 Xiaolin Lan1Qipeng Zhang1Ruqing Zeng1Fang Liang1Jiaman Li1
Xiaolin Lan1Qipeng Zhang1Ruqing Zeng1Fang Liang1Jiaman Li1 Gan Li1Feng Li2
Gan Li1Feng Li2 Yaqiong Ye1*
Yaqiong Ye1* Mengmeng Zhao1*
Mengmeng Zhao1*- 1Guangdong Provincial Key Laboratory of Animal Molecular Design and Precise Breeding, School of Animal Science and Technology, Foshan University, Foshan, Guangdong, China
- 2Center of Animal Epidemic Disease Prevention Control, Chongzuo, Guangxi, China
1 Introduction
Akabane disease (AKAD), also known as Akabane encephalomyelitis, is an arthropod-borne infectious disease caused primarily by Akabane virus (AKAV), characterized by abortion, premature birth, stillbirth, congenital joint contractures, and arthrogryposis-hydranencephaly syndrome (AH) in cattle and sheep (1, 2). Additionally, AKAV can infect avian embryos, mice, and hamsters, leading to death. AKAV is widespread in Africa, the Middle East, East Asia, Southeast Asia, and Australia (3–5). Between 1972 and 1975, a serious outbreak of Akabane disease broke out in Japan, with the original strain JaGAr39 isolated from mosquitoes in Japan in 1959, resulting in over 31,000 cases of abortion, stillbirth, congenital joint diseases, and AH syndrome cases (6). In 1974, AKAV-induced AH syndrome was prevalent in Australia, when more than 8,000 confirmed cases were reported (7). In 1998, AKAV was first isolated from mosquitoes in Shanghai, China, and named the SH-1 strain (8). The global prevalence of AKAD exhibits distinct regional, periodic, and seasonal patterns (9) and can spread across species, causing significant economic losses to livestock industries in endemic regions and posing a long-term threat to animal health and sustainable farming practices. As the envelope glycoprotein of AKAV, Gc not only serves as a crucial target for the immune system of vertebrate hosts, eliciting strong specific immune responses, but also plays a decisive role in various important biological properties of AKAV, such as pathogenicity, neutralizing activity, hemagglutination ability (10–12). It is crucial to study the key aspects of the AKAV Gc gene to improve the economic viability of livestock farming and ensure its stable development.
2 Materials and methods
2.1 The dataset
From the GenBank database on the NCBI website, a total of 147 AKAV Gc strains sequences were selected, comprising 61 strains from Japan, 39 from the United States, 35 from China, 2 from Israel, 9 from South Korea, and 1 from Turkey, spanning lineages I-V (see Supplementary Table 1). These strains encompass different years from 1959 to 2023, including the majority of AKAV strains from China. The genetic variation of the AKAV Gc gene sequence over the past 64 years has helped to analyze the evolution of AKAV Gc and provide a theoretical basis for AKAV prevention and control. Out of the 147 AKAV strains, 45 strains were carefully selected for further Gc sequence analysis, representing strains from lineages I-V, including reference strains, vaccine strains, and epidemic strains, to ensure a comprehensive analysis of genetic variations in the AKAV Gc gene (Table 1).
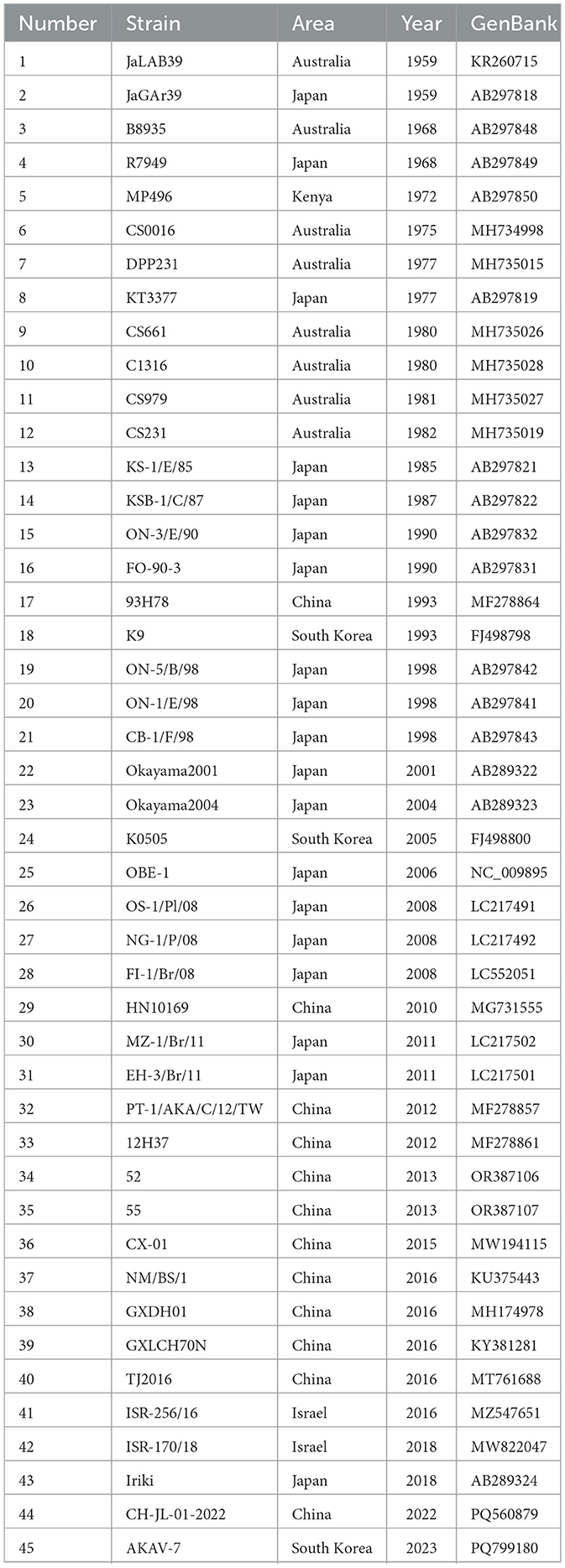
Table 1. Information of 45 selected AKAV strains.
2.2 Analysis of Akabane virus gene sequences
The nucleotide similarity of 45 strains selected from 147 strains of AKAV Gc was analyzed. These 45 AKAV strains cover classical strains and vaccine strains in different countries and different periods. The nucleotide homology of Gc gene was analyzed from NCBI website (https://www.ncbi.nlm.nih.gov/) by Clustal W method in MegAlign function of DNAStar software (version 7.0, Madison, WI). The phylogenetic analysis of 147 AKAV Gc sequences (Supplementary Table 1) was carried out by using the maximum likelihood method (ML) with 1,000 Bootstrap repeated sampling of MEGA software (7th edition, Mega Limited, Auckland, New Zealand). Subsequently, these sequences were annotated with a network tool called interactive tree of life.
3 Descriptive results
In this study, 45 representative isolates were systematically selected from the initial 147 strains. These isolates covered a wide range of time (1959–2023) and geography, including Japan, China, South Korea, Australia, and Israel. They also represented five known genetic families (I to V). Key reference strains like the prototype strain JaGAr39, vaccine strain OBE-1, and genetically distinct isolates such as MP496 were included for a comprehensive analysis. The phylogenetic tree, based on Gc protein sequences, displayed significant genetic diversity. Amino acid homology similarity ranged from 69.2% to 100% (refer to Figure 1). The maximum likelihood phylogenetic tree was utilized to analyze the relationship between these strains further, highlighting conservation and variation (refer to Figure 2).

Figure 1. Forty-five representative AKAV strains (Table 1) were selected to obtain Gc individual sequences for nucleotide homology analysis. The nucleotide homology of Gc sequences was analyzed using the Clustal W algorithm in DNAStar software (version 7.0, Madison, WI).
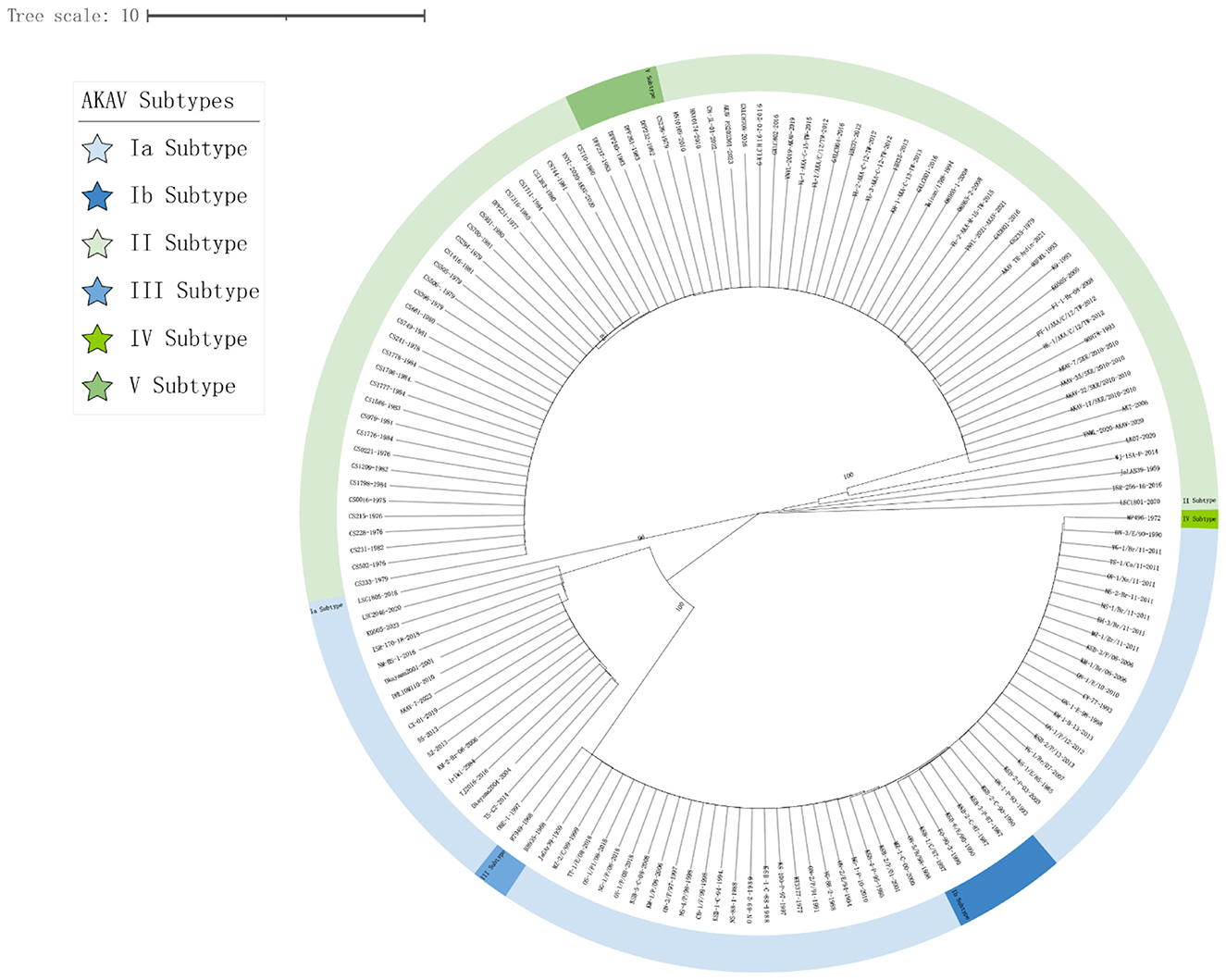
Figure 2. Phylogenetic analysis of AKAV Gc genes (Supplementary Table 1). Clustal W alignment was conducted using the MegAlign feature in DNAStar software (version 7.0). Subsequently, the maximum likelihood method with 1,000 bootstrap replicates was performed using MEGA software (version 11.0). The generated trees were visualized and annotated using the online “The Interactive Tree of Life” (https://itol.embl.de, accessed on 20 May 2025) software. The five subtypes (I–V) of AKAV, Ia, Ib, II, III, IV, and V, are labeled.
Notably, the Kenyan strain MP496 formed a unique and deeply branched pedigree (pedigree IV), showing stark differences from all isolates in Southeast Asia and Australia. Its nucleotide homology with other AKAV isolates ranged from 69.2% to 69.8%, with the highest similarity to R7949 at 69.8% and the lowest with OBE-1 and TJ2016 at 69.2%.
In contrast, pedigree I included strains like CY-77 from Taiwan Province, Iriki, and ON-1/E/98 from Japan, while pedigree II encompassed vaccine strains OBE-1, JaGAr39, and KT3377, forming a major Asian branch. Australian isolates B8935 and R7949 clustered together into lineage III, underlining the impact of geographical isolation on AKAV evolution. Isolates from the same country and year displayed high nucleotide homology (>99%). For instance, Chinese isolates 52 and 55 exhibited a 99.8% homology, while Japanese isolates JaGAr39 and JaLAB39 showed 100% similarity, suggesting a shared ancestral origin. Australian isolates B8935, CS0016, and CS231, collected from 1968 to 1982, formed a closely related phylogenetic group with 97.1%-99.5% nucleotide homology. The Korean strain AKAV-7 shared 83.1%-85.7% nucleotide homology with the Chinese strain PT-1-AKA-C-12-TW.
Between 2010 and 2023, East Asian isolates from China, South Korea, and Japan formed a highly homologous group, with nucleotide homology exceeding 97%. For example, the Korean strain AKAV-7 had a 99.5% homology with Japanese strains EH-3-Br-11 and MZ-1-Br-11, indicating recent cross-border transmission of closely related virus populations. These results underscore the combined impacts of geographical isolation and time evolution on AKAV genetic diversity.
The comprehensive analysis of similarity and diversity among these AKAV strains has unveiled the genetic evolution of the AKAV Gc gene across different regions and time points. By comparing the amino acid sequences of these strains, a better understanding of the population structure and transmission dynamics of AKAV is gained. High similarity between specific strains may suggest shared transmission pathways within the same region or time period, while lower similarity could indicate genetic differentiation and evolution across different regions or time points. Furthermore, through phylogenetic analysis, the phylogenetic relationships and classification among different subtypes of AKAV can be further elucidated. This aids in comprehending the geographical distribution and pathogenic differences among various subtypes, providing crucial insights for tailored disease control strategies. The associations between different subtypes or strains may be linked to various factors such as host population structure, environmental influences, and viral transmission pathways. Combining the results of amino acid similarity and phylogenetic analysis allows for a more comprehensive understanding of the genetic diversity and transmission pathways of AKAV. This helps predict the transmission potential of different subtypes or strains, assess the risk of virus epidemics, and provide guidance for future disease surveillance and control measures. Through in-depth exploration of genetic variations and evolutionary mechanisms of the AKAV Gc gene, better preparation can be made for potential epidemic threats, safeguarding the health and sustainable development of the livestock industry. These research findings provide crucial scientific foundations for understanding the transmission, pathogenesis, and epidemiology of AKAV, offering robust support for future disease prevention and control efforts.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
Author contributions
XL: Writing – original draft. QZ: Writing – original draft. RZ: Writing – original draft. FaL: Writing – original draft. JL: Writing – original draft. GL: Writing – original draft. FeL: Writing – review & editing. YY: Writing – review & editing. MZ: Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This research was supported by Key Research and Development Special Project of Henan Province (251111113600), Key Scientific and Technological Grants in Henan Province (24210211031), Guangdong Provincial Department of Education's distinctive innovation initiative (2023KTSCX128), and the National Natural Science Foundation of China (31902279).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Gen AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fvets.2025.1715626/full#supplementary-material
References
1. De Regge N. Akabane, Aino and Schmallenberg virus-where do we stand and what do we know about the role of domestic ruminant hosts and Culicoides vectors in virus transmission and overwintering? Curr Opin Virol. (2017) 27:15–30. doi: 10.1016/j.coviro.2017.10.004
2. Qiao J, Chen C, Ren Y. Research progress on Akabane disease. J Tarim Univ. (2008) 103–6. doi: 10.1063/1.2837055
3. Taylor WP, Mellor PS. The distribution of Akabane virus in the Middle East. Epidemiol Infect. (1994) 113:175–85. doi: 10.1017/S0950268800051591
4. Jun Q, Qingling M, Zaichao Z, Kuojun C, Jingsheng Z, Minxing M, et al. serological survey of akabane virus infection in cattle and sheep in northwest China. Trop Anim Health Prod. (2012) 44:1817–20. doi: 10.1007/s11250-012-0168-3
5. Kono R, Hirata M, Kaji M, Goto Y, Ikeda S, Yanase T, et al. Bovine epizootic encephalomyelitis caused by Akabane virus in southern Japan. BMC Vet Res. (2008) 4:20. doi: 10.1186/1746-6148-4-20
6. Brenner J, Yanase T, Kato T, Yaakobi S, Khinich E, Paz R, et al. Serological evidence suggests that several Simbu serogroup viruses circulated in Israel. Vet Ital. (2019) 55:81–9. doi: 10.12834/VetIt.1397.7622.2
7. Coverdale OR, Cybinski DH, St George TD. Congenital abnormalities in calves associated with Akabane virus and Aino virus. Aust Vet J. (1978) 54:151–2. doi: 10.1111/j.1751-0813.1978.tb05538.x
8. Yadav P, Shete A, Bondre V, Patil D, Kokate P, Chaudhari S, et al. Isolation and characterization of Oya virus a member of Simbu serogroup, family Bunyaviridae, isolated from Karnataka, India. Infect Genet Evol: J Mol Epidemiol Evol Genet Infect Dis. (2016) 44:122–6. doi: 10.1016/j.meegid.2016.06.049
9. Li C, Xue F, Wang K, Wang Y, Ma X. Research progress on the epidemiology and prevention of Akabane disease. Adv Vet Med. (2023) 44:96–100. doi: 10.16437/j.cnki.1007-5038.2023.06.022
10. Yanase T, Yoshida K, Ohashi S, Kato T, Tsuda T. Sequence analysis of the medium RNA segment of three Simbu serogroup viruses, Akabane, Aino, and Peaton viruses. Virus Res. (2003) 93:63–9. doi: 10.1016/S0168-1702(03)00066-2
11. Xu S, Tong T, Bai Y, Wang Q, Zhang W, Sun Q, et al. Eukaryotic expression and antigenicity detection of Akabane virus G1 gene. Chin J Prev Vet Med. (2008) 440–4.
Keywords: Akabane disease, Akabane virus, Gc gene, Gc gene genetic variation, phylogeny
Citation: Lan X, Zhang Q, Zeng R, Liang F, Li J, Li G, Li F, Ye Y and Zhao M (2025) Genetic evolution analysis of AKAV Gc gene. Front. Vet. Sci. 12:1715626. doi: 10.3389/fvets.2025.1715626
Received: 29 September 2025; Accepted: 29 October 2025;
Published: 13 November 2025.
Edited by:
Jun Ji, Nanyang Normal University, ChinaReviewed by:
Guanmin Zheng, Henan Agricultural University, ChinaXinliang Fu, Zhongkai University of Agriculture and Engineering, China
Copyright © 2025 Lan, Zhang, Zeng, Liang, Li, Li, Li, Ye and Zhao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mengmeng Zhao, bWVuZ21lbmd6aGFvMjAyMUBmb3N1LmVkdS5jbg==; Yaqiong Ye, Y244NzQ0NjJAMTYzLmNvbQ==