (58, 59). Taking these results into account, we suggest an important immunomodulatory role of the lncRNA_2340.1 in MAP-infected cows with diffuse lesions by suppressing HOXA6 expression and enhancing apoptosis in the clinical state of MAP infection. Interestingly, the lncRNA_2340.1 shared 84.7% homology with the human HOXA-AS3 lncRNA. Dysregulation of the HOXA-AS3 lncRNA contributes to the development of multiple cancer types (60). Finally, in the comparison of cows with diffuse vs. focal lesions, the lncRNA_145550.3 was downregulated (log2 fold = −2.836), and although it was predicted to interact with the ENSBTAT00000078506, the interaction was not statistically significant (p-value = 0.848). Interestingly, we found that the lncRNA_145550.3 shared 81.6% of homology with the human lncRNA TCONS-00241516.
The enrichment analysis of QTLs revealed that the DE lncRNAs identified in cows with focal lesions vs. controls were located in two QTLs previously associated with clinical mastitis (p-value 0.045). Previous studies found that the incidence of mastitis, PTB, and the coexistence of both infections was significant (61). Therefore, these QTLs could make the cows more susceptible to clinical mastitis and PTB. In the cows with diffuse lesions vs. controls, the DE lncRNAs overlapped with QTL previously associated with HDL cholesterol levels, bovine tuberculosis, bovine leukemia virus, and MAP susceptibility. If the identified DE lncRNAs have a fundamental role in regulation due to their interactions with transcripts nearby, this might provide key insights for the development of breeding programs for PTB resistance.
5 Conclusion
Using RNA-Seq data from ICV, the primary site of MAP infection, a total of 1,434 lncRNAs were identified, 899 of them were novel. Among the lncRNAs dysregulated in cattle with PTB-associated lesions, seven were linked to genes DE in ICV such as MTMR9, RGMB, HOXA6, ENSBTAT00000037068, ENSBTAT00000084572, and ENSBTAT00000078506. While some DE lncRNAs (ENSBTAG00000050406 and lncRNA_2340.1) activate genes with roles in apoptosis, host immune response, and inflammation, it is also conceivable that MAP utilizes some host lncRNAs (lncRNA1086.1) to block the induction of anti-bacterial genes and apoptosis. Interestingly, two of the DE lncRNAs (lncRNA_1086.1 and lncRNA_2340.1) were homologous with the human PRNCR1 and HOXA-AS3 lncRNAs that act as oncogenes or tumor suppressors, respectively. The identified lncRNAs were located in regions previously associated with MAP susceptibility and other bovine diseases such as clinical mastitis, bovine tuberculosis, and bovine leukemia virus, suggesting that the identified lncRNAs might have a conserved function in response to MAP infection and potentially to other pathogens as well. Future research should focus on elucidating the specific mechanism by which the identified lncRNAs regulate gene expression and disease pathogenesis.
Data availability statement
The datasets presented in this study are deposited in the Gene Expression Omnibus repository, accession number GSE137395.
Ethics statement
The animal study was approved by Servicio Regional de Investigación y Desarrollo Agroalimentario (SERIDA). The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
GB-B: Investigation, Visualization, Methodology, Data curation, Writing – original draft. VA: Writing – review & editing, Formal analysis, Supervision, Software, Methodology, Data curation, Investigation. ÁC: Formal analysis, Methodology, Supervision, Software, Writing – review & editing, Validation, Conceptualization, Resources. MA-H: Project administration, Funding acquisition, Resources, Conceptualization, Supervision, Writing – review & editing, Investigation.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This research was funded by MCIN/AEI/10.13039/501100011033 and ERDF “A way of making Europe,” grants RTI2018-094192-R-C21 and PID2021-122197OR-C21 to MA-H.
Acknowledgments
Technical and human support provided by the DIPC Supercomputing Center is gratefully acknowledged. We are grateful to Kyle P. Hearn for the careful editing of the manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Generative AI statement
The authors declare that no Gen AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fvets.2025.1601267/full#supplementary-material
SUPPLEMENTARY FIGURE 1 | Two-graph receiver operating characteristic curve (ROC) to determine the optimal coding probability cutoff value. Dashed black lines represent the threshold chosen by FEELnc codpot as the point that maximizes sensitivity (blue lines) and specificity (red lines) of prediction of lncRNAs.
SUPPLEMENTARY TABLE 1 | Summary statistics of the LGRM step.
SUPPLEMENTARY TABLE 2 | QTL enrichment analysis.
References
1. Ott, SL, Wells, SJ, and Wagner, BA. Herd-level economic losses associated with Johne’s disease on US dairy operations. Prev Vet Med. (1999) 40:179–92. doi: 10.1016/S0167-5877(99)00037-9
2. Rasmussen, P, Barkema, HW, Mason, S, Beaulieu, E, and Hall, DC. Economic losses due to Johne’s disease (paratuberculosis) in dairy cattle. J Dairy Sci. (2021) 104:3123–43. doi: 10.3168/jds.2020-19381
3. Nielsen, SS, and Toft, N. A review of prevalences of paratuberculosis in farmed animals in Europe. Prev Vet Med. (2009) 88:1–14. doi: 10.1016/j.prevetmed.2008.07.003
4. Dow, CT. Warm, sweetened milk at the twilight of immunity - Alzheimer’s disease - inflammaging, insulin resistance, M. Paratuberculosis and immunosenescence. Front Immunol. (2021) 12:714179. doi: 10.3389/fimmu.2021.714179
5. Juste, RA, Elguezabal, N, Pavón, A, Garrido, JM, Geijo, M, Sevilla, I, et al. Association between Mycobacterium avium subsp. paratuberculosis DNA in blood and cellular and humoral immune response in inflammatory bowel disease patients and controls. Int J Infect Dis. (2009) 13:247–54. doi: 10.1016/j.ijid.2008.06.034
6. Pierce, ES. Could Mycobacterium avium subspecies paratuberculosis cause Crohn’s disease, ulcerative colitis… And colorectal cancer? Inf Agents Cancer. (2018) 13:1–6. doi: 10.1186/s13027-017-0172-3
7. Bermudez, LE, Petrofsky, M, Sommer, S, and Barletta, RG. Peyer’s patch-deficient mice demonstrate that Mycobacterium avium subsp. paratuberculosis translocates across the mucosal barrier via both M cells and enterocytes but has inefficient dissemination. Infect Immun. (2010) 78:3570–7. doi: 10.1128/IAI.01411-09
8. Khare, S, Lawhon, SD, Drake, KL, Nunes, JES, Figueiredo, JF, Rossetti, CA, et al. Systems biology analysis of gene expression during in vivo Mycobacterium avium paratuberculosis enteric colonization reveals role for immune tolerance. PLoS One. (2012) 7:e42127. doi: 10.1371/journal.pone.0042127
9. Rees, WD, Lorenzo-Leal, AC, Steiner, TS, and Bach, H. Mycobacterium avium subspecies paratuberculosis infects and replicates within human monocyte-derived dendritic cells. Microorganisms. (2020) 8:994. doi: 10.3390/microorganisms8070994
11. Whitlock, RH, and Buergelt, C. Preclinical and clinical manifestations of Paratuberculosis (including pathology). Vet Clin N Am Food Anim Pract. (1996) 12:345–56. doi: 10.1016/S0749-0720(15)30410-2
12. González, J, Geijo, MV, García-Pariente, C, Verna, A, Corpa, JM, Reyes, LE, et al. Histopathological classification of lesions associated with natural Paratuberculosis infection in cattle. J Comp Pathol. (2005) 133:184–96. doi: 10.1016/j.jcpa.2005.04.007
13. Sweeney, RW. Pathogenesis of Paratuberculosis. Vet Clin N Am Food Anim Pract. (2011) 27:537–46. doi: 10.1016/j.cvfa.2011.07.001
14. Koets, AP, Eda, S, and Sreevatsan, S. The within host dynamics of Mycobacterium avium ssp. paratuberculosis infection in cattle: where time and place matter. Vet Res. (2015) 46:61. doi: 10.1186/s13567-015-0185-0
15. Whitlock, RH, Wells, SJ, Sweeney, RW, and Van Tiem, J. ELISA and fecal culture for paratuberculosis (Johne’s disease): sensitivity and specificity of each method. Vet Microbiol. (2000) 77:387–98. doi: 10.1016/S0378-1135(00)00324-2
16. Rearick, D, Prakash, A, McSweeny, A, Shepard, SS, Fedorova, L, and Fedorov, A. Critical association of ncRNA with introns. Nucleic Acids Res. (2011) 39:2357–2366. doi: 10.1093/nar/gkq1080
17. Gruber, AJ, and Zavolan, M. Alternative cleavage and polyadenylation in health and disease. Nat Rev Genet. (2019) 20:599–614. doi: 10.1038/s41576-019-0145-z
18. Bocchetti, M, Scrima, M, Melisi, F, Luce, A, Sperlongano, R, Caraglia, M, et al. LncRNAs and immunity: coding the immune system with noncoding oligonucleotides. Int J Mol Sci. (2021) 22:1741. doi: 10.3390/ijms22041741
19. Ruan, X, Li, P, Chen, Y, Shi, Y, Pirooznia, M, Seifuddin, F, et al. In vivo functional analysis of non-conserved human lncRNAs associated with cardiometabolic traits. Nat Commun. (2020) 11:45. doi: 10.1038/s41467-019-13688-z
20. Johnsson, P, Lipovich, L, Grandér, D, and Morris, KV. Evolutionary conservation of long non-coding RNAs; sequence, structure, function. Biochim Biophys Acta Gen Subj. (2014) 1840:1063–71. doi: 10.1016/j.bbagen.2013.10.035
21. Mukherjee, N, Calviello, L, Hirsekorn, A, de Pretis, S, Pelizzola, M, and Ohler, U. Integrative classification of human coding and noncoding genes through RNA metabolism profiles. Nat Struct Mol Biol. (2017) 24:86–96. doi: 10.1038/nsmb.3325
22. Cabili, MN, Trapnell, C, Goff, L, Koziol, M, Tazon-Vega, B, Regev, A, et al. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. (2011) 25:1915–27. doi: 10.1101/gad.17446611
23. Koufariotis, LT, Chen, Y-PP, Chamberlain, A, Vander Jagt, C, and Hayes, BJ. A catalogue of novel bovine long noncoding RNA across 18 tissues. PLoS One. (2015) 10:e0141225. doi: 10.1371/journal.pone.0141225
24. Taniue, K, and Akimitsu, N. The functions and unique features of LncRNAs in Cancer development and tumorigenesis. Int J Mol Sci. (2021) 22:632. doi: 10.3390/ijms22020632
25. Asselstine, V, Medrano, JF, Muniz, MMM, Mallard, BA, Karrow, NA, and Cánovas, A. Novel lncRNA regulatory elements in milk somatic cells of Holstein dairy cows associated with mastitis. Commun Biol. (2024) 7:98. doi: 10.1038/s42003-024-05764-y
26. Billerey, C, Boussaha, M, Esquerré, D, Rebours, E, Djari, A, Meersseman, C, et al. Identification of large intergenic non-coding RNAs in bovine muscle using next-generation transcriptomic sequencing. BMC Genomics. (2014) 15:499. doi: 10.1186/1471-2164-15-499
27. Huang, W, Long, N, and Khatib, H. Genome-wide identification and initial characterization of bovine long non-coding RNAs from EST data. Anim Genet. (2012) 43:674–82. doi: 10.1111/j.1365-2052.2012.02325.x
28. Qu, Z, and Adelson, DL. Bovine ncRNAs are abundant, primarily intergenic, conserved and associated with regulatory genes. PLoS One. (2012) 7:e42638. doi: 10.1371/journal.pone.0042638
29. Tong, C, Chen, Q, Zhao, L, Ma, J, Ibeagha-Awemu, EM, and Zhao, X. Identification and characterization of long intergenic noncoding RNAs in bovine mammary glands. BMC Genomics. (2017) 18:468. doi: 10.1186/s12864-017-3858-4
30. Weikard, R, Hadlich, F, and Kuehn, C. Identification of novel transcripts and noncoding RNAs in bovine skin by deep next generation sequencing. BMC Genomics. (2013) 14:789. doi: 10.1186/1471-2164-14-789
31. Bao, Y, Wu, S, Yang, T, Wang, Z, Wang, Y, Jiang, X, et al. Analysis of long non-coding RNA expression profile of bovine monocyte-macrophage infected by Mycobacterium avium subsp. paratuberculosis. BMC Genomics. (2022) 23:768. doi: 10.1186/s12864-022-08997-5
32. Gupta, P, Peter, S, Jung, M, Lewin, A, Hemmrich-Stanisak, G, Franke, A, et al. Analysis of long non-coding RNA and mRNA expression in bovine macrophages brings up novel aspects of Mycobacterium avium subspecies paratuberculosis infections. Sci Rep. (2019) 9:1571. doi: 10.1038/s41598-018-38141-x
33. Marete, A, Ariel, O, Ibeagha-Awemu, E, and Bissonnette, N. Identification of Long non-coding RNA isolated from naturally infected macrophages and associated with bovine Johne’s disease in Canadian Holstein using a combination of neural networks and logistic regression. Front Vet Sci. (2021) 8:639053. doi: 10.3389/fvets.2021.639053
34. Alonso-Hearn, M, Canive, M, Blanco-Vazquez, C, Torremocha, R, Balseiro, A, Amado, J, et al. RNA-Seq analysis of ileocecal valve and peripheral blood from Holstein cattle infected with Mycobacterium avium subsp. paratuberculosis revealed dysregulation of the CXCL8/IL8 signaling pathway. Sci Rep. (2019) 9:14845. doi: 10.1038/s41598-019-51328-0
35. Blanco-Vázquez, C, Alonso-Hearn, M, Juste, RA, Canive, M, Iglesias, T, Iglesias, N, et al. Detection of latent forms of Mycobacterium avium subsp. paratuberculosis infection using host biomarker-based ELISAs greatly improves paratuberculosis diagnostic sensitivity. PLoS One. (2020) 15:e0236336. doi: 10.1371/journal.pone.0236336
36. Cánovas, A, Rincón, G, Bevilacqua, C, Islas-Trejo, A, Brenaut, P, Hovey, RC, et al. Comparison of five different RNA sources to examine the lactating bovine mammary gland transcriptome using RNA-sequencing. Sci Rep. (2014) 4:5297. doi: 10.1038/srep05297
37. Cardoso, TF, Quintanilla, R, Castelló, A, González-Prendes, R, Amills, M, and Cánovas, Á. Differential expression of mRNA isoforms in the skeletal muscle of pigs with distinct growth and fatness profiles. BMC Genomics. (2018) 19:145. doi: 10.1186/s12864-018-4515-2
38. Bolger, AM, Lohse, M, and Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. (2014) 30:2114–20. doi: 10.1093/bioinformatics/btu170
39. Dobin, A, Davis, CA, Schlesinger, F, Drenkow, J, Zaleski, C, Jha, S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. (2013) 29:15–21. doi: 10.1093/bioinformatics/bts635
40. Liao, Y, Smyth, GK, and Shi, W. The R package Rsubread is easier, faster, cheaper and better for alignment and quantification of RNA sequencing reads. Nucleic Acids Res. (2019) 47:e47–7. doi: 10.1093/nar/gkz114
41. Love, MI, Huber, W, and Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. (2014) 15:550. doi: 10.1186/s13059-014-0550-8
42. Fonseca, PAS, Suárez-Vega, A, Marras, G, and Cánovas, Á. GALLO: an R package for genomic annotation and integration of multiple data sources in livestock for positional candidate loci. Gigascience. (2020) 9:1–9. doi: 10.1093/gigascience/giaa149
43. Hu, Z-L, Park, CA, Wu, X-L, and Reecy, JM. Animal QTLdb: an improved database tool for livestock animal QTL/association data dissemination in the post-genome era. Nucleic Acids Res. (2013) 41:D871–9. doi: 10.1093/nar/gks1150
44. Fitzgerald, KA, and Caffrey, DR. Long noncoding RNAs in innate and adaptive immunity. Curr Opin Immunol. (2014) 26:140–6. doi: 10.1016/j.coi.2013.12.001
45. Jachowicz, JW, Strehle, M, Banerjee, AK, Blanco, MR, Thai, J, and Guttman, M. Xist spatially amplifies SHARP/SPEN recruitment to balance chromosome-wide silencing and specificity to the X chromosome. Nat Struct Mol Biol. (2022) 29:239–49. doi: 10.1038/s41594-022-00739-1
46. Gupta, SK, Maclean, PH, Ganesh, S, Shu, D, Buddle, BM, Wedlock, DN, et al. Detection of microRNA in cattle serum and their potential use to diagnose severity of Johne’s disease. J Dairy Sci. (2018) 101:10259–70. doi: 10.3168/jds.2018-14785
47. Gao, Y, Cao, J, Han, B, and Sun, D. Preliminary exploration of mRNA, lncRNA, and miRNA expressions in the bovine jejunum unveils novel aspects of Mycobacterium avium subspecies paratuberculosis infections. BMC Genomics. (2025) 26:108. doi: 10.1186/s12864-025-11299-1
48. Jiang, C, Li, Y, Zhao, Z, Lu, J, Chen, H, Ding, N, et al. Identifying and functionally characterizing tissue-specific and ubiquitously expressed human lncRNAs. Oncotarget. (2016) 7:7120–33. doi: 10.18632/oncotarget.6859
49. Zou, J, Chang, S-C, Marjanovic, J, and Majerus, PW. MTMR9 increases MTMR6 enzyme activity, stability, and role in apoptosis. J Biol Chem. (2009) 284:2064–71. doi: 10.1074/jbc.M804292200
50. Bardhan, A, Banerjee, A, Basu, K, Pal, DK, and Ghosh, A. PRNCR1: a long non-coding RNA with a pivotal oncogenic role in cancer. Hum Genet. (2022) 141:15–29. doi: 10.1007/s00439-021-02396-8
51. Ghafouri-Fard, S, Khoshbakht, T, Hussen, BM, Baniahmad, A, Taheri, M, and Salimi, A. A review on the role of PRNCR1 in human disorders with an especial focus on cancer. Pathol Res Prac. (2022) 237:154026. doi: 10.1016/j.prp.2022.154026
52. Li, J, Ye, L, Kynaston, HG, and Jiang, WG. Repulsive guidance molecules, novel bone morphogenetic protein co-receptors, are key regulators of the growth and aggressiveness of prostate cancer cells. Int J Oncol. (2011) 40:544–50. doi: 10.3892/ijo.2011.1251
53. Ye, L, Kynaston, H, and Jiang, W. Bone metastasis in prostate cancer: molecular and cellular mechanisms (review). Int J Mol Med. (2007) 20:103–11. doi: 10.3892/ijmm.20.1.103
54. Xia, Y, Cortez-Retamozo, V, Niederkofler, V, Salie, R, Chen, S, Samad, TA, et al. Dragon (repulsive guidance molecule b) inhibits IL-6 expression in macrophages. J Immunol. (2011) 186:1369–76. doi: 10.4049/jimmunol.1002047
55. Feng, Y, Zhang, T, Wang, Y, Xie, M, Ji, X, Luo, X, et al. Homeobox genes in cancers: from carcinogenesis to recent therapeutic intervention. Front Oncol. (2021) 11:770428. doi: 10.3389/fonc.2021.770428
56. Mallo, M. Reassessing the role of Hox genes during vertebrate development and evolution. Trends Genet. (2018) 34:209–17. doi: 10.1016/j.tig.2017.11.007
57. Nam, J, and Nei, M. Evolutionary change of the numbers of Homeobox genes in bilateral animals. Mol Biol Evol. (2005) 22:2386–94. doi: 10.1093/molbev/msi229
58. Lin, J, Zhu, H, Hong, L, Tang, W, Wang, J, Hu, H, et al. Coexpression of HOXA6 and PBX2 promotes metastasis in gastric cancer. Aging. (2021) 13:6606–24. doi: 10.18632/aging.202426
59. Wu, S, Wu, F, and Jiang, Z. Effect of HOXA6 on the proliferation, apoptosis, migration and invasion of colorectal cancer cells. Int J Oncol. (2018) 52:2093–100. doi: 10.3892/ijo.2018.4352
60. Yao, Q, Wang, C, Wang, Y, Zhang, X, Jiang, H, and Chen, D. The integrated comprehension of lncRNA HOXA-AS3 implication on human diseases. Clin Transl Oncol. (2022) 24:2342–50. doi: 10.1007/s12094-022-02920-w
Keywords: long non-coding RNAs, paratuberculosis, transcriptomics, RNA-Seq, biomarkers, gene regulation
Citation: Badia-Bringué G, Asselstine V, Cánovas Á and Alonso-Hearn M (2025) Genome-wide long non-coding RNA expression profile and its regulatory role in the ileocecal valve from Mycobacterium avium subsp. paratuberculosis-infected cattle. Front. Vet. Sci. 12:1601267. doi: 10.3389/fvets.2025.1601267
Edited by:
Shi-Yi Chen, Sichuan Agricultural University, ChinaReviewed by:
Diana Giannuzzi, University of Padua, ItalySamanta Mecocci, University of Perugia, Italy
Copyright © 2025 Badia-Bringué, Asselstine, Cánovas and Alonso-Hearn. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marta Alonso-Hearn, bWFsb25zb0BuZWlrZXIuZXVz