 Lingyu He
Lingyu He Nwai Oo Khine
Nwai Oo Khine Jeongmin Song
Jeongmin Song Celine Loubière
Celine Loubière Patrick Butaye
Patrick Butaye- 1Jockey Club College of Veterinary Medicine and Life Sciences, City University of Hong Kong, Kowloon, Hong Kong SAR, China
- 2Department of Microbiology and Immunology, Cornell University College of Veterinary Medicine, Cornell University, Ithaca, NY, United States
- 3Faculty of Veterinary Medicine, Department of Pathobiology, Pharmacology and Zoological Medicine, Ghent University, Merelbeke, Belgium
Strangles, caused by the host-adapted Streptococcus equi subsp. equi (S. equi), imposes significant welfare and economic losses on the equine industry worldwide. Understanding its genomic features, virulence-associated genes (VAGs), antimicrobial resistance (AMR) and mobile genetic elements (MGEs) is essential for disease control and vaccine development. This study aimed to characterize the accessory genome composition, geographic distribution of VAGs and MGEs, and AMR profiles of S. equi by a large-scale genomic analysis of global publicly available S. equi sequences. All publicly available S. equi sequences in the Sequence Read Archive (SRA) database were retrieved and assembled. A total of 552 high-quality assemblies were obtained for further analysis. The strains originated from five continents (North/South America, Europe, Asia and Oceania). The geographical distribution of VAGs (analyzed using an in-house Streptococcus equi virulence factor database), antibiotic resistance gene (ARG) profiles, and the contribution of MGEs to S. equi VAGs were analyzed in this study. The results revealed that S. equi exhibited a closed pangenome with 1,661 core and 982 accessory genes. Among 71 identified VAGs, 40 were core VAGs, while accessory VAGs showed significant geographic variations, especially in nutritional/metabolic factor genes and exotoxin genes. No acquired ARGs were detected except a single qacG gene encoding resistance to quaternary ammonium compounds. This study revealed a functional specialization of MGEs, where prophages carry superantigen genes (speH, speI) and the hyaluronidase gene hylP; genomic islands (GIs) harbor iron acquisition genes (eqb cluster) and the virD4 gene encoding the T4SS coupling protein; and integrative conjugative elements (ICEs) carry the heme metabolism cluster (htsA, shp) and streptolysin S-associated genes (sagA, sagD). The geographic variation of VAGs suggests regional adaptive pressures and supports genome streamlining in S. equi. In conclusion, S. equi exhibits a closed and streamlined genome, characteristic of host-adapted bacteria. There is a minimal acquisition of ARGs while key VAGs are retained. Prophages, GIs, and ICEs play specialized roles in VAG distribution. These findings provide insights into prioritizing VAGs for strangles vaccine development and surveillance of antigenic variation to mitigate vaccine escape.
1 Introduction
Strangles, caused by Streptococcus equi subspecies equi (S. equi), is a widespread infectious disease of horses that poses significant welfare and economic costs worldwide (1). The disease is characterized by pyrexia, acute swelling and subsequent abscess formation of the submandibular and retropharyngeal lymph nodes (2). Approximately 10% of recovered horses become persistent carriers harboring S. equi in the guttural pouches without clinical signs of disease. These are so-called “silent carriers” (3). S. equi is a host-adapted β-hemolytic bacterium of the Lancefield group C streptococcal that evolved from the broader-host-range subspecies Streptococcus equi subspecies zooepidemicus (S. zooepidemicus) (4). Streptococcus equi subspecies share >80% DNA sequence homology with Lancefield group A Streptococcus pyogenes (S. pyogenes) (5), a major human pathogen. Genomic studies have revealed that S. equi has undergone significant genome specialization and decay, driven by its persistent infection in horses (6). This evolution toward host restriction was characterized by the loss and acquisition of various genetic elements, especially virulence-associated genes (VAGs) and mobile genetic elements (MGEs) (6).
The multicomponent subunit vaccine against strangles, Strangvac, is composed of eight antigens (7). These components are CNE, a collagen-binding protein that mediates adhesion to the host extracellular matrix (8); EAG, which binds host proteins like IgG and albumin for immune evasion (9); three collagen-like proteins (SclC, SclF, SclI) that are immunogenic during infection (10); two surface proteins, SEQ_0402 and SEQ_0256, with SEQ_0402 (Eq8) being implicated in virulence (11); and IdeE, a secreted IgG-degrading enzyme that disrupts antibody-mediated immunity (12). In addition to these, other VAGs are critical to S. equi pathogenicity. One of the key virulence factors of S. equi is the M-like protein (SeM) encoded by fbp, a primary cell wall-associated protein and a protective antigen (13). The fbp gene has also been used as a target for the molecular detection and differentiation of S. equi from its closely related subspecies S. zooepidemicus (14). Furthermore, other VAGs in the accessory genome of S. equi, such as the superantigen-encoding genes speH (seeH), speI (seeI), speK (seeL), and speL (seeM), are important to the ability of this host-restricted pathogen to cause lymph node abscess and strangles (5, 15). A deeper understanding of the distribution and function of these diverse virulence factors is therefore essential for advancing vaccine development and disease surveillance.
Although previous studies have characterized the core genome of S. equi strains from 18 countries (16), there is no complete database of all VAGs of S. equi, leaving critical gaps in the geographic variations of its accessory genome. In addition, the evolutionary pressure causing accessory genome adaptation and the geographical variations have not been systematically investigated. Therefore, a comprehensive analysis of 552 high-quality S. equi genomes spanning five continents (1965–2023) was conducted. Our study reveals the accessory genome structure of S. equi and provides valuable information for vaccine design strategies and acquired antimicrobial resistance (AMR) surveillance. The analysis of the accessory genome of S. equi provides additional insights into the genomic decay and specialization model in this host-adapted pathogen (6).
2 Materials and methods
2.1 Strain collection, genome assembly and pan-genome analysis
Publicly available S. equi whole genome sequencing raw read samples (n = 985) were downloaded from the Sequence Read Archive (SRA) database of National Center for Biotechnology Information (NCBI) in July 2024 with keywords “Streptococcus equi subsp. equi.” The complete genome of Se4047, isolated from a case of strangles in the UK in 1990, was used as the reference (5). De novo assembly was conducted using the normal mode of Unicycler (v0.5.0) with default parameters (17), quality was assessed with QUAST (v5.2.0) (18). Genome assembly metrics derived from QUAST were obtained for all strains to validate genome quality. To ensure high-quality assemblies for downstream analysis, stringent quality control criteria were applied. Assemblies were excluded if they met any of the following criteria: (i) N50 < 30,000 bp, (ii) NG50 < 30,000 bp, (iii) number of contigs > 200, (iv) total assembly length < 2,000,000 bp, (v) longest contig length < 80,000 bp, or (vi) genome fraction < 90% (19). These thresholds were established to remove assemblies with poor contiguity, excessive fragmentation, or incomplete genome coverage. The genome sequences were annotated using Prokka (v1.14.6) (20). The analysis was run specifying the genus as Streptococcus, enabling the use of genus-specific database, and providing the custom in-house Streptococcus_VFDB as an external protein source. The “metagenome” mode was also used to account for potential gene fragments at contig ends. The resulting GFF files were used for pan-genome analysis with Roary (v3.12.0) to determine the core and accessory genome components (21). Roary was executed with MAFFT for core gene alignment, disabling paralog splitting, and using a default BLASTp identity threshold. Core genes were defined as genes present in ≥99% of genomes, soft core genes present in 95–99% of genomes, shell genes present in 15–95% of genomes, and cloud genes present in <15% of genomes. Pan-genome accumulation curves for 552 S. equi genomes were generated from 100 random permutations. To quantitatively assess its nature, the mean pan-genome curve was fitted to a power-law function (P(n) = c * n^γ, P(n) is the pan-genome size for n genomes.) using the non-linear least squares (nls) function in R. The pan-genome was considered closed if the growth exponent γ was close to zero.
2.2 Streptococcus_VFDB database construction and VAGs detection
Putative VAGs in Streptococcus equi subspecies were systematically identified through a dual approach. First, a comprehensive literature review was conducted to curate a list of potential VAGs reported in S. equi and its closely related pathogen S. zooepidemicus. The literature search was performed using PubMed and Google Scholar databases up to November 2024. Search term was “Streptococcus equi.” All original research articles, reviews, and relevant genomic studies reporting genes implicated in pathogenicity were considered. Second, the Virulence Factor Database entries (VFDB)1 for the genus Streptococcus were included. The combined list was formatted into a custom BLAST database (hereafter referred to as the in-house Streptococcus_VFDB) as a search-ready BLAST database for ABRicate (v1.0.1; see Supplementary Table S1 for the complete list of curated VAGs in the in-house Streptococcus_VFDB database).2 VAG presence was assessed using ABRicate with default alignment criteria (with ≥80% identity and ≥80% coverage).
2.3 Identification of ARGs
Acquired ARGs and chromosomal mutations known to confer resistance were identified using ABRicate (v1.0.1) with the ResFinder database (March 2025) with default parameters and the Comprehensive Antibiotic Resistance Database—Resistance Gene Identifier (CARD-RGI) (v6.0.3) tool (minimum identity ≥95%) (22).
2.4 Identification of MGEs
Prophage regions were detected using PHASTEST (v3.0) (23). Integrative conjugative elements (ICEs) and integrative mobilizable elements (IMEs) were identified using ICEscreen (v1.3.2) and ICEfinder2 (v2.0) (24, 25). BLAST comparisons were performed against all known ICE sequences from the ICEberg database (v2.0) to identify and classify the detected ICE elements (26). Genomic islands (GIs) were identified using IslandViewer4 (27), which incorporates IslandPath-DIMOB (28), IslandPick (27) and SIGI-HMM (29). Regions identified by at least two algorithms were recognized as a GI. All the bioinformatics tools were used with default parameters.
2.5 Prophage sequence similarity analysis
To refine the characterization of prophages detected in the assembled S. equi draft genomes, the nucleotide sequences identified by PHASTEST (v3.0) were aligned against reference sequences of known prophages ΦSeq1, ΦSeq2, ΦSeq3, and ΦSeq4 from the S. equi Se4047 genome (NCBI Reference Sequence: NC_012471.1). Pairwise global alignments were performed using the Needleman-Wunsch algorithm as implemented in the EMBOSS needle tool (v6.6.0.0) with default parameters (30). Nucleotide identity percentages were calculated to assess sequence similarity and aid in classifying prophages within the S. equi collection in the present study.
2.6 Identification of MGE-associated ARGs and VAGs
The sequences corresponding to identified prophages and ICEs were extracted from the assembled genomes using custom Python scripts. Comparative analysis to identify MGEs carrying ARGs and VAGs was performed by BLAST searching MGE draft sequences against the CARD database in CARD-RGI tool (v6.0.3; ≥ 80% identity) and in-house Streptococcus_VFDB databases [BLASTn (dc-megablast), ≥ 80% identity and ≥ 80% coverage], respectively.
2.7 Statistical analysis
Statistical analysis and visualization were performed using R (version 4.4.2) with the FSA (v0.9.6), car (v3.1.3), dunn.test (v1.3.6), multcompView packages (v0.1.10), ggplot2 (v3.5.1) and ComplexHeatmap (v2.22.0). The heat maps were drawn using ChiPlot.3 For comparisons among multiple groups, data were first assessed for normality using the Shapiro-Wilkinson test and for homogeneity of variances using Levene’s test. The Kruskal-Wallis rank sum test was used to evaluate differences across groups that had a non-normal distribution. When the Kruskal-Wallis test indicated statistically significant differences (p < 0.05), post-hoc pairwise comparisons were conducted using Dunn’s test with Bonferroni correction to adjust for multiple testing. Adjusted p-values were used to determine statistical significance, with thresholds defined as follows: p < 0.05 (*), p < 0.01 (**), and p < 0.001 (***). Bar charts display means with error bars representing 95% confidence intervals (CIs).
3 Results
3.1 Sequences included and geographic distribution of S. equi
From the 985 S. equi genome sequences in the SRA database of NCBI (collected from 1965 to 2023), 552 high-quality S. equi assemblies could finally be included in the study. Detailed metadata for these 552 strains are present in Supplementary Table S2. The strains originated from 18 countries across five continents: North America (n = 261), Europe (n = 149), Asia (n = 109), Oceania (n = 19), and South America (n = 14; Figure 1).
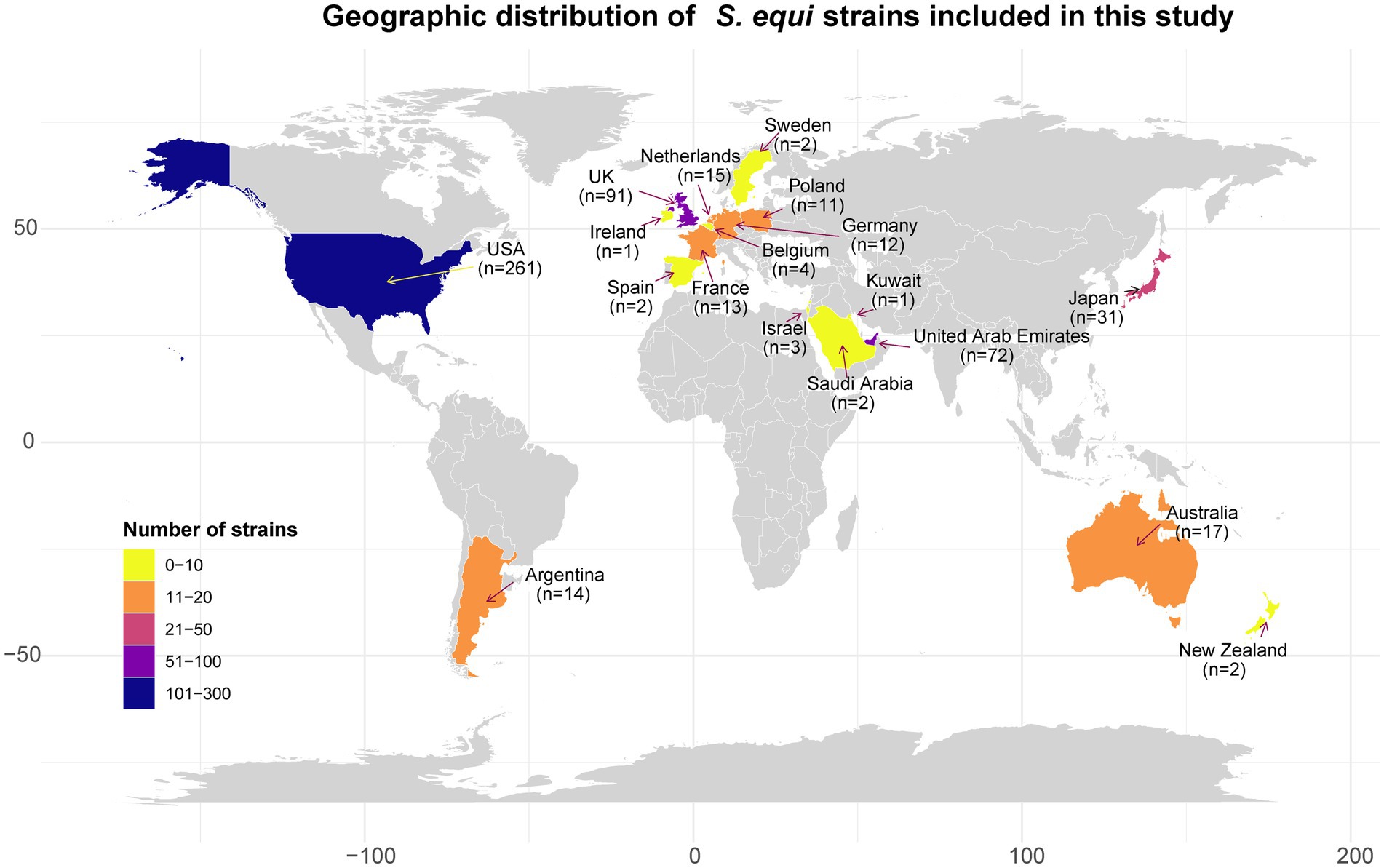
Figure 1. The geographical distribution of 552 S. equi strains examined in this study. Countries are colored according to the number of strains. The x-axis and y-axis represent longitude and latitude, respectively.
3.2 Genomic features of S. equi and pangenome analysis
The average size and GC content were 2,088,082 bp and 41.3%, respectively (Supplementary Table S3). Pan-genome analysis revealed 1,661 core genes and 982 accessory genes (Figure 2A). The pan-genome accumulation curve rapidly reached a plateau, and yielded a growth exponent (γ) of 0.047 (Figure 2B), indicating S. equi possesses a closed pan-genome.
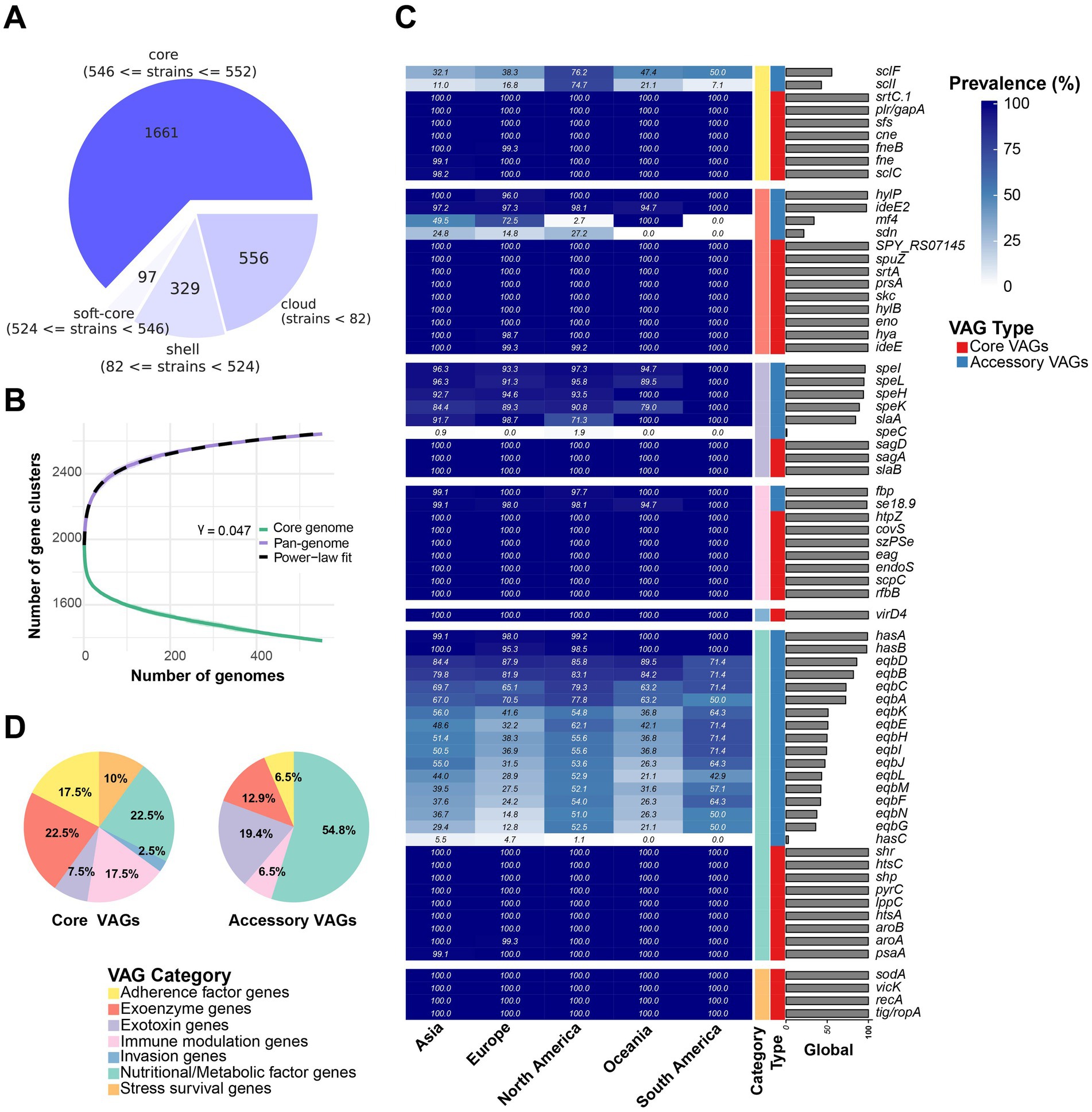
Figure 2. Pangenome analysis and VAG distribution pattern overview. (A) Pangenome analysis of 552 S. equi strains. The pie chart shows the number of protein-coding genes in the core, soft-core, shell, and cloud of the pangenome of 552 S. equi strains. The core genome is defined as genes present in 99–100% of 552 S. equi strains. Soft core, shell and cloud genomes are defined as those present in 95–99%, 15–95% and 0–15% of the strains, respectively. (B) Pan- and core-genome accumulation curves. The core-genome curve (green) and pan-genome curve (purple) show the number of gene clusters as genomes are sequentially added. The pan-genome curve was fitted to a power-law model (dashed line), yielding a γ of 0.047. Shaded areas indicate the interquartile range from 100 permutations. (C) Accessory VAGs prevalence in each continent and at the global level. (D) Different categories of core and accessory VAG proportion.
3.3 Accessory VAGs displayed geographical variation
A total of 71 VAGs in the 552 S. equi genomes were found (Figure 2C; Supplementary Table S4). Functional annotation and clustering of these 71 VAGs classified them into seven distinct functional groups: nutritional/metabolic factor genes, exoenzyme genes, immune modulation genes, exotoxin genes, adherence factor genes, stress survival genes, and invasion factor genes. Among these, 40 VAGs were core VAGs, being present in more than 99% of strains across all functional groups. All invasion genes (n = 1) and stress survival genes (n = 4) were core VAGs. The hyaluronate lyase encoding gene hylP, the IgG endopeptidase encoding gene ideE2, the superantigen encoding genes speI, speL, speK, and speH, the H factor binding protein encoding gene se18.9, the M-like protein SeM encoding gene fbp, and the hyaluronic acid capsule biosynthesis components encoding has operon genes hasA and hasB were largely conserved (Figure 2C). A total of 31 VAGs were less present and were part of only five functional groups, most of them were nutrition/metabolic factor genes (54.8%, 17/31; Figure 2D).
Some accessory VAGs exhibited distinct geographic distribution patterns (Figure 3). The adherence factor genes sclF and sclI were found to be more present in strains from North America, detected in 76.2% (199/261) and 74.7% (195/261) of the strains, respectively. The exoenzyme gene mf4 was more present in Asia, Europe, and in all strains from Oceania, but less common in North America (2.7%, 7/261) and absent in South America (0%, 0/14). Meanwhile, the Phospholipase A2 toxin (PLA2) SlaA gene slaA was less prevalent in North American strains, 71.3% (186/261) strains, compared to over 90% presence in strains from the other four continents (Figure 2C). The pyrogenic mitogen SpeC encoding gene speC, which was found to be carried by prophage SF370.1 from S. pyogenes SF370 (31), was detected in five North American strains and one Asia strain. Among the strains analyzed, the eqb gene locus was present in a majority of North and South American strains (more than 50%), while its presence was lower in strains from other continents (below 40%). Conversely, 54 strains completely lacked the equibactin siderophore system (eqbA–N; Figures 2B, 3). Within the eqb gene locus, the eqbA-D genes exhibited higher conservation compared to eqbE-N (Figure 3).
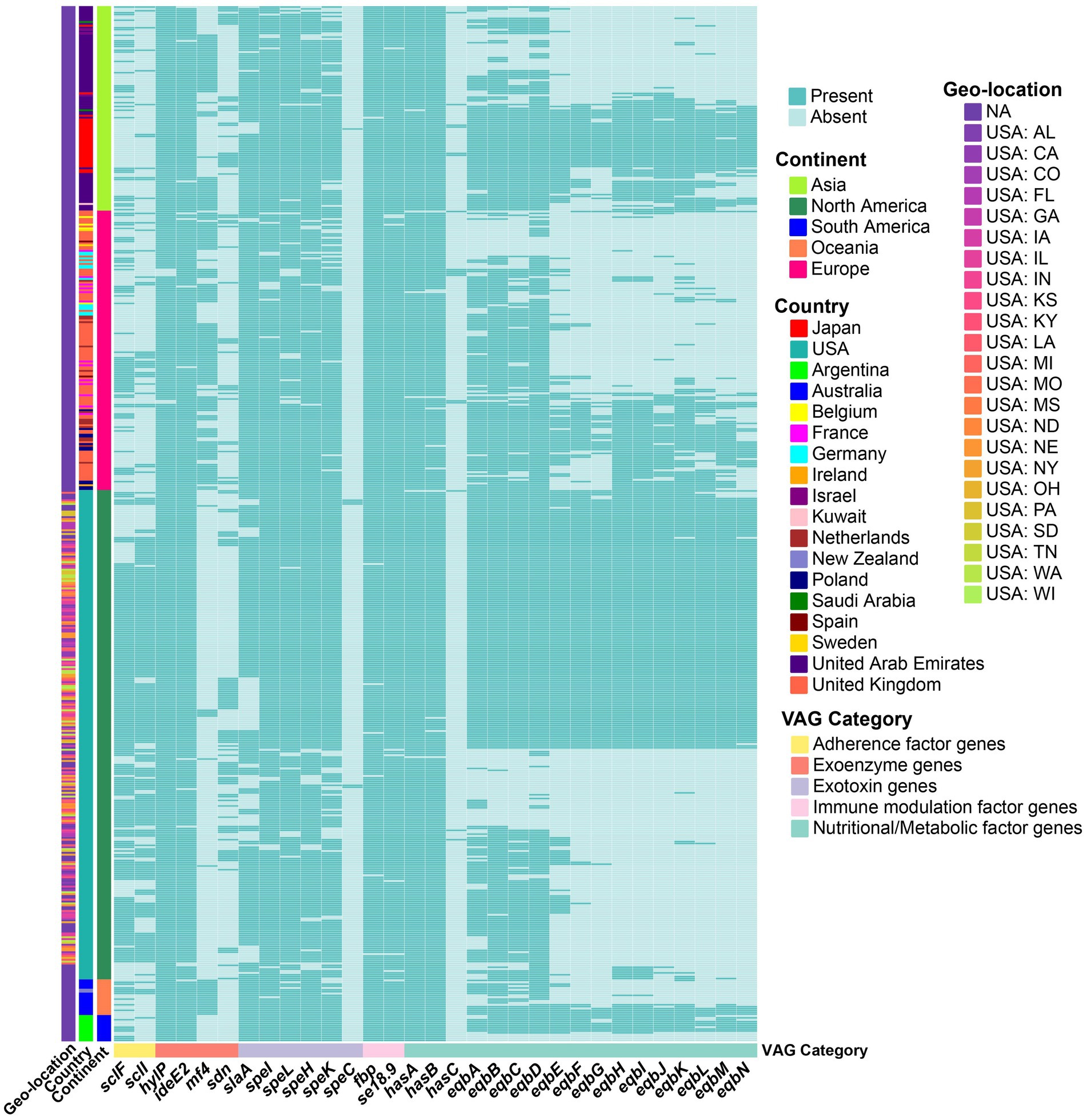
Figure 3. Heatmap showing the presence (green) or absence (light green) of accessory VAGs in S. equi strains from different continents. Each row represents a strain, grouped by continent (left annotation), country, and geo-location (additional left annotations). Each column represents a VAG, annotated by functional category. Strains are clustered based on gene presence/absence profiles. Country and geo-location annotations indicate the specific country and, for USA strains, the state of origin.
3.4 ARGs in S. equi
No penicillin-binding protein (PBP) mutations or other β-lactam resistance determinants were detected, consistent with our finding of an overall lack of acquired ARGs in the analyzed strains. In one isolate from South America, resistance was fond against quaternary ammonium disinfectants, through the qacG gene.
3.5 Identification of the MGEs in S. equi
Each of the 552 genomes contained 3 to 8 prophages, most of which were intact (Figure 4A). Pairwise comparisons showed that both North America and Oceania had significantly lower intact prophage counts compared to Asia (North America: p = 3.49e-07; Oceania: p = 0.0008) and Europe (North America: p = 2.58e-07; Oceania: p = 0.001; Figure 4A). In total, 21 different prophages were identified among the S. equi strains in our study (Figure 5). One PHAGE_Strept_315.2 was found in nearly all strains (531/552) across all continents (Asia: 97.2%, South America: 14/14, Oceania: 19/19, Europe: 91.3%, North America: 98.1%; Figure 4B). PHAGE_Strept_315.6 was the second most prevalent and present in 80.2% (443/552) of the strains. Other prophages were present in about 40% or lower of the strains (Figure 4B). South American strains exhibited the lowest prophage diversity (only seven high-frequency prophages) with PHAGE_Strept_315.2 and PHAGE_Strept_315.6 dominating (>85%, 14/14 and 12/14, respectively). Notably, the prophage PHAGE_Strept_P9 was detected at a higher frequency in North American strains (42.1%, 110/261) compared to the global average frequency (30.2%, 167/552; Figure 4B).
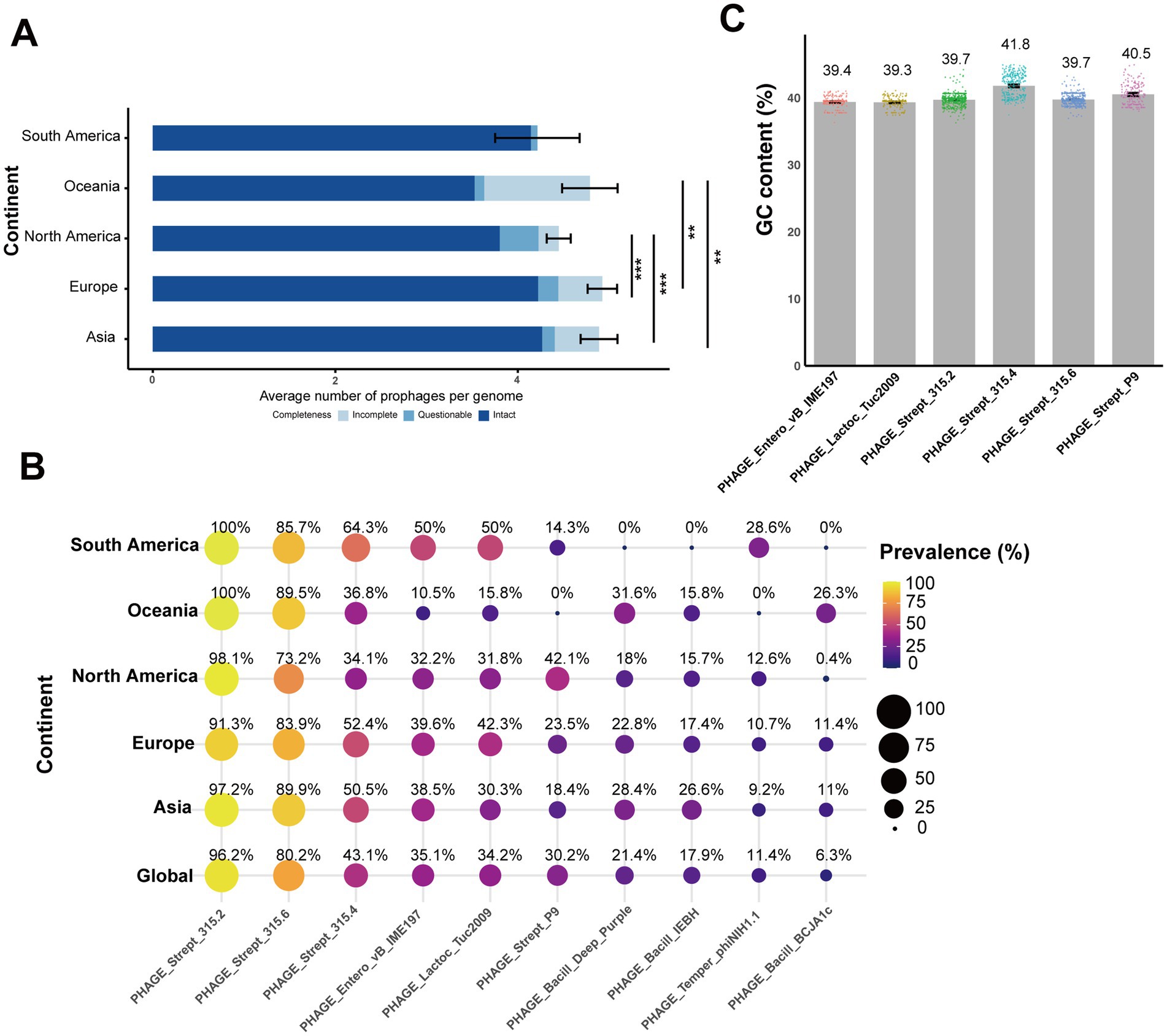
Figure 4. Diversity and distribution of prophages in S. equi across continents. (A) The integrity of prophages in S. equi from different continents. Stacked bar plots show the average number of intact, questionable, and incomplete prophages per genome for each continent. The average values are displayed as colored segments in each bar (blue: intact, light blue: questionable, gray blue: incomplete). (B) Bubble plot showing the prevalence of the top 10 prophages in S. equi strains across different continents and globally. Bubble size and color indicate the percentage of isolates carrying that prophage. Prevalence values are displayed inside each bubble. Prophages are ordered by their global prevalence. (C) Bar plot showing the average GC content (%) of the six most prevalent prophages in S. equi strains. Individual data points are shown as colored dots for each prophage. Bar charts display means with error bars representing 95% confidence intervals. Adjusted p-values were used to determine statistical significance, with thresholds defined as follows: p < 0.01 (**) and p < 0.001 (***).
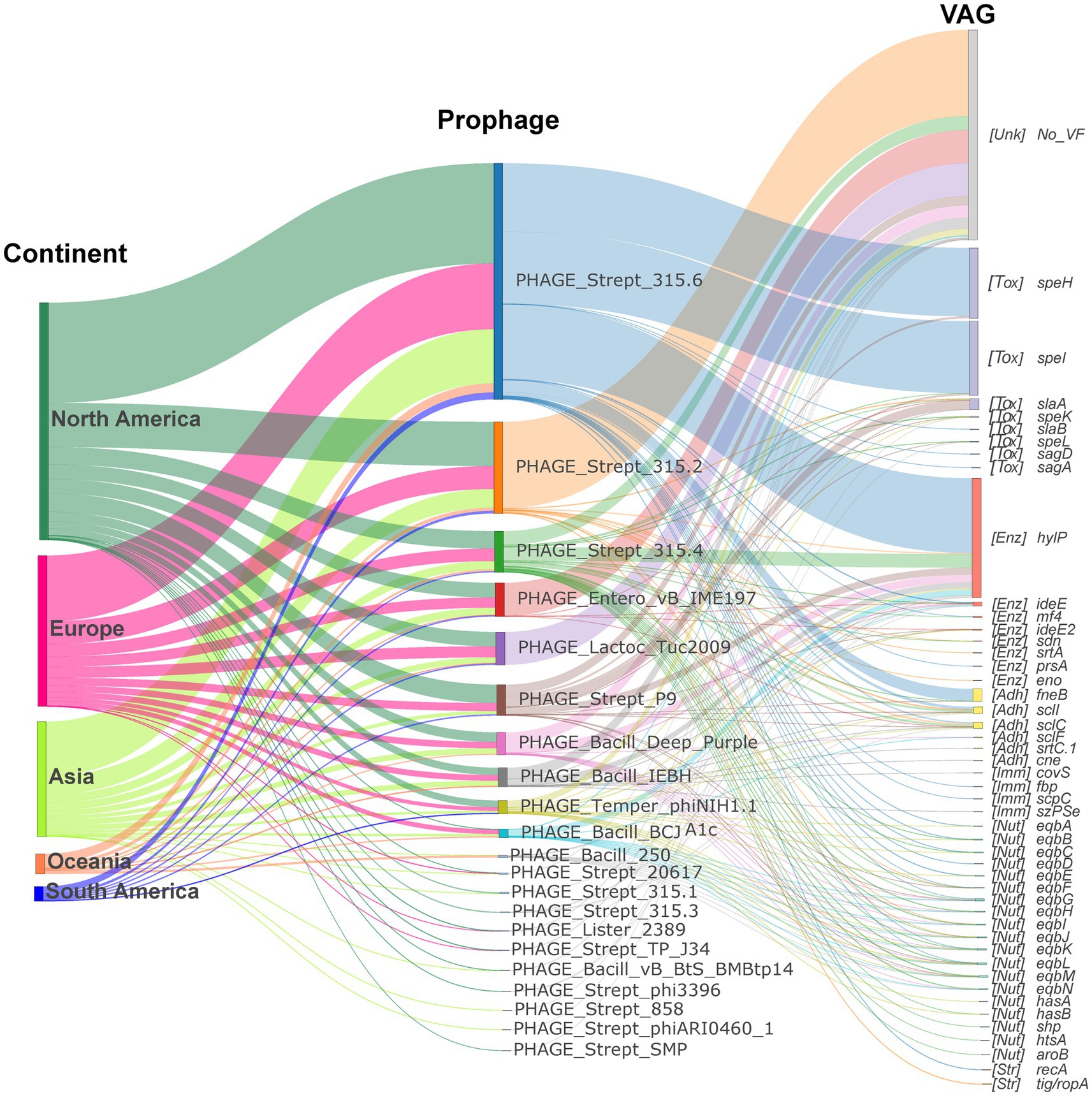
Figure 5. Contribution of the prophages to S. equi VAG distribution. Sankey diagram illustrating the prophage-encoding VAGs flow from continent to prophage to VAG. The left nodes represent continents, the middle nodes represent prophages, and the right nodes represent VAGs. Node and link colors indicate continent, prophage, and VAG category, respectively. VAG nodes are labeled with functional category abbreviations: Adh (Adherence factor gene), Enz (Exoenzyme gene), Tox (Exotoxin gene), Imm (Immune modulation gene), Inv (Invasion gene), Nut (Nutritional/Metabolic factor gene), Str (Stress survival gene).
The mean GC content of these prophages (Figure 4C) ranged from 39.3 to 41.8%, with PHAGE_Strept_315.4 exhibiting the highest average GC content (41.8% ± 1.72), slightly exceeding the genomic background (41.3%). Other common prophages, including PHAGE_Strept_315.2, PHAGE_Strept_315.6, and PHAGE_Strept_P9, showed mean GC contents of approximately 39.7, 39.7, and 40.5%, respectively (Figure 4C), indicating consistent nucleotide composition within prophage populations in S. equi.
Global pairwise sequence comparisons showed that PHAGE_Strept_315.6, PHAGE_Strept_P9, and PHAGE_Strept_315.2 exhibited nucleotide identities greater than 70% with the well-characterized ΦSeq prophages ΦSeq4, ΦSeq3, and ΦSeq2, respectively, suggesting these three prophages potentially correspond to ΦSeq4, ΦSeq3, and ΦSeq2 previously described in S. equi (5).
No intact or partial IMEs were detected across the entire dataset. However, the results of ICEscreen (v1.3.2) showed that all strains harbored two partial ICEs classified as type IV secretion system-type (T4SS-type) based on the conserved presence of VirB4, an ATPase essential for T4SS assembly and function. BLAST comparisons against all known ICE sequences from the ICEberg database (v2.0) (26) revealed that only four strains AQSNo.6 (Asia), AQSNo.12 (Asia), PA17110CY230005 (North America), and Pl0969 (Europe) exhibited ICE sequences with homology (>70% identity) to ICEse2, which encodes for a non-ribosomal peptide synthetase system in S. equi (32). In contrast, the remaining strains did not contain any homologous ICEs that were in the ICEberg database. To further validate the presence of ICESe2, an additional screening was performed using ICEfinder2 (v2.0) directly. However, consistent with the initial analyses, ICEfinder2 did not identify any ICESe2 elements among the strains (data not shown). The GC content of ICE-associated sequences was also examined. The average GC content of ICE sequences varied slightly among the geographic regions, ranging from 41.9% in Oceania to 43.1% in North and South America (Figure 6A). Notably, these ICE GC contents are slightly higher than the average GC content of the 552 S. equi genomes (41.3%). Pairwise comparisons showed that ICEs in strains from North America exhibited significantly higher GC content compared to those from both Europe (p = 0.0004) and Oceania (p = 0.04), indicating geographic variation in ICE nucleotide composition (Figure 6A).
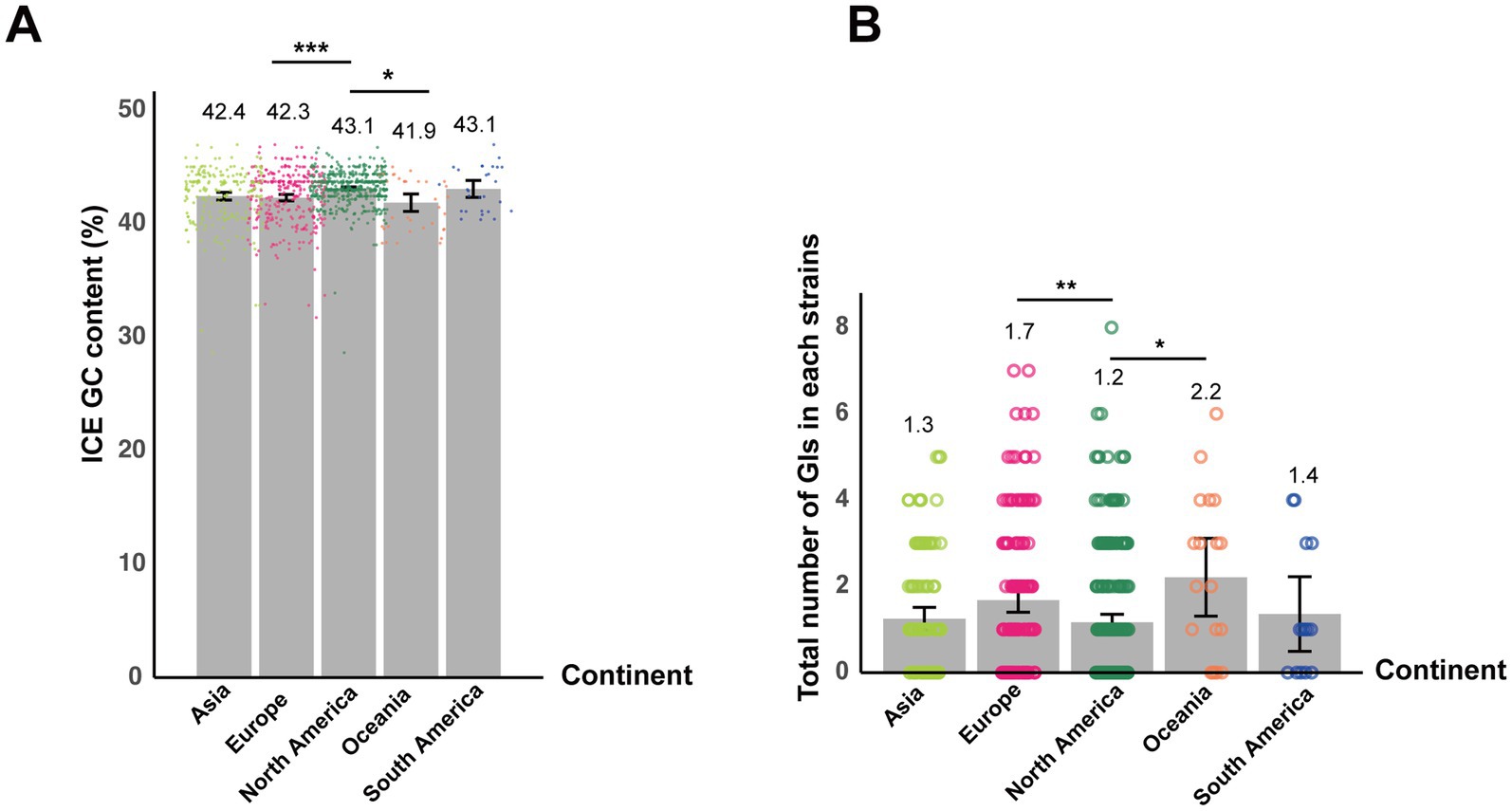
Figure 6. Geographic variation in ICE GC content and GI abundance among S. equi strains. (A) Bar plot showing the mean GC content of ICEs in S. equi strains from different continents. Individual data points are shown as colored dots. (B) Bar plot showing the average number of GIs per S. equi strain across different continents. Individual data points are shown as colored dots. Bar charts display means with error bars representing 95% confidence intervals. Adjusted p-values were used to determine statistical significance, with thresholds defined as follows: p < 0.05 (*), p < 0.01 (**), and p < 0.001 (***).
GIs were detected in 319 of the 552 S. equi strains, with each draft genome harboring 0–8 GIs (Figure 6B). The average number of GIs per genome varied among continents. Strains from North America exhibited significantly lower mean GI count than Oceania (p = 0.04) and Europe (p = 0.003, respectively; Figure 6B).
3.6 Contribution of the MGEs to S. equi VAG distributions
To understand the mechanisms underlying the observed VAG distribution, the contribution of the MGEs to S. equi VAG distribution was investigated. It revealed that prophages, ICEs and GIs serve as primary carriers of VAGs in S. equi.
Although PHAGE_Strept_315.2 was the most common prophage, it did not always carry the highest number of VAGs, whereas PHAGE_Strept_315.6, PHAGE_Strept_315.4, and PHAGE_Strept_P9 were the dominant VAG carriers despite their lower prevalence (Figure 4B; Supplementary Figure S1). As shown in Figure 5, prophage-encoding VAGs predominantly comprised exotoxin genes, exoenzyme genes and adhesion genes. The hyaluronidase gene hylP was carried by various prophages, predominantly PHAGE_Strept_315.6, PHAGE_Strept_315.4, PHAGE_Strept_P9, PHAGE_Bacill_Deep_Purple, PHAGE_Bacill_IEBH and PHAGE_Bacill_BCJA1c (Figure 5). PHAGE_Strept_315.6 primarily carried the superantigen genes speI/speH and hylP (Supplementary Figure S1; Figure 5) while PHAGE_Strept_P9 carried the exotoxin gene slaA (Supplementary Figure S1).
Significant differences were observed in the number of pathogenicity islands between continents (p = 0.0024), with North America having significantly fewer pathogenicity islands compared to Europe and Oceania (adjusted p = 0.0006 and 0.0266, respectively; Figure 7). The main VAGs encoded by pathogenicity islands were eqbA-D and virD4 (Figure 7).
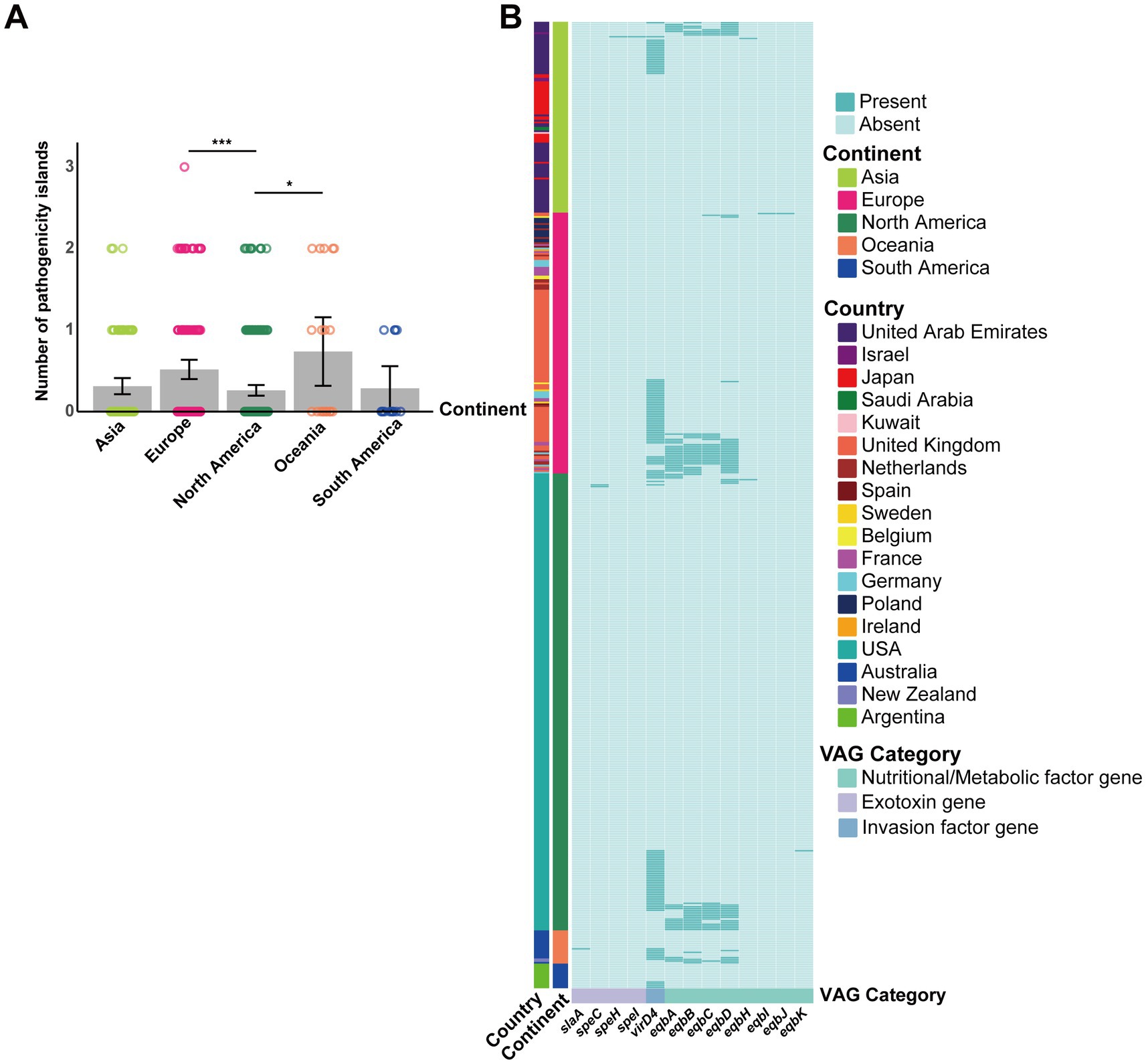
Figure 7. Contribution of GIs to S. equi VAG distribution. (A) Bar plot showing the average number of pathogenicity islands per S. equi strain across different continents. Individual data points are shown as colored dots. (B) Heatmap showing the presence (green) or absence (light green) of VAGs encoded by GIs in S. equi strains from different continents. Each row represents a strain, grouped by continent (left annotation). Each column represents a VAG, annotated by functional category. Strains are clustered based on gene presence/absence profiles.
The partial ICEs carried the sfs gene, which encodes a fibronectin-binding protein (Fn-binding protein). ICEs also carried streptolysin S-associated genes, sagA and sagD, mainly in strains from North America. Nutritional and metabolic factor genes htsA and shp, responsible for heme acquisition and iron uptake, were core genes in S. equi (Figure 2C), though some were also carried by ICEs (Figure 8).
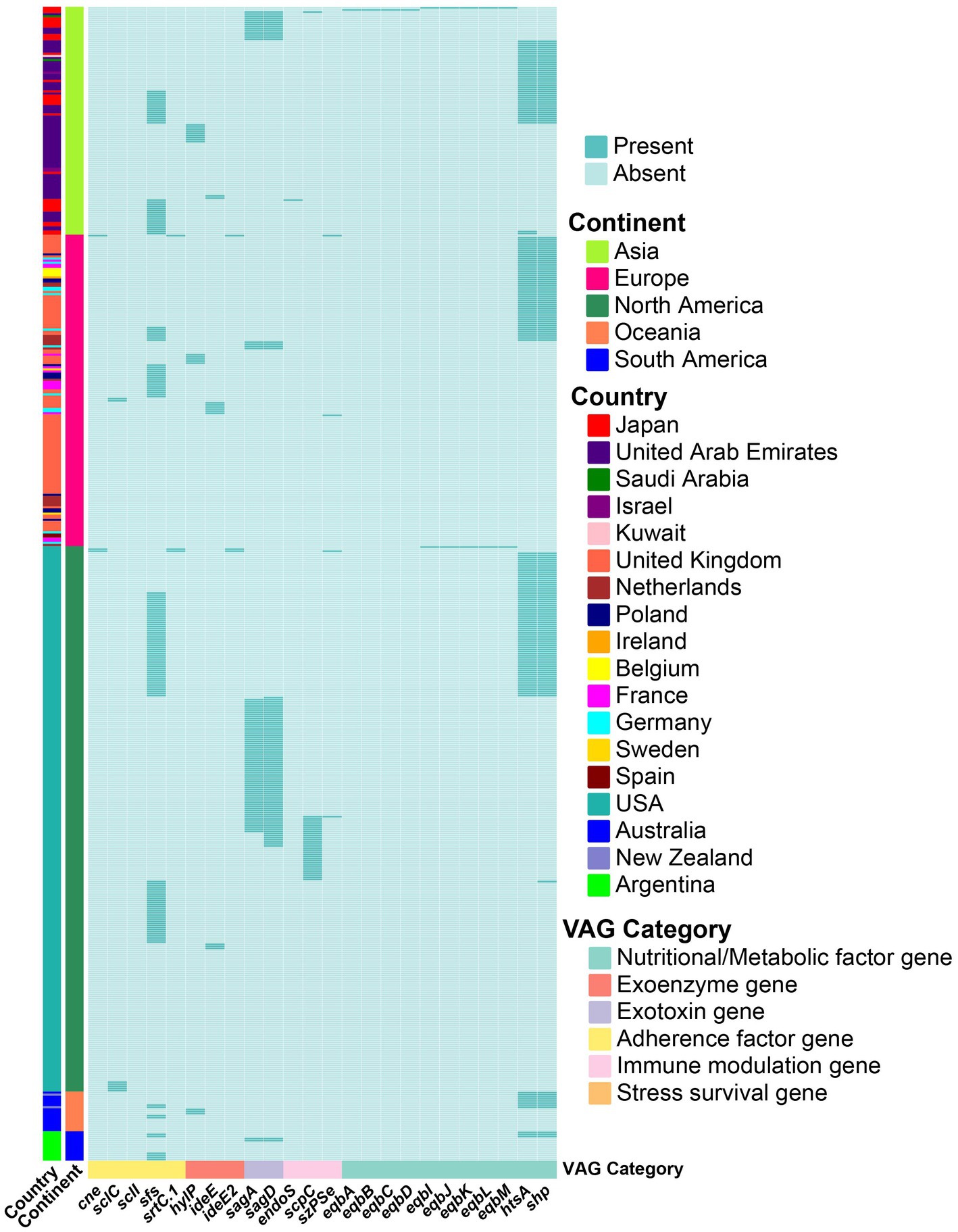
Figure 8. Contribution of the ICEs to S. equi VAG distribution. Heatmap showing the presence (green) or absence (light green) of VAGs encoded by ICEs in S. equi strains from different continents. Each row represents a strain, grouped by continent (left annotation). Each column represents a VAG, annotated by functional category. Strains are clustered based on gene presence/absence profiles.
4 Discussion
Our study indicates that S. equi exhibits a closed pangenome structure, typical for host-adapted bacteria like Bordetella parapertussis (B. parapertussis), Bordetella pertussis (B. pertussis), as well as all mycoplasma species as the extreme example of genome reductive evolution, which purge non-essential MGEs while retaining niche-critical genes (33, 34). Our findings provide evidence that host-adapted bacteria undergo genome streamlining, resulting in smaller and more clonal genomes (6, 35, 36).
Some accessory VAGs, which are also present in the Strangvac vaccine, exhibit significant geographic variation. This regional divergence is critical because vaccines impose selective pressure on bacteria, particularly in host-restricted species (37). Such selective pressure can drive the emergence of vaccine escape variants, as observed with serotype replacement and capsule switching of vaccine serotypes to nonvaccine types (NVTs) in S. pneumoniae following vaccination (38–40). These observations highlight a potential blind spot in current S. equi surveillance strategies, which often overlook the dynamic accessory genome (6, 41). Therefore, integrating the accessory genome analysis is necessary for proper surveillance of pathogens. It will enable the assessment of the efficacy of the antigens in vaccines and the early detection of potential vaccine escape variants.
Experimentally validated virulence factors of S. equi critical for pathogenicity include immune evasion (SeM, Se18.9, SzPse, slaA, superantigens speL and speK, EAG) (6, 9, 42–47), antibody disruption (ideE and ideE2) (48, 49), adhesion (CNE, FNEB, and SclC) (7, 10, 50, 51), capsule formation (hasA, hasB, and hasC) (52–54) and iron acquisition (eqb cluster) (32). Genes encoding CNE, SzPse, FNEB, SclC, EAG, ideE were identified as core VAGs that are universally conserved across the 552 strains. Although genes encoding SeM, Se18.9, superantigens SpeL, SpeK, SpeH and SpeI, as well as capsule biosynthesis genes hasA and hasB were classified as accessory VAGs in the present study, they showed broad conservation across all continents. Only genes encoding SlaA and the iron acquisition cluster eqb displayed notable geographical differences. This finding supports the genome streamlining hypothesis that host-adapted bacteria lose unnecessary genes over time, resulting in smaller, more clonal genomes that retain only essential virulence and fitness genes for specialized niches (35).
speL, speK, speH and speI encode superantigens that cause nonspecific T cell activation and release of proinflammatory cytokines, playing essential roles in severe streptococcal infections (55). Such immune overreaction is linked to streptococcal toxic shock syndrome (STSS) caused by S. pyogenes in humans (56). These exotoxins also contribute to the pathogenicity in S. equi and S. zooepidemicus (57, 58). Our study revealed that speL, speK, speH, and speI were widely distributed across S. equi strains, indicating that they are critical for the immune evasion strategies employed during S. equi infections. Furthermore, T cell receptor-binding deficient superantigens have shown promise as vaccine adjuvants by enhancing antigen presentation (59). These findings suggest that the conserved superantigen-encoding genes may serve as potential antigenic targets for the development of a strangles vaccine. However, the role of superantigens in the pathogenesis of S. equi remains largely unknown, and experimental validation is required to assess their immunogenicity and protective efficacy.
slaA and slaB are two homologous phospholipase A2 (PLA2) toxins encoding genes, found in both S. equi and S. zooepidemicus. slaB has been shown to be present in all strains of both species, while slaA is only in 31% of S. zooepidemicus strains and in all S. equi strains (26/26) (5). However, our results showed slaA was less prevalent in North American strains and more conserved in strains from other continents. PLA2 toxins were shown to be non-essential in the pathogenesis of strangles in a susceptible natural host (46). However, they can disrupt host cell membranes (Type II exotoxins) and their downregulation could mitigate excessive host tissue damage, helping S. equi evade immune clearance and promote long-term colonization (60). Persistent S. equi strains often lose some virulence genes, which may be part of an adaptive genomic streamlining process during chronic infection (6). Consistent with this, the lower conservation of the slaA gene in North American strains in our study suggests such an adaptation may occur in this region.
Streptococcus equi are almost always encapsulated. Non-encapsulated mutants have been shown to be less virulent (52, 53). Capsule synthesis is controlled by hasA (hyaluronate synthase), hasB (UDP-glucose dehydrogenase) and hasC (UDP-glucose pyrophosphorylase). While deletions in either hasA or hasB result in loss of capsule synthesis and virulence, deletions in hasC do not lead to capsule loss (54, 61). Our findings showed that hasA and hasB were present in nearly all strains, whereas hasC was rarely present, providing further support for genome reduction in host-restricted pathogens.
The iron uptake systems of S. equi facilitate the development of lymph node abscesses (6, 32). Among these systems, equibactin (EqbA-N), a siderophore produced via the Nonribosomal peptide synthetases (NRPS), plays a major role (32, 62). However, our results showed that this gene cluster was absent in a multitude of strains. This finding aligned with previous reports of microevolution in persistently infected guttural pouches, characterized by deletions or amplifications, including the loss of the entire equibactin locus in some S. equi subpopulations (6). Large genomic deletions are hallmark indicators of genome reduction as bacteria adapt to more restricted niches (63–65). This reductive evolution involves shedding non-essential genes, which may include those encoding products analogous to host molecules that bacteria can exploit (66). Our results revealed that although the equibactin locus was absent in many strains of S. equi, the heme acquisition genes (shp, shr, htsA) were core genes. The results suggested that in strains lacking the equibactin locus, the core heme system may compensate for iron uptake, reflecting adaptation to fluctuating iron availability during infection likely due to changing niches in the respiratory tract. Similar findings of siderophore production dynamics have been observed during B. pertussis infection in mice (67). The coexistence of multiple iron acquisition systems not only compensates for variable iron availability but also enhances pathogenic fitness, as demonstrated in Klebsiella pneumoniae (K. pneumoniae), where the classical K. pneumoniae (cKp) strains typically encode genes for 1–2 siderophores, while hypervirulent K. pneumoniae (hvKp) strains encode genes for four siderophores (enterobactin, yersiniabactin, salmochelin, aerobactin), enhancing their superior tissue dissemination and hypervirulence (68). Whether this also applies to S. equi remains to be determined.
No acquired ARGs were detected, except for a resistance gene qacG against the quaternary ammonium compounds in a single South American strain (Arg0107). This absence of ARGs is unexpected, given the widespread resistance reported in other equine bacteria (69). However, a low prevalence of acquired ARGs is also observed in other highly specialized veterinary pathogens, such as Mycoplasmata spp. and Actinobacillus pleuropneumoniae (70, 71), suggesting that the specific lifestyle of pathogen may limit its capacity for horizontal gene transfer. As an obligate equine pathogen, S. equi has a reduced number of pili and other sortase-processed proteins on its surface compared to opportunistic pathogens like S. zooepidemicus, which may restrict the number of niches it can occupy (5, 72). This niche restriction, combined with its colonization pattern that avoids the nasopharynx where microbial exchange is common, further isolates S. equi from the broader bacterial gene pool, thus restricting its interaction with other bacteria and the subsequent acquisition of ARGs (73). Further microbiome studies are necessary to validate this hypothesis. Further microbiome studies are warranted to validate this hypothesis. It should also be considered that alternative mechanisms may contribute to the observed phenomena. Given the seemingly genome reduction, a decreased capacity for acquiring foreign DNA might also play a role. These possibilities require further investigation.
MGEs play a significant role in horizontal gene transfer (HGT) that leads to the dissemination of ARGs and VAGs and shapes the genome, often conferring selective advantages (74–78). Our study revealed that prophages, GIs, and ICEs play only a role in VAG distribution but not in ARG dissemination in S. equi, indicating that AMR does not pose a selective advantage for this bacterium. This finding may be explained by the unique lifestyle of the S. equi, whereby it hides in abscesses as well as in the guttural pouches where the diffusion of antibiotics is minimal.
A widespread prevalence of intact prophages was observed across all strains. They frequently carry superantigen genes (speH, speI) and the hyaluronate lyase encoding gene hylP. Specifically, prophages PHAGE_Strept_315.6, PHAGE_Strept_315.4 and PHAGE_Strept_P9 served as primary reservoirs that share a close relationship with phages of S. pyogenes (79, 80). Our analysis revealed that the GC content of common prophages closely matches the overall GC content of S. equi genomes, indicating that these prophages are well-adapted to S. equi and have been stable components of the bacterial genome. This finding is consistent with the role of bacteriophages as major elements in shaping S. equi evolution away from its ancestor, S. zooepidemicus (79).
While previous studies have confirmed the conserved insertion sites of ΦSeq1-4 and ICESe2 (5, 32, 81, 82), no evidence of these elements were detected in the present study. Although prophages corresponding to ΦSeq2, ΦSeq3, and ΦSeq4 were identified, their carriage of key VAGs like slaA, speK, and speL showed a mosaic pattern inconsistent with previous reports. For example, the slaA gene was predominantly found on PHAGE_Strept_P9 (ΦSeq3-like prophage), contrary to its known association with ΦSeq2 (73). However, these discrepancies may be caused by the problematic assemblies of short-read sequencing of regions with lots of repeats, which are typical around prophages and ICEs. These elements were potentially distributed across multiple contigs. It may also cause failures to identify MGEs if hallmark genes used for their detection are deleted, rearranged, or mutated (83). Therefore, whole-genome sequencing approaches incorporating long-read data, closed genomes, or targeted PCR validation are needed to reconcile these observations conclusively.
In conclusion, our analysis of 552 global S. equi genomes provides a comprehensive framework for understanding its accessory genome and genome adaptation. While acquired antibiotic resistance remains absent, geographic variations in MGEs and VAGs were observed. Our findings support the concept of “genome streamlining,” where this host-adapted pathogen sheds non-essential genes while retaining specialized genes. The geographic patterns likely arise from complex interactions involving host adaptation, and the ecological factors underlying these patterns remain to be determined. Future studies employing functional genomics, and long-read sequencing data will be necessary to clarify these dynamics. Our work provides a high-resolution map of S. equi VAGs, which is crucial for future vaccine strategies. While core antigens remain primary targets, the observed regional variations in key VAGs highlight the importance of ongoing genomic surveillance to monitor antigenic drift and inform the timely update of vaccine formulations (Table 1), thereby maintaining efficacy against evolving local pathogen populations.

Table 1. Potential vaccine antigens to be included in a regional vaccine.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
Author contributions
LH: Visualization, Validation, Formal analysis, Data curation, Methodology, Writing – original draft, Investigation. NK: Writing – review & editing, Methodology. JS: Supervision, Writing – review & editing. CL: Writing – review & editing, Supervision. PB: Writing – review & editing, Funding acquisition, Conceptualization, Resources, Supervision, Project administration, Methodology.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This study was supported by grant 9380166 from City University of Hong Kong, grant GSP246 from the Research Talent Hub Hong Kong and a grant from the Hong Kong Jockey Club Foundation, JC STEM Lab of Integrated Microbial Genomics (Project No. 2025–0037).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that Gen AI was used in the creation of this manuscript. Generative AI was used solely for language polishing and refinement of the manuscript, without involvement in study design, data collection, analysis, interpretation, or the core content creation of the manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fvets.2025.1721958/full#supplementary-material
Footnotes
References
1. Boyle, AG, Timoney, JF, Newton, JR, Hines, MT, Waller, AS, and Buchanan, BR. Streptococcus equi infections in horses: guidelines for treatment, control, and prevention of strangles-revised consensus statement. J Vet Intern Med. (2018) 32:633–47. doi: 10.1111/jvim.15043
2. Wilkins, PA, Lascola, KM, Woolums, AR, Bedenice, D, Giguère, S, Boyle, AG, et al. Chapter 31 - diseases of the respiratory system In: BP Smith, DC Metre, and N Pusterla, editors. Large animal internal medicine (sixth edition). St. Louis (MO): Mosby (2020). 559.
3. Waller, AS. New perspectives for the diagnosis, control, treatment, and prevention of strangles in horses. Vet Clin North Am Equine Pract. (2014) 30:591–607. doi: 10.1016/j.cveq.2014.08.007
4. Timoney, JF. The pathogenic equine streptococci. Vet Res. (2004) 35:397–409. doi: 10.1051/vetres:2004025
5. Holden, MT, Heather, Z, Paillot, R, Steward, KF, Webb, K, Ainslie, F, et al. Genomic evidence for the evolution of Streptococcus equi: host restriction, increased virulence, and genetic exchange with human pathogens. PLoS Pathog. (2009) 5:e1000346. doi: 10.1371/journal.ppat.1000346
6. Harris, SR, Robinson, C, Steward, KF, Webb, KS, Paillot, R, Parkhill, J, et al. Genome specialization and decay of the strangles pathogen, Streptococcus equi, is driven by persistent infection. Genome Res. (2015) 25:1360–71. doi: 10.1101/gr.189803.115
7. Robinson, C, Frykberg, L, Flock, M, Guss, B, Waller, AS, and Flock, JI. Strangvac: a recombinant fusion protein vaccine that protects against strangles, caused by Streptococcus equi. Vaccine. (2018) 36:1484–90. doi: 10.1016/j.vaccine.2018.01.030
8. Lannergard, J, Frykberg, L, and Guss, B. Cne, a collagen-binding protein of Streptococcus equi. FEMS Microbiol Lett. (2003) 222:69–74. doi: 10.1016/S0378-1097(03)00222-2
9. Jiang, X, Ma, X, Su, L, Zhang, B, He, Z, and Su, Y. Analysis of distinct variants of immunoglobulin G binding protein Eag on humoral immunity and bacterial clearance of Streptococcus equi subspecies equi. Microb Pathog. (2025) 205:107726. doi: 10.1016/j.micpath.2025.107726
10. Karlstrom, A, Jacobsson, K, and Guss, B. Sclc is a member of a novel family of collagen-like proteins in Streptococcus equi subspecies equi that are recognised by antibodies against Sclc. Vet Microbiol. (2006) 114:72–81. doi: 10.1016/j.vetmic.2005.10.036
11. Tscheschlok, L, Venner, M, Steward, K, Bose, R, Riihimaki, M, and Pringle, J. Decreased clinical severity of strangles in weanlings associated with restricted seroconversion to optimized Streptococcus equi ssp equi assays. J Vet Intern Med. (2018) 32:459–64. doi: 10.1111/jvim.15037
12. Timoney, JF, Yang, J, Liu, J, and Merant, C. Idee reduces the bactericidal activity of equine neutrophils for Streptococcus equi. Vet Immunol Immunopathol. (2008) 122:76–82. doi: 10.1016/j.vetimm.2007.10.017
13. Meehan, M, Lynagh, Y, Woods, C, and Owen, P. The fibrinogen-binding protein (Fgbp) of Streptococcus equi subsp. equi additionally binds IgG and contributes to virulence in a mouse model. Microbiology (Reading). (2001) 147:3311–22. doi: 10.1099/00221287-147-12-3311
14. Kelly, C, Bugg, M, Robinson, C, Mitchell, Z, Davis-Poynter, N, Newton, JR, et al. Sequence variation of the Sem gene of Streptococcus equi allows discrimination of the source of strangles outbreaks. J Clin Microbiol. (2006) 44:480–6. doi: 10.1128/JCM.44.2.480-486.2006
15. Proft, T, Webb, PD, Handley, V, and Fraser, JD. Two novel superantigens found in both group a and group C Streptococcus. Infect Immun. (2003) 71:1361–9. doi: 10.1128/IAI.71.3.1361-1369.2003
16. Mitchell, C, Steward, KF, Charbonneau, ARL, Walsh, S, Wilson, H, Timoney, JF, et al. Globetrotting strangles: the unbridled national and international transmission of Streptococcus equi between horses. Microb Genom. (2021) 7:1–14. doi: 10.1099/mgen.0.000528
17. Wick, RR, Judd, LM, Gorrie, CL, and Holt, KE. Unicycler: resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput Biol. (2017) 13:e1005595. doi: 10.1371/journal.pcbi.1005595
18. Gurevich, A, Saveliev, V, Vyahhi, N, and Tesler, G. Quast: quality assessment tool for genome assemblies. Bioinformatics. (2013) 29:1072–5. doi: 10.1093/bioinformatics/btt086
19. Chen, TW, Gan, RC, Chang, YF, Liao, WC, Wu, TH, Lee, CC, et al. Is the whole greater than the sum of its parts? De novo assembly strategies for bacterial genomes based on paired-end sequencing. BMC Genomics. (2015) 16:648. doi: 10.1186/s12864-015-1859-8
20. Seemann, T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. (2014) 30:2068–9. doi: 10.1093/bioinformatics/btu153
21. Page, AJ, Cummins, CA, Hunt, M, Wong, VK, Reuter, S, Holden, MT, et al. Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics. (2015) 31:3691–3. doi: 10.1093/bioinformatics/btv421
22. Alcock, BP, Huynh, W, Chalil, R, Smith, KW, Raphenya, AR, Wlodarski, MA, et al. Card 2023: expanded curation, support for machine learning, and Resistome prediction at the comprehensive antibiotic resistance database. Nucleic Acids Res. (2023) 51:D690–9. doi: 10.1093/nar/gkac920
23. Wishart, DS, Han, S, Saha, S, Oler, E, Peters, H, Grant, JR, et al. Phastest: faster than Phaster, better than Phast. Nucleic Acids Res. (2023) 51:W443–50. doi: 10.1093/nar/gkad382
24. Lao, J, Lacroix, T, Guedon, G, Coluzzi, C, Payot, S, Leblond-Bourget, N, et al. Icescreen: a tool to detect Firmicute Ices and Imes, isolated or enclosed in composite structures. NAR Genom Bioinform. (2022) 4:1–12. doi: 10.1093/nargab/lqac079
25. Liu, M, Li, X, Xie, Y, Bi, D, Sun, J, Li, J, et al. Iceberg 2.0: an updated database of bacterial integrative and conjugative elements. Nucleic Acids Res. (2019) 47:D660–5. doi: 10.1093/nar/gky1123
26. Bi, D, Xu, Z, Harrison, EM, Tai, C, Wei, Y, He, X, et al. Iceberg: a web-based resource for integrative and conjugative elements found in Bacteria. Nucleic Acids Res. (2012) 40:D621–6. doi: 10.1093/nar/gkr846
27. Bertelli, C, Laird, MR, Williams, KP, Simon Fraser University Research Computing GLau, BY, Hoad, G, et al. Islandviewer 4: expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res. (2017) 45:W30–5. doi: 10.1093/nar/gkx343
28. Bertelli, C, and Brinkman, FSL. Improved Genomic Island predictions with Islandpath-Dimob. Bioinformatics. (2018) 34:2161–7. doi: 10.1093/bioinformatics/bty095
29. Waack, S, Keller, O, Asper, R, Brodag, T, Damm, C, Fricke, WF, et al. Score-based prediction of Genomic Islands in prokaryotic genomes using hidden Markov models. BMC Bioinformatics. (2006) 7:142. doi: 10.1186/1471-2105-7-142
30. Madeira, F, Madhusoodanan, N, Lee, J, Eusebi, A, Niewielska, A, Tivey, ARN, et al. The Embl-Ebi job dispatcher sequence analysis tools framework in 2024. Nucleic Acids Res. (2024) 52:W521–5. doi: 10.1093/nar/gkae241
31. Canchaya, C, Desiere, F, McShan, WM, Ferretti, JJ, Parkhill, J, and Brussow, H. Genome analysis of an inducible prophage and prophage remnants integrated in the Streptococcus pyogenes strain Sf370. Virology. (2002) 302:245–58. doi: 10.1006/viro.2002.1570
32. Heather, Z, Holden, MT, Steward, KF, Parkhill, J, Song, L, Challis, GL, et al. A novel streptococcal integrative conjugative element involved in Iron acquisition. Mol Microbiol. (2008) 70:1274–92. doi: 10.1111/j.1365-2958.2008.06481.x
33. Parkhill, J, Sebaihia, M, Preston, A, Murphy, LD, Thomson, N, Harris, DE, et al. Comparative analysis of the genome sequences of Bordetella pertussis, Bordetella parapertussis and Bordetella bronchiseptica. Nat Genet. (2003) 35:32–40. doi: 10.1038/ng1227
34. Belcher, T, Dubois, V, Rivera-Millot, A, Locht, C, and Jacob-Dubuisson, F. Pathogenicity and virulence of Bordetella pertussis and its adaptation to its strictly human host. Virulence. (2021) 12:2608–32. doi: 10.1080/21505594.2021.1980987
35. Moran, NA, and Plague, GR. Genomic changes following host restriction in Bacteria. Curr Opin Genet Dev. (2004) 14:627–33. doi: 10.1016/j.gde.2004.09.003
36. Citti, C, and Blanchard, A. Mycoplasmas and their host: emerging and re-emerging minimal pathogens. Trends Microbiol. (2013) 21:196–203. doi: 10.1016/j.tim.2013.01.003
37. Mikucki, A, and Kahler, CM. Microevolution and its impact on Hypervirulence, antimicrobial resistance, and vaccine escape in Neisseria meningitidis. Microorganisms. (2023) 11:1–31. doi: 10.3390/microorganisms11123005
38. Weinberger, DM, Malley, R, and Lipsitch, M. Serotype replacement in disease after pneumococcal vaccination. Lancet. (2011) 378:1962–73. doi: 10.1016/S0140-6736(10)62225-8
39. Singleton, RJ, Hennessy, TW, Bulkow, LR, Hammitt, LL, Zulz, T, Hurlburt, DA, et al. Invasive pneumococcal disease caused by nonvaccine serotypes among Alaska native children with high levels of 7-valent pneumococcal conjugate vaccine coverage. JAMA. (2007) 297:1784–92. doi: 10.1001/jama.297.16.1784
40. Coffey, TJ, Enright, MC, Daniels, M, Morona, JK, Morona, R, Hryniewicz, W, et al. Recombinational exchanges at the capsular polysaccharide biosynthetic locus lead to frequent serotype changes among natural isolates of Streptococcus pneumoniae. Mol Microbiol. (1998) 27:73–83.
41. McGlennon, A, Waller, A, Verheyen, K, Slater, J, Grewar, J, Aanensen, D, et al. Surveillance of strangles in UK horses between 2015 and 2019 based on laboratory detection of Streptococcus equi. Vet Rec. (2021) 189:e948. doi: 10.1002/vetr.948
42. Boschwitz, JS, and Timoney, JF. Characterization of the Antiphagocytic activity of equine fibrinogen for Streptococcus equi subsp. equi. Microb Pathog. (1994) 17:121–9.
43. Boschwitz, JS, and Timoney, JF. Inhibition of C3 deposition on Streptococcus equi subsp. equi by M protein: a mechanism for survival in equine blood. Infect Immun. (1994) 62:3515–20.
44. Timoney, JF, Suther, P, Velineni, S, and Artiushin, SC. The Antiphagocytic activity of Sem of Streptococcus equi requires capsule. J Equine Sci. (2014) 25:53–6. doi: 10.1294/jes.25.53
45. Tiwari, R, Qin, A, Artiushin, S, and Timoney, JF. Se18.9, an anti-phagocytic factor H binding protein of Streptococcus equi. Vet Microbiol. (2007) 121:105–15. doi: 10.1016/j.vetmic.2006.11.023
46. Lopez-Alvarez, MR, Salze, M, Cenier, A, Robinson, C, Paillot, R, and Waller, AS. Immunogenicity of phospholipase a(2) toxins and their role in Streptococcus equi pathogenicity. Vet Microbiol. (2017) 204:15–9. doi: 10.1016/j.vetmic.2017.04.002
47. Paillot, R, Robinson, C, Steward, K, Wright, N, Jourdan, T, Butcher, N, et al. Contribution of each of four Superantigens to Streptococcus equi-induced Mitogenicity, gamma interferon synthesis, and immunity. Infect Immun. (2010) 78:1728–39. doi: 10.1128/IAI.01079-09
48. Lannergard, J, and Guss, B. Idee, an igg-endopeptidase of Streptococcus equi ssp. equi. FEMS Microbiol Lett. (2006) 262:230–5. doi: 10.1111/j.1574-6968.2006.00404.x
49. Hulting, G, Flock, M, Frykberg, L, Lannergard, J, Flock, JI, and Guss, B. Two novel igg endopeptidases of Streptococcus equi. FEMS Microbiol Lett. (2009) 298:44–50. doi: 10.1111/j.1574-6968.2009.01698.x
50. Lannergard, J, Flock, M, Johansson, S, Flock, JI, and Guss, B. Studies of fibronectin-binding proteins of Streptococcus equi. Infect Immun. (2005) 73:7243–51. doi: 10.1128/IAI.73.11.7243-7251.2005
51. Waller, A, Flock, M, Smith, K, Robinson, C, Mitchell, Z, Karlstrom, A, et al. Vaccination of horses against strangles using recombinant antigens from Streptococcus equi. Vaccine. (2007) 25:3629–35. doi: 10.1016/j.vaccine.2007.01.060
52. Anzai, T, Timoney, JF, Kuwamoto, Y, Fujita, Y, Wada, R, and Inoue, T. In vivo pathogenicity and resistance to phagocytosis of Streptococcus equi strains with different levels of capsule expression. Vet Microbiol. (1999) 67:277–86.
53. Galan, JE, and Timoney, JF. Mucosal nasopharyngeal immune responses of horses to protein antigens of Streptococcus equi. Infect Immun. (1985) 47:623–8.
54. Walker, JA, and Timoney, JF. Construction of a stable non-mucoid deletion mutant of the Streptococcus equi pinnacle vaccine strain. Vet Microbiol. (2002) 89:311–21. doi: 10.1016/s0378-1135(02)00205-5
55. Sahr, A, Former, S, Hildebrand, D, and Heeg, K. T-cell activation or Tolerization: the yin and Yang of bacterial Superantigens. Front Microbiol. (2015) 6:1153. doi: 10.3389/fmicb.2015.01153
56. Proft, T, Sriskandan, S, Yang, L, and Fraser, JD. Superantigens and streptococcal toxic shock syndrome. Emerg Infect Dis. (2003) 9:1211–8. doi: 10.3201/eid0910.030042
57. Commons, RJ, Smeesters, PR, Proft, T, Fraser, JD, Robins-Browne, R, and Curtis, N. Streptococcal Superantigens: categorization and clinical associations. Trends Mol Med. (2014) 20:48–62. doi: 10.1016/j.molmed.2013.10.004
58. Rash, NL, Robinson, C, DeSouza, N, Nair, S, Hodgson, H, Steward, K, et al. Prevalence and disease associations of Superantigens Szef, Szen and Szep in the S. Zooepidemicus population and possible functional redundancy of Szef. Res Vet Sci. (2014) 97:481–7. doi: 10.1016/j.rvsc.2014.09.001
59. Radcliff, FJ, Loh, JM, Ha, B, Schuhbauer, D, McCluskey, J, and Fraser, JD. Antigen targeting to major histocompatibility complex class II with streptococcal mitogenic exotoxin Z-2 M1, a superantigen-based vaccine carrier. Clin Vaccine Immunol. (2012) 19:574–86. doi: 10.1128/CVI.05446-11
60. Murakami, M, Yamamoto, K, Miki, Y, Murase, R, Sato, H, and Taketomi, Y. The roles of the secreted phospholipase a(2) gene family in immunology. Adv Immunol. (2016) 132:91–134. doi: 10.1016/bs.ai.2016.05.001
61. Ashbaugh, CD, Alberti, S, and Wessels, MR. Molecular analysis of the capsule gene region of group a Streptococcus: the Hasab genes are sufficient for capsule expression. J Bacteriol. (1998) 180:4955–9.
62. Nygaard, TK, Liu, M, McClure, MJ, and Lei, B. Identification and characterization of the heme-binding proteins Seshp and Sehtsa of Streptococcus equi subspecies equi. BMC Microbiol. (2006) 6:82. doi: 10.1186/1471-2180-6-82
63. Losada, L, Ronning, CM, DeShazer, D, Woods, D, Fedorova, N, Kim, HS, et al. Continuing evolution of Burkholderia mallei through genome reduction and large-scale rearrangements. Genome Biol Evol. (2010) 2:102–16. doi: 10.1093/gbe/evq003
64. Price, EP, Sarovich, DS, Mayo, M, Tuanyok, A, Drees, KP, Kaestli, M, et al. Within-host evolution of Burkholderia pseudomallei over a twelve-year chronic carriage infection. MBio. (2013) 4:1–10. doi: 10.1128/mBio.00388-13
65. Armbruster, CR, Marshall, CW, Garber, AI, Melvin, JA, Zemke, AC, Moore, J, et al. Adaptation and genomic Erosion in fragmented Pseudomonas aeruginosa populations in the sinuses of people with cystic fibrosis. Cell Rep. (2021) 37:109829. doi: 10.1016/j.celrep.2021.109829
66. Wixon, J. Featured organism: reductive evolution in bacteria: Buchnera sp., rickettsia prowazekii and Mycobacterium leprae. Comp Funct Genomics. (2001) 2:44–8. doi: 10.1002/cfg.70
67. Brickman, TJ, Hanawa, T, Anderson, MT, Suhadolc, RJ, and Armstrong, SK. Differential expression of Bordetella pertussis Iron transport system genes during infection. Mol Microbiol. (2008) 70:3–14. doi: 10.1111/j.1365-2958.2008.06333.x
68. Choby, JE, Howard-Anderson, J, and Weiss, DS. Hypervirulent Klebsiella pneumoniae - clinical and molecular perspectives. J Intern Med. (2020) 287:283–300. doi: 10.1111/joim.13007
69. Kabir, A, Lamichhane, B, Habib, T, Adams, A, El-Sheikh Ali, H, Slovis, NM, et al. Antimicrobial resistance in equines: a growing threat to horse health and beyond-a comprehensive review. Antibiotics (Basel). (2024) 13:1–49. doi: 10.3390/antibiotics13080713
70. Korneenko, E, Rog, I, Chudinov, I, Lukina-Gronskaya, A, Kozyreva, A, Belyaletdinova, I, et al. Antibiotic resistance and viral co-infection in children diagnosed with pneumonia caused by Mycoplasma pneumoniae admitted to Russian hospitals during October 2023-February 2024. BMC Infect Dis. (2025) 25:363. doi: 10.1186/s12879-025-10712-0
71. Archambault, M, Harel, J, Goure, J, Tremblay, YD, and Jacques, M. Antimicrobial susceptibilities and resistance genes of Canadian isolates of Actinobacillus pleuropneumoniae. Microb Drug Resist. (2012) 18:198–206. doi: 10.1089/mdr.2011.0150
72. Beres, SB, Sesso, R, Pinto, SW, Hoe, NP, Porcella, SF, Deleo, FR, et al. Genome sequence of a Lancefield group C Streptococcus zooepidemicus strain causing epidemic nephritis: new information about an old disease. PLoS One. (2008) 3:e3026. doi: 10.1371/journal.pone.0003026
73. Waller, AS, Paillot, R, and Timoney, JF. Streptococcus equi: a pathogen restricted to one host. J Med Microbiol. (2011) 60:1231–40. doi: 10.1099/jmm.0.028233-0
74. Carraro, N, Rivard, N, Burrus, V, and Ceccarelli, D. Mobilizable Genomic Islands, different strategies for the dissemination of multidrug resistance and other adaptive traits. Mob Genet Elem. (2017) 7:1–6. doi: 10.1080/2159256X.2017.1304193
75. Delavat, F, Miyazaki, R, Carraro, N, Pradervand, N, and van der Meer, JR. The hidden life of integrative and conjugative elements. FEMS Microbiol Rev. (2017) 41:512–37. doi: 10.1093/femsre/fux0008
76. Estrada, AA, Gottschalk, M, Gebhart, CJ, and Marthaler, DG. Comparative analysis of Streptococcus suis genomes identifies novel candidate virulence-associated genes in north American isolates. Vet Res. (2022) 53:23. doi: 10.1186/s13567-022-01039-8
77. Bellanger, X, Payot, S, Leblond-Bourget, N, and Guedon, G. Conjugative and Mobilizable Genomic Islands in Bacteria: evolution and diversity. FEMS Microbiol Rev. (2014) 38:720–60. doi: 10.1111/1574-6976.12058
78. Xie, O, Zachreson, C, Tonkin-Hill, G, Price, DJ, Lacey, JA, Morris, JM, et al. Overlapping Streptococcus pyogenes and Streptococcus dysgalactiae subspecies equisimilis household transmission and Mobile genetic element exchange. Nat Commun. (2024) 15:3477. doi: 10.1038/s41467-024-47816-1
79. Tiwari, R, Artiushin, S, and Timoney, JF. P9, a temperate bacteriophage of Streptococcus equi. Int Congr Ser. (2006) 1289:165–8. doi: 10.1016/j.ics.2005.11.086
80. Beres, SB, Sylva, GL, Barbian, KD, Lei, B, Hoff, JS, Mammarella, ND, et al. Genome sequence of a serotype M3 strain of group a Streptococcus: phage-encoded toxins, the high-virulence phenotype, and clone emergence. Proc Natl Acad Sci USA. (2002) 99:10078–83. doi: 10.1073/pnas.152298499
81. Wilson, HJ, Dong, J, van Tonder, AJ, Ruis, C, Lefrancq, N, McGlennon, A, et al. Progressive evolution of Streptococcus equi from Streptococcus equi subsp. zooepidemicus and adaption to equine hosts. Microb Genom. (2025) 11:1–13. doi: 10.1099/mgen.0.001366
82. Rotinsulu, DA, Ewers, C, Kerner, K, Amrozi, A, Soejoedono, RD, Semmler, T, et al. Molecular features and antimicrobial susceptibilities of Streptococcus equi ssp. equi isolates from strangles cases in Indonesia. Vet Sci. (2023) 10:1–15. doi: 10.3390/vetsci10010049
83. Casjens, S. Prophages and bacterial genomics: what have we learned so far? Mol Microbiol. (2003) 49:277–300. doi: 10.1046/j.1365-2958.2003.03580.x
Keywords: horse, strangles, virulence-associated genes, antibiotic resistance genes, mobile genetic elements
Citation: He L, Khine NO, Song J, Loubière C and Butaye P (2025) Geographic diversity of the Streptococcus equi subsp. equi accessory genome: implications for vaccines and global surveillance. Front. Vet. Sci. 12:1721958. doi: 10.3389/fvets.2025.1721958
Edited by:
Roswitha Merle, Free University of Berlin, GermanyReviewed by:
Hongxuan He, Chinese Academy of Sciences (CAS), ChinaBin Xu, Jiangsu Academy of Agricultural Sciences, China
Copyright © 2025 He, Khine, Song, Loubière and Butaye. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lingyu He, bGluZ3l1aGUyLWNAbXkuY2l0eXUuZWR1Lmhr