 Liyang Li
Liyang Li Wen Feng
Wen Feng- Department of Clinical Veterinary Medicine, College of Veterinary Medicine, Huazhong Agricultural University, Wuhan, China
Introduction: Feline herpesvirus type 1 (FHV-1) is a primary pathogen causing feline upper respiratory tract diseases (FURTD), but its impact on the upper respiratory tract microbiota remains unclear. This study aimed to evaluate the impact of FHV-1 infection on the upper respiratory tract microbiota by comparing the microbial composition between FHV-1-positive group with FHV-1-negative group.
Methods: The microbial diversity in the upper respiratory tract of FHV-1-positive cats (n = 8) were analyzed using 16S rRNA high-throughput sequencing, and then this diversity was compared with that in healthy FHV-1-negative controls (n = 4).
Results: Sequencing results showed that FHV-1 infection significantly increased microbial diversity (Shannon index: 5.55 ± 0.17 vs. 5.30 ± 0.11, p < 0.05; Simpson index: 0.95 ± 0.01 vs. 0.94 ± 0.00, p < 0.01) and altered community structure, as indicated by beta diversity analysis. At the phylum level, Actinobacteriota showed significantly higher relative abundance in the FHV-1-positive group than in the FHV-1-negative control group (p < 0.05). For the genus level, Porphyromonas and Bergeyella were significantly less abundant in FHV-1-positive group versus FHV-1-negative healthy control group (p < 0.05). Linear discriminant analysis effect size (LEfSe) identified Prevotellaceae as a biomarker for the FHV-1-positive group.
Discussion: This study provided the first evidence that FHV-1 infection significantly alters the diversity and composition of the upper respiratory tract microbiota in cats. These microbiota changes were likely to play an important role in the pathogenesis of FURTD and offer new targets for the development of microbiota-based therapeutic strategies.
1 Introduction
Feline herpesvirus type 1 (FHV-1) is one of the most prevalent etiological agents of feline upper respiratory tract disease (FURTD), and has a global endemic presence (1). Epidemiological investigations demonstrate that the global prevalence of FHV-1 infection ranges from 20.9 to 55.26% in domestic cat populations, with significantly higher infection rates observed in high-density environments such as multi-cat households and animal shelters (2–4). Infected cats typically manifest classic symptoms, including conjunctivitis, rhinitis, and tracheitis (5). Severe cases may progress to corneal ulcers, pneumonia, or even death, posing a substantial threat to the global cat breeding industry and feline pet health (6, 7). Existing studies have demonstrated that cats infected with FHV-1 will maintain lifelong viral persistence (2). Following primary infection, FHV-1 typically establishes latency in the trigeminal ganglia and mandibular nerves of affected cats without causing clinical manifestations. However, viral reactivation and subsequent replication may occur during periods of immunosuppression or environmental stress, leading to recurrent clinical disease (8).
The upper respiratory microbiota, serving as the first line of defense against pathogens, engages in intricate symbiotic interactions with the host (9). Research in human medicine has demonstrated that respiratory viral infections can significantly disrupt microbial diversity, promote the overgrowth of opportunistic pathogens, and exacerbate tissue damage and systemic inflammation (10). Similarly, studies on feline calicivirus (FCV) infection have linked oral dysbiosis to elevated levels of pro-inflammatory cytokines, including IL-1β and TNF-α (11). However, whether FHV-1 infection similarly disrupts the upper respiratory microbiota and contributes to disease progression remains unclear. Despite advancements in understanding FHV-1 infection mechanisms and its interaction with host immunity, the dynamic changes in the upper respiratory microbiota following FHV-1 infection, key microbial taxa, and the mechanisms of microbe-immune interactions remain undefined (12).
This study aimed to compare the microbial diversity between FHV-1-positive and healthy cats, identify differentially abundant taxa and potential biomarkers, and explore the clinical implications of these findings for FURTD management. By addressing these objectives, our research endeavors to provide novel theoretical foundations and innovative strategies for clinical diagnosis and treatment.
2 Materials and methods
2.1 Sample collection and grouping
All samples for this study were derived from an epidemiological survey of feline upper respiratory tract disease conducted in Wuhan between 2021 and 2024, encompassing a total of 1,427 cases (Supplementary material 1). From this dataset, we selected 8 FHV-1-positive samples based on stringent inclusion criteria, ensuring all specimens were obtained from adult British Shorthair cats exhibiting upper respiratory symptoms, housed in single-cat household, and having received routine vaccinations. The cohort maintained a balanced gender distribution (4 males and 4 females), with laboratory confirmation via RT-qPCR confirming FHV-1 positivity while excluding co-infections with feline calicivirus (FCV), Mycoplasma felis (Mf), Chlamydia felis (Cf), and Bordetella bronchiseptica (Bb). Additionally, four FHV-1-negative control samples were included, sourced from asymptomatic adult British Shorthairs under identical conditions (single-cat households, vaccinated, 2 males and 2 females), with RT-qPCR verification confirming the absence of FHV-1, FCV, Mf, Cf, and Bb. All samples were collected from animals that had not received antimicrobial treatment within the preceding week (Table 1).
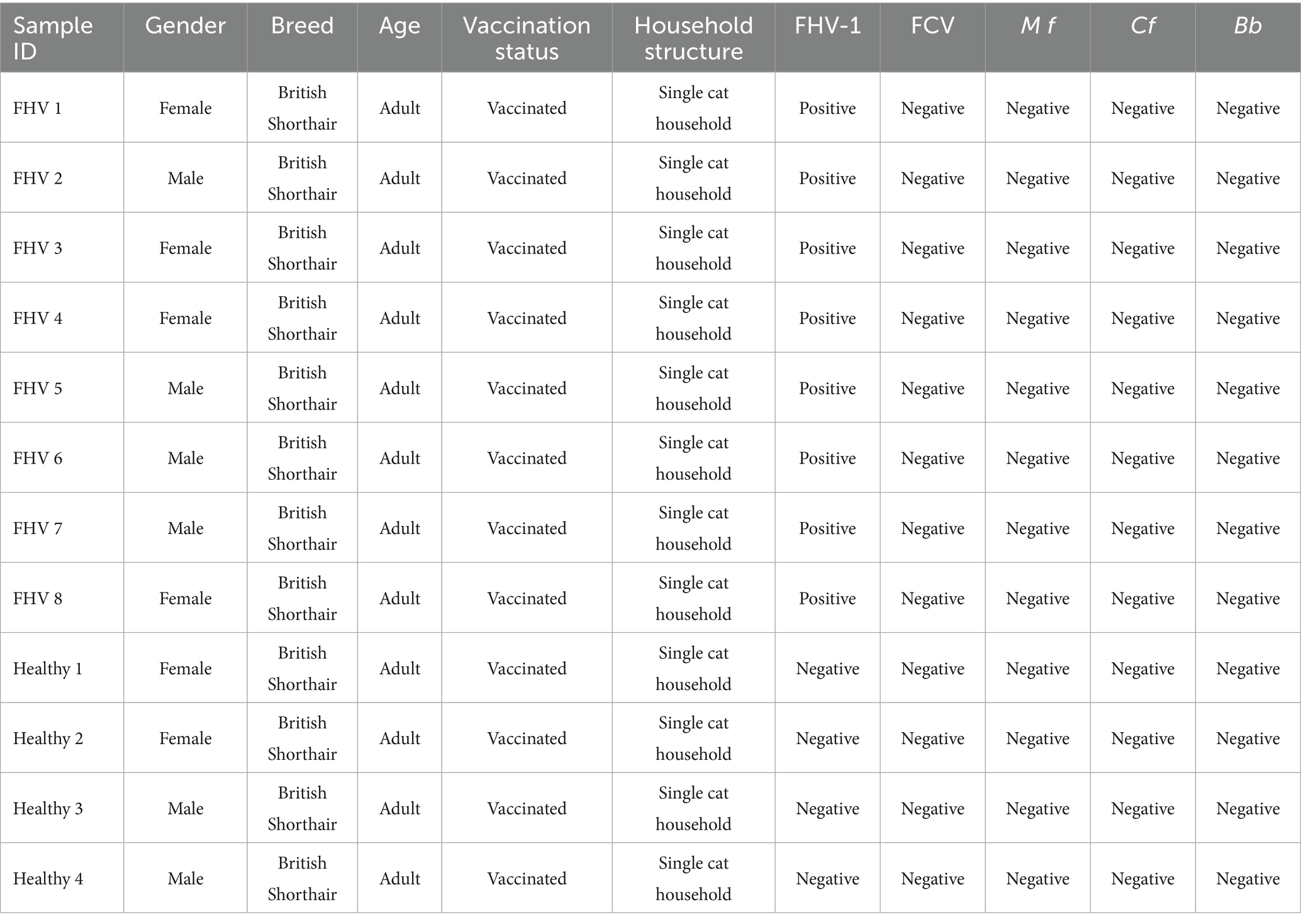
Table 1. Basic information of the samples.
The sterile swabs were inserted transnasally to the nasopharynx (depth adjusted by weight: 4–5 cm for <3 kg, 5–7 cm for ≥3 kg), rotated three times against the posterior pharyngeal wall, and immediately preserved in RNA/DNA stabilization solution at −80°C. All procedures complied with the Guide for the Care and Use of Laboratory Animals (National Research Council, 2011) and were approved by the Institutional Animal Care and Use Committee of HZAU (protocol code: HZAUCA-2025-0038). Owners provided written consent for sample collection and data use, including potential risks and benefits.
2.2 High-throughput 16S rRNA gene sequencing
Total DNA was extracted using the TGuide S96 Magnetic Soil/Feces DNA Kit (Tiangen Biotech, China) with negative controls to exclude contamination (sterile ultrapure water) according to manufacturer’s instructions. The quality and quantity of the extracted DNA were examined using electrophoresis on a 1.8% agarose gel, and DNA concentration and purity were determined with NanoDrop 2000 UV–Vis spectrophotometer (Thermo Scientific, USA). The full-length 16S rRNA gene were amplified with primer pairs 27F: AGRGTTTGATYNTGGCTCAG and 1492R: TASGGHTACCTTGTTASGACTT. Both the forward and reverse 16S primers were tailed with sample-specific PacBio barcode sequences to allow for multiplexed sequencing. We chose to use barcoded primers because this reduces chimera formation as compared to the alternative protocol in which primers are added in a second PCR reaction. The KOD One PCR Master Mix (TOYOBOLife Science, Japan) was used to perform 25 cycles of PCR amplification, with initial denaturation at 95°C for 2 min, followed by 25 cycles of denaturation at 98°C for 10 s, annealing at 55°C for 30 s, and extension at 72°C for 1 min 30 s, and a final step at 72°C for 2 min. The total of PCR amplicons was purified with VAHTSTM DNA Clean Beads (Vazyme, China) and quantified using the Qubit dsDNA HS Assay Kit and Qubit 3.0 Fluorometer (Thermo Fisher Scientific, USA). After the individual quantification step, amplicons were pooled in equal amounts. SMRTbell libraries were prepared from the amplified DNA by SMRTbell Express Template Prep Kit 2.0 according to the manufacturer’s instructions (Pacific Biosciences, USA). Purified SMRTbell libraries from the pooled and barcoded samples were sequenced on a PacBio Sequel II platform (Beijing Biomarker Technologies, China) using Sequel II binding kit 2.0.
2.3 Bioinformatics analysis
The bioinformatics analysis of this study was performed with the aid of the BMKCloud.1 The raw reads generated from sequencing were filtered and demultiplexed using the SMRT Link software (version 8.0) with the minPasses ≥5 and minPredicted Accuracy ≥0.9, in order to obtain the circular consensus sequencing (CCS) reads. Subsequently, the lima (version 1.7.0) was employed to assign the CCS sequences to the corresponding samples based on their barcodes. CCS reads containing no primers and those reads beyond the length range (1,200–1,650 bp) were discarded through the recognition of forward and reverse primers and quality filtering using the Cutadapt (version 2.7) quality control process. The UCHIME algorithm (v8.1) was used in detecting and removing chimera sequences to obtain the clean reads. Sequences with similarity > 97% were clustered into the same operational taxonomic unit (OTU) by USEARCH (v10.0), and the OTUs < 2 in all samples were filtered.
Clean reads then were conducted on feature classification to output ASVs (amplicon sequence variants) by DADA2, and the ASVs < 2 in all samples were filtered. Taxonomy annotation of the OTUs/ASVs was performed based on the Naive Bayes classifier in QIIME2 using the SILVA database (release 138.1) with a confidence threshold of 70%. The Alpha diversity was calculated and displayed by the QIIME2 and R software, respectively. Beta diversity was determined to evaluate the degree of similarity of microbial communities from different samples using QIIME. The Permutational Multivariate Analysis of Variance (PERMANOVA) analyses were performed using the vegan package in R, with visualizations generated in Python. Prior to the above analysis, rarefaction curves and rank abundance curves were generated to assess the sequencing depth. Furthermore, differential taxa identification between groups was performed using Linear discriminant analysis effect size (LEfSe) analysis with a Linear Discriminant Analysis (LDA) score threshold of 2.0 and a p-value cutoff of < 0.05, combined with Wilcoxon rank-sum tests to validate group-specific differences in individual taxonomic units. For dominant genera with relatively high abundance (relative abundance > 1%), further validation was conducted using ANOVA tests for normally distributed data or Kruskal-Wallis tests for non-normally distributed data. p-values were corrected using the Benjamini-Hochberg method to control the false discovery rate.
3 Results
3.1 Analysis of sequencing sequence and OTUs number
Sequencing results showed that the FHV-1-positive group obtained an average of 51,679.375 effective CCS, while the FHV-1-negative group obtained an average of 54,363.25 effective CCS (Table 2). The rarefaction curve results indicated that all curves approached a saturation trend, suggesting that the number of species in this environment would not increase significantly with the increase in sequencing depth (Figures 1A,B). At 97% sequence similarity threshold, microbial community analysis identified 272 shared OTUs between FHV-1-positive group and FHV-1-negative group. FHV-1-positive group harbored 120 unique OTUs, whereas FHV-1-negative group contained 14 unique OTUs (Figures 1C,D). These findings demonstrate significant disparities in microbial diversity between the two groups, with FHV-1-positive group exhibiting remarkably higher microbial diversity than FHV-1-negative group.
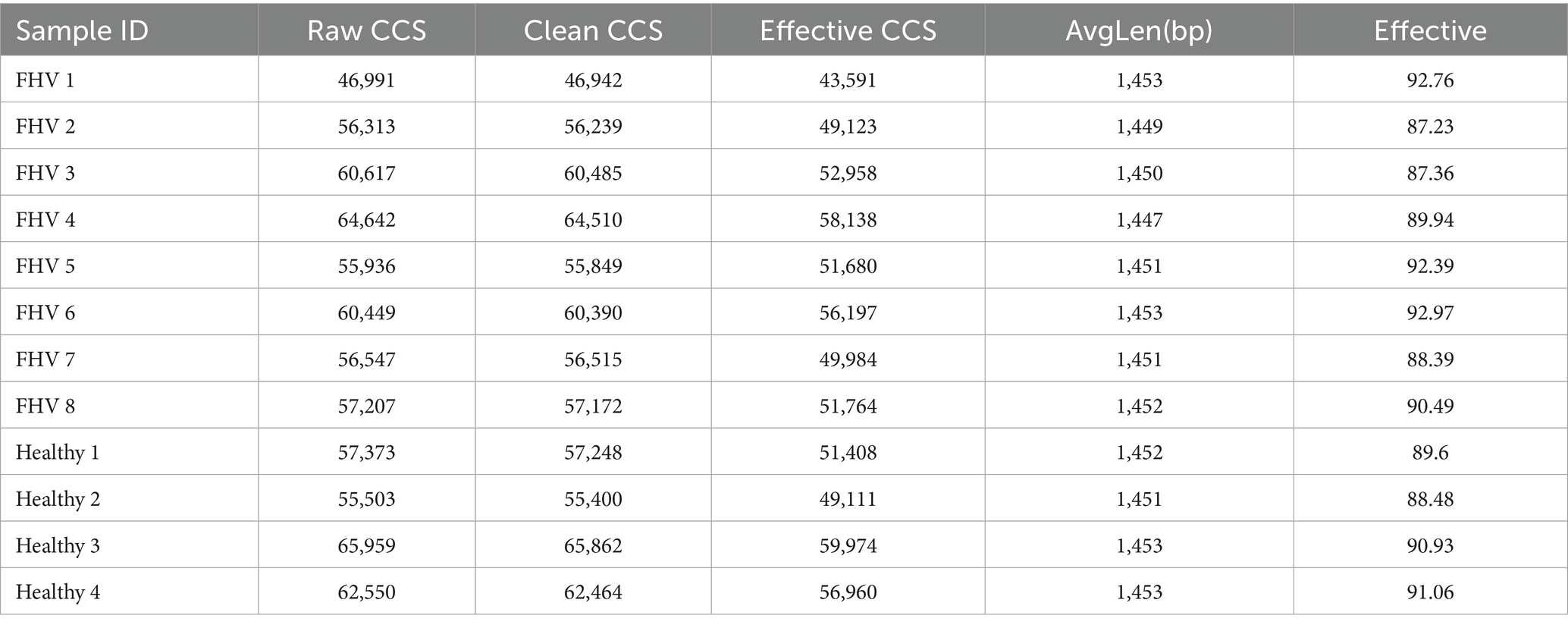
Table 2. Statistics of sample sequencing data processing results.
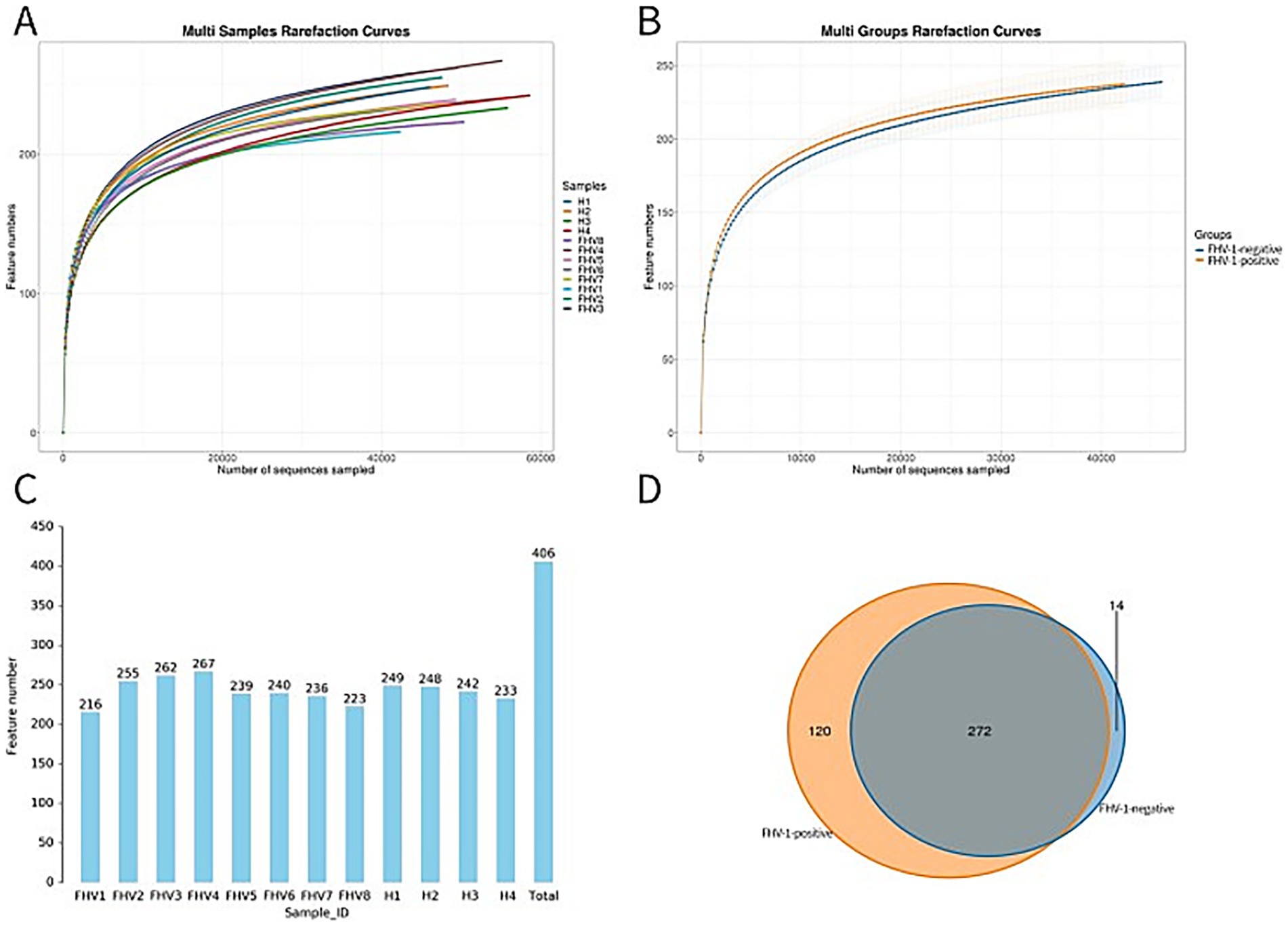
Figure 1. Display of OTU clustering results. (A) Multi samples rarefaction curves. (B) Multi groups rarefaction curves. (C) Number of OTUs in each sample. (D) Venn diagram of OTU distribution between FHV-1-positive and FHV-1-negative cats.
3.2 Alpha diversity
Alpha diversity analysis demonstrated significant microbial community differences between groups (Figure 2). FHV-1-positive group exhibited significantly higher Shannon (p < 0.05) and Simpson indices (p < 0.01) compared to FHV-1-negative group, indicating enhanced microbial diversity in infected individuals. However, no significant intergroup differences were observed in Chao1 (p > 0.05) or ACE indices (p > 0.05), suggesting comparable species richness between groups (Table 3).
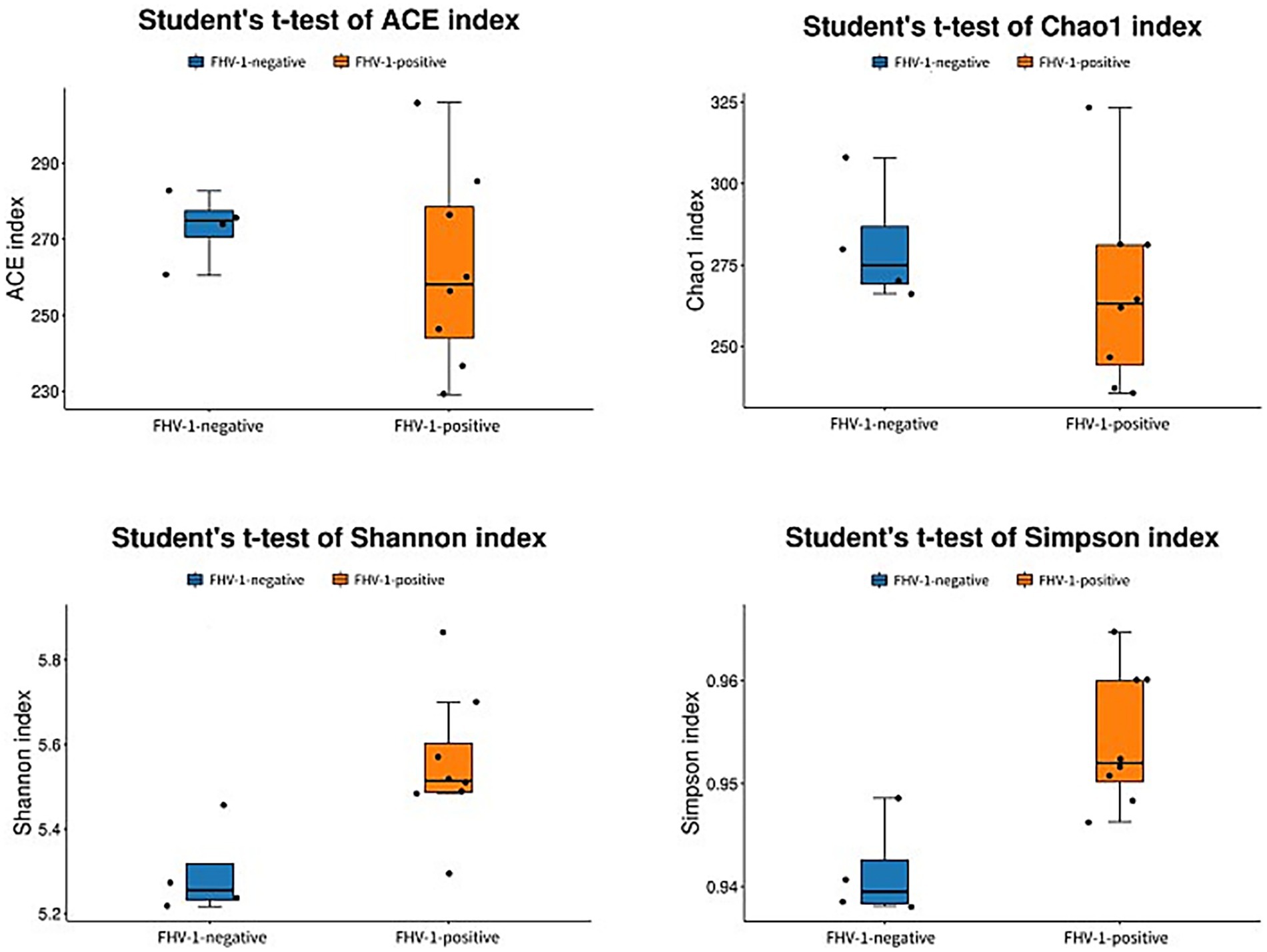
Figure 2. Analyses diagram of alpha diversity index.
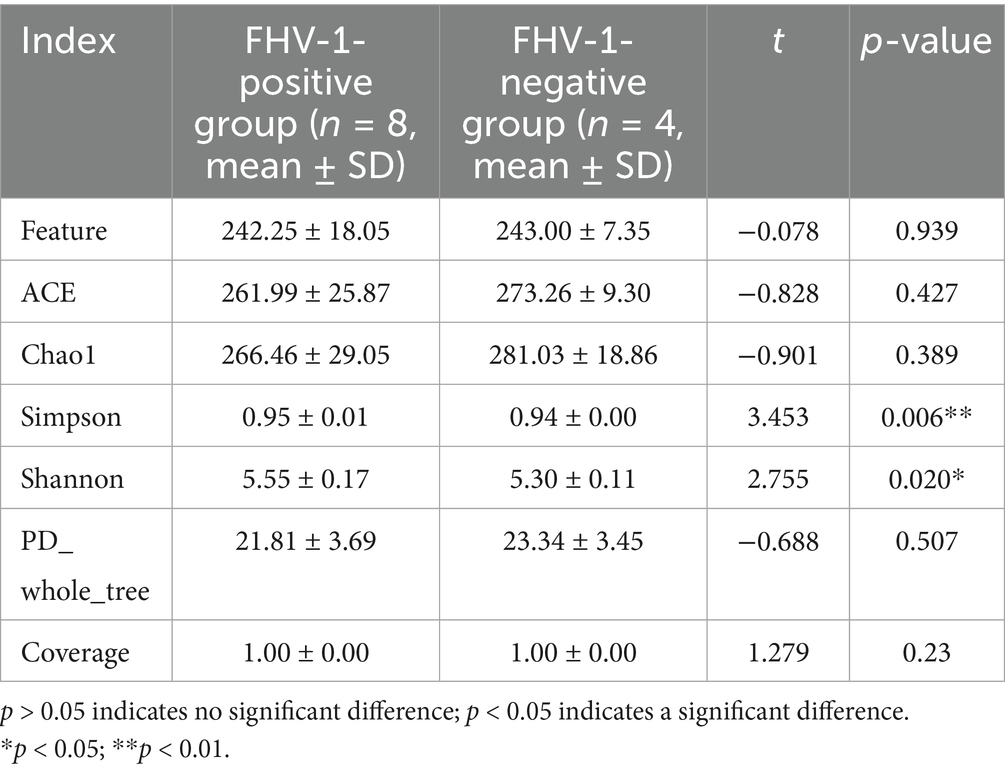
Table 3. Analysis of differences in alpha diversity indices.
3.3 Beta diversity
Principal component analysis (PCA) and Principal coordinates analysis (PCoA) revealed distinct microbial community structures between groups (Figures 3A,B). The FHV-1-negative group showed tight clustering with minimal intra-group variation, while the FHV-1-positive group exhibited significantly greater dispersion, indicating substantial compositional dissimilarities.
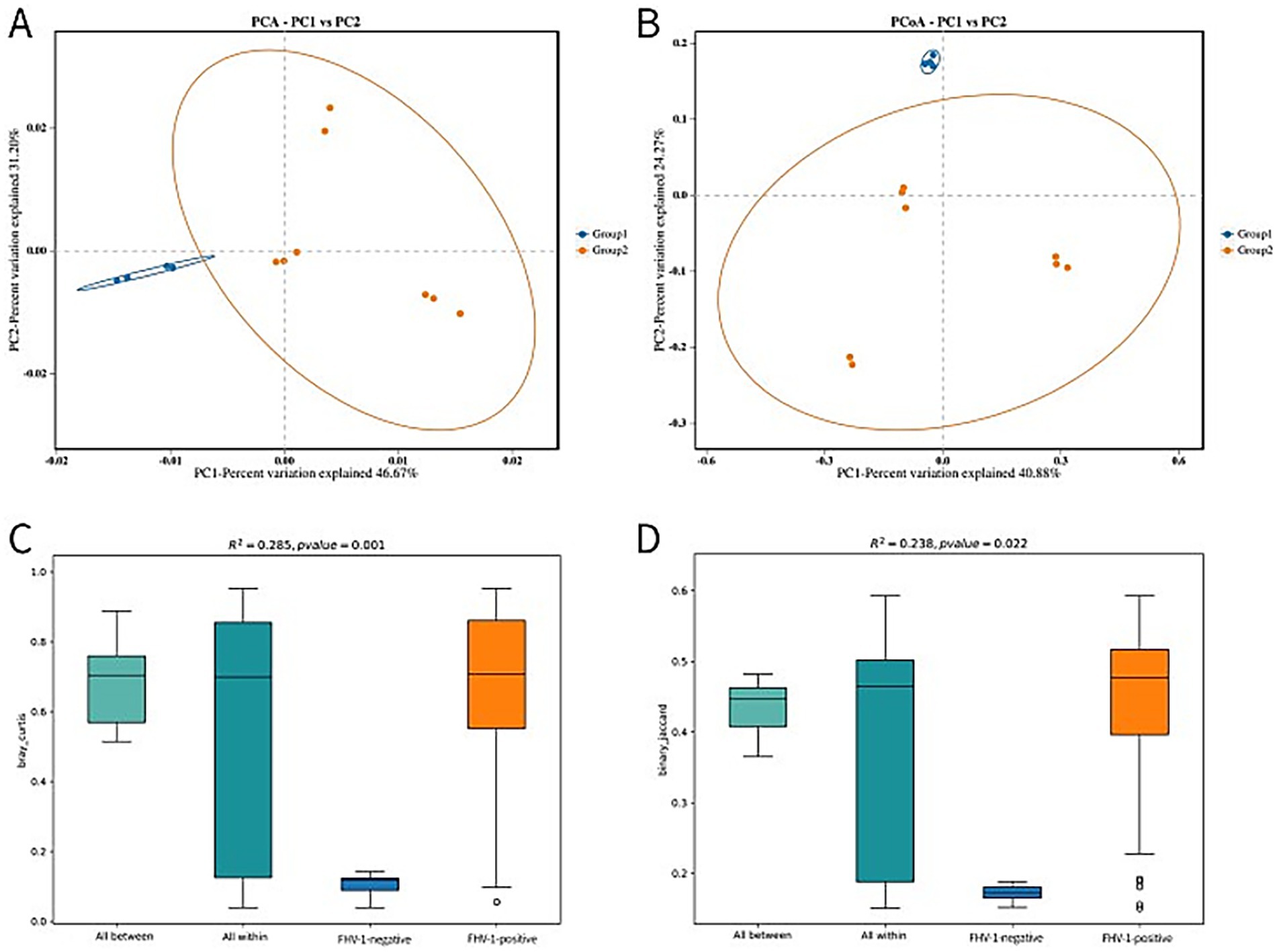
Figure 3. Beta diversity analysis plots. (A) PCA analysis plot. The first two principal components account for 46.67% (PC1) and 31.20% (PC2) of the total variation. (B) PCoA analysis plot. The first two principal components account for 40.88% (PC1) and 24.27% (PC2) of the total variation. (C) PERMANOVA analysis box plot (based on Bray-Curtis distance). (D) PERMANOVA analysis box plot (based on binary Jaccard distance).
The PERMANOVA analysis results demonstrated differential sensitivity of distance metrics in detecting microbial community variation. Analysis using Bray–Curtis dissimilarity showed highly significant differences (R2 = 0.285, p = 0.001) (Figure 3C), whereas the binary Jaccard metric yielded more modest but still statistically significant separation (R2 = 0.238, p = 0.022) (Figure 3D). A larger R2 indicates a higher degree of explanatory power of group partitioning for the observed differences, reflecting greater divergence between groups, while a p-value < 0.05 signifies high reliability of the test results.
3.4 Taxonomic composition and differential abundance
The top 10 phyla in terms of species abundance are Bacteroidota, Firmicutes, Proteobacteria, Fusobacteriota, Spirochaetota, Patescibacteria, Desulfobacterota, Synergistota, Campylobacterota, and Actinobacteriota (Figures 4A,B). Among them, the abundance of Actinobacteriota in the FHV-1-positive group is significantly higher than that in the FHV-1-negative group (0.34% ± 0.05% vs. 0.17% ± 0.03%, p = 0.0433) (Table 4).
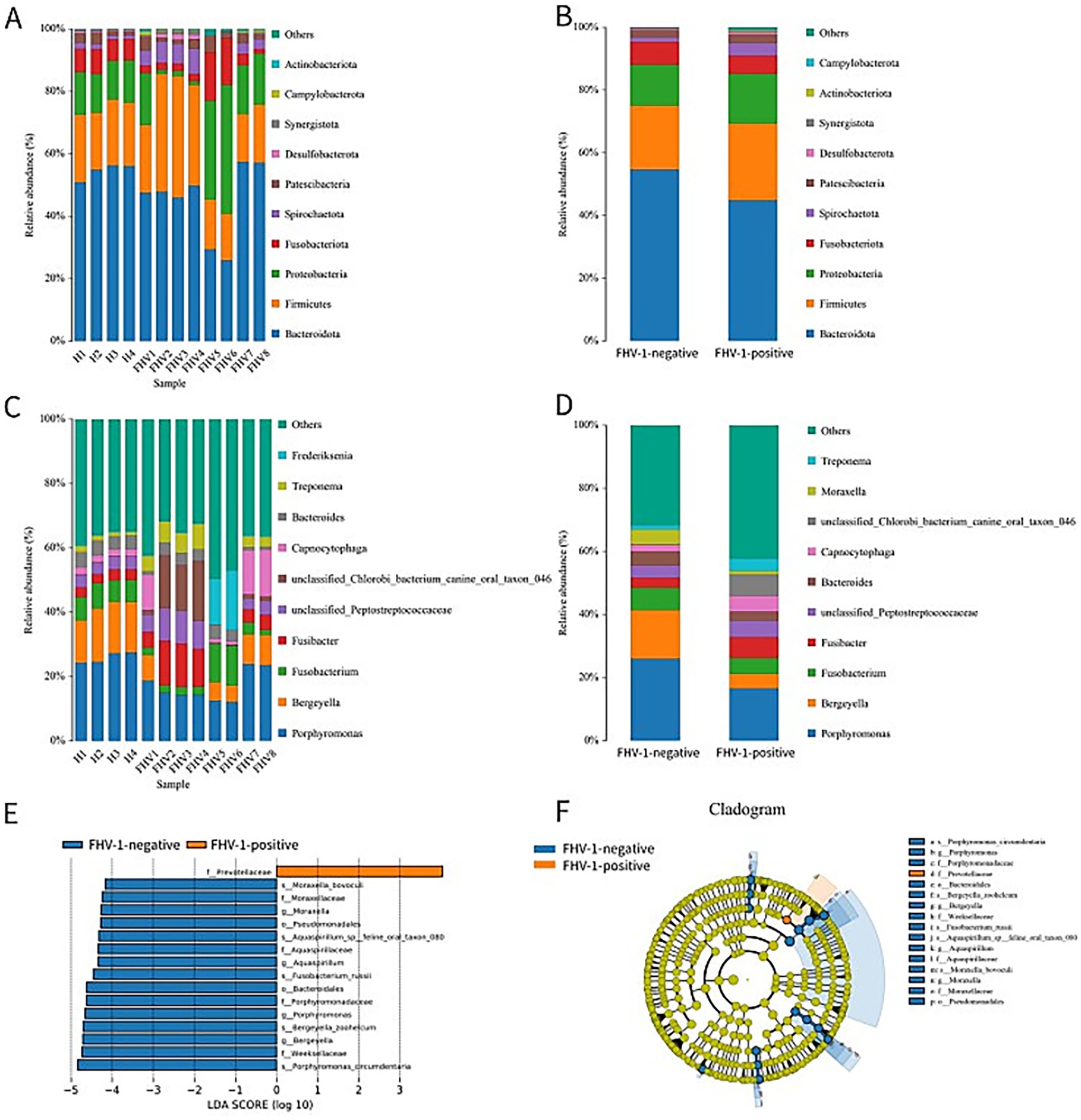
Figure 4. Species classification composition diagrams. (A) Species distribution diagram of each sample at the phylum level. (B) Inter-group species distribution diagram of FHV-1-positive and FHV-1-negative at the phylum level. (C) Species distribution diagram of each sample at the genus level. (D) Inter-group species distribution diagram of FHV-1-positive and FHV-1-negative at the genus level. (E) LDA scores of significant taxa (LDA > 4, p < 0.05). (F) Cladogram showing phylogenetic distribution of differentially abundant taxa (colored by enriched group; yellow = non-significant).
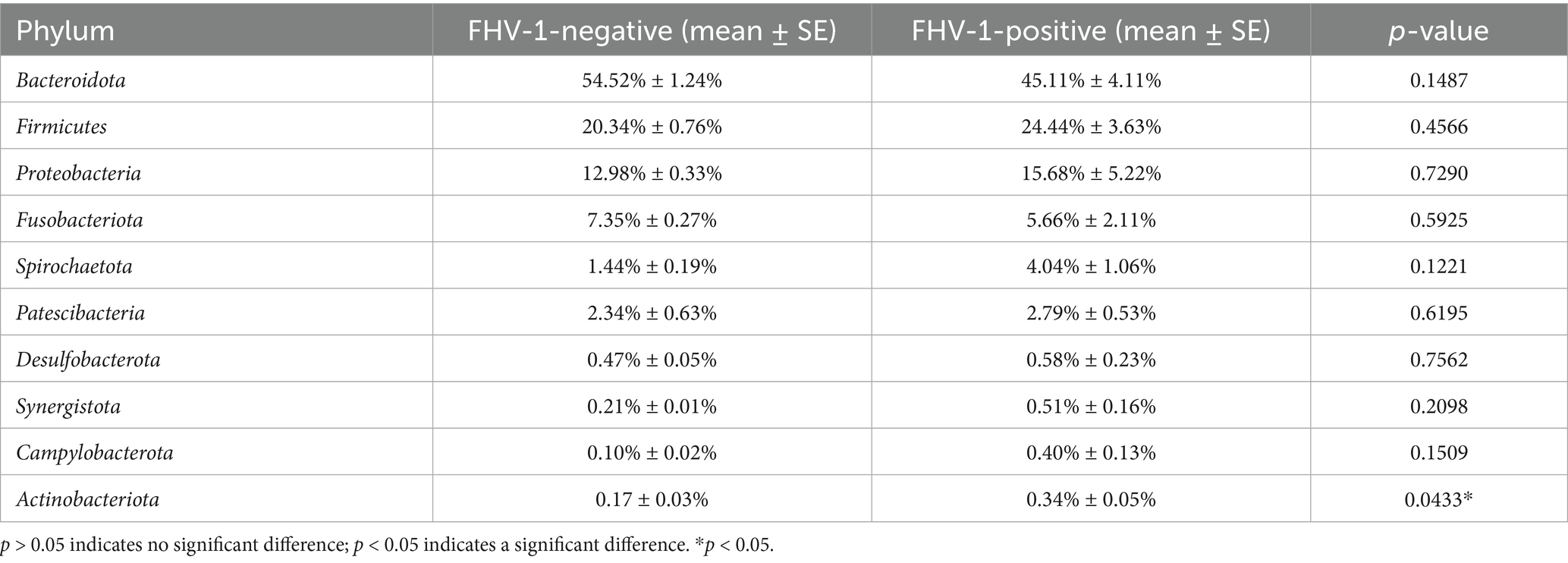
Table 4. Analysis of relative abundance of microbiota at phylum level between two groups of samples.
The top 10 genera in terms of species abundance are Porphyromonas, Bergeyella, Fusobacterium, Fusibacter, unclassified_Peptostreptococcaceae, unclassified_Chlorobi_bacterium_canine_oral_taxon_046, Capnocytophaga, Bacteroides, Treponema, and Frederiksenia (Figures 4C,D). At the genus level, Porphyromonas (16.77% ± 1.66% vs. 25.88% ± 0.86%, p = 0.0043), Bergeyella (4.68% ± 1.45% vs. 15.38% ± 0.76%, p = 0.0006) showed marked reductions in the FHV-1-positive group (Table 5). These reductions may disrupt commensal microbiota-mediated immune surveillance, potentially facilitating secondary infections.
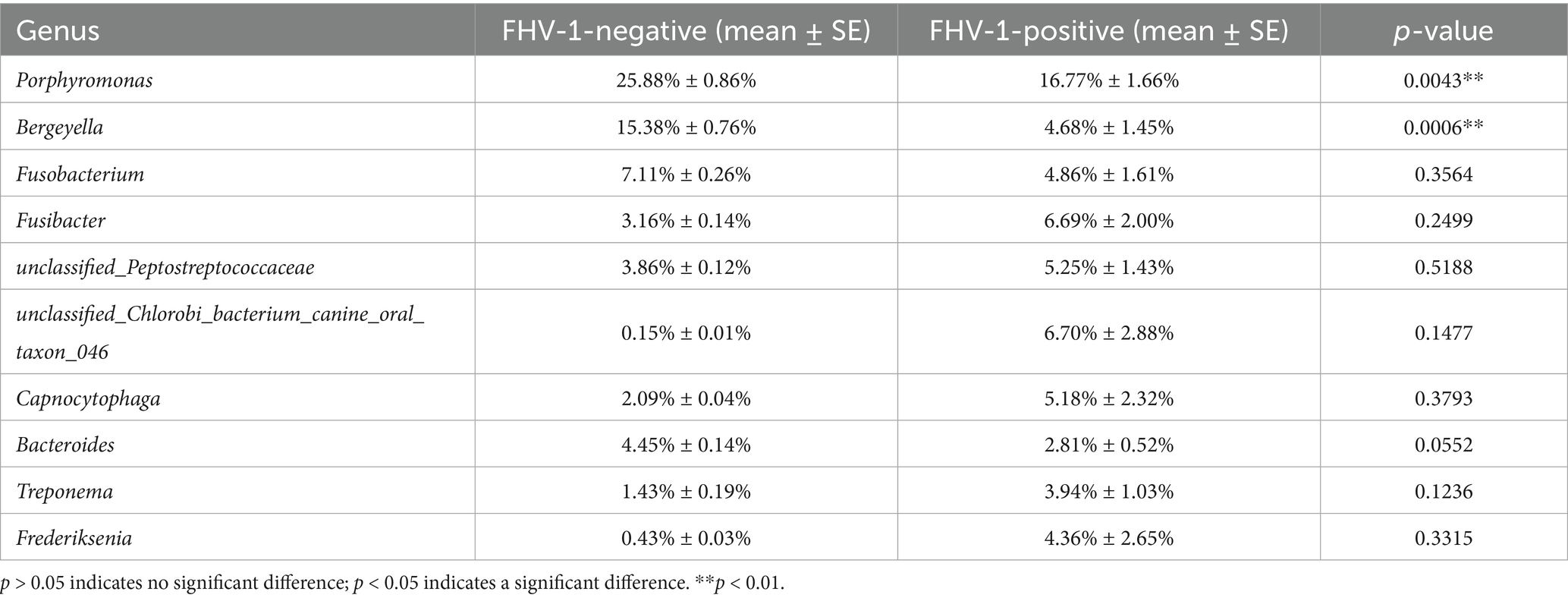
Table 5. Analysis of relative abundance of microbiota at genus level between two groups of samples.
LEfSe analysis revealed that the family Prevotellaceae (LDA score> 4, p < 0.05) was significantly enriched in the FHV-1-positive group compared to FHV-1-negative group (Figure 4E), with its taxonomic hierarchy further illustrated in the cladogram (Figure 4F).
4 Discussion
Our study revealed a statistically significant elevation in microbial alpha diversity in FHV-1-positive cats, as demonstrated by increased Shannon (p = 0.02) and Simpson (p = 0.006) indices compared to their FHV-1-negative counterparts. This observed shift in microbial diversity likely results from the complex interplay between virus, host, and microbiota dynamics (13). Specifically, FHV-1-induced mucosal damage may disrupt colonization resistance mechanisms, thereby facilitating opportunistic pathogen invasion and proliferation. Furthermore, the virus-mediated immunosuppression appears to compromise host microbial regulation, permitting expansion of typically commensal or low-abundance bacterial species. We hypothesize that the increased alpha diversity in FHV-1-positive cats may be related to the risk of secondary infection. Previous studies have shown that viral infections can disrupt mucosal barriers and increase microbial diversity, thereby promoting secondary bacterial colonization (14). To minimize sampling bias, all samples were collected from the same anatomical site using sterile swabs, and samples were only included if collected within 24 h of clinical symptom onset to reduce the impact of disease progression on microbial composition.
The beta diversity analysis clearly showed a significant alteration in the microbial community structure in FHV-1-positive group. The PCA, PCoA and PERMANOVA analysis results indicated that the microbiota of the FHV-1-positive group was more dispersed and distinct from that of the FHV-1-negative group. This structural change suggests that FHV-1 infection remodels the upper respiratory tract microbiota, potentially affecting its normal functions.
The significant increase in the abundance of Actinobacteriota at the phylum level in the FHV-1-positive group is particularly noteworthy. Actinobacteriota are known to produce a variety of bioactive compounds, including antibiotics and immunomodulatory substances (15). In the context of FHV-1 infection, the increased abundance of Actinobacteriota might represent an adaptive response of the microbiota to combat the viral infection (16). Conversely, the significant reduction in the abundance of Porphyromonas and Bergeyella at the genus level in the FHV-1-positive is also of great interest. The altered microenvironment caused by FHV-1 infection, such as changes in pH, oxygen concentration, or the availability of nutrients, may no longer be favorable for their growth and survival (17). However, the reduction in these genera that are potentially pathogenic does not necessarily guarantee a beneficial outcome. It could disrupt the normal ecological balance of the microbiota, leading to the overgrowth of other opportunistic pathogens (18, 19).
The identification of Prevotellaceae as a biomarker for the FHV-1-positive group by LEfSe analysis provides a new perspective on the role of specific microbial families in FHV-1 infection. Prevotellaceae have been associated with various physiological and pathological processes in the respiratory tract (20, 21). In this study, its increased abundance in the infected group suggests that it may play a role in the response to FHV-1 infection. It could be involved in modulating the host immune response, competing with other pathogens for resources, or adapting to the altered microenvironment caused by the virus (22, 23).
FHV-1 infection drives microbial community changes through a cascade of interconnected biological events. The virus initially damages mucosal epithelial cells through cytolytic replication, compromising both physical barriers and chemical defenses like mucins and antimicrobial peptides (24). Simultaneously, FHV-1 orchestrates a complex immune modulation, first suppressing pro-inflammatory cytokines to evade detection before triggering excessive inflammation. This immunological manipulation disrupts the host’s ability to regulate microbial populations through multiple pathways—impairing phagocytic clearance, altering nutrient competition dynamics, and modifying the availability of critical resources like iron (25). The virus further influences bacterial communities by depleting host cellular resources and modifying surface receptor expression, thereby reshaping the metabolic landscape and adhesion sites within the mucosal environment. These multifaceted interactions between viral pathogenesis, host responses, and microbial ecology collectively explain the observed shifts in both diversity and composition of the respiratory microbiota (26).
5 Conclusion
This study provided the first evidence that FHV-1 infection significantly alters the diversity and composition of the upper respiratory tract microbiota in cats. These microbiota changes were likely to play an important role in the pathogenesis of FURTD and offer new targets for the development of microbiota-based therapeutic strategies.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding author.
Ethics statement
The animal studies were approved by The Institutional Animal Care and Use Committee of Huazhong Agricultural University. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent was obtained from the owners for the participation of their animals in this study.
Author contributions
LL: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Project administration, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. WF: Conceptualization, Investigation, Supervision, Writing – original draft, Writing – review & editing. LB: Conceptualization, Investigation, Software, Writing – original draft, Writing – review & editing. GD: Formal analysis, Funding acquisition, Project administration, Resources, Validation, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. The study was funded under the special fund for Small Animal Diseases of Huazhong Agricultural University, Wuhan, China.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The authors declare that no Gen AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fvets.2025.1663056/full#supplementary-material
Footnotes
References
1. Bergmann, M, Ballin, A, Schulz, B, Dörfelt, R, and Hartmann, K. Treatment of acute viral feline upper respiratory tract infections. Tierarztl Prax Ausg K Kleintiere Heimtiere. (2019) 47:98–109. doi: 10.1055/a-0870-0801
2. Cavalheiro, JB, Echeverria, JT, Ramos, CAN, and Babo-Terra, VJ. Frequency of feline herpesvirus 1 (Fhv-1) in domestic cats from Campo Grande, MS, Brazil. An Acad Bras Cienc. (2023) 95:e20221010. doi: 10.1590/0001-3765202320221010
3. Kim, S, Cheng, Y, Fang, Z, Zhongqi, Q, Weidong, Y, Yilmaz, A, et al. First report of molecular epidemiology and phylogenetic characteristics of feline herpesvirus (Fhv-1) from naturally infected cats in Kunshan, China. Virol J. (2024) 21:115. doi: 10.1186/s12985-024-02391-1
4. Płoneczka-Janeczko, K, Bierowiec, K, Bania, J, Kiełbowicz, M, and Kiełbowicz, Z. Felid herpesvirus 1 (Fhv 1) carriers among urban breeding facilities in Wroclaw (Poland). Berl Munch Tierarztl Wochenschr. (2014) 127:243–6. doi: 10.2376/0005-9366-127-243
5. Gaskell, R, Dawson, S, Radford, A, and Thiry, E. Feline Herpesvirus. Vet Res. (2007) 38:337–54. doi: 10.1051/vetres:2006063
6. Gould, D. Feline herpesvirus-1: ocular manifestations, diagnosis and treatment options. J Feline Med Surg. (2011) 13:333–46. doi: 10.1016/j.jfms.2011.03.010
7. Vaz, PK, Job, N, Horsington, J, Ficorilli, N, Studdert, MJ, Hartley, CA, et al. Low genetic diversity among historical and contemporary clinical isolates of felid herpesvirus 1. BMC Genomics. (2016) 17:704. doi: 10.1186/s12864-016-3050-2
8. Gaskell, RM, and Povey, RC. Re-excretion of feline viral rhinotracheitis virus following corticosteroid treatment. Vet Rec. (1973) 93:204–5. doi: 10.1136/vr.93.7.204
9. Gao, J, Yi, X, and Wang, Z. The application of multi-omics in the respiratory microbiome: progresses, challenges and promises. Comput Struct Biotechnol J. (2023) 21:4933–43. doi: 10.1016/j.csbj.2023.10.016
10. Smail, SW, Albarzinji, N, Salih, RH, Taha, KO, Hirmiz, SM, Ismael, HM, et al. Microbiome dysbiosis in Sars-Cov-2 infection: implication for pathophysiology and management strategies of Covid-19. Front Cell Infect Microbiol. (2025) 15:1537456. doi: 10.3389/fcimb.2025.1537456
11. Fontes, AC, Vieira, MC, Oliveira, M, Lourenço, L, Viegas, C, Faísca, P, et al. Feline calicivirus and natural killer cells: a study of its relationship in chronic gingivostomatitis. Vet World. (2023) 16:1708–13. doi: 10.14202/vetworld.2023.1708-1713
12. Tian, J, Liu, Y, Liu, X, Sun, X, Zhang, J, and Qu, L. Feline herpesvirus 1 Us3 blocks the type I interferon signal pathway by targeting interferon regulatory factor 3 dimerization in a kinase-independent manner. J Virol. (2018) 92:e00047-18. doi: 10.1128/jvi.00047-18
13. Li, H, Wu, X, Zeng, H, Chang, B, Cui, Y, Zhang, J, et al. Unique microbial landscape in the human oropharynx during different types of acute respiratory tract infections. Microbiome. (2023) 11:157. doi: 10.1186/s40168-023-01597-9
14. Hanada, S, Pirzadeh, M, Carver, KY, and Deng, JC. Respiratory viral infection-induced microbiome alterations and secondary bacterial pneumonia. Front Immunol. (2018) 9:2640. doi: 10.3389/fimmu.2018.02640
15. de Córdoba-Ansón, PF, Linares-Ambohades, I, Baquero, F, Coque, TM, and Pérez-Cobas, AE. The respiratory tract microbiome and human health. Microb Biotechnol. (2025) 18:e70147. doi: 10.1111/1751-7915.70147
16. Kumpitsch, C, Koskinen, K, Schöpf, V, and Moissl-Eichinger, C. The microbiome of the upper respiratory tract in health and disease. BMC Biol. (2019) 17:87. doi: 10.1186/s12915-019-0703-z
17. Synowiec, A, Dąbrowska, A, Pachota, M, Baouche, M, Owczarek, K, Niżański, W, et al. Feline herpesvirus 1 (Fhv-1) enters the cell by receptor-mediated endocytosis. J Virol. (2023) 97:e0068123. doi: 10.1128/jvi.00681-23
18. Portilho, FVR, Nóbrega, J, de Almeida, BO, Mota, AR, de Paula, CL, Listoni, FJP, et al. Microbial complexity of oral cavity of healthy dogs identified by mass spectrometry and next-generation sequencing. Animals (Basel). (2023) 13:2467. doi: 10.3390/ani13152467
19. Sturgeon, A, Stull, JW, Costa, MC, and Weese, JS. Metagenomic analysis of the canine oral cavity as revealed by high-throughput pyrosequencing of the 16s Rrna gene. Vet Microbiol. (2013) 162:891–8. doi: 10.1016/j.vetmic.2012.11.018
20. Hope, CK, Strother, M, Creber, HK, and Higham, SM. Lethal photosensitisation of prevotellaceae under anaerobic conditions by their endogenous porphyrins. Photodiagn Photodyn Ther. (2016) 13:344–6. doi: 10.1016/j.pdpdt.2015.07.173
21. Zhang, L, Liu, Y, Zheng, HJ, and Zhang, CP. The oral microbiota may have influence on oral cancer. Front Cell Infect Microbiol. (2019) 9:476. doi: 10.3389/fcimb.2019.00476
22. Cuevas-Sierra, A, Riezu-Boj, JI, Guruceaga, E, Milagro, FI, and Martínez, JA. Sex-specific associations between gut prevotellaceae and host genetics on adiposity. Microorganisms. (2020) 8:938. doi: 10.3390/microorganisms8060938
23. Luo, Y, Zhou, Y, Huang, P, Zhang, Q, Luan, F, Peng, Y, et al. Causal relationship between gut prevotellaceae and risk of Sepsis: a two-sample Mendelian randomization and clinical retrospective study in the framework of predictive, preventive, and personalized medicine. EPMA J. (2023) 14:697–711. doi: 10.1007/s13167-023-00340-6
24. Megremis, S, Constantinides, B, Xepapadaki, P, Yap, CF, Sotiropoulos, AG, Bachert, C, et al. Respiratory eukaryotic virome expansion and bacteriophage deficiency characterize childhood asthma. Sci Rep. (2023) 13:8319. doi: 10.1038/s41598-023-34730-7
25. Loh, JS, Mak, WQ, Tan, LKS, Ng, CX, Chan, HH, Yeow, SH, et al. Microbiota-gut-brain axis and its therapeutic applications in neurodegenerative diseases. Signal Transduct Target Ther. (2024) 9:37. doi: 10.1038/s41392-024-01743-1
Keywords: feline herpesvirus type 1, upper respiratory tract, microbiota, diversity, 16S rRNA sequencing
Citation: Li L, Feng W, Bi L and Deng G (2025) Feline herpesvirus type 1 infection alters the diversity of upper respiratory tract microbiota in cats. Front. Vet. Sci. 12:1663056. doi: 10.3389/fvets.2025.1663056
Edited by:
Mengmeng Zhao, Foshan University, ChinaReviewed by:
Mohamed Zeineldin, Animal and Plant Health Inspection Service (USDA), United StatesZhenwei Bi, Jiangsu Academy of Agricultural Sciences (JAAS), China
Copyright © 2025 Li, Feng, Bi and Deng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ganzhen Deng, ZGd6QG1haWwuaHphdS5lZHUuY24=