 Jieru Wang1†
Jieru Wang1† Lanshu Bi2†Hong chen1
Lanshu Bi2†Hong chen1 Xiaopeng Li1Abliz Khamili3Waili Kurban4Chenchen Yang1Peng Niu1
Xiaopeng Li1Abliz Khamili3Waili Kurban4Chenchen Yang1Peng Niu1 Fei Huang1Di Fang1,5
Fei Huang1Di Fang1,5 Chunmei Han1,5*
Chunmei Han1,5* Qinghua Gao1,5*
Qinghua Gao1,5*- 1College of Animal Science and Technology, Tarim University, Alar, China
- 2Xinjiang Bazhou Animal Husbandry Work Station, Bazhou, China
- 3Bureau of Agriculture and Rural Development, Ruoqiang County, Bazhou, China
- 4Agricultural Development Centre, Ruoqiang County, Bazhou, China
- 5Key Laboratory of Livestock and Forage Resources Utilization Around Tarim, Ministry of Agriculture and Rural Areas, Alar, China
As a premium indigenous sheep breed endemic to Tarim Basin, Xinjiang, China, Lop sheep demonstrate remarkable adaptability to extreme arid, high-temperature and sandstorm-prone environments, with some ewes exhibiting exceptional prolificacy. To elucidate the genetic regulation of this reproductive trait, we integrated whole-genome resequencing and transcriptome sequencing, conducting genome-wide association study on 110 Lop ewes with particular emphasis on the regulatory role in litter size determination of MMP16 gene. Through multi-dimensional investigations including qRT-PCR, immunohistochemistry, ovarian granulosa cells culture, and transcriptome sequencing, we revealed that MMP16 gene significantly influences folliculogenesis and ovulation by modulating extracellular matrix (ECM) remodeling and PI3K-AKT signaling pathway activation. Our research systematically elucidates genetic variations associated with prolificacy in Lop sheep and deciphers the biological function of MMP16 gene, providing novel insights into the genetic architecture of ovine reproductive traits. The findings establish a theoretical foundation for molecular marker-assisted breeding and the exploitation of superior germplasm resources.
1 Introduction
Sheep constitute a cornerstone of global livestock production systems, and their reproductive performance serves as the primary constraint that directly affects production efficiency and economic outcomes of the industry (1). Litter size is a critical reproductive trait in sheep (2–4). Enhancing this trait can significantly increase the number of sheep (5–7), enabling sustainable provision of high-quality lamb, wool, and dairy products to meet growing market demands. Lop sheep, a superior indigenous breed originating from the Tarim Basin, exhibits exceptional adaptation to extreme climatic conditions, robust genetic stability, and pronounced disease resistance. Therefore, investigating the genetic mechanisms underlying their fecundity is essential for refining breeding programs and advancing ovine reproductive efficiency.
Several important genes influencing sheep litter size have been identified, such as BMPR1B, BMP15, and GDF9, which can regulate the synthesis and secretion of hormones related to follicular growth and ovulation in sheep (8, 9). However, litter size in different sheep breeds is regulated by distinct major genes. For example, a mutation in the PIK3CD gene influences the litter size of Small Tail Han sheep by affecting its expression level (10), while a 24-bp indel in the AHR gene is associated with litter size and the number of live lambs in Australian White sheep (11). Genetic studies of sheep litter size have concentrated on the biological mechanisms underlying follicular development and ovulation, particularly how key genes modulate dominant follicle selection and ovulation rate, suggesting that follicular development directly influences ewe lambing potential.
In female mammals, follicular development is a key component of the reproductive cycle, whereby the number of mature follicles directly dictates the ovulation rate (12). Ovarian granulosa cells (GCs) surround oocytes to form follicular structures, which play pivotal roles in regulating follicular growth, hormone secretion, oocyte maturation, and embryonic development (13–15), and their proliferation and differentiation capacities directly determine the growth potential of follicles (16, 17). Additionally, apoptosis of GCs directly induces follicular atresia (18), which decreases ovulation rate and thereby directly impacts litter size. Thus, ovarian GCs serve as an ideal in vitro model for investigating sheep fecundity.
Traditional breeding methods exhibit a long cycle and high variability in outcomes (19), whereas molecular marker-assisted breeding techniques enable precise screening of genotypes associated with litter size control at the DNA molecular level, improving the accuracy of selection and shortening the reproductive cycle (20, 21). Recent advancements in biotechnology have propelled high-throughput sequencing technology to become an indispensable tool for discovering superior traits in genetics research. It not only reveals the genetic basis of sheep fecundity but also provides new molecular markers and breeding strategies for sheep breeding (6, 22–24). Chantepie identified SNPs significantly associated with sheep litter size using ovine 50 k SNP chip (25). Zhang obtained key genes related to sheep tail type and wool quality traits through joint analysis of genome-wide association study (GWAS) and selective sweep (26). Pokharel performed integrated mRNA-miRNA analysis to identify candidate genes associated with litter size in Finn sheep and Texel sheep (27). Additionally, as the cost of whole genome sequencing, the gold standard for GWAS, continues to decrease (28–30), GWAS has become increasingly widely applied in sheep populations. Xiang reveals five new genes (MAP3K1, ANKRD55, ABCB1, MEF2C, and TRNAW-CCA-87) related to growth hormone and energy metabolism that are significantly associated with sheep body weight traits through whole genome sequencing and GWAS analysis (31). By capitalizing on its strengths in genome-wide coverage, robust statistical power, and data-driven methodologies, genome-wide association studies (GWAS) have significantly advanced the detailed dissection of the polygenic genetic architecture underlying complex traits (32–36).
In southern Xinjiang, China, we identified two Lop sheep populations characterized by high litter size and lamb survival rate, and proceeded to conduct the following studies using these populations as the research subjects: (1) Resequenced 110 Lop sheep and identified genetic variants significantly associated with litter size through GWAS. (2) Analyzed the expression profiles of the MMP16 gene in different tissues, follicular cells, and performed ovarian spatiotemporal localization analysis to explore its role in follicular development. (3) Conducted the vitro model of overexpressing and interfering with MMP16 in GCs to investigate its effects on follicular development. Our study reveals the key regulatory role of the MMP16 gene in the formation of high fecundity traits in Lop sheep through the integration of population genetics, gene function validation, and molecular regulatory mechanism analysis. The findings have not only provided a theoretical basis for deciphering the genetic mechanisms underlying high fecundity in sheep but also laid a scientific foundation for developing molecular marker based breeding strategies for elite breeds.
2 Materials and methods
2.1 Animal care
Animal experiments were approved by the Animal Ethics Committee of the College of Animal Science and Technology of Tarim University (No. DTU 20230126).
2.2 Animal samples and tissues collection
Two populations of Lop sheep were sourced from agricultural facilities in Weili and Ruoqiang county, Xinjiang, China. All subjects were maintained under standardized husbandry conditions, receiving identical nutritional regimens and environmental parameters. No significant intergroup differences were observed in phenotypic metrics including mean age, body mass, somatometric measurements (such as body length, chest circumference), or other physiological characteristics. Whole blood samples were collected via jugular venipuncture using 5 mL K2EDTA anticoagulant tubes. Collected specimens were promptly aliquoted and preserved at −20 °C for subsequent analysis.
The sheep were divided into the single-lamb group (n = 3) and the multi-lamb group (n = 3) according to the record of litter size for three consecutive years. After estrus synchronization, sheep in the same physiological state underwent deep anesthesia through intravenous injection of 3% pentobarbital sodium solution (Solarbio, Beijing, China). Postmortem, reproductive neuroendocrine tissues (hypothalamus, pituitary) and reproductive tissues (ovary, uterus, fallopian tube) were surgically excised under aseptic conditions. Tissue specimens were dissected into 50–100 mg fragments using sterile surgical instruments, and flash-frozen in liquid nitrogen within 2 min of excision to preserve RNA integrity.
Fresh ovine ovaries were aseptically harvested and transported in phosphate-buffered saline pre-warmed to 37 °C, supplemented with antibiotic supplementation (100 IU/mL penicillin and 50 mg/mL streptomycin), and maintained at physiological osmolarity.
2.3 DNA extraction and genotyping
Genomic DNA was extracted from blood samples using the magnetic bead method according to the manufacturer’s instructions for the Blood DNA extraction Kit (TIANGEN, Beijing, China). The integrity of the extracted DNA was detected by 1% agarose gel, and the concentration of the DNA was determined using NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, Waltham, United States). The qualified DNA was submitted to Beijing Novogene Co., Ltd. for 10 × whole-genome resequencing.
The TruSeq DNA PCR-Free kit was used to construct a whole-genome sequencing library with an insertion fragment size of approximately 350 bp. The libraries were sequenced on the Illumina HiSeq 2000 platform, with each fragment generating a paired end read of 100 bp. FastQC software1 was used to evaluate the quality of the sequencing data. Fastp software was used for quality control of sequencing data (37), using default parameters. BWA-MEM algorithm was used to compare the sequencing data with the sheep reference genome (UI_Ramb_v2.0/GCF_016772045.1). The MarkDuplicates module in Picard Tools software was used to remove PCR duplicates. GATK 4.0 module HaplotypeCaller was used for mutation identification. Plink was used for quality control, and the single nucleotide polymorphisms (SNPs) were removed if the loss rate was less than 90%, the minor allele frequency was lower than 0.05, or the Hardy–Weinberg equilibrium was less than <10−6.
2.4 Genome-wide association studies
The linear mixed model is widely used in genome-wide association study because it can correct the population structure and the complex relationships within the population. The model is as follows:
where y is the litter size, α includes vectors of fixed effects (parity, years, and seasons), β is the vector of random effects, μ is the vector of permanent environmental effects, e is the vector of residual effects, and W, X, and Z are the correspondent incidence matrices of α, β, and μ, respectively.
2.5 Total RNA extraction, cDNA synthesis and quantitative real-time polymerase chain reaction
Total RNA was isolated using TRIzol reagent (Invitrogen, Carlsbad, United States) and the concentration of RNA were assessed by measuring absorbance at 260 nm and 280 nm using a spectrophotometer (Thermo, Waltham, MA, United States). Additionally, the integrity of RNA was assessed through 1.5% agarose gel electrophoresis. The cDNA was synthesized using a PrimeScript RT reagent kit (TaKaRa, Beijing, China). qPCR was performed using ChamQ Universal SYBR qPCR MasterMix (Vazyme, Nanjing, China). According to the manufacturer’s instructions, the amplification was carried out in a reaction system of 15 μL, and primer information was shown in Table 1.
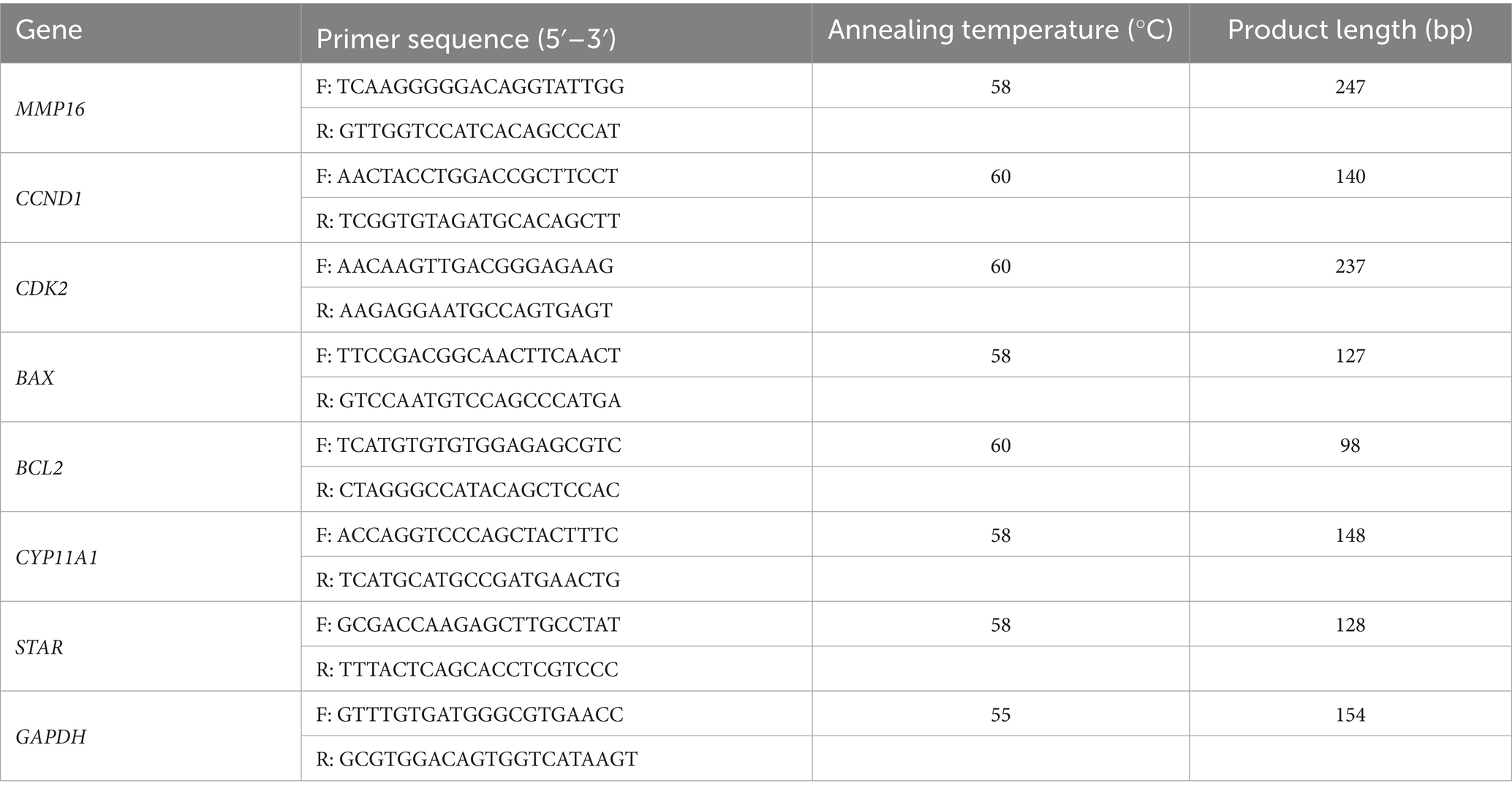
Table 1. Primer information.
2.6 Immunohistochemistry
Ovine ovarian tissues were fixed by immersion in 4% paraformaldehyde for 24 h, progressively dehydrated through a graded ethanol series, embedded in paraffin blocks, and sectioned into 5 μm-thick slices. Sections were mounted on glass slides for subsequent histological processing. Tissue sections were deparaffinized, rehydrated through a graded ethanol series, and washed with PBS three times for 5 min each. Following PBS washing, tissue sections were probed with rabbit anti-MMP16 primary antibody (Affinity Biosciences, Changzhou, China), horizontally positioned in a humidity-controlled incubation chamber, and maintained at 4 °C for 12–16 h. Secondary antibodies were applied and incubated at room temperature for 10 min. Nuclei were counterstained with hematoxylin (2–3 min), rinsed under running tap water for chromatin visualization, dehydrated through ascending ethanol concentrations and cleared in xylene.
2.7 Isolation and culture of sheep ovarian granulosa cells
Ovaries were sequentially washed three times with sterile saline solution containing antibiotics (100 U/mL penicillin, 50 μg/mL streptomycin), with 1–2 min cycles per wash. Following superficial adipose tissue excision with aseptic forceps, specimens were processed through two subsequent sterile saline rinsing cycles under laminar airflow conditions. Ovarian follicular dissection was conducted under aseptic conditions within a sterile culture dish employing microsurgical scalpels, utilizing controlled digital pressure to facilitate intact follicular antrum evacuation. The aspirated follicular fluid was transferred to 15 mL conical centrifuge tubes and subsequently subjected to centrifugation at 1,500 g for 5 min. Supernatant was carefully decanted, retaining the granulosa cell-enriched pellet. Pelleted cells were resuspended in 1 mL complete growth medium (DMEM supplemented with 10% FBS). Cultures were maintained at 37 °C in a humidified 5% CO₂ incubator for subsequent expansion.
2.8 Cell transfection
Ovine GCs were plated in 6-well culture plates containing 2 mL of DMEM (Gibco, New York, United States) per well, and maintained at 37 °C in a humidified 5% CO2 atmosphere. When the cells reached 70–80% confluence, the original culture medium was discarded, and the cells were washed twice with 1 mL of PBS. The cells in each well were transfected with 100 pmol of siRNA or 4 μg lenti-MMP16 according to the manufacturer’s protocol for Lipofectamine 2000 (Invitrogen, Carlsbad, United States).
2.9 Cell proliferation test
Inoculated GCs into 96-well plates. After transfection, 10% CCK-8 solution was added to each well in accordance with the CCK-8 instructions. After culturing in the cell culture incubator, the absorbance at 450 nm was determined using an microplate reader.
EdU incorporation was analyzed using the EdU Cell Proliferation Kit (RiboBio, Guangzhou, China) following the manufacturer’s standardized protocol. According to the manufacturer’s instructions, cells were sequentially processed as follows: initial incubation with EdU solution for 2 h at 37 °C, followed by two washes with PBS. Subsequently, cells were fixed with 4% paraformaldehyde in PBS for 30 min at room temperature. After fixation, samples underwent two additional PBS washes. Permeabilization was then performed using 0.5% Triton X-100 solution (100 μL per sample) for 10 min at ambient temperature. Add 100 μL of staining reaction solution to each well and incubate at room temperature on a shaker for 30 min. Then add 100 μL reaction solution to each well and incubate at room temperature for 30 min. Finally, wash three times with PBS and place the cells under a fluorescence microscope for observation.
2.10 TUNEL
Fixed cells were gently aspirated and subjected to a single PBS rinse. Add PBS containing 0.3% Triton X-100 (Beyotime, Shanghai, China) to cells and incubate at room temperature for 5 min. Wash twice with PBS, add 100 μL of TUNEL (TdT enzyme and fluorescent labeling solution) detection solution, and incubate for 60 min. Wash with PBS three times, seal the slides and observe under a fluorescence microscope. Post-incubation specimens underwent three PBS washing cycles, mounted under coverslips using antifade medium, and imaged via laser scanning confocal microscopy with standardized emission settings.
2.11 Protein extraction and western blot
The pelleted cell was loaded onto a spin column, and 200 μL cell lysis buffer was added. After incubation at room temperature for 2 min, the mixture was centrifuged at 16,000 r/min for 2 min. The flow-through was immediately placed on ice, and the spin column was discarded to complete protein extraction. Protein concentrations were measured and adjusted to equal levels, followed by denaturation in boiling water. The 12% separating gel and 5% stacking gel were prepared using an SDS-PAGE gel kit for protein electrophoresis. Following SDS-PAGE, the gel region containing the target protein was excised and transferred onto a PVDF membrane. The membrane was blocked with blocking buffer for 60 min, then incubated with primary antibody (Affinity biosciences, Cincinnati, United States) at room temperature for 60 min. Subsequently, secondary antibody (Epizyme, Shanghai, China) was applied and incubated at room temperature for 60 min, with GAPDH serving as the internal control protein and imaging was performed using a ChemiDoc chemiluminescence detection system.
2.12 Transcriptome sequencing
After overexpressing the MMP16 gene in GCs, transcriptome sequencing was performed. Using CASAVA base calling, the image data of sequencing fragments obtained from high-throughput sequencers were converted into sequence data in FASTQ format. These files underwent filtration to remove raw data, sequencing error rate verification, and GC content distribution evaluation, ultimately yielding clean reads for subsequent analysis. The clean reads were aligned to the reference genome using HISAT2 software to rapidly and accurately obtain read localization information on the reference genome (38). Quantitative analysis was conducted using the featureCounts tool from the subread software package (39). Subsequently, statistical analysis of expression data was performed to identify genes with significantly different expression levels across sample conditions. The clusterProfiler software was employed for Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis of differentially expressed genes (DEGs), aiming to identify major affected biological functions or pathways (40).
2.13 Statistical analysis
All experimental procedures were performed in triplicate biological replicates, and cycle threshold (CT) values were calculated using the 2−ΔΔct method. SPSS 24.0 was used to analyze whether the difference was significant by one-way ANOVA analyses. The difference was considered significant at p < 0.05, and * represents p < 0.05, ** represents p < 0.01 and ns represents no significant difference.
3 Results
3.1 Sequencing quality analysis and quality control
Whole-genome resequencing was conducted on 110 Lop sheep, yielding a total raw sequencing data output of 5,236.34 Gb. The sequencing depth ranged from a minimum of 4.32 × to a maximum of 11.55×, with an average depth of 7.45 × and an average coverage of 98.20% (Q20 ≥ 98.17%, Q30 ≥ 96.71%). The alignment rate to the reference genome exceeded 99.4%. Variants were annotated primarily to intergenic regions of coding genes, totaling 8,497,913, followed by intronic regions of coding genes, with 4,875,735 variants. After quality control, 37,772,316 SNPs were retained for further analysis.
3.2 Genome-wide association study
To identify genetic variants associated with reproductive traits in Lop sheep, we conducted GWAS using genotype data from 110 individuals and phenotypic records of litter size. The Manhattan plot revealed multiple candidate loci associated with litter size, including genes such as MMP16, BMP1, STK3, EXT1, and GRIP1 (Figure 1). These genes are functionally implicated in reproductive processes, including follicular development and maturation, cellular proliferation and apoptosis, and litter size regulation (41–43) (Table 2). Notably, a cluster of SNPs was identified within the MMP16 gene on chromosome 9, suggesting its potential role as a key genetic determinant of reproductive performance.
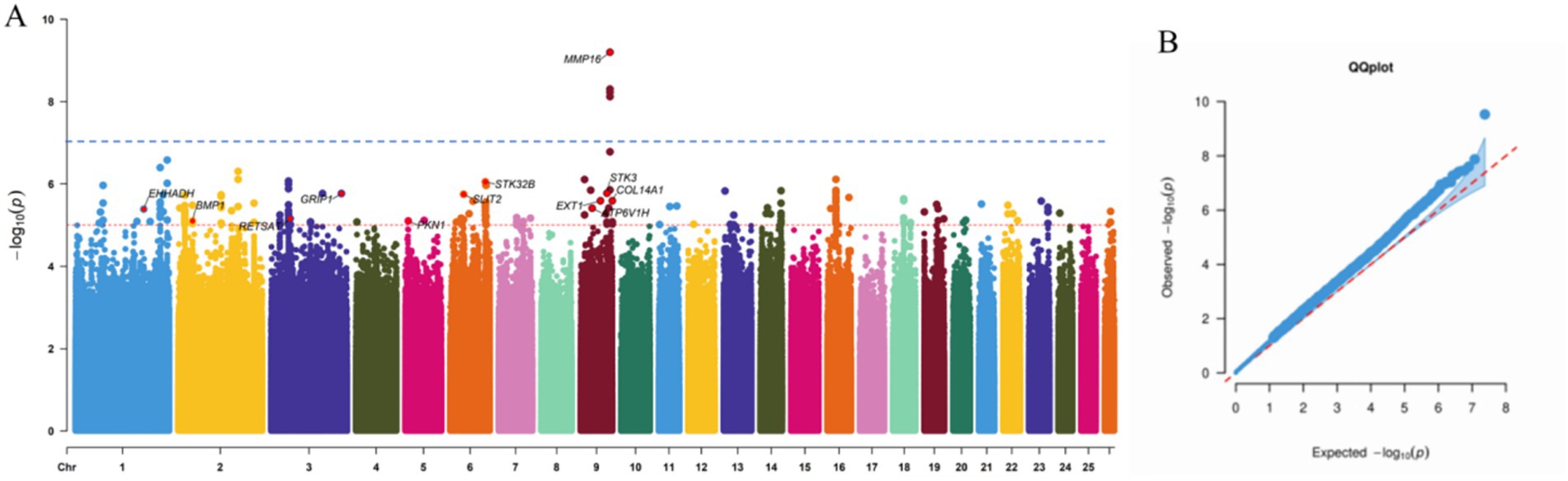
Figure 1. Manhattan and quantile-quantile plot of SNP GWAS. (a) The blue line corresponds to the 5% chromosome-wide significance threshold using a Bonferroni correction (10−7). The rede line corresponds to a suggestive chromosome-wide threshold of 10−5. (b) Quantile-quantile plot of GWAS.
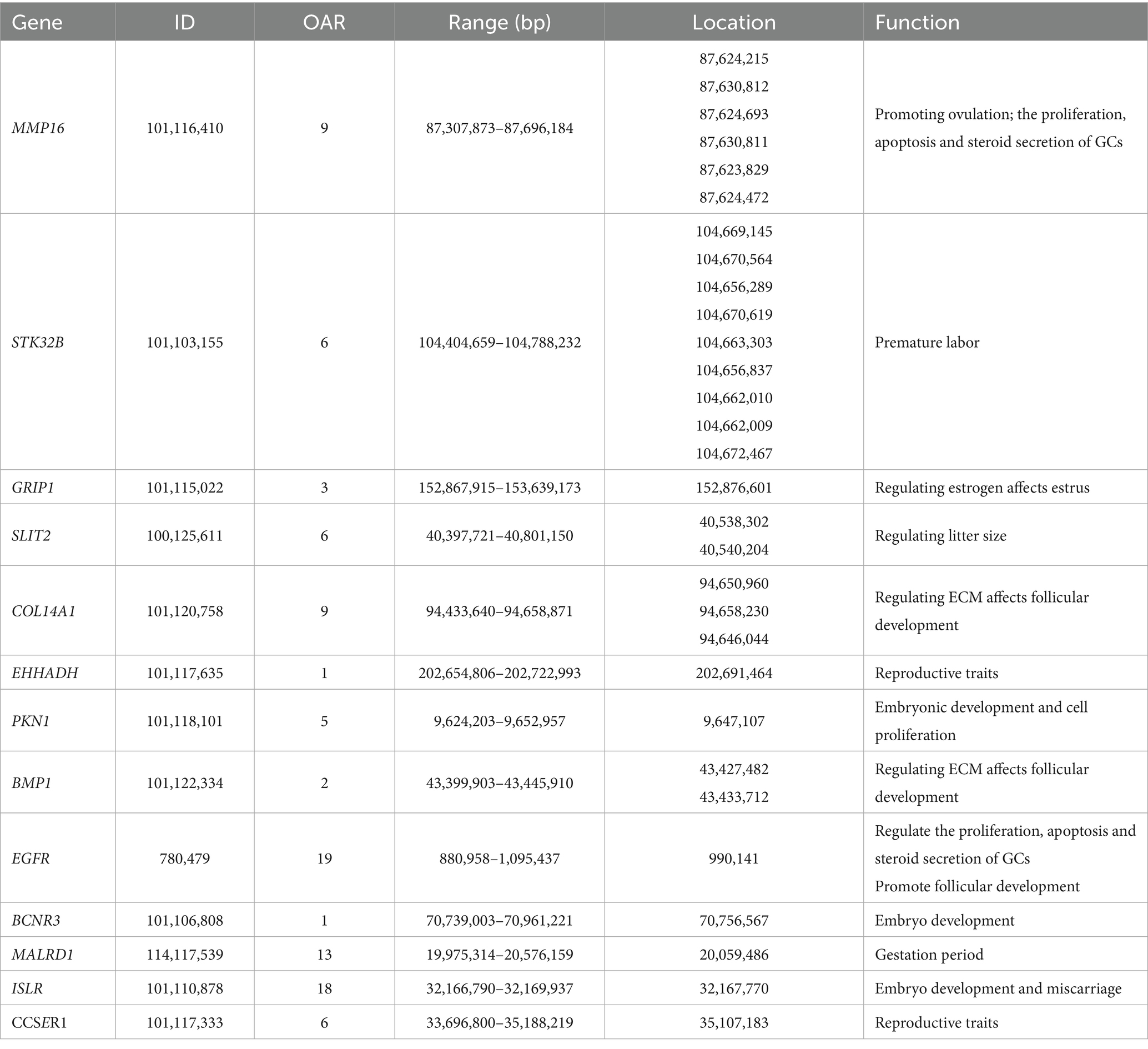
Table 2. The information of candidate genes identified by GWAS.
3.3 Expression of MMP16 gene in different tissues and localization in ovary
To investigate the role of MMP16 in sheep reproduction, we used qPCR and immunohistochemistry to analyze the expression of MMP16 in different tissues and the localization in sheep ovary. We examined the relative expressions of MMP16 in ovary, uterus, fallopian tube, hypothalamus and pituitary of single-lamb and multi-lamb (Figure 2). The results showed that MMP16 gene expressed in all five tissues of both the single-lamb and multi-lamb sheep, with the highest expression in ovary (Figure 2a). The expression of MMP16 in the ovary and uterus of the multi-lamb sheep was significantly higher than that in the single-lamb sheep (p < 0.01), but the expression in the fallopian tube was significantly lower than that in the single-lamb sheep (p < 0.01) (Figure 2b). The MMP16 exhibited stage-specific expression patterns in ovarian follicles, with transcript levels progressively increasing throughout folliculogenesis. The highest expression was observed in the corpus luteum, while large follicles demonstrated significantly elevated MMP16 expression compared to medium and small follicles (p < 0.05).
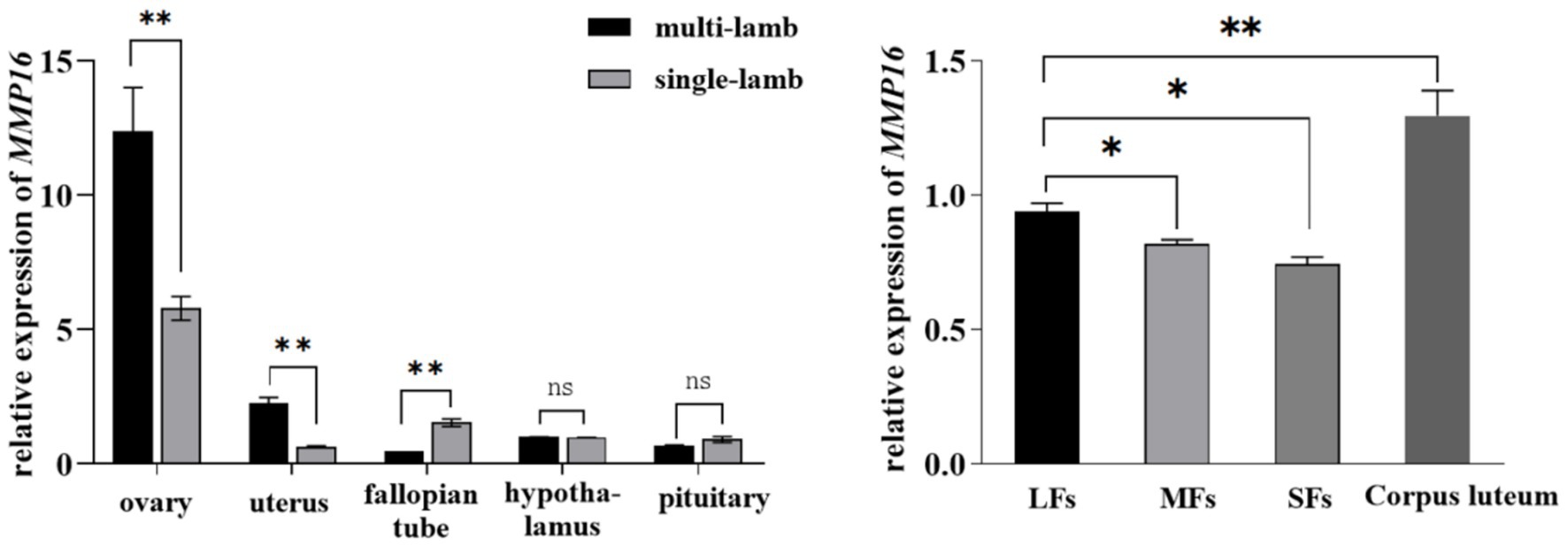
Figure 2. Expression of MMP16 in different tissues and follicles. (a) Relative expression of MMP16 in ovary, uterus, fallopian tube, hypothalamus and pituitary of single-lamb and multi-lamb sheep. (b) The relative expression levels of MMP16 in corpus luteum, large (LFs), medium (MFs), and small (SFs) follicles. *denotes significant, **denotes highly significant.
The results of IHC also demonstrated stage-specific localization of MMP16 during ovine folliculogenesis (Figure 3). MMP16 was located at different developmental stages of follicles, fills the entire follicular fluid in mature follicles, and was located in oocyte, GCs, cumulus cells, theca cells and corpus luteum. These results suggest that the MMP16 gene plays a role in ovine ovarian follicle maturation, ovulation and luteal formation.
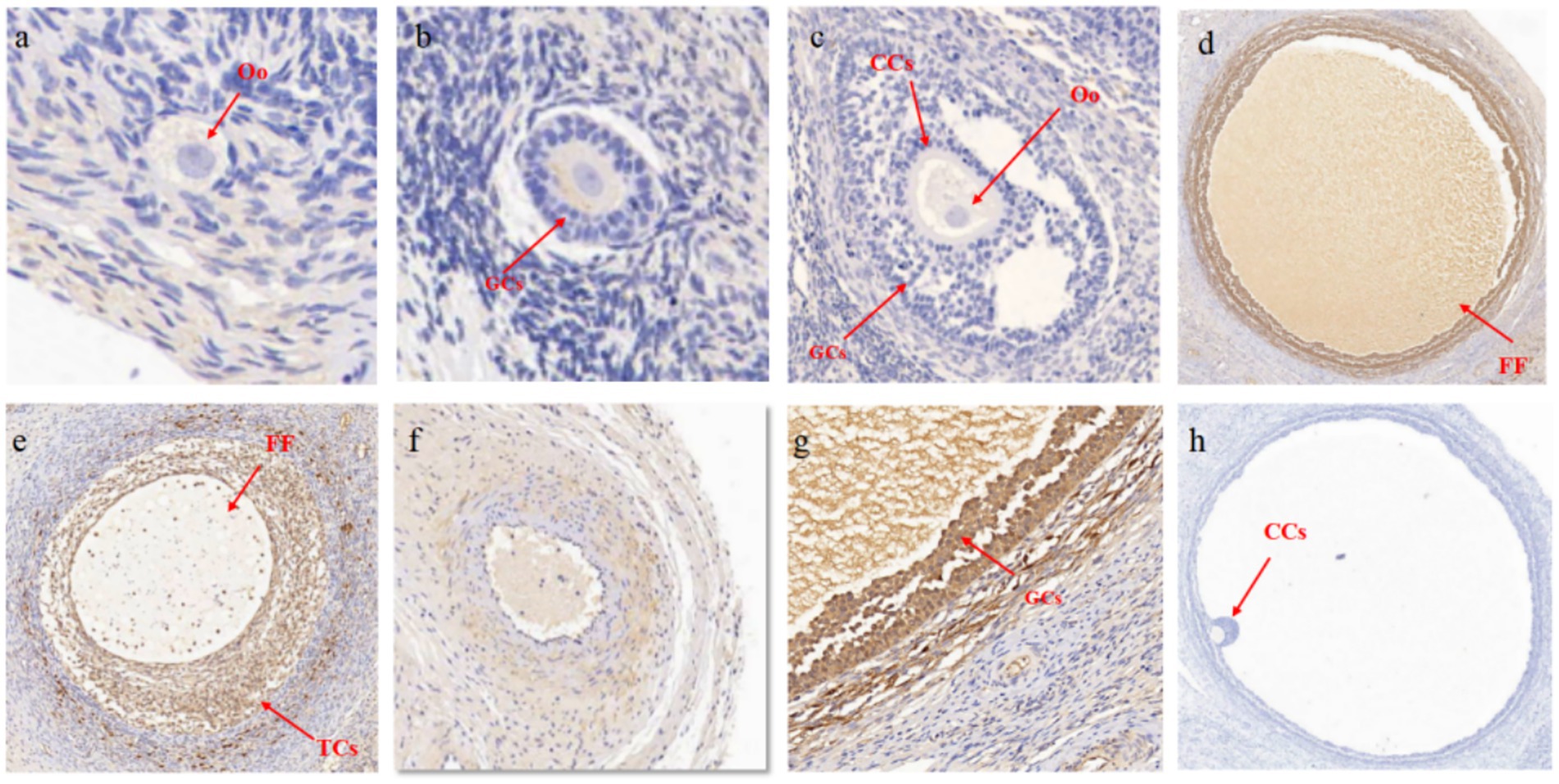
Figure 3. Localization of MMP16 in ovaries. (a) Primordial follicles, 50 μM. (b) Primary follicle, 100 μM. (c) Secondary follicles, 200 μM. (d) Mature follicles, 500 μM. (e) Large coelomic follicles, 500 μM. (f) corpus luteum, 200 μM. (g) GCs, 100 μM. (h) Negative control, 500 μM. CCs, cumulus cells; Oo, oocyte; TCs, theca cells; FF, follicular fluid.
3.4 Validation of MMP16 overexpression and interference efficiency in GCs
The qPCR and western blot analyses revealed that transfection with the lentiviral plasmid significantly upregulated MMP16 mRNA expression levels compared to the vector (p < 0.01), while concurrently demonstrating a marked increase in MMP16 protein expression (Figure 4a). After transfection of the three groups of si-RNA, the results showed that compared with the NC group, si-RNA2 significantly down-regulated the expression of MMP16 gene in GCs (p < 0.01), and si-RNA3 significantly down-regulated the expression of MMP16 gene in GCs (p < 0.05). There was no significant change in MMP16 gene expression in si-RNA1 (p > 0.05) (Figure 4b). Western blot analysis demonstrated that the expression level of MMP16 protein was significantly down-regulated in the si-RNA2. Based on these findings, si-RNA2 was selected for subsequent functional studies investigating GCs.
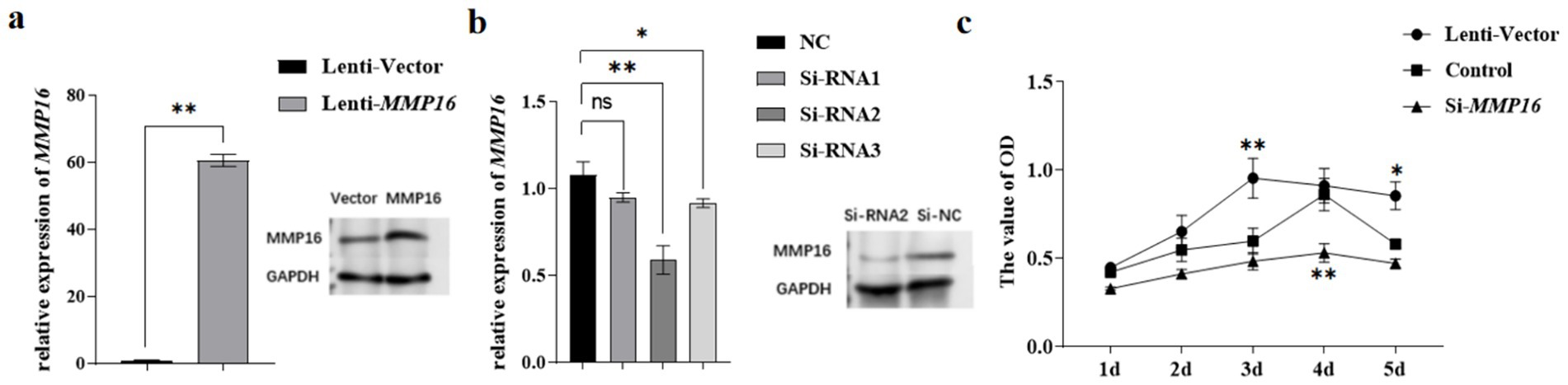
Figure 4. Validation of MMP16 overexpression and interference efficiency in GCs. (a) The overexpression efficiency of MMP16 in GCs was detected by qPCR and western blot. (b) The interference efficiency of MMP16 in GCs was detected by qPCR and western blot. (c) CCK-8 measured cell viability after overexpression/interference with MMP16. *denotes significant, **denotes highly significant.
3.5 The effects of MMP16 on the viability and proliferation of GCs
To assess the effects of MMP16 on the viability and proliferation of GCs, we employed qRT-PCR, CCK-8 assay, EdU, and TUNEL analysis. The CCK-8 assay demonstrated that the MMP16 increased the viability of GCs on day 3 compared to the vector group (p < 0.01), while interfering with the expression of MMP16 yielded opposite results (Figure 4c). The Si-RNA2 interfered with the expression of MMP16 in GCs induced progressive suppression of activity, with culminating in statistically significant decreased on day 4 (p < 0.01).
Furthermore, the EdU analysis indicated that the MMP16 significantly promoted the proliferation of GCs compared to the vector group, whereas interfering with the expression of MMP16 had the opposite effect. Overall, these results suggested that MMP16 could increase the viability and promote the proliferation of GCs. TUNEL analysis was used to detect the effect of MMP16 on the apoptosis of GCs (Figure 5). The results showed that overexpression of MMP16 inhibited the apoptosis of GCs (p < 0.05), while interference with MMP16 promoted the apoptosis of GCs (p < 0.01). In order to further explore the effects of MMP16 on proliferation and apoptosis of GCs, qPCR was used to detect the expressions of proliferation-related genes CCND1 and CDK2, apoptosation-related genes BAX and BCL2. The qPCR results revealed that MMP16 overexpression significantly increased the expression of CCND1, CDK2, and BCL2 gene (p < 0.01), and significantly reduced the expression of BAX (p < 0.01) (Figure 5b and Figure 6b). Apoptosis of GCs was assayed using TUNEL after transfection with MMP16 (Figures 6a, c).
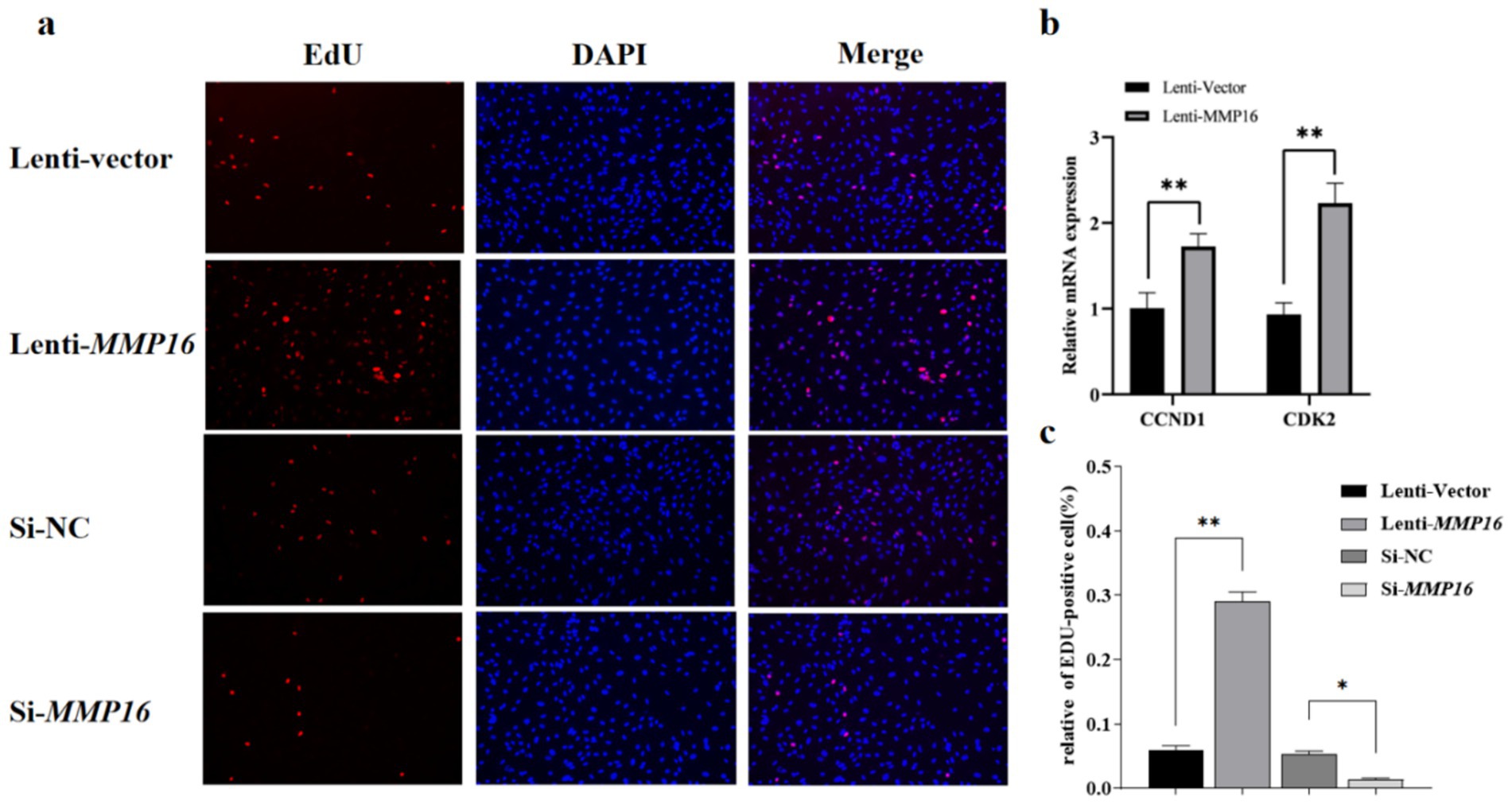
Figure 5. Effects of overexpression and interference of MMP16 on the proliferation ability of sheep GCs. (a,c) Proliferation of GCs was assayed using EdU assays after transfection with MMP16. (b) The relative expression levels of CCND1 and CDK2 after transfection with MMP16. *denotes significant, **denotes highly significant.
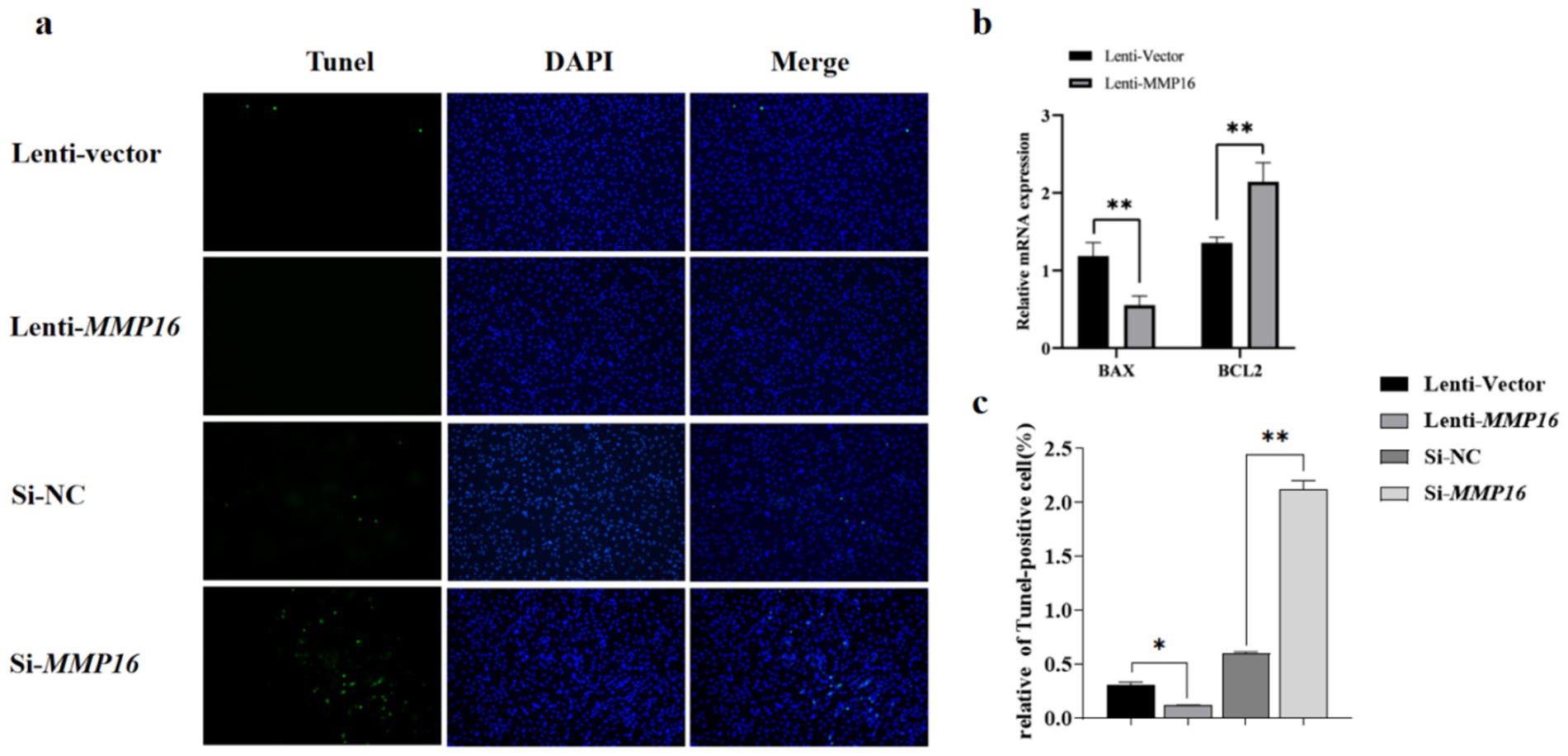
Figure 6. Effects of overexpression and interference of MMP16 on the apoptosis ability of sheep GCs. (a,c) Apoptosis of GCs was assayed using TUNEL after transfection with MMP16. (b) The relative expression levels of BAX and BCL2 after transfection with MMP16. *denotes significant, **denotes highly significant.
3.6 Impact of MMP16 overexpression and interference on the steroid hormone levels of GCs and the expression of associated genes
We examined the impact of MMP16 overexpression and interference after 48 h on the expression of steroid synthesis-related genes and hormone levels (Figure 7). The influence of overexpression of MMP16 on E2 concentration was significantly higher than that of vector (p < 0.05) (Figure 7a), and on P4 concentration was significantly lower than that of vector (p < 0.05) (Figure 7b), while interference of MMP16 had the opposite effect. The mRNA expressions level of STAR and CYP11A1, key enzymes in steroid hormone synthesis pathway, significantly increased compared with the vector group (p < 0.05 or p < 0.01) (Figure 7c). These results suggest that MMP16 may regulate the expression of steroid synthesis-related genes.
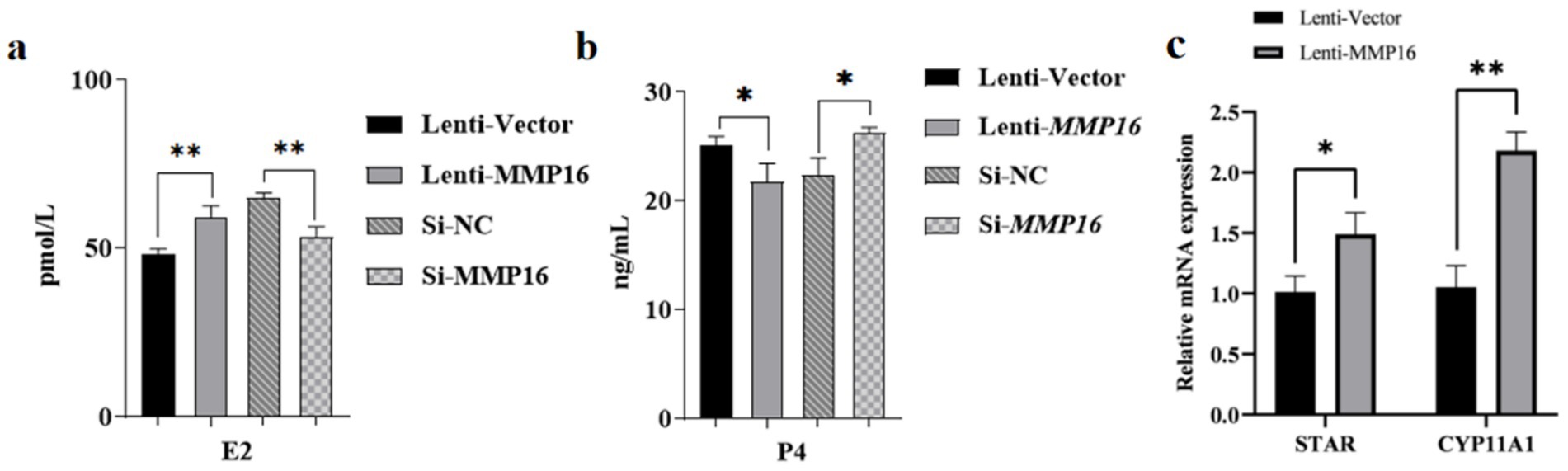
Figure 7. The effects of MMP16 overexpression and interference on steroid hormones and related genes. (a) The concentration of E2 after transfection with MMP16. (b) The concentration of P4 after transfection with MMP16. (c) The relative expression levels of STAR and CYP11A1 after transfection with MMP16. *denotes significant, **denotes highly significant.
3.7 Transcriptome sequencing analysis
To investigate further regulatory mechanisms of MMP16, we performed transcriptome sequencing to identify potential signaling pathways after MMP16 overexpression (Figure 8). The sequencing results showed that a total of 296,607,618 raw reads were obtained. After quality control, a total of 292,873,450 clean reads were obtained. The volcano plot depicting DEGs revealed that the transcript levels of 920 genes were significantly up-regulated (Padj < 0.05), while 474 genes exhibited significant down-regulation (Padj < 0.05) (Figure 8a). Subsequent KEGG analysis of the DEGs unveiled that these genes were notably enriched in pathways associated with the ECM-receptor interaction as well as the PI3K-Akt signaling pathway (Figure 8b). Furthermore, transcriptome sequencing detected the expressions of CCND1, CDK2, BCL2, STAR, and CYP11A1 gene significantly increased, and the expression of BAX significantly decreased, which was consistent with our qPCR results.
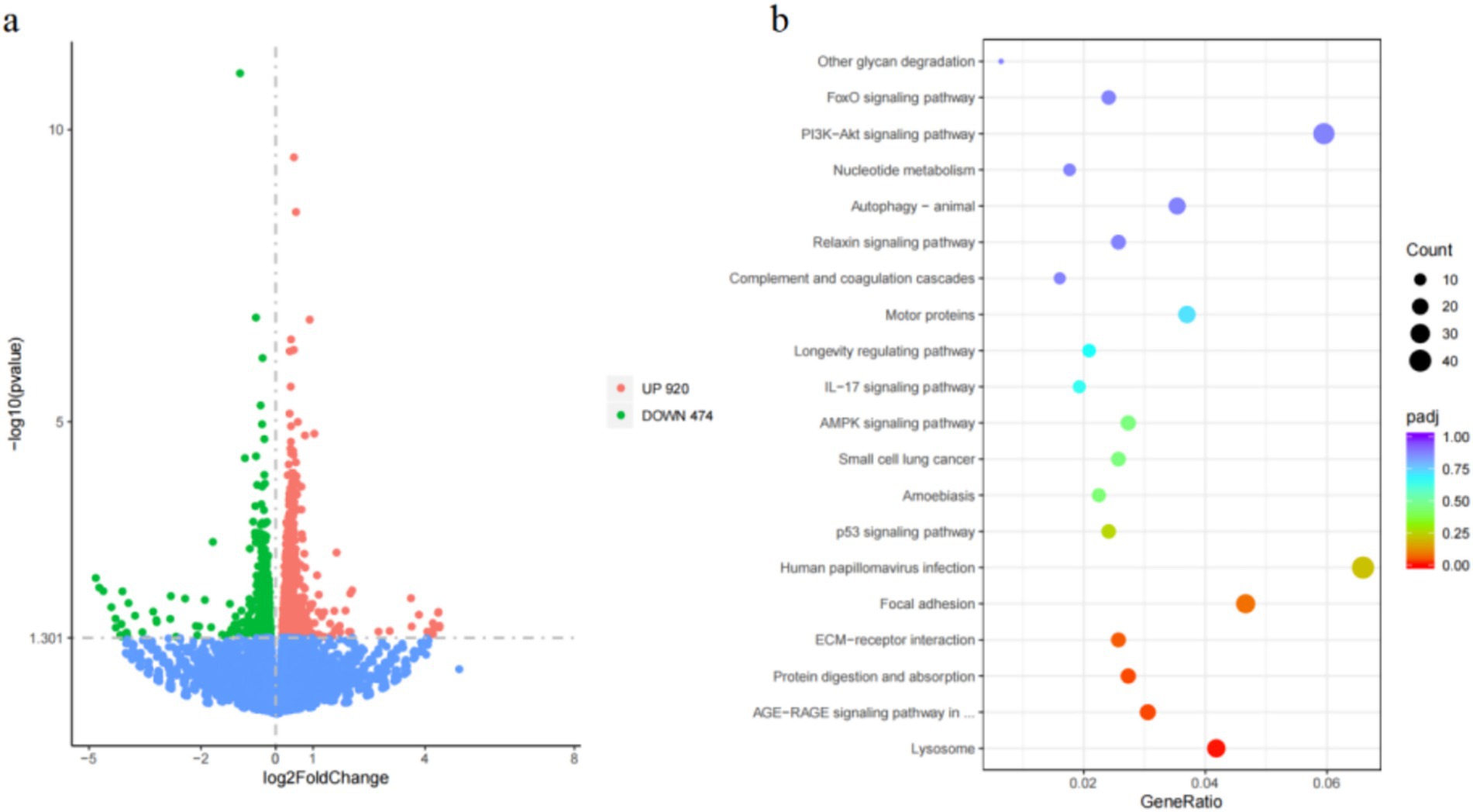
Figure 8. Differential gene identification and enrichment analysis results. (a) Differential gene volcano map after MMP16 overexpression in sheep ovarian granulosa cells. (b) Bubble map of KEGG enrichment analysis.
3.8 The expression of related genes in the PI3K-AKT signaling pathway
The mRNA expression levels of COL4A1, ITGA3, ITGB5, PI3K, AKT, CCND1, CDKN1A, BCL2L1 and CREB3 in the PI3K-AKT signaling pathway were detected by qPCR and western blot (Figure 9). The results showed that the expression levels of these related genes significantly increased after overexpression of MMP16 (Figure 9a), which was consistent with the result of protein expression (Figure 9b).
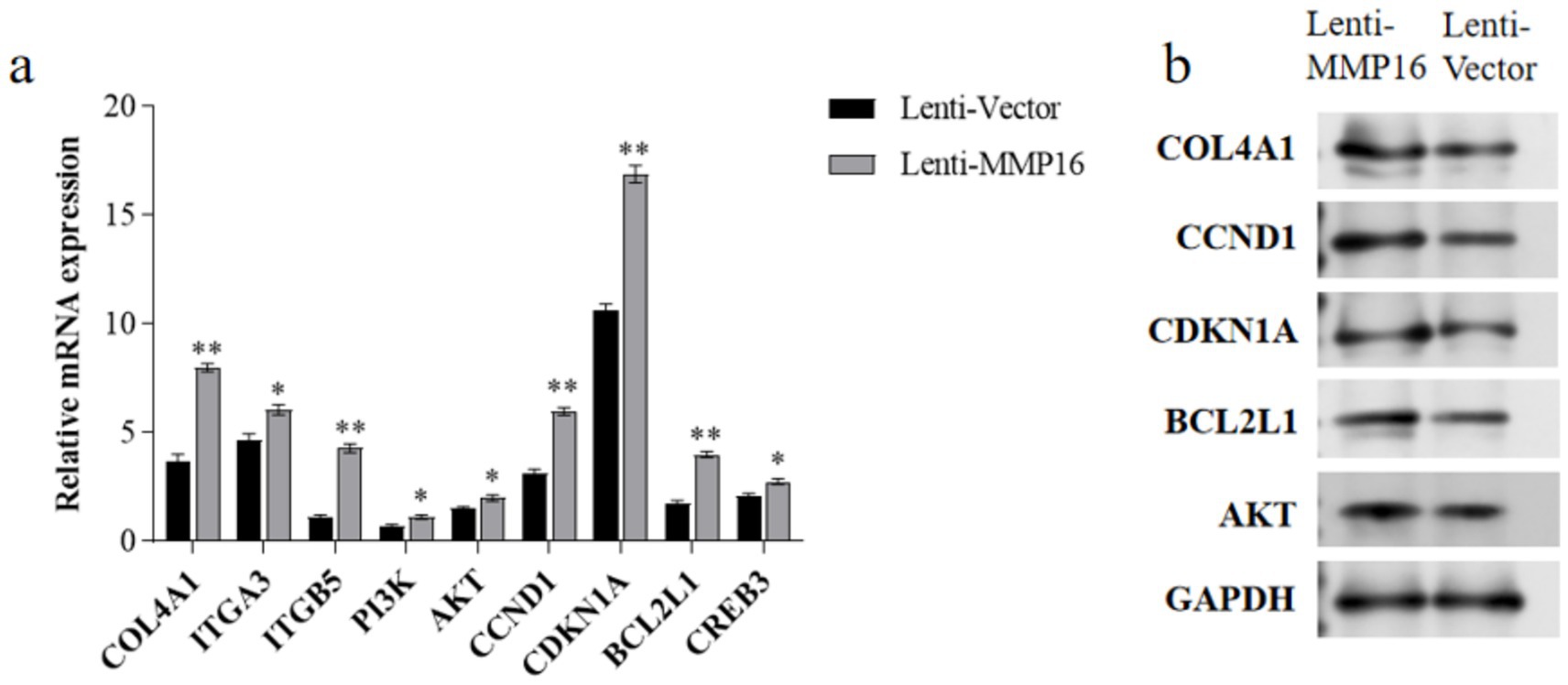
Figure 9. The expressions of related genes in the PI3K-AKT signaling pathway. (a) The relative mRNA expressions of related genes in the PI3K-AKT signaling pathway. (b) The protein expressions of related genes in the PI3K-AKT signaling pathway. *denotes significant, **denotes highly significant.
4 Discussion
Genetic improvement of litter size in livestock represents a promising strategy for enhancing production efficiency. Through genome-wide association study, we identified key candidate genes associated with litter size traits in Lop sheep, and further elucidated the molecular regulatory mechanism of MMP16 in GCs. These findings provide novel insights into the genetic architecture underlying prolificacy in sheep and offer valuable references for understanding mammalian reproductive efficiency.
Reproductive processes are inherently complex, with quantitative traits such as ovulation rate and litter size being governed by the cumulative effects of major-effect mutations and minor-effect loci within a polygenic regulatory framework. BMP1 was initially identified in osseous extracts demonstrating ossification-inducing activity at ectopic sites (44), and subsequent investigations revealed its critical role in modulating GCs proliferation and apoptosis, thereby participating in the physiological process of follicular selection (45). The estrogen receptor coactivator GRIP1 is functionally conserved in ovarian tissues across sheep, bovine, and porcine species, and its expression level governs the transcriptional activation of estrogen-responsive genes, thereby modulating systemic hormonal responsiveness in target reproductive tissues (46). The expression of EGFR was identified in sheep ovarian follicles at different periods, with melatonin demonstrating modulatory effects on its expression levels, suggesting EGFR may exert stage-specific regulatory roles during follicular development (47, 48). SLIT2 expressed in ovary, with its expression demonstrating luteal phase-specific upregulation (13). Notably, this spatiotemporal expression pattern was negatively regulated by human chorionic gonadotropin and cortisol, and pharmacological inhibition of the SLIT-ROBO signaling enhanced luteal cell migration capacity and reduced apoptotic rate (49). These results suggest that SLIT may be involved in ovarian development by controlling the migration or apoptosis of cells.
MMP16, a membrane-type matrix metalloproteinase (MT-MMP), critically regulates extracellular matrix (ECM) homeostasis through spatiotemporal proteolytic remodeling, positioning it as a key modulator in both physiological tissue morphogenesis and pathological conditions such as cancer metastasis and fibrosis (50–53). Within the female reproductive axis, MMP16 serves as a pivotal modulator of folliculogenesis through dynamic ECM proteolysis. Its spatiotemporal remodeling of basement membrane components orchestrates critical transitions including follicular selection, ovulation, and corpus luteum formation, while simultaneously modulating atresia via granulosa cell apoptosis regulation (54). MMP16 exhibits its highest expression levels in the ovary, with a progressive increase observed throughout follicular development, ultimately reaching peak expression in the corpus luteum, which implicates MMP16 in orchestrating key reproductive processes including folliculogenesis, ovulation, and luteal formation. Comparative analysis revealed significantly higher MMP16 expression levels in ovarian tissues of multi-lamb compared to single-lamb, suggesting its potential regulatory role in litter size through follicular development optimization. Previous study has established ECM components as predominant proteolytic substrates for collagenases and gelatinases, with GCs specific expression of these proteases being essential for follicular wall ECM degradation and subsequent ovulation facilitation through targeted matrix remodeling (55). During the ovulation period of rats and humans, MMP16 is regulated by chorionic gonadotropin, which upregulates the proteolytic activity within the follicles, thereby driving the occurrence of follicular ovulation (56). Furthermore, some studies have also found that in mouse GCs, the increased expression of MT-MMPs and TIMP1 lead to an increase in MMP2 (55). Our study similarly demonstrated that MMP16 overexpression significantly upregulated MMP2 expression based on transcriptome sequencing data. In follicles, MMP2 and MMP9 proteins predominantly localized to follicular tissue remodeling sites, with their mRNA and protein levels positively correlating with follicular diameter (57). Synthesized and secreted by GCs and theca cells of small antral follicles, these proteases are hypothesized to participate in dominant follicle selection, given that follicular dominance is established during the antral stage (58). Falkowski unveiled an additional regulatory mechanism where in the MMP14-TIMP1 complex activates theca/granulosa-derived MMP2 upon migratory cumulus-oocyte complex, triggering rapid apical follicular rupture and oocyte extrusion (59).
Furthermore, we observed that MMP16 overexpression was significantly associated with enhanced PI3K-AKT pathway activity, suggesting its potential to promote ovine follicular maturation and ovulation via activation of this key intracellular signaling cascade. The PI3K-AKT pathway, a well-characterized signaling pathway, plays pivotal roles in diverse physiological processes including cellular proliferation, differentiation, apoptosis, and metabolic regulation. Recent studies have established its essential involvement throughout folliculogenesis, spanning primordial follicle recruitment, GCs proliferation, corpus luteum survival, and oocyte maturation (60). Within the PI3K-AKT pathway, coordinated upregulation of the cell cycle regulator CCND1 and downregulation of CDKN1A collectively drive GCs proliferation, a finding corroborated by EdU assay results. CYP11A1, a key enzyme regulating steroid hormone biosynthesis in GCs, modulates follicular development and ovulation through hormonal dynamics (61). Notably, our study demonstrates that MMP16 significantly upregulates CYP11A1 expression, implicating its functional role in potentiating GCs steroidogenic activity. Concurrently, AMPK, a crucial intracellular energy sensor, maintains cellular energy homeostasis and regulates metabolic processes. Within the reproductive system, AMPK activity alterations exert profound implications on follicular development and ovulation. Specifically, FSH-activated PI3K/AKT and AMPK pathways play critical roles in orchestrating GCs mitotic progression and cell cycle regulation (62). MMP16 overexpression effectively suppresses AMPK phosphorylation, thereby diminishing its activity, which likely facilitates follicular maturation and ovulation (63). In summary, by inhibiting AMPK activity and activating the PI3K-AKT pathway, MMP16 promotes GCs proliferation, suppresses apoptosis, and regulates steroid hormone synthesis. These integrated effects collectively enhance ovulation efficiency and litter size.
5 Conclusion
This study integrated population genetics analysis and functional validation to elucidate the critical role of the MMP16 gene in establishing high fecundity traits in Luobo sheep. Genome-wide association study identified MMP16 as significantly associated with litter size variation. Tissue-specific expression profiling demonstrated predominant ovarian expression of MMP16, with peak levels localized to the corpus luteum and mature follicles. IHC analysis further confirmed MMP16 protein expression in granulosa and theca cells, indicating its involvement in follicular development, maturation and ovulation regulation. Functional investigations revealed that MMP16 overexpression enhances GCs proliferation, suppresses apoptosis, and modulates ECM remodeling through PI3K-AKT pathway activation concurrent with AMPK phosphorylation inhibition. Coordinated upregulation of steroidogenic genes further substantiated its role in follicular maturation. These findings provide both a theoretical breakthrough in understanding the genetic basis of ovine hyperprolificacy and a scientific foundation for precision breeding strategies to enhance sheep production efficiency.
Data availability statement
The datasets analyzed during the current study are available, China National Center for Bioinformation/Beijing Institute of Genomics, Chinese Academy of Sciences repository, accession number: GVM000916.
Ethics statement
The animal studies were approved by the Animal Ethics Committee of the College of Animal Science and Technology of Tarim University (No. DTU 20230126). The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent was obtained from the owners for the participation of their animals in this study.
Author contributions
JW: Writing – review & editing, Investigation, Writing – original draft, Visualization. LB: Writing – original draft, Visualization, Investigation, Writing – review & editing. HC: Writing – original draft, Software. XL: Visualization, Writing – review & editing, Conceptualization. AK: Project administration, Resources, Writing – review & editing. WK: Writing – review & editing, Resources, Project administration. CY: Writing – review & editing, Methodology, Writing – original draft. PN: Writing – original draft, Formal Analysis, Writing – review & editing. FH: Writing – original draft, Software, Visualization. DF: Writing – review & editing. CH: Conceptualization, Project administration, Funding acquisition, Writing – review & editing, Writing – original draft, Supervision. QG: Conceptualization, Project administration, Supervision, Writing – review & editing, Funding acquisition, Writing – original draft.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This research was funded by the Bazhou Comprehensive Experiment Station of the Autonomous Region’s Sheep Industry System (Grant No. XJARS-09-17), GWAS-based screening of candidate genes for polyembryony traits in Xinjiang Lop sheep (Grant No. BSGJSYS202304), and Southwards Innovation Team for Collegiate Utilisation of High Quality Sheep Germplasm Resources in the Circum-Tarim Region (Grant No. 2019CB010).
Acknowledgments
We thank the computations in this paper were run on the bioinformatics computing platform of Tarim University. We thank the team of teachers Zhao Chunjiang and Liu Yu from China Agricultural University for their help. We would like to thank everyone who contributed towards the article. We would like to express our gratitude to the Autonomous Region Sheep Industry System Ba-zhou Comprehensive Experimental Station Project for its funding support.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The authors declare that no Gen AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Footnotes
References
1. Muthusamy, M, Akinsola, O, Pal, P, Akinsola, OM, Ramasamy, C, Ramasamy, S, et al. Comparative genomic insights into adaptation, selection signatures, and population dynamics in indigenous Indian sheep and foreign breeds. Front Genet. (2025) 16:1621960. doi: 10.3389/fgene.2025.1621960
2. Farrell, L, Creighton, P, Bohan, A, McGovern, F, and McHugh, N. Bio-economic modelling of sheep meat production systems with varying flock litter size using field data. Animal. (2022) 16:100640. doi: 10.1016/j.animal.2022.100640
3. Bao, J, Xiong, J, Huang, J, Yang, P, Shang, M, and Zhang, L. Genetic diversity, selection signatures, and genome-wide association study identify candidate genes related to litter size in Hu sheep. Int J Mol Sci. (2024) 25:9397. doi: 10.3390/ijms25179397
4. Ma, XF, Liu, AJ, Zheng, Z, Hu, BX, Zhi, YX, Liu, C, et al. Resolving and functional analysis of Rna editing sites in sheep ovaries and associations with litter size. Animal. (2024) 18:101342. doi: 10.1016/j.animal.2024.101342
5. Cui, W, Wang, H, Li, J, Lv, D, Xu, J, Liu, M, et al. Sheep litter size heredity basis using genome-wide selective analysis. Reprod Domest Anim. (2024) 59:e14689. doi: 10.1111/rda.14689
6. Tao, L, He, X, Wang, X, di, R, and Chu, M. Litter size of sheep (Ovis aries): inbreeding depression and homozygous regions. Genes. (2021) 12:109. doi: 10.3390/genes12010109
7. Hernández-Montiel, W, Martínez-Núñez, MA, Ramón-Ugalde, JP, Román-Ponce, SI, Calderón-Chagoya, R, and Zamora-Bustillos, R. Genome-wide association study reveals candidate genes for litter size traits in Pelibuey sheep. Animals. (2020) 10:434. doi: 10.3390/ani10030434
8. Qi, MY, Xu, LQ, Zhang, JN, Li, MO, Lu, MH, and Yao, YC. Effect of the Booroola fecundity (FecB) gene on the reproductive performance of ewes under assisted reproduction. Theriogenology. (2020) 142:246–50. doi: 10.1016/j.theriogenology.2019.10.038
9. Wang, J, Liu, Y, Guo, S, di, R, Wang, X, He, X, et al. Polymorphisms of the Bmpr1B, Bmp15 and Gdf9 fecundity genes in four Chinese sheep breeds. Arch Anim Breed. (2024) 67:51–60. doi: 10.5194/aab-67-51-2024
10. He, X, Wei Wang, XD, and Chu, M. Association between single-nucleotide polymorphism in Pik3cd gene and litter size in small tail Han sheep. Anim Biotechnol. (2023) 34:3337–42. doi: 10.1080/10495398.2022.2140059
11. Hongying Ren, QZ, Tang, Q, Xue, T, Wang, Q, Hongwei, X, and Pan, C. A 24-bp indel within the sheep Ahr gene is associated with litter size. Anim Biotechnol. (2022) 33:1533–8. doi: 10.1080/10495398.2021.1914071
12. Wang, H, Feng, X, Muhatai, G, and Wang, L. Expression profile analysis of sheep ovary after superovulation and estrus synchronisation treatment. Vet Med Sci. (2022) 8:1276–87. doi: 10.1002/vms3.783
13. De Figueiredo, JR, Cadenas, J, De Lima, LF, et al. Advances in in vitro folliculogenesis in domestic ruminants. Anim Reprod. (2020) 16:52–65. doi: 10.21451/1984-3143-Ar2018-123
14. Heng, D, Wang, Q, Ma, X, Tian, Y, Xu, K, Weng, X, et al. Role of Oct4 in the regulation of Fsh-induced granulosa cells growth in female mice. Front Endocrinol. (2019) 10:915. doi: 10.3389/fendo.2019.00915
15. Lai, T, Chen, H, Wu, P, Lai, TH, Chen, HT, Wu, PH, et al. The presence of Tgfβ3 in human ovarian intrafollicular fluid and its involvement in thromboxane generation in follicular granulosa cells through a canonical Tgfβri, Smad2/3 signaling pathway and Cox-2 induction. Int J Mol Sci. (2024) 25:5558. doi: 10.3390/ijms25105558
16. Liu, YX, Zhang, Y, Li, YY, Liu, XM, Wang, XX, Zhang, CL, et al. Regulation of follicular development and differentiation by intra-ovarian factors and endocrine hormones. Front Biosci. (2019) 24:983–93. doi: 10.2741/4763
17. Alam, MH, and Miyano, T. Interaction between growing oocytes and granulosa cells in vitro. Reprod Med Biol. (2020) 19:13–23. doi: 10.1002/rmb2.12292
18. Li, M, Liang, W, Zhu, C, and Qin, S. Smad4 mediates Bmf involvement in sheep granulosa cell apoptosis. Gene. (2022) 817:146231. doi: 10.1016/j.gene.2022.146231
19. Zhang, C l, Zhang, J, Tuersuntuoheti, M, Zhou, W, Han, Z, Li, X, et al. Landscape genomics reveals adaptive divergence of indigenous sheep in different ecological environments of Xinjiang, China. Sci Total Environ. (2023) 904:166698. doi: 10.1016/j.scitotenv.2023.166698
20. Yue, L, Li, W, Pan, Y, Lan, X, Zhang, Q, and Pan, C. Polymorphism of Gtf2A1 gene is associated with litter size in sheep. Theriogenology. (2023) 208:194–200. doi: 10.1016/j.theriogenology.2023.06.004
21. Akhmet, N, Zhu, L, Song, J, Akhatayeva, Z, Zhang, Q, Su, P, et al. Exploring the sheep Mast4 gene variants and their associations with litter size. Animals. (2024) 14:591. doi: 10.3390/ani14040591
22. Gholizadeh, M, and Esmaeili-Fard, SM. Meta-analysis of genome-wide association studies for litter size in sheep. Theriogenology. (2022) 180:103–12. doi: 10.1016/j.theriogenology.2021.12.025
23. Zhu, M, Li, P, Wu, W, Zheng, W, Huang, J, Tulafu, H, et al. The genetic characterization of germplasm and identification of the litter size trait associated candidate genes in Dexin mutton and fine-wool sheep. Front Genet. (2024) 15:1457634. doi: 10.3389/fgene.2024.1457634
24. Tao, L, He, XY, Wang, FY, Pan, LX, Wang, XY, Gan, SQ, et al. Identification of genes associated with litter size combining genomic approaches in Luzhong mutton sheep. Anim Genet. (2021) 52:545–9. doi: 10.1111/age.13078
25. Chantepie, L, Bodin, L, Sarry, J, Woloszyn, F, Plisson-Petit, F, Ruesche, J, et al. Genome-wide identification of a regulatory mutation in Bmp15 controlling prolificacy in sheep. Front Genet. (2020) 11:585. doi: 10.3389/fgene.2020.00585
26. Zhang, J, Zhang, CL, Li, X, Yang, R, Zhou, W, Han, Z, et al. Genetic analysis of key agronomic traits of local sheep breeds in Xinjiang, China. Int J Biol Macromol. (2024) 280:135869. doi: 10.1016/j.ijbiomac.2024.135869
27. Pokharel, K, Peippo, J, Honkatukia, M, Seppälä, A, Rautiainen, J, Ghanem, N, et al. Integrated ovarian mrna and mirna transcriptome profiling characterizes the genetic basis of prolificacy traits in sheep (Ovis aries). BMC Genomics. (2018) 19:104. doi: 10.1186/s12864-017-4400-4
28. Aherrahrou, N, Tairi, H, and Aherrahrou, Z. Genomic privacy preservation in genome-wide association studies: taxonomy, limitations, challenges, and vision. Brief Bioinform. (2024) 25:bbae356. doi: 10.1093/bib/bbae356
29. Giacomini, KM, Yee, SW, Mushiroda, T, Weinshilboum, RM, Ratain, MJ, and Kubo, M. Genome-wide association studies of drug response and toxicity: an opportunity for genome medicine. Nat Rev Drug Discov. (2017) 16:70–13. doi: 10.1038/nrd.2016.234
30. Tam, V, Patel, N, Turcotte, M, Bossé, Y, Paré, G, and Meyre, D. Benefits and limitations of genome-wide association studies. Nat Rev Genet. (2019) 20:467–84. doi: 10.1038/s41576-019-0127-1
31. Xiang, X, Peng, C, Cao, D, Chen, Z, Jin, H, Nie, S, et al. Whole genome sequencing reveals that five genes are related to Bw trait in sheep. Animal. (2024) 18:101282. doi: 10.1016/j.animal.2024.101282
32. Arzik, Y, Kizilaslan, M, Behrem, S, Piel, LMW, White, SN, Çınar, MU, et al. Exploring Genetic Factors Associated with Moniezia spp. Tapeworm Resistance in Central Anatolian Merino Sheep via GWAS Approach. Animals (Basel) (2025). 15:812. doi: 10.3390/ani15060812
33. Jiang, J, Cao, Y, Shan, H, Wu, J, Song, X, and Jiang, Y. The GWAS analysis of body size and population verification of related SNPs in Hu sheep. Front Genet. (2021) 12:642552. doi: 10.3389/fgene.2021.642552
34. Wang, H, Li, C, Li, J, Zhang, R, An, X, Yuan, C, et al. Genomic selection for weaning weight in alpine merino sheep based on Gwas prior marker information. Animals. (2024) 14:1904. doi: 10.3390/ani14131904
35. Li, C, Li, J, Wang, H, Zhang, R, An, X, Yuan, C, et al. Genomic selection for live weight in the 14th month in alpine merino sheep combining Gwas information. Animals. (2023) 13:3516. doi: 10.3390/ani13223516
36. Duijvesteijn, N, Van Der Werf, JHJ, and Kinghorn, BP. Segregation Gwas to linearize a non-additive locus with incomplete penetrance: an example of horn status in sheep. Genet Sel Evol. (2024) 56:61. doi: 10.1186/s12711-024-00928-0
37. Chen, S, Zhou, Y, Chen, Y, and Gu, J. Fastp: an ultra-fast all-in-one Fastq preprocessor. Bioinformatics. (2018) 34:i884–90. doi: 10.1093/bioinformatics/bty560
38. Kim, D, Paggi, JM, Park, C, Bennett, C, and Salzberg, SL. Graph-based genome alignment and genotyping with Hisat2 and Hisat-genotype. Nat Biotechnol. (2019) 37:907–15. doi: 10.1038/s41587-019-0201-4
39. Liao, Y, Smyth, GK, and Shi, W. The R package Rsubread is easier, faster, cheaper and better for alignment and quantification of Rna sequencing reads. Nucleic Acids Res. (2019) 47:e47–7. doi: 10.1093/nar/gkz114
40. Wu, T, Hu, E, Xu, S, Chen, M, Guo, P, Dai, Z, et al. Clusterprofiler 4.0: a universal enrichment tool for interpreting omics data. Innovation. (2021) 2:100141. doi: 10.1016/j.xinn.2021.100141
41. Ocłoń, E, and Hrabia, A. miRNA expression profile in chicken ovarian follicles throughout development and miRNA-mediated MMP expression. Theriogenology. (2021) 160:116–27. doi: 10.1016/j.theriogenology.2020.11.004
42. Chen, J, Liu, F, Wu, J, Yang, Y, He, J, Wu, F, et al. Effect of Stk3 on proliferation and apoptosis of pancreatic cancer cells via Pi3K/Akt/mtor pathway. Cell Signal. (2023) 106:110642. doi: 10.1016/j.cellsig.2023.110642
43. Canty-Laird, E, Carré, GA, Mandon-Pépin, B, Kadler, KE, and Fabre, S. First evidence of bone morphogenetic protein 1 expression and activity in sheep ovarian Follicles1. Biol Reprod. (2010) 83:138–46. doi: 10.1095/biolreprod.109.082115
44. Wozney, JM, Rosen, V, Celeste, AJ, Mitsock, LM, Whitters, MJ, Kriz, RW, et al. Novel regulators of bone formation: molecular clones and activities. Science. (1988) 242:1528–34. doi: 10.1126/science.3201241
45. Lei, X, Cui, K, Li, Z, Su, J, Jiang, J, Zhang, H, et al. Bmp-1 participates in the selection and dominance of buffalo follicles by regulating the proliferation and apoptosis of granulosa cells. Theriogenology. (2016) 85:999–1012. doi: 10.1016/j.theriogenology.2015.11.011
46. Hlaing, M, Nam, K, Lou, J, Pope, WF, and Nephew, KP. Evidence for expression of estrogen receptor cofactor messenger ribonucleic acid in the ovary and uterus of domesticated animals (sheep, cow and pig). Life Sci. (2001) 68:1427–38. doi: 10.1016/S0024-3205(01)00937-7
47. Tian, X, Wang, F, Zhang, L, He, C, Ji, P, Wang, J, et al. Beneficial effects of melatonin on the in vitro maturation of sheep oocytes and its relation to melatonin receptors. Int J Mol Sci. (2017) 18:834. doi: 10.3390/ijms18040834
48. Wang, W, Lv, J, Duan, H, Ding, Z, Zeng, J, Lv, C, et al. Regulatory role of melatonin on epidermal growth factor receptor, type I collagen α1 chain, and caveolin 1 in granulosa cells of sheep antral follicles. Anim Sci J. (2022) 93:e13760. doi: 10.1111/asj.13760
49. Dickinson, RE, Hryhorskyj, L, Tremewan, H, Hogg, K, Thomson, AA, McNeilly, AS, et al. Involvement of the Slit/Robo pathway in follicle development in the fetal ovary. Reproduction. (2010) 139:395–407. doi: 10.1530/Rep-09-0182
50. Li, Y, Wang, Y, Yu, L, Sun, C, Cheng, D, Yu, S, et al. miR-146b-5p inhibits glioma migration and invasion by targeting Mmp16. Cancer Lett. (2013) 339:260–9. doi: 10.1016/j.canlet.2013.06.018
51. Yang, R, Xu, J, Hua, X, Tian, Z, Xie, Q, Li, J, et al. Overexpressed miR-200a promotes bladder cancer invasion through direct regulating dicer/miR-16/Jnk2/Mmp-2 axis. Oncogene. (2020) 39:1983–96. doi: 10.1038/s41388-019-1120-z
52. Jo, W, Kim, M, Oh, J, Kim, CS, Park, C, Yoon, S, et al. Microrna-29 ameliorates fibro-inflammation and insulin resistance in Hif1α-deficient obese adipose tissue by inhibiting Endotrophin generation. Diabetes. (2022) 71:1746–62. doi: 10.2337/db21-0801
53. Suenaga, M, Mashima, T, Kawata, N, Dan, S, Seimiya, H, and Yamaguchi, K. Preclinical analysis and clinical validation to identify biomarkers of regorafenib efficacy in patients with metastatic colorectal cancer. J Clin Oncol. (2024) 42:171–1. doi: 10.1200/Jco.2024.42.3_suppl.171
54. Fata, JE, Ho, ATV, Leco, KJ, Moorehead, RA, and Khokha, R. Cellular turnover and extracellular matrix remodeling in female reproductive tissues: functions of metalloproteinases and their inhibitors. Cell Mol Life Sci. (2000) 57:77–95. doi: 10.1007/s000180050500
55. Curry, JTE, and Osteen, KG. The matrix metalloproteinase system: changes, regulation, and impact throughout the ovarian and uterine reproductive cycle. Endocr Rev. (2003) 24:428–65. doi: 10.1210/er.2002-0005
56. Puttabyatappa, M, Jacot, TA, Al-Alem, LF, Rosewell, KL, Duffy, DM, Brännström, M, et al. Ovarian membrane-type matrix metalloproteinases: induction of Mmp14 and Mmp16 during the periovulatory period in the rat, macaque, and human. Biol Reprod. (2014) 91:34. doi: 10.1095/biolreprod.113.115717
57. Asadzadeh, R, Khosravi, S, Zavareh, S, Ghorbanian, MT, Paylakhi, SH, and Mohebbi, SR. Vitrification affects the expression of matrix metalloproteinases and their tissue inhibitors of mouse ovarian tissue. Int J Reprod Biomed. (2016) 14:173–80.
58. Zhu, G, Kang, L, Wei, Q, et al. Expression and regulation of Mmp1, Mmp3, and Mmp9 in the chicken ovary in response to gonadotropins, sex hormones, and Tgfb1. Biol Reprod. (2014) 90:57. doi: 10.1095/biolreprod.113.114249
59. Falkowski, K, Bielecka, E, Thøgersen, IB, Bocheńska, O, Płaza, K, Kalińska, M, et al. Kallikrein-related peptidase 14 activates zymogens of membrane type matrix metalloproteinases (Mt-Mmps)-a CleavEx based analysis. Int J Mol Sci. (2020) 21:4383. doi: 10.3390/ijms21124383
60. Makker, A, Goel, MM, and Mahdi, AA. Pi3K/Pten/Akt and Tsc/mtor signaling pathways, ovarian dysfunction, and infertility: an update. J Mol Endocrinol. (2014) 53:R103–18. doi: 10.1530/Jme-14-0220
61. Hu, MC, Hsu, HJ, Guo, IC, and Chung, B-c. Function of Cyp11a1 in animal models. Mol Cell Endocrinol. (2004) 215:95–100. doi: 10.1016/j.mce.2003.11.024
62. Dong, J, Guo, C, Yang, Z, Wu, Y, and Zhang, C. Follicle-stimulating hormone alleviates ovarian aging by modulating mitophagy- and Glycophagy-based energy metabolism in hens. Cells. (2022) 11:3270. doi: 10.3390/cells11203270
Keywords: lob sheep, whole-genome resequencing, litter size, MMP16, ovarian granulosa cells
Citation: Wang J, Bi L, chen H, Li X, Khamili A, Kurban W, Yang C, Niu P, Huang F, Fang D, Han C and Gao Q (2025) Genome-wide association study reveals candidate genes for litter size and function validation of MMP16 gene in lop sheep. Front. Vet. Sci. 12:1682236. doi: 10.3389/fvets.2025.1682236
Edited by:
Xiangdong Ding, China Agricultural University, ChinaReviewed by:
Xiaoyun He, Chinese Academy of Agricultural Sciences, ChinaBuying Han, Qinghai University, China
Copyright © 2025 Wang, Bi, chen, Li, Khamili, Kurban, Yang, Niu, Huang, Fang, Han and Gao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chunmei Han, Y2h1bm1laWhhbjIyNEAxNjMuY29t; Qinghua Gao, Z3FoZGt5QDEyNi5jb20=
†These authors have contributed equally to this work