 Jiayang Li1,2†
Jiayang Li1,2† Hong Zhang3†Songhua Long2†Wenting Li2
Hong Zhang3†Songhua Long2†Wenting Li2 Tuhong Wang2Jian Yu4Ying Zhou5Shuo Zou6
Tuhong Wang2Jian Yu4Ying Zhou5Shuo Zou6 Hongjian Zhu1*
Hongjian Zhu1* Jianping Xu7*
Jianping Xu7* Yi Cheng2,8*
Yi Cheng2,8*- 1Hunan Provincial Key Laboratory for Biology and Control of Plant Diseases and Insect Pests, Hunan Agricultural University, Changsha, Hunan, China
- 2Institute of Bast Fiber Crops and Center of Southern Economic Crops, Chinese Academy of Agricultural Sciences, Changsha, Hunan, China
- 3Shenzhen Noposion Crop Science Co., Ltd., Shenzhen, Guangdong, China
- 4Xishuangbanna Dai Autonomous Prefecture Tea Industry Development Service Center, Jinghong, Yunnan, China
- 5Institute of Agricultural Sciences of Xishuangbanna Prefecture of Yunnan Province, Jinghong, Yunnan, China
- 6Changsha Agricultural and Rural Bureau, Changsha, Hunan, China
- 7Department of Biology, McMaster University, Hamilton, ON, Canada
- 8Hunan Provincial Key Laboratory of the Traditional Chinese Medicine Agricultural Biogenomics, Changsha Medical University, Changsha, China
Endophytic bacteria within plant tissues play crucial roles in plant health, stress tolerance, and contribute to the metabolite diversity of host plants. Cannabis sativa L. is an economically significant plant, with industrial hemp (IH) and medicinal Cannabis (MC) being the two main cultivars. However, the composition and functional traits of their endophytic bacterial communities in roots and leaves are not well understood. In this study, DNA metabarcode sequencing were employed to compare the bacterial communities between IH and MC. Significant differences were observed in the root and leaf niches. IH roots were enriched with stress-tolerant bacteria, while MC roots showed higher levels of biofilm-forming bacteria. In leaves, differences were even more pronounced, particularly in the abundance of Gram-negative bacteria, potential pathogens, stress-tolerant bacteria, and biofilm-forming bacteria. PICRUSt2 functional predictions revealed differences in nitrogen metabolism and secondary metabolite biosynthesis pathways in different cultivars and niches, while FAPROTAX analysis highlighted variations in carbon, nitrogen, and sulfur cycling functions. These findings underscore the distinct roles of bacterial communities in regulating plant health, stress responses, and metabolic processes in different niches and cultivars, providing insights for improving cultivation practices and plant resilience.
1 Introduction
Cannabis sativa L. is a versatile crop widely utilized in fiber production (Panthapulakkal and Sain, 2007), pharmaceutical development (Atakan, 2012), and construction materials (Zimniewska, 2022). Cultivars of this species are commonly divided into two types, industrial hemp (IH) and medical Cannabis (MC). IH is primarily grown for its high yield of fiber and seed (Ahmed et al., 2022), while MC is distinguished by its active compounds, particularly cannabidiol (CBD), making it highly valuable in the medical (Crescente et al., 2023; Song et al., 2023) and recreational fields. Recent advances in the biological study of C. sativa L. and its symbiotic microorganisms (Islam et al., 2023; Law et al., 2020) have underscored the importance of endophytic bacteria in promoting plant growth, enhancing disease resistance, and augmenting environmental adaptability (Zhuang et al., 2021).
Endophytic bacteria are microbial communities that colonize the internal tissues (Guzmán-Guzmán et al., 2023) of plants without apparent disease symptoms (Lundberg et al., 2012). Endophytic bacteria engage in intricate symbiotic interactions with the host plants, where they contribute to plant health through multiple mechanisms. These include directly facilitating nutrient uptake by promoting nitrogen fixation and phosphate solubilization, as well as indirectly enhancing plant immunity (Xiong et al., 2024) and improving resilience to environmental stressors such as drought and pathogens (Balthazar et al., 2022). Pseudomonas in C. sativa L. have been reported to promote plant growth via phytohormone production and biocontrol activity. Additionally, these bacteria may influence the biosynthesis of key secondary metabolites such as tetrahydrocannabinol (THC) and cannabidiol (CBD) by modulating plant metabolic pathways (Balthazar et al., 2022).
The composition of endophytic bacteria is largely shape by host genotype (Liu et al., 2022; Wang Y.C. et al., 2022), environmental conditions (Santoyo, 2022), and tissue-specific niches (Zhang et al., 2022). Roots and leaves support distinct microbial assemblages due to differences in nutrient availability (Francioli et al., 2018; Potter et al., 2023), plant defense strategies (Lu et al., 2025), and microbial colonization routes (Zeng et al., 2023). Root-associated endophytes are primarily influenced by rhizodeposition (Xu et al., 2020), a process in which plant roots exude organic acids, flavonoids, and terpenoids that selectively recruit beneficial bacteria. For instance, Pseudomonas (Orozco-Mosqueda et al., 2018) enhances plant health through antimicrobial compound production, Bacillus (Igiehon and Babalola, 2018) promotes growth via phytohormone synthesis, and Rhizobium (Xiao et al., 2017) facilitates nitrogen fixation. In contrast, leaf endophytes are shaped by phyllosphere chemistry and environmental exposure (Chen et al., 2020). Microbial recruitment occurs through airborne deposition, insect-mediated transmission (Li et al., 2022), and cuticle adhesion (Sohrabi et al., 2023). Adaptations such as UV resistance and biofilm formation enable these microbes to persist in dynamic aerial environments. Despite growing interest, research on the composition and functional roles of endophytic bacterial communities in C. sativa L. remain limited, necessitating further studies to unravel the underlying mechanisms shaping these microbial communities (Chen et al., 2024; Xiao et al., 2023).
In Cannabis, several studies have explored the composition and ecological functions of associated microbial communities. Research has shown that bacterial communities colonizing the roots of industrial hemp contribute significantly to plant growth promotion (Taghinasab and Jabaji, 2020; Wei et al., 2021). Endophytic microorganisms can enhance plant growth by synthesizing phytohormones such as indole-3-acetic acid (IAA) (Etesami and Glick, 2024), facilitating nitrogen fixation (Yan et al., 2024), solubilizing phosphate (Chen et al., 2021), and producing siderophores (Schalk, 2025). In addition to promoting plant growth, these microbes enhance abiotic stress tolerance by producing osmoprotectants (e.g., proline, trehalose) (Anand et al., 2023), antioxidants (e.g., catalase, superoxide dismutase) (Hwang et al., 2022), and modulating stress-responsive gene expression (Jan et al., 2024). Furthermore, root- and leaf-associated microbiomes contribute to disease resistance via multiple strategies, including the synthesis of antifungal and antibacterial metabolites, the induction of systemic resistance (ISR) (Watts et al., 2023), and the priming of plant immune responses (Pal et al., 2022).
The metabolomic and physiological differences among Cannabis varieties play a crucial role in shaping their associated microbial communities (Lobato et al., 2024). The production of cannabinoids, terpenoids, and flavonoids can exert selective pressures on bacterial colonization and community composition. High-THC cultivars, for example, have been shown to host distinct microbial communities compared to fiber-type hemp, suggesting that secondary metabolites can influence microbial assembly (Ahmad et al., 2024). These findings highlight the complex interplay between Cannabis secondary metabolism and microbial ecology, with potential implications for optimizing plant health and productivity.
This study aims to explore the differences in the composition and function of endophytic bacterial communities between IH and MC, with the ultimate goal of potentially enhancing their respective desired phenotypes by manipulating their endophytic bacteria. The specific objectives are: (1) to compare the bacterial communities present in the roots and leaves of both industrial hemp and medicinal Cannabis through DNA metabarcode sequencing; (2) to identify bacterial taxa that show differential abundance between the two types, which could serve as potential biomarkers; and (3) to perform a predictive functional analysis using PICRUSt2 and FAPROTAX to reveal functional distinctions within the bacterial communities associated with each cultivar type. This study advances the understanding of plant-microbe interactions by revealing distinct microbial signatures in industrial hemp and medicinal Cannabis, emphasizing their ecological and functional significance. The findings lay a foundation for microbiome-based crop improvement strategies, including enhancing plant resilience, optimizing growth conditions, and implementing microbiome-informed cultivation practices tailored for industrial and medicinal applications.
2 Materials and methods
2.1 Sample collection
In this study, all root and leaf samples (root samples: 112, leaf samples: 79) were collected from eight locations across six provinces in China where Cannabis is cultivated (Table 1). All sampling was conducted during the flowering period of the hemp plants. Field samples were transported to the laboratory on dry ice to preserve their integrity. Upon arrival, the samples were rinsed 3–4 times with sterile water until the roots were clean, and the leaf surfaces were free of soil. The cleaned samples underwent surface sterilization by sequential immersion in 75% ethanol for 30 s, 1% sodium hypochlorite solution for 4 min, and 95% ethanol for 10 s, followed by three rinses with sterile distilled water (Li et al., 2023; Misaghi and Donndelinger, 1990). After sterilization, the samples were cut into small pieces and stored at −80°C for subsequent microbial DNA extraction.
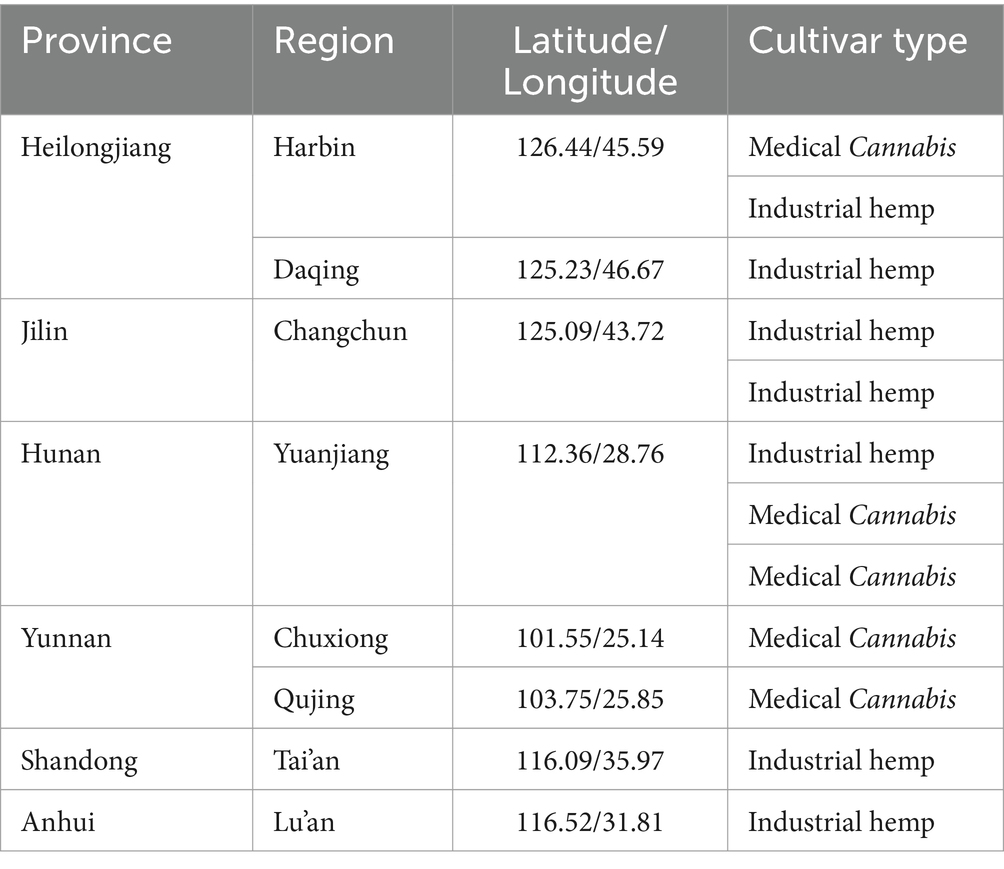
Table 1. Sample information.
2.2 DNA extraction, sequencing, and processing
The total genomic DNA of each endophytic microbial community was extracted following the protocol by the E.Z.N.A.® Soil DNA Kit (Omega Bio-tek, Norcross, GA, United States). The extracted DNA’s quality and quantity were evaluated by 1% agarose gel electrophoresis and a NanoDrop 2000 spectrophotometer (Thermo Scientific, United States). The primers 799F (5′-AACMGGATTAGATAC CCKG-3′) and 1392R (5′-ACGGGCGGTGTGTRC-3′) were selected for the first round of PCR amplification of the V5–V7 variable region, and the primers 799F (5′-AACMGGATTAGATACCCKG-3′) and 1193R (5′-ACGTCATCCCCACCTTCC-3′) were selected for the second round of PCR amplification of the V5–V7 variable region. Library construction of the purified PCR products was performed using the NEXTFLEX Rapid DNA-Seq Kit, following these steps: (1) adapter ligation, (2) bead-based selection to remove self-ligated adapter fragments, (3) PCR amplification to enrich the library template, and (4) bead-based purification to obtain the final library. High-throughput sequencing was then conducted on the Illumina NextSeq 2000 platform (Shanghai Majorbio Bio-Pharm Technology Co., Ltd.) (Bouchez et al., 2016). The raw sequencing data have been submitted to the NCBI Sequence Read Archive (Re: PRJNA1169634, Le: PRJNA1172293).
2.3 Bioinformatics analysis
The raw reads were quality-controlled using fastp (version 0.19.6) by trimming low-quality bases (Q < 20) with a sliding window of 50 bp and removing reads shorter than 50 bp or containing N bases. The processed paired-end reads were then merged using FLASH software (version 1.2.11) with a minimum overlap length of 10 bp and a maximum mismatch ratio of 0.2. The DADA2 plugin within the Qiime2 pipeline was employed to denoise the quality-controlled and merged sequences. Sequences identified as chloroplast or mitochondrial were excluded from all samples. To minimize the influence of sequencing depth on subsequent Alpha and Beta diversity analyses, the sequencing depth was rarefied to maintain an average sequence coverage of 98.00% for each sample. Taxonomic classification of OTUs was carried out using the Naive Bayes classifier, based on the Silva 16S rRNA gene database (v. 138) within Qiime2 (Edgar, 2013).
2.4 Statistical analyses
All statistical analysis was conducted using the Shanghai MajorBio Cloud Platform.1 The functional potential of the microbial communities was predicted using PICRUSt2 and FAPROTAX. For diversity analysis, alpha diversity indices (Sobs, Shannon, ACE) were calculated using Mothur (version 1.30.1), and beta diversity was assessed using the Bray-Curtis distance metric, followed by PCoA analysis in QIIME (version 1.17). Significant differences in community composition between groups were evaluated using the ANOSIM group difference test. To identify significant differences in dominant bacterial genera between groups, the Wilcoxon rank-sum test was applied. Additionally, LEfSe (LDA score > 3.5) was used to identify biomarker taxa differentiating the groups. Source tracking analysis was conducted using FEAST (Shenhav et al., 2019).
3 Results
3.1 Endophytic bacterial differences within and between medicinal Cannabis and industrial hemp from Harbin
To investigate the differences in endophytic microbial communities in different hemp varieties, we first compared the root (Re) and leaf (Le) endophytic bacterial communities of MC (HRBA) and IH (HRBB) cultivated in the same geographic area, around the city of Harbin in Northeast China. The results showed that the diversity of endophytic bacteria in the roots was significantly higher than that in the leaves. Overall, while there was limited difference in bacterial richness and diversity between HRBA_Re and HRBB_Re, principal coordinate analysis (PCoA) revealed significant differences between the communities of HRBA_Re and HRBB_Re (p < 0.05). In contrast, while the Shannon and ACE indices showed significant differences between HRBA_Le and HRBB_Le (Supplementary Table S1). The community structure of HRBA_Le and HRBB_Le were very similar to each other (Figure 1A).
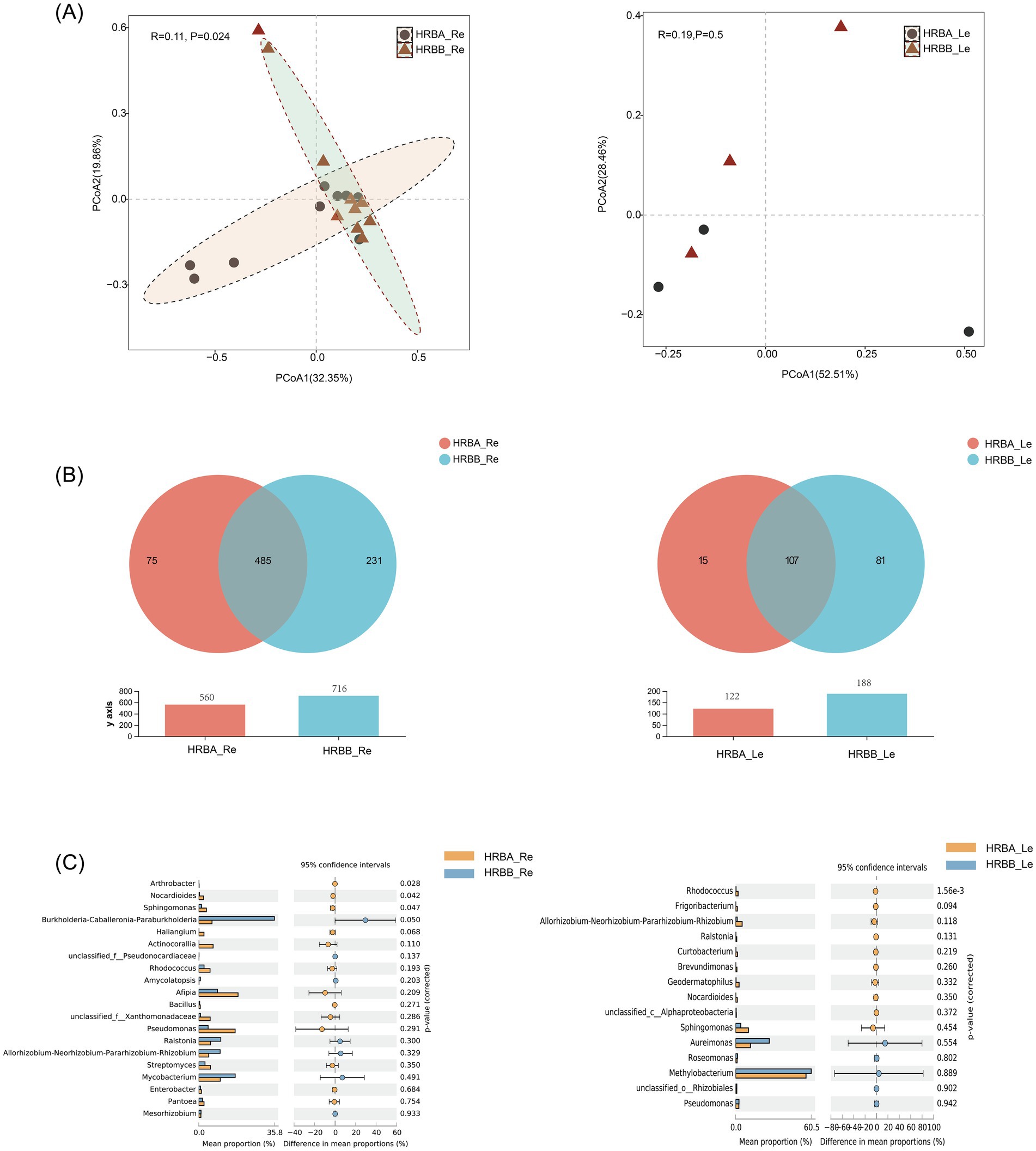
Figure 1. Endophytic bacterial community diversity of HRBA and HRBB. (A) Principal component analysis (PCoA) of root and leaf endophytic bacterial communities. (B) Venn diagram of endophytic bacteria. (C) Differential analysis of endophytic bacterial genera.
Venn diagram analysis indicated that HRBA_Re and HRBB_Re shared 485 OTUs, with 75 OTUs unique to HRBA_Re and 231 OTUs unique to HRBB_Re (Figure 1B). The Wilcoxon rank-sum test revealed significant differences at the genus level, showing that the relative abundances of Arthrobacter, Nocardioides, and Sphingomonas were significantly higher in HRBA_Re compared to HRBB_Re (Figure 1C). For the endophytic bacterial communities in leaves, HRBA_Le and HRBB_Le shared 107 OTUs, with 15 OTUs unique to HRBA_Le and 81 OTUs unique to HRBB_Le (Figure 1B). Additionally, the relative abundance of Rhodococcus was significantly higher in HRBA_Le compared to HRBB_Le (Figure 1C). Together, these results revealed both similarities and differences in endophytic bacterial communities between IH and MC in both roots and leaves.
3.2 Diversity of endophytic bacteria in industrial hemp and medicinal Cannabis across China
We further compared the endophytic bacterial communities between IH and MC cultivars across China. The diversity and richness of endophytic bacteria in the roots and leaves were assessed using Sobs, Shannon, and Ace indices. Overall, the results showed that the diversity and richness of endophytic bacteria were significantly higher in roots than in leaves (Table 2, p < 0.05). However, no significant differences were observed between IH_Re and MC_Re. Between IH_Le and MC_Le, the Shannon index was significantly higher in IH_Le than in MC_Le, whereas the ACE index exhibited the opposite trend.
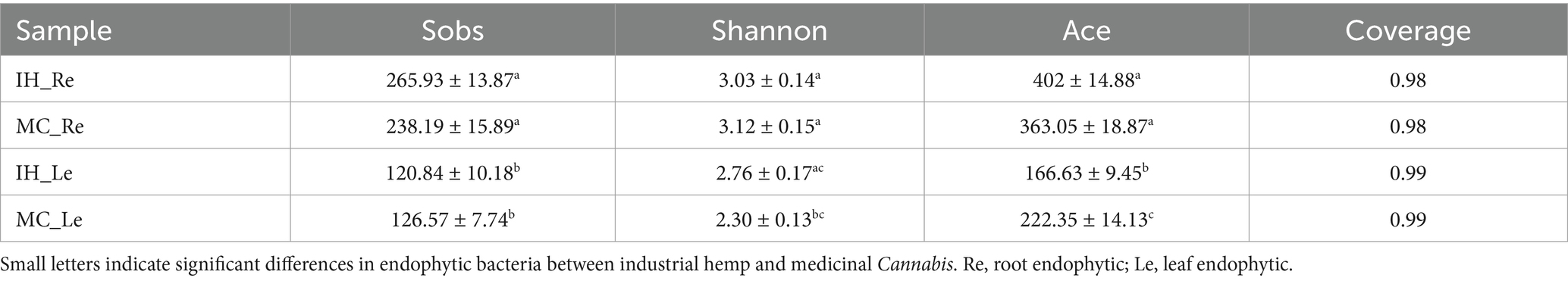
Table 2. Diversity indices of endophytic bacteria in industrial hemp (IH) and medicinal Cannabis (MC).
Additionally, at the OTU level, partial least squares discriminant analysis (PLS-DA) and PCoA were used to evaluate the similarity of the endophytic bacterial communities. The results showed a significant difference between the endophytic bacterial communities of MC and IH (Figures 2A,B, p < 0.01), with clear separation in their clustering patterns (Figures 2C,D). In addition, the differences in endophytic bacteria were more pronounced in the leaves (R = 0.1915) than in the roots (R = 0.0528).
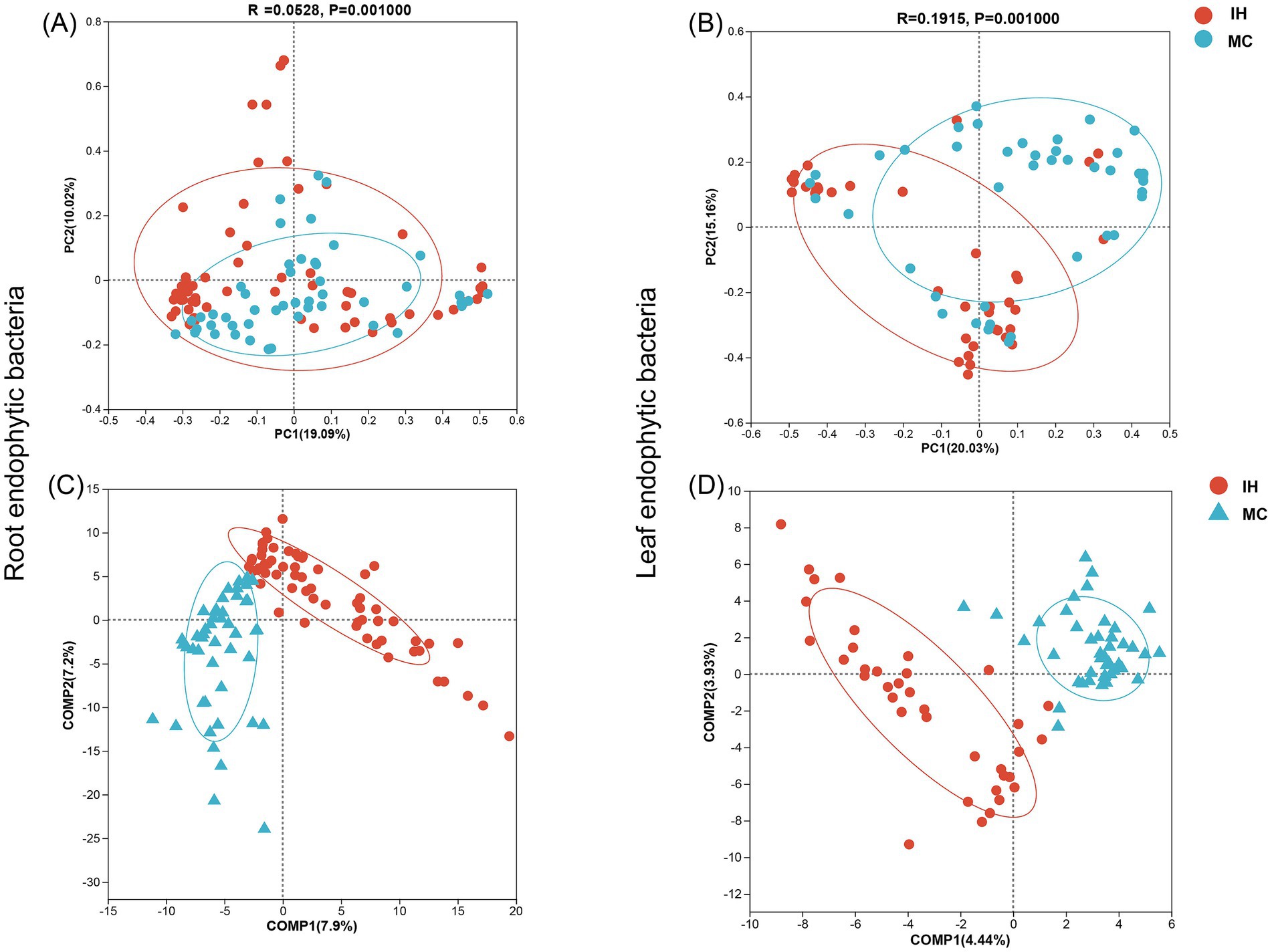
Figure 2. Endophytic bacterial community diversity of industrial hemp and medicinal Cannabis. (A,B) Principal component analysis (PCoA) of root and leaf endophytic bacterial communities. (C,D) Partial least squares discriminant analysis (PLS-DA) of root and leaf endophytic bacterial communities.
The findings indicate that while the endophytic bacterial communities in the roots of IH and MC cultivars exhibit minimal differences, significant variations were observed in the bacterial communities of the leaves, suggesting cultivar-specific influences on bacterial composition in leaf tissues.
3.3 Composition of the endophytic bacteria in industrial hemp and medical Cannabis
The dominant endophytic bacterial communities in the roots and leaves of industrial hemp (IH) and medicinal Cannabis (MC) were analyzed. At the class level (Figures 3A,D), Actinobacteria, Gammaproteobacteria, Alphaproteobacteria, and Bacilli are the dominant endophytic bacterial classes (relative abundance > 1%) in both IH and MC. Additionally, IH_Re contains another dominant class, Polyangia (3.68%). MC_Le has a higher relative abundance of Clostridia (1.30%) and Bacteroidia (1.01%).
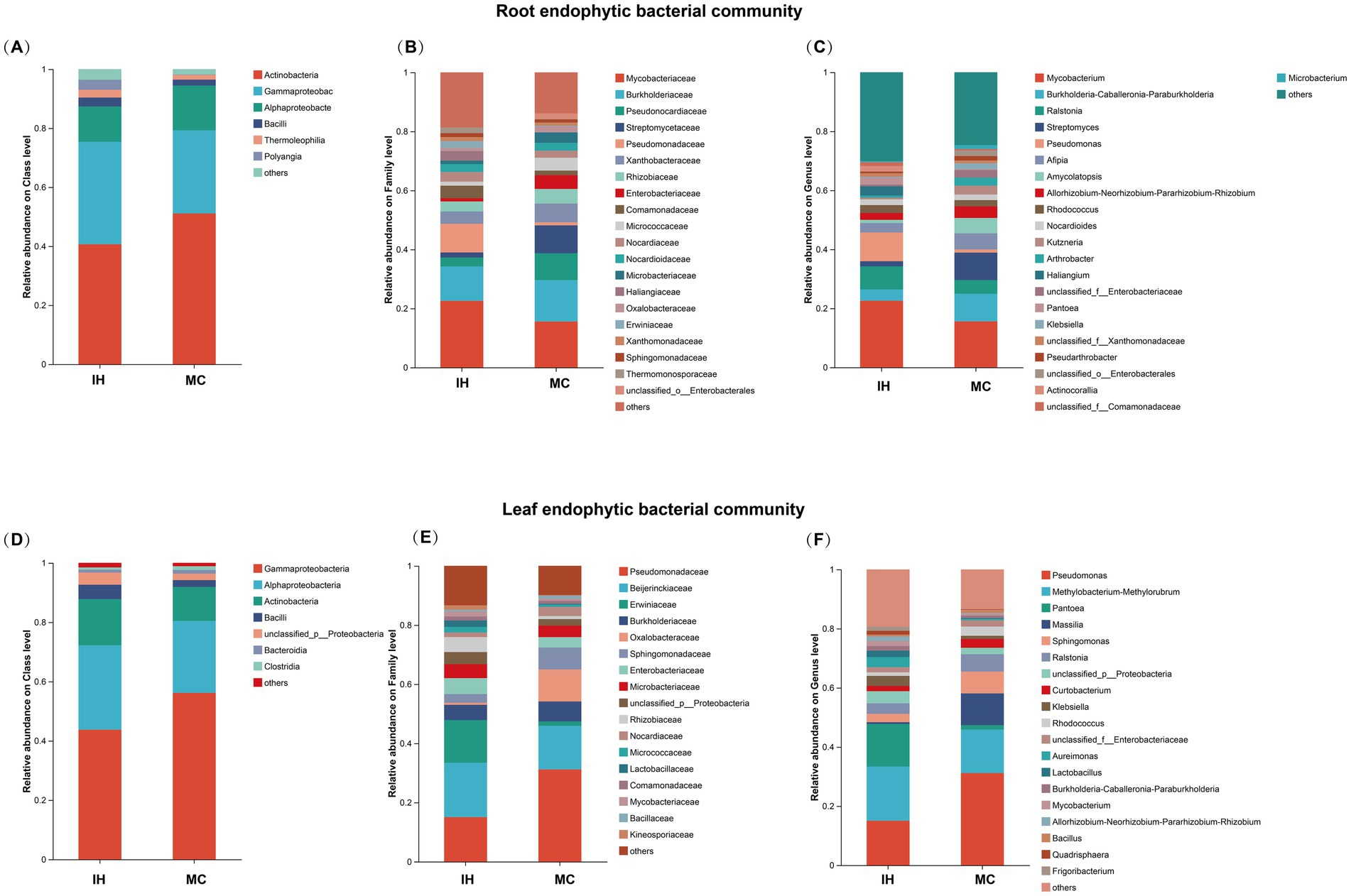
Figure 3. Endophytic bacterial community compositions of industrial hemp and medicinal Cannabis. (A–C) The relative abundance of dominant root endophytic bacteria. (D–F) The relative abundance of dominant leaf endophytic bacteria.
At the family level (Figure 3B), the dominant families in the root endophytic communities of IH_Re and MC_Re revealed notable differences in their relative abundances. For example, Mycobacteriaceae was more abundant in IH_Re (22.57%) than in MC_Re (15.57%), whereas Burkholderiaceae exhibited a higher relative abundance in MC_Re (14.05%) compared to IH_Re (11.68%). Furthermore, Pseudonocardiaceae (IH: 2.97%, MC: 9.08%) and Streptomycetaceae (IH: 1.75%, MC: 9.46%) were more prominent in MC_Re, whereas Pseudomonadaceae was significantly more abundant in IH_Re (9.74%) than in MC_Re (1.04%). Similarly, the dominant families in the leaf endophytic communities also revealed distinct distribution patterns (Figure 3E). For instance, Pseudomonadaceae had a higher relative abundance in MC_Le (31.18%) than in IH_Le (15.04%), whereas Erwiniaceae was significantly more abundant in IH_Le (14.41%) compared to MC_Le (1.50%). Moreover, Oxalobacteraceae exhibited a higher relative abundance in MC_Le (10.79%) than in IH_Le (0.68%).
At the genus level, IH and MC have 12 and 15 predominant root endophytic bacterial genera, respectively (Figure 3C), with specific relative abundances detailed in Supplementary Table S2. Notable differences in the composition of predominant endophytic bacterial genera are observed between IH and MC. For instance, Haliangium and Actinocorallia are predominant in IH, while Burkholderia-Caballeronia-Paraburkholderia, Streptomyces, Klebsiella, Amycolatopsis, Arthrobacter, Kutzneria, Pseudarthrobacter and Microbacterium are dominant in MC.
Additionally, 15 and 10 predominant leaf endophytic bacterial genera were identified in IH and MC (Figure 3F), respectively. The composition of predominant bacterial genera in the leaves also differs markedly, with Pseudomonas, Pantoea, and Massilia being the representative genera (Supplementary Table S2).
The observed differences in bacterial compositions between IH and MC suggest that cultivar plays a significant role in shaping the structure of endophytic bacterial communities in both roots and leaves.
3.4 Shared and unique bacteria in industrial hemp and medical Cannabis
The comparison of endophytic bacterial communities in the roots and leaves of industrial hemp (IH) and medicinal Cannabis (MC) revealed distinct patterns of bacterial diversity and potential biomarkers. The Venn diagram showed that IH_Re and MC_Re shared 938 OTUs, while 55 OTUs were unique to IH_Re and 25 OTUs were unique to MC_Re (Figure 4A). The dominant bacterial genera exhibited a degree of host preference. Analysis of the top 10 dominant endophytic genera using the Wilcoxon rank-sum test revealed that the relative abundances of Pseudomonas (p < 0.05) and Haliangium (p < 0.001) were significantly higher in IH_Re than in MC_Re (Figure 4B). Furthermore, linear discriminant analysis effect size (LEfSe, LDA > 3.5) and random forest (RF) were used to identify differential bacteria between industrial hemp and medical Cannabis roots. Combining the results of both methods (Figures 4C,D), five biomarker taxa were identified: IH_Re: Actinocorallia, BD1-7 clade; MC_Re: Streptomyces, Amycolatopsis, Afipia.
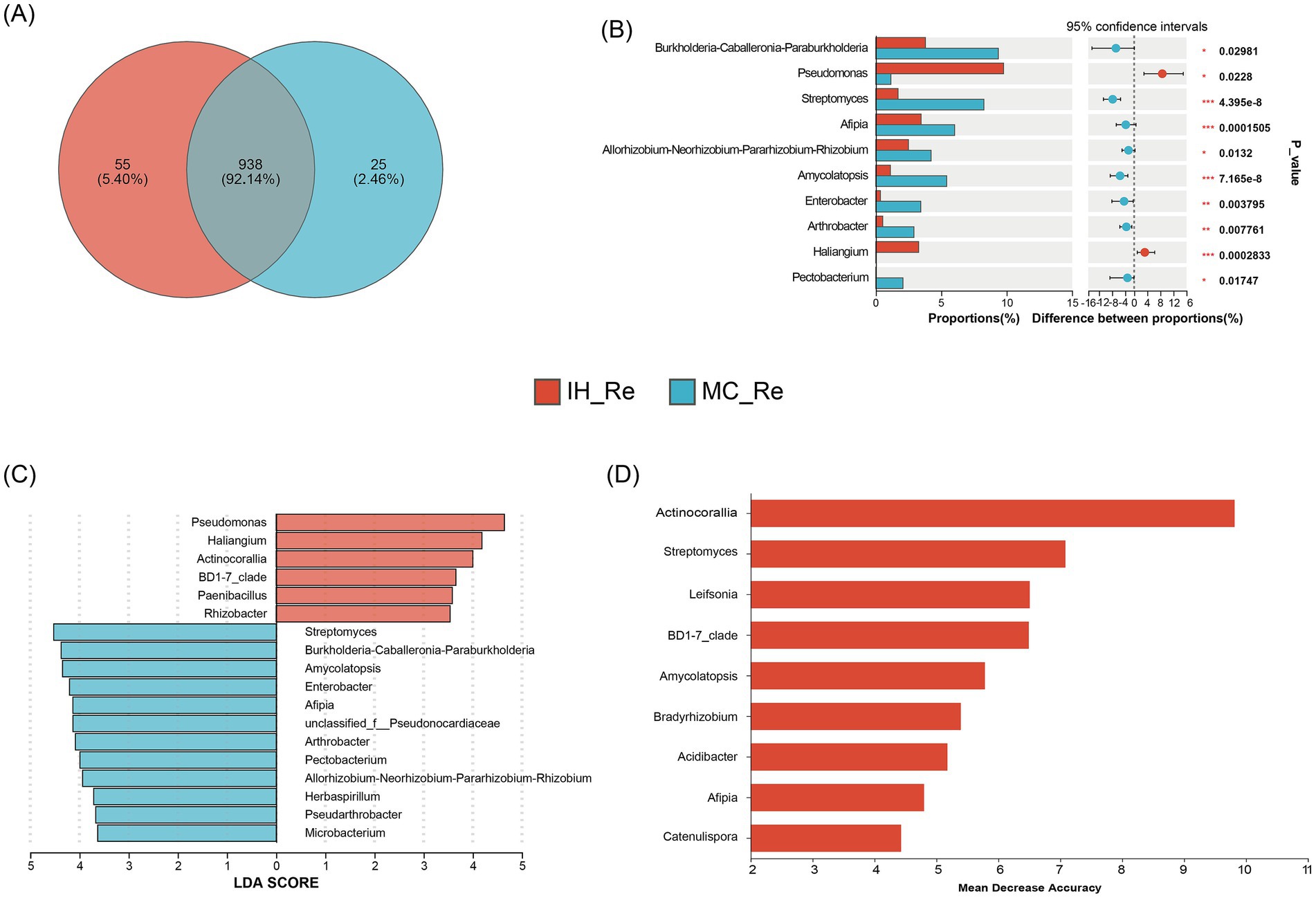
Figure 4. Differential root endophytic bacteria identification between industrial hemp and medical Cannabis. (A) Venn diagram of root endophytic bacteria. (B) Differential analysis of root endophytic bacterial genera. *0.01 < p ≤ 0.05; **0.001 < p ≤ 0.01; ***p ≤ 0.001. (C) Graphical summary at the genus in group sample type of biomarkers by LEfSe. (D) Significant features identified by random forest.
In the leaves of industrial hemp and medical Cannabis, 292 OTUs were shared, with 3 unique OTUs identified in IH_Le and 20 unique OTUs in MC_Le (Figure 5A). A significance analysis of the top 10 dominant endophytic bacterial genera in the leaves showed that five of the 10 genera exhibited significant differences (Figure 5B). For instance, the relative abundance of Pantoea was significantly lower in MC compared to IH, while the relative abundances of the remaining genera were significantly higher in MC than in IH. Additionally, five potential biomarker taxa were identified in the comparison between IH_Le and MC_Le: IH_Le: Frigoribacterium, Allorhizobium-Neorhizobium-Pararhizobium-Rhizobium, Klenkia; MC_Le: Rhodococcus, Ralstonia (Figures 5C,D).
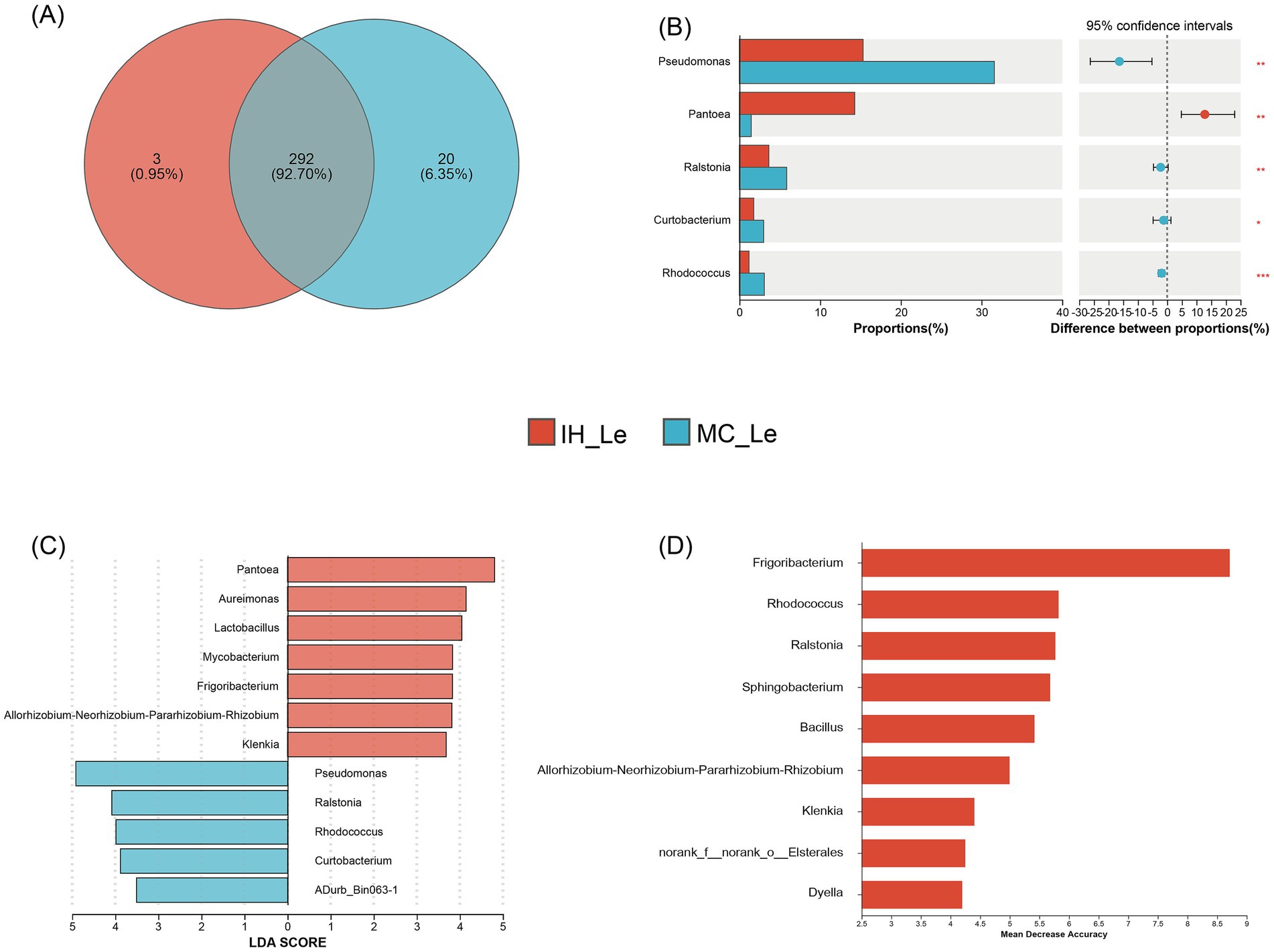
Figure 5. Differential leaf endophytic bacteria of industrial hemp and medical Cannabis. (A) Venn diagram of leaf endophytic bacteria. (B) Differential analysis of leaf endophytic bacterial genera. *0.01 < p ≤ 0.05; **0.001 < p ≤ 0.01; ***p ≤ 0.001. (C) Graphical summary at the genus in group sample type of biomarkers by LEfSe. (D) Significant features identified by random forest.
Additionally, source tracking analysis was performed on the endophytic bacteria of IH and MC. In IH, 44.01% of the endophytic bacteria in the leaves were shown to be likely originated from the roots, while the remaining 55.99% was classified as unknown. In contrast, only 17.98% of the endophytic bacteria in the leaves of MC were inferred as derived from the roots, with the remaining 82.02% being categorized as unknown (Figure 6).
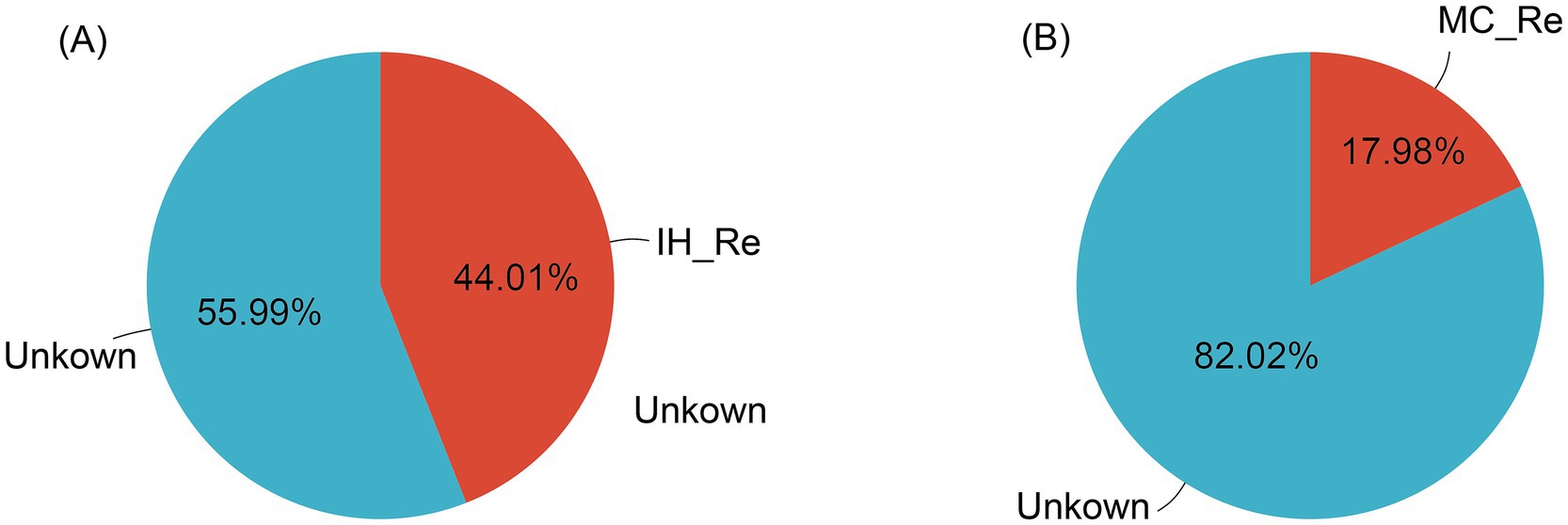
Figure 6. Source tracking analysis of leaf endophytic bacteria. (A) Industrial hemp. (B) Medicinal Cannabis.
These results highlight the cultivar-specific characteristics of endophytic bacterial communities, with significant differences in both root and leaf microbiota, suggesting varying ecological features and host-specific influences on bacterial composition.
3.5 Function prediction of the endophytic bacteria in industrial hemp and medical Cannabis
Functional prediction analyses were conducted using PICRUSt2 and FAPROTAX to compare the functional microbial communities across the root and leaf niches, as well as between the IH and MC cultivars. The Wilcoxon rank-sum test was used to identify significant differences in functional categories. The results revealed distinct metabolic pathways between the two niches, with notable differences in key metabolic processes.
In the Re (Figure 7A), pathways related to the citric acid cycle (TCA cycle), fatty acid degradation, and oxidative phosphorylation were more prevalent, indicating enhanced energy metabolism and oxidative capacity. In contrast, pathways involved in secondary metabolite biosynthesis (such as sulfur metabolism, porphyrin and purine metabolism) were more abundant in the Le, suggesting a higher abundance of microbial communities associated with environmental adaptability and secondary metabolite production.
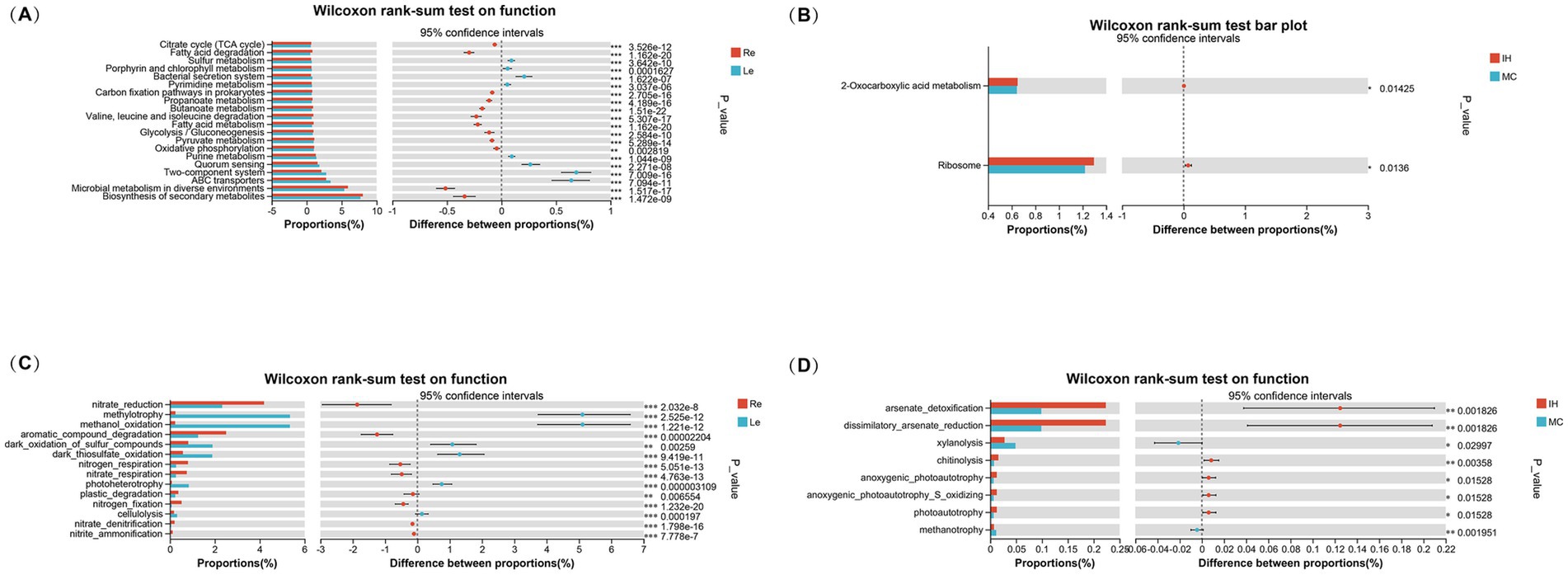
Figure 7. Functional prediction of endophytic bacteria in the roots and leaves of industrial hemp and medicinal Cannabis. (A) Functional prediction of endophytic bacteria in the roots and leaves based on PICRUSt2. (B) Functional prediction of endophytic bacteria in different cultivars based on PICRUSt2. (C) Functional prediction of endophytic bacteria in the roots and leaves based on FAPROTAX. (D) Functional prediction of endophytic bacteria in different cultivars based on FAPROTAX.
When comparing the IH and MC cultivars, significant differences were observed in only two functional categories: ribosome function and 2-oxocarboxylic acid metabolism among the top 30 functions (Figure 7B).
FAPROTAX-based predictions further examined the sulfur, nitrogen, hydrogen, and carbon cycles. Functional differences were observed between the ecological niches (Figure 7C). Eight plant growth-related functions, such as nitrate reduction, aromatic compound degradation, nitrogen respiration, nitrate respiration, plastic degradation, nitrogen fixation, nitrate denitrification, and nitrite ammonification, were significantly enriched in the roots. In contrast, the leaves were enriched in functions related to methylotrophy, methanol oxidation, photoheterotrophy, dark thiosulfate oxidation, dark sulfur compound oxidation, and cellulolysis.
Additional revealing eight significant functional differences between the cultivars. IH was enriched in microbial communities associated with arsenate detoxification and dissimilatory arsenate reduction. Additionally, chitinolysis, anoxygenic photoautotrophy, anoxygenic_photoautotrophy_S_oxidizing, and photoautotrophy were significantly more abundant in IH compared to MC. Conversely, xylanolysis and methanotrophy were significantly enriched in MC (Figure 7D).
In summary, the result highlights the complex and dynamic nature of plant-associated microbiomes, with functional differences driven by both plant genotype and ecological niches. Understanding these functional pathways can provide insights into how microbial communities contribute to plant growth, stress responses, and overall plant health.
4 Discussion
4.1 Factors shaping the Cannabis endophytic bacterial composition
The plant microbiome is shaped by multiple biotic and abiotic factors, including soil properties (Sokol et al., 2022), plant cultivars (Matsumoto et al., 2021), environmental conditions (Zhou et al., 2024), and root exudates (Sasse et al., 2018). Soil microbiomes serve as the primary reservoir from which plants recruit their endophytic and rhizosphere-associated microorganisms (Berendsen et al., 2012). Differences in bacterial communities between Industrial hemp (IH) and Medical Cannabis (MC) may partially stem from the distinct soil environment in which these plants were cultivated. Soil properties such as Ph (Zhang et al., 2025), nutrient availability (Yan et al., 2025) and organic matter content (Yan et al., 2023) can influence microbial community composition and functional potential, thereby affecting the microbial taxa that ultimately colonize plant tissues (Philippot et al., 2024).
Plant cultivar plays a critical role in microbial selection, particularly in shaping root associated microbiomes. Previous studies have demonstrated that different cultivars can selectively enrich specific microbial taxa due to variations in root exudates (Yue et al., 2023) and plant immune responses (Wang Y. Y. et al., 2022). The differences observed in the bacteria of IH and MC grown in the same region suggest that host genotype influences microbial recruitment beyond the soil microbiome’s initial composition.
4.2 Soil influence on bacterial community composition
Our findings indicate that differences in bacterial community composition between IH and MC may, in part, be driven by the distinct soil microbiomes associated with their respective cultivation environments. The variation in dominant bacterial families between IH_Re and MC_Re suggests that microbial recruitment from the soil is a major factor shaping the endophytic microbiome. Soil properties can exert selective pressures on microbial populations, affecting their colonization potential in plant tissues (Duan et al., 2023; Su et al., 2023; Wang et al., 2023).
While the direct impact of soil on leaf endophytic microbiomes is less pronounced, source tracking analysis revealed that a proportion of the endophytic bacteria in leaves originated from the roots, indicating a potential indirect influence. The significantly higher proportion of root-derived bacteria in IH_Le compared to MC_Le suggests that industrial hemp may facilitate more efficient microbial transfer from roots to leaves. This difference could be attributed to host-specific factors such as xylem microbiome transport (Hamaoka et al., 2022), phyllosphere conditions, or systemic signaling mechanisms that influence microbial colonization (Copeland et al., 2015). Future studies incorporating soil physicochemical analysis, metagenomic sequencing, and microbial tracking at multiple plant compartments would provide further insights into the extent of soil influence on the Cannabis microbiome, including its indirect effects on aboveground tissues.
4.3 Cultivar—dependent bacterial recruitment in a shared environment
In our study, some IH and MC were cultivated in the same geographic region. However, our results revealed significant differences in their endophytic bacterial communities, particularly in the roots. This suggests that host genetic factors play a crucial role in shaping microbial recruitment beyond the influence of the surrounding soil. The observed differences may stem from variations in root exudate composition (Pang et al., 2021), plant immune responses (Castrillo et al., 2017), and physiological traits (Carrión et al., 2019) unique to each cultivar. Root exudates serve as key mediators in microbiome assembly, selectively attracting or repelling specific bacterial taxa (Ji et al., 2023). Previous studies have demonstrated that plant genotypes can influence the quantity and composition of exuded organic compounds, thereby shaping distinct microbial communities even when plants share the same soil (He et al., 2024; Zhang et al., 2019).
In our study, HRBA_Re and HRBB_Re exhibited significant differences in their bacterial communities despite comparable levels of alpha diversity. The enrichment of genera such as Arthrobacter, Nocardioides, and Sphingomonas in HRBA_Re suggests that IH may favor the recruitment of bacteria with potential roles in stress tolerance, biodegradation, or nutrient cycling. Conversely, the significant differentiation in OTUs between HRBA_Re and HRBB_Re indicates that MC recruits a distinct set of bacterial, possibly due to differences in root physiology or interactions with soil microbes.
Interestingly, the leaf microbiota of IH and MC were more similar in composition, with fewer significant differences in dominant taxa. This suggests that while roots play a primary role in shaping the initial endophytic community through direct interactions with the soil, the microbial assembly in leaves may be influenced by factors such as phyllosphere environmental conditions or systemic plant signaling, which could lead to greater homogenization across cultivars. The significantly higher abundance of Rhodococcus in HRBA_Le may reflect differences in host physiology or specific plant-microbe interactions that favor certain taxa in the phyllosphere.
Overall, these findings highlight the complex interplay between plant genetics and environmental factors in microbiome assembly. While soil provides a microbial reservoir, host-specific factors ultimately exert selective pressures that shape the final composition of the endophytic community.
4.4 Functional adaptations of endophytic bacterial communities in industrial hemp and medicinal Cannabis
The functional adaptations of endophytic bacterial communities in industrial hemp (IH) and medicinal Cannabis (MC) are shaped by both plant genotype (Hawkes et al., 2021), plant physiology and niche factors (Van Bel et al., 2021). The observed functional differences between IH and MC reflect the divergent ecological niches these cultivars occupy and their distinct physiological roles. Industrial hemp, generally cultivated for its fiber and seeds, often grows in a wider variety of environments, including soils with higher stress levels (e.g., heavy metal contamination or nutrient-poor conditions). This may explain the enrichment of microbial pathways related to arsenate detoxification and dissimilatory arsenate reduction in IH. Such pathways are known to be crucial for the survival of microbes in contaminated soils, suggesting that IH-associated microbiota play a critical role in mitigating environmental stressors. Studies have shown that plants growing in polluted environments often harbor microbiomes with functional traits that enhance stress tolerance (Singh et al., 2023) and pollutant degradation (Tian et al., 2024). In contrast, MC, known for its high resin content and therapeutic properties, shows a functional enrichment in carbon metabolism pathways such as xylanolysis and methanotrophy, which may be linked to the production of bioactive secondary metabolites like THC and CBD. This suggests that MC’s microbiome may support the plant’s metabolic needs for secondary metabolite synthesis, which is vital for its medicinal value. This functional divergence between IH and MC underlines the adaptive role of microbial communities in supporting plant health and productivity in different growing conditions.
The functional differences observed between the root and leaf niches further emphasize that ecological factors drive microbial community specialization. The root was notably enriched in nitrogen-related functions such as nitrate reduction, nitrogen fixation, and nitrate denitrification, all of which are essential for nutrient acquisition and establishing beneficial plant-microbe symbioses in the root zone. These findings align with studies highlighting the role of root-associated microbiota in nitrogen cycling, critical for plant growth and productivity (Pang and Xu, 2024). Conversely, the leaf exhibited a higher abundance of pathways involved in secondary metabolite biosynthesis, including methylotrophy and methanol oxidation, which are linked to the plant’s defense mechanisms and adaptation to oxidative stress (Liu et al., 2023). This suggests that while root microbiota primarily facilitate nutrient cycling and plant growth, leaf microbiota are more involved in environmental stress response, pathogen defense, and plant immunity regulation. These results emphasize the dual roles of microbial communities in plant growth and defense, and highlight the importance of both plant genotype and ecological niches in shaping microbiome functionality.
4.5 Limitations of functional predictions from 16S rRNA data
Although 16S rRNA gene sequencing is a valuable tool for characterizing microbial community structure, its application in functional predictions has several limitations (Curry et al., 2022). 16S data primarily provide taxonomic information, and functional predictions are based on known genome databases. However, 16S rRNA genes are highly conserved across species, limiting their ability to capture functional diversity within microbial genomes (Pan et al., 2023). This limitation can lead to inaccurate or incomplete functional predictions, particularly for species with limited genomic data. Additionally, 16S data do not account for non-coding regions or functional genes critical to microbial metabolism and ecological interactions, which are essential for a comprehensive understanding of microbial roles.
Moreover, functional predictions derived from 16S rRNA data are constrained by the completeness of current databases and the challenge of capturing dynamic, environment-specific functions. Microbial community functions are influenced not only by taxonomy but also by factors such as environmental conditions, gene expression, and plant genotype. Therefore, while 16S data can reveal microbial community composition, it often fails to provide a complete picture of functional activity, particularly in specific ecological niches. To overcome these limitations, integrating other high-throughput omics techniques, such as metagenomics or transcriptomics, is essential for generating more accurate and comprehensive functional profiles.
5 Conclusion
This study enhances our understanding of plant-microbe interactions by identifying distinct microbial signatures in industrial hemp (IH) and medicinal Cannabis (MC), highlighting their ecological and functional implications. Significant differences were observed between the root and leaf microbiomes, emphasizing the crucial roles of both plant genotype and ecological niche in shaping microbial communities. In the roots, Actinocorallia and the BD1-7 clade were significantly enriched in IH, while MC roots had a higher abundance of Streptomyces, Amycolatopsis, and Afipia. In the leaves, key biomarker taxa such as Allorhizobium-Neorhizobium-Pararhizobium-Rhizobium, Frigoribacterium, Klenkia were enriched in IH, whereas Rhodococcus and Ralstonia were more abundant in MC. Functional prediction analysis revealed significant differences in metabolic pathways related to nitrogen metabolism, secondary metabolite biosynthesis, and stress tolerance between IH and MC. These findings suggest that microbes in roots play a key role in nitrogen cycling and stress management, while leaves bacteria likely influence the production of bioactive secondary metabolites crucial for the plant’s medicinal properties.
These findings underscore the importance of both plant genotype and ecological niche in shaping microbial communities. While plant genotype has long been recognized as a key driver of microbiome structure, this study demonstrates that environmental factors—such as soil type, nutrient availability, and ecological niche (root vs. leaf)—are equally critical in determining microbial community composition and function. This research provides a more comprehensive understanding of plant-microbe interactions, highlighting how microbial communities in different niches contribute to nutrient cycling, stress tolerance, and secondary metabolite production. The future studies integrating metagenomics and metabolomics will be valuable in further elucidating the molecular mechanisms underlying these plant-microbiome interactions.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: https://www.ncbi.nlm.nih.gov/, PRJNA1169634 and https://www.ncbi.nlm.nih.gov/, PRJNA1172293.
Author contributions
JL: Software, Writing – original draft. HZha: Data curation, Writing – review & editing. SL: Investigation, Writing – review & editing. WL: Methodology, Writing – review & editing. TW: Investigation, Writing – review & editing. JY: Investigation, Writing – review & editing. YZ: Investigation, Writing – review & editing. SZ: Investigation, Methodology, Writing – review & editing. HZhu: Data curation, Writing – review & editing. JX: Data curation, Writing – review & editing. YC: Funding acquisition, Investigation, Methodology, Supervision, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported in part by the Agricultural Science and Technology Innovation Program (CAAS-ASTIP-BFC), the Hunan Provincial Key Laboratory of the TCM Agricultural Biogenomics, and the Natural Science Foundation of Hunan Province (2021JJ40648), and the Shenzhen Sustainable Development Special Project (KCXFZ20230731100700001).
Conflict of interest
HZha is employed by Shenzhen Noposion Crop Science Co., Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Generative AI statement
The authors declare that no Gen AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/ARTICLES/10.3389/fmicb.2025.1524703/full#supplementary-material
SUPPLEMENTARY FIGURE S1 | The rarefaction curve of industrial hemp (IH) and medicinal Cannabis (MC).
Footnotes
References
Ahmad, W., Coffman, L., Ray, R. L., Balan, V., Weerasooriya, A., and Khan, A. L. (2024). Microbiome diversity and variations in industrial hemp genotypes. Sci. Rep. 14:29560. doi: 10.1038/s41598-024-79192-7
Ahmed, A., Islam, M. Z., Mahmud, M. S., Sarker, M. E., and Islam, M. R. (2022). Hemp as a potential raw material toward a sustainable world: a review. Heliyon 8:e08753. doi: 10.1016/j.heliyon.2022.e08753
Anand, U., Pal, T., Yadav, N., Singh, V. K., Tripathi, V., Choudhary, K. K., et al. (2023). Current scenario and future prospects of Endophytic microbes: promising candidates for abiotic and biotic stress Management for Agricultural and Environmental Sustainability. Microb. Ecol. 86, 1455–1486. doi: 10.1007/s00248-023-02190-1
Atakan, Z. (2012). Cannabis, a complex plant: different compounds and different effects on individuals. Ther Adv Psychopharmacol 2, 241–254. doi: 10.1177/2045125312457586
Balthazar, C., Joly, D. L., and Filion, M. (2022). Exploiting beneficial Pseudomonas spp. for Cannabis production. Front. Microbiol. 12:833172. doi: 10.3389/fmicb.2021.833172
Berendsen, R. L., Pieterse, C. M. J., and Bakker, P. (2012). The rhizosphere microbiome and plant health. Trends Plant Sci. 17, 478–486. doi: 10.1016/j.tplants.2012.04.001
Bouchez, T., Blieux, A. L., Dequiedt, S., Domaizon, I., Dufresne, A., Ferreira, S., et al. (2016). Molecular microbiology methods for environmental diagnosis. Environ. Chem. Lett. 14, 423–441. doi: 10.1007/s10311-016-0581-3
Carrión, V. J., Perez-Jaramillo, J., Cordovez, V., Tracanna, V., de Hollander, M., Ruiz-Buck, D., et al. (2019). Pathogen-induced activation of disease-suppressive functions in the endophytic root microbiome. Science 366:606. doi: 10.1126/science.aaw9285
Castrillo, G., Teixeira, P., Paredes, S. H., Law, T. F., de Lorenzo, L., Feltcher, M. E., et al. (2017). Root microbiota drive direct integration of phosphate stress and immunity. Nature 543:513. doi: 10.1038/nature21417
Chen, X. L., Li, L. L., and He, Y. H. (2024). Epiphytic and endophytic bacteria on Camellia oleifera phyllosphere: exploring region and cultivar effect. BMC Ecol. Evol. 24:62. doi: 10.1186/s12862-024-02240-3
Chen, T., Nomura, K., Wang, X. L., Sohrabi, R., Xu, J., Yao, L. Y., et al. (2020). A plant genetic network for preventing dysbiosis in the phyllosphere. Nature 580:653. doi: 10.1038/s41586-020-2185-0
Chen, J. Q., Zhao, G. Y., Wei, Y. H., Dong, Y. H., Hou, L. Y., and Jiao, R. Z. (2021). Isolation and screening of multifunctional phosphate solubilizing bacteria and its growth-promoting effect on Chinese fir seedlings. Sci. Rep. 11:9081. doi: 10.1038/s41598-021-88635-4
Copeland, J. K., Yuan, L. J., Layeghifard, M., Wang, P. W., and Guttman, D. S. (2015). Seasonal community succession of the Phyllosphere microbiome. Mol. Plant-Microbe Interact. 28, 274–285. doi: 10.1094/mpmi-10-14-0331-fi
Crescente, G., Minervini, G., Spagnuolo, C., and Moccia, S. (2023). Cannabis bioactive compound-based formulations: new perspectives for the Management of Orofacial Pain. Molecules 28:106. doi: 10.3390/molecules28010106
Curry, K. D., Wang, Q., Nute, M. G., Tyshaieva, A., Reeves, E., Soriano, S., et al. (2022). Emu: species-level microbial community profiling of full-length 16S rRNA Oxford Nanopore sequencing data. Nat. Methods 19:845. doi: 10.1038/s41592-022-01520-4
Duan, P. P., Fu, R. T., Nottingham, A. T., Domeignoz-Horta, L. A., Yang, X. Y., Du, H., et al. (2023). Tree species diversity increases soil microbial carbon use efficiency in a subtropical forest. Glob. Chang. Biol. 29, 7131–7144. doi: 10.1111/gcb.16971
Edgar, R. C. (2013). UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 10:996. doi: 10.1038/nmeth.2604
Etesami, H., and Glick, B. R. (2024). Bacterial indole-3-acetic acid: a key regulator for plant growth, plant-microbe interactions, and agricultural adaptive resilience. Microbiol. Res. 281:127602. doi: 10.1016/j.micres.2024.127602
Francioli, D., Schulz, E., Buscot, F., and Reitz, T. (2018). Dynamics of soil bacterial communities over a vegetation season relate to both soil nutrient status and plant growth phenology. Microb. Ecol. 75, 216–227. doi: 10.1007/s00248-017-1012-0
Guzmán-Guzmán, P., Kumar, A., de los Santos-Villalobos, S., Parra-Cota, F. I., Orozco-Mosqueda, M. D., Fadiji, A. E., et al. (2023). Trichoderma species: our best fungal allies in the biocontrol of plant diseases-a review. Plants-Basel 12:432. doi: 10.3390/plants12030432
Hamaoka, K., Aoki, Y., Takahashi, S., Enoki, S., Yamamoto, K., Tanaka, K., et al. (2022). Diversity of endophytic bacterial microbiota in grapevine shoot xylems varies depending on wine grape-growing region, cultivar, and shoot growth stage. Sci. Rep. 12:15772. doi: 10.1038/s41598-022-20221-8
Hawkes, C. V., Kjoller, R., Raaijmakers, J. M., Riber, L., Christensen, S., Rasmussen, S., et al. (2021). Extension of plant phenotypes by the foliar microbiome. Annual Rev Plant Biol 72, 823–846. doi: 10.1146/annurev-arplant-080620-114342
He, X. M., Wang, D. N., Jiang, Y., Li, M., Delgado-Baquerizo, M., McLaughlin, C., et al. (2024). Heritable microbiome variation is correlated with source environment in locally adapted maize varieties. Nat Plants 10, 598–617. doi: 10.1038/s41477-024-01654-7
Hwang, H. H., Chien, P. R., Huang, F. C., Yeh, P. H., Hung, S. H. W., Deng, W. L., et al. (2022). A plant Endophytic bacterium Priestia megaterium StrainBP-R2 isolated from the halophyte Bolboschoenus planiculmis enhances plant growth under salt and drought stresses. Microorganisms 10:2047. doi: 10.3390/microorganisms10102047
Igiehon, N. O., and Babalola, O. O. (2018). Rhizosphere microbiome modulators: contributions of nitrogen fixing Bacteria towards sustainable agriculture. Int. J. Environ. Res. Public Health 15:574. doi: 10.3390/ijerph15040574
Islam, M. M., Rengel, Z., Storer, P., Siddique, K. H. M., and Solaiman, Z. M. (2023). Microbial consortium inoculant and rock mineral fertiliser differentially improved yield and nutrient uptake of industrial hemp (Cannabis sativa L.) varieties. Ind. Crop. Prod. 197:116599. doi: 10.1016/j.indcrop.2023.116599
Jan, S. S., Khan, N. A., Asaf, S., Shahzad, R., Lubna, M. I., Bilal, S., et al. (2024). Consortium of Endophytic Bacillus australimaris CK11 and Staphylococcus epidermidis CK9 from Commiphora gileadensis mediates tomato resilience to combined salinity, heat, and drought stresses. J. Plant Growth Regul. 24, 1–24. doi: 10.1007/s00344-024-11394-9
Ji, N. N., Liang, D., Clark, L., Sacks, E. J., and Kent, A. D. (2023). Host genetic variation drives the differentiation in the ecological role of the native Miscanthus root-associated microbiome. Microbiome 11:216. doi: 10.1186/s40168-023-01646-3
Law, A. D., McNees, C. R., and Moe, L. A. (2020). The microbiology of hemp retting in a controlled environment: steering the hemp microbiome towards more consistent Fiber production. Agronomy 10:492. doi: 10.3390/agronomy10040492
Li, Y. T., Qi, G. F., Xie, Z. Q., Li, B. L., Wang, R., Tan, J., et al. (2023). The Endophytic root microbiome is different in healthy and Ralstonia solanacearum-infected plants and is regulated by a consortium containing beneficial Endophytic Bacteria. Microbiol Spectr. 11:e0203122. doi: 10.1128/spectrum.02031-22
Li, P. D., Zhu, Z. R., Zhang, Y. Z., Xu, J. P., Wang, H. K., Wang, Z. Y., et al. (2022). The phyllosphere microbiome shifts toward combating melanose pathogen. Microbiome 10:56. doi: 10.1186/s40168-022-01234-x
Liu, J., Abdelfattah, A., Wasserman, B., Wisniewski, M., Droby, S., Fazio, G., et al. (2022). Contrasting effects of genotype and root size on the fungal and bacterial communities associated with apple rootstocks. Hortic. Res. 9:uhab013. doi: 10.1093/hr/uhab013
Liu, H. W., Wang, J. T., Delgado-Baquerizo, M., Zhang, H. Y., Li, J. Y., and Singh, B. K. (2023). Crop microbiome responses to pathogen colonisation regulate the host plant defence. Plant Soil 488, 393–410. doi: 10.1007/s11104-023-05981-0
Lobato, C., de Freitas, J. M., Habich, D., Kogl, I., Berg, G., and Cernava, T. (2024). Wild again: recovery of a beneficial Cannabis seed endophyte from low domestication genotypes. Microbiome 12:239. doi: 10.1186/s40168-024-01951-5
Lu, T., Li, J. J., Xiao, E. Z., Zhong, H. R., Deng, J. M., Ma, L., et al. (2025). Assemblage of root-associated microbiome contributes to disparate of two rice under aluminum stress. Plant Physiol. Biochem. 220:109539. doi: 10.1016/j.plaphy.2025.109539
Lundberg, D. S., Lebeis, S. L., Paredes, S. H., Yourstone, S., Gehring, J., Malfatti, S., et al. (2012). Defining the core Arabidopsis thaliana root microbiome. Nature 488:86. doi: 10.1038/nature11237
Matsumoto, H., Fan, X. Y., Wang, Y., Kusstatscher, P., Duan, J., Wu, S. L., et al. (2021). Bacterial seed endophyte shapes disease resistance in rice. Nat. Plants 7, 60–72. doi: 10.1038/s41477-020-00826-5
Misaghi, I. J., and Donndelinger, C. R. (1990). Endophytic bacteria in symptom-free cotton plants. Phytopathology 80, 808–811. doi: 10.1094/Phyto-80-808
Orozco-Mosqueda, M. D., Rocha-Granados, M. D., Glick, B. R., and Santoyo, G. (2018). Microbiome engineering to improve biocontrol and plant growth-promoting mechanisms. Microbiol. Res. 208, 25–31. doi: 10.1016/j.micres.2018.01.005
Pal, G., Saxena, S., Kumar, K., Verma, A., Sahu, P. K., Pandey, A., et al. (2022). Endophytic Burkholderia: multifunctional roles in plant growth promotion and stress tolerance. Microbiol. Res. 265:127201. doi: 10.1016/j.micres.2022.127201
Pan, P. P., Gu, Y. C., Sun, D. L., Wu, Q. L. L., and Zhou, N. Y. (2023). Microbial diversity biased estimation caused by Intragenomic heterogeneity and interspecific conservation of 16S rRNA genes. Appl. Environ. Microbiol. 89:e0210822. doi: 10.1128/aem.02108-22
Pang, Z. Q., Chen, J., Wang, T. H., Gao, C. S., Li, Z. M., Guo, L. T., et al. (2021). Linking plant secondary metabolites and plant microbiomes: a review. Front. Plant Sci. 12:621276. doi: 10.3389/fpls.2021.621276
Pang, Z. Q., and Xu, P. (2024). Probiotic model for studying rhizosphere interactions of root exudates and the functional microbiome. ISME J. 18:wrae223. doi: 10.1093/ismejo/wrae223
Panthapulakkal, S., and Sain, M. (2007). Injection-molded short hemp fiber/glass fiber-reinforced polypropylene hybrid composites—mechanical, water absorption and thermal properties. J. Appl. Polym. Sci. 103, 2432–2441. doi: 10.1002/app.25486
Philippot, L., Chenu, C., Kappler, A., Rillig, M. C., and Fierer, N. (2024). The interplay between microbial communities and soil properties. Nat. Rev. Microbiol. 22, 226–239. doi: 10.1038/s41579-023-00980-5
Potter, T. S., Anacker, B. L., Churchill, A. C., and Bowman, W. D. (2023). Plant species' influence on rhizosphere microbial communities depends on N availability. Plant Soil 491, 681–696. doi: 10.1007/s11104-023-06148-7
Santoyo, G. (2022). How plants recruit their microbiome? New insights into beneficial interactions. J. Adv. Res. 40, 45–58. doi: 10.1016/j.jare.2021.11.020
Sasse, J., Martinoia, E., and Northen, T. (2018). Feed your friends: do Plant exudates shape the root microbiome? Trends Plant Sci. 23, 25–41. doi: 10.1016/j.tplants.2017.09.003
Schalk, I. J. (2025). Bacterial siderophores: diversity, uptake pathways and applications. Nat. Rev. Microbiol. 23, 24–40. doi: 10.1038/s41579-024-01090-6
Shenhav, L., Thompson, M., Joseph, T. A., Briscoe, L., Furman, O., Bogumil, D., et al. (2019). FEAST: fast expectation-maximization for microbial source tracking. Nat. Methods 16:627. doi: 10.1038/s41592-019-0431-x
Singh, A., Mazahar, S., Chapadgaonkar, S. S., Giri, P., and Shourie, A. (2023). Phyto-microbiome to mitigate abiotic stress in crop plants. Front. Microbiol. 14:1210890. doi: 10.3389/fmicb.2023.1210890
Sohrabi, R., Paasch, B. C., Liber, J. A., and He, S. Y. (2023). Phyllosphere microbiome. Annu. Rev. Plant Biol. 74, 539–568. doi: 10.1146/annurev-arplant-102820-032704
Sokol, N. W., Slessarev, E., Marschmann, G. L., Nicolas, A., Blazewicz, S. J., Brodie, E. L., et al. (2022). Life and death in the soil microbiome: how ecological processes influence biogeochemistry. Nat. Rev. Microbiol. 20, 415–430. doi: 10.1038/s41579-022-00695-z
Song, C., Saloner, A., Fait, A., and Bernstein, N. (2023). Nitrogen deficiency stimulates cannabinoid biosynthesis in medical cannabis plants by inducing a metabolic shift towards production of low-N metabolites. Ind. Crop. Prod. 202:116969. doi: 10.1016/j.indcrop.2023.116969
Su, J. M., Wang, Y. Y., Bai, M., Peng, T. H., Li, H. S., Xu, H. J., et al. (2023). Soil conditions and the plant microbiome boost the accumulation of monoterpenes in the fruit of Citrus reticulata 'Chachi'. Microbiome 11:61. doi: 10.1186/s40168-023-01504-2
Taghinasab, M., and Jabaji, S. (2020). Cannabis microbiome and the role of endophytes in modulating the production of secondary metabolites: An overview. Microorganisms 8:355. doi: 10.3390/microorganisms8030355
Tian, K., Zhang, Y., Yao, D. D., Tan, D., Fu, X. J., Chen, R. H., et al. (2024). Synergistic interactions in core microbiome Rhizobiales accelerate 1,4-dioxane biodegradation. J. Hazard. Mater. 476:135098. doi: 10.1016/j.jhazmat.2024.135098
Van Bel, M., Fisher, A. E., Ball, L., Columbus, J. T., and Berlemont, R. (2021). Phylosymbiosis in the rhizosphere microbiome extends to nitrogen cycle functional potential. Microorganisms 9:2476. doi: 10.3390/microorganisms9122476
Wang, Y. C., Hussain, M., An, X. C., Jiang, X. J., and Mao, R. Q. (2022). Assessing the bacterial communities composition from differently treated agarwood via 16S rRNA gene Metabarcoding. Life-Basel 12:1697. doi: 10.3390/life12111697
Wang, Y. Y., Wang, X. L., Sun, S., Jin, C. Z., Su, J. M., Wei, J. P., et al. (2022). GWAS, MWAS and mGWAS provide insights into precision agriculture based on genotype-dependent microbial effects in foxtail millet. Nat. Commun. 13:5913. doi: 10.1038/s41467-022-33238-4
Wang, C., Wang, X., Zhang, Y., Morrissey, E., Liu, Y., Sun, L. F., et al. (2023). Integrating microbial community properties, biomass and necromass to predict cropland soil organic carbon. ISME Commun 3:86. doi: 10.1038/s43705-023-00300-1
Watts, D., Palombo, E. A., Castillo, A. J., and Zaferanloo, B. (2023). Endophytes in agriculture: potential to improve yields and tolerances of agricultural crops. Microorganisms 11:1276. doi: 10.3390/microorganisms11051276
Wei, G. F., Ning, K., Zhang, G. Z., Yu, H. B., Yang, S. M., Dai, F., et al. (2021). Compartment niche shapes the assembly and network of Cannabis sativa associated microbiome. Front. Microbiol. 12:714993. doi: 10.3389/fmicb.2021.714993
Xiao, X., Chen, W. M., Zong, L., Yang, J., Jiao, S., Lin, Y. B., et al. (2017). Two cultivated legume plants reveal the enrichment process of the microbiome in the rhizocompartments. Mol. Ecol. 26, 1641–1651. doi: 10.1111/mec.14027
Xiao, J., Sun, Y., He, Y., Tang, X. F., Yang, S. D., and Huang, J. Y. (2023). Comparison of Rhizospheric and Endophytic bacterial compositions between netted and oriental melons. Microbiol. Spectrum 11:e0402722. doi: 10.1128/spectrum.04027-22
Xiong, X. Q., Zeng, J., Ning, Q., Liu, H. Q., Bu, Z. G., Zhang, X., et al. (2024). Ferroptosis induction in host rice by endophyte OsiSh-2 is necessary for mutualism and disease resistance in symbiosis. Nat. Commun. 15:5012. doi: 10.1038/s41467-024-49099-y
Xu, Y., Ge, Y., Song, J. X., and Rensing, C. (2020). Assembly of root-associated microbial community of typical rice cultivars in different soil types. Biol. Fertil. Soils 56, 249–260. doi: 10.1007/s00374-019-01406-2
Yan, S. S., Jiang, H. W., Li, J. W., Yan, C., Ma, C. M., Zhang, Z. X., et al. (2023). Effect of short-term organic matter returns on soil organic carbon fractions, phosphorus fractions and microbial Community in Cold Region of China. Agronomy-Basel 13:2805. doi: 10.3390/agronomy13112805
Yan, G. Y., Luo, X., Liu, G. C., Huang, B. B., Wang, H. L., Sun, X. Y., et al. (2024). Nitrogen addition affects tree trait expression by altering endophytic microbe diversity. Plant Soil. 24, 1–15. doi: 10.1007/s11104-024-07103-w
Yan, X. L., Zhou, L. Y., Chen, Y. M., Liu, R. Q., Guo, L. Q., Li, N., et al. (2025). Ectomycorrhizal fungi explain more variation in rhizosphere nutrient availability than root traits in temperate forests. Appl. Soil Ecol. 207:105923. doi: 10.1016/j.apsoil.2025.105923
Yue, H., Yue, W. J., Jiao, S., Kim, H., Lee, Y. H., Wei, G. H., et al. (2023). Plant domestication shapes rhizosphere microbiome assembly and metabolic functions. Microbiome 11:70. doi: 10.1186/s40168-023-01513-1
Zeng, Q., Zhao, Y., Shen, W., Han, D. J., and Yang, M. M. (2023). Seed-to-seed: plant Core vertically transmitted microbiota. J. Agric. Food Chem. 71, 19255–19264. doi: 10.1021/acs.jafc.3c07092
Zhang, J. W., Guo, Z. Y., Liu, J., Pan, X. Z., Huang, Y. N., Cui, X. D., et al. (2025). Neutral pH induces complex and stable soil microbial networks in agricultural ecosystems. Plant Soil. doi: 10.1007/s11104-024-07195-4
Zhang, J. Y., Liu, Y. X., Zhang, N., Hu, B., Jin, T., Xu, H. R., et al. (2019). NRT1.1B is associated with root microbiota composition and nitrogen use in field-grown rice. Nat. Biotechnol. 37:676. doi: 10.1038/s41587-019-0104-4
Zhang, X. X., Ma, Y. N., Wang, X., Liao, K. J., He, S. W., Zhao, X., et al. (2022). Dynamics of rice microbiomes reveal core vertically transmitted seed endophytes. Microbiome 10:216. doi: 10.1186/s40168-022-01422-9
Zhou, H. P., Shi, H. F., Yang, Y. Q., Feng, X. X., Chen, X., Xiao, F., et al. (2024). Insights into plant salt stress signaling and tolerance. J. Genet. Genomics 51, 16–34. doi: 10.1016/j.jgg.2023.08.007
Zhuang, L. B., Li, Y., Wang, Z. S., Yu, Y., Zhang, N., Yang, C., et al. (2021). Synthetic community with six Pseudomonas strains screened from garlic rhizosphere microbiome promotes plant growth. Microb. Biotechnol. 14, 488–502. doi: 10.1111/1751-7915.13640
Keywords: bacterial diversity, community composition, Cannabis sativa L., community differences, functional prediction
Citation: Li J, Zhang H, Long S, Li W, Wang T, Yu J, Zhou Y, Zou S, Zhu H, Xu J and Cheng Y (2025) DNA metabarcode analyses reveal similarities and differences in plant microbiomes of industrial hemp and medicinal Cannabis in China. Front. Microbiol. 16:1524703. doi: 10.3389/fmicb.2025.1524703
Edited by:
Mubasher Hussain, Guangdong Pest control Technology Group, ChinaReviewed by:
Setu Bazie Tagele, University of California, Riverside, United StatesJosé Luis Aguirre-Noyola, National Autonomous University of Mexico, Mexico
Zhiqiang Pang, Chinese Academy of Sciences (CAS), China
Afeez Adesina Adedayo, Western Illinois University, United States
Copyright © 2025 Li, Zhang, Long, Li, Wang, Yu, Zhou, Zou, Zhu, Xu and Cheng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hongjian Zhu, aG9uZ2ppYW42MkAxNjMuY29tJianping Xu,anB4dUBtY21hc3Rlci5jYQ==; Yi Cheng, Y2hlbmd5aUBjYWFzLmNu
†These authors share first authorship