 MingXiang Li
MingXiang Li Min Wang3
Min Wang3 GuiYing Hao
GuiYing Hao- 1College of Animal Science, Xichang University, Xichang, China
- 2Key Laboratory of Animal Epidemic Disease Detection and Prevention in Panxi District, Sichuan, China
- 3Academy of Agricultural Sciences of Liangshan Yi Autonomous Prefecture, Xichang, China
- 4Agricultural and Rural Bureau of Liangshan Yi Autonomous Prefecture, Xichang, China
Introduction: Porcine reproductive and respiratory syndrome (PRRS), caused by porcine reproductive and respiratory syndrome virus (PRRSV), occurs frequently in China, and severely hinders the healthy development of the pig farming industry.
Methods: To determine the genetic diversity and epidemiological characteristics of PRRSV strains in Sichuan Province, we collected 499 clinical samples suspected of PRRSV infection from 101 pig farms in 19 cities from 2023 to 2024.
Results and Discussion: Among the 499 samples, 162 were positive for PRRSV, with a total prevalence of 32.46% according to RT-qPCR. Among the 101 pig farms, 55 were positive farm, resulting in a rate of 54.46%. Further analysis of the complete ORF5 gene sequences of 56 PRRSV strains revealed that they could be classified into six lineages: PRRSV-1, lineage 8 (HP-PRRSV), lineage 5 (Classical PRRSV), lineage 1.8 (NADC30-like strain), lineage 1.5 (NADC34-like strain), and lineage 3.5 (QYYZ-like strain). Notably, both the lineage 8 and PRRSV-1 strain were detected in the same sample, indicating the presence of mixed infection. This study revealed the coexistence of multiple lineages of PRRSV in Sichuan Province, with the lineage 1.8 emerging as the predominant epidemic lineage. The concurrent prevalence of multiple lineages underscores the importance of selecting matching vaccines on the basis of locally prevalent strains and the need for continuous epidemiological monitoring of PRRSV.
1 Introduction
PRRSV is one of the most economically devastating pathogens in swine production worldwide (1, 2). The disease is highly contagious, and is characterized by reproductive failure in sows (including abortions, stillbirths, and weak neonates) and severe respiratory distress in growing pigs (manifesting as dyspnea, fever, and high mortality) (3, 4). Despite extensive control efforts, PRRSV remains difficult to manage due to its rapid genetic evolution, sophisticated immune evasion mechanisms, and the limited cross-protection offered by current vaccines, including modified live virus (MLV) strains (5).
PRRSV is an enveloped, positive-sense RNA virus belonging to the order Nidovirales, family Arteriviridae, with a genome of approximately 15 kb (5–8). The viral genome contains at least 10 open reading frames (ORFs), among which ORF5 encodes the major envelope glycoprotein GP5, a highly variable antigenic target that serves as a key marker for PRRSV genotyping and molecular epidemiology (9, 10). Consequently, ORF5 sequence analysis has become a cornerstone for tracking PRRSV genetic diversity and classifying circulating strains (11). PRRSV is divided into two distinct genotypes: PRRSV-1 (European, Type I) and PRRSV-2 (North American, Type II), each genotype containing multiple genetic lineages (12, 13). The emergence of highly pathogenic PRRSV (HP-PRRSV, lineage 8) in 2006, marked by a 30-amino-acid deletion in non-structural proteins 2 (NSP2), triggered a nationwide outbreak with abortion rates of 30–50% in sows and mortality rates of 20–100% in piglets (14, 15). Currently, PRRSV-2 is phylogenetically classified into five major lineages in China: lineage 1.8 (NADC30-like), lineage 1.5 (NADC34-like), lineage 3 (QYYZ and GM2), lineage 5 (VR2332 and BJ-4), and lineage 8 (CH-1a and JXA1) (16, 17). The epidemiology of PRRSV in China has undergone significant shifts over time. Recent surveillance data indicate that PRRSV-2 still predominates, with lineage 1.5 and 1.8 remaining the most prevalent (18).
In contrast, PRRSV-1 strains, although less common, have shown increasing genetic diversification in China. These strains are classified into four lineages: BJEU06-1-like, Amervac-like, HKEU16-like, and NMEU09-1-like, with BJEU06-1-like emerging as the dominant lineage (58.1% by ORF5 analysis; 60% by whole-genome analysis) (19–21). Notably, three new PRRSV-1 lineages have been identified since 2018, suggesting accelerated viral evolution in recent years. While PRRSV-1 is traditionally considered less virulent, recent evidence indicates increasing pathogenicity, necessitating close monitoring (22).
Given the enduring threat posed by PRRSV to swine production, this study investigated the current molecular epidemiology of PRRSV in Sichuan Province from 2023 to 2024. We screened 499 clinical samples by RT-qPCR and sequenced the ORF 5 gene of 56 representative PRRSV-positive isolates. Through phylogenetic and molecular analyses, we evaluated the genetic diversity and evolutionary patterns of the virus. Our results offer valuable insights for improving regional strategies to control PRRSV.
2 Materials and methods
2.1 Sample collection and processing
From 2023 to 2024, we collected 499 clinical samples (lung tissue, lymph nodes, placental tissue, and whole blood) from 101 swine farms across 19 prefecture-level cities in Sichuan Province (Bazhong City, Chengdu City, Dazhou City, Deyang City, Ganzi Prefecture, Guang’an City, Guangyuan City, Leshan City, Liangshan Prefecture, Luzhou City, Meishan City, Mianyang City, Nanchong City, Neijiang City, Suining City, Ya’an City, Yibin City, Zigong City, and Ziyang City). All samples were collected from pigs exhibiting clinical symptoms of PRRSV infection, including elevated body temperature (>40°C) and reduced appetite. The samples were transported on ice and maintained under cold chain conditions until processing (see Supplementary Table S1).
For preparation, 0.5 g of each individual tissue sample was homogenized in sterile PBS (1:5 w/v) in 1.5 mL microcentrifuge tubes, subjected to three freeze–thaw cycles (−80°C), and centrifuged at 12,000×g for 10 min at 4°C. The clarified supernatant was either processed immediately or stored at −80°C.
2.2 RNA extraction and cDNA synthesis
Total RNA was extracted using the TRIzol reagent (Life Technologies, Carlsbad, USA). The RNA quality was assessed by spectrophotometry (NanoDrop), with A260/A280 ratios ranging from 1.8–2.0 considered acceptable. First-strand cDNA was synthesized from 500 ng of total RNA using random priming (Vazyme, Nanjing, CN), following the manufacturer’s protocol. The cDNA products were stored at −20°C until further use. For comparison, the live attenuated vaccine strain TJM-F92 (Zoetis Biology Co., Ltd., Girona, Spain) and PBS were used as positive and negative controls, respectively.
2.3 PRRSV detection and amplification of the complete ORF5 gene
To detect PRRSV in clinical samples, real-time PCR was performed using the detection kit for PRRS produced by Vazyme Biotech Co., Ltd. (Vazyme, Nanjing, China), following the manufacturer’s instructions. Amplification was carried out using the LightCycler 96 Real-Time PCR Detection System (LightCycler® 96 System, Roche, USA).
The primers used to amplify complete ORF5 gene are summarized in Table 1. The amplification reaction system (50 μL in the total system) was constructed as follows: 25 μL of 2 × Phanta Flash Master Mix (Dye Plus) (Vazyme, Nanjing, CN), 2.5 μL of forward and reverse primers, 5 μL of cDNA template, and 15 μL of DEPC water. Amplification was performed under the following reaction conditions: 5 min at 94°C for 1 cycle; 30 cycles of 30 s at 94°C, 30 s at 58°C, and 1 min at 72°C; 10 min of incubation at 72°C for 1 cycle; and a final hold at 4°C. The PCR products were purified using a gel extraction kit (Vazyme, Nanjing, China), then cloned into the pUC-19 vector (Vazyme, Nanjing, China) following the manufacturer’s protocol, and subsequently transformed into DH5α competent cells (Vazyme, Nanjing, China). Three clones of each PCR product were then analyzed via Sanger sequencing by Sangon BioTech Co., Ltd. (Chengdu, CN). Finally, the assembled sequences were generated utilizing SeqMan software (Version 7.0; DNASTAR, USA).
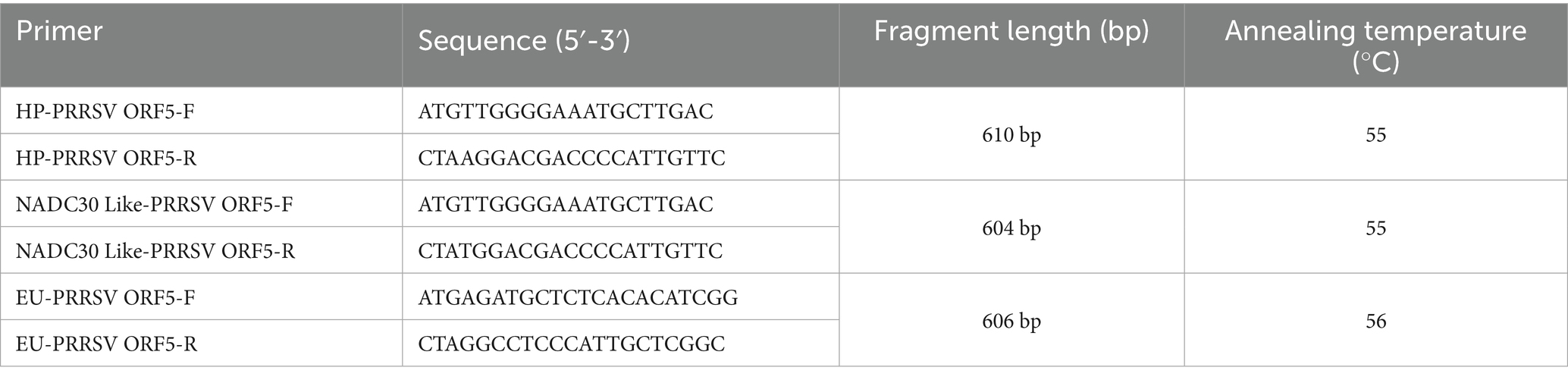
Table 1. ORF5 whole gene amplification primers.
2.4 Phylogenetic and molecular characterization
For comparative analysis, we included 56 representative PRRSV reference sequences retrieved from the GenBank database. Detailed information is provided in Table 2. Sequence alignment was performed using a two-step approach: (1) Nucleotide (nt) and deduced amino acid (aa) sequences were initially aligned using the Clustal W algorithm implemented in MegAlign (DNASTAR), followed by (2) manual refinement to ensure proper codon alignment. Phylogenetic reconstruction was conducted using Molecular Evolutionary Genetics Analysis (MEGA) software version 7.0, employing the maximum likelihood method with the General Time Reversible model incorporating gamma distribution and invariant sites (GTR + G + I), which was determined as the optimal substitution model through model testing. Branch support was assessed with 1,000 bootstrap replicates. The resulting phylogenetic tree was visualized and annotated using iTOL v6.9.1 (Interactive Tree of Life). For structural characterization, the GP5 protein sequences were analyzed using the ClustalW program in DNAStar v7.0 to identify conserved domains and mutation patterns.
Table 2. The reference strain information of PRRSV.
3 Results
3.1 The lineage 1.8 is the dominant epidemic lineage in the Sichuan Province from 2023 to 2024
This study examined 499 clinical samples and detected a total of 162 PRRSV-positive samples with an overall prevalence of 32.46%. The prevalence was 34.11% (88/258) in 2023 and 30.70% (74/241) in 2024, showing a slight downward trend. Regional distribution analysis (Table 3, Figure 1) revealed significant differences in prevalence among 19 prefectures, specifically, Bazhong City (17.65%), Chengdu City (34.09%), Dazhou City (26.32%), Deyang City (32.00%), Ganzi Prefecture (18.18%), Guang’an City (36.36%), Guangyuan City (25.93%), Leshan City (30.30%), Liangshan Prefecture (11.76%), Luzhou City (38.24%), Meishan City (41.46%), Mianyang City (34.38%), Nanchong City (28.00%), Neijiang City (31.03%), Suining City (45.00%), Ya’an City (37.50%), Yibin City (37.84%), Zigong City (31.58%), and Ziyang City (30.43%).
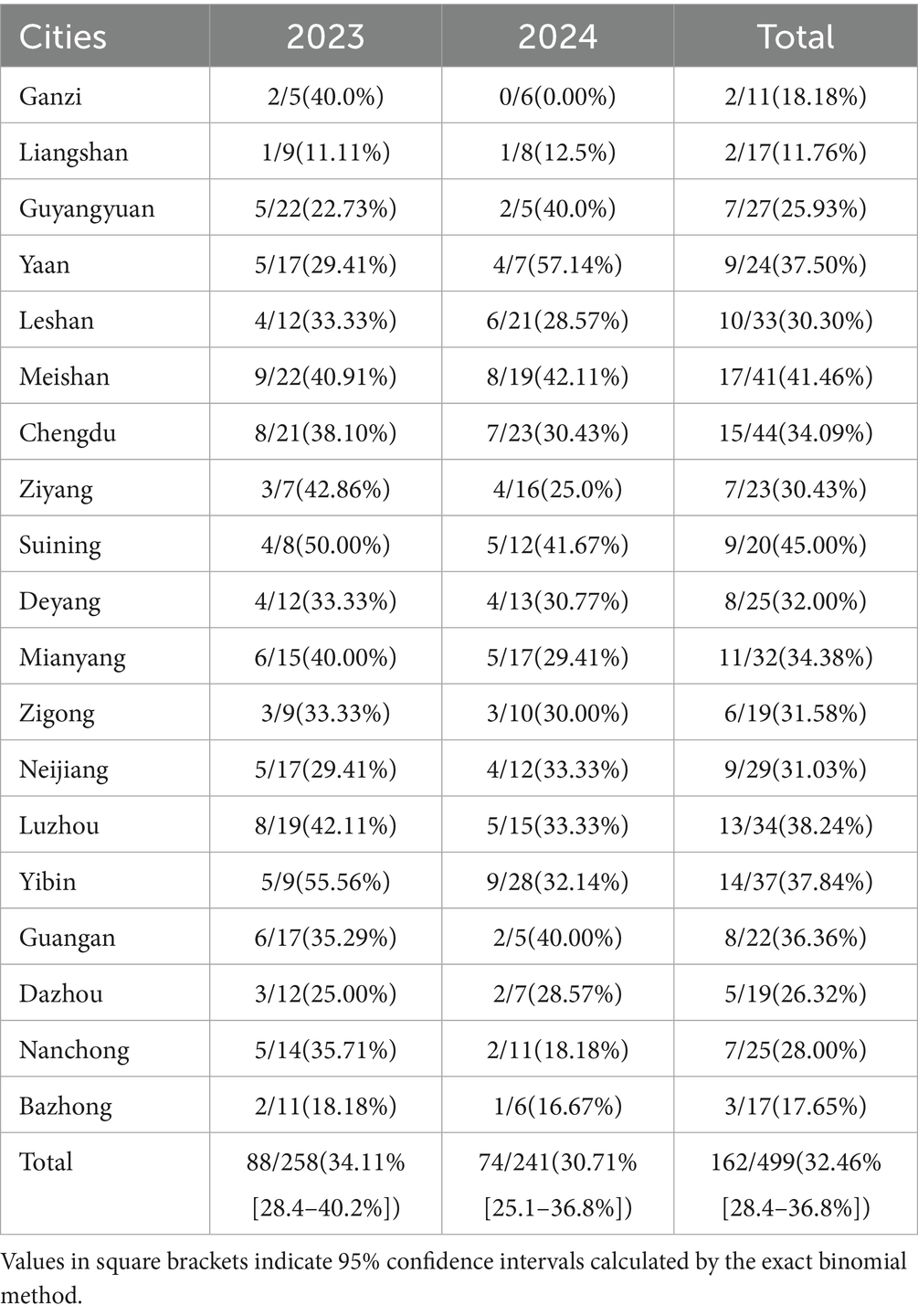
Table 3. Information on samples collected in Sichuan province between 2023 and 2024 and results of PRRSV detection.
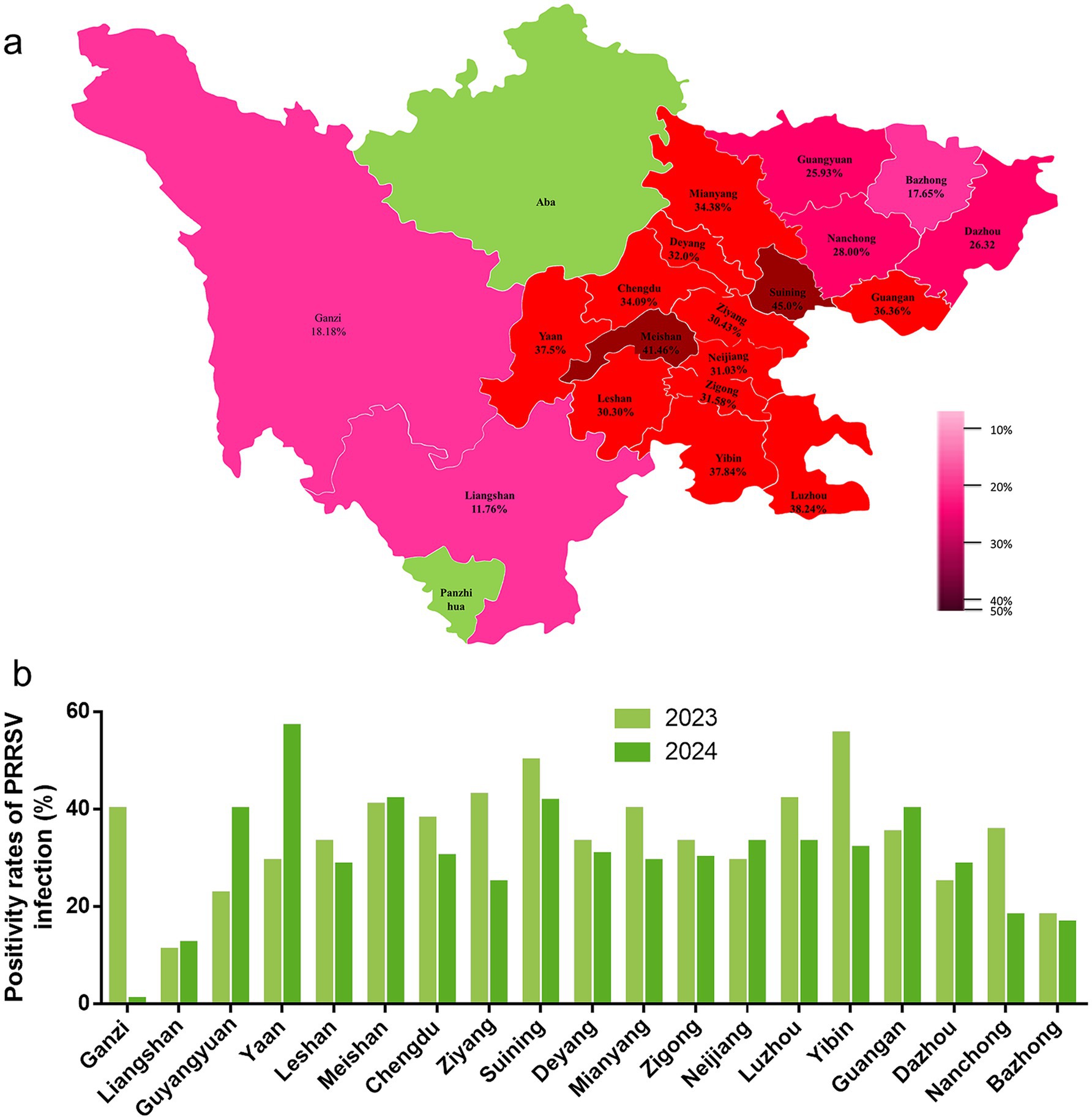
Figure 1. Distribution of PRRSV detection rate in Sichuan Province. (a) The distribution of PRRSV detection rates across 19 regions in Sichuan Province from 2023 to 2024. The pink areas represent the regions involved in testing, while the green areas indicate those not participating in the detection, and the numbers indicate the PRRSV prevalence in different cities. (b) The prevalence from 2023 to 2024 in 19 regions, where light green represents 2023 and dark green represents 2024.
In terms of seasonal epidemic characteristics, the prevalence in spring 2024 increased slightly compared with that in 2023, but it was lower in summer and autumn. Analysis of prevalence among different pig populations showed that weaning piglets had the highest prevalence (>40%), followed by growers and finishers pigs (16–20%), whereas no positive samples were detected in boar semen. Among the 101 tested pig farms, 55 were positive for PRRSV, with a farm-level prevalence of 54.46% (Figure 2).
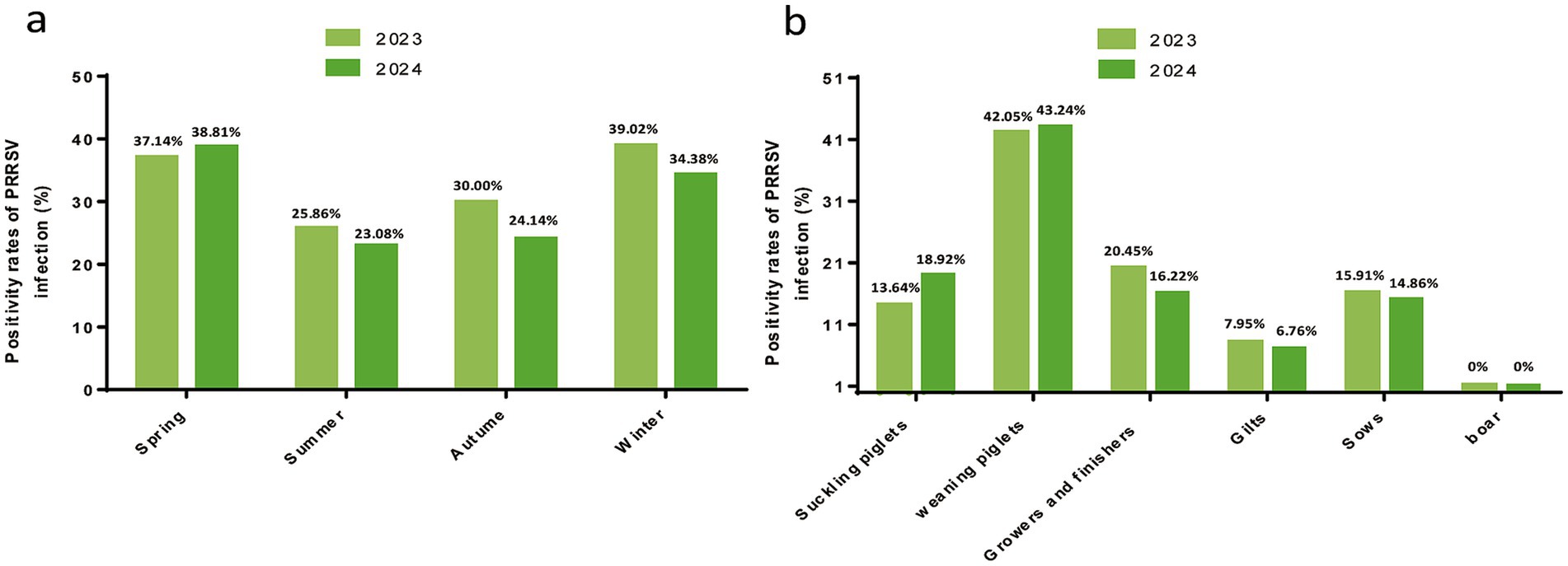
Figure 2. Detection of PRRSV infection in pigs in Sichuan Province. (a) PRRSV positivity rates in different seasons. Seasons were defined as winter (December–February), spring (March–May), summer (June–August), and autumn (September–November). The corresponding prevalences were labeled in the histogram. (b) PRRSV positivity in pigs at different swine herds.
The results of the genotype analysis revealed that among the 162 positive samples, the NADC30-like strain accounted for the highest proportion (44.44%, 72/162), followed by the classical PRRSV strain (23.46%, 38/162). The detection rates of HP-PRRSV (11.73%, 19/162), the NADC34-like strain (6.79%, 11/162), and the recombinant QYYZ-like strain (5.56%, 9/162) were relatively low. Notably that PRRSV-1 was detected only sporadically in local areas, with a prevalence of 8.02% (13/162). In particular, both the HP-PRRSV and PRRSV-1 were detected simultaneously in the same sample from a diseased pig farm, suggesting the presence of mixed infection. Epidemiological distribution characteristics revealed the coexistence of multiple strains and mixed infections with different genotypes, with the NADC30-like strain (lineage 1.8) being the predominant epidemic strain, and PRRSV-1 infections exhibiting sporadic characteristics.
3.2 Sequencing and phylogenetic analysis
To delve deeper into the genetic variation characteristics of PRRSV from 2023 to 2024, this study selected 1–2 positive samples from each of 55 PRRSV-positive farms for sequencing. As a result, 56 high-quality sequences were obtained for genetic evolution analysis, including 6 sequences of PRRSV-1 strains and 50 sequences of PRRSV-2 strains.
Analysis of the ORF5 sequences of the 6 PRRSV-1 strains (with a gene length of 606 bp) revealed nt homology of 94.9–99.7% among the strains. The nt homologies with the reference strains BJEU06 and NMEU09 were 85.3–85.5% and 83.8–84.3%, respectively, and 83.8–84.8% with previously isolated strains from Sichuan. Phylogenetic tree analysis indicated that the strains isolated in this study clustered with the early Sichuan isolate SC-2020-1, forming a new subgroup (Figure 3b).
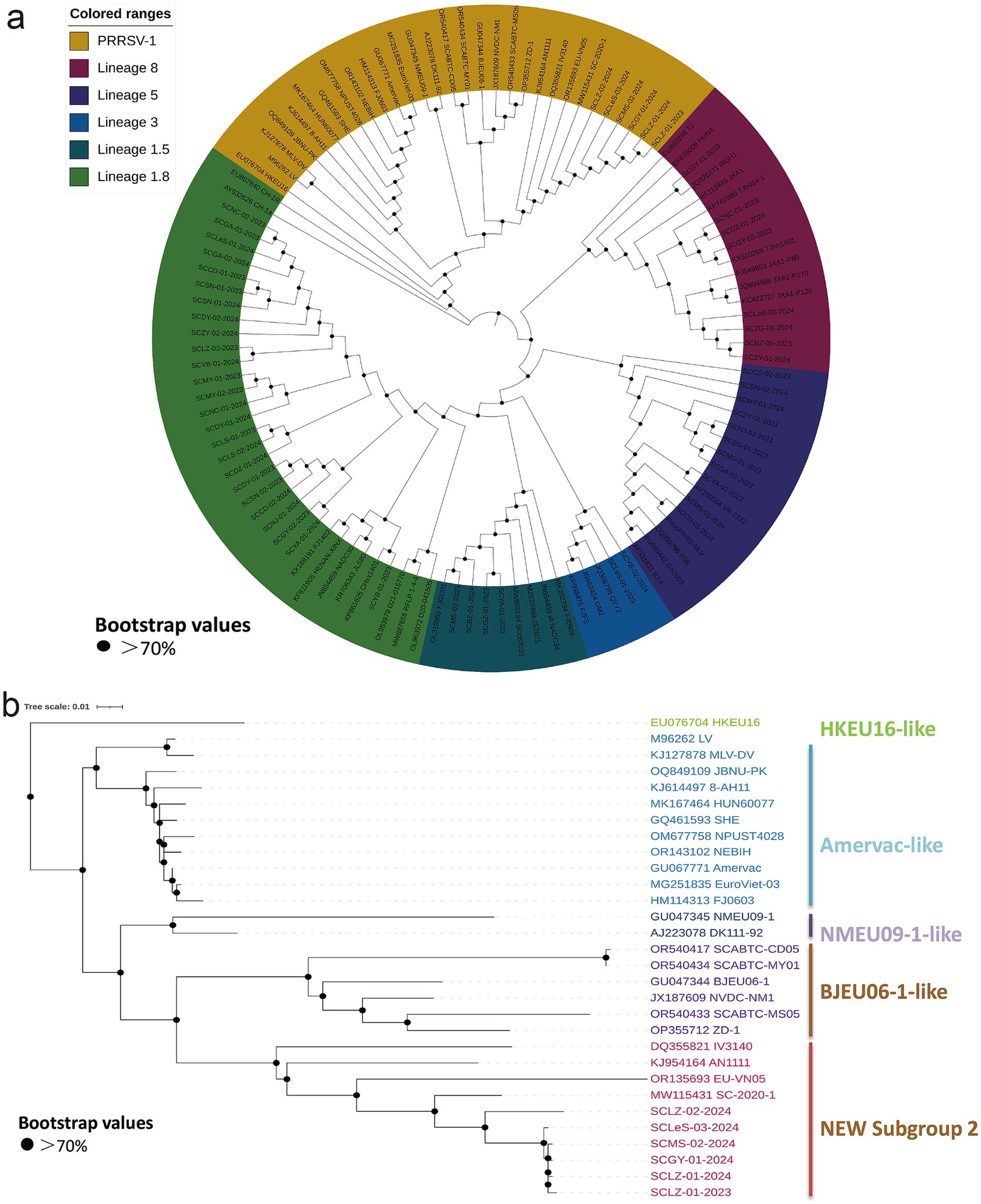
Figure 3. Phylogenetic analysis based on PRRSV ORF5 gene sequences. The phylogenetic tree was constructed using the Neighbor-Joining method in MEGA 7 software. The GTR + G + I model was adopted as the best-fit substitution model, and 1,000 bootstrap replicates were performed. (a) Genetic evolution analysis was conducted on 56 sequences obtained in this study and reference sequences, classifying PRRSV into six categories: PRRSV-1 (brown), lineage 1.8 (light green), lineage 1.5 (light blue), lineage 3 (dark blue), lineage 5 (purple), and lineage 8 (pink). (b) Evolutionary analysis of six PRRSV-1 sequences obtained in this study and reference strains revealed that the six sequences formed a new lineage.
The remaining 50 strains all belonged to PRRSV-2, with nt lengths ranging from 600 to 603 bp. Specifically, 11 strains clustered with the Classical strain, sharing a nt homology of 97.2–100%. Eight sequences grouped with the HP-PRRSV strains, presented nt homology of 98–99.5%. Twenty-five strains clustered with NADC30-like strains, exhibiting nucleotide (nt) homology ranging from 86.4 to 100%. Notably, 15 out of 25 strains harbored a three-nucleotide deletion at positions 94–96 in their sequences. Four strains were grouped with NADC34-like strain, with nt homology ranging from 93.9 to 96.2%. Finally, two strains clustered with the recombinant strain QYYZ-like, demonstrating nt homology of 89.2–92% (Figure 3a).
3.3 Deduced amino acid sequence analysis of the GP5 protein
First, we analyzed the aa homology of six sequences of PRRSV-1with reference strains. The homology among these six sequences ranged from 92.1 to 99%, whereas the homology with reference strains was between 85.1 and 91.6% (Figure 4a). Among them, SCLZ-02-2024 presented the highest homology with SC-2020-1. Furthermore, by comparing the aa sequences of the six PRRSV-1 sequences with those of representative strains from Sichuan (SCABTC-MY01, SCABTC-CD05, SCABTC-MS05, and SC-2020-1), we identified notable variations clustered in three key regions. Specifically, in the signal peptide region, there are seven characteristic mutation sites: 6 K → 6 T, 7 L → 7S/L, 16C → 16Y, 17F → 17C, 20 L → 20F, 23 L → 23S, and 28S → 28F. Within Hypervariable Region 1 (HVR1), a mutation was observed at position 56 (N → A). In Hypervariable Region 2 (HVR2), mutations occurred at positions 101 (A/V → I/T) and 103–106 (CDE/N → YGE). Additionally, a significant aa mutation was identified at position 170 (E → D) in the B-cell epitope 3 region. Three characteristic aa mutations were detected in T-cell epitope 2: 116 (A/T → V), 119 (F → L), and 123 (V → I) (Figure 5a).

Figure 4. Analysis of PRRSV aa homology differences using the MegAlign program in DNAstar software; (a) The aa homology of PRRSV-1 GP5 protein; (b) The aa homology of GP5 proteins from different lineages in PRRSV-2.
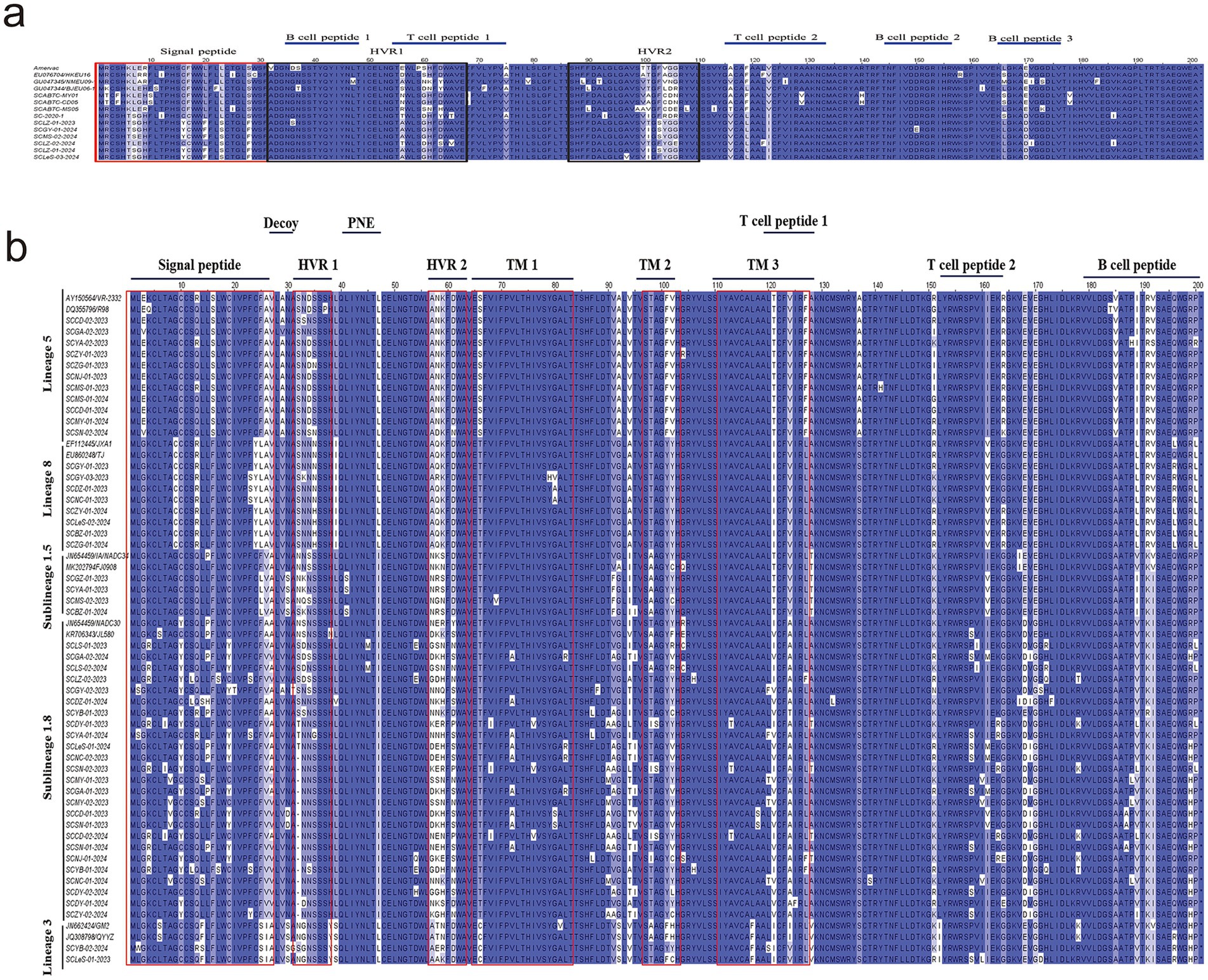
Figure 5. aa analysis of PRRSV GP5 protein (using Jalview-2 for multiple amino acid sequence alignment). (a) The aa variability of PRRSV-1 GP5 protein was analyzed, where the red box indicates the signal peptide region, the black box marks the hypervariable regions 1 and 2, and the T-cell and B-cell epitopes are identified with blue lines on the graph; (b) The aa variability of PRRSV-1 GP5 protein was conducted. The red boxes identify the signal peptide region, hypervariable regions 1 and 2, and transmembrane regions 1, 2, and 3. The T-cell and B-cell epitopes are labeled above the figure.
Regarding the diversity of PRRSV-2 strains, we conducted aa mutation analysis for different lineages separately. Within lineage 1.5, the aa homology between the reference strain sequence and the isolated strain ranged from 91 to 93.5% (Figure 4b). Notably, there was an aa mutation at position 25 (F → L) in the signal peptide region, and two aa mutations at positions 189 (V → I) and 192 (V → I) in the B-cell epitope (Figure 5b).
In lineage 1.8, the strains isolated in this study shared 84.1–93% aa homology with those of NADC30 and JL580 (Figure 4b). There were 78 aa mutation sites, with 15 strains showing a deletion of one aa at position 32. Mutations were present at positions 4 (K → R), 15 (P → L), 19 (C → Y), and 26 (A → V) in the signal peptide, as well as at positions 151 (K → R) and 159 (V → I) in T-cell epitope 2. Additionally, there were five aa mutations in the B-cell epitope located at positions 187 (T → A), 188 (P → L), 189 (V → L), 199 (R → H), and 200 (P → L) (Figure 5b).
Within lineage 3, the aa homology between the isolated strains and the reference strains ranged from 92.5 to 94% (Figure 4b). The analysis of aa differences revealed mutations in both strains. Specifically, in T-cell epitope 1, there was a mutation at aa site 124 (I → V). For T-cell epitope 2, the SCLeS-01-2023 strain presented mutations at positions 151 (K → R) and 158 (P → S), whereas the SCYB-02-2024 strain presented three unique mutation sites in the signal peptide region at positions 2 (L → W) and 13–14 (QF → RS) (Figure 5b).
In lineage 5, the strains in this study shared 95–99% aa homology with those of the reference strain, but they exhibited the fewest aa mutation sites. A prominent mutation occurred at position 151 (R/G → I) within T-cell epitope 2. Compared with the reference strains TJ and WUH1 in lineage 8, the eight strains in this study shared the homology of 95.9–99.1% (Figure 4b). Additionally, there were relatively few aa mutation sites. Specifically, within the signal peptide region, only the SCGY-03-2023, SCDZ-01-2023, and SCNC-01-2023 strains exhibited mutations. In addition, the mutation was observed at the 23rd aa position (F → S), a mutation at the T-cell epitope 2 position 151 (R → K), and individual aa mutations at the B-cell epitope 196 (Figure 5b).
Furthermore, we compared five lineages within PRRSV-2 and identified specific mutation sites. Lineage 1 was characterized by a distinct mutation at position 47 (I → L) compared with other genealogies. Lineage 3 contained multiple aa mutations that differ from other genealogies, including mutations at positions 25 (S → F/L), 26 (I → A/V), 39 (S → L/I), 66 (C → T/S), 92 (S → A/G), 117 (F → L), and 128 (V → T/A). In lineage 5, aa mutations at positions 16 (S → F), 66 (S → T), 137 (A → S), and 185 (V → A) differed from those in the other four lineages. Lineage 8 differed primarily from other lineages at aa position 9 (C → G) (Figure 5b).
4 Discussion
PRRS is an infectious disease that has the greatest impact on the economic benefits of the pig farming industry in China (4, 23). It has been prevalent for more than 20 years. Despite widespread vaccination efforts, the emergence of new strains due to their extreme variability, high genetic diversity, and genetic recombination has weakened the cross-protection effects of conventional vaccines, leading to the widespread and difficult-to-control prevalence of PRRSV in China. Long-term epidemiological investigations enable the dynamic monitoring and tracking of genetic variations in PRRSV, facilitating the development of targeted prevention strategies to control the spread and transmission of the virus. This study analyzed the prevalence and strain variation characteristics of PRRSV in pig populations based on the ORF5 gene from 19 regions of Sichuan Province between 2023 and 2024. The results provide insights into the latest genetic variations in PRRSV and inform more reasonable measures to control PRRSV.
In this study, among the 499 clinical samples collected, 162 were positive for PRRSV, with a prevalence of 32.46% (162/499). This rate is lower than the previously reported PRRSV prevalence in Southwest China from 2012 to 2020 and from 2021 to 2023 (52.32%, 282/539; 39.74%, 643/1618) (24, 25), indicating a downward trend in the prevalence of PRRSV in this region in recent years. Notably, PRRSV also has a relatively low prevalence in other regions of China. For example, it is reported that the PRRSV prevalence on farms in Guangxi was 19.57% (3); in Henan and Shanxi, the prevalence was only 16.99% (26). A study conducted in Central China reported a prevalence of 23.98% (27); the prevalence in Northern China was only 25.26% (28), while the prevalence in the Northeast was ranged from 17.54 to 53.33% (29). These data indicate that the prevalence of PRRSV differs across regions and that prevalence vigilance and strengthened monitoring are needed to control its transmission.
The analysis of seasonal patterns revealed that PRRSV is detectable year-round, with winter showing the highest prevalence at 39.02% in 2023, followed by spring at 38.81% in 2024. This epidemiological trend aligns with previous research findings (30), although it differs from a study that identified spring as the season with the highest prevalence (31). PRRSV outbreaks, while not strictly seasonal, often correlate with specific seasons. Colder months, particularly winter and early spring, are associated with increased viral activity and prevalence. This seasonal variation could be influenced by environmental conditions, farming practices, and viral stability across temperatures. Colder weather enhances the environmental stability of the virus and may increase pig stress levels, potentially increasing their susceptibility to infection. Low temperatures also favor virus transmission (32).
When considering different pig stages, weaning piglets exhibited the highest prevalence at 42.59% (69/162), with no positive cases detected among boars and a prevalence of 7.41% (12/162) among gilts. The elevated prevalence in nursery pigs can be attributed to two primary factors: increased infection risk due to waning maternally derived antibodies and immature cellular immunity, peaking 3–6 weeks postweaning, and persistent PRRSV infection resulting from high-density housing and poor ventilation in nursery facilities. Conversely, the low prevalence among replacement sows is attributed to the acclimatization of incoming PRRSV strains and vaccination, which reduces transmission risk within the herd. These findings underscore the increased susceptibility of piglets and nursery pigs, highlighting the need for enhanced protective measures during these critical stages. Recent studies have indicated that the “All In/All Out” (AIAO) weaning strategy can effectively reduce the circulation of PRRSV during the nursery phase, possibly by decreasing cross-infection among pigs of different ages. Delayed weaning or mixing of piglets (such as keeping frail piglets and mixing them with newly weaned piglets) significantly increases the likelihood of virus transmission (33). It is worth noting that there are variations in prevalence rates across different regions, with lower rates observed in mountainous areas primarily due to the effect of geographical isolation.
The phylogenetic tree constructed on the basis of PRRSV-1 and PRRSV-2 sequences (Figure 3) demonstrated that 50 PRRSV-2 strains from Sichuan Province can be classified into five lineages (lineages 1.5, 1.8, 3, 5, and 8). Among these lineages, lineage 1.8 (NADC30-like strain) represents the dominant circulating lineage (accounting for 42.86%), which aligns with the recent PRRSV epidemic trend in China. The characteristic 131-aa deletion in the nsp2 protein of this gene may enhance its replicative fitness (21). The co-circulation of multiple genes is consistent with epidemiological reports from other regions of China and globally (34, 35). Furthermore, within the same region, the prevalent virus strains are gradually changing. From 2012 to 2020, lineage 8 were dominant in Sichuan (24), whereas a notable transition to lineage 1.8 occurred between 2021 and 2023, indicating a significant alteration in circulating genealogies (25). Recent epidemiological surveys of PRRSV revealed that the prevalent lineage in major pig-farming regions of China have gradually shifted from lineage 8 to lineage 1.8. In a few areas, such as Henan and Shanxi Provinces, the lineages 1.8 and 1.5 respectively, predominantly circulate (26), with multiple lineages co-circulating. In Vietnam, however, the prevalent strain of PRRSV remains primarily lineage 8 (36). These variations may reflect differences in vaccination strategies or swine production systems, warranting further investigation. Unexpectedly, we detected concurrent lineage 8 and PRRSV-1 in one farm, potentially linked to cross-regional swine introduction. Furthermore, studies have reported a higher detection rate of PRRSV-1 in China’s original swine farms than in slaughterhouses, indicating that the source of PRRSV-1 in China may still be linked to breed introduction. These findings suggest two key points: (1) Appropriate measures need to be taken for the prevention and control of PRRSV-1, (2) detection during exotic breed introduction and isolated acclimatization are highly essential. These findings clarify the complex epidemiological landscape of PRRSV, emphasizing the need for vaccine development based on local circulating strain characteristics and the formulation of precise regional control strategies. Continuous molecular epidemiological monitoring can establish a dynamic early warning system for circulating strains, providing a scientific basis for vaccine updates and the optimization of prevention and control systems.
The GP5 protein of PRRSV, which serves as the main target of the host immune response, is crucial for vaccine development and investigations into viral pathogenesis mechanisms (37). This protein contains the most prevalent neutralizing epitopes, encompassing a variety of T-cell (119–127 aa and 151–159 aa) and B-cell epitopes (36–52 aa and 168–198 aa) (38, 39). An examination of the amino acid sequences of the GP5 protein across 56 PRRSV strains revealed 78 mutations in the lineage 1.8 (Figure 5b). Notable differences were found in the areas of the signal peptide, B-cell epitopes, and T-cell epitopes among lineages. Specific mutations can act as molecular markers for lineage differentiation; for example, lineage 5 shows distinct amino acid changes at positions 16 (S → F), 66 (S → T), 137 (A → S), and 185 (V → A) compared with the other four lineages. In lineage 8, the main differentiation from other lineages occurs at amino acid position 9 (C → G). Lineage 1 is defined by a unique mutation at position 47 (I → L) in contrast to other genealogies. Lineage 3 displays several amino acid variations that set it apart from other lineages, including changes at positions 25 (S → F/L), 26 (I → A/V), 39 (S → L/I), 66 (C → T/S), 92 (S → A/G), 117 (F → L), and 128 (V → T/A). Furthermore, the emergence or loss of previously identified N-glycosylation sites on the GP5 protein has been noted. Research has suggested that alterations in these glycosylation sites can either increase or reduce a strain’s virulence (40, 41). In our investigation, 15 sequences exhibited aa deletions at position 32 in lineage 1.8. Nevertheless, earlier studies indicated deletions primarily at positions 33 or 34 (42–45), presenting a discrepancy with our results but agreeing with findings by Zhang (46). The role of these deleted aa in immune evasion and the virulence of the strain is still uncertain, and future research utilizing reverse genetics may be pursued. Furthermore, research has shown that mutations at positions R13 and 151R are associated with increased strain virulence (47). In this investigation, five of sequences exhibited mutations at position 151 in lineage 1.8, whereas no mutations were detected at position 13. Potential effect of these mutation sites on virulence needs further investigation.
In conclusion, we acquired both the prevalence rates and the molecular characteristics of PRRSV across 19 areas within Sichuan Province. At present, the lineage 1.8 continues to be the most common lineage, yet the increasing prevalence of PRRSV-1 required further investigation and ongoing monitoring. Additionally, distinct mutations and deletions in the 32 aa of PRRSV GP5 have been noted various strains, contributing to the genetic variation of PRRSV. These findings provide crucial information and insights that improve our understanding of epidemiology and could assist in understanding the prevalence, genetic features, and the development of detection kits and vaccines, and ultimately help establish effective prevention and control strategies for PRRSV.
Data availability statement
The datasets generated in this study are available and the sequences have been deposited in the GenBank database under the Accession numbers PV832371-PV832426.
Ethics statement
The animal study was approved by the Ethics Committee of Xichang University (Permit Number: XCUSW-2022-006). The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
MXL: Writing – review & editing, Investigation, Writing – original draft, Funding acquisition, Data curation, Formal analysis. MW: Methodology, Investigation, Writing – original draft. HL: Writing – review & editing, Investigation. KLZ: Supervision, Writing – review & editing. ZQH: Writing – original draft, Investigation. WXB: Writing – original draft, Investigation. GYH: Resources, Supervision, Writing – review & editing. GWY: Writing – review & editing, Funding acquisition, Supervision.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by the Liangshan Academic and Technical Leader Training Fund in 2024 (Grant No. NO2024(21)); and the Liangshan Prefecture Science and Technology Program Project (Grant No. 23CGZH0004); and the Open Project of the State Key Laboratory of Animal Biotech Breeding (Grant No. 2025SKLAB6-07); and the Agriculture and Rural Bureau livestock and poultry disease purification demonstration project of Liangshan Prefecture Yi Autonomous Prefecture (Grant No. LSNY2024020).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The authors declare that no Gen AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fvets.2025.1618962/full#supplementary-material
References
1. Zhang, J, Bai, J, Sun, Y, Liu, X, Gao, Y, Wang, X, et al. Comparison of pathogenicity of different subgenotype porcine reproductive and respiratory syndrome viruses isolated in China. Microb Pathog. (2022) 168:105607. doi: 10.1016/j.micpath.2022.105607
2. Nieuwenhuis, N, Duinhof, TF, and van Nes, A. Economic analysis of outbreaks of porcine reproductive and respiratory syndrome virus in nine sow herds. Vet Rec. (2012) 170:225. doi: 10.1136/vr.100101
3. Fang, K, Liu, S, Li, X, Chen, H, and Qian, P. Epidemiological and genetic characteristics of porcine reproductive and respiratory syndrome virus in South China between 2017 and 2021. Front Vet Sci. (2022) 9:853044. doi: 10.3389/fvets.2022.853044
4. Zhang, Z, Li, Z, Li, H, Yang, S, Ren, F, Bian, T, et al. The economic impact of porcine reproductive and respiratory syndrome outbreak in four Chinese farms: based on cost and revenue analysis. Front Vet Sci. (2022) 9:1024720. doi: 10.3389/fvets.2022.1024720
5. Benfield, DA, Nelson, E, Collins, JE, Harris, L, Goyal, SM, Robison, D, et al. Characterization of swine infertility and respiratory syndrome (SIRS) virus (isolate ATCC VR-2332). J Vet Diagn Invest. (1992) 4:127–33. doi: 10.1177/104063879200400202
6. Evans, AB, Loyd, H, Dunkelberger, JR, van Tol, S, Bolton, MJ, Dorman, KS, et al. Antigenic and biological characterization of ORF2-6 variants at early times following PRRSV infection. Viruses. (2017) 9:113. doi: 10.3390/v9050113
7. Kappes, MA, and Faaberg, KS. PRRSV structure, replication and recombination: origin of phenotype and genotype diversity. Virology. (2015) 479-480:475–86. doi: 10.1016/j.virol.2015.02.012
8. Ma, J, Ma, L, Yang, M, Wu, W, Feng, W, and Chen, Z. The function of the PRRSV-host interactions and their effects on viral replication and propagation in antiviral strategies. Vaccines (Basel). (2021) 9:364. doi: 10.3390/vaccines9040364
9. Lunney, JK, Fang, Y, Ladinig, A, Chen, N, Li, Y, Rowland, B, et al. Porcine reproductive and respiratory syndrome virus (PRRSV): pathogenesis and interaction with the immune system. Annu Rev Anim Biosci. (2016) 4:129–54. doi: 10.1146/annurev-animal-022114-111025
10. Do, VT, Dao, HT, and Hahn, TW. Generation of a cold-adapted PRRSV with a nucleotide substitution in the ORF5 and numerous mutations in the hypervariable region of NSP2. J Vet Sci. (2020) 21:e85. doi: 10.4142/jvs.2020.21.e85
11. Luo, Q, Zheng, Y, Zhang, H, Yang, Z, Sha, H, Kong, W, et al. Research progress on glycoprotein 5 of porcine reproductive and respiratory syndrome virus. Animals. (2023) 13:813. doi: 10.3390/ani13050813
12. Brinton, MA, Gulyaeva, AA, Balasuriya, UBR, Dunowska, M, Faaberg, KS, Goldberg, T, et al. ICTV virus taxonomy profile: Arteriviridae 2021. J Gen Virol. (2021) 102:001632. doi: 10.1099/jgv.0.001632
13. Wensvoort, G, Terpstra, C, Pol, JM, ter Laak, EA, Bloemraad, M, de Kluyver, EP, et al. Mystery swine disease in the Netherlands: the isolation of Lelystad virus. Vet Q. (1991) 13:121–30. doi: 10.1080/01652176.1991.9694296
14. Han, GW, Lei, KX, Xu, HL, and He, F. Genetic characterization of a novel recombinant PRRSV2 from lineage 8, 1 and 3 in China with significant variation in replication efficiency and cytopathic effects. Transbound Emerg Dis. (2020) 67:1574–84. doi: 10.1111/tbed.13491
15. Tian, K, Yu, X, Zhao, T, Feng, Y, Cao, Z, Wang, C, et al. Emergence of fatal PRRSV variants: unparalleled outbreaks of atypical PRRS in China and molecular dissection of the unique hallmark. PLoS One. (2007) 2:e526. doi: 10.1371/journal.pone.0000526
16. Yim-Im, W, Anderson, TK, Paploski, IAD, VanderWaal, K, Gauger, P, Krueger, K, et al. Refining PRRSV-2 genetic classification based on global ORF5 sequences and investigation of their geographic distributions and temporal changes. Microbiol Spectr. (2023) 11:e0291623. doi: 10.1128/spectrum.02916-23
17. Chen, N, Ye, M, Huang, Y, Li, S, Xiao, Y, Li, X, et al. Identification of two porcine reproductive and respiratory syndrome virus variants sharing high genomic homology but with distinct virulence. Viruses. (2019) 11:875. doi: 10.3390/v11090875
18. Zhao, HZ, Wang, FX, Han, XY, Guo, H, Liu, CY, Hou, LN, et al. Recent advances in the study of NADC34-like porcine reproductive and respiratory syndrome virus in China. Front Microbiol. (2022) 13:950402. doi: 10.3389/fmicb.2022.950402
19. Gong, B, Xu, H, Sun, Q, Li, C, Xiang, L, Zhao, J, et al. Dissecting genetic diversity and evolutionary trends of Chinese PRRSV-1 based on whole-genome analysis. Transbound Emerg Dis. (2024) 2024:9705539. doi: 10.1155/2024/9705539
20. Chen, N, Liu, Q, Qiao, M, Deng, X, Chen, X, and Sun, M. Whole genome characterization of a novel porcine reproductive and respiratory syndrome virus 1 isolate: genetic evidence for recombination between Amervac vaccine and circulating strains in mainland China. Infect Genet Evol. (2017) 54:308–13. doi: 10.1016/j.meegid.2017.07.024
21. Li, C, Xu, H, Zhao, J, Gong, B, Sun, Q, Xiang, L, et al. Epidemiological investigation and genetic evolutionary analysis of PRRSV-1 on a pig farm in China. Front Microbiol. (2022) 13:1067173. doi: 10.3389/fmicb.2022.1067173
22. Jeong, CG, Nazki, S, Kim, SC, Khatun, A, Noh, YH, Lee, DU, et al. Comparison of the pathogenicity of porcine reproductive and respiratory syndrome virus (PRRSV)-1 and PRRSV-2 in pregnant sows. Arch Virol. (2022) 167:425–39. doi: 10.1007/s00705-021-05303-8
23. Guo, J, Liu, Z, Tong, X, Wang, Z, Xu, S, Chen, Q, et al. Evolutionary dynamics of type 2 porcine reproductive and respiratory syndrome virus by whole-genome analysis. Viruses. (2021) 13:2469. doi: 10.3390/v13122469
24. Zhao, J, Xu, Z, Xu, T, Zhou, Y, Li, J, Deng, H, et al. Molecular characterization of the Nsp2 and ORF5s of PRRSV strains in Sichuan China during 2012-2020. Animals (Basel). (2022) 12:3309. doi: 10.3390/ani12233309
25. Huang, B, Xu, T, Luo, Z, Deng, L, Jian, Z, Lai, S, et al. Prevalence and genetic diversity of PRRSV in Sichuan province of China from 2021 to 2023: evidence of an ongoing epidemic transition. Virology. (2024) 600:110213. doi: 10.1016/j.virol.2024.110213
26. Zhao, YY, Ma, X, Chen, XM, Song, YP, Zheng, LL, Ma, SJ, et al. Molecular detection and genetic characteristics of porcine reproductive and respiratory syndrome virus in Central China. Microb Pathog. (2024) 197:107024. doi: 10.1016/j.micpath.2024.107024
27. Zhai, W, Yu, S, Zhang, P, Lin, Y, Ge, S, Zhang, T, et al. Epidemiology and genetic characteristics of porcine reproductive and respiratory syndrome virus in the Hunan and Hebei provinces of China. Vet Sci. (2023) 10:63. doi: 10.3390/vetsci10010063
28. Jian, Y, Lu, C, Shi, Y, Kong, X, Song, J, and Wang, J. Genetic evolution analysis of PRRSV ORF5 gene in five provinces of northern China in 2024. BMC Vet Res. (2025) 21:242. doi: 10.1186/s12917-025-04679-y
29. Li, C, Zhao, J, Li, W, Xu, H, Gong, B, Sun, Q, et al. Prevalence and genetic evolution of porcine reproductive and respiratory syndrome virus in commercial fattening pig farms in China. Porcine Health Manag. (2024) 10:5. doi: 10.1186/s40813-024-00356-y
30. Tousignant, SJP, Perez, AM, Lowe, JF, Yeske, PE, and Morrison, RB. Temporal and spatial dynamics of porcine reproductive and respiratory syndrome virus infection in the United States. Am J Vet Res. (2015) 76:70–6. doi: 10.2460/ajvr.76.1.70
31. Yuan, N, Yang, Z, Lv, F, Dou, L, Li, X, Zhao, B, et al. Molecular epidemiology and genetic evolution of porcine reproductive and respiratory syndrome virus in northern China during 2021-2023. Viruses. (2025) 17:85. doi: 10.3390/v17010085
32. Lim, S, Perez, AM, and Kanankege, KST. Modeling the seasonal variation of windborne transmission of porcine reproductive and respiratory syndrome virus between swine farms. Viruses. (2023) 15:1765. doi: 10.3390/v15081765
33. Heiselberg, PR, Kristensen, CS, Kvisgaard, LK, and Larsen, LE. Impact of weaning procedures on porcine reproductive and respiratory syndrome virus (PRRSV) circulation in the nursery section. BMC Vet Res. (2025) 21:206. doi: 10.1186/s12917-025-04623-0
34. Li, P, Shen, Y, Wang, T, Li, J, Li, Y, Zhao, Y, et al. Epidemiological survey of PRRS and genetic variation analysis of the ORF5 gene in Shandong Province, 2020-2021. Front Vet Sci. (2022) 9:987667. doi: 10.3389/fvets.2022.987667
35. Yin, B, Qi, S, Sha, W, Qin, H, Liu, L, Yun, J, et al. Molecular characterization of the Nsp2 and ORF5 (ORF5a) genes of PRRSV strains in nine provinces of China during 2016-2018. Front Vet Sci. (2021) 8:605832. doi: 10.3389/fvets.2021.605832
36. Li, G, Li, Y, He, C, Liu, X, Lv, C, Liu, K, et al. Sequence analysis of the GP5 protein of porcine reproductive and respiratory syndrome virus in Vietnam from 2007 to 2023. Front Microbiol. (2024) 15:1475208. doi: 10.3389/fmicb.2024.1475208
37. Fang, L, Jiang, Y, Xiao, S, Niu, C, Zhang, H, and Chen, H. Enhanced immunogenicity of the modified GP5 of porcine reproductive and respiratory syndrome virus. Virus Genes. (2006) 32:5–11. doi: 10.1007/s11262-005-5839-y
38. Ostrowski, M, Galeota, JA, Jar, AM, Platt, KB, Osorio, FA, and Lopez, OJ. Identification of neutralizing and nonneutralizing epitopes in the porcine reproductive and respiratory syndrome virus GP5 ectodomain. J Virol. (2002) 76:4241–50. doi: 10.1128/JVI.76.9.4241-4250.2002
39. Young, JE, Dvorak, CMT, Graham, SP, and Murtaugh, MP. Isolation of porcine reproductive and respiratory syndrome virus GP5-specific, neutralizing monoclonal antibodies from Hyperimmune sows. Front Immunol. (2021) 12:638493. doi: 10.3389/fimmu.2021.638493
40. Akter, F, Roychoudhury, P, Dutta, TK, Subudhi, PK, Kumar, S, Gali, JM, et al. Isolation and molecular characterization of GP5 glycoprotein gene of Betaarterivirus suid 2 from Mizoram, India. Virusdisease. (2021) 32:748–56. doi: 10.1007/s13337-021-00735-x
41. Wei, Z, Lin, T, Sun, L, Li, Y, Wang, X, Gao, F, et al. N-linked glycosylation of GP5 of porcine reproductive and respiratory syndrome virus is critically important for virus replication in vivo. J Virol. (2012) 86:9941–51. doi: 10.1128/JVI.07067-11
42. Zhou, L, Kang, R, Xie, B, Tian, Y, Wu, X, Lv, X, et al. Identification of a novel recombinant type 2 porcine reproductive and respiratory syndrome virus in China. Viruses. (2018) 10:151. doi: 10.3390/v10040151
43. Sun, YF, Yu, H, Jiang, X, Ma, JF, Xu, CQ, Yu, XX, et al. Novel ORF5 deletion of NADC30-like porcine reproductive and respiratory syndrome viruses circulating in northern China from 2016 to 2018. J Vet Diagn Invest. (2020) 32:928–32. doi: 10.1177/1040638720954543
44. Jiang, ZY, Cai, RJ, Li, Y, and Li, CL. Genetic variation analysis of PRRSV GP5 gene in Guangdong Province from 2014 to 2016. GDAgric Sci. (2016) 43:90–5.
45. Fan, YF, Bai, J, and Jiang, P. Genetic variation analysis of GP5 gene of porcine reproductive and respiratory syndrome virus epidemic strain in Shandong province. J Domest Anim Ecol. (2017) 38:63–7.
46. Zhang, J. Genetic variation of GP5 gene of porcine reproductive and respiratory syndrome virus and establishment of double fluorescence quantitative PCR detection method. Ph.D. thesis, Nanjing Agricultural University, Nanjing, China (2020)
Keywords: porcine reproductive and respiratory syndrome virus (PRRSV), phylogenetic analysis, lineage 1.8, PRRSV-1, ORF5
Citation: Li MX, Wang M, Li H, Zhou KL, Hu ZQ, Bai WX, Hao GY and Yan GW (2025) Prevalence and genetic diversity of porcine reproduction and respiration syndrome viruses in Sichuan Province from 2023–2024. Front. Vet. Sci. 12:1618962. doi: 10.3389/fvets.2025.1618962
Edited by:
Camila Hamond, University of Connecticut, United StatesReviewed by:
Jana Kvicerova, University of South Bohemia in České Budějovice, CzechiaMengmeng Zhao, Foshan University, China
Copyright © 2025 Li, Wang, Li, Zhou, Hu, Bai, Hao and Yan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: GuangWen Yan, eWd3ZHdnQDEyNi5jb20=