 Young-Jae Si1†
Young-Jae Si1† Sun-Hak Lee2,3†Dong-Ju Kim1Kwanghee Lee1Min-a Lee1
Sun-Hak Lee2,3†Dong-Ju Kim1Kwanghee Lee1Min-a Lee1 Dong-Yeop Lee2
Dong-Yeop Lee2 Ye-Ram Seo2,3Hyesung Jeong1Suwoong Lee1
Ye-Ram Seo2,3Hyesung Jeong1Suwoong Lee1 Dong-Hun Lee2*
Dong-Hun Lee2*- 1Wildlife Disease Research Team, National Institute of Wildlife Disease Control and Prevention, Gwangju, Republic of Korea
- 2Wildlife Health Laboratory, College of Veterinary Medicine, Konkuk University, Seoul, Republic of Korea
- 3Avian Disease Laboratory, College of Veterinary Medicine, Konkuk University, Seoul, Republic of Korea
1 Introduction
The A/goose/Guangdong/1/1996 (Gs/GD) lineage of highly pathogenic avian influenza (HPAI) viruses initially identified in China in 1996 and have evolved over subsequent decades, characterized by accumulation of point mutations and multiple reassortment events with low pathogenic avian influenza (LPAI) viruses (1, 2). Particularly those of the H5N1 subtype within clade 2.3.4.4b have emerged as a significant threat to poultry, wild birds, and mammals worldwide (3). Since their widespread dissemination in wild bird population in the early 2020s, these viruses have caused multiple sporadic infections in mammals, including small carnivores, marine mammals, cattle, and humans (3–6). These cross-species transmissions, often linked to the consumption of infected birds or exposure to contaminated environments, have raised significant concerns about the zoonotic potential of clade 2.3.4.4b H5N1 and its capacity to evolve into a strain with pandemic potential.
Since its initial detection in 2014 with the H5N8 subtype in wild birds, multiple subtypes of clade 2.3.4.4b highly pathogenic avian influenza viruses (HPAIVs) have caused recurrent outbreaks in South Korea including H5N1, H5N6, and H5N8 (7). Notable epidemics occurred during 2021–2022, with 44 cases primarily of H5N1 identified in wild birds (7), and 2022–2023, with 174 wild bird cases, highlighting its persistent circulation wild waterfowl population (8). These outbreaks underscore the role of wild birds in introduction and spread of the virus across South Korea, posing ongoing challenges to poultry industry and public health. In addition, although HPAIV infection in domestic cats were reported in 2016 and 2023 (9–11), no instances of wild mammalian infection were documented in South Korea during these epidemics.
Here, we report the first documented case of H5N1 HPAI in a wild mammal in South Korea, identified in a leopard cat (Prionailurus bengalensis). On March 18, 2025, a wild leopard cat discovered moribund near a freshwater reservoir in Hwasun County of Jeollanam-do Province and submitted to the National Institute of Wildlife Disease Control and Prevention (NIWDC) of Korea. We isolated the H5N1 virus from this leopard cat, sequenced, and assessed its evolutionary history and molecular markers indicative of mammalian adaptation.
2 Materials and methods
2.1 Sample collection and virus isolation
On March 18, 2025, a wild leopard cat was found moribund near the reservoir in Hwasun County, Jeollanam-do Province, South Korea (GPS coordinates ≈ 35°03′N, 126°59′E). The wild leopard cat transported to the Jeollanamdo wild animal rescue center and died within a few hours. The carcass was submitted to the biosafety level 3 facility of NIWDC and organs including brain, trachea, and lung were collected. Samples were placed in phosphate-buffered saline with 400 mg/ml gentamicin, homogenized by vortexing, and filtered using a 0.45-μm Minisart Syringe Filter (Sartorius, Göttingen, Germany) following centrifugation at 3,000 rpm for 10 min. Filtered supernatant was inoculated into 10-day-old specific-pathogen-free embryonated chicken eggs and incubated at 37°C for 72 h. Allantoic fluids were harvested and tested for hemagglutination activity (HA) using 0.5% chicken red blood cells. RNA was extracted from tissue samples and HA-positive allantoic fluids using the Maxwell RSC simply RNA Tissue Kit (Promega, Madison, WI, USA) and screened for influenza A matrix and H5 genes via real-time reverse transcription-PCR (rRT-PCR), following established protocols (12).
2.2 Whole genome sequencing and sequence analysis
Complementary DNA was synthesized using the SuperScript III First-Strand Synthesis System (Invitrogen, Carlsbad, CA, USA), and the eight gene segments were amplified with AccuPrime Pfx DNA Polymerase (Invitrogen, Carlsbad, CA, USA), as previously described (13). DNA libraries were prepared using the Illumina DNA Prep Kit (Illumina, San Diego, CA, USA) and sequenced on the Illumina MiSeq platform (paired-end 150 bp). Raw reads were trimmed using BBDuk (v38.84) with a minimum quality threshold of 30 (14), assembled de novo with SPAdes (v3.15.5). Trimmed reads were mapped to the top BLAST result from the GISAID EpiFlu database using Minimap2 (v2.24). Consensus sequences were generated using Geneious Prime software and deposited in GISAID Epiflu database (EPI_ISL_20051149).
2.3 Phylogenetic and mutation analysis
The top 250 hits for each segment query were retrieved and sequences with high identity (ranging from 99.5 to 99.9%, depending on the segment) were removed using CD-hit (15). We also included genome sequences of four H5N1 HPAIVs [A/Wild_Duck/Korea/24WF364-8P/2024, A/Eurasian_wigeon/Korea/24WF382-7P/2024, A/Vulture/Korea/24WC103/2024, and A/Bean_goose/Korea/24WC196/2025] which were isolated from wild birds during the winter season of 2024-2025, all of which have been deposited in GISAID with their respective accession numbers (EPI_ISL_20051150, 19832581-19832583). Phylogenetic trees were constructed for each gene segment using RAxML v8.0 with the general time reversible model and 1,000 bootstrap replicates (16). Interactive Tree of Life (iTOL) was used to visualize the tree of each gene (17). A Bayesian relaxed-clock phylogeny of the hemagglutinin (HA) gene was reconstructed using BEAST version 1.10.4 (18), employing the Hasegawa-Kishino-Yano substitution model with an uncorrelated log-normal distribution and a Gaussian Markov Random Field (GMRF) Bayesian skyride coalescent prior (19). The Markov Chain Monte Carlo (MCMC) process was executed in parallel across three chains, each comprising 50 million iterations, with results combined after a 10% burn-in. All parameters achieved effective sample sizes (ESS) >200 and were assessed using TRACER v1.5 (http://tree.bio.ed.ac.uk/software/tracer/) (20). A maximum clade credibility (MCC) tree was generated using TreeAnnotator and visualized with FigTree v1.4.4 (http://tree.bio.ed.ac.uk/software/figtree/).
Molecular markers of mammalian adaptation, pathogenicity, and drug resistance were identified using the FluMut tool (21). In the FluMut analysis, in addition to the sequence isolated from the leopard cat, seven of clade 2.3.4.4b HPAI H5N1 viruses were additionally analyzed including viruses reported in infected mammals in the United States (4), viruses isolated from domestic cats in Korea in 2023 (9), and viruses isolated from wild birds during the 2024–2025 winter season (22).
3 Descriptive results
3.1 Isolation and genome sequencing of the virus
The brain and trachea from the submitted leopard cat tested positive for influenza A virus via chicken embryonated egg inoculation and rRT-PCR. The isolated virus, designated A/Leopard Cat/Korea/24WM130/2025(H5N1) (hereafter 24WM130) yielded 288,814 NGS reads, enabling assembly of complete coding genome sequences across all eight influenza virus segments.
3.2 Genome analysis
The virus was identified as HPAIV based on the presence of multiple basic amino acids at the HA proteolytic cleavage site (PLREKRRKR/G) (23). All gene segments of the 24WM130 virus clustered with clade 2.3.4.4b H5N1 HPAIVs isolated from wild birds in South Korea during the 2024–2025 winter season, showing close genetic relatedness and a likely origin from infected wild birds through predation or scavenging (Supplementary Figure 1). We previously reported two genotypes of H5N1 clade 2.3.4.4b viruses in October 2024, the genotype 1 and 2, represented by A/Northern pintail/Korea/24WC025/2024 virus and A/Mandarin duck/Korea/24WS005-2/2024 virus, respectively (22). Genotype 1 possessed a G2d-lineage HA gene and a genome constellation identical to strains circulating in Japan during 2023–2024. Genotype 2 carried a G2c-lineage HA gene, neuraminidase (NA) and M genes from H5Nx clade 2.3.4.4b viruses circulating in 2022–2024, and internal genes from LPAIVs in the East Asian–Australasian flyway. The 24WM130 virus had a G2d-lineage HA gene, while its remaining segments closely matched those of genotype 2, indicating a reassortant virus derived from early HPAI outbreaks in October 2024 (22) (Figure 1B). These reassortment events are likely driven by the high density and mobility of migratory birds, which promote co-infection and gene exchange. In the Bayesian phylogenetic analysis of the HA gene, the 24WM130 virus clustered with clade 2.3.4.4b H5N1 HPAIVs from wild birds in South Korea during December 2024–February 2025 and supported by a high posterior probability (0.99; Figure 1A). Their tMRCA was estimated to be August 2024 (95% BCI: April 24–November 20, 2024), suggesting that these viruses originated most likely during the late breeding season of waterfowl in Eurasia.
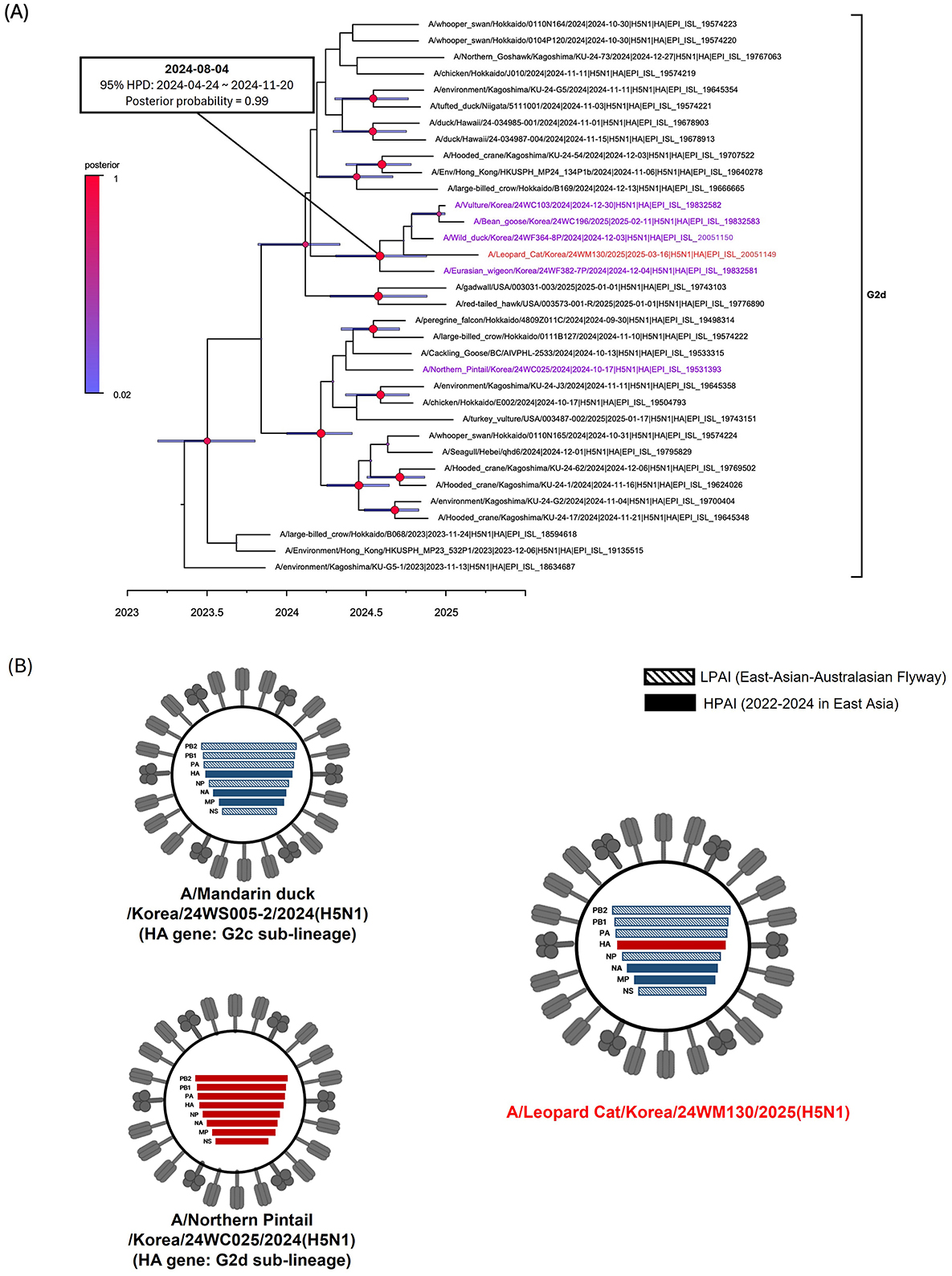
Figure 1. Phylogenetic analysis and genotypes of clade 2.3.4.4b H5N1 HPAI virus found in a wild leopard cat (Prionailurus bengalensis) in South Korea, March 2025. (A) Time-scaled Maximum clade credibility phylogeny constructed using the hemagglutinin gene sequence of G2d subgroup of H5N1 HPAI virus. Red taxa marked with circles represent the H5N1 isolate from the leopard cat in South Korea. Purple taxa indicate the H5N1 HPAI viruses isolated from wild birds during the 2024–2025 winter season. Node bars represent 95% HPD of the node height with a posterior probability >0.5. Node size and color correspond to posterior probability values. The horizontal axis represents the decimal year. The estimated time of the most recent common ancestry with 95% highest posterior density (HPD) is displayed on the node of cluster. (B) Schematic representation of the genotypes of H5N1 viruses from South Korea, 2024–2025. Bars represent eight gene segments of the avian influenza virus in the following order (top to bottom): polymerase basic 2, polymerase basic 1, polymerase acidic, hemagglutinin, nucleoprotein, neuraminidase, matrix, and non-structural. Different bar colors indicate different virus origins estimated from maximum-likelihood phylogenetic trees.
Leopard cat is an endangered, solitary, and opportunistic mesocarnivore species native to South Korea and other parts of Asia. Their solitary lifestyle, coupled with the absence of additional HPAI detections in other mammals, suggests that this outbreak was most likely a sporadic event. The leopard cat's ecological overlap with wild birds and free-ranging mammals raises concerns about potential disease spillovers and their role as an intermediate species in future pandemics (24–26).
Among the 36 mammalian adaptation markers identified in 24WM130, three mutations (Table 1), I292V in PB2, D154N in HA, and D74N in NS, were unique compared to other H5N1 viruses identified in mammals. The I292V in PB2 and D74N in NS substitutions have been associated with increased polymerase activity in mammalian hosts and enhanced virulence in mice. The D154N in HA has been reported to increase binding affinity to α2,6-linked sialic acid receptors (27, 28). Notably, the 24WM130 virus did not possess the E627K or D701N in PB2 which were previously identified in clade 2.3.4.4b HPAI H5N1 viruses isolated from domestic cats in South Korea and M631L in PB2 which was a unique mutation found in dairy cows in the U.S. The HPAI H5N1 viruses from wild birds in Korea during the same wintering season also had 35 mammalian adaptation markers out of 36 detected in the 24WM130, highlighting the high potential for clade 2.3.4.4b H5N1 viruses in wild birds to spillover to mammalian hosts. The D74N substitution, detected exclusively in the 24WM130 virus and absent from closely related avian viruses in Korea during 2024–2025, suggests it may have emerged in the leopard cat.
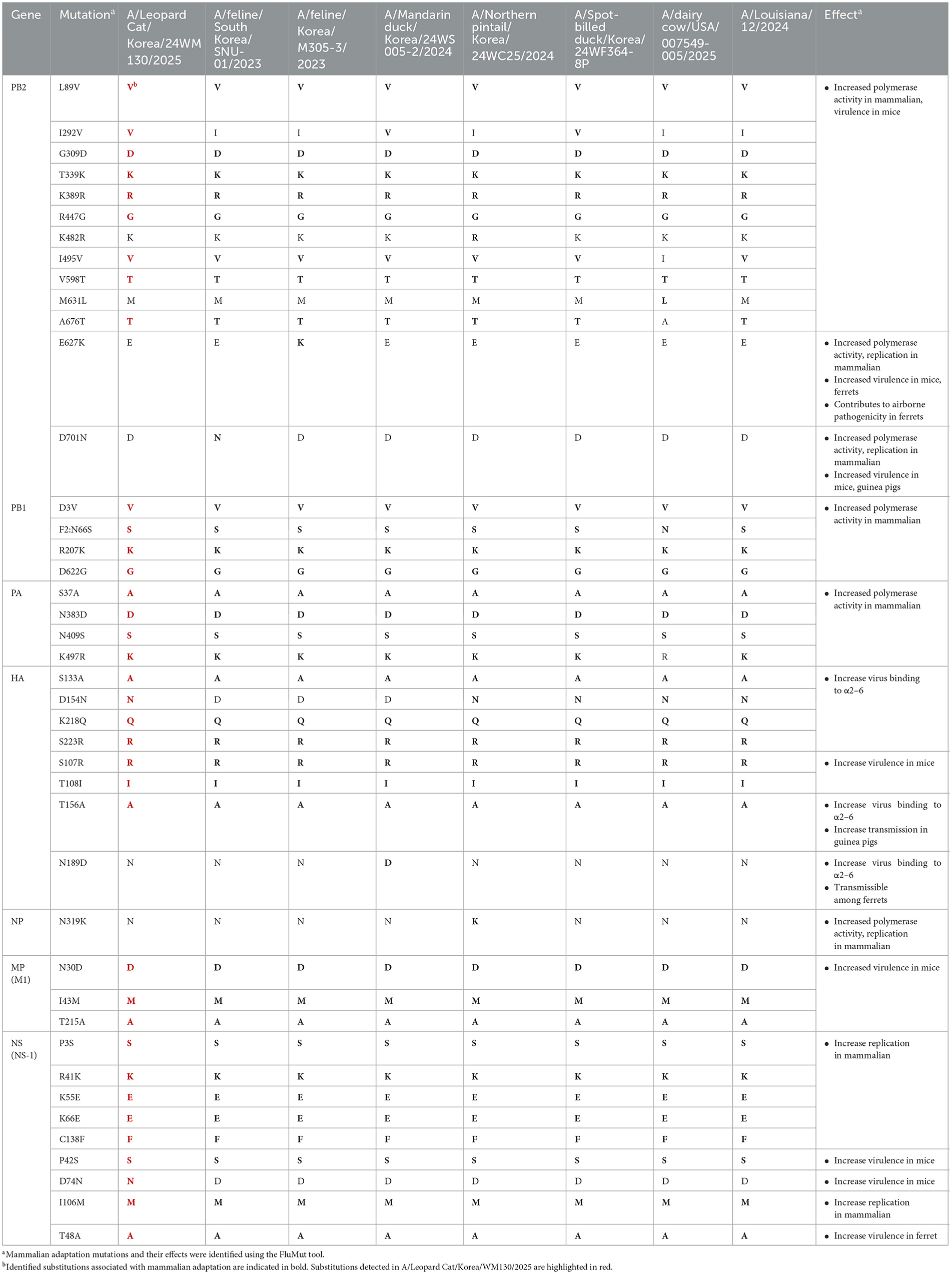
Table 1. Comparison of mammalian adaptation markers among A/Leopard Cat/Korea/WM130/2025, other mammalian-derived strains, and clade 2.3.4.4b highly pathogenic H5N1 viruses isolated from Korean wild birds in the same season.
4 Conclusion
The identification of clade 2.3.4.4b H5N1 in a wild leopard cat in South Korea highlights the evolving epidemiology of HPAI and the need for expanded surveillance in wild mammals. Although infections in domestic cats were reported in South Korea in 2016 and 2023 (9–11), this case represents the first confirmed detection of HPAI H5N1 in a wild mammalian species in the country. Given the role of wild birds in the spread and maintenance of HPAIVs, the significant presence of mammalian adaptation markers in viruses circulating in wild bird populations is of great concern, particularly for potential widespread dissemination and interspecies transmissions. Genomic data from this study, shared via GISAID, will support ongoing efforts to track viral spread and assess zoonotic risks. Enhanced monitoring of predator-prey interactions and mammalian populations near avian habitats is recommended to mitigate future spillover events. Further in-depth genomic and phenotypic analyses are warranted to better understand the pathobiological characteristics and zoonotic potential of this virus.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
Ethics statement
Ethical approval was not required for the study involving animals in accordance with the local legislation and institutional requirements because it involved a naturally deceased wild animal (carcass), and no live animal experimentation was conducted.
Author contributions
Y-JS: Writing – review & editing, Resources, Formal analysis. S-HL: Investigation, Writing – review & editing, Writing – original draft, Formal analysis, Data curation, Methodology. D-JK: Resources, Formal analysis, Writing – review & editing. KL: Writing – review & editing, Resources. M-aL: Resources, Writing – review & editing. D-YL: Investigation, Writing – original draft, Formal analysis. Y-RS: Data curation, Formal analysis, Investigation, Writing – original draft. HJ: Funding acquisition, Resources, Writing – review & editing. SL: Funding acquisition, Writing – review & editing, Resources. D-HL: Conceptualization, Supervision, Writing – original draft, Investigation, Funding acquisition, Writing – review & editing, Data curation.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This research was financially supported by a grant from the National Institute of Wildlife Disease Control and Prevention (NIWDC), Ministry of Environment, Republic of Korea (grant number 2025-006).
Acknowledgments
We thank the GISAID Initiative for providing access to reference sequences. This paper was also supported by the KU Research Professor Program of Konkuk University.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Gen AI was used in the creation of this manuscript.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fvets.2025.1638067/full#supplementary-material
Supplementary Figure 1 | Maximum-likelihood tree constructed using the complete coding nucleotide sequences of (A) polymerase basic protein 2, (B) polymerase basic protein 1, (C) polymerase acidic protein, (D) hemagglutinin protein, (E) nucleoprotein, (F) neuraminidase protein, (G) matrix protein, and (H) non-structural protein. Red taxa marked with circles represent the H5N1 isolate from the leopard cat (Prionailurus bengalensis) in South Korea. Purple taxa indicate the H5N1 HPAI viruses isolated from wild birds during the 2024–2025 winter season. Squares denote HPAI viruses detected in wild birds in South Korea in October 2024. Background shading indicates HPAI isolates in blue. Numerical values at the nodes represent bootstrap support values (%) based on 1,000 replicates. Bootstrap values below 70% are not shown.
References
2. Alexander D. “Highly pathogenic avian influenza (fowl plague).” In: OIE OIE Manual of standards for diagnostic tests and vaccines List A and B diseases of mammals birds and bees 3rd, ed. Office International des Epizooties: Paris (1996). p. 155–60.
3. Xie Z, Yang J, Jiao W, Li X, Iqbal M, Liao M, et al. Clade 2.3. 4.4 B highly pathogenic avian influenza H5n1 viruses: knowns, unknowns, and challenges. J Virol. (2025) 99:e00424-25. doi: 10.1128/jvi.00424-25
4. Nguyen T-Q, Hutter C, Markin A, Thomas M, Lantz K, Killian ML, et al. Emergence and interstate spread of highly pathogenic avian influenza a (H5n1) in dairy cattle. Biorxiv. (2024). doi: 10.1101/2024.05.01.591751
5. Leguia M, Garcia-Glaessner A, Muñoz-Saavedra B, Juarez D, Barrera P, Calvo-Mac C, et al. Highly pathogenic avian influenza a (H5n1) in marine mammals and seabirds in Peru. Nat Commun. (2023) 14:5489. doi: 10.1038/s41467-023-41182-0
6. Burrough ER, Magstadt DR, Petersen B, Timmermans SJ, Gauger PC, Zhang J, et al. Highly pathogenic avian influenza a (H5n1) clade 2.3. 4.4 B virus infection in domestic dairy cattle and cats, United States, 2024. Emerg Infect Dis. (2024) 30:1335. doi: 10.3201/eid3007.240508
7. Lee S-H, Kwon J-H, Youk S, Lee S-W, Lee D-H, Song C-S. Epidemiology and pathobiology of H5nx highly pathogenic avian influenza in South Korea (2003–2024): a comprehensive review. Vet Q. (2025) 45:23–38. doi: 10.1080/01652176.2025.2498918
8. Seo Y-R, Cho AY, Si Y-J, Lee S-I, Kim D-J, Jeong H. Evolution and spread of highly pathogenic avian influenza a(H5n1) clade 2.3.4.4b virus in wild birds, South Korea, 2022–2023. Emerg Infect Dis. (2024) 30:299–309. doi: 10.3201/eid3002.231274
9. Lee K, Yeom M, Vu TTH, Do H-Q, Na W, Lee M, et al. Characterization of highly pathogenic avian influenza a (H5n1) viruses isolated from cats in South Korea, 2023. Emerg Microbes Infect. (2024) 13:2290835. doi: 10.1080/22221751.2023.2290835
10. Lee K, Lee E-K, Lee H, Heo G-B, Lee Y-N, Jung J-Y, et al. Highly pathogenic avian influenza a (H5n6) in domestic cats, South Korea. Emerg Infect Dis. (2018) 24:2343. doi: 10.3201/eid2412.180290
11. Kang Y-M, Heo G-B, An S-H, Lee H, Park E, Cha RM, et al. Highly pathogenic avian influenza a (H5n1) virus infection in cats, South Korea, 2023. Emerg Infect Dis. (2024) 30:2510. doi: 10.3201/eid3012.240154
12. Spackman E. Avian influenza virus detection and quantitation by real-time Rt-Pcr. Methods Mol Biol. (2020) 2123:137–48. doi: 10.1007/978-1-0716-0346-8_11
13. Lee D-H. “Complete genome sequencing of influenza a viruses using next-generation sequencing.” In: Animal Influenza Virus. Springer: New York (2020). p. 69–79. doi: 10.1007/978-1-0716-0346-8_6
14. Bushnell B. “Bbmap: a fast, accurate, splice-aware aligner.” In: Conference: 9th Annual Genomics of Energy & Environment Meeting, Walnut Creek, CA, March 17–20 (2014).
15. Fu L, Niu B, Zhu Z, Wu S, Li W. Cd-Hit: accelerated for clustering the next-generation sequencing data. Bioinformatics. (2012) 28:3150–2. doi: 10.1093/bioinformatics/bts565
16. Stamatakis A. Raxml version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. (2014) 30:1312–3. doi: 10.1093/bioinformatics/btu033
17. Letunic I, Bork P. Interactive tree of life (Itol) V5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. (2021) 49:W293–W6. doi: 10.1093/nar/gkab301
18. Suchard MA, Weiss RE, Sinsheimer JS. Bayesian selection of continuous-time Markov Chain evolutionary models. Mol Biol Evol. (2001) 18:1001–13. doi: 10.1093/oxfordjournals.molbev.a003872
19. Minin VN, Bloomquist EW, Suchard MA. Smooth skyride through a rough skyline: bayesian coalescent-based inference of population dynamics. Mol Biol Evol. (2008) 25:1459–71. doi: 10.1093/molbev/msn090
20. Rambaut A, Drummond AJ, Xie D, Baele G, Suchard MA. Posterior summarization in bayesian phylogenetics using tracer 17. Syst Biol. (2018) 67:901–4. doi: 10.1093/sysbio/syy032
21. Giussani E, Sartori A, Salomoni A, Cavicchio L, de Battisti C, Pastori A, et al. Flumut: a tool for mutation surveillance in highly pathogenic H5n1 genomes. Virus Evol. (2025) 11:veaf011. doi: 10.1093/ve/veaf011
22. Si Y-J, Kim D-J, Lee S-H, Seo Y-R, Jeong H, Lee S, et al. New incursions of H5n1 clade 2.3. 4.4 B highly pathogenic avian influenza viruses in wild birds, South Korea, October 2024. Front Vet Sci. (2025) 11:1526118. doi: 10.3389/fvets.2024.1526118
23. Lee D-H, Torchetti MK, Killian ML, Brown I, Swayne DE. Genome sequences of haemagglutinin cleavage site predict the pathogenicity phenotype of avian influenza virus: statistically validated data for facilitating rapid declarations and reducing reliance on in vivo testing. Avian Pathol. (2024) 53:242–6. doi: 10.1080/03079457.2024.2317430
24. Choi T-Y, Kwon H-S, Woo D-G, Park C-H. Habitat selection and management of the leopard cat (prionailurus bengalensis) in a rural area of Korea. Korean J Environ Ecol. (2012) 26:322–32.
25. Suzán G, Ceballos G. The role of feral mammals on wildlife infectious disease prevalence in two nature reserves within Mexico City limits. J Zoo Wildl Med. (2005) 36:479–84. doi: 10.1638/04-078.1
26. Vanak AT, Gompper ME. Interference competition at the landscape level: the effect of free-ranging dogs on a native mesocarnivore. J Appl Ecol. (2010) 47:1225–32. doi: 10.1111/j.1365-2664.2010.01870.x
27. Suttie A, Deng Y-M, Greenhill AR, Dussart P, Horwood PF, Karlsson EA. Inventory of molecular markers affecting biological characteristics of avian influenza a viruses. Virus Genes. (2019) 55:739–68. doi: 10.1007/s11262-019-01700-z
Keywords: HPAI H5N1, wild leopard cat, South Korea, clade 2.3.4.4b, mammalian adaptation, zoonotic potential
Citation: Si Y-J, Lee S-H, Kim D-J, Lee K, Lee M-a, Lee D-Y, Seo Y-R, Jeong H, Lee S and Lee D-H (2025) First detection of clade 2.3.4.4b H5N1 highly pathogenic avian influenza virus in a wild leopard cat (Prionailurus bengalensis) in South Korea. Front. Vet. Sci. 12:1638067. doi: 10.3389/fvets.2025.1638067
Received: 30 May 2025; Accepted: 30 June 2025;
Published: 29 July 2025.
Edited by:
Iryna Goraichuk, Agricultural Research Service (USDA), United StatesReviewed by:
Victor C. Huber, University of South Dakota, United StatesIrene Iglesias, National Institute for Agricultural and Food Research and Technology, Spain
Judith Oguzie, University of Texas Medical Branch, United States
Copyright © 2025 Si, Lee, Kim, Lee, Lee, Lee, Seo, Jeong, Lee and Lee. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dong-Hun Lee, ZG9uZ2h1bmxlZUBrb25rdWsuYWMua3I=
†These authors have contributed equally to this work