 Xiang yu Li1†Piao Piao Yang1†Yunzhu Tang1†XiaoFeng Li1Xuejiao Jia1Mengqi Liu1
Xiang yu Li1†Piao Piao Yang1†Yunzhu Tang1†XiaoFeng Li1Xuejiao Jia1Mengqi Liu1 Yonggang Li1*
Yonggang Li1* Wei Zhao1,2*
Wei Zhao1,2*- 1College of Basic Medical Sciences, Jinzhou Medical University, Jinzhou, China
- 2Liaoning Province Key Laboratory of Human Phenome Research, Jinzhou, China
Background: Shigella spp. are Gram-negative enteropathogens responsible for bacillary dysentery (shigellosis) in humans and animals. While the primary reservoirs of Shigella are historically recognized as humans and non-human primates, emerging evidence indicates an expanding host range encompassing diverse animal species. This study systematically characterizes a Shigella sp. strain isolated from diarrheic suckling mice.
Methods: Diarrheic suckling mice intestinal contents were collected and analyzed using 16S rRNA sequencing technology. Shigella sp. JZ001 strain was isolated, purified, and identified. Bacterial morphology was examined by transmission electron microscopy. Carbon source utilization profiles were determined using BIOLOG GENIII MicroPlates™. Short-chain fatty acid (SCFA) composition was quantified by gas chromatography–mass spectrometry (GC–MS) following methyl-tert-butyl ether extraction. Polar lipid analysis was performed through two-dimensional thin-layer chromatography with detection using 10% ethanolic molybdatophosphoric acid. Whole genome sequencing was performed using a hybrid approach combining Illumina NovaSeq and Nanopore MinION platforms. Phylogenetic reconstruction was performed using maximum-likelihood algorithms implemented in MEGA.
Results: Rotavirus-SA11 infected suckling mice exhibited watery feces 4 days post-infection. Intestinal contents were analyzed using 16S rRNA sequencing technology, which revealed a significant increase in the genus Shigella in the RV-infected group. A Shigella strain was isolated and purified from the intestinal contents and designated as JZ001. Polyphasic characterization confirmed that strain JZ001 belongs to the Enterobacteriaceae family and is phylogenetically closest to Shigella spp. The genome consists of a single circular chromosome (5,329,126 bp; G + C content 50.65%) and one plasmid (1,09829 bp, G + C content 40.14%). Comparative genomic analysis identified 24 Salmonella pathogenicity island (SPI)-like regions. Substrate assimilation tests demonstrated metabolic versatility, including utilization of D-Maltose, D-Trehalose, α-D-Glucose, D-Mannose, D-Fructose, D-Galactose and Propionic Acid. Cellular fatty acid methyl ester (FAME) analysis identified predominant components as: Dodecanoic acid (12:0; 11.08%), Tetradecanoic acid (14:0; 12.00%), Hexadecanoic acid (16:0; 21.55%), cycloheptadecanoic acid (17:0 cyclo; 13.99%), Dodecanal (12:0 aldehyde; 22.32%). The polar lipid profile consisted of diphosphatidylglycerol, phosphatidylglycerol, and phosphatidylethanolamine, with notable absence of aminolipids and aminophospholipids.
Conclusion: First study of murine Shigella JZ001 offers insights into genomic plasticity and host adaptation mechanisms. The genomic dataset lays foundation for host-pathogen studies and facilitates developing murine models for therapy evaluation.
Introduction
Shigellosis is an acute inflammatory bowel disease characterized by bloody mucoid diarrhea, pyrexia, and abdominal cramping, typically presenting as a self-limiting gastrointestinal infection. The etiological agent, Shigella spp., are Gram-negative, non-sporulating bacilli within the family Enterobacteriaceae (1). Human-pathogenic Shigella strains are taxonomically classified into four species: S. dysenteriae (serogroup A), S. flexneri (serogroup B), S. boydii (serogroup C), and S. sonnei (serogroup D) (2). Clinical manifestations in humans include colonic mucosal ulceration, acute-phase responses (leukocytosis, elevated C-reactive protein), and electrolyte imbalance secondary to secretory diarrhea (3). In contrast, the pathophysiological presentation of shigellosis in non-human hosts remains poorly characterized (4). The natural hosts of Shigella are typically humans and other primates, but it has been shown that the host range of Shigella has expanded to many animals (5–9). Animal Shigella infection is becoming a major threat to animal health and a potential zoonotic risk, posing dangers of cross-species transmission to humans. However, limited information is available regarding the genetic background of Shigella strains isolated from animals. Previous reports indicate that mice are naturally resistant to oral challenge with Shigella (10). The lack of a suitable animal model has significantly hindered our understanding of Shigella pathogenesis. To date, no murine-derived Shigella strains have been reported, particularly with respect to their isolation and whole-genome architecture.
In this study, we isolated a novel Shigella strain (designated JZ001) from diarrheic laboratory-bred suckling mice and conducted systematic pheno-genomic characterization. Through comparative whole-genome sequencing and functional annotation, we revealed close phylogenetic relationships with human clinical isolates. These findings offer critical insights into the evolutionary trajectory of Shigella across host species boundaries and provide foundational data for developing murine infection models to study zoonotic transmission dynamics.
Methods
Collection and treatment of sample and 16S rRNA sequencing
Three-day-old suckling mice were divided into two groups: Rotavirus SA11 infected group and uninfected controls group. The RV-infected group was orally administered 50 μL of RV strain SA-11 at a concentration of 106 PFU/mL, while the uninfected control group received 50 μL of phosphate-buffered saline (PBS) (11). All mice were euthanized 4 days post-RV infection, at the time point when the most severe diarrheal symptoms were observed. The procedures for the care and use of animals were approved by the Committee on Laboratory Animal Ethics of Jinzhou Medical University, and all applicable institutional and governmental regulations concerning the ethical use of animals were followed (Approval ID: 2019014).
16S rRNA gene sequencing and analysis were performed as described in our previous study (11). Briefly, genomic DNA was extracted from mouse fecal samples. The V3–V4 region of the bacterial 16S ribosomal RNA gene was amplified using primers 342F and 806R, and sequenced on the Illumina MiSeq platform (Illumina, Inc., San Diego, CA, USA). The sequencing reads were assembled and used for subsequent 16S analysis. Based on phylotypic annotation, the relative abundances of the top 10 genera were calculated, and star-plot histograms were generated for each sample at the genus level. Phylotypes with relatively high abundances and their proportions could be examined across different taxonomic levels (12, 13).
Fecal samples were also diluted with sterile PBS and streaked onto Salmonella–Shigella (SS) agar plates. Twenty bacterial colonies were randomly selected for PCR detection and sequencing analysis. Colonies testing positive for Shigella spp. were subsequently re-cultured on SS agar for further characterization.
Culture media, bacterial isolation and cultivation
The faecal specimens were streaked onto selective agars for the isolation of Shigella spp. as described previously (14). The Shigella isolates were identified biochemically using triple-sugar iron, lysine iron, motility-indole-ornithine, and Simmons citrate agars. Colonies appearing after incubation for 2, 5 and 10 days were picked and re-streaked on agar plates of same media. Bacterial purity was evaluated by by assessing colony morphology, 16S rRNA gene and genome sequencing.
Cell morphology observation and chemotaxonomic determinations
Single colonies of Shigella were inoculated and cultured overnight at 37 °C. 1 mL of the resulting culture was harvested, and bacterial cells were collected by centrifugation at 8,000 × g for 10 min at 4 °C. Subsequently, 10 μL of the resuspended bacterial suspension was dropped onto a copper grid. 10 μL of 1.5% phosphotungstic acid solution (pH 6.8–7.0) was added to the grid for staining, with the staining process lasting 90 s. Following staining, the grid was air-dried at room temperature for 15–20 min and finally observed under a transmission electron microscope (HT7800, HITACHI).
Carbon sources utilization was determined using the 96-well BIOLOG GENIII MicroPlate (OmniLog Data Collection Software, Identification System version 2.3, BIOLOG), which contained 95 different carbon substrates. Bacteria strains were cultured in TSA medium for 24 h at 30 °C, after which the cells were harvested. Cellular fatty acids were then extracted and methylated according to the standard midi protocol by Agilent 6,890 N (Fatty Acid Methyl Esters, Microbial ID, Sherlock Version 6.0B). Identification was performed by SHERLOCK® Microbial Identification System (Microbial ID, American). Short-chain fatty acids (SCFAs) were measured using GC–MS. For extraction, 1 mL of culture was mixed with 1 mL of ethyl acetate, and the upper layer was collected for GC–MS analysis. The separation of polar lipids was performed using two-dimensional thin-layer chromatography (TLC) on silica gel coated plates, 10×10 cm; Merck. The detection of total lipds was accomplished by employing a solution of 10% ethanolic molybdate and phosphoric acid (Sigma). Aminolipids were detected using a 0.4% solution of ninhydrin (Sigma) in butanol. The presence of phospholipids was detected using a Zinzadze reagent (a molybdenum blue spray reagent with a concentration of 1.3%, obtained from Sigma). The identification of glycolipids was accomplished through the utilization of a 0.5% α-naphthol sulphuric acid reagent.
Phylogenetic tree construction
Phylogenetic analysis was performed using the ClustalW alignment tool and the Molecular Evolutionary Genetic Analysis (MEGA X) software1 (15).
Whole genome sequencing and analysis
Genomic DNA was extracted using the Bacteria DNA Kit (Tiangen Biotech Co., Ltd., Beijing, China). DNA integrity and purity were assessed using 1% agarose gel electrophoresis. The Qubit 4.0 Fluorometer and NanoDrop One spectrophotometer (both from Thermo Fisher Scientific, Waltham, USA) were used to determine DNA concentration and purity.
DNA template libraries were constructed using the BluePippin system, with concentration measured by Qubit 4.0 and average fragment size evaluated using the Agilent 4,200 system (Agilent Technologies, Santa Clara, CA) (16). Genome component prediction included coding genes, repetitive sequences, non-coding RNAs, and prophages. Gene prediction was performed using Glimmer3 (17). Transfer RNAs (tRNAs) were predicted using tRNAscan-SE (version 1.4; http://lowelab.ucsc.edu/tRNAscan-SE/), while ribosomal RNAs (rRNAs) were identified using rRNAmmer (version 1.2; http://www.cbs.dtu.dk/services/RNAmmer/). Small RNAs (sRNAs) were annotated by comparison with the Rfam database and filtered using the cmsearch program (18–20).
Prophage prediction was carried out using PHAST_finder (version 2.1) (21), and genomic islands were identified with IslandPath-DIMOB (22). CRISPR sequences were detected using CRISPRdigger (23).
Functional annotation of predicted genes was performed using whole-genome BLAST searches against the NR (Non-Redundant Protein), COG (Clusters of Orthologous Groups), Swiss-Prot, KEGG (Kyoto Encyclopedia of Genes and Genomes), and GO (Gene Ontology) databases. Prediction of secreted proteins was conducted using SignalP (24), LipoP (25), TMHMM (26), and PSORTb (27). Pathogen-host interaction genes were annotated using the PHI database (28), and virulence factors were identified using the VFDB (29).
Based on genome assembly, coding gene prediction, non-coding RNA analysis, and functional annotations, the complete genome map was visualized using Circos software (30).
The complete genome sequence of strain JZ001 has been submitted to the National Center for Biotechnology Information (NCBI) under BioProject number PRJNA1238207.
Comparative genomic analysis
Ten publicly available genomes of Shigella sp.—including GCF_002290485.1, GCF_022354085.1, GCF_013374815.1, GCF_002950275.1, GCF_002950135.1, GCF_002950295.1, GCF_002950215.1, GCF_000754175.1, GCF_002950155.1, and GCF_002950055.1—were utilized for comparative genomic analysis with strain JZ001. Orthologous Average Nucleotide Identity Software Tool (OAT) (31) was employed to ascertain the average nucleotide identity (ANI) of 16S rRNA, with the objective of identifying bacterial genomic relatives. ANI > 95% indicates that the two genomes belong to the same species. MUMmer software (Version 3.23) was then employed to compare the Shigella Sp JZ001 genome with the reference genome (32), and the collinearity relationship between the genomes was determined. Subsequently, gene family analysis was conducted using the OrthoFinder program with protein sequences from 11 genomes as input files (33).
Culture preservation
Bacterial strains were cultured in liquid medium for 2 days. The cultures were stored in the laboratory by the addition of an equal volume of 65% (v/v) glycerol (1 mL), and then stored at −80 °C for long-term preservation. All type strains assigned by this study were deposited at the Agricultural Culture Collection of China (ACCC) under the number ACCC 64336.
Results
Isolation Shigella in mice
In this study, intestinal microbiota analysis was conducted on previously collected samples from rotavirus-infected diarrheic suckling mice (11). As shown in Figures 1A,B, the fecal microbiota of rotavirus-infected mice was dominated by Lactobacillus spp. and Shigella spp. Notably, the relative abundance of Shigella spp. in the rotavirus-infected group was markedly higher than in the uninfected control group (Figure 1B). The detailed relative abundance values of all microbial taxa identified in the community composition analysis are provided in Supplementary Table S1.
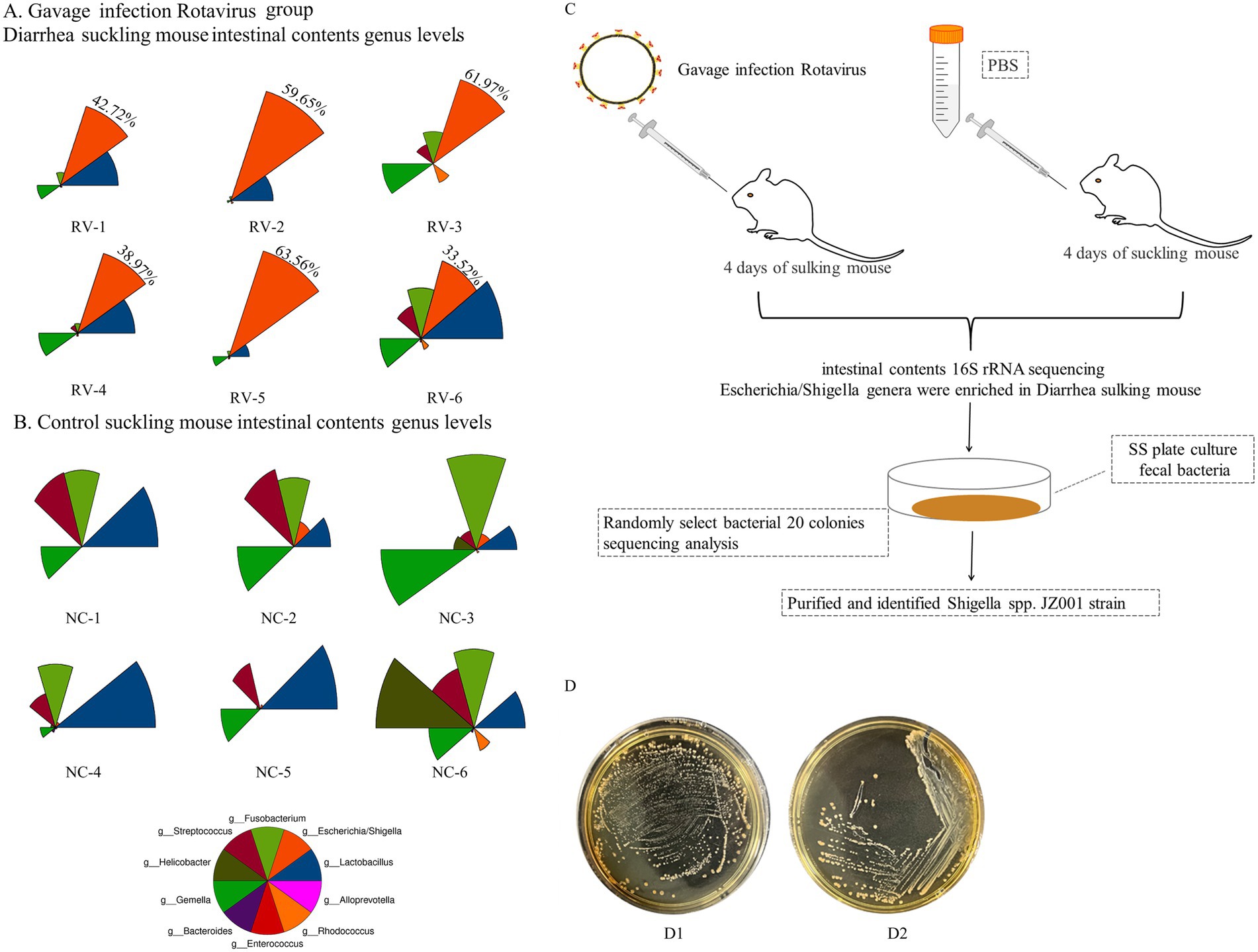
Figure 1. (A,B) Show the results of intestinal microbiota analysis on fecal samples from rotavirus-infected diarrheic suckling mice (RV group) and age-matched control mice (NC group). (A) The abundance of bacterial phylotypes in the gut microbiota of fecal samples from diarrheic mice (RV group), The red color sector represents Shigella spp., and its relative percentage values are displayed in the red sector area; (B) control mice (NC group). The relative abundances of the top 10 bacterial phylotypes at the genus level via star plots (Supplementary Table S1). Star plots visualize the relative abundance of individual phylotypes within each sample: each sector, distinguished by a unique color, corresponds to one genus-level phylotype, and the radius of each sector is proportional to the relative abundance of the corresponding phylotype—with a longer radius indicating a higher relative abundance. (C) Schematic diagram of isolation method for Shigella spp. in mice. (D) D1 Mice fecal culture on SS agar plate; D2 Shigella spp. Culture in SS plate.
Based on these findings, diarrheal fecal samples were first cultured on SS agar. Twenty randomly selected colonies were analyzed, and two were identified as Shigella spp. (Figure 1C). Subsequently, targeted isolation was conducted using Shigella-selective agar, and colonies confirmed as Shigella sp. JZ001 by PCR and sequence analysiswere selected for pure culture (Figure 1D).
Bacterial growth and cell morphology
After 24 h of cultivation on standard agar medium, Shigella strain JZ001 formed semi-transparent, smooth-surfaced, circular, gray-white colonies. Examination of cell morphology (Figure 2) revealed that JZ001 cells were rod-shaped and lacked flagella.
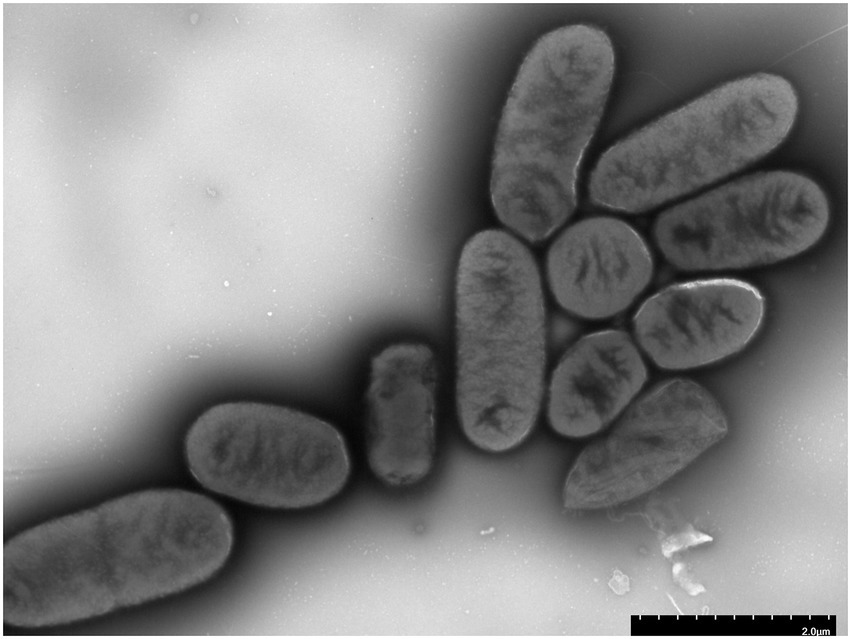
Figure 2. Representative transmission electron microphotographs (TEM) of JZ001 strain Overnight culture of each strains were collected and stained by with 1.5% phosphotungstic acid for 90 s and examined under TEM. Magnification = 15,000.
Physiological and biochemical characterization
The assimilation of 95 carbon sources by strain JZ001 was assessed, with the results summarized in Table 1. The strain metabolized 64 of the 95 carbon sources tested, showing a broad metabolic versatility in carbon source utilization. Monosaccharides and disaccharides were particularly favored. Notably, JZ001 was able to assimilate D-maltose, D-trehalose, α-D-flucose, D-mannose, D-fructose, D-galactose, and propionic acid, demonstrating a versatile metabolic profile. The JZ001 strain was capable of growth under both acidic (pH 5) and weakly acidic (pH 6) conditions. It also grew stably in the presence of 1, 4, and 8% NaCl and exhibited tolerance or reduced sensitivity to most antibiotics and metabolic inhibitors. Notably, the strain demonstrated resistance to rifampicin and vancomycin, among other antibiotics.
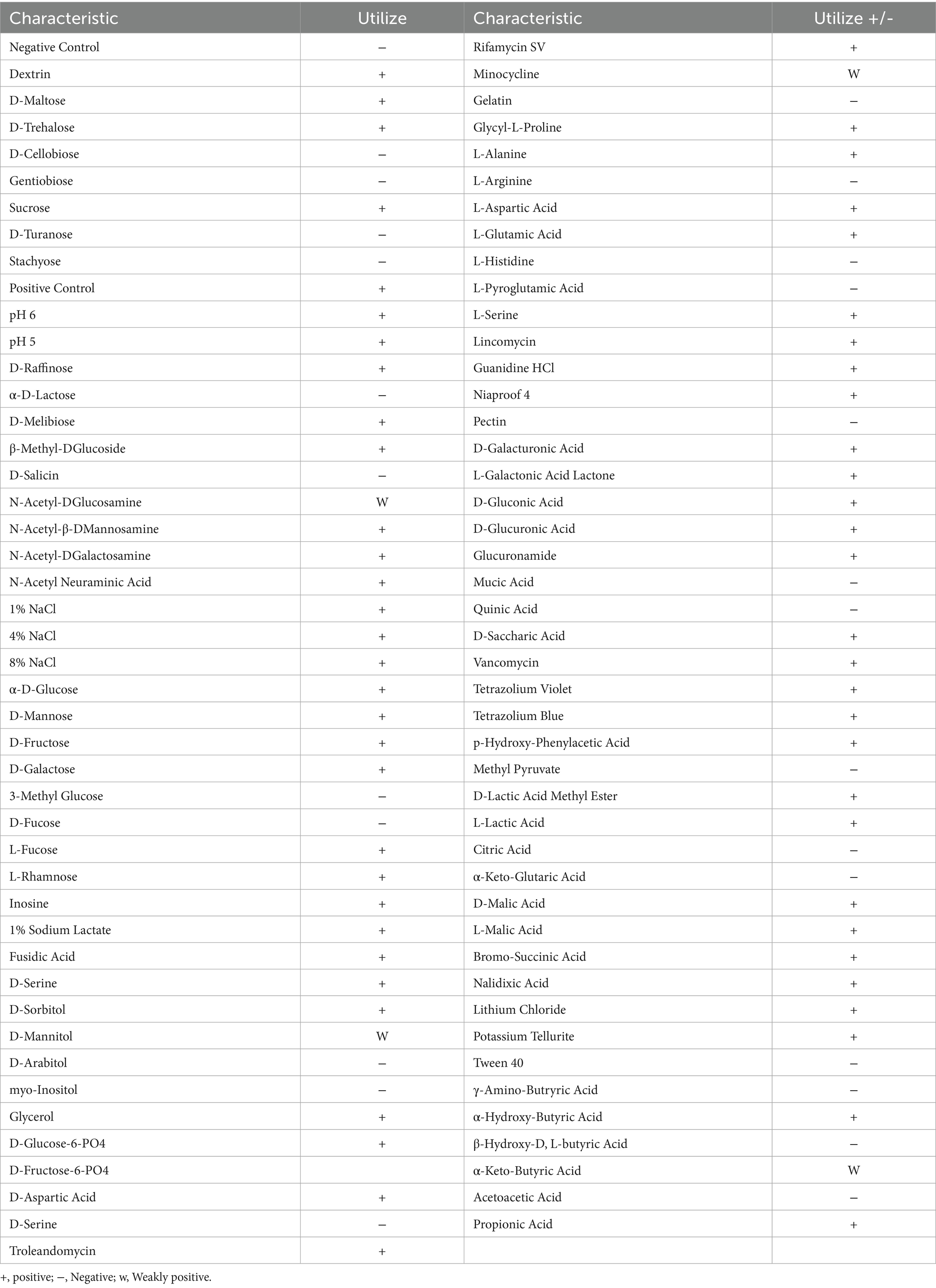
Table 1. Utilization of the carbon sources on BIOLOG GENIII MicroPlate.
Cellular fatty acid and polar lipid profiling
The cellular fatty acid and polar lipid profiles of strain JZ001 were determined (Table 2; Figure 3). Using a 10% threshold to define predominant fatty acids, JZ001 exhibited the following: 12:0 (11.08%), 14:0 (12.00%), 16:0 (21.55%), 17:0 cyclo (13.99%), 12:0 aldehydes (22.32%), and an unknown component (10.95%). Polar lipid profiling revealed the presence of diphosphatidylglycerol, phosphatidylglycerol, phosphatidylethanolamine, and aminolipids. However, aminophospholipids were not detected (Figure 3). No novel fatty acid species, previously unreported in human Shigella strains, were detected in the JZ001 strain. Its core fatty acid profile remains consistent with the genus-level characteristics of Shigella (e.g., dominated by 16:0 and 17:0 cyclo).
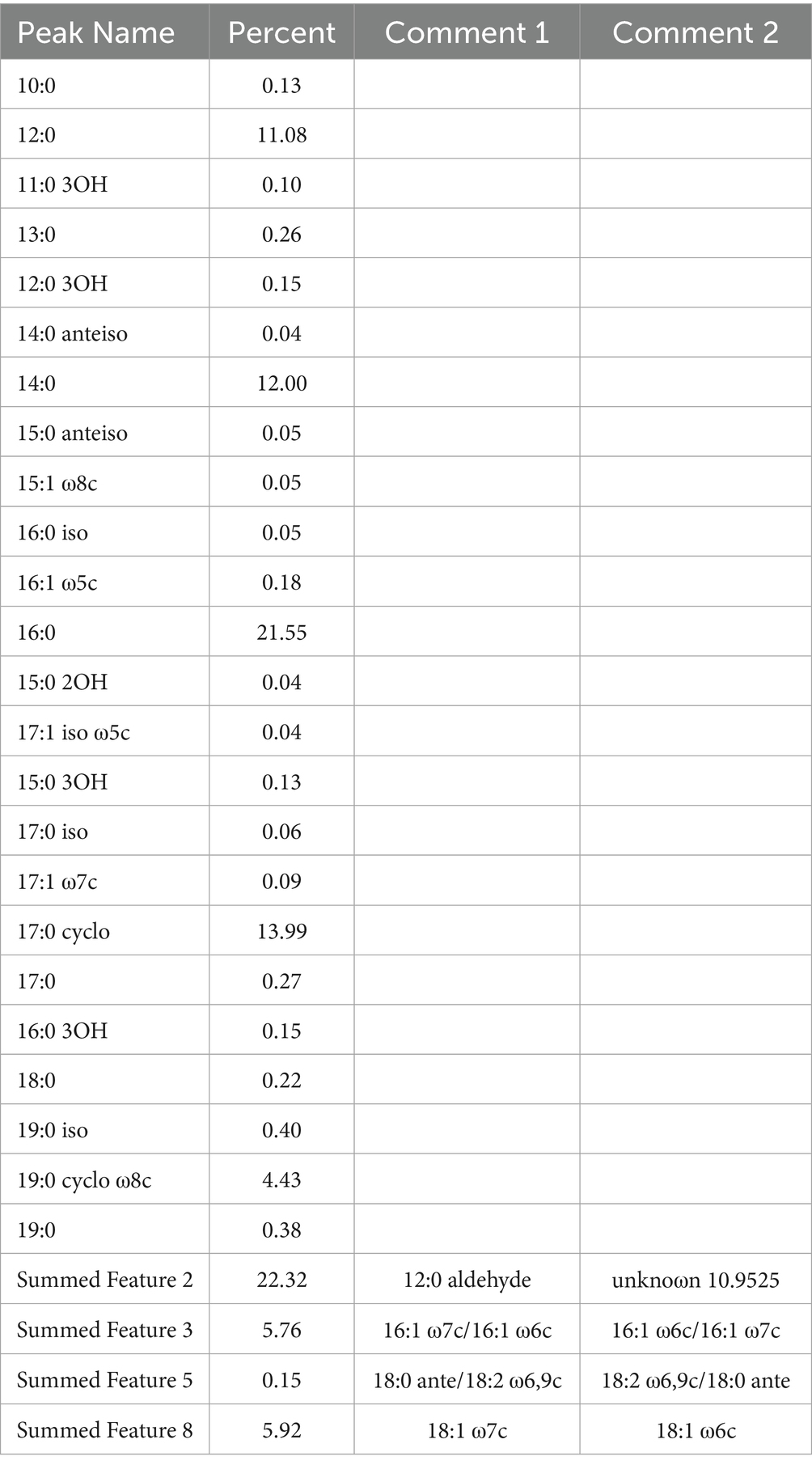
Table 2. Cellular fatty acids and polar acid compositions.
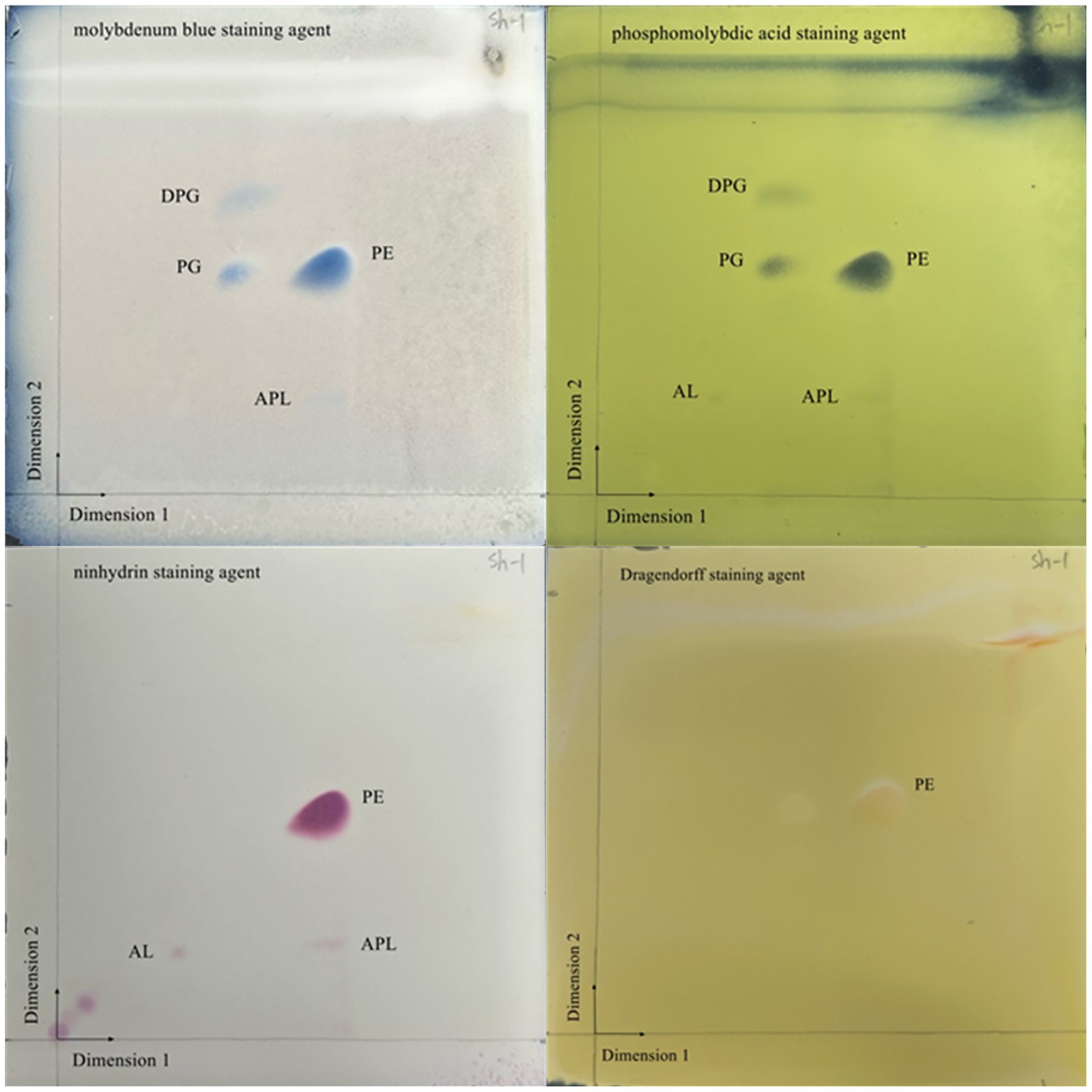
Figure 3. Two-dimensional TLC plate of polar lipids extracted from JZ001 strain. The plate was sprayed with 10% (v/v) molybdophosphoric aicd to show all polar lipids present. DPG, diphosphatidylglycerol; PG, phosphatidylglycerol; PE, phosphatidylethanolamine; AL, aminolipid; APL, aminophospholipid.
General features of the JZ001 genome
Whole-genome sequencing of strain JZ001 produced 8,361,226 clean reads, totaling 1,249,861,482 base pairs. The average read length was 12,933.9 bp, with the longest read measuring 22,023 bp. The total assembled sequence length reached 1,761,547,250 bp, corresponding to 98.82% genome coverage, ensuring high sequencing accuracy. Raw data have been deposited in the NCBI database under BioProject number PRJNA1238207.
Genome assembly resulted in two gap-free, circular contigs. The larger contig, with a total length of 5,219,297 bp (Figure 4A; Table 3), corresponds to the chromosomal replicon. The smaller contig measured 109,829 bp and likely represents the virulence plasmid (Figure 4B).
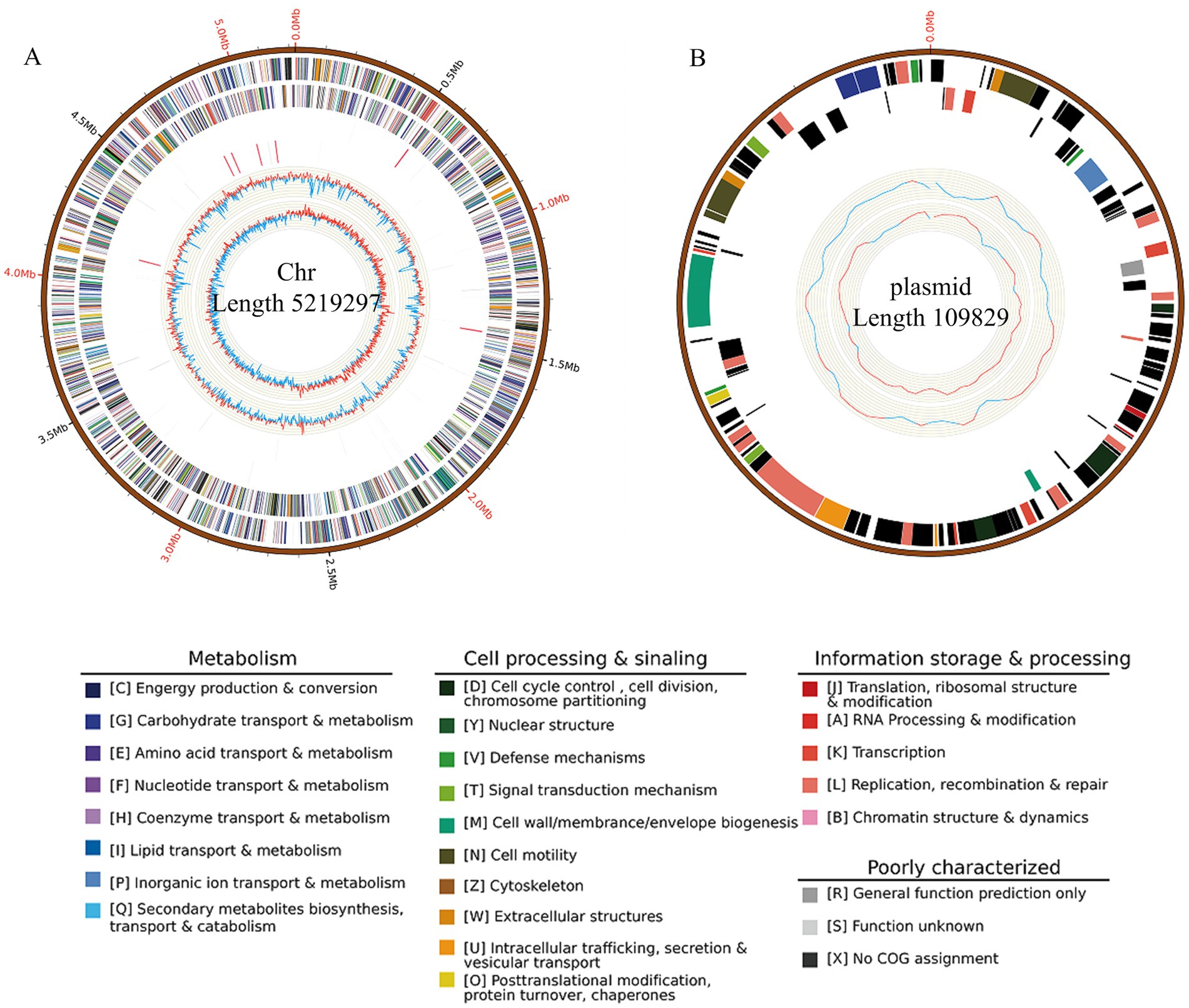
Figure 4. Circular map of the JZ001 genome. The genome is composed of a chromosome and one plasmid. The outermost ring represents the scale marks of the genome. The two following rings within the scale ring depict coding regions (CDSs) in the forward (blue) and reverse (yellow) strand. Moving toward the center, the next rings depict the rRNA (red) in the forward and reverse strand, followed by rings showing the tRNAs (black) in the forward and reverse strands. The innermost ring shows the GC content. The figure was generated by Circos software.
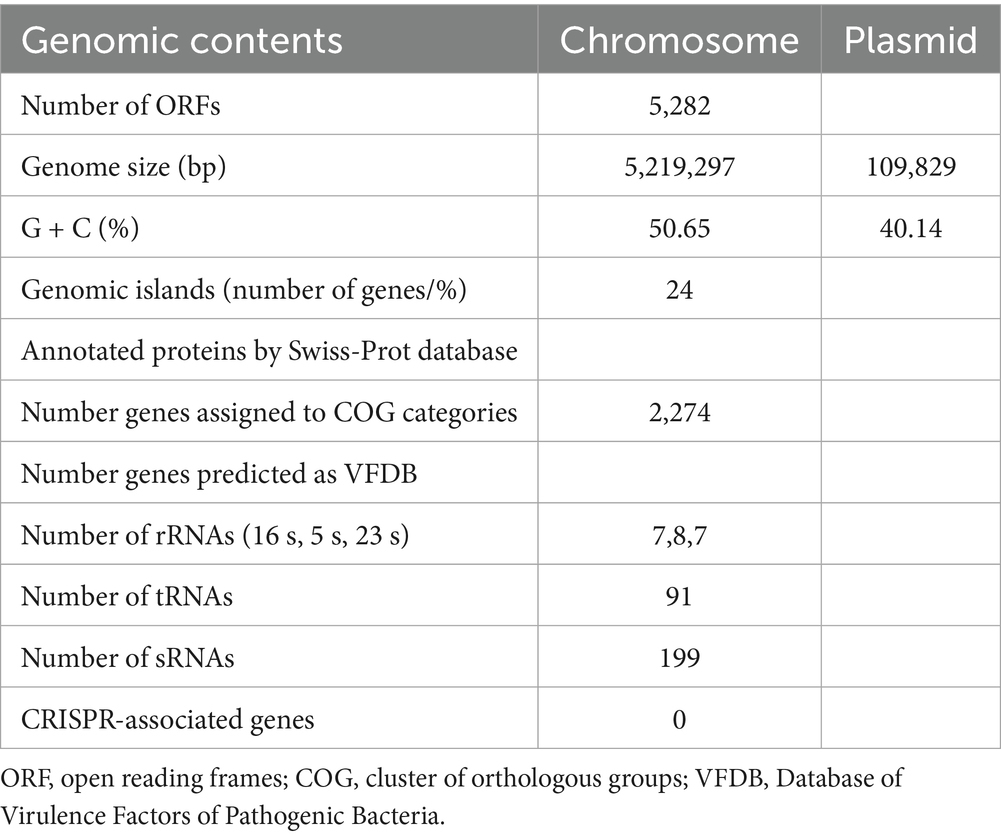
Table 3. Basic genome information of JZ001.
Mobile genetic elements
To further elucidate the genomic basis of adaptation and virulence, mobile genetic elements in JZ001 were analyzed. The chromosome of JZ001 was predicted to contain 24 genomic islands (GIs), designated GI1–GI24 (Table 4). Several of these GIs were associated with virulence and included SPI-1, SPI-3, SPI-5, SPI-8, SPI-11, SPI-15, SPI-18, and SPI-21. Their structures and functions are illustrated in Figure 5. GI 21 of this strain harbors the cidA and tib genes, whereas GI 18 contains four core genes: rfbA, rfbC, gndA, and ugd. GI 5 carries cidA, hrpB, tibA, and cah. GI 3 is enriched in multiple genes related to virulence and metabolism, including cidA, cyaB, and pic. Here, cyaB acts as a core effector gene contributing to pathogenicity, while picA and picB encode serine proteases. GI 1 predominantly carries the cidA and ehaG genes.
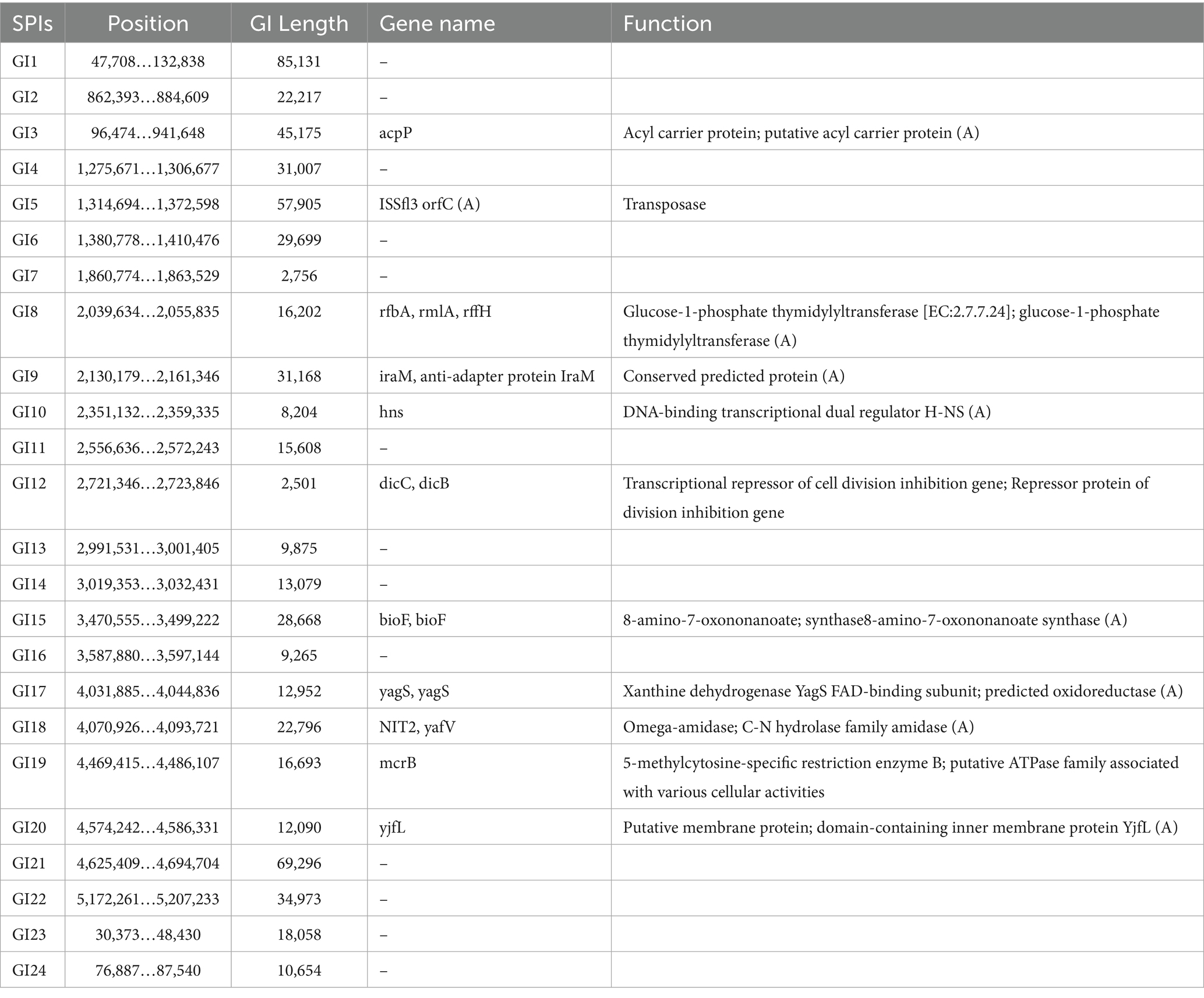
Table 4. Predicated pathogenicity islands (SPIs) in ZJ001.
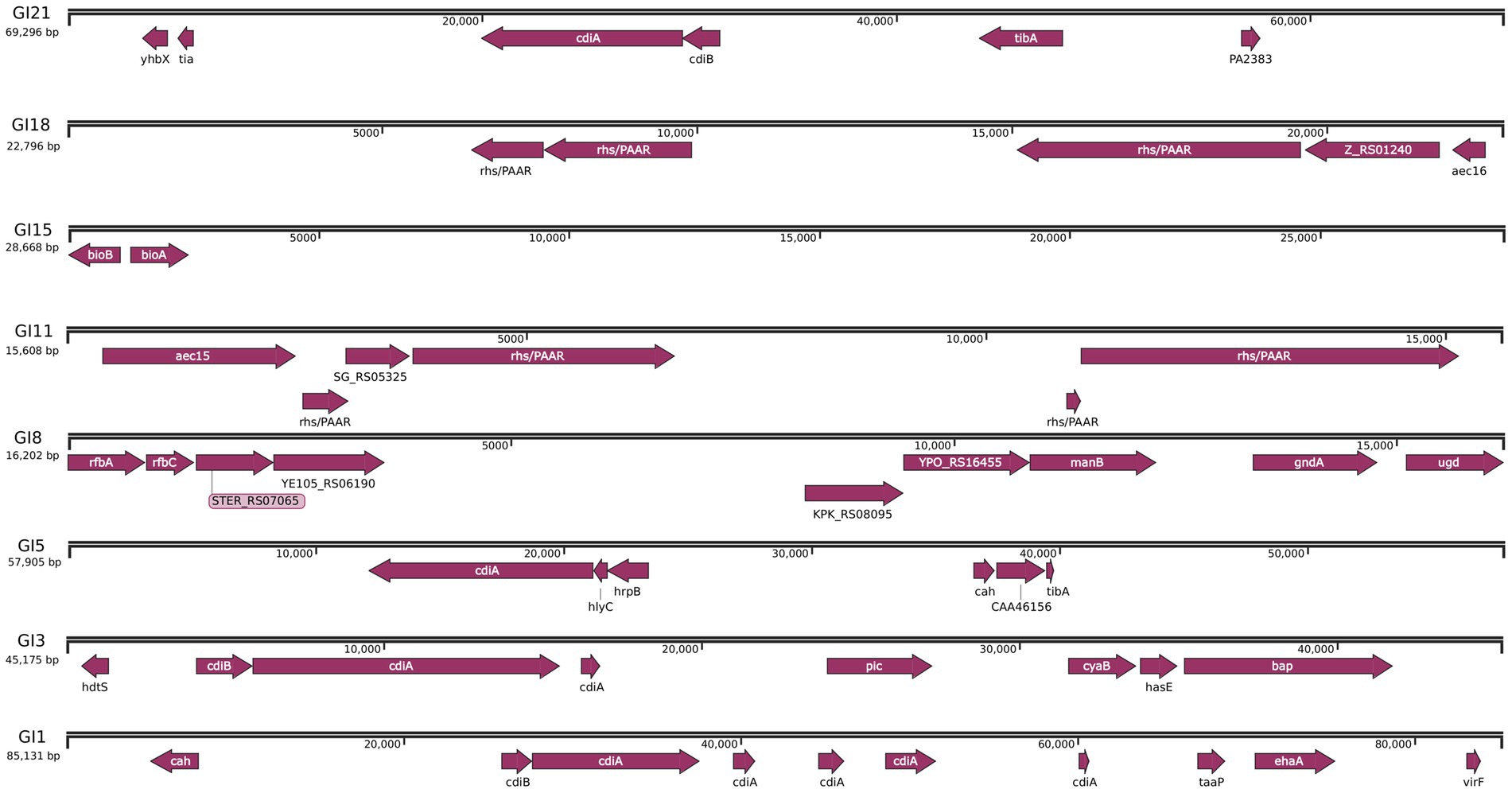
Figure 5. Detailed mapping pathogenicity islands and plasmid in JZ001 genome. Schematic representation of the genes carried within the eight SPIs predicated in the genome of JZ001. The grey boxes represented the range of SPIs, and the brown boxes showed the CDS encoded in these SPIs regions.
Prophages temperate phage genomes integrated into the bacterial chromosome are known to enhance bacterial adaptability and adhesion. PHAST analysis identified eight prophage regions in JZ001, totaling 368,421 bp, with an average length of 46,052.62 bp. Details of these prophages, including length, genomic position, number of CDSs, and GC content, are presented in Table 5.
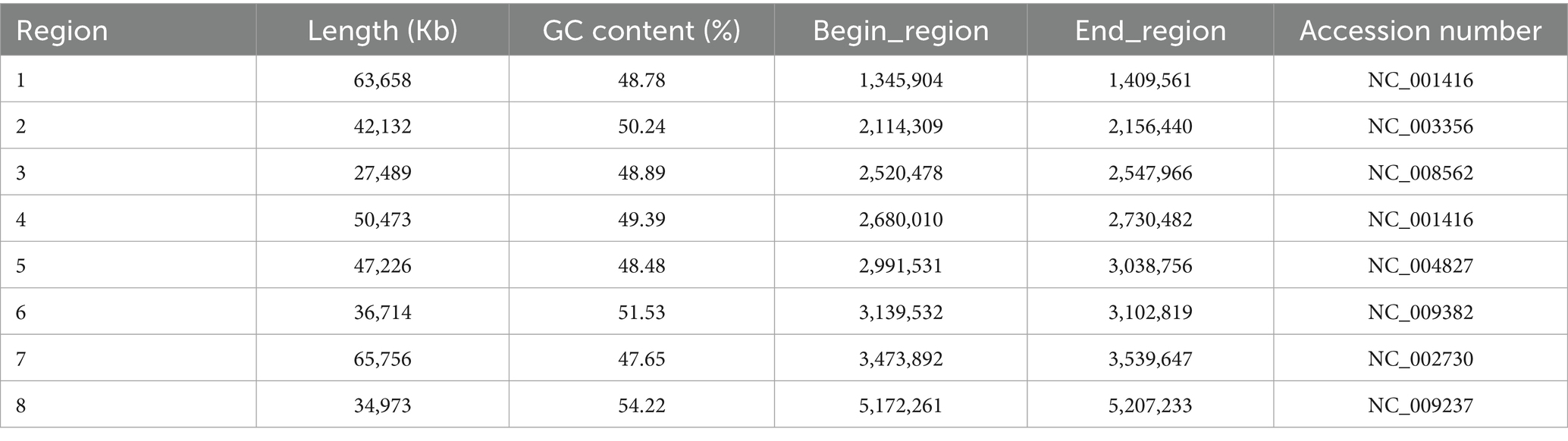
Table 5. Prophage content of Shigella sp. (JZ001) analyzed using PHAST tool.
Comparative genome analysis
Whole-genome alignment was performed between strain JZ001 and ten publicly available Shigella spp. genomes. Given that no mouse-derived Shigella strain data is currently accessible in public data repositories; these ten genomes were selected from the ATCC database of human strains and chosen with 1–2 representative strains per Shigella serogroup to encompass taxonomic diversity. Homologous genomic blocks shared among these strains were identified, indicating a high degree of similarity (Figure 6; Table 6). ANI (Average Nucleotide Identity) analysis revealed that JZ001 had the highest similarity to Shigella sonnei strain SE6-1 (ANI > 99%). Phylogenetic analysis based on 16S rRNA sequences using the neighbor-joining method revealed that the strains clustered according to their serotypes. Notably, strain JZ001 exhibited close phylogenetic relatedness to Shigella sonnei (GenBank accession: MZ540766.1) (Figure 7).
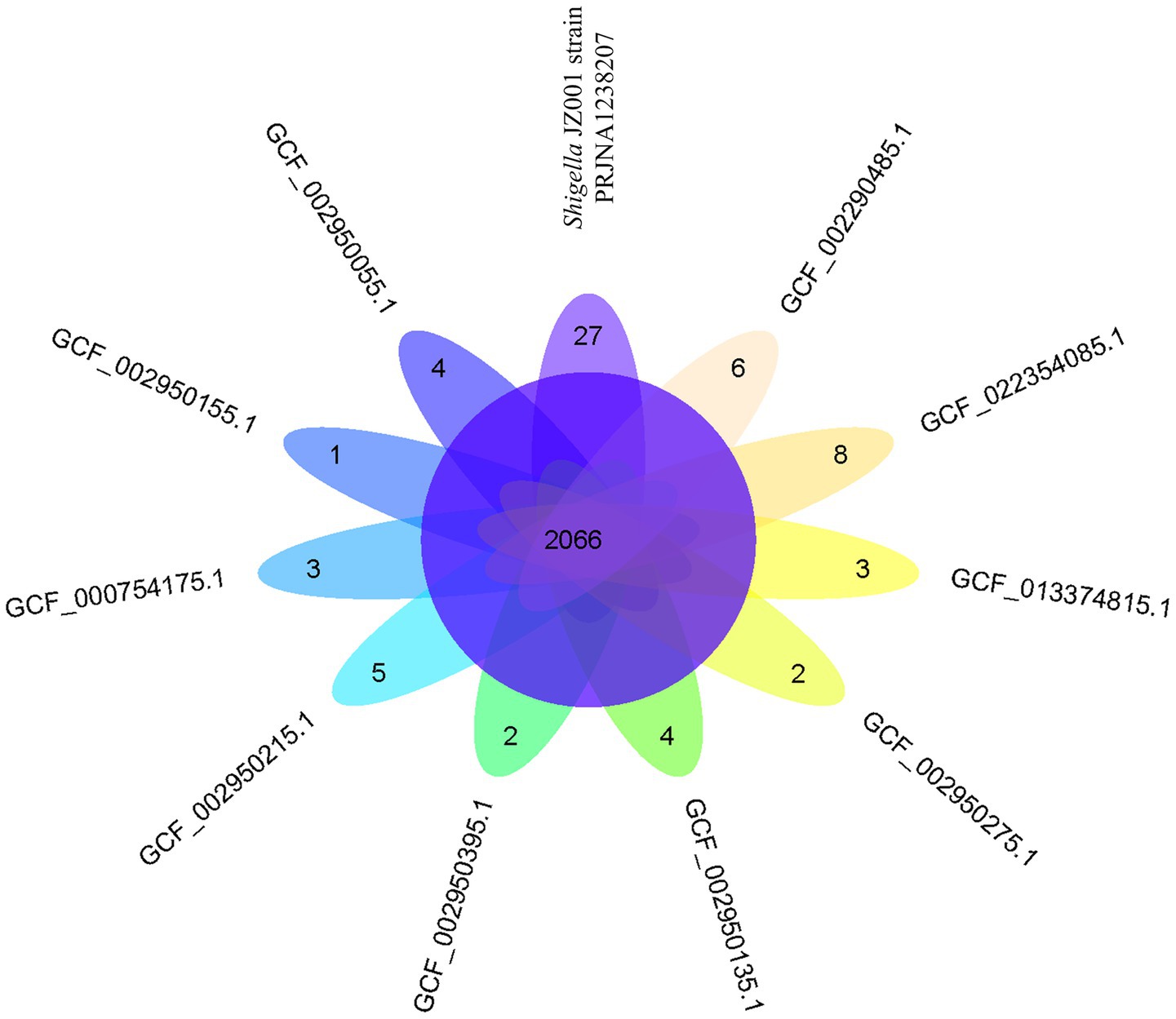
Figure 6. Venn diagram showing the number of genes of orthologous CDSs shared between ten strains and Shigella JZ001 strain (PRJNA1238207).
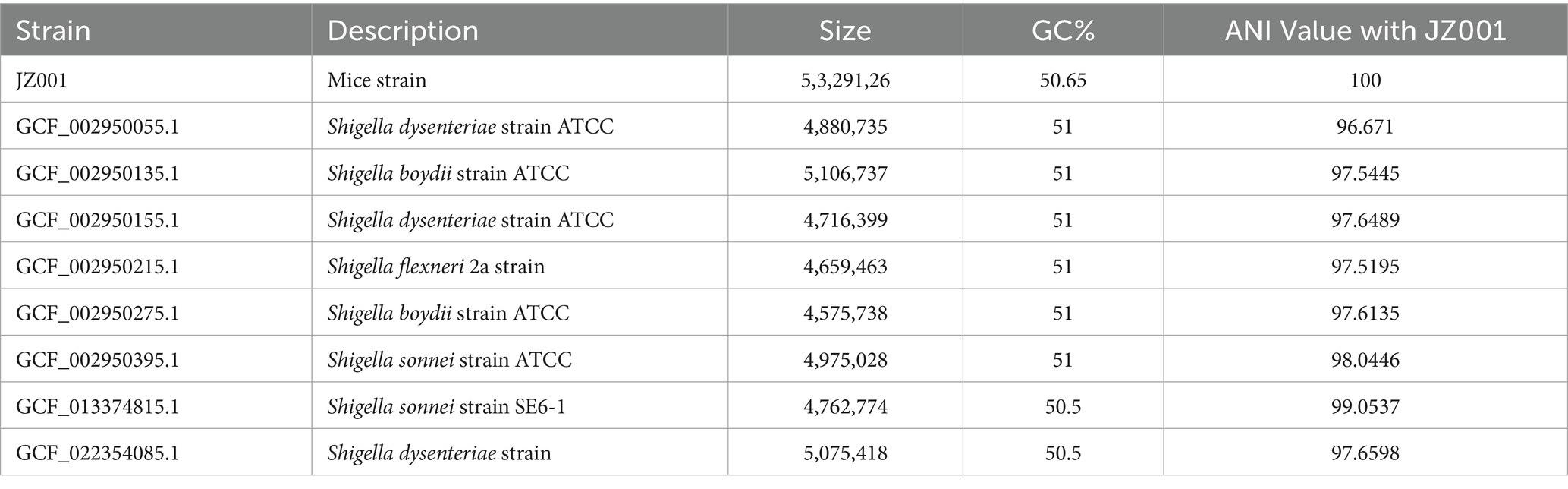
Table 6. Basic features of ten strains and ANI value between these strains with JZ001.
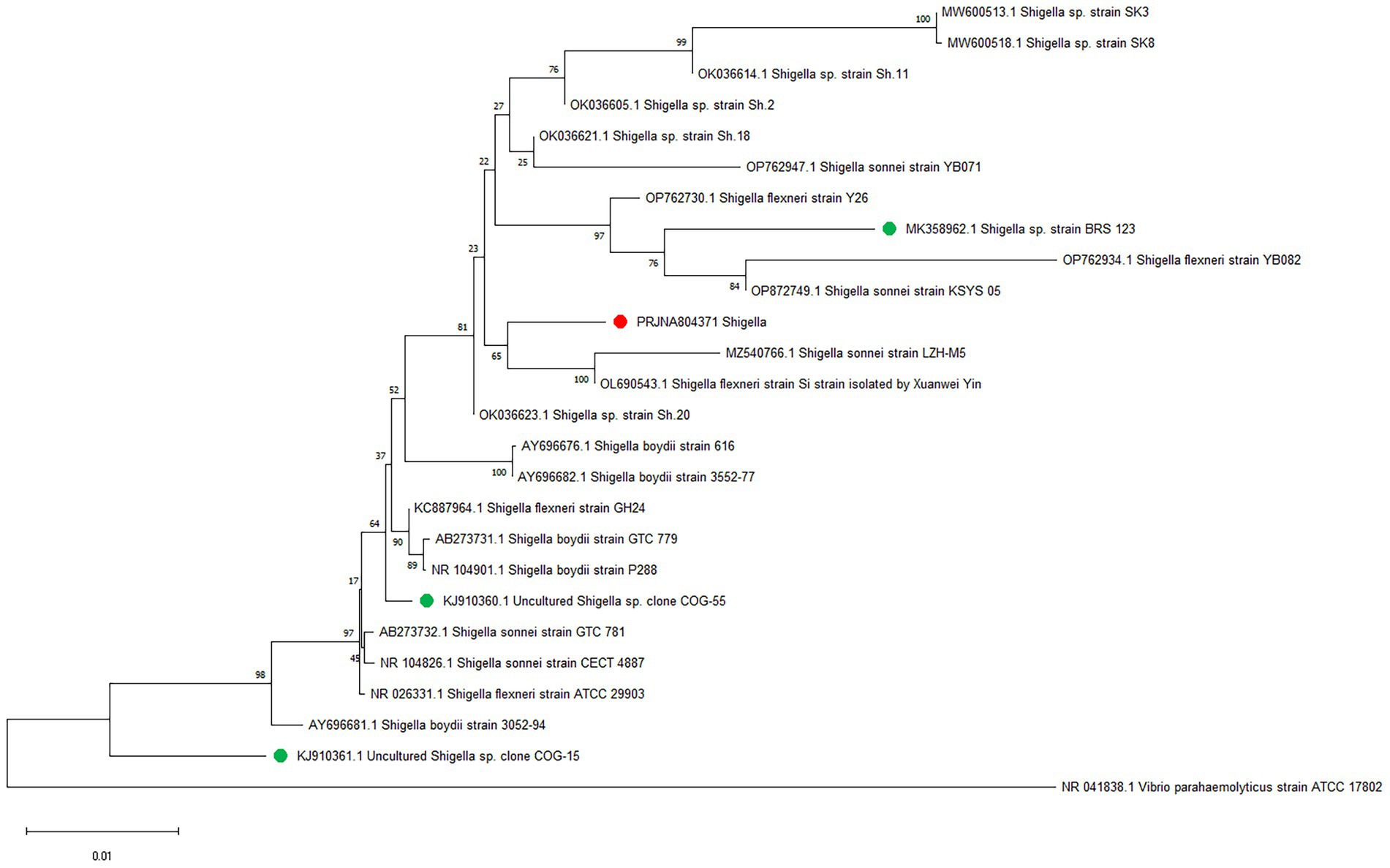
Figure 7. Phylogenetic tree depicting the relationship between JZ001 and 25 reference strains constructed based on the genomic BLAST in NCBI.
Discussion
Shigella spp., are a group of Gram-negative enterobacteria characterized by a lack of flagellar motility and an inability to ferment lactose (2). Shigella has no known animal reservoirs and is therefore considered to be human specific. Previous studies have primarily used mice to model infection human shigellosis infection, using intraperitoneal challenge with Shigella to mimic human bacillary dysentery (4).
Although mice are used to model Shigella infectious diseases, they are not susceptible to Shigella (34, 35). Mouse gut microbiota provides a protective barrier against human Shigella (36). Some Shigella effectors required for colonization specifically target human proteins and are often unable to interact with the mouse analogs (36). Therefore, the isolation of murine Shigella and the use of mouse Shigella to establish an infection model may provide more favorable model tools for subsequent research on the biological properties, pathogenicity, and other aspects of Shigella (4). However, we found no reports on the complete genome and biological characteristics of shigellosis in mice. In this research we isolate a murine Shigella sp. JZ001, for the first time, and sequenced the complete genome of and analysed the biological characteristics of the murine Shigella spp.
Human-derived Shigella strains primarily colonize the colonic mucosa, where glucose serves as the dominant carbon source in their ecological niche (37, 38). These isolates universally lack the ability to metabolize D-maltose and D-trehalose (39), and they typically cannot efficiently metabolize D-fructose or D-galactose. Moreover, most human-derived Shigella strains do not ferment D-mannose (40), with only a few serotypes showing weak metabolic activity toward this substrate.
In contrast, strain JZ001isolated from mice in the present study has retained and optimized multiple carbon metabolic pathways through natural selection. Beyond α-D-glucose, JZ001 can metabolize D-maltose, D-trehalose, D-mannose, D-fructose, D-galactose, and propionic acid. This suggests that JZ001 possesses a more versatile monosaccharide metabolic system and a broader range of metabolic substrates. Notably, it can utilize not only carbohydrates but also short-chain fatty acids (e.g., propionic acid) as alternative carbon and energy sources.
Additional key phenotypes of JZ001 include tolerance to acidic conditions and high osmotic stress, traits that enhance its ability to traverse the gastric acid barrier and increase its survival in feces. Importantly, the JZ001 strain exhibits a distinct antimicrobial resistance profile, characterized by resistance to rifampicin and vancomycin. Early literature reports have demonstrated that Shigella flexneri and Shigella sonnei isolates of human origin were 100% susceptible to rifampicin (41). However, in recent years, rifampicin-resistant strains of human-derived Shigella have also been documented, which are attributed to genetic mutations (42). In contrast, the intrinsic resistance of the JZ001 strain to vancomycin is consistent with the well-established vancomycin resistance phenotype of human-derived Shigella strains reported in previous studies (43).
As core components of the cell membrane, polar lipids and fatty acids can influence critical bacterial characteristics such as virulence, environmental adaptability, and drug susceptibility (37). The major lipidomic difference between strain JZ001 and previously reported human Shigella strains is the absence of phosphatidylserine (PS) in JZ001. This absence represents a distinguishing signature feature of the strain. Future experiments will compare the virulence of JZ001 with that of human Shigella strains using cell invasion assays and animal models, with the aim of elucidating the molecular mechanisms underlying this difference and its biological significance.
The pathogenesis of Shigella spp. is strictly dependent on the virulence plasmid, which encodes several factors that are essential for invasion and subversion of host defenses (44). Shigella genomes naturally harbor hundreds of insertion sequences (IS), and their genes are frequently inactivated either through IS-mediated disruption or IS-mediated genome rearrangement. In addition, the insertion sequences (IS) elements in Shigella have been shown to contribute to the antibiotic resistance and pathogen evolution (45, 46).
Strain JZ001 harbors functional genes within its genomic islands (GIs). As shown in Figure 5, genomic island GI21 contains cidA and tibA. Previous studies have reported that the cidA gene encodes a contact-dependent inhibition (CDI) protein (47), which suppresses the growth of other commensal bacteria in the intestine through intercellular contact, thereby providing Shigella with an ecological advantage in gut microbiota competition. The adhesion encoded by the tibA gene enhances bacterial adherence to intestinal epithelial cells, facilitating infection and colonization (48).
Genomic island GI18 contains three core genes rfbC, gndA, and ugd, that contribute to bacterial metabolic survival and structural resistance. Specifically, rfbC is involved in lipopolysaccharide (LPS) biosynthesis, a key component of the Shigella cell wall that increases resistance to host immune pressures in the intestine (49). gndA encodes glucose-6-phosphate dehydrogenase, which generates NADPH for bacterial survival via the pentose phosphate pathway (PPP) (38). However, the virulence potential of strain JZ001 requires further experimental validation, including comparative studies of virulence-associated genes between murine- and human-derived Shigella strains. Based on the findings of this study, we hypothesize that the core genomic islands of strain JZ001 carry functional genes related to adhesion, barrier penetration, virulence regulation, and metabolic adaptation, which may represent key genetic elements enabling pathogenicity in suckling mouse hosts.
Comparative genomic analysis revealed that JZ001 shares >99% average nucleotide identity (ANI) with Shigella sonnei strain SE6-1 (MZ540766.1), strongly supporting its tentative classification as S. sonnei. ANI is widely used to assess evolutionary relationships and species boundaries, with values >95% indicating that two genomes belong to the same species (50, 51). JZ001 and S. sonnei clearly exceed this threshold, providing robust genomic evidence to classify JZ001 as a member of S. sonnei. A notable limitation, however, is the difference in host origin: JZ001 was isolated from mice, whereas all reference genomes (including SE6-1) are human-derived. Because Shigella is traditionally regarded as a human-specific pathogen, and there are currently no literature reports on naturally occurring Shigella isolates from mice, it remains unclear whether JZ001 represents a mouse-adapted variant of S. sonnei. Future studies should compare the transcriptomic profiles of JZ001 during murine infection with those of human-derived S. sonnei strains to identify host-specific gene expression signatures.
In conclusion, the genome sequence reported here represents the first complete genome sequence of a mouse-derived Shigella strain. It provides a high-quality reference genome that will be extremely valuable for transcriptomics, differential expression analysis, studies of molecular pathogenesis, and investigations into genome evolution. Subsequent studies will establish a suckling mouse infection model for strain JZ001 to enable dynamic monitoring of microbial alterations, host inflammatory responses, and disease outcomes. In addition, gene knockout and transcriptomic analyses will be performed to determine whether JZ001 exhibits pathogenicity in mice.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: https://www.ncbi.nlm.nih.gov/, PRJNA1238207.
Ethics statement
The animal study was approved by Ethics Committee of Jinzhou Medical University (Approval ID: 2019014). The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
XYL: Investigation, Writing – original draft, Data curation, Formal analysis. PY: Data curation, Formal analysis, Investigation, Writing – original draft. YT: Formal analysis, Investigation, Writing – original draft. XFL: Data curation, Methodology, Software, Writing – original draft. XJ: Data curation, Methodology, Formal analysis, Writing – original draft. ML: Formal analysis, Investigation, Writing – original draft. YL: Resources, Supervision, Writing – review & editing. WZ: Resources, Supervision, Funding acquisition, Investigation, Project administration, Writing – original draft.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by Liaoning Province Science and Technology Plan Joint Fund Project, China (No. 2023-MSLH-046) and Open Project of the Key Laboratory of Medical Molecular Virology, Ministry of Education/Health Commission/School of Medicine, Fudan University in 2024 (No. MV-2024002).
Acknowledgments
We thank The Institute of Biological Anthropology at Jinzhou Medical University provides financial support.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The authors declare that no Gen AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fvets.2025.1686554/full#supplementary-material
Footnotes
References
1. Bengtsson, RJ, Simpkin, AJ, Pulford, CV, Low, R, Rasko, DA, Rigden, DJ, et al. Pathogenomic analyses of Shigella isolates inform factors limiting shigellosis prevention and control across LMICs. Nat Microbiol. (2022) 7:251–61. doi: 10.1038/s41564-021-01054-z
2. Baker, S, and The, HC. Recent insights into Shigella. Curr Opin Infect Dis. (2018) 31:449–54. doi: 10.1097/qco.0000000000000475
3. GBD Diarrhoeal Diseases Collaborators. Estimates of global, regional, and National Morbidity, mortality, and aetiologies of diarrhoeal diseases: a systematic analysis for the global burden of disease study 2015. Lancet Infect Dis. (2017) 17:909–48. doi: 10.1016/s1473-3099(17)30276-1
4. Alphonse, N, and Odendall, C. Animal models of shigellosis: a historical overview. Curr Opin Immunol. (2023) 85:102399. doi: 10.1016/j.coi.2023.102399
5. Maurelli, AT, Routh, PR, Dillman, RC, Ficken, MD, Weinstock, DM, Almond, GW, et al. Shigella infection as observed in the experimentally inoculated domestic pig, Sus scrofa domestica. Microb Pathog. (1998) 25:189–96. doi: 10.1006/mpat.1998.0230
6. Onyango, DM, Wandili, S, Kakai, R, and Waindi, EN. Isolation of Salmonella and Shigella from fish harvested from the Winam gulf of Lake Victoria, Kenya. J Infect Dev Ctries. (2009) 3:99–104. doi: 10.3855/jidc.56
7. Shi, R, Yang, X, Chen, L, Chang, HT, Liu, HY, Zhao, J, et al. Pathogenicity of Shigella in chickens. PLoS One. (2014) 9:e100264. doi: 10.1371/journal.pone.0100264
8. Zhu, Z, Shi, Y, Zhou, X, Li, B, and Zhang, J. Molecular characterization of fluoroquinolone and/or cephalosporin resistance in Shigella Sonnei isolates from yaks. BMC Vet Res. (2018) 14:177. doi: 10.1186/s12917-018-1500-6
9. Zhu, Z, Wang, W, Cao, M, Zhu, Q, Ma, T, Zhang, Y, et al. Virulence factors and molecular characteristics of Shigella flexneri isolated from calves with Diarrhea. BMC Microbiol. (2021) 21:214. doi: 10.1186/s12866-021-02277-0
10. Brunner, K, Samassa, F, Sansonetti, PJ, and Phalipon, A. Shigella-mediated immunosuppression in the human gut: subversion extends from innate to adaptive immune responses. Hum Vaccin Immunother. (2019) 15:1317–25. Epub 2019419. doi: 10.1080/21645515.2019.1594132
11. Zhao, W, Yu, ML, Tao, X, Cheng, MH, Liu, CC, Liu, Y, et al. Analysis of the intestinal microbial community altered during rotavirus infection in suckling mice. Virol J. (2021) 18:254. doi: 10.1186/s12985-021-01727-5
12. Wang, W, Cao, J, Li, JR, Yang, F, Li, Z, and Li, LX. Comparative analysis of the gastrointestinal microbial communities of Bar-headed goose (Anser indicus) in different breeding patterns by high-throughput sequencing. Microbiol Res. (2016) 182:59–67. doi: 10.1016/j.micres.2015.10.003
13. Yasuda, K, Oh, K, Ren, B, Tickle, TL, Franzosa, EA, Wachtman, LM, et al. Biogeography of the intestinal mucosal and Lumenal microbiome in the Rhesus macaque. Cell Host Microbe. (2015) 17:385–91. doi: 10.1016/j.chom.2015.01.015
14. Panchalingam, S, Antonio, M, Hossain, A, Mandomando, I, Ochieng, B, Oundo, J, et al. Diagnostic microbiologic methods in the GEMS-1 case/control study. Clin Infect Dis. (2012) 55 Suppl 4:S294–302. doi: 10.1093/cid/cis754
15. Kumar, S, Stecher, G, Li, M, Knyaz, C, and Tamura, K. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol. (2018) 35:1547–9. doi: 10.1093/molbev/msy096
16. Chin, CS, Alexander, DH, Marks, P, Klammer, AA, Drake, J, Heiner, C, et al. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat Methods. (2013) 10:563–9. doi: 10.1038/nmeth.2474
17. Berlin, K, Koren, S, Chin, CS, Drake, JP, Landolin, JM, and Phillippy, AM. Assembling large genomes with single-molecule sequencing and locality-sensitive hashing. Nat Biotechnol. (2015) 33:623–30. doi: 10.1038/nbt.3238
18. Lowe, TM, and Eddy, SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. (1997) 25:955–64. doi: 10.1093/nar/25.5.955
19. Lagesen, K, Hallin, P, Rødland, EA, Staerfeldt, HH, Rognes, T, and Ussery, DW. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. (2007) 35:3100–8. doi: 10.1093/nar/gkm160
20. Petersen, TN, Brunak, S, von Heijne, G, and Nielsen, H. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods. (2011) 8:785–6. doi: 10.1038/nmeth.1701
21. Arndt, D, Marcu, A, Liang, Y, and Wishart, DS. PHAST, PHASTER and PHASTEST: tools for finding prophage in bacterial genomes. Brief Bioinform. (2019) 20:1560–7. doi: 10.1093/bib/bbx121
22. Bertelli, C, and Brinkman, FSL. Improved Genomic Island predictions with Islandpath-Dimob. Bioinformatics. (2018) 34:2161–7. doi: 10.1093/bioinformatics/bty095
23. Ge, R, Mai, G, Wang, P, Zhou, M, Luo, Y, Cai, Y, et al. CRISPRdigger: detecting CRISPRs with better direct repeat annotations. Sci Rep. (2016) 6:32942. doi: 10.1038/srep32942
24. Almagro Armenteros, JJ, Tsirigos, KD, Sønderby, CK, Petersen, TN, Winther, O, Brunak, S, et al. SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat Biotechnol. (2019) 37:420–3. doi: 10.1038/s41587-019-0036-z
25. Juncker, AS, Willenbrock, H, Von Heijne, G, Brunak, S, Nielsen, H, and Krogh, A. Prediction of lipoprotein signal peptides in gram-negative Bacteria. Protein Sci. (2003) 12:1652–62. doi: 10.1110/ps.0303703
26. Krogh, A, Larsson, B, von Heijne, G, and Sonnhammer, EL. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol. (2001) 305:567–80. doi: 10.1006/jmbi.2000.4315
27. Yu, NY, Wagner, JR, Laird, MR, Melli, G, Rey, S, Lo, R, et al. PSORTb 3.0: improved protein subcellular localization prediction with refined localization subcategories and predictive capabilities for all prokaryotes. Bioinformatics. (2010) 26:1608–15. doi: 10.1093/bioinformatics/btq249
28. Urban, M, Cuzick, A, Seager, J, Wood, V, Rutherford, K, Venkatesh, SY, et al. PHI-base: the pathogen-host interactions database. Nucleic Acids Res. (2020) 48:D613–20. doi: 10.1093/nar/gkz904
29. Chen, L, Yang, J, Yu, J, Yao, Z, Sun, L, Shen, Y, et al. VFDB: a reference database for bacterial virulence factors. Nucleic Acids Res. (2005) 33:D325–8. doi: 10.1093/nar/gki008
30. Krzywinski, M, Schein, J, Birol, I, Connors, J, Gascoyne, R, Horsman, D, et al. Circos: an information aesthetic for comparative genomics. Genome Res. (2009) 19:1639–45. doi: 10.1101/gr.092759.109
31. Lee, I, Ouk Kim, Y, Park, SC, and Chun, J. OrthoANI: an improved algorithm and software for calculating average nucleotide identity. Int J Syst Evol Microbiol. (2016) 66:1100–3. doi: 10.1099/ijsem.0.000760
32. Marçais, G, Delcher, AL, Phillippy, AM, Coston, R, Salzberg, SL, and Zimin, A. MUMmer4: a fast and versatile genome alignment system. PLoS Comput Biol. (2018) 14:e1005944. doi: 10.1371/journal.pcbi.1005944
33. Emms, DM, and Kelly, S. OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. (2019) 20:238. doi: 10.1186/s13059-019-1832-y
34. Freter, R. Experimental enteric Shigella and Vibrio infections in mice and guinea pigs. J Exp Med. (1956) 104:411–8. doi: 10.1084/jem.104.3.411
35. McGuire, CD, and Floyd, TM. Studies on experimental shigellosis. I. Shigella infections of normal mice. J Exp Med. (1958) 108:269–76. doi: 10.1084/jem.108.2.269
36. Mitchell, PS, Roncaioli, JL, Turcotte, EA, Goers, L, Chavez, RA, Lee, AY, et al. NAIP-NLRC4-deficient mice are susceptible to shigellosis. eLife. (2020) 9:9. doi: 10.7554/eLife.59022
37. Tinevez, JY, Arena, ET, Anderson, M, Nigro, G, Injarabian, L, André, A, et al. Shigella-mediated oxygen depletion is essential for intestinal mucosa colonization. Nat Microbiol. (2019) 4:2001–9. doi: 10.1038/s41564-019-0525-3
38. Waligora, EA, Fisher, CR, Hanovice, NJ, Rodou, A, Wyckoff, EE, and Payne, SM. Role of intracellular carbon metabolism pathways in Shigella flexneri virulence. Infect Immun. (2014) 82:2746–55. doi: 10.1128/iai.01575-13
39. Ito, H, Kido, N, Arakawa, Y, Ohta, M, Sugiyama, T, and Kato, N. Possible mechanisms underlying the slow lactose fermentation phenotype in Shigella Spp. Appl Environ Microbiol. (1991) 57:2912–7. doi: 10.1128/aem.57.10.2912-2917.1991
40. Gross, RJ, Thomas, LV, Cheasty, T, Rowe, B, and Lindberg, AA. Four new provisional serovars of Shigella. J Clin Microbiol. (1989) 27:829–31. doi: 10.1128/jcm.27.5.829-831.1989
41. Galushko, NA, D'Iachenko, AG, Chemich, ND, and D'Iachenko, PA. Antibiotic resistance of Shigellae and rationale for Etiotropic therapy of Shigella infections. Zh Mikrobiol Epidemiol Immunobiol. (2005) 2:71–5.
42. Ye, C, Lan, R, Xia, S, Zhang, J, Sun, Q, Zhang, S, et al. Emergence of a new multidrug-resistant serotype X variant in an epidemic clone of Shigella flexneri. J Clin Microbiol. (2010) 48:419–26. doi: 10.1128/JCM.00614-09
43. Fernandes, MM, Ivanova, K, Hoyo, J, Pérez-Rafael, S, Francesko, A, and Tzanov, T. Nanotransformation of vancomycin overcomes the intrinsic resistance of gram-negative Bacteria. ACS Appl Mater Interfaces. (2017) 9:15022–30. doi: 10.1021/acsami.7b00217
44. Buchrieser, C, Glaser, P, Rusniok, C, Nedjari, H, D'Hauteville, H, Kunst, F, et al. The virulence plasmid Pwr100 and the repertoire of proteins secreted by the type iii secretion apparatus of Shigella flexneri. Mol Microbiol. (2000) 38:760–71. doi: 10.1046/j.1365-2958.2000.02179.x
45. Wei, J, Goldberg, MB, Burland, V, Venkatesan, MM, Deng, W, Fournier, G, et al. Complete genome sequence and comparative genomics of Shigella flexneri serotype 2a strain 2457T. Infect Immun. (2003) 71:2775–86. doi: 10.1128/iai.71.5.2775-2786.2003
46. Prosseda, G, Di Martino, ML, Campilongo, R, Fioravanti, R, Micheli, G, Casalino, M, et al. Shedding of genes that interfere with the pathogenic lifestyle: the Shigella model. Res Microbiol. (2012) 163:399–406. doi: 10.1016/j.resmic.2012.07.004
47. Halvorsen, TM, Schroeder, KA, Jones, AM, Hammarlöf, D, Low, DA, Koskiniemi, S, et al. Contact-dependent growth inhibition (CDI) systems deploy a large family of polymorphic Ionophoric toxins for inter-bacterial competition. PLoS Genet. (2024) 20:e1011494. doi: 10.1371/journal.pgen.1011494
48. Mitra, S, Saha, DR, Pal, A, Niyogi, SK, Mitra, U, and Koley, H. Hemagglutinating activity is directly correlated with colonization ability of Shigellae in suckling mouse model. Can J Microbiol. (2012) 58:1159–66. doi: 10.1139/w2012-095
49. Rajakumar, K, Jost, BH, Sasakawa, C, Okada, N, Yoshikawa, M, and Adler, B. Nucleotide sequence of the rhamnose biosynthetic operon of Shigella Flexneri 2a and role of lipopolysaccharide in virulence. J Bacteriol. (1994) 176:2362–73. doi: 10.1128/jb.176.8.2362-2373.1994
50. Hernández-Salmerón, JE, and Moreno-Hagelsieb, G. Fastani, mash and dashing equally differentiate between Klebsiella species. PeerJ. (2022) 10:e13784. doi: 10.7717/peerj.13784
Keywords: Shigella spp., diarrheic suckling mice, virulence, whole genome sequencing, comparative genomic analysis
Citation: Li Xy, Yang PP, Tang Y, Li XF, Jia X, Liu M, Li Y and Zhao W (2025) Whole genome analysis of Shigella sp. JZ001: a novel strain isolated from diarrheic suckling mice. Front. Vet. Sci. 12:1686554. doi: 10.3389/fvets.2025.1686554
Edited by:
Faham Khamesipour, Ministry of Health and Medical Education, IranReviewed by:
Domonkos Sváb, HUN-REN Veterinary Medical Research Institute, HungaryRimi Chowdhury, Miami University, United States
Copyright © 2025 Li, Yang, Tang, Li, Jia, Liu, Li and Zhao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yonggang Li, bHlnam9AaG90bWFpbC5jb20=; Wei Zhao, emhhb3dlaS12QGp6bXUuZWR1LmNu
†These authors share first authorship