 Lucilla Cucco1Elisa Albini1Francesca Blasi1Serenella Orsini1Alessandro Fiorucci1Annalisa Dettori1
Lucilla Cucco1Elisa Albini1Francesca Blasi1Serenella Orsini1Alessandro Fiorucci1Annalisa Dettori1 Sara Petrin2
Sara Petrin2 Arianna Peruzzo2Silvano Salaris2Marta Paniccià1
Arianna Peruzzo2Silvano Salaris2Marta Paniccià1 Giovanni Pezzotti1
Giovanni Pezzotti1 Francesca Romana Massacci1*
Francesca Romana Massacci1* Chiara Francesca Magistrali1,3
Chiara Francesca Magistrali1,3- 1Istituto Zooprofilattico Sperimentale dell'Umbria e delle Marche "Togo Rosati", Perugia, Italy
- 2Istituto Zooprofilattico Sperimentale delle Venezie, Padova, Italy
- 3Istituto Zooprofilattico Sperimentale della Lombardia e dell'Emilia Romagna "Bruno Ubertini", Brescia, Italy
Streptococcus canis, a multi-host pathogen commonly isolated from dogs and cats has been occasionally reported in severe cases of human infection. This study aimed to explore the genetic diversity, antimicrobial resistance (AMR), and pathogenicity of S. canis isolates collected between 2004–2021, in Italy. Fifty-five S. canis isolates from clinical cases in domestic animals were investigated for susceptibility to antibiotics and then characterized for sequence type (ST), virulence profile, and antimicrobial-resistant genes through whole genome sequencing (WGS). All isolates were susceptible to beta-lactams, while frequently exhibiting resistance to lincosamides, chlortetracyclines, and macrolides. Six out of 55 isolates of S. canis, all collected between 2020 and 2021, were multi-drug resistant (MDR). The most common AMR gene in the dataset was lmrP conferring resistance for streptogramin, tetracycline, macrolide, streptogramin A, and lincosamide. Other determinants of AMR were the tet genes. Twenty-one distinct STs were identified, with ST9 being the most prevalent in our collection. Regarding the virulence genes, forty-three isolates were positive for the ssp-5 gene, which encodes an agglutinin receptor. Comparison with other 46 S. canis genomes available in public repositories revealed that the Italian isolates clustered by the S. canis M-like (SCM) protein gene and ST and did not group according to their host, area, or year of origin. In conclusion, our study underscores the susceptibility of Italian S. canis isolates to beta-lactam antibiotics, which remain the first line of defense in managing infections. In Italy, ST9 represents the predominant clone of this pathogen. Despite the diversity in species of origin and the various STs identified, our findings confirm that S. canis has not adapted to different ecological niches and corroborate the accidental pathogenic nature of human cases.
1 Introduction
Streptococcus canis is a β-hemolytic Streptococcus species from the Lancefield Group G, typically colonizing the skin and mucous membranes of asymptomatic dogs and cats (1, 2). S. canis can cause a variety of infections in these animals, including skin and soft tissue infections, and, although rare, more severe diseases such as ulcerative keratitis, necrotizing fasciitis, septicemia, endocarditis, respiratory disease, genital, and urinary infections (3–8). Less frequently, S. canis has been found in other wild and domestic mammalian hosts: in cattle is a rare but contagious agent of mastitis (3, 9). S. canis is also a zoonotic agent, with the first infection in humans described in 1998. Since then, a growing number of cases have been reported in humans, including severe cases of bacteremia, osteomyelitis, endocarditis, and pneumonia (10–12). Human infections are generally a consequence of exposure to dogs, or, less frequently, to cats, and occur after bite wounds, superficial ulcers, or cellulitis (12). Molecular epidemiological investigations have revealed significant genomic overlap between animal and human isolates, suggesting direct interspecies transmission (5, 13). Moreover, S. canis infection is rare in the setting of neonatal sepsis; however, it can lead to high morbidity and mortality as reported recently (14).
Therapy against S. canis infection is based on antimicrobials, with penicillins identified as the first-line antibiotic class in animals and humans (15). The development of AMR in S. canis has been reported, but the mechanisms of resistance are not yet fully characterized (16–18). S. canis has been reported to show low resistance rates to quinolones (7.0%) and from 5.6 to 39.7% for tetracyclines (15). Conversely, the species is considered highly susceptible to beta-lactams (15). This assumption has recently been challenged by reports of beta-lactam-resistant isolates from animals emerging in Japan (19).
Despite some research efforts, S. canis remains less studied compared to other streptococcal species, and many aspects of its epidemiology and virulence are still not well understood (3–5, 20). Two genotyping methods, a multi-locus sequence typing (MLST) scheme (21) and a scheme based on the allelic variations of the SCM protein gene (17), were used to investigate the diversity of S. canis population. Nevertheless, a consensus on the preferred method for S. canis typing was not reached, complicating the understanding of the population structure of this pathogen (13, 21–23). More recently, these two systems were compared to core genome typing based on data from WGS (5). This comparison highlighted that both MLST and SCM typing schemes lack in describing the diversity within the S. canis population, probably because they both analyze small fragments of the bacterial genome. By contrast, core genome analysis based on WGS provides a more comprehensive understanding of the epidemiology of this pathogen (5, 13). The application of WGS could potentially address many of the current knowledge gaps regarding S. canis, particularly in terms of its population structure, evolution, and host specificity.
Here we use whole genome sequencing of S. canis isolates collected between 2004 and 2021 from clinical cases in domestic animals in Italy to investigate genetic diversity, antimicrobial resistance, and pathogenicity.
2 Materials and methods
2.1 Bacterial isolates and species identification
We investigated 55 isolates collected from dogs (n = 25), cats (n = 13), cattle (n = 3), and a hedgehog (n = 1) collected in Italy from 2004 to 2021 (Supplementary Table S1). All the isolates originated from individual cases of clinical disease. To the best of our knowledge, they were not epidemiologically linked, as they were collected from different animals, in different geographical locations, at different years, and from different veterinary clinics or diagnostic laboratories. In our collection, we included the reference strain of Culture Collection University of Gothenburg (CCUG): Streptococcus canis CCUG 27668. The samples were cultured on 5% sheep blood agar (Biolife Italiana Srl, Milan, Italy) at 5% CO2, 37°C for 24–48 h. Suspected β-hemolytic colonies were selected, and confirmed as belonging to the genus Streptococcus by Matrix-Assisted Laser Desorption Ionization-Time of Flight Mass Spectrometry (MALDI-TOF MS) (Bruker Daltonics GmbH, Germany).
Isolates were identificated at specie level by the 16S rRNA gene sequencing. DNA was extracted using QIAamp® DNA Mini Kit (Qiagen, Hilden, Germany) following the manufacturer’s instruction and used to perform the PCR reaction with universal primers 27F (5′-AGAGTTTGATCCTGGCTCAG-3′) and 1492R (5′-TACGGYTACCTTGTTACGACTT-3′) (24), containing 10 μL of 5x Taq buffer, 1.5 mM MgCl2, 200 μM dNTPs, 1 U of Taq DNA polymerase (Promega Corporation, Wisconsin, USA), 0.2 μM of primers and 10 ng of DNA template, brought up to a final volume of 50 μL with ultra-pure water. The reactions were performed on a thermocycler (Eppendorf, Hamburg, Germany) under these conditions: 4 min at 96°C, followed by 30 cycles of 1 min at 94°C, 1 min at 56°C and 1 min at 72°C, and a final extension step at 72°C for 10 min. Aliquots of 5 μL of each reaction were analyzed on 1% (w/v) agarose gel in TBE buffer.
The PCR products were purified using High Pure PCR Product Purification Kit (Roche, Basel, Switzerland), sequenced with specific primers using BigDye™ Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, Massachusetts, USA) in 3500 Genetic Analyzer (Applied Biosystems Massachusetts, USA). The DNA sequences were analyzed using BioEdit sequence alignment tool and compared with the sequences deposited in the National Center for Biotechnology Information (NCBI)-GenBank database using the BLAST alignment tool.1 The isolates were identified unambiguously, with ≥ 98.7% similarity to the 16S rRNA sequence of the corresponding strain.
2.2 Antimicrobial susceptibility testing
We assessed MICs using a commercial MIC panel (BOP06F, Sensititre; Trek Diagnostic Systems Inc.) according to the manufacturer’s instructions. Susceptibility to erythromycin was assessed using Etest strips (Liofilchem, Roseto degli Abruzzi, Italy), with a tested concentration range of 0.016–256 μg/mL. Streptococcus pneumoniae ATCC 49619 was used as a quality control strain. The MIC values for chlortetracycline, penicillin, trimethoprim/sulfamethoxazole, and erythromycin were interpreted using the breakpoints recommended by the Clinical Laboratory Standards Institute M100 Ed. 34th (25). MIC values for ampicillin, clindamycin, ceftiofur, and enrofloxacin were interpreted according to the breakpoints from CLSI Vet01S, Ed. 7th edition (26). Based on the clinical breakpoints, the isolates were classified as susceptible (S), intermediate (I), or resistant (R). An isolate was classified as multi-resistant when it was resistant to at least three antibiotic classes representing third-generation cephalosporins (ceftiofur), penicillin (ampicillin and penicillin), lincosamides (clindamycin), fluoroquinolones (enrofloxacin), sulfonamides (trimethoprim/sulfamethoxazole), macrolides (erythromycin), and tetracycline (chlortetracycline) (27). For gentamicin and florfenicol, MIC values were interpreted using the epidemiological cut-off (ECOFF) criteria established by EUCAST.2 When using the ECOFF, the isolates were categorized as wild-type (WT) or non-wild-type (nWT). According to EUCAST definitions, “wild-type” (WT) isolates are those with MICs at or below the epidemiological cutoff value (ECOFF), representing populations without acquired or mutational resistance mechanisms, whereas “non-wild-type” (NWT) isolates exhibit MICs above the ECOFF, indicating the likely presence of such resistance mechanisms.
2.3 Whole genome sequencing
In order to investigate ST, virulence profile, and antimicrobial resistant genes, the 55 S. canis isolates were whole genome sequenced. Each DNA was then quantified with the Qubit fluorometer (QubitTM DNA HS Assay, Thermo Fisher Scientific Inc.). Libraries were prepared using the Nextera XT Library Prep kit (Illumina Inc., San Diego, CA) and then sequenced on an Illumina NextSeq 550 platform to generate 300 bp paired-end reads.
2.4 Bioinformatic analysis
Illumina reads were trimmed and checked for quality using Fastp v0.19.5 (28) with default parameters. The metrics used for reads quality assessment were: number of contigs, mean values for N50 and L50 and GC% values (Supplementary Table S2). The reads were assembled using SPAdes genome assembler v3.11.1 (29), checked for quality assessment of draft genome sequences with QUAST v5.0.2 (30), and annotated using Prokka v1.14.6 (31). The resulting general feature formats (GFFs) produced by Prokka were analyzed with Roary v3.11.3 (32) to obtain a core genome alignment. In silico multi-locus (ML) ST analysis was performed by submitting sequences to S. canis MLST database to obtain allele number and ST. The new allele sequences or STs were submitted to the database curator.3
Antibiotic-resistance genes were analyzed with ABRicate,4 using ResFinder, considering only genes with a ≥95% coverage and ≥99% identity.
Our investigation of quinolone resistance focused on parE, gyrB, parC, and gyrA mutations. parE, gyrB, parC, and gyrA sequences of the 55 isolates were manually aligned running MUSCLE online5 and using Streptococcus canis HL_98_2 (GenBank NZ_CP053789.1) as reference.
Virulence genes were searched by BLASTN v2.13.0+ creating a database of 19 previously described genes (5) and using a ≥90% coverage and ≥20% identity.
In order to compare our isolates to the ones available in the literature, we downloaded the 46 genomes referring to the S. canis (5), in the Sequence Read Archive (SRA) database. The dataset included 26 isolates from dogs, 11 from humans, 6 from cats, 2 from cattle, and 1 from seal. Those selected isolates were originated from UK (41), South Korea (4) and USA (1). The 46 genomes were compared with the 55 isolates of our study creating a maximum likelihood (ML) phylogenetic tree (FastTree 2.1.11) (33) of the core genome of S. canis isolates. The tree was manually annotated using iTOL (v.6, https://itol.embl.de/, accessed date: September 2, 2024).
We classified SCM 1-15 using BLASTN and a database of SCM coding sequences previously described by Pagnossin et al. (5). For the classification, we chose an identity percentage >98% and a coverage percentage >70% (5). The nucleotide sequences of the M protein gene of our S. canis isolates were aligned with the nucleotide sequences of M proteins available in the literature using MUSCLE (5).
3 Results
3.1 AMR phenotypes and genotypes
The distribution of S. canis isolates according to antibiotic MIC values is shown in Table 1. The isolates were classified as resistant to chlortetracycline (18/55, 32.7%), clindamycin (6/55, 10.9%) and to erythromycin (6/55, 10.9%). Forty-five isolates out of 55 (81.8%) were intermediate for enrofloxacin. Moreover, 41/55 (74.5%) isolates were considered as non wild-type for gentamicin. Multi-resistance was detected in 6/55 (10.9%) isolates of S. canis, showing simultaneous resistance to erythromycin, chlortetracycline and clindamycin. Those isolates, collected between 2020 and 2021, belonged to dogs (4), and cats (2). The most common AMR gene in the dataset was lmrP (55/55) conferring resistance for streptogramin, tetracycline, macrolide, streptogramin A, lincosamide. Other determinants of AMR were the tet genes (Table 2, Figure 1).
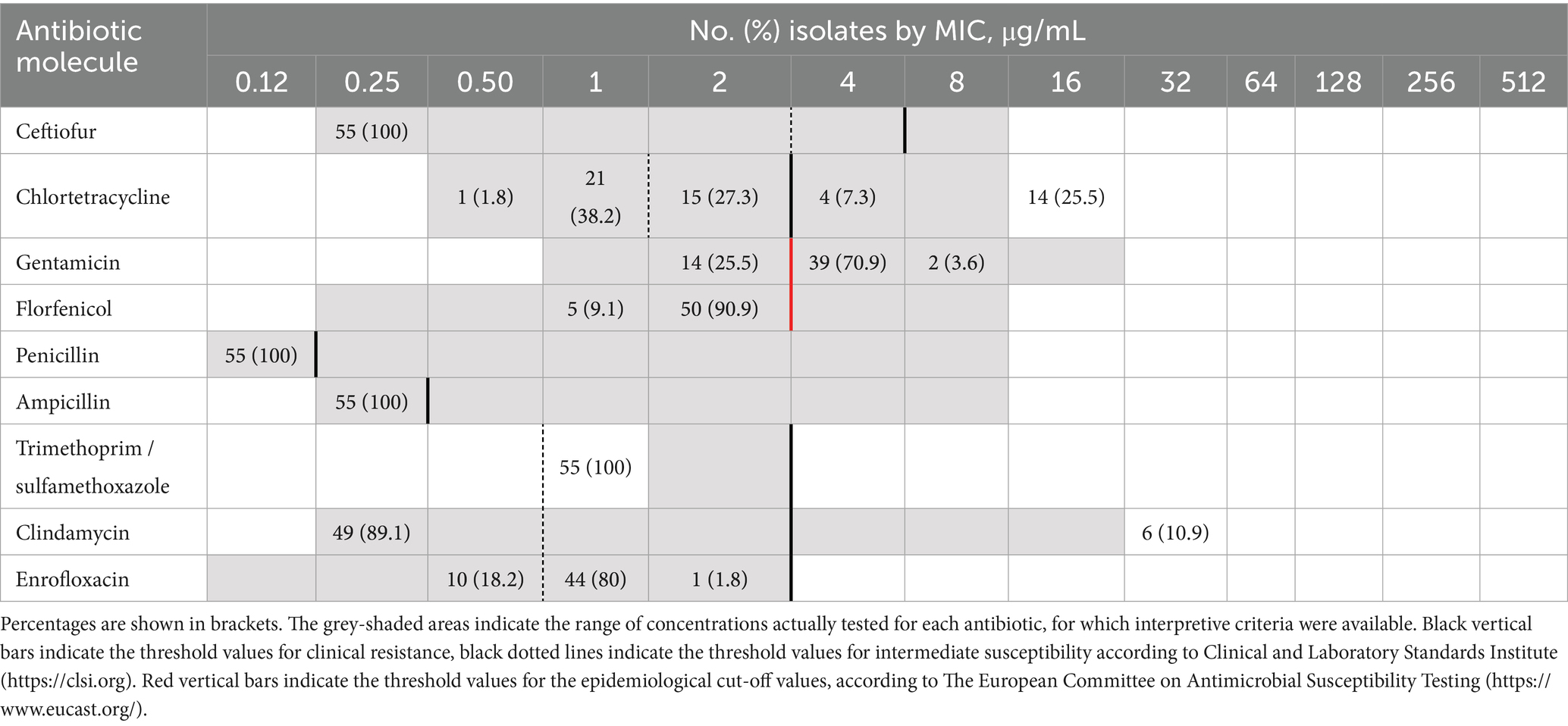
Table 1. Distribution of MIC (minimum inhibitory concentration) values among the 55 S. canis isolates tested using a commercial MIC panel (BOP06F, Sensititre; Trek Diagnostic Systems Inc.).
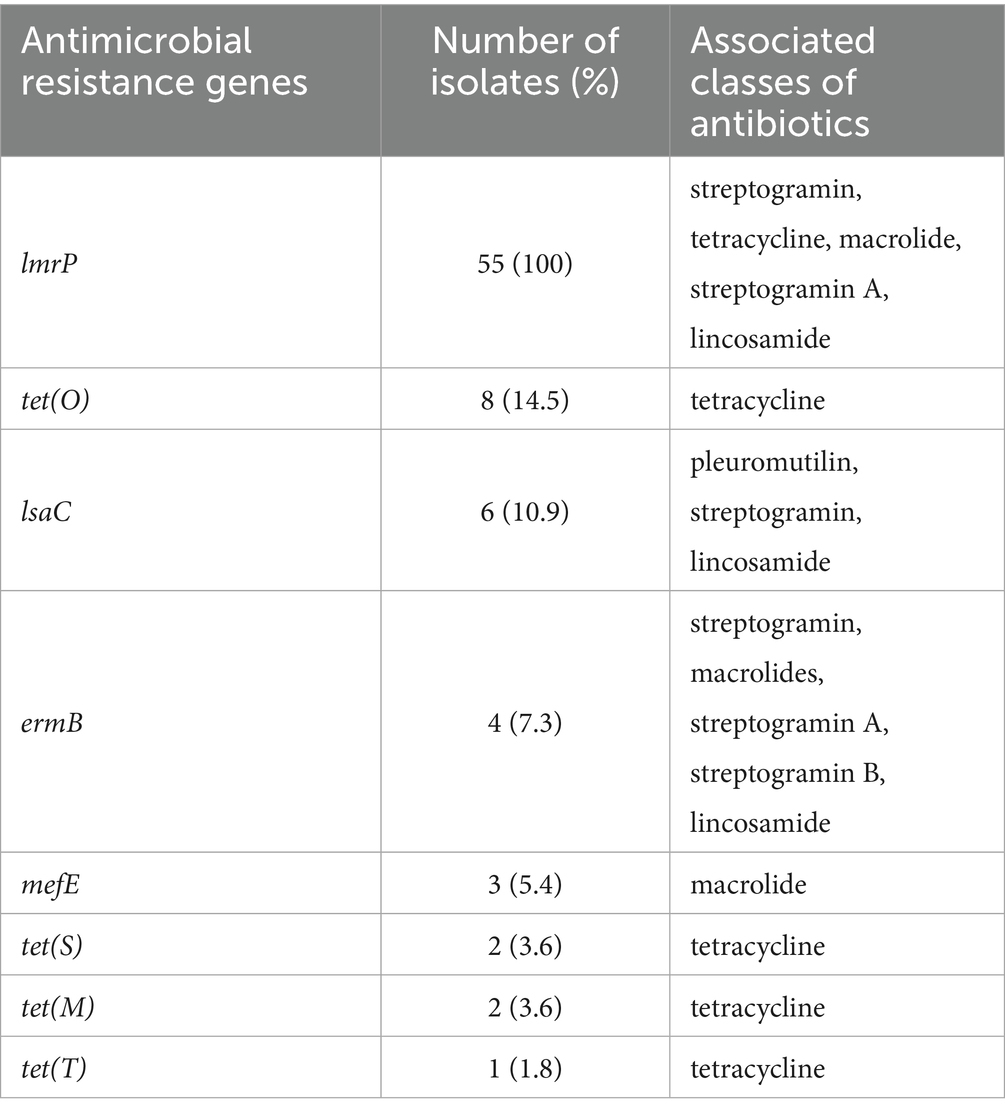
Table 2. Antimicrobial resistance genes and their associated classes of antibiotics, as indicated by the Comprehensive Antibiotic Resistance Database (CARD; https://card.mcmaster.ca/home), identified in 55 Streptococcus canis isolates.
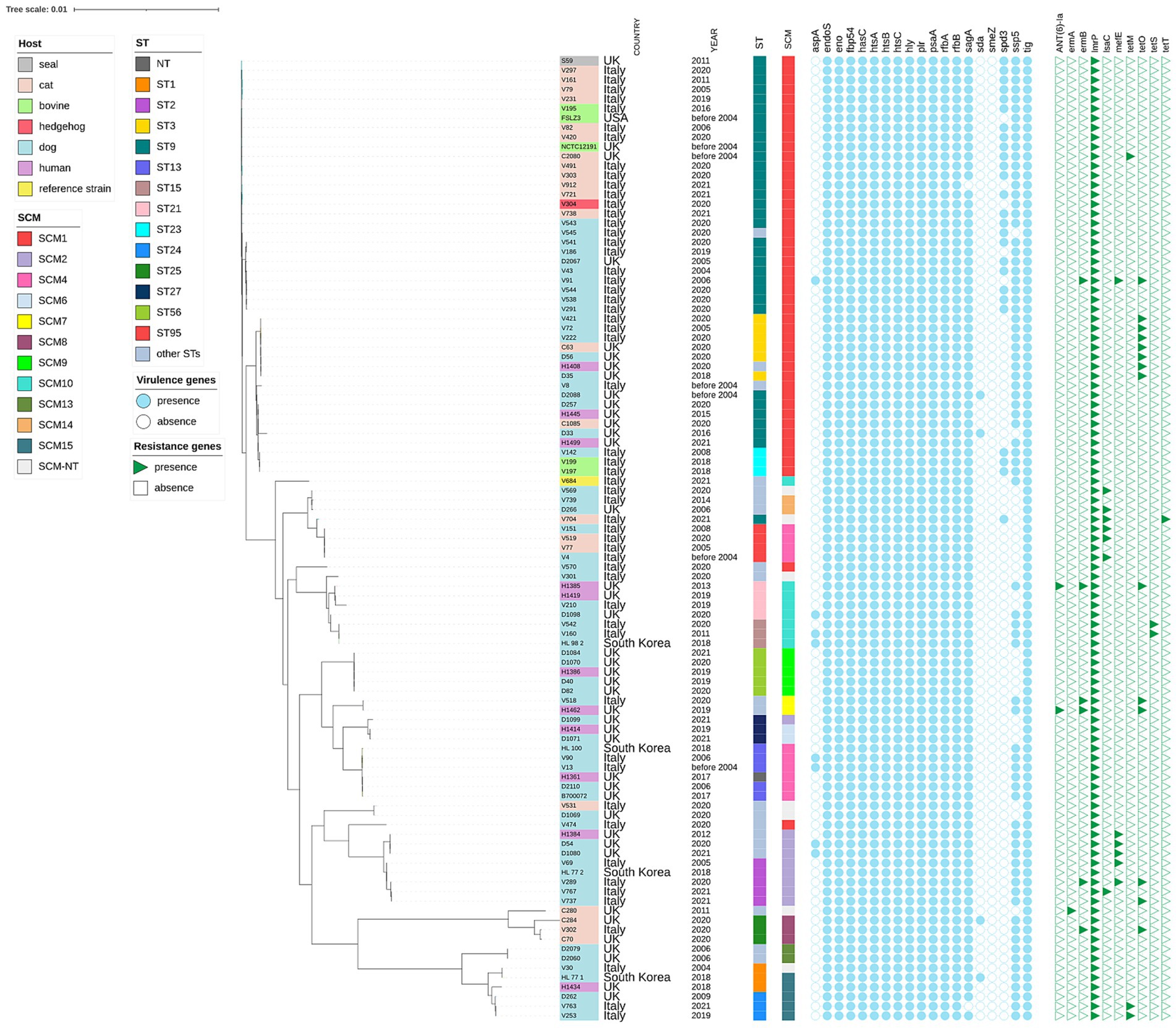
Figure 1. Maximum likelihood phylogenetic tree based on core genome alignment of 101 Streptococcus canis isolates collected from various countries and hosts. The tree was constructed and annotated using the iTOL interactive interface (https://itol.embl.de). For each isolate, the sequence type (ST), SCM group, virulence genes, and antibiotic resistance genes are indicated. The isolate labels are color-coded according to the host of origin (e.g., dog, cat, seal, human, bovine, hedgehog), enabling immediate visual correlation between host specificity and phylogenetic clustering.
None of the isolates harbored the mutations associated to a reduced susceptibility to quinolones, namely Ser81/Glu85 in gyrA, Gly408 in gyrB, Ser67/Asp71 in parC or Asp438 in parE (34) (Supplementary Table S3).
3.2 Genomic analysis
The mean length of the 55 assemblies was 2,099,785 (min 1,892,070; max 2,486,022) with an average number of contigs of 76 (min 45; max 162). The mean values for N50 and L50 were 84,699 (min 29,080; max 155,133) and 9 (min 5; max 23). GC% value ranged between 39.52 and 38.82 (Supplementary Table S2). Twenty-one distinct STs were identified, with ST9 being the most prevalent, accounting for 38.2% (n = 21) of our collection (Supplementary Table S4). Both ST95 and ST2 were also common, each representing 7.3% (n = 4) of the samples. Eight new STs were identified as ST91-ST101 (ID289-ID299).6
The distribution of putative virulence genes was investigated in the 55 S. canis isolates and we described the presence of 18 out of 19 virulence genes as reported in Supplementary Table S1 and Figure 1. Twelve of these genes (63.2%) were detected in each isolate. Forty-three isolates (78.8%) of our collection were positive for the carriage of the ssp-5 gene, which encodes an agglutinin receptor. None of our isolates were positive for smeZ.
The characterization of the allelic variations of the SCM gene across the collection is provided in Supplementary Table S1 and Figure 1.
3.3 Phylogenetic analysis
Comparison with other S. canis genomes available in public repositories revealed that the Italian isolates clustered by the SCM and ST. Some STs belonged to one SCM allele alone: e.g. ST1 to SCM15, ST3 to SCM1, ST15 to SCM10, ST23 to SCM1 (Figure 1, Supplementary Table S1). The agreement among phylogenetic methods was not complete: in the ML phylogenetic tree, ST3, ST23, ST61, ST94, and ST101 clustered together with ST9, while all of them were classified as SCM1. This ST9-SCM1 phylogenetic cluster was the largest in our collection. Regardless of the typing method, the phylogenetic tree indicated that the 101 S. canis isolates did not group according to their host, area or year of origin. Human isolates belonged to different STs, harbored various SCM types and were scattered along the phylogenetic tree (Figure 1).
4 Discussion
S. canis is a significant pathogen in both canines and cattle and is increasingly recognized as an emerging zoonotic agent. Despite its clinical importance, there remains a scarcity of comprehensive data regarding the epidemiology, antibiotic susceptibility, and virulence mechanisms of this bacterium. Our study addresses this gap by presenting data on S. canis isolates collected from various animal hosts over a span of 17 years in Italy. Additionally, we conducted comparative genomic analyses between our S. canis isolates and publicly available genomes from human cases and different geographical regions, thereby providing new insights into the genetic diversity of this microorganism.
Regarding the analysis of virulence characteristics, we identified 18 out of the 19 virulence genes previously described in the literature (5). Several genes such as eno, fbp54, hasC, hyl, plr, rfbA, rfbB, which were identified in our collection, are recognized as components of the S. pyogenes core genome, which further supports the close evolutionary relationship between S. canis and S. pyogenes (35). hasC is part of the has operon, which is responsible for the synthesis of hyaluronic acid. In fact, in S. pyogenes, the capsule composed of hyaluronic acid has a composition analogous to that of hyaluronic acid found in human connective tissue, which contributes to the low immunogenicity of the bacterium in the host (36). In the genus Streptococcus, fbp54 encodes a surface protein capable of binding to fibrinogen and fibronectin, thus being involved in adhesion mechanisms (37). Forty-three isolates were positive for the ssp-5 gene, being one of the most prevalent virulence genes in our collection. The gene ssp-5 codes for an agglutinin receptor and is responsible for adhesion and colonization of Streptococcus to different substrates inside the host (38). Notably, ssp-5 has been identified in other Streptococcus species, such as S. suis and S. canis, both of which are associated with zoonotic transmission from animals to humans. Its presence is strongly correlated with increased pathogenicity, making it a critical factor in cross-species infections and a valuable target for surveillance and therapeutic interventions (5, 38).
Moreover, S. canis, like the majority of species within this genus, is generally susceptible to the beta-lactam class of antibiotics (4, 39, 40). A recent study has reported reduced susceptibility to penicillin-G in S. canis isolates from dogs in Japan, with this resistance attributed to amino acid substitutions in penicillin-binding proteins (15). In contrast, all isolates in our collection exhibited full susceptibility to beta-lactams, including penicillin, ampicillin, and ceftiofur. This finding reinforces the continued efficacy of beta-lactams as the first-line treatment for S. canis infections in Italy. Our isolates frequently exhibited resistance to lincosamides, tetracyclines, and macrolides consistent with previous reports (4, 5, 41–43). Resistance to tetracyclines was detected in approximately one-third of our collection, a proportion aligning with other studies, where it ranges from 30–40% (4, 21, 44, 45). This resistance was associated with the presence of tet genes, with tet(O) being the most prevalent, followed by tet(M), tet(S), and tet(T). While the presence of tet(O) and tet(M) is well documented in the literature, the detection of tet(S) and tet(T) is relatively rare (5, 46). Notably, all Italian ST15 isolates harbored tet(S), and all ST3 was positive for tet(O).
Resistance to macrolides and lincosamides, likely attributable to the MLSB phenotype (macrolides, lincosamides, and streptogramin B group), was detected in six isolates. This resistance was generally associated with the presence of the lmrP and ermB determinants (4, 42). Additionally, up to 80% of the isolates were categorized as non-susceptible to enrofloxacin according to the CLSI breakpoints for veterinary pathogens, which classify MIC values of 1–2 μg/mL as intermediate. In S. canis, resistance to quinolones is generally associated with substitutions in the parC (Ser67/Asp71), gyrA (Ser81/Glu85) and parE (Asp438) sequences (47). None of these substitutions were detected in our collection (Supplementary Table S3). It is noteworthy that the aforementioned substitutions are typically coupled with MIC values for quinolones higher than 2 μg/mL, while our isolates exhibited MIC values equal or below 2 μg/mL (47).
A total of 6 out of 55 S. canis isolates (10.9%) obtained from the bacterial collection between 2020 and 2021 exhibited a multi-drug resistant phenotype. Of these, four isolates originated from canine hosts, three of which belonged to ST2, while the remaining two were derived from cats. The emergence of MDR strains within S. canis is a growing concern, as resistance to multiple antibiotic classes can severely limit therapeutic options for treating infections in companion animals. The observed association of MDR with ST2 strains may indicate clonal expansion or selective pressure within this lineage. From a clinical standpoint, MDR S. canis infections may result in prolonged illness, treatment failure, or increased reliance on last-resort antimicrobials. Moreover, the potential zoonotic transmission of resistant S. canis strains from pets to humans—particularly immunocompromised individuals—poses a notable public health risk, as companion animals can act as reservoirs and vectors for antimicrobial-resistant bacteria (13). These findings underscore the importance of routine antimicrobial susceptibility monitoring and the implementation of prudent antibiotic use policies in veterinary practice.
There is no standard reference technique for the phylogenetic analysis of S. canis; therefore, three methods were utilized in parallel: core genome analysis, MLST and SCM sequences analysis. For the phylogenetic analysis, publicly available genomes of S. canis were also included, even though their number was quite limited. The data confirm that ST9 of S. canis is a dominant sequence type in Italy, consistent with previous studies in other European countries, such as Portugal and Germany (48). ST9 is characterized by the presence of allele 1 of the S. canis M-like protein (SCM1), a recognized virulence factor of this bacterium. Similarly, ST21 was associated with the production of SCM allele 10, as previously reported by Fukushima et al. (49), but this association is not exclusive, as the same variant was found in ST15. The phylogenetic analysis of the core genome revealed that the isolates did not cluster based on their species of origin. For instance, isolates belonging to ST9 originated from diverse sources, including dogs, cats, cattle, hedgehogs, and seals, yet were placed within the same clusters in the phylogenetic tree. Human-origin isolates did not form separate clusters but were included within the same clusters as canine, feline, and bovine isolates. The comparison among the three methods highlights an incomplete agreement between MLST typing and the core genome analysis, as shown by the presence of multiple STs in the same cluster. Genomic analyses, including multilocus sequence typing, confirm the zoonotic origin of these infections and illustrate genetic recombination events with Streptococcus dysgalactiae, enhancing its virulence and adaptability (21). ST9 was isolated from a patient with bacteremia, while previous animal studies had consistently identified ST9 in dogs suffering from dermatitis and wound infections (22). This suggests that certain STs are predisposed to cross-species infection and may possess enhanced virulence factors, such as the scm gene.
The SCM classification system groups scm alleles based on sequence similarity into three major categories: Group I (alleles 1–7), Group II (alleles 8–15), and SCM-NT (non-typeable). Group I includes classical alleles typically associated with less invasive strains, while Group II encompasses novel variants such as allele 10, which has been linked to increased intracellular invasion and potential virulence, particularly in strains belonging to ST21 and ST15 (49). SCM-NT strains either lack detectable scm sequences or express untypeable variants, and their role in pathogenesis is still under investigation. This grouping provides a molecular framework for epidemiological and virulence profiling of S. canis, particularly in zoonotic contexts (49, 50). We did not observe a clustering between SCM group 1 isolates and SCM group 2 isolates in the phylogenetic tree. As already noted by Pagnossin et al. (5), the analysis of data from WGS offers higher discrimination as compared to SCM or MLST analysis. Our study provides S. canis genomes from novel geographical regions, periods, and hosts, thereby offering new opportunities to compare this pathogen diversity across various ecological niches.
The high genetic similarity of S. canis isolates from different hosts and tissues confirms the generalist nature of this pathogen and its lack of adaptation to specific host species, in agreement with findings by other authors (5, 13). A limitation of this study is the relatively small number of S. canis isolates analyzed. However, the strains were collected over a broad temporal span (2004–2021) and from diverse host species and geographic regions, which enhances the relevance of the observed phylogenetic patterns and allows for insights into potential long-term and cross-host transmission dynamics. Regardless of genetic lineage, S. canis seems capable of cross-species transmission. This genomic evidence is supported by the nature of infections reported in cattle, where before spreading among lactating cows, S. canis infection usually originates from cats or dogs having access to the barn (9). More importantly, S. canis infections in humans are primarily attributed to a close contact, often through bites, with dogs and cats (4).
In conclusion, our study underscores the susceptibility of Italian S. canis isolates to beta-lactams antibiotics, which remain the first line of defense in managing infections. In Italy, ST9 represents the predominant clone of this pathogen. Despite the diversity in species of origin and the various sequence types identified, our findings confirm that S. canis has not adapted to different ecological niches, corroborating the accidental pathogenic nature of human cases.
Data availability statement
The data presented in the study are deposited in the National Center for Biotechnology Information SRA [BioProject ID PRJNA1175870 (accession numbers SAMN44373913—SAMN44373967)].
Author contributions
LC: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Software, Writing – original draft, Writing – review & editing. EA: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Writing – original draft, Writing – review & editing. FB: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Writing – original draft, Writing – review & editing. SO: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Writing – original draft, Writing – review & editing. AF: Data curation, Formal analysis, Methodology, Writing – original draft, Writing – review & editing. AD: Formal analysis, Investigation, Methodology, Writing – original draft, Writing – review & editing. SP: Data curation, Formal analysis, Investigation, Methodology, Software, Writing – original draft, Writing – review & editing. AP: Data curation, Formal analysis, Investigation, Methodology, Software, Writing – original draft, Writing – review & editing. SS: Data curation, Formal analysis, Investigation, Methodology, Software, Writing – original draft, Writing – review & editing. MP: Conceptualization, Formal analysis, Investigation, Supervision, Validation, Writing – original draft, Writing – review & editing. GP: Conceptualization, Formal analysis, Investigation, Supervision, Validation, Writing – original draft, Writing – review & editing. FM: Conceptualization, Data curation, Formal analysis, Investigation, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. CM: Conceptualization, Formal analysis, Funding acquisition, Investigation, Project administration, Resources, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by the Italian Ministry of Health (grant number: RC 007/2020 IZSUM).
Acknowledgments
We sincerely thank Dr. Massimiliano Orsini for his invaluable contributions to this project. His dedication and passion for research were truly inspiring. His untimely passing is a great loss, and we dedicate this work to his memory.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The authors declare that no Gen AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fvets.2025.1645885/full#supplementary-material
Footnotes
1. ^www.ncbi.nlm.nih.gov/BLAST
3. ^https://pubmlst.org/organisms/Streptococcus-canis
4. ^https://github.com/tseemann/abricate
References
1. Devriese, LA, Hommez, J, Kilpper-Balz, R, and Schleifer, KH. Streptococcus canis sp. nov.: a species of group G streptococci from animals. Int J Syst Bacteriol. (1986) 36:422–5. doi: 10.1099/00207713-36-3-422
2. Fukushima, Y, Tsuyuki, Y, Goto, M, Yoshida, H, and Takahashi, T. Species identification of β-hemolytic streptococci from diseased companion animals and their antimicrobial resistance data in Japan (2017). Jpn J Infect Dis. (2019) 72:94–8. doi: 10.7883/yoken.JJID.2018.231
3. Hassan, AA, Akineden, Ö, and Usleber, E. Identification of Streptococcus canis isolated from milk of dairy cows with subclinical mastitis. J Clin Microbiol. (2005) 43:1234–8. doi: 10.1128/JCM.43.3.1234-1238.2005
4. Pagnossin, D, Smith, A, Oravcová, K, and Weir, W. Streptococcus canis, the underdog of the genus. Vet Microbiol. (2022) 273:109524. doi: 10.1016/j.vetmic.2022.109524
5. Pagnossin, D, Weir, W, Smith, A, Fuentes, M, Coelho, J, and Oravcova, K. Streptococcus canis genomic epidemiology reveals the potential for zoonotic transfer. Microb Genomics. (2023) 9:1–12. doi: 10.1099/mgen.0.000974
6. Prescott, JF, Mathews, K, Gyles, CL, Matsumiya, L, Miller, C, Rinkhardt, N, et al. Canine streptococcal toxic shock syndrome in Ontario: an emerging disease? Can Vet J. (1995) 36:486–7. Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC1687011/
7. Matsuu, A, Kanda, T, Sugiyama, A, Murase, T, and Hikasa, Y. Mitral stenosis with bacterial myocarditis in a cat. J Vet Med Sci. (2007) 69:1171–4. doi: 10.1292/jvms.69.1171
8. Kruger, EF, Byrne, BA, Pesavento, P, Hurley, KF, Lindsay, LL, and Sykes, JE. Relationship between clinical manifestations and pulsed-field gel profiles of Streptococcus canis isolates from dogs and cats. Vet Microbiol. (2010) 146:167–71. doi: 10.1016/j.vetmic.2010.04.026
9. Eibl, C, Baumgartner, M, Urbantke, V, Sigmund, M, Lichtmannsperger, K, Wittek, T, et al. An outbreak of subclinical mastitis in a dairy herd caused by a novel Streptococcus canis sequence type (ST55). Animals. (2021) 11:550. doi: 10.3390/ani11020550
10. Amsallem, M, Iung, B, Bouleti, C, Armand-Lefevre, L, Eme, A-L, Touati, A, et al. First reported human case of native mitral infective endocarditis caused by Streptococcus canis. Can J Cardiol. (2014) 30:1462.e1–2. doi: 10.1016/j.cjca.2014.07.013
11. Lacave, G, Coutard, A, Troché, G, Augusto, S, Pons, S, Zuber, B, et al. Endocarditis caused by Streptococcus canis: an emerging zoonosis? Infection. (2016) 44:111–4. doi: 10.1007/s15010-015-0809-3
12. Wang, MS, Huaringa, ME, Feld, LN, Ochiai, K, Whelan, TE, and Frazier, NM. Streptococcus canis native aortic valve endocarditis linked to cat exposure: a case report and review. J Community Hosp Intern Med Perspect. (2024) 14:91–5. doi: 10.55729/2000-9666.1318
13. Richards, VP, Zadoks, RN, Pavinski Bitar, PD, Lefébure, T, Lang, P, Werner, B, et al. Genome characterization and population genetic structure of the zoonotic pathogen, Streptococcus canis. BMC Microbiol. (2012) 12:1–16. doi: 10.1186/1471-2180-12-293
14. Saleh, E, O’Neal, A, Torres, E, Vargas, L, Rodriguez, M, and Chaudhary, S. Streptococcus canis rapidly progressive and fatal neonatal Sepsis in a term infant. Pediatr Infect Dis J. (2025) 44:e161–5. doi: 10.1097/INF.0000000000004723
15. Imanishi, I, Iyori, K, Také, A, Asahina, R, Tsunoi, M, Hirano, R, et al. Antibiotic-resistant status and pathogenic clonal complex of canine Streptococcus canis-associated deep pyoderma. BMC Vet Res. (2022) 18:395. doi: 10.1186/s12917-022-03482-3
16. Fulde, M, Rohde, M, Polok, A, Preissner, KT, Chhatwal, GS, and Bergmann, S. Cooperative plasminogen recruitment to the surface of Streptococcus canis via M protein and enolase enhances bacterial survival. MBio. (2013) 4:e00629–12. doi: 10.1128/mBio.00629-12
17. Fulde, M, Rohde, M, Hitzmann, A, Preissner, KT, Nitsche-Schmitz, DP, Nerlich, A, et al. SCM, a novel M-like protein from Streptococcus canis, binds (mini)-plasminogen with high affinity and facilitates bacterial transmigration. Biochem J. (2011) 434:523–35. doi: 10.1042/BJ20101121
18. Bergmann, S, Eichhorn, I, Kohler, TP, Hammerschmidt, S, Goldmann, O, Rohde, M, et al. SCM, the M protein of Streptococcus canis binds immunoglobulin G. Front Cell Infect Microbiol. (2017) 7:80. doi: 10.3389/fcimb.2017.00080
19. Kawara, Y, Goto, M, Maeda, T, Yoshida, H, Tsuyuki, Y, and Takahashi, T. Seven draft genome sequences of Streptococcus canis strains, revealing reduced penicillin-G susceptibility. Microbiol Resour Announc. (2024) 13:e0021924. doi: 10.1128/mra.00219-24
20. Sykes, JE, Kittleson, MD, Pesavento, PA, Byrne, BA, MacDonald, KA, and Chomel, BB. Evaluation of the relationship between causative organisms and clinical characteristics of infective endocarditis in dogs: 71 cases (1992-2005). J Am Vet Med Assoc. (2006) 228:1723–34. doi: 10.2460/javma.228.11.1723
21. Pinho, MD, Matos, SC, Pomba, C, Lübke-Becker, A, Wieler, LH, Preziuso, S, et al. Multilocus sequence analysis of Streptococcus canis confirms the zoonotic origin of human infections and reveals genetic exchange with Streptococcus dysgalactiae subsp. equisimilis. J Clin Microbiol. (2013) 51:1099–109. doi: 10.1128/JCM.02912-12
22. Taniyama, D, Abe, Y, Sakai, T, Kikuchi, T, and Takahashi, T. Human case of bacteremia caused by Streptococcus canis sequence type 9 harboring the scm gene. IDCases. (2017) 7:48–52. doi: 10.1016/j.idcr.2017.01.002
23. Ahmad, Y, Gertz, REJ, Li, Z, Sakota, V, Broyles, LN, Van Beneden, C, et al. Genetic relationships deduced from emm and multilocus sequence typing of invasive Streptococcus dysgalactiae subsp. equisimilis and S. canis recovered from isolates collected in the United States. J Clin Microbiol. (2009) 47:2046–54. doi: 10.1128/JCM.00246-09
24. Miller, CS, Handley, KM, Wrighton, KC, Frischkorn, KR, Thomas, BC, and Banfield, JF. Short-read assembly of full-length 16S amplicons reveals bacterial diversity in subsurface sediments. PLoS One. (2013) 8:e56018. doi: 10.1371/journal.pone.0056018
25. CLSI. CLSI M100 – Performance Standards for Antimicrobial Susceptibility Testing – 34th Edition. Clin Lab Stand Inst (2024)
26. CLSI. CLSI VET01S – Performance Standards for Antimicrobial Disk and Diluition Susceptibility Tests for Bacteria Isolated From Animals – 7th Edition. Clin Lab Stand Inst (2024)
27. Magiorakos, AP, Srinivasan, A, Carey, RB, Carmeli, Y, Falagas, ME, Giske, CG, et al. Multidrug-resistant, extensively drug-resistant and pandrug-resistant bacteria: an international expert proposal for interim standard definitions for acquired resistance. Clin Microbiol Infect. (2012) 18:268–81. doi: 10.1111/j.1469-0691.2011.03570.x
28. Chen, S, Zhou, Y, Chen, Y, and Gu, J. Fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. (2018) 34:i884–90. doi: 10.1093/bioinformatics/bty560
29. Prjibelski, A, Antipov, D, Meleshko, D, Lapidus, A, and Korobeynikov, A. Using SPAdes De novo assembler. Curr Protoc Bioinforma. (2020) 70:e102. doi: 10.1002/cpbi.102
30. Quast, C, Pruesse, E, Yilmaz, P, Gerken, J, Schweer, T, Yarza, P, et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. (2013) 41:D590–6. doi: 10.1093/nar/gks1219
31. Seemann, T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. (2014) 30:2068–9. doi: 10.1093/bioinformatics/btu153
32. Page, AJ, Cummins, CA, Hunt, M, Wong, VK, Reuter, S, Holden, MTG, et al. Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics. (2015) 31:3691–3. doi: 10.1093/bioinformatics/btv421
33. Price, MN, Dehal, PS, and Arkin, AP. FastTree 2 – approximately maximum-likelihood trees for large alignments. PLoS One. (2010) 5:e9490. doi: 10.1371/journal.pone.0009490
34. Fukushima, Y, Oshiteru, M, Yukie, K, Yuzo, T, Haruno, Y, Tetsuya, M, et al. Draft genome sequence of blood-origin Streptococcus canis strain FU149, isolated from a dog with necrotizing soft tissue infection. Microbiol Resour Announc. (2020) 9. doi: 10.1128/mra.00737-20
35. Lefébure, T, Richards, VP, Lang, P, Pavinski-Bitar, P, and Stanhope, MJ. Gene repertoire evolution of Streptococcus pyogenes inferred from phylogenomic analysis with Streptococcus canis and Streptococcus dysgalactiae. PLoS One. (2012) 7:e37607. doi: 10.1371/journal.pone.0037607
36. DeWinter, LM, Low, DE, and Prescott, JF. Virulence of Streptococcus canis from canine streptococcal toxic shock syndrome and necrotizing fasciitis. Vet Microbiol. (1999) 70:95–110. doi: 10.1016/S0378-1135(99)00128-5
37. Courtney, HS, Li, Y, Dale, JB, and Hasty, DL. Cloning, sequencing, and expression of a fibronectin/fibrinogen-binding protein from group a streptococci. Infect Immun. (1994) 62:3937–46. doi: 10.1128/iai.62.9.3937-3946.1994
38. Lee, IPA, and Andam, CP. Frequencies and characteristics of genome-wide recombination in Streptococcus agalactiae, Streptococcus pyogenes, and Streptococcus suis. Sci Rep. (2022) 12:1515. doi: 10.1038/s41598-022-04995-5
39. Fuursted, K, Stegger, M, Hoffmann, S, Lambertsen, L, Andersen, PS, Deleuran, M, et al. Description and characterization of a penicillin-resistant Streptococcus dysgalactiae subsp. equisimilis clone isolated from blood in three epidemiologically linked patients. J Antimicrob Chemother. (2016) 71:3376–80. doi: 10.1093/jac/dkw320
40. Moyaert, H, Morrissey, I, De Jong, A, El Garch, F, Klein, U, Ludwig, C, et al. Antimicrobial susceptibility monitoring of bacterial pathogens isolated from urinary tract infections in dogs and cats across Europe: ComPath results. Microb Drug Resist. (2017) 23:391–403. doi: 10.1089/mdr.2016.0110
41. El Garch, F, Youala, M, Simjee, S, Moyaert, H, Klee, R, Truszkowska, B, et al. Antimicrobial susceptibility of nine udder pathogens recovered from bovine clinical mastitis milk in Europe 2015–2016: VetPath results. Vet Microbiol. (2020) 245:108644. doi: 10.1016/j.vetmic.2020.108644
42. Oh, SI, Kim, JW, Kim, J, So, B, Kim, B, and Kim, HY. Molecular subtyping and antimicrobial susceptibility of Streptococcus dysgalactiae subspecies equisimilis isolates from clinically diseased pigs. J Vet Sci. (2020) 21:e57. doi: 10.4142/jvs.2020.21.e57
43. Rojo-Bezares, B, Toca, L, Azcona-Gutiérrez, JM, Ortega-Unanue, N, Toledano, P, and Sáenz, Y. Streptococcus dysgalactiae subsp. equisimilis from invasive and non-invasive infections in Spain: combining epidemiology, molecular characterization, and genetic diversity. Eur J Clin Microbiol Infect Dis. (2021) 40:1013–21. doi: 10.1007/s10096-020-04119-9
44. Galpérine, T, Cazorla, C, Blanchard, E, Boineau, F, Ragnaud, J-M, and Neau, D. Streptococcus canis infections in humans: retrospective study of 54 patients. J Infect. (2007) 55:23–6. doi: 10.1016/j.jinf.2006.12.013
45. Lyskova, P, Vydrzalova, M, and Mazurova, J. Identification and antimicrobial susceptibility of bacteria and yeasts isolated from healthy dogs and dogs with otitis externa. J Vet Med A Physiol Pathol Clin Med. (2007) 54:559–63. doi: 10.1111/j.1439-0442.2007.00996.x
46. Stefańska, I, Kwiecień, E, Kizerwetter-Świda, M, Chrobak-Chmiel, D, and Rzewuska, M. Tetracycline, macrolide and lincosamide resistance in Streptococcus canis strains from companion animals and its genetic determinants. Antibiotics. (2022) 11. doi: 10.3390/antibiotics11081034
47. Fukushima, Y, Tsuyuki, Y, Goto, M, Yoshida, H, and Takahashi, T. Novel quinolone nonsusceptible Streptococcus canis strains with point mutations in quinolone resistance-determining regions and their related factors. Jpn J Infect Dis. (2020) 73:242–9. doi: 10.7883/yoken.JJID.2019.392
48. Pinho, MD, Foster, G, Pomba, C, MacHado, MP, Baily, JL, Kuiken, T, et al. Streptococcus canis are a single population infecting multiple animal hosts despite the diversity of the universally present M-like protein SCM. Front Microbiol. (2019) 10:1–10. doi: 10.3389/fmicb.2019.00631
49. Fukushima, Y, Takahashi, T, Goto, M, Yoshida, H, and Tsuyuki, Y. Novel diverse sequences of the Streptococcus canis M-like protein (SCM) gene and their prevalence in diseased companion animals: association of their alleles with sequence types. J Infect Chemother. (2020) 26:908–15. doi: 10.1016/j.jiac.2020.04.004
Keywords: Streptococcus canis , antibiotic resistance, virulence, zoonosis, epidemiology
Citation: Cucco L, Albini E, Blasi F, Orsini S, Fiorucci A, Dettori A, Petrin S, Peruzzo A, Salaris S, Paniccià M, Pezzotti G, Massacci FR and Magistrali CF (2025) Genomic analysis of Streptococcus canis from different hosts in Italy 2004–2021: diversity, antimicrobial resistance, and virulence profiles. Front. Vet. Sci. 12:1645885. doi: 10.3389/fvets.2025.1645885
Edited by:
Ihab Habib, United Arab Emirates University, United Arab EmiratesReviewed by:
Mihaela Niculae, University of Agricultural Sciences and Veterinary Medicine of Cluj-Napoca, RomaniaNattinee Kittiwan, Department of Livestock Development, Thailand
Copyright © 2025 Cucco, Albini, Blasi, Orsini, Fiorucci, Dettori, Petrin, Peruzzo, Salaris, Paniccià, Pezzotti, Massacci and Magistrali. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Francesca Romana Massacci, ZnIubWFzc2FjY2lAaXpzdW0uaXQ=