 Lan Yang
Lan Yang Han Gao
Han Gao Dacheng Liu*
Dacheng Liu*- College of Veterinary Medicine, Inner Mongolia Agricultural University, Hohhot, China
Inflammatory bowel disease (IBD), comprising Crohn’s disease (CD) and ulcerative colitis (UC), is a chronic, relapsing inflammatory disorder of the gastrointestinal tract with multifactorial etiology. The etiology and pathogenesis of ulcerative colitis are diverse, so it is crucial to explore the pathogenesis through animal models. Selecting appropriate animal models is critical for advancing our understanding of UC pathogenesis and therapeutic strategies. This review discusses various UC animal models and compares their characteristics in relation to disease mechanisms and therapeutic research.
Introduction
Ulcerative colitis (UC) is a chronic inflammatory disease of the colon that typically begins in the rectum and progresses proximally in a continuous pattern, affecting the mucosal layer of the colon (1). Patients usually present with abdominal pain, diarrhea, and bloody stools, with the latter being a hallmark of UC. The development of appropriate animal models is essential to understand the pathogenesis of UC and evaluate new treatments.
Exploration of the pathogenesis of disease
Experimental models of ulcerative colitis can simulate the development and progression process of the disease, enable detailed investigation of the role and interrelationship of immune response, intestinal microbiota, genetic susceptibility and environmental factors in the pathogenesis of UC. For example, dextran sulfate sodium DSS—induced UC models allow researchers to study pathological changes in intestinal tissue and cytokine responses to better understand mucosal barrier disruption and inflammatory activation (1, 2). The neurotransmitters secreted by intestinal neurons, such as 5-hydroxytryptamine, also modulate mucosal barrier function and contribute to gut inflammation (3).
Facilitating drug development
The animal models of ulcerative colitis can be constructed to provide an effective platform for drug development, which can be used to test the efficacy and safety of various new drugs, accelerating the drug development process, and improving the success rate of development. For example, a model of ulcerative colitis established by a team from Kyoto University in Japan using human induced pluripotent stem cell-derived colonoid organs provides a promising platform for testing therapeutics such as tofacitinib (4).
Toward precision medicine
Different animals or patients with ulcerative colitis may have individual differences. Animal models can simulate distinct UC subtypes and disease stages, offering valuable insights for designing personalized treatment strategies. This approach supports the development of precision medicine aimed at improving treatment efficacy and patient prognosis.
Pathogenesis of ulcerative colitis
Environmental factors
In some developed countries, the incidence of UC has shown a significant increasing trend, which may indicate that environmental triggers are an important influencing factor in the development of UC (5). Lifestyle changes, dietary inconsistencies, and use of certain medications such as nonsteroidal antiinflammatory drugs (NSAIDs) and antibiotics are considered contributing factors to UC (6). Interestingly, smoking has been observed to reduce the incidence of UC, possibly due to the effects of carbon monoxide and nicotine on mucosal immune responses (7). In addition, there is evidence that the pathogenic factors of IBD show a clear negative correlation with breastfeeding methods, reproduction modes, and individual animal factors (8).
Gut microbiota
The gut microbiome is a relatively complex community that lives in symbiosis with its host in the gastrointestinal tract. The development of multiple diseases may be associated with dysbiosis of the gut microbiome. For example, dysbiosis of the gut microbiota can damage the intestinal mucosal barrier, disrupting intestinal permeability, enabling pathogenic microorganisms to exert inflammatory effects through the barrier, and thus leading to the development of UC. Moreover, an imbalance in the gut microbiota can cause some harmful bacteria to proliferate massively. This has been linked to chronic inflammation and the release of pro-inflammatory factors, increasing the risk of developing UC. For patients with UC, the most striking change is an imbalance in the gut microbiota, characterized by a decrease in the abundance of Firmicutes and Bacteroidetes (9). In patients with UC, the damage of the intestinal mucosal layer causes bacterial invasion of the submucosa and massive proliferation of bacteria in the submucosa, leading to inflammation (10, 11). The inflammation results in further damage to the mucosal layer of the intestinal wall, more bacteria invade and cause the submucosal layer to rupture, producing a vicious cycle (10). The Human Microbiome Project found that the levels of short chain fatty acids (SCFAs), Ruminococcaceae and Lachnospiraceae in the gut of UC patients was markedly reduced, and the abundance of pro-inflammatory microorganisms, such as Enterobacteriaceae, was significantly increased (12, 13). Therefore, the imbalance of gut microbiota is a major influencing factor in the development of UC.
Genetic factors
While genetic susceptibility contributes to UC development, no single mutation fully accounts for disease onset, as the recombinant protein IL23 receptor (IL23R), the intracellular receptor NOD2 and the leukocyte antigen HLA have the strongest genetic effects (14). Relevant studies have shown that the development of UC is associated with genetic susceptibility, and the incidence of relatives and twins of UC patients is significantly higher than that of other normal population. These studies have helped us to better understand the pathogenesis of UC. Mayberry and other scholars found in 1980 that the children and siblings of UC patients had a 30-fold higher risk of developing UC later compared with normal people (15). Monsén et al. (16) also found a significantly higher incidence of UC in immediate family members of UC patients compared with the others in their families. To explore the genetic factors of UC, a genome-wide association study (GWAS) or meta-analysis was performed to formulate relevant immunoarrays to pinpoint the relevant susceptibility loci of UC, which can help us identify relevant genetic variations in IBD and UC (14, 17, 18).
Abnormal immune response
A hallmark of UC histology is the presence of a ‘crypt abscess’, which is formed by neutrophil infiltration into the colonic crypt (6). During the inflammatory process, neutrophils first pass through the colonic epithelium and eventually undergo apoptosis in the crypts of the colon (19). Neutrophils may survive the inflammation by the hypoxia-inducible factor HIF-1α, but prolonged neutrophil survival may lead to excessive release of reactive oxygen species and pro-inflammatory mediators (20–22). Calprotectin is present in neutrophils. It is released through the patient’s blood or feces and serologically reacts with the patient’s perinuclear anti-neutrophil antibodies (23–25). Thus, after the onset of the disease, inflammatory neutrophils and monocytes and their pro-inflammatory factors generate an inflammatory response that promotes the pathological process (6, 26). Such an environment contributes to the subsequent neutrophils, monocytes, maintenance of their functional and survival efficiency, and affects the patient’s ability to resolve the inflammation and to redress their homeostasis (27, 28).
Mitochondrial abnormalities
The latest research on ulcerative colitis is a direct exploration of the intestinal mucosa (29, 30). Some experts found that the expression of genes responsible for energy production in mitochondria (gene encoding mitochondrial oxidative phosphorylation components) was significantly reduced, which suggests that abnormalities in mitochondria can lead to the development of ulcerative colitis (6, 27). Mitochondria are special membrane-bound intracellular compartments that are considered the “powerhouse of the cell,” as they produce a large amount of ATP that is used to power the cell’s biochemical reactions. Previous studies have demonstrated that the mitochondria are involved in the inflammatory response (31, 32), and that loss of mitochondrial function or mitochondrial dysfunction is also an important factor in the development of ulcerative colitis. The studies on mitochondrial function have found that mitochondria are usually located in a specific environment (33, 34), and mitochondrial dysfunction may trigger oxidative stress, reduce ATP production, and release mitochondrial DNA, which acts as a danger-associated molecular pattern (DAMP), amplifying inflammation (35–37). The above studies help us understand the relationship between mitochondria and ulcerative colitis and find a new approach to treat ulcerative colitis, for example, anti-oxidative stress therapy targeting the mitochondria of UC patients.
Extraintestinal manifestations of UC
Types of extraintestinal manifestations
Extraintestinal manifestations (EIMs) are the main factors that lead to complications and decrease the chance of survival in patients (38). Kilic et al. (39) categorized EIMs into two groups: those directly associated with intestinal inflammation (e.g., arthritis, iritis) and those considered independent autoimmune manifestations (e.g., primary sclerosing cholangitis).
Characteristics of extraintestinal manifestations
Due to cross-reactivity of antigens, the gut microbiota may trigger systemic immune responses that affect extraintestinal sites such as skin and joints (40). Disruption of the intestinal barrier allows bacterial antigens to trigger systemic immune responses, including cross-reactivity with skin and joint epitopes (41). The generation of autoimmune responses is directly correlated with immune susceptibility related to leukocyte antigens (42). In UC, prolonged course of the disease, reduced quality of life, intestinal fibrosis, and increased incidence and mortality are strongly associated with untreated EIMs. The gut is the preferred site for the treatment of EIMs. Active treatment of EIMs in early-stage UC has now been shown to be effective in preventing catastrophic outcomes.
UC model construction methods and evaluation criteria
Chemically induced models
DSS-induced UC model
DSS is a polyanionic derivative that appears as a white powder, is odorless, and gives a clear solution when dissolved in water. It is one of the most common and effective drugs for UC induction (2). In DSS-induced intestinal inflammation generally occurs in the lamina propria of the intestine (43). Sulfated polysaccharides in DSS exert a toxic effect on the colonic epithelium, which disrupts the epithelial cell barrier of the intestine, leads to epithelial injury and loss of mucosal barrier integrity and induces immune responses (44). DSS can induce UC by affecting host DNA replication, inhibiting the overproliferation of intestinal epithelial cells, enhancing the release of inflammatory factors, and disrupting the balance of the gut microbiome (45). The partial pathogenic factors are shown in Table 1.
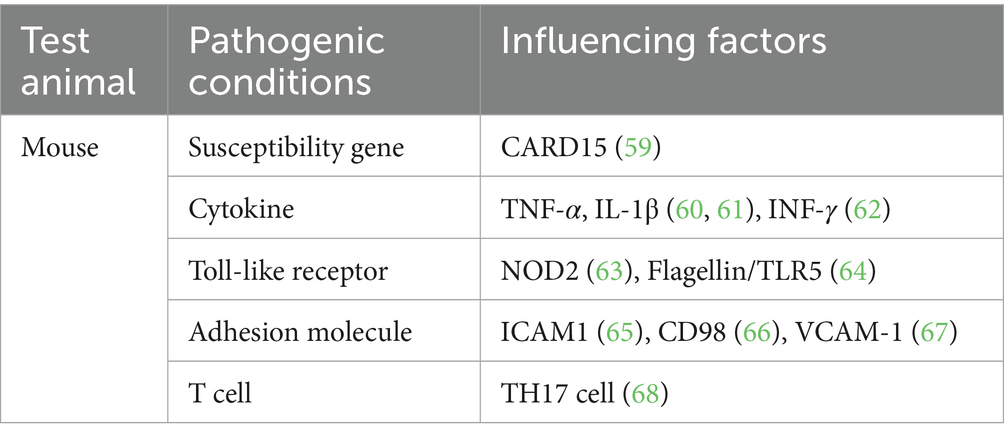
Table 1. Pathogenic factors of DSS-induced UC.
Generally speaking, high molecular weight DSS does not lead to colitis, whereas low molecular weight (5 KDa) DSS induces inflammation (43). DSS is not easily soluble in water, so DSS is added to distilled water heated to 37°C in advance. The DSS aqueous solution should be stirred thoroughly with a sterilized glass rod to ensure that DSS is completely dissolved before administration. If DSS is not sufficiently dissolved, the undissolved salt will block the mouth of the bottle, resulting in difficulty in drug administration or insufficient drug intake of the animals, and thus failure of the model construction. It is also necessary to check the clarity of the liquid before administration. Cloudiness may indicate microbial contamination; Fresh DSS solution should be prepared under sterile conditions. In this case, the bottle should be cleaned or replaced with a new one to prepare new DSS aqueous solution. This is very important. Careless check may lead to wrong test results. While the test group is administered the drug, the control group should be given the same quality of water.
During the test period, some points are worth paying special attention to, as follows.
• On Day 0, the mice in the administration group and the control group should be marked with a marking pen. This is to avoid potential inaccuracy of the subsequent test results, as earmarking may cause stress in the animals, affecting their normal feeding and drinking.
• To avoid fighting between the test mice (which may cause their injuries and thus affect the test results), the mice with gentle disposition should be selected and reared in separate cages to participate in the test.
• The administration method needs to be determined. Rectal administration may lead to stress reaction in mice, intragastric administration may lead to death of mice due to incorrect operation, and free drinking may lead to insufficient drug intake. Therefore, the determination of the administration method is an important factor influencing the test results.
• During the first 3 days of the test, the weight of the mice in the administration group may increase, and start to gradually decrease with the occurrence of bloody stools. Since there is no clear requirement on when to sacrifice the mice, the investigators can determine when to sacrifice the mice for the subsequent test according to the weight loss or the severity of bloody stools (46).
TNBS-induced UC model
2,4,6-Trinitrobenzenesulfonic acid (TNBS) is a hapten that becomes immunogenic after binding to host proteins. TNBS induces an immune response in the host when it binds to the host. The model induced with TNBS is characterized by short modeling time, reproducibility, and simplicity of induction. TNBS is a typical chemical substance used to induce ulcerative colitis models. The mechanism of TNBS as an inducer is as follows: TNBS acts as a hapten, penetrating ethanol-damaged mucosa and binding to host proteins to form neoantigens that trigger a Th1-dominant immune response (47). This model is an immune response mediated primarily by the helper T cell TH1, characterized by infiltration of CD4 + cells, neutrophils, and macrophages, which ultimately leads to transmural colitis (43).
When TNBS is used as an inducer, ethanol is commonly used as its solvent, and the administration is generally intrarectal. Ethanol can first disrupt the intestinal mucosal barrier and assist TNBS in causing intestinal inflammatory injury (48, 49). However, there is no consensus among scholars on the choice of TNBS and ethanol doses. Park et al. (50) used 0.8 mL of 5% TNBS as an inducer and 50% ethanol solution as a solvent, while Isik et al. (51) used 1 mL of 3% TNBS as an inducer and 40% ethanol as a solvent.
Induction of UC by gene knockout technology
The use of genetic engineering technologies for model establishment is an important turning point in the research of UC (52). In the 1990s, Kühn R and his team were the first to propose the construction of a colitis model by knocking out the gene that interferes with the production of IL-10 in the mouse. The emergence of this technology was reported as the establishment of the first spontaneous colitis mouse model (53). Polymorphisms in the IL-10 gene are associated with increased UC susceptibility (54), and genetic polymorphism at the IL-10 gene locus promotes the development of ulcerative colitis (55). Anderson C et al. demonstrated that IL-10 gene knockout mice would develop spontaneous colitis 3 months after knockout (56), with inflammation concentrated in the duodenum, proximal jejunum, and ascending colon (53). The IL-10 gene knockout-induced colitis model is mainly characterized by massive infiltration of macrophages, neutrophils, and lymphocytes (53). Some scholars found that the mice subject to knockout of MUC2 and IL-10 genes showed shortened time to disease and more severe symptoms compared with the mice receiving knockout of IL10 gene. Such a model constructed by double-knockout technology is suitable for the study of UC. Dual knockout mice lacking MUC2 and IL-10 developed severe colitis, growth retardation, bloody diarrhea, and intense mucosal inflammation at 4 weeks of age – very similar to human UC. Thus, the model established by this technology was able to mimic the clinical symptoms of UC patients (57). In addition, deletion of the IL-10 gene in T-cells, especially in regulatory T-cells, contributes to the development of colitis. Therefore, IL-10 plays a critical role in intestinal homeostasis (58). Other UC-relevant models include oxazolone-induced colitis, which models Th2-dominated inflammation, and adoptive T-cell transfer models, which are valuable for dissecting chronic adaptive immune responses.
Model evaluation criteria
The UC models constructed through different methods have different evaluation criteria, as shown in Table 2.
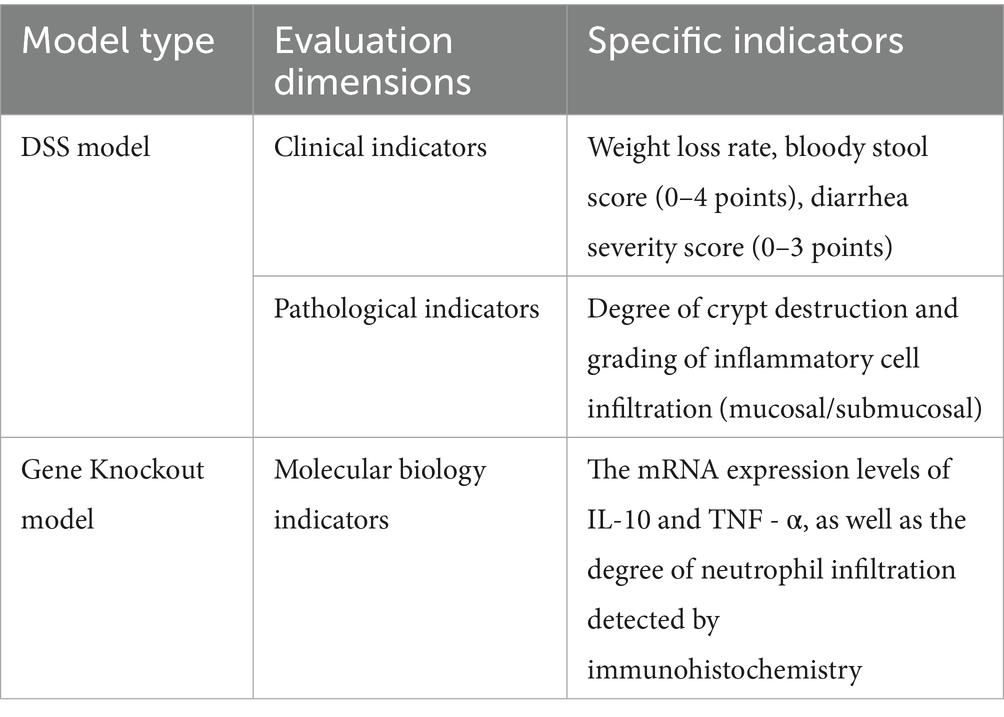
Table 2. Evaluation indicators for different UC models.
Limitations and future directions
At present, despite their utility, current UC animal models have important limitations due to differences in induction mechanisms, species biology, and lack of standardization. For example, chemically induced models still rely on exogenous chemical substances to damage animal intestinal mucosa, which is different from the natural onset of UC. For instance, DSS-induced models rely on chemical injury to the epithelium and produce short-term acute inflammation, whereas UC in humans is typically chronic and relapsing. Acute inflammation is its main pathological feature, while the pathological process of UC is usually chronic and recurrent; TNBS requires ethanol pre-treatment to facilitate haptenation, resulting in a Th1-dominated response more typical of Crohn’s disease than UC, which is confined to the mucosa and associated with Th2/Th17 profiles. In addition, the degree of inflammation development in chemically induced models is greatly affected by external factors such as drug concentration, animal species, and gender. Different methodologies also lead to significant differences in the results, and the standardization level is relatively insufficient.
Therefore, based on the limitations of existing UC models, in the future, we need to consider multiple factors and precision when constructing UC pathological models. A more comprehensive modeling approach could combine chemical induction with gene editing and microbiota manipulation to better recapitulate the pathogenic cascade---from epithelial barrier disruption to immune dysregulation and microbial imbalance.
At present, the UC model has laid a certain foundation for exploring the pathogenesis and related drug development, but it still has certain limitations. Further integration of biotechnology—including gene editing, organoid systems, and microbiome transplantation—may improve model fidelity and enable more accurate simulation of UC pathogenesis. This, in turn, could accelerate translation from model systems to clinical therapies.
Conclusion
Against the background of the development of new drugs and the prevention and intervention of such diseases, the development and refinement of UC models remains central to preclinical research and drug discovery. Despite their widespread use in human medicine, these models remain underutilized in veterinary research. Improved model systems can advance translational applications across species.
Author contributions
LY: Data curation, Investigation, Methodology, Writing – original draft. HG: Methodology, Resources, Writing – original draft. DL: Supervision, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by University Basic Scientific Research Business Expenses Project-Young Teachers Research Ability Enhancement Fund Project, 2023 Financial Funds (BR230118), Inner Mongolia Autonomous Region Science and Technology Project (2022YFDZ0051) and the Scientific Research Project of Colleges and Universities in Inner Mongolia Autonomous region (BR22-11-17).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The authors declare that no Gen AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Jiang, X, Chen, X, Dong, R, Wang, J, Pan, Y, and Cao, Y. Establishment of a mouse model of inflammatory bowel disease using dextran sulfate sodium. Adv Clin Exp Med. (2023) 32:563–73. doi: 10.17219/acem/156647
2. Yang, C, and Merlin, D. Unveiling colitis: a journey through the dextran sodiumsulfate-induced model. Inflamm Bowel Dis. (2024) 30:844–53. doi: 10.1093/ibd/izad312
3. Liu, N, Sun, S, Wang, P, Sun, Y, Hu, Q, and Wang, X. The mechanism of secretion and metabolism of gut-derived 5-hydroxytryptamine. Int J Mol Sci. (2021) 22:7931. doi: 10.3390/ijms22157931
4. Yokoi, F, Deguchi, S, Watanabe, Y, and Takayama, K. Establishment of an ulcerative colitis model using colon organoids derived from human induced pluripotent stem cells. iScience. (2024) 27:111049. doi: 10.1016/j.isci.2024.111049
5. Kuenzig, ME, Fung, SG, Marderfeld, L, Mak, JWY, Kaplan, GG, Ng, SC, et al. Twenty-first century trends in the global epidemiology of pediatric-onset inflammatory bowel disease: systematic review. Gastroenterology. (2022) 162:1147–1159.e4. doi: 10.1053/j.gastro.2021.12.282
6. Porter, RJ, Kalla, R, and Ho, GT. Ulcerative colitis: recent advances in the understanding of disease pathogenesis. F1000Res. (2020) 9:F1000 Faculty Rev-294. doi: 10.12688/f1000research.20805.1
7. Sheikh, SZ, Hegazi, RA, Kobayashi, T, Onyiah, JC, Russo, SM, Matsuoka, K, et al. An anti-inflammatory role for carbon monoxide and heme oxygenase-1 in chronic Th2-mediated murine colitis. J Immunol. (2011) 186:5506–13. doi: 10.4049/jimmunol.1002433
8. Tavakoli, P, Vollmer-Conna, U, Hadzi-Pavlovic, D, and Grimm, MC. A review of inflammatory bowel disease: a model of microbial, immune and neuropsychological integration. Public Health Rev. (2021) 42:1603990. doi: 10.3389/phrs.2021.1603990
9. Nishida, A, Inoue, R, Inatomi, O, Bamba, S, Naito, Y, and Andoh, A. Gut microbiota in the pathogenesis of inflammatory bowel disease. Clin J Gastroenterol. (2018) 11:1–10. doi: 10.1007/s12328-017-0813-5
10. Ghouri, YA, Tahan, V, and Shen, B. Secondary causes of inflammatory bowel diseases. World J Gastroenterol. (2020) 26:3998–4017. doi: 10.3748/wjg.v26.i28.3998
11. Gomaa, EZ. Human gut microbiota/microbiome in health and diseases: a review. Antonie Van Leeuwenhoek. (2020) 113:2019–40. doi: 10.1007/s10482-020-01474-7
12. Duvallet, C, Gibbons, SM, Gurry, T, Irizarry, RA, and Alm, EJ. Meta-analysis of gut microbiome studies identifies disease-specific and shared responses. Nat Commun. (2017) 8:1784. doi: 10.1038/s41467-017-01973-8
13. He, C, Wang, H, Liao, W, Liao, WD, Peng, C, Shu, X, et al. Characteristics of mucosa-associated gut microbiota during treatment in Crohn’s disease. World J Gastroenterol. (2019) 25:2204–16. doi: 10.3748/wjg.v25.i18.2204
14. Annese, V. Genetics and epigenetics of IBD. Pharmacol Res. (2020) 159:104892. doi: 10.1016/j.phrs.2020.104892
15. Mayberry, JF, Rhodes, J, and Newcombe, RG. Familial prevalence of inflammatory bowel disease in relatives of patients with crohn’s disease. Br Med J. (1980) 280:84. doi: 10.1136/bmj.280.6207.84
16. Monsén, U, Broström, O, Nordenvall, B, Sörstad, J, and Hellers, G. Prevalence of inflammatory bowel disease among relatives of patients with ulcerative colitis. Scand J Gastroenterol. (1987) 22:214–8. doi: 10.3109/00365528708991882
17. Broekema, RV, Bakker, OB, and Jonkers, IH. A practical view of fine-mapping and gene prioritization in the post-genome-wide association era. Open Biol. (2020) 10:190221. doi: 10.1098/rsob.190221
18. Jans, D, and Cleynen, I. Genetic variation in IBD: progress, clues to pathogenesis and possible clinical utility. Hum Genet. (2023) 142:669–82. doi: 10.1007/s00439-023-02521-9
19. El-Zimaity, H, Shaffer, SR, Riddell, RH, Pai, RK, and Bernstein, CN. Beyond neutrophils for predicting relapse and remission in ulcerative colitis. J Crohns Colitis. (2023) 17:767–76. doi: 10.1093/ecco-jcc/jjac178
20. Islam, SMT, Won, J, Khan, M, Mannie, MD, and Singh, I. Hypoxia-inducible factor-1 drives divergent immunomodulatory functions in the pathogenesis of autoimmune diseases. Immunology. (2021) 164:31–42. doi: 10.1111/imm.13335
21. O’Sullivan, KM, and Holdsworth, SR. Neutrophil extracellular traps: a potential therapeutic target in MPO-ANCA associated casculitis? Front Immunol. (2021) 12:635188. doi: 10.3389/fimmu.2021.635188
22. Taylor, CT, and Colgan, SP. Regulation of immunity and inflammation by hypoxia in immunological niches. Nat Rev Immunol. (2017) 17:774–85. doi: 10.1038/nri.2017.103
23. Angelidou, I, Chrysanthopoulou, A, Mitsios, A, Arelaki, S, Arampatzioglou, A, Kambas, K, et al. REDD1/autophagy pathway is associated with neutrophil-driven IL-1β inflammatory response in active ulcerative colitis. J Immunol. (2018) 200:3950–61. doi: 10.4049/jimmunol.1701643
24. Dinallo, V, Marafini, I, Di Fusco, D, Laudisi, F, Franzè, E, Di Grazia, A, et al. Neutrophil extracellular traps sustain inflammatory signals in ulcerative colitis. J Crohns Colitis. (2019) 13:772–84. doi: 10.1093/ecco-jcc/jjy215
25. Kaur, H, Kasapoglu, M, Yadavalli, R, Nawaz, S, Althwanay, A, AlEdani, E, et al. Inflammatory bowel disease and its association with perinuclear antineutrophil cytoplasmic antibodies: a systematic review. Cureus. (2024) 16:e57872. doi: 10.7759/cureus.57872
26. Friedrich, M, Pohin, M, and Powrie, F. Cytokine networks in the pathophysiology of inflammatory bowel disease. Immunity. (2019) 50:992–1006. doi: 10.1016/j.immuni.2019.03.017
27. Ben-Arosh, H, and Avraham, R. Tissue-specific macrophage immunometabolism. Curr Opin Immunol. (2023) 84:102369. doi: 10.1016/j.coi.2023.102369
28. Na, YR, Stakenborg, M, Seok, SH, and Matteoli, G. Macrophages in intestinal inflammation and resolution: a potential therapeutic target in IBD. Nat Rev Gastroenterol Hepatol. (2019) 16:531–43. doi: 10.1038/s41575-019-0172-4
29. Denson, LA, Curran, M, McGovern, DPB, Koltun, WA, Duerr, RH, Kim, SC, et al. Challenges in IBD research: precision medicine. Inflamm Bowel Dis. (2019) 25:S31–9. doi: 10.1093/ibd/izz078
30. Haberman, Y, Karns, R, Dexheimer, PJ, Schirmer, M, Somekh, J, Jurickova, I, et al. Ulcerative colitis mucosal transcriptomes reveal mitochondriopathy and personalized mechanisms underlying disease severity and treatment response. Nat Commun. (2019) 10:38. doi: 10.1038/s41467-018-07841-3
31. VanPortfliet, JJ, Chute, C, Lei, Y, Shutt, TE, and West, AP. Mitochondrial DNA release and sensing in innate immune responses. Hum Mol Genet. (2024) 33:R80–91. doi: 10.1093/hmg/ddae031
32. Vianello, C, Cocetta, V, Caicci, F, Boldrin, F, Montopoli, M, Martinuzzi, A, et al. Interaction between mitochondrial DNA variants and mitochondria/endoplasmic reticulum contact sites: a perspective review. DNA Cell Biol. (2020) 39:1431–43. doi: 10.1089/dna.2020.5614
33. Ho, GT, Aird, RE, Liu, B, Boyapati, RK, Kennedy, NA, Dorward, DA, et al. MDR1 deficiency impairs mitochondrial homeostasis and promotes intestinal inflammation. Mucosal Immunol. (2018) 11:120–30. doi: 10.1038/mi.2017.31
34. Rath, S, Sharma, R, Gupta, R, Ast, T, Chan, C, Durham, TJ, et al. MitoCarta3.0: an updated mitochondrial proteome now with sub-organelle localization and pathway annotations. Nucleic Acids Res. (2021) 49:D1541–7. doi: 10.1093/nar/gkaa1011
35. Boyapati, RK, Dorward, DA, Tamborska, A, Kalla, R, Ventham, NT, Doherty, MK, et al. Mitochondrial DNA is a pro-inflammatory damage-associated molecular pattern released during active IBD. Inflamm Bowel Dis. (2018) 24:2113–22. doi: 10.1093/ibd/izy095
36. Lan, A, Guerbette, T, Andriamihaja, M, Magnin, B, Bordet, M, Ferron, PJ, et al. Mitochondrial remodeling and energy metabolism adaptations in colonic crypts during spontaneous epithelial repair after colitis induction in mice. Free Radic Biol Med. (2023) 205:224–33. doi: 10.1016/j.freeradbiomed.2023.06.007
37. Pérez-Treviño, P, Velásquez, M, and García, N. Mechanisms of mitochondrial DNA escape and its relationship with different metabolic diseases. Biochim Biophys Acta Mol basis Dis. (2020) 1866:165761. doi: 10.1016/j.bbadis.2020.165761
38. Sange, AH, Srinivas, N, Sarnaik, MK, Modi, S, Pisipati, Y, Vaidya, S, et al. Extra-intestinal manifestations of inflammatory bowel disease. Cureus. (2021) 13:e17187. doi: 10.7759/cureus.17187
39. Kilic, Y, Kamal, S, Jaffar, F, Sriranganathan, D, Quraishi, MN, and Segal, JP. Prevalence of extraintestinal manifestations in inflammatory bowel disease: a systematic review and meta-analysis. Inflamm Bowel Dis. (2024) 30:230–9. doi: 10.1093/ibd/izad061
40. Taurog, JD, Richardson, JA, Croft, JT, Simmons, WA, Zhou, M, Fernández-Sueiro, JL, et al. The germfree state prevents development of gut and joint inflammatory disease in HLA-B27 transgenic rats. J Exp Med. (1994) 180:2359–64. doi: 10.1084/jem.180.6.2359
41. Vavricka, SR, Greuter, T, and Zeitz, J. Extraintestinal manifestations in chronic inflammatory bowel diseases. Ther Umsch. (2019) 75:281–5. doi: 10.1024/0040-5930/a001004
42. Roussomoustakaki, M, Satsangi, J, Welsh, K, Louis, E, Fanning, G, Targan, S, et al. Genetic markers may predict disease behavior in patients with ulcerative colitis. Gastroenterology. (1997) 112:1845–53. doi: 10.1053/gast.1997.v112.pm9178675
43. Baydi, Z, Limami, Y, Khalki, L, Zaid, N, Naya, A, Mtairag, EM, et al. An update of research animal models of inflammatory bowel disease. Sci World J. (2021) 2021:2021: 7479540. doi: 10.1155/2021/7479540
44. Fan, L, Zuo, S, Tan, H, Hu, J, Cheng, J, Wu, Q, et al. Preventive effects of pectin with various degrees of esterification on ulcerative colitis in mice. Food Funct. (2020) 11:2886–97. doi: 10.1039/c9fo03068a
45. Wu, Y, Ran, L, Yang, Y, Gao, X, Peng, M, Liu, S, et al. Deferasirox alleviates DSS-induced ulcerative colitis in mice by inhibiting ferroptosis and improving intestinal microbiota. Life Sci. (2023) 314:121312. doi: 10.1016/j.lfs.2022.121312
46. Cai, W, Pierzynowska, K, Stiernborg, M, Xu, J, Nilsson, IAK, Svensson, U, et al. Multispecies synbiotics alleviate dextran sulfate sodium (DSS)-induced colitis: effects on clinical scores, intestinal pathology, and plasma biomarkers in male and female mice. Clin Nutr ESPEN. (2024) 63:74–83. doi: 10.1016/j.clnesp.2024.06.011
47. Hu, Y, Tang, J, Xie, Y, Xu, W, Zhu, W, Xia, L, et al. Gegen Qinlian decoction ameliorates TNBS-induced ulcerative colitis by regulating Th2/Th1 and Tregs/Th17 cells balance, inhibiting NLRP3 inflammasome activation and reshaping gut microbiota. J Ethnopharmacol. (2024) 328:117956. doi: 10.1016/j.jep.2024.117956
48. Katsandegwaza, B, Horsnell, W, and Smith, K. Inflammatory bowel disease: a review of pre-clinical murine models of human disease. Int J Mol Sci. (2022) 23:9344. doi: 10.3390/ijms23169344
49. Pang, J, Ding, J, Zhang, L, Zhang, Y, Yang, Y, Bai, X, et al. Effect of recombinant serine protease from adult stage of Trichinella spiralis on TNBS-induced experimental colitis in mice. Int Immunopharmacol. (2020) 86:106699. doi: 10.1016/j.intimp.2020.106699
50. Park, SY, Ku, SK, Lee, ES, and Kim, JA. 1,3-Diphenylpropenone ameliorates TNBS-induced rat colitis through suppression of NF-κB activation and IL-8 induction. Chem Biol Interact. (2012) 196:39–49. doi: 10.1016/j.cbi.2012.02.002
51. Isik, F, Akbay, TT, Yarat, A, Tunali Akbay, T, Genc, Z, Pisiriciler, R, et al. Protective effects of black cumin (Nigella sativa) oil on TNBS-induced experimental colitis in rats. Dig Dis Sci. (2011) 56:721–30. doi: 10.1007/s10620-010-1333-z
52. Sharma, TT, Rabizadeh, RR, Prabhakar, VS, Bury, MI, and Sharma, AK. Evolving experimental platforms to evaluate ulcerative colitis. Adv Biol. (2022) 6:e2200018. doi: 10.1002/adbi.202200018
53. Kühn, R, Löhler, J, Rennick, D, Rajewsky, K, and Müller, W. Interleukin-10-deficient mice develop chronic enterocolitis. Cell. (1993) 75:263–74. doi: 10.1016/0092-8674(93)80068-p
54. Liu, M, Yuan, W, and Park, S. Association between IL-10 rs3024505 and susceptibility to inflammatory bowel disease: a systematic review and meta-analysis. Cytokine. (2022) 149:155721. doi: 10.1016/j.cyto.2021.155721
55. Uvarova, AN, Zheremyan, EA, Ustiugova, AS, Murashko, MM, Bogomolova, EA, Demin, DE, et al. Autoimmunity-associated SNP rs3024505 disrupts STAT3 binding in B cells, leading to IL10 dysregulation. Int J Mol Sci. (2024) 25:10196. doi: 10.3390/ijms251810196
56. Anderson, CA, Boucher, G, Lees, CW, Franke, A, D'Amato, M, Taylor, KD, et al. Meta-analysis identifies 29 additional ulcerative colitis risk loci, increasing the number of confirmed associations to 47. Nat Genet. (2011) 43:246–52. doi: 10.1038/ng.764
57. Sluis, MVD, Bouma, J, Vincent, A, van der Sluis, M, Velcich, A, Carraway, KL, et al. Combined defects in epithelial and immunoregulatory factors exacerbate the pathogenesis of inflammation: mucin 2-interleukin 10-deficient mice. Lab Investig. (2008) 88:634–42. doi: 10.1038/labinvest.2008.28
58. Shohan, M, Dehghani, R, Khodadadi, A, Dehnavi, S, Ahmadi, R, Joudaki, N, et al. Interleukin-22 and intestinal homeostasis: protective or destructive? IUBMB Life. (2020) 72:1585–602. doi: 10.1002/iub.2295
59. Dorofeyev, AE, Dorofeyeva, AA, Kiriyan, EA, Rassokhina, OA, and Dynia, YZ. Genetic polymorphism in patients with early and late onset of ulcerative colitis. Wiad Lek. (2020) 73:87–90. doi: 10.36740/WLek202001116
60. Souza, RF, Caetano, MAF, Magalhães, HIR, and Castelucci, P. Study of tumor necrosis factor receptor in the inflammatory bowel disease. World J Gastroenterol. (2023) 29:2733–46. doi: 10.3748/wjg.v29.i18.2733
61. Hu, J, Huang, H, Che, Y, Ding, C, Zhang, L, Wang, Y, et al. Qingchang Huashi formula attenuates DSS-induced colitis in mice by restoring gut microbiota-metabolism homeostasis and goblet cell function. J Ethnopharmacol. (2021) 266:113394. doi: 10.1016/j.jep.2020.113394
62. Wu, M, Wang, Q, Huang, B, Wu, MM, Wang, QM, Huang, BY, et al. Dioscin ameliorates murine ulcerative colitis by regulating macrophage polarization. Pharmacol Res. (2021) 172:105796. doi: 10.1016/j.phrs.2021.105796
63. Al-Bderee, NMH, Al-Saad, NFN, Al-Imari, MJ, Mudhaher Habbeb, S, and Mizal Azoz, A. Genetic polymorphisms of NOD2 and ATG16L1 in different types of digestive tract inflammation. Arch Razi Inst. (2023) 78:493–8. doi: 10.22092/ARI.2022.359754.2473
64. Luo, S, Deng, X, Liu, Q, Pan, Z, Zhao, Z, Zhou, L, et al. Emodin ameliorates ulcerative colitis by the flagellin-TLR5 dependent pathway in mice. Int Immunopharmacol. (2018) 59:269–75. doi: 10.1016/j.intimp.2018.04.010
65. Liang, Z, Hu, X, Lin, R, Tang, Z, Ye, Z, Mao, R, et al. Identification of shared gene signatures and molecular mechanisms between chronic kidney disease and ulcerative colitis. Front Immunol. (2023) 14:1078310. doi: 10.3389/fimmu.2023.1078310
66. Ma, L, Ma, Y, Gao, Q, Liu, S, Zhu, Z, Shi, X, et al. Mulberry leaf lipid nanoparticles: a naturally targeted CRISPR/Cas9 oral delivery platform for alleviation of colon diseases. Small. (2024) 20:e2307247. doi: 10.1002/smll.202307247
67. Cremin, M, Tay, EXY, Ramirez, VT, Murray, K, Nichols, RK, Brust-Mascher, I, et al. TRPV1 controls innate immunity during Citrobacter rodentium enteric infection. PLoS Pathog. (2023) 19:e1011576. doi: 10.1371/journal.ppat.1011576
Keywords: Crohn’s disease, ulcerative colitis, animal models, etiology, pathogenesis
Citation: Yang L, Gao H and Liu D (2025) Advance on establishment of pathological model of ulcerative colitis. Front. Vet. Sci. 12:1618260. doi: 10.3389/fvets.2025.1618260
Edited by:
Matthew Lanza, Penn State Milton S. Hershey Medical Center, United StatesReviewed by:
Baoqing Guo, Iowa State University, United StatesYinghua Zhang, Northeast Agricultural University, China
Copyright © 2025 Yang, Gao and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dacheng Liu, bm1nbGRjQDE2My5jb20=
†These authors have contributed equally to this work