Abstract
The ongoing outbreak of Coronavirus disease 2019 infection achieved pandemic status on March 11, 2020. As of September 8, 2020 it has caused over 890,000 mortalities world-wide. Coronaviral infections are enabled by potent immunoevasory mechanisms that target multiple aspects of innate immunity, with severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) able to induce a cytokine storm, impair interferon responses, and suppress antigen presentation on both MHC class I and class II. Understanding the immune responses to SARS-CoV-2 and its immunoevasion approaches will improve our understanding of pathogenesis, virus clearance, and contribute toward vaccine and immunotherepeutic design and evaluation. This review discusses the known host innate immune response and immune evasion mechanisms driving SARS-CoV-2 infection and pathophysiology.
Introduction
In December 2019, the outbreak of novel severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) – the causative agent of Coronavirus disease 2019 (COVID-19) – began in Wuhan, Hubei Province, China, from where it spread rapidly. On March 11, 2020 the World Health Organization declared COVID-19 a global pandemic (1, 2). COVID-19 is not the first severe respiratory disease outbreak caused by coronaviruses (CoVs) in humans, with two epidemic diseases – severe acute respiratory syndrome (SARS-CoV) in 2003–2004 and Middle East respiratory syndrome (MERS–CoV) in 2012 – caused by similar zoonotic transmission of CoVs (3). The primary symptoms of COVID-19 are similar to those of SARS-CoV and MERS-CoV: fever, fatigue, dry cough, discomfort in the upper chest, occasional diarrhea, and dyspnea. Severe cases exhibit secondary infections, cytokine-storm driven sepsis, and multi-organ failure (4, 5). COVID-19 patients primarily develop pneumonia, lymphopenia, and feature pulmonary ground glass opacity on chest CT (6, 7).
Coronaviruses are in the Coronaviridae family, enveloped viruses with a positive-sense single-stranded RNA genome ranging from 26 to 32 kb in size (8). In humans CoV infections are common, with four CoVs (229E, NL63, OC43, and HKU1) causing ∼10% of common cold cases (4). The infection of human cells by CoVs is mediated by interactions between envelope-anchored spike glycoprotein (S-protein) of CoV with one of two host cell receptors: angiotensin-converting enzyme 2 (ACE2) or CD147 (9, 10). The S-protein consists of two subunits: S1 which functions as the receptor-binding domain (RBD), and S2 which drives the fusion of the viral membrane with the host cell membrane (11). Spike glycoprotein activation and viral entry is mediated by cleavage of the S protein by the host transmembrane protease, serine 2 (TMPRSS2) (12, 13). Sequencing of SARS-CoV-2 from patients revealed that it shares 79.6% homology to SARS-CoV, 50% MERS-CoV, and 96% to bat SARS-like CoV at the whole genome level (12, 14). The RBD of SARS-CoV-2 is derived from a pangolin-infecting CoV and exhibits a 10-fold increase in affinity between the RBD and ACE2 compared to SARS-CoV, further consistent with ACE2 as the prominent receptor for SARS-CoV-2 (15). This increase in receptor affinity was generated by the recombination of the pangolin CoV and a bat SARS-like CoV within the RBD region, a characteristic which could lead to a more efficient cell entry (16). Interestingly, crytal structure evaluation by cryo-electron microscopy (Cryo-EM) showed that SARS-CoV-2 RBD is biased toward the lying state conformation, which reduces receptor binding by burying the RBD within the spike protein trimer. In contrast, the SARS-CoV RBD is mostly in the exposed “standing up” state which favors receptor binding (17–19). This bias toward the lying state may favor SARS-CoV-2 immune evasion by masking the RBD domain from neutralizing antibodies. There have been reports that SARS-CoV-2 can also gain entry into cells via CD147, but the importance of this pathway for viral entry, and the concordant receptor-binding motifs, remain largely unelucidated (10).
Less well understood than SARS-CoV-2’s biology is it resulting immune responses, immunopathology, and immune evasion mechanisms. Understanding these responses will be vital for the development of immunotherapies or vaccines against COVID-19 (1, 20). Coronaviruses are adept at manipulating immune responses and interfere with the interferon (IFN) pathway, with several structural proteins (M and N) and non-structural protein (NSP1 and NSP3) from SARS-CoV and MERS-CoV acting as interferon antagonists (21). These CoVs also interfere with pattern recognition receptor (PRR) signaling such as Toll-like receptors (TLRs) and retinoic acid-inducible gene I (RIG-I) like receptors (22), and generate a strong inflammatory response (23, 24). This drives a non-productive inflammation, resulting in a cytokine storm and disseminated damage to the host, while avoiding induction of an anti-viral interferon response. Indeed, an early study of 41 COVID-19 patients identified increased levels of pro-inflammatory cytokines including IL-2 and IL-7, with more severe disease producing elevated G-CSF, MCP-1, MIP-1α, IP-10, and TNF-α (25). These pro-inflammatory cytokines drive an influx of neutrophils and other myeloid cells into the lung, producing a strong local inflammatory response and significant immunopathology (26). This is consistent with SARS and MERS, indicating that a cytokine storm and lymphopenia play a crucial role in the COVID-19 pathogenesis (25, 27, 28). In addition to manipulating cytokines, CoVs also manipulate other immunological processes including antigen presentation (29). In this review we discuss the major innate immunological pathways involved in responses to CoV infection, and the mechanisms used by SARS-CoV-2 and related CoVs to overcome these defenses.
Interferons and Cytokines
The innate immune system is an evolutionary conserved set of cellular and chemical defenses critical for the recognition and restriction of pathogens, and for the subsequent activation of an adaptive immune response (21). Innate immunity is initiated by pathogen-associated molecular patterns, evolutionarily conserved molecular structures specific to pathogens that act as ligands for PRRs. Ligand binding triggers signaling pathways that coverage on transcription factors including NF-κB, IRF3, and AP-1, which synergistically promote type I interferon (IFN-I) production. These cytokines act in a paracrine fashion on neighboring cells via the IFN-α/β receptor (IFNAR) and induce expression of interferon-stimulated genes (ISGs) (30). Interferon-stimulated genes are an essential component of innate antiviral defense, acting to both limit viral entry and restricting viral replication after a virus enters a host cell (31, 32). Interferon-stimulated gene expression is driven predominantly by IFNAR-mediated activation of the Jak/STAT pathway, resulting in binding of STAT1 homodimers and STAT1/2 heterodimers to the promoter region of ISGs.
Severe acute respiratory syndrome coronavirus 2 has evolved multiple mechanisms to manipulate this key antiviral response. It was recently demonstrated that ACE2 is an ISG, suggesting that SARS-CoV-2 may exploit IFN-driven ACE2 upregulation to enhance infection (31). Furthermore, CoVs including SARS-CoV and MERS-CoV encode multiple proteins which antagonize IFN signaling. These functions represent a key anti-immune mechanism of SARS-like CoV’s, and are critical for the viral manipulation of the innate immune response and promoting early viral pathogenesis (33, 34). Because SARS-CoV-2 may utilize ISG’s to enhance infectivity, and because it is unclear whether the IFN response restricts SARS-CoV-2 replication, it is unknown if IFN-directed therapies will be beneficial to COVID-19 patients. A recent study found that COVID-19 patients who failed to produce IFN-α experienced more severe clinical outcomes (35). In addition, IFN-α potently inhibits the replication of SARS-CoV-2 in vitro (36). The National Health Commission of China has proposed guidelines for the treatment of SARS-CoV-2, which includes aerosolized recombinant IFN-α, based on the observation that IFN-α inhibits SARS-CoV replication in vitro (37, 38). At the time of this writing only one retrospective analysis of these treatment guidelines has been published, identifying a modest benefit of IFN-α in combination with lopinavir, ritonavir, and ribavirin (39). The combinatorial use of these drugs makes it difficult to assess the role of IFN-α alone, but these data suggest that induction of the IFN-I pathway produces a beneficial response in SARS-CoV-2 patients. In addition to IFN-I, type III IFNs (IFN-λ) exhibit more potent antiviral functions than IFN-α in treating influenza infection, without activating inflammation and tissue damage induced by IFN-α (40, 41). On the other hand, it has been shown that IFN-λ treatment could inhibit bacterial uptake by neutrophils in the lung during influenza superinfection, suggesting that IFN-λ may increase susceptibility to lower repiratory tract infection, potentially increasing the risk of super-infection of COVID-19 patients to super-infection with other pathogens (42, 43).
Similar to type I IFNs, IFNs-λ is decreased during COVID-19 infection (44), and IFN-λ hinders SARS-CoV-2 replication in vitro in human intestinal epithelial cells (45). Interestingly, IFN-λ is present in lower airways in COVID-19 patients where it mediates antiproliferative effects in during the repair of the lung epithelium. This antiprolifertative effect occurs via p53 induction, but due to its slowing of epithelial repair, may increase the risk of life-threating bacterial superinfections in the lung during both influenza and COVID-19 infection (46, 47). Thus, timing and duration are critical parameters of IFN action and in COVID-19 patients who contract a secondary bacterial infection, it might be crucial to intervene with recombinant interferons when virus is in upper airways. Later in disease, when inflammation is increased in the lower airways, it may be beneficial to block signaling cascades initiated by interferons and other inflammatory cytokines in order to control superinfecting pathogens.
The mechanisms used by CoVs to manipulate the IFN response can be divided into three categories: (1) avoidance, where the virus protects itself from recognition by PRRs, (2) IFN induction suppression, where the virus inhibits the transcription of interferons (48), and (3) IFN signaling suppression, where viral proteins inhibit IFNAR signaling (49). The viral membrane (M) protein, nucleocapsid (N) protein, and the non-structural proteins NSP1, NSP3b, NSP4a, NSP4b, NSP15, play crucial roles in modulation of the host immune response (50). The SARS-CoV N protein modulates signaling though the TGF-β receptor by forming a complex with Smad3. This enhances Smad3/p300 transcription, driving lung fibrosis. This interaction also prevents Smad3 from complexing with Smad4, thereby enhancing the survival of infected cells by antagonizing TGF-β-sensitized apoptosis (51). Moreover, SARS-CoV and MERS-CoV hide their RNA genome from host detection by cytosolic (e.g., RIG-I) and endosomal (e.g., TLR3/7) PRRs by replicating in double membrane vesicles that excludes these PRRs (52, 53). Indeed, both the induction of IFN-α or β and the ability to restrict both MERS-CoV and SARS-CoV infection requires TLR3 signaling, and SARS-CoV is able to antagonize the TLR signaling pathway via its papain-like protease (PLpro) (54, 55). Finally, during viral replication the SARS-CoV and MERS-CoV N protein catalytically modifies host proteins through SUMOylation and ubiquitination, via interactions with the host proteins hUbc9 and TRIM25, respectively (22, 56). While the targets of N protein-mediated SUMOylation remains unclear, N protein-targeted ubiquitination targets RIG-I for proteasomal degradation, thus depleting the cell of a critical virus-detecting PRR (22). The N proteins of SARS-CoV and SARS-CoV-2 are 90% conserved, suggesting that these immune avoidance activities are likely conserved in SARS-CoV-2 (Supplementary Figure 1). Thus, through manipulating multiple components of the host’s PRR system, CoVs are able to limit or avoid activation of many host anti-viral processes.
Analysis of MERS-CoV patients with differing severity has demonstrated significantly lower IFN-I responses in patients who succumb to infection versus to those who recover, highlighting the importance of IFN induction (57). It has been shown that the IFN pathway is inhibited by SARS-CoV-2 to an extent similar to the inhibition by MERS-CoV and SARS-CoV, thereby impairing both innate T cell antiviral responses (58–60). While the specific molecular pathways mediating the suppression of IFN induction in SARS-CoV-2 have not been fully elucidated, these mechanisms are well understood in SARS-CoV and MERS and are likely conserved (Figure 1). Severe acute respiratory syndrome coronavirus NSP1 promotes the degradation of IFN-β mRNA (61), while ORF6 disrupts IFN induction by preventing the transport of IRF3 and STAT1 into the nucleus (62; 63). This occurs via two mechanisms, with ORF6 and ORF3b reducing IFNAR signaling by disrupting STAT1 nuclear import and promoting STAT1 proteolytic degradation, respectively (62, 64). ORF4a interacts with dsRNA and the RIG-I like receptor cofactor PACT, inhibiting IFN induction (65, 66). ORF4b blocks IFN induction by binding to both TBK1 and IKKε (67, 68). MERS-CoV’s ORF4b, 5, and M proteins have been shown to prevent nuclear translocation of IRF3 (69). At least some of these pathways are conserved in SARS-CoV-2, with a recent study demonstrating that SARS-CoV-2 ORF3b is a potent IFN antagonist, which suppresses IFN induction more efficiently than the SARS-CoV ortholog (70). Notably, a recent study reported that ORF3b is one of the most common antibody-recognized antigens during early stage COVID-19 infection (71), suggesting that ORF3b is highly expressed in the acute stage of the infection and may represent an immunodominant epitope. NSP13, NSP14, NSP15, and ORF6 have been suggested to function as IFN antagonists by suppressing IRF3 nuclear localization (72). Additionally, SARS-CoV-2 M protein through interacting with RIG-I/MDA-5-MAVS signaling pathway inhibits the production of type I and III IFNs (73). As discussed above, the SARS and MERS PLpro is a deubiquitinating/deISGylating enzyme (74), which enhances immunoevasion by downregulating IFN-β transcription (75). The PLpro proteins of SARS-CoV and SARS-CoV-2 are 76% conserved, suggesting that this activity is likely conserved in SARS-CoV-2 (Supplementary Figure 2).
FIGURE 1
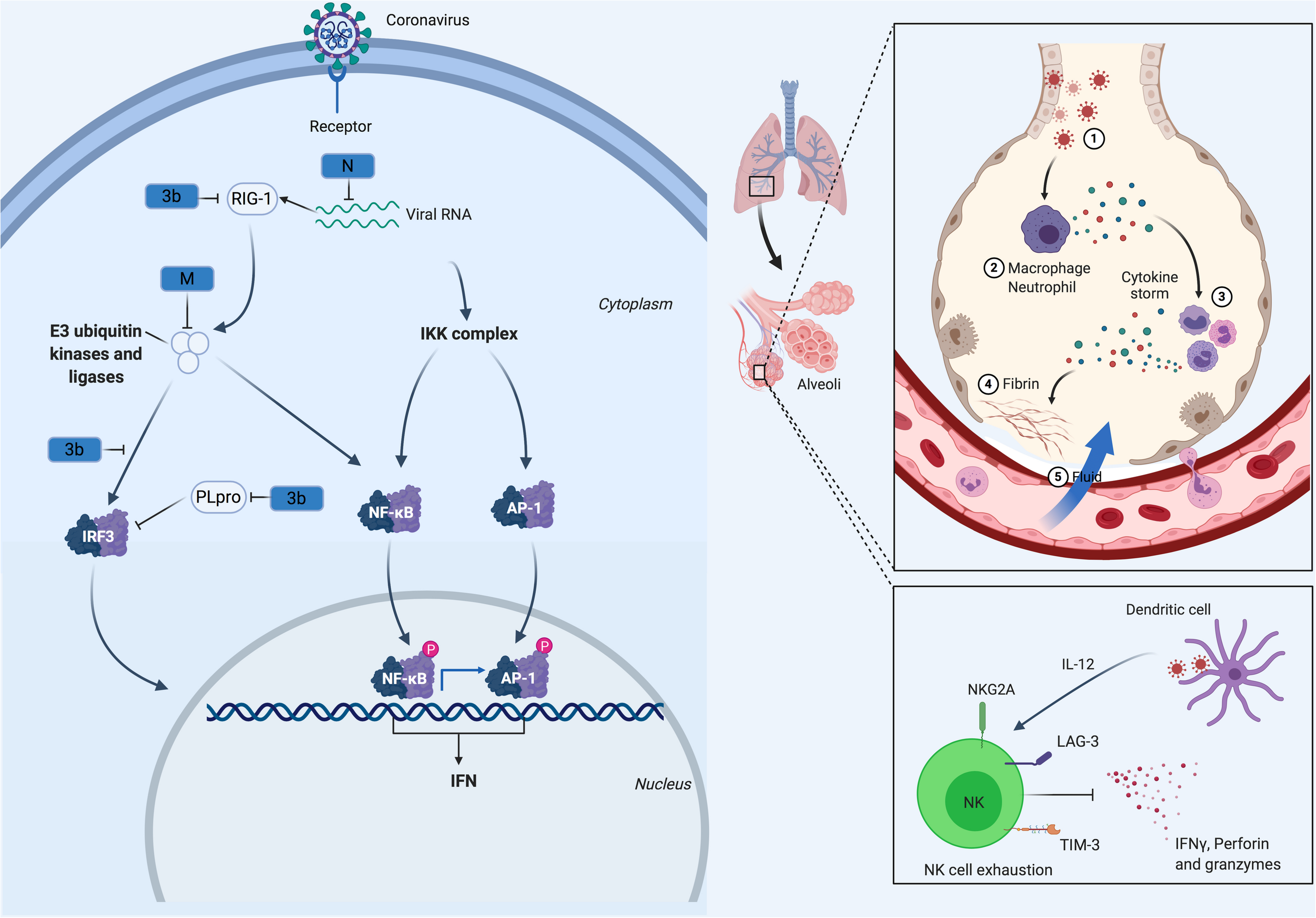
CoV Innate Immune Evasion. The innate immune response to CoV’s is activated upon detection of viral pathogen-associated molecular patterns, such as double-stranded RNA, via host PRRs such as RIG-I. Following viral recognition, transcription factors including NF-κB, AP-1 and IRF3 are activated and translocate to the nucleus where they induce the expression of interferons. Both MERS-CoV and SARS-CoV, through their M, N, non-structural proteins (NSP1, 3b, 4a, 4b, 5, 6), and PLpro, have developed mechanisms to interfere with these signaling pathways. This alters the cytokine secretion profile of infected cells to enhance the recruitment of myeloid immune cells over NK cells, which in turn produce more cytokines, creating a cycle of inflammation that damages the lung. Many of these processes are likely conserved in SARS-CoV-2.
COVID-19 patients experience a cytokine storm that drives much of the pathophysiology of the disease. Both pro-and anti-inflammatory circulating cytokines increase in SARS-CoV-2 patients, with cytokines including G-CSF, TNFα, MCP1, IL-10, IL-2 and IL-7 becoming elevated in critically ill patients (76, 77). IL-6 is thought to be a major driver of pathology, with the highest levels observed in non-survivors and critically ill COVID-19 patients (78, 79). The mechanism leading to elevated IL-6 in severe COVID-19 is not currently clear, but is likely driven through activation of virus-specific PRRs. However, other pathways are likely involved in patients with severe disease. For example, SARS-CoV triggers the production of oxidized phospholipids, which then drives IL-6 production via TLR4 (80). The upregulation of IL-6 is an established effect of TLR4 signaling, which occurs via NF-κB and MAPK signaling pathways (81). Finally, the N protein of SARS-CoV-2 induces the expression of IL-6 in infected airway epithelium by binding to the NF-κB regulatory elements of the IL-6 promoter (82). Clearly, SARS-CoV-2 pathology is driven by an unbalanced cytokine response, although the viral processes driving these responses remain to be fully elucidated.
Pattern Recognition Receptor Evasion
In addition to modulating IFN-I and IL-6 responses, CoVs also engage in immune evasion through limiting PRR activation. Coronaviruses replicate in a double-membrane structure derived from the endoplasmic reticulum (52). The endoplasmic reticulum lacks PRR’s, and as such, this compartment secludes the replicating virus from both cytosolic PRRs such as RIG-I and from endosomal PRRs such as TLR3 and TLR7 (83–85). In addition to physically excluding PRRs from the site of viral replication, CoVs suppress PRR signaling directly. As discussed above, coronaviral N-proteins direct the host cell ubiquitination machinery to target the cytosolic RNA sensor RIG-I, leading to RIG-I degradation (22). Coronaviruses also suppress TLR signaling; for example, the MERS-CoV spike protein induces expression of the negative TLR regulator IRAK-M in macrophages through activating the dipeptidyl-peptidase 4 receptor (86). Likewise, SARS-CoV induces the broad dysregulation of TLR-associated signaling molecules in infected macrophages, although the specific mechanism of this dysregulation and its net effect on TLR signaling has not been established (87). While these activities have not yet been reported in SARS-CoV-2, the high degree of conservation between it, MERS-CoV, and SARS-CoV indicates that some manipulation of the PRR system likely occurs during SARS-CoV-2 infection.
Neutrophils
Neutrophilia is an early indicator of SARS-CoV-2 infection, although it is unclear whether this increase is due to the release of the marginated neutrophil pool versus the release of bone marrow-derived cells (88–90). While the extent to which neutrophils are responsible for COVID-19 pathophysiology is unclear, significant neutrophil infiltration has been reported in autopsied COVID-19 patients (91, 92). Infiltration is not the only mechanism by which neutrophils may cause pathology on COVID-19 patients. Indeed, pathological effects of neutrophil extracellular traps (NETs) have been identified in a variety of inflammatory conditions including thrombosis, sepsis, and respiratory failure (93–95). Neutrophil extracellular traps are comprised of extracellular DNA fibers, histones, microbicidal proteins, proteases such as neutrophil elastase, and oxidant enzymes such as myeloperoxidase, that are released by neutrophils in response to many infectious agents. If not regulated properly, NETs initiate and propagate inflammation and thrombosis (96–98).
For the first time, a recent study showed that NETs released by neutrophils contributes to organ damage and mortality in COVID-19 patients (99). This is consistent with another recent study that identified markers of NET release, including myeloperoxidase-DNA and citrullinated histone H3, in COVID-19 patients, with the sera of these patients potently inducing NETosis of control neutrophils in vitro (100). The high levels of IL-6 observed in COVID-19 patients is likely a driver of this NETosis, as in other inflammatory diseases IL-6 induces the systemic release of NETs (101, 102). Additional triggers of NETosis include virus-damaged epithelial cells (103, 104), activated endothelial cells (105), activated platelets (106, 107), and inflammatory cytokines such as IL-1β (108, 109). Transcriptional analysis of bronchoalveolar lavage fluid and peripheral blood mononuclear cells from COVID-19 patients showed that elevated levels of CXCL2 and CXCL8 contributes to the recruitment of neutrophils to the lung, aggravating the inflammatory response (110). Moreover, activated neutrophils express properdin, factor B, and C3, thus driving complement activation (111), a marker of severe COVID-19 disease (112, 113). Consistent with a role for NETs in COVID-19 immunopathology, a small clinical trial demonstrated a protective effect of dipyridamole, an FDA approved drug which inhibits NETosis via blocking signaling through adenosine A2A receptors (93, 114).
NK Cells
Natural killer (NK) cells are essential to the control of viral infections, and functional impairment of NK cells correlate with persistence of SARS-CoV-2 (115, 116). Reduced peripheral blood NK cells numbers is frequently observed in severe COVID-19 patients (89, 116–118). Killer-immunoglobulin like receptors (KIRs) which are expressed on plasma membrane of NK cells alongside CD16 play crucial roles in NK cells licensing and their subsequent cytotoxic functions (119). In peripheral blood, NK cells expressing KIRs and CD16 are significantly decreased in SARS-CoV and SARS-CoV-2 infection, suggesting either impaired maturation of NK cells or migration of circulating NK cells into the peripheral tissues of SARS-CoV-2 patients (120, 121). Moreover, lower NK cell numbers correlate with higher IL-6 plasma concentrations in SARS-CoV-2 infection (117, 120). In vitro, IL-6 and soluble IL-6 receptor impair perforin and granzyme B production by healthy donor NK cells, which could be restored by treatment with the IL-6R inhibitor tocilizumab (122). Furthermore, secretion of cytokines such as IL-12 by macrophages and dendritic cells (DCs) could promote NK cell proliferation, cytotoxicity, survival, and IFN-γ production (123, 124). The later may occur through IFN-λ-mediated IL-12 production by macrophages (125), suggesting that early IFN-γ production and NK cell stimulation may act to limit SARS-CoV-2 infection.
The expression of inhibitory receptor NKG2A on NK cells results in the functional exhaustion of NK cells in chronic viral infection and cancer (126, 127). It is reported that NK cells become functionally exhausted in SARS-CoV-2 patients, as evidenced by increased NKG2A expression and decreased expression of CD107a, IFN-γ, IL-2 and granzyme B in NK cells (76, 116). Additionally, recent studies reported upregulated expression of the genes encoding inhibitory receptors including TIM3 and LAG3 in NK cells from COVID-19 patients (60, 128). The implications of this high incidence of exhausted NK cells in SARS-CoV-2 patients remain unelucidated. While NK cell numbers and activity appear to be diminished in COVID-19 patients, anti-S protein antibodies are capable of inducing NK-cell mediated antibody-dependent cell cytotoxicity in an in vitro model of SARS-CoV-2 infection, indicating that normally functional NK cells should contribute to protective immunity against SARS-CoV-2 (129).
Macrophages
Macrophages are one of the primary drivers of innate immunity in response to CoV infection, with macrophage activity driving both inflammation, and much of the pathology, in COVID-19 patients (130). Macrophages in the lung and upper respiratory tract act as sentinel cells and are among the first immune cells to encounter incoming virions. In response, these macrophages can limit early viral replication through initiating a IFN-I response, as well as through initiating an inflammatory response to recruit additional immune cells (131). While this inflammatory response is required to initiate immune responses against SARS-CoV-2, excess inflammation in the form of a cytokine storm contributes to the mortality associated with COVID-19 (132). The lung has at least two distinct macrophage populations - interstitial macrophages and alveolar macrophages (AMs) (125), and there are at least two subsets of interstitial macrophages – of which, the nerve-and airway-associated macrophages (NAMs) may be particularly important for restricting inflammation in response to CoV’s. Nerve-and airway-associated macrophages are highly divergent from AMs in their gene expression profile and tissue localization. Nerve-and airway-associated macrophages are located in the lung interstitia, closely associated with innervating nervous tissue, and have a different ontology and growth factor dependence than AMs (133). During influenza infection NAMs are critical in limiting viral-induced inflammation, whereas AMs are pro-inflammatory and required for viral clearance. These findings indicate that AMs are anti-viral and pro-inflammatory, whereas NAMs are and act to prevent excessive and damaging inflammation. Importantly, during influenza NAMs suppress IL-6 production, indicating that NAMs may be a critical control point in determining IL-6 levels, and thus may regulate the cytokine storm in COVID-19 patients (133).
Consistent with these observations, distinct macrophage populations were identified in the lungs of COVID-19 patients, and while this study did not investigate NAMs, it did identify enrichment of anti-inflammatory monocyte-derived (FCN1high) macrophages in patients with mild disease, while resident pro-fibrotic (SPP1high) and inflammatory AM (FAPB4+) populations dominated in patients with severe COVID-19 (134). Combined, these studies demonstrate that macrophage polarization and the relative proportion of macrophage subtypes is a major factor in COVID-19 severity.
Macrophages can be infected by SARS-CoV-2 (130), indicating that SARS-CoV-2 directly manipulates macrophages to evade immunity. It is unclear what effect SARS-CoV-2 infection has on macrophage function, but infection of macrophages by other CoVs is known to induce altered functional states. Macrophages infected by MERS-CoV express high levels of major histocompatibility complex I (MHC I), CD80 and CD86, but lack major histocompatibility complex II (MHC II), indicating that MHC II presentation is impaired by MERS-CoV (58, 135). Consistently, MHC II downregulation was recently demonstrated in monocytes and B cells from COVID-19 patients (128). Moreover, monocyte HLA-DR is downregulated in severe COVID-19 patients, with expression patially restored by an IL-6 inhibitor (136). The mechanism of MHC II downregulation is not completely understood, but is due in-part to changes in the epigenetic landscape of the infected cells (137). Indeed, the epigenetic downregulation of MHC II is a mechanism shared by other CoVs, for example, the human CoV-EMC also downregulates MHC II via epigenetic reprograming (29). However, epigenetic reprograming of antigen presentation is not universal, and for example, is not a feature of SARS-CoV despite this virus also limiting antigen presentation on MHC II (29).
The recently published interactome of SARS-CoV-2 provides some potential insights into mechanisms by which this virus may interfere with macrophage function (138). The blockade of interferon signaling, described above, would limit ISG expression in macrophages, including cytokine-induced MHC II expression (139). The SARS-CoV-2 protein Nsp5 interacts with the epigenetic regulator histone deacetylase 2 (HDAC2), which regulates MHC II expression and cytokine production (138, 140, 141). While it is unknown whether SARS-CoV-2 inhibits or enhances HDAC2 activity, this interaction indicates a potential direct modulation of the cytokine storm and antigen presentation. Nsp13 and ORF8 of SARS-CoV-2 interacts with multiple components of the Golgi trafficking system and may utilize this as a mechanism to restrict MHC export to the cell surface. Indeed, a recent study demonstrated that ORF8 of SARS-CoV-2 can directly bind to MHC I molecules at endoplasmic reticulum and redirect them to autolysosomes for degradation (142). This is a common approach used by viruses to limit antigen presentation; for example, the HIV Nef protein restricts antigen presentation on MHC I by redirecting MHC trafficking toward the Golgi (143, 144). Lastly, SARS-CoV-2’s Nsp10 interacts with the endocytosis regulator AP2, a critical regulator of MHC II trafficking to antigen loading compartments (138, 145, 146). While these immunoevasory mechanisms remain largely theoretical, the ability of SARS-CoV-2 to infect macrophages and interact with proteins central to macrophage function suggests a potent ability to modulate macrophage activity, and through this, the systemic immune response.
Dendritic Cells
Dendritic cells are key players in antigen presentation, cytokine production, priming specific T cell responses, and a loss of DCs function could lead to delayed immune responses in COVID-19 patients (147). Previous studies demonstrated that SARS-CoV infects DCs, resulting in poor antiviral cytokine expression with an upregulation of inflammatory chemokines including MIP-1α (148). SARS-CoV also enhanced pro-inflammatory cytokine (IL-6 and IL-12) production by DCs in response to secondary activation signals by bacterial LPS, further contributing to a damaging inflammatory response (149). Moreover, plasmacytoid dendritic cells (pDCs) have been identified as a subset of DCs able to secrete large amounts of IFN I after contact with CoVs (150), although this is not universal – MERS-CoV infection induces significantly higher type I and III IFNs production by pDCs than does SARS-CoV (151). While the role of pDCs in COVID-19 remains largely unexplored, the high levels of IFN-Is produced by these cells suggests a protective role.
Severe acute respiratory syndrome coronavirus 2 infection appears to target DCs directly – a recent study identified reduces DC frequency and functionally in COVID-19 patients, and a concordant impairment in the subsequent activation of T cells. Thus, in addition to further driving the cytokine storm, SARS-CoV-2 may infect DCs to limit DC maturation, and thus suppress T cell-mediated responses (44, 152, 153). This may represent a critical juncture in disease progression, as interference of T cell activation by modulating DC maturation could account for the lack of long-lasting humoral immunity and other defects in adaptive immunity associated with CoV infection. While infection of DCs appears to play an important role in driving the cytokine storm and modulating T cell responsiveness to SARS-CoV-2, the specific mechanisms used by the virus to alter DC function remains to be investigated.
Trained Immunity: A Defense Against COVID-19?
Recent studies have shown that innate immune populations may possess a memory phenotype, termed Trained Immunity (TRIM), wherein innate immune cells undergo mitochondrial, metabolic, and epigenetic reprograming following exposure to a pathogen (154), making the cell more responsive to subsequent pathogen exposures. Unlike with adaptive immunity, this “training” primes the cell to respond in an enhanced manner not only to the initial pathogen, but also to other pathogens encountered during the multi-week period over which TRIM lasts (154). TRIM can be effective against viral pathogens – for example, aerosolized bacterial lysates enhanced innate immune responses and increase survival against influenza A and other respiratory viruses (155, 156).
The induction of TRIM by Bacillus Calmette-Guerin (BCG) and other vaccines has been extensively demonstrated (157, 158). A recent study demonstrated that BCG administration to low-weight newborns reduced mortalities from infectious diseases by 43% over the neonatal period (159). It is tempting to postulate that the decreased incidence and rate of complications of COVID-19 reported in children (160) may be partially attributed to the frequent vaccinations – and therefore the frequent induction of TRIM – that occurs during routine childhood vaccination schedules. Encouraging studies have shown a correlation between universal BCG and influenza vaccination policies and reduced mortality rates in COVID-19 infection (161, 162), and while these studies are not directly indicative of a causal relationship between these vaccinations and protection against SARS-CoV-2, they suggest that using existing approved vaccines to induce TRIM may be a viable approach to limit the negative effects of COVID-19 (157, 158). Consequentially, a number of studies are investigating whether BCG and MMR vaccines can attenuate COVID-19 pathology (163–166).
While existing vaccines represent the quickest avenue to leverage TRIM as a preventative measure for COVID-19, other compounds may provide superior protection. For example, β-glucan polysaccharides found in the cell wall of bacteria, yeast and fungi, are known potent initiators of TRIM (167). β-glucans exert antiviral effects and decrease the severity of several respiratory viruses (168, 169). Orally administered β-glucan traffics into lymph nodes and spleen, where they activate DCs, leading to the expansion and activation of antigen-specific T cells and enhance T cell production of effector cytokines such as IFN-γ (170). This enhanced T cell activity results from β-glucan induced IFN-β production by DCs, which enhances the production of IFN-γ and Granzyme-B by CD8+ T cells (171). While β-glucans are commonly consumed by humans in foods prepared with yeast, care needs to be taken when considering their use for inducing TRIM in SARS-CoV-2 patients, as some studies have shown that β-glucans enhance M1 polarization of AMs – a phenomenon that may enhance the cytokine storm induced by SARS-CoV-2 (172, 173).
Future Prospects and Conclusion
The rapid spread of SARS-CoV-2 has become a global concern. Currently, there are no approved drugs or vaccines to treat human CoVs, but recent advances in our understanding of the immune response and immune evasion mechanisms of CoV’s opens up many therapeutic avenues. These include mechanisms for limiting viral entry and replication, promoting viral clearance, and inducing productive anti-CoV immune responses. Investigating how SARS-CoV-2 modifies gene expression in innate immune cells will be crucial to identifying immune mechanisms that could be modulated to improve patient outcomes. Addressing IFN evasion mechanisms and preventing viral immune evasion may contribute to enhancing viral clearance and lessening immunopathology. While a major driver of the cytokine storm in COVID-19 patients, IL-6 has both pro- and anti-inflammatory properties, giving it a complex role in COVID-19 pathology. Inhibition of IL-6 signaling and elucidation of the mechanism which elevates IL-6 in patients will help to find new potential strategies to reduce pathology during COVID-19 infection. Finally, newly available tools such as next generation sequencing will provide key information on the clinical features of the disease and potential targets for the development of drugs and vaccines. While an effective treatment for COVID-19 remains elusive, this large array of tools and knowledge should enable the rapid development of preventative and therapeutic treatments for this newly emerged disease.
Statements
Author contributions
NT prepared the main body of the manuscript, with assistance from ST. NH performed the bioinformatics analysis. BH supervised the project and assisted in the authoring and revision of the manuscript. All authors contributed to the article and approved the submitted version.
Funding
The work in BH’s laboratory was funded by the Canadian Institute for Health Research Operating Grant (Project Grant 406738) and Ontario Ministry of Research and Innovation Early Researcher Award.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2020.580641/full#supplementary-material
References
1.
PrompetcharaEKetloyCPalagaT.Immune responses in COVID-19 and potential vaccines: lessons learned from SARS and MERS epidemic.Asian Pac J Allergy Immunol. (2020) 38:1–9.
2.
ZhuNZhangDWangWLiXYangBSongJet alA novel coronavirus from patients with pneumonia in China, 2019.N Engl J Med. (2020) 382:727–33.
3.
De WitEvan DoremalenNFalzaranoDMunsterVJ.SARS and MERS: recent insights into emerging coronaviruses.Nat Rev Microbiol. (2016) 14:523–34. 10.1038/nrmicro.2016.81
4.
PaulesCIstonHDFauciAS.Coronavirus Infections—more than just the common cold.JAMA. (2020) 323:707–8. 10.1001/jama.2020.0757
5.
WangTDuZZhuFCaoZAnYGaoYet alComorbidities and multi-organ injuries in the treatment of COVID-19.Lancet. (2020) 395:e52. 10.1016/s0140-6736(20)30558-4
6.
ChanJF-WYuanSKokK-HToKK-WChuHYangJet alA familial cluster of pneumonia associated with the 2019 el coronavirus indicating person-to-person transmission: a study of a family cluster.Lancet. (2020) 395:514–23. 10.1016/s0140-6736(20)30154-9
7.
WuFZhaoSYuBChenY-MWangWSongZ-Get alA new coronavirus associated with human respiratory disease in China.Nature. (2020) 579:265–9.
8.
SuSWongGShiWLiuJLaiACKZhouJet alEpidemiology, genetic recombination, and pathogenesis of coronaviruses.Trends Microbiol. (2016) 24:490–502. 10.1016/j.tim.2016.03.003
9.
PerlmanSNetlandJ.Coronaviruses post-SARS: update on replication and pathogenesis.Nat Rev Microbiol. (2009) 7:439–50. 10.1038/nrmicro2147
10.
WangKChenWZhouY-SLianJ-QZhangZDuPet alSARS-CoV-2 invades host cells via a el route: CD147-spike protein.bioRxiv. (2020). [Preprint]. 10.1101/2020.03.14.988345
11.
BonaviaAZelusBDWentworthDETalbotPJHolmesKV.Identification of a receptor-binding domain of the spike glycoprotein of human coronavirus HCoV-229E.J Virol. (2003) 77:2530. 10.1128/jvi.77.4.2530-2538.2003
12.
HoffmannMKleine-WeberHSchroederSKrügerNHerrlerTErichsenSet alSARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and Is blocked by a clinically proven protease inhibitor.Cell [Internet]. (2020) 181:271–80.e8. 10.1016/j.cell.2020.02.052
13.
ShullaAHeald-SargentTSubramanyaGZhaoJPerlmanSGallagherTA.Transmembrane serine protease is linked to the severe acute respiratory syndrome coronavirus receptor and activates virus entry.J Virol. (2011) 85:873. 10.1128/jvi.02062-10
14.
LuRZhaoXLiJNiuPYangBWuHet alGenomic characterisation and epidemiology of 2019 el coronavirus: implications for virus origins and receptor binding.Lancet. (2020) 395:565–74. 10.1016/s0140-6736(20)30251-8
15.
XiaoKZhaiJFengYZhouNZhangXZouJ-Jet alIsolation of SARS-CoV-2-related coronavirus from Malayan pangolins.Nature [Internet]. (2020) 583:286–9. 10.1038/s41586-020-2313-x
16.
LiXGiorgiEEichannMHFoleyBXiaoCKongX-Pet alEmergence of SARS-CoV-2 through recombination and strong purifying selection.bioRxiv. (2020). [Preprint]. 10.1101/2020.03.20.000885
17.
YuanYCaoDZhangYMaJQiJWangQet alCryo-EM structures of MERS-CoV and SARS-CoV spike glycoproteins reveal the dynamic receptor binding domains.Nat Commun. (2017) 8:15092.
18.
LetkoMziAMunsterV.Functional assessment of cell entry and receptor usage for SARS-CoV-2 and other lineage B betacoronaviruses.Nat Microbiol. (2020) 5:562–9. 10.1038/s41564-020-0688-y
19.
OuXLiuYLeiXLiPMiDRenLet alCharacterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV.Nat Commun. (2020) 11:1620.
20.
TaefehshokrNBaradaranBBaghbanzadehATaefehshokrS.Promising approaches in cancer immunotherapy.Immunobiology. (2020) 225:151875. 10.1016/j.imbio.2019.11.010
21.
FungTSLiuDX.Human coronavirus: host-pathogen interaction.Annu Rev Microbiol. (2019) 73:529–57. 10.1146/annurev-micro-020518-115759
22.
HuYLiWGaoTCuiYJinYLiPet alThe Severe acute respiratory syndrome coronavirus nucleocapsid inhibits type I interferon production by interfering with TRIM25-Mediated RIG-I ubiquitination.J Virol. (2017) 91:e2143–2116.
23.
CatanzaroMFagianiFRacchiMCorsiniEGovoniSLanniC.Immune response in COVID-19: addressing a pharmacological challenge by targeting pathways triggered by SARS-CoV-2.Signal Transduct Target Ther. (2020) 5:84–84.
24.
de ckenMDhaliwalKDanielsenACGautronASDominguez-VillarM.TLR7 and TLR8 activate distinct pathways in monocytes during RNA virus infection.Sci Signal. (2019) 12:eaaw1347. 10.1126/scisignal.aaw1347
25.
HuangCWangYLiXRenLZhaoJHuYet alClinical features of patients infected with 2019 el coronavirus in Wuhan, China.Lancet. (2020) 395:497–506.
26.
VabretNBrittonGJGruberCHegdeSKimJKuksinMet alImmunology of COVID-19: current state of the science.Immunity [Internet]. (2020) 52:910–41.
27.
MahallawiWHKhabourOFZhangQMakhdoumHMSulimanBA.MERS-CoV infection in humans is associated with a pro-inflammatory Th1 and Th17 cytokine profile.Cytokine. (2018) 104:8–13. 10.1016/j.cyto.2018.01.025
28.
WongCKLamCWKWuAKLIpWKLeeNLSChanIHSet alPlasma inflammatory cytokines and chemokines in severe acute respiratory syndrome.Clin Exp Immunol. (2004) 136:95–103.
29.
JossetLMenacheryVDGralinskiLEAgnihothramSSovaPCarterVSet alCell host response to infection with el human coronavirus EMC predicts potential antivirals and important differences with SARS coronavirus.mBio. (2013) 4:e00165.
30.
LiGFanYLaiYHanTLiZZhouPet alCoronavirus infections and immune responses.J Med Virol. (2020) 92:424–32.
31.
ZieglerCGKAllonSJNyquistSKMbanoIMiaoVNCaoYet alSARS-CoV-2 receptor ACE2 is an interferon-stimulated gene in human airway epithelial cells and is enriched in specific cell subsets across tissues.Cell. (2020) 181:1016–35.e19.
32.
BaileyCCZhongGHuangI-CFarzanM.IFITM-family proteins: the cell’s first line of antiviral defense.Annu Rev Virol. (2014) 1:261–83. 10.1146/annurev-virology-031413-085537
33.
BékésMRutWKasperkiewiczPMulderMPCOvaaHDragMet alSARS hCoV papain-like protease is a unique Lys48 linkage-specific di-distributive deubiquitinating enzyme.Biochem J. (2015) 468:215–26. 10.1042/bj20141170
34.
MielechAMKilianskiABaez-SantosYMMesecarADBakerSC.MERS-CoV papain-like protease has deISGylating and deubiquitinating activities.Virology. (2014) 450–451:64–70. 10.1016/j.virol.2013.11.040
35.
Trouillet-AssantSVielSGaydAPonsSRichardJ-CPerretMet alType I IFN immunoprofiling in COVID-19 patients.J Allergy Clin Immunol [Internet]. (2020) 146:206–8.e2.
36.
LokugamageKGHageASchindewolfCRajsbaumRMenacheryVD.SARS-CoV-2 is sensitive to type I interferon pretreatment.bioRxiv. (2020). [Preprint]. 10.1101/2020.03.07.982264
37.
MantloEBukreyevaNuyamaJPaesslerSHuangC.Antiviral activities of type I interferons to SARS-CoV-2 infection.Antiviral Res. (2020) 179:104811. 10.1016/j.antiviral.2020.104811
38.
StockmanLJBellamyRGarnerP.SARS: systematic review of treatment effects.PLoS Med. (2006) 3:e343. 10.1371/journal.pmed.0030343
39.
YuanJZouRZengLKouSLanJLiXet alThe correlation between viral clearance and biochemical outcomes of 94 COVID-19 infected discharged patients.Inflamm Res [Internet]. (2020) 69:599–606. 10.1007/s00011-020-01342-0
40.
DavidsonSMcCabeTMCrottaSGadHHHesselEMBeinkeSet alIFNλ is a potent anti-influenza therapeutic without the inflammatory side effects of IFNα treatment.EMBO Mol Med. (2016) 8:1099–112. 10.15252/emmm.201606413
41.
GalaniIETriantafylliaVEleminiadouE-EKoltsidaOStavropoulosAManioudakiMet alInterferon-λ mediates non-redundant front-line antiviral protection against influenza virus infection without compromising host fitness.Immunity (2017) 46:875–90.e6.
42.
PlanetPJParkerDCohenTSSmithHLeonJDRyanCet alLambda interferon restructures the nasal microbiome and increases susceptibility to Staphylococcus aureus superinfection Schneewind O, editor.mBio. (2016) 7:e1939–1915.
43.
RichHEMcCourtCCZhengWQMcHughKJRobinsonKMWangJet alInterferon lambda inhibits bacterial uptake during influenza superinfection Bäumler AJ, editor.Infect Immun. (2019) 87:e114–9.
44.
Blanco-MeloDNilsson-PayantBELiuW-CUhlSHoaglandDMøllerRet alImbalanced host response to SARS-CoV-2 drives development of COVID-19.Cell (2020) 181:1036–45.e9.
45.
StaniferMLKeeCCorteseMZuanCMTrianaSMukenhirnMet alCritical role of type III interferon in controlling SARS-CoV-2 infection in human intestinal epithelial cells.Cell Rep [Internet]. (2020) 32:107863. 10.1016/j.celrep.2020.107863
46.
MajorJCrottaSLlorianMMcCabeTMGadHHPriestnallSLet alType I and III interferons disrupt lung epithelial repair during recovery from viral infection.Science. (2020) 369:712. 10.1126/science.abc2061
47.
BroggiAGhoshSSpositoBSpreaficoRBalzariniFLo CascioAet alType III interferons disrupt the lung epithelial barrier upon viral recognition.Science. (2020) 369:706. 10.1126/science.abc3545
48.
HallerOKochsGWeberF.The interferon response circuit: induction and suppression by pathogenic viruses.Virology. (2006) 344:119–30. 10.1016/j.virol.2005.09.024
49.
RodriguezJJParisienJ-PHorvathCM.Nipah virus V protein evades alpha and gamma interferons by preventing STAT1 and STAT2 activation and nuclear accumulation.J Virol. (2002) 76:11476–83. 10.1128/jvi.76.22.11476-11483.2002
50.
ShokriSMahmoudvandSTaherkhaniRFarshadpourF.Modulation of the immune response by Middle East respiratory syndrome coronavirus.J Cell Physiol. (2019) 234:2143–51. 10.1002/jcp.27155
51.
ZhaoXNichollsJMChenY-G.Severe Acute respiratory syndrome-associated coronavirus nucleocapsid protein interacts with smad3 and modulates transforming growth factor-β signaling.J Biol Chem. (2008) 283:3272–80. 10.1074/jbc.m708033200
52.
KnoopsKKikkertMWormSHEZevenhoven-DobbeJCvan der MeerYKosterAJet alSARS-coronavirus replication is supported by a reticulovesicular network of modified endoplasmic reticulum.PLoS Biol. (2008) 6:e226–226. 10.1371/journal.pbio.0060226
53.
SnijderEJvan der MeerYZevenhoven-DobbeJOnderwaterJJMvan der MeulenJKoertenHKet alUltrastructure and origin of membrane vesicles associated with the severe acute respiratory syndrome coronavirus replication complex.J Virol. (2006) 80:5927–40. 10.1128/jvi.02501-05
54.
LiS-WWangC-YJouY-JHuangS-HHsiaoL-HWanLet alSARS coronavirus papain-like protease inhibits the TLR7 Signaling pathway through removing Lys63-linked polyubiquitination of TRAF3 and TRAF6.Int J Mol Sci. (2016) 17:678. 10.3390/ijms17050678
55.
ToturaALWhitmoreAAgnihothramSSchäferAKatzeMGHeiseMTet alToll-like receptor 3 signaling via TRIF contributes to a protective innate immune response to severe acute respiratory syndrome coronavirus infection.mBio. (2015) 6:e638–615.
56.
FanZZhuoYTanXZhouZYuanJQiangBet alSARS-CoV nucleocapsid protein binds to hUbc9, a ubiquitin conjugating enzyme of the sumoylation system.J Med Virol. (2006) 78:1365–73. 10.1002/jmv.20707
57.
FaureEPoissyJGoffardAFournierCKipnisETitecatMet alDistinct immune response in two MERS-CoV-Infected patients: can we go from bench to bedside?PLoS One. (2014) 9:e88716. 10.1371/journal.pone.0088716
58.
ChannappanavarRFehrARZhengJWohlford-LenaneCAbrahanteJEMackMet alIFN-I response timing relative to virus replication determines MERS coronavirus infection outcomes.J Clin Invest. (2019) 130:3625–39. 10.1172/jci126363
59.
ChannappanavarRFehrARVijayRMackMZhaoJMeyerholzDKet alDysregulated type I interferon and inflammatory monocyte-macrophage responses cause lethal pneumonia in SARS-CoV-infected mice.Cell Host Microbe. (2016) 19:181–93. 10.1016/j.chom.2016.01.007
60.
HadjadjJYatimNBarnabeiLCorneauABoussierJPereHet alImpaired type I interferon activity and exacerbated inflammatory responses in severe Covid-19 patients.medRxiv. (2020). [Preprint]. 10.1101/2020.04.19.20068015
61.
KamitaniWNarayananKHuangCLokugamageKIkegamiTItoNet alSevere acute respiratory syndrome coronavirus nsp1 protein suppresses host gene expression by promoting host mRNA degradation.Proc Natl Acad Sci USA. (2006) 103:12885. 10.1073/pnas.0603144103
62.
FriemanMYountBHeiseMKopecky-BrombergSAPalesePBaricRS.Severe acute respiratory syndrome coronavirus ORF6 antagonizes STAT1 function by sequestering nuclear import factors on the rough endoplasmic reticulum/golgi membrane.J Virol. (2007) 81:9812. 10.1128/jvi.01012-07
63.
SpiegelMPichlmairAtínez-SobridoLCrosJGarcía-SastreAHallerOet alInhibition of beta interferon induction by severe acute respiratory syndrome coronavirus suggests a two-step model for activation of interferon regulatory factor 3.J Virol. (2005) 79:2079. 10.1128/jvi.79.4.2079-2086.2005
64.
Kopecky-BrombergSAtínez-SobridoLFriemanMBaricRAPaleseP.Severe acute respiratory syndrome coronavirus open reading frame (ORF) 3b, ORF 6, and nucleocapsid proteins function as interferon antagonists.J Virol. (2007) 81:548–57. 10.1128/jvi.01782-06
65.
SiuK-LYeungMLKokK-HYuenK-SKewCLuiP-Yet alMiddle east respiratory syndrome coronavirus 4a protein is a double-stranded RNA-binding protein that suppresses PACT-induced activation of RIG-I and MDA5 in the innate antiviral response.J Virol. (2014) 88:4866–76. 10.1128/jvi.03649-13
66.
NiemeyerDZillingerTMuthDZieleckiFHorvathGSulimanTet alMiddle East respiratory syndrome coronavirus accessory protein 4a is a type I interferon antagonist.J Virol. (2013) 87:12489–95. 10.1128/jvi.01845-13
67.
YangYYeFZhuNWangWDengYZhaoZet alMiddle East respiratory syndrome coronavirus ORF4b protein inhibits type I interferon production through both cytoplasmic and nuclear targets.Sci Rep. (2015) 5:17554.
68.
MatthewsKLColemanCMvan der MeerYSnijderEJFriemanMB.The ORF4b-encoded accessory proteins of Middle East respiratory syndrome coronavirus and two related bat coronaviruses localize to the nucleus and inhibit innate immune signalling.J Gen Virol. (2014) 95(Pt 4):874–82. 10.1099/vir.0.062059-0
69.
YangYZhangLGengHDengYHuangBGuoYet alThe structural and accessory proteins M, ORF 4a, ORF 4b, and ORF 5 of Middle East respiratory syndrome coronavirus (MERS-CoV) are potent interferon antagonists.Protein Cell. (2013) 4:951–61. 10.1007/s13238-013-3096-8
70.
KonnoYKimuraIUriuKFukushiMIrieTKoyanagiYet alSARS-CoV-2 ORF3b is a potent interferon antagonist whose activity is further increased by a naturally occurring elongation variant.bioRxiv. (2020). [Preprint]. 10.1101/2020.05.11.088179
71.
HachimAKavianNCohenCAChinAWChuDKMokCKPet alBeyond the Spike: identification of viral targets of the antibody response to SARS-CoV-2 in COVID-19 patients.medRxiv. (2020). [Preprint]. 10.1101/2020.04.30.20085670
72.
YuenC-KLamJ-YWongW-MMakL-FWangXChuHet alSARS-CoV-2 nsp13, nsp14, nsp15 and orf6 function as potent interferon antagonists.Emerg Microbes Infect. (2020) 9:1418–28. 10.1080/22221751.2020.1780953
73.
ZhengYZhuangM-WHanLZhangJNanM-LGaoCet alSevere acute respiratory syndrome coronavirus 2 (SARS-CoV-2) membrane (M) protein inhibits type I and III interferon production by targeting RIG-I/MDA-5 signaling.bioRxiv. (2020). [Preprint]. 10.1101/2020.07.26.222026
74.
LindnerHAFotouhi-ArdakaniNLytvynVLachancePSuleaTMénardR.The papain-like protease from the severe acute respiratory syndrome coronavirus is a deubiquitinating enzyme.J Virol. (2005) 79:15199. 10.1128/jvi.79.24.15199-15208.2005
75.
Bailey-ElkinBAKnaapRCMJohnsonGGDaleboutTJNinaberDKvan KasterenPBet alCrystal structure of the Middle East respiratory syndrome coronavirus (MERS-CoV) papain-like protease bound to ubiquitin facilitates targeted disruption of deubiquitinating activity to demonstrate its role in innate immune suppression.J Biol Chem. (2014) 289:34667–82. 10.1074/jbc.m114.609644
76.
ChenGWuDGuoWCaoYHuangDWangHet alClinical and immunological features of severe and moderate coronavirus disease 2019.J Clin Invest. (2020) 130:2620–9. 10.1172/jci137244
77.
LiuYYanL-MWanLXiangT-XLeALiuJ-Met alViral dynamics in mild and severe cases of COVID-19.Lancet Infect Dis. (2020) 20:656–7. 10.1016/s1473-3099(20)30232-2
78.
ChenXZhaoBQuYChenYXiongJFengYet alDetectable serum severe acute respiratory syndrome coronavirus 2 viral load (RNAemia) is closely correlated with drastically elevated interleukin 6 level in critically Ill patients with coronavirus disease 2019.Clin Infect Dis [Internet]. (2020) 17:ciaa449. 10.1093/cid/ciaa449
79.
RuanQYangKWangWJiangLSongJ.Clinical predictors of mortality due to COVID-19 based on an analysis of data of 150 patients from Wuhan, China.Intensive Care Med. (2020) 46:846–8. 10.1007/s00134-020-05991-x
80.
ImaiYKubaKNeelyGGYaghubian-MalhamiRPerkmannTvan LooGet alIdentification of oxidative stress and toll-like receptor 4 signaling as a key pathway of acute lung injury.Cell. (2008) 133:235–49.
81.
NyatiKKMasudaKZamanMM-UDubeyPKMillrineDChaliseJPet alTLR4-induced NF-κB and MAPK signaling regulate the IL-6 mRNA stabilizing protein Arid5a.Nucleic Acids Res. (2017) 45:2687–703. 10.1093/nar/gkx064
82.
ZhangXWuKWangDYueXSongDZhuYet alNucleocapsid protein of SARS-CoV activates interleukin-6 expression through cellular transcription factor NF-κB.Virology. (2007) 365:324–35. 10.1016/j.virol.2007.04.009
83.
ChengGZhongJChungJChisariFV.Double-stranded DNA and double-stranded RNA induce a common antiviral signaling pathway in human cells.Proc Natl Acad Sci USA. (2007) 104:9035–40. 10.1073/pnas.0703285104
84.
ChiuY-HMacMillanJBChenZJ.RNA polymerase III detects cytosolic DNA and induces type I interferons through the RIG-I pathway.Cell. (2009) 138:576–91. 10.1016/j.cell.2009.06.015
85.
JohnsenIBNguyenTTRingdalMTryggestadAMBakkeOLienEet alToll-like receptor 3 associates with c-Src tyrosine kinase on endosomes to initiate antiviral signaling.EMBO J. (2006) 25:3335–46. 10.1038/sj.emboj.7601222
86.
Al-QahtaniAALyroniKAznaourovaMTseliouMAl-AnaziMRAl-AhdalMNet alMiddle east respiratory syndrome corona virus spike glycoprotein suppresses macrophage responses via DPP4-mediated induction of IRAK-M and PPARγ.Oncotarget. (2017) 8:9053–66. 10.18632/oncotarget.14754
87.
HuWYenY-TSinghSKaoC-LWu-HsiehBA.SARS-CoV regulates immune function-related gene expression in human monocytic cells.Viral Immunol. (2012) 25:277–88. 10.1089/vim.2011.0099
88.
RodriguezLPekkarinenPTadepallyLKTanZRosatCCPouCet alSystems-level immunomonitoring from acute to recovery phase of severe COVID-19.medRxiv. (2020). [Preprint]. 10.1101/2020.06.03.20121582
89.
SongC-YXuJHeJ-QLuY-Q.COVID-19 early warning score: a multi-parameter screening tool to identify highly suspected patients.medRxiv. (2020). [Preprint]. 10.1101/2020.03.05.20031906
90.
ZhangBZhouXZhuCFengFQiuYFengJet alImmune phenotyping based on neutrophil-to-lymphocyte ratio and IgG predicts disease severity and outcome for patients with COVID-19.medRxiv. (2020). [Preprint]. 10.1101/2020.03.12.20035048.
91.
FoxSEAkmatbekovAHarbertJLLiGBrownJQVanderHRS.Pulmonary and cardiac pathology in Covid-19: the first autopsy series from new orleans.medRxiv (2020). [Preprint]. 0.1101/2020.04.06.20050575
92.
YaoXLiTHeZPingYLiuHYuSet al[A pathological report of three COVID-19 cases by minimal invasive autopsies].Zhonghua Bing Li Xue Za Zhi. (2020) 49:411–7.
93.
AliRAGandhiAAMengHYalavarthiSVreedeAPEstesSKet alAdenosine receptor agonism protects against NETosis and thrombosis in antiphospholipid syndrome.Nat Commun. (2019) 10:1916.
94.
GrailerJJCanningBAKalbitzMHaggadoneMDDhondRMAndjelkovicAVet alCritical role for the NLRP3 inflammasome during acute lung injury.J Immunol. (2014) 192:5974. 10.4049/jimmunol.1400368
95.
XuJZhangXPelayoRMonestierMAmmolloCTSemeraroFet alExtracellular histones are major mediators of death in sis.Nat Med. (2009) 15:1318–21. 10.1038/nm.2053
96.
Narayana MoorthyANarasarajuTRaiPPerumalsamyRTanKBWangSet alIn vivo and in vitro studies on the roles of neutrophil extracellular traps during secondary pneumococcal pneumonia after priy pulmonary influenza infection.Front Immunol. (2013) 4:56–56.
97.
SaffarzadehMJuenemannCQueisserMALochnitGBarretoGGaluskaSPet alNeutrophil extracellular traps directly induce epithelial and endothelial cell death: a predominant role of histones.PLoS One. (2012) 7:e32366. 10.1371/journal.pone.0032366
98.
TwaddellSHBainesKJGraingeCGibsonPG.The emerging role of neutrophil extracellular traps in respiratory disease.CHEST. (2019) 156:774–82. 10.1016/j.chest.2019.06.012
99.
BarnesBJAdroverJMBaxter-StoltzfusABorczukACools-LartigueJCrawfordJMet alTargeting potential drivers of COVID-19: neutrophil extracellular traps.J Exp Med [Internet]. (2020) 217:e20200652.
100.
ZuoYYalavarthiSShiHGockmanKZuoMMadisonJAet alNeutrophil extracellular traps in COVID-19.JCI Insight [Internet]. (2020) 5:11.
101.
MehtaPMcAuleyDFBrownMSanchezETattersallRSMansonJJ.COVID-19: consider cytokine storm syndromes and immunosuppression.Lancet. (2020) 395:1033–4. 10.1016/s0140-6736(20)30628-0
102.
MerzaMHartmanHRahmanMHwaizRZhangERenströmEet alNeutrophil extracellular traps induce trypsin activation, inflammation, and tissue damage in mice with severe acute pancreatitis.Gastroenterology. (2015) 149:1920–31.e8.
103.
CortjensBde BoerOJde JongRAntonisAFSabogal PiñerosYSLutterRet alNeutrophil extracellular traps cause airway obstruction during respiratory syncytial virus disease.J Pathol. (2016) 238:401–11. 10.1002/path.4660
104.
NarasarajuTYangESamyRPNgHHPohWPLiewA-Aet alExcessive neutrophils and neutrophil extracellular traps contribute to acute lung injury of influenza pneumonitis.Am J Pathol. (2011) 179:199–210. 10.1016/j.ajpath.2011.03.013
105.
GuptaAKJoshiMBPhilippovaMErnePHaslerPHahnSet alActivated endothelial cells induce neutrophil extracellular traps and are susceptible to NETosis-mediated cell death.S Lett. (2010) 584:3193–7. 10.1016/j.febslet.2010.06.006
106.
ClarkSRMaACTavenerSAMcDonaldBGoodarziZKellyMMet alPlatelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in tic blood.Nat Med. (2007) 13:463–9. 10.1038/nm1565
107.
McDonaldBDavisRPKimS-JTseMEsmonCTKolaczkowskaEet alPlatelets and neutrophil extracellular traps collaborate to promote intravascular coagulation during sis in mice.Blood. (2017) 129:1357–67. 10.1182/blood-2016-09-741298
108.
Meher AkshayaKSpinosaMDavisJPPopeNLaubachVESuGet alNovel role of IL (Interleukin)-1β in neutrophil extracellular trap formation and abdominal aortic aneurysms.Arterioscler Thromb Vasc Biol. (2018) 38:843–53. 10.1161/atvbaha.117.309897
109.
SilPWicklumHSurellCRadaB.Macrophage-derived IL-1β enhances monosodium urate crystal-triggered NET formation.Inflamm Res. (2017) 66:227–37. 10.1007/s00011-016-1008-0
110.
XiongYLiuYCaoLWangDGuoMJiangAet alTranscriptomic characteristics of bronchoalveolar lavage fluid and peripheral blood mononuclear cells in COVID-19 patients.Emerg Microbes Infect. (2020) 9:761–70. 10.1080/22221751.2020.1747363
111.
de BontCMBoelensWCPruijnGJM.NETosis, complement, and coagulation: a triangular relationship.Cell Mol Immunol. (2019) 16:19–27. 10.1038/s41423-018-0024-0
112.
GaoTHuMZhangXLiHZhuLLiuHet alHighly pathogenic coronavirus N protein aggravates lung injury by MASP-2-mediated complement over-activation.medRxiv. (2020). [Preprint]. 10.1101/2020.03.29.20041962
113.
LlitjosJ-FLeclercMChochoisCMonsallierJ-MRamakersMAuvrayMet alHigh incidence of venous thromboembolic events in anticoagulated severe COVID-19 patients.J Thromb Haemost JTH. (2020) 18:1743–6. 10.1111/jth.14869
114.
LiuXLiZLiuSSunJChenZJiangMet alPotential therapeutic effects of dipyridamole in the severely ill patients with COVID-19.Acta Pharm Sin B. (2020) 20:1205–15. 10.1016/j.apsb.2020.04.008
115.
ZhangCWangXLiSTwelkmeyerTWangWZhangSet alNKG2A is a NK cell exhaustion checkpoint for HCV persistence.Nat Commun. (2019) 10:1507.
116.
ZhengMGaoYWangGSongGLiuSSunDet alFunctional exhaustion of antiviral lymphocytes in COVID-19 patients.Cell Mol Immunol. (2020) 17:533–5. 10.1038/s41423-020-0402-2
117.
WangWHeJLiePHuangLWuSYongpingLet alThe definition and risks of cytokine release syndrome-Like in 11 COVID-19-Infected Pneumonia critically ill patients: disease characteristics and retrospective analysis.medRxiv. (2020). [Preprint]. 10.1101/2020.02.26.20026989
118.
YuLTongYShenGFuALaiYZhouXet alImmunodepletion with hypoxemia: a potential high risk subtype of coronavirus disease 2019.medRxiv. (2020). [Preprint]. 10.1101/2020.03.03.20030650
119.
SivoriSVaccaPDel ZottoGMunariEMingariMCMorettaL.Human NK cells: surface receptors, inhibitory checkpoints, and translational applications.Cell Mol Immunol. (2019) 16:430–41. 10.1038/s41423-019-0206-4
120.
WangFNieJWangHZhaoQXiongYDengLet alCharacteristics of peripheral lymphocyte subset alteration in COVID-19 pneumonia.J Infect Dis. (2020) 221:1762–9. 10.1093/infdis/jiaa150
121.
XiaC-QXuL-LWangZQinZ-QTongZ-HHuangK-Wet alThe involvement of natural killer cells in the pathogenesis of severe acute respiratory syndrome.Am J Clin Pathol. (2004) 121:507–11. 10.1309/wpk7-y2xk-nf4c-bf3r
122.
CifaldiLPrencipeGCaielloIBracagliaCLocatelliFDe BenedettiFet alInhibition of natural killer cell cytotoxicity by interleukin-6: implications for the pathogenesis of macrophage activation syndrome.Arthritis Rheumatol. (2015) 67:3037–46. 10.1002/art.39295
123.
FerlazzoGPackMThomasDPaludanCSchmidDStrowigTet alDistinct roles of IL-12 and IL-15 in human natural killer cell activation by dendritic cells from secondary lymphoid organs.Proc Natl Acad Sci USA. (2004) 101:16606–11. 10.1073/pnas.0407522101
124.
tín-FontechaAThomsenLLBrettSGerardCLippMLanzavecchiaAet alInduced recruitment of NK cells to lymph nodes provides IFN-γ for TH1 priming.Nat Immunol. (2004) 5:1260–5. 10.1038/ni1138
125.
de GroenRABoltjesAHouJLiuB-SMcPheeFFriborgJet alIFN-λ-mediated IL-12 production in macrophages induces IFN-γ production in human NK cells.Eur J Immunol. (2015) 45:250–9. 10.1002/eji.201444903
126.
AndréPDenisCSoulasCBourbon-CailletCLopezJArnouxTet alAnti-NKG2A mAb is a checkpoint inhibitor that promotes anti-tumor immunity by unleashing both T and NK cells.Cell. (2018) 175:1731–43.e13.
127.
LiFWeiHWeiHGaoYXuLYinWet alBlocking the natural killer cell inhibitory receptor NKG2A increases activity of human natural killer cells and clears hepatitis B virus infection in mice.Gastroenterology. (2013) 144:392–401. 10.1053/j.gastro.2012.10.039
128.
WilkAJRustagiAZhaoNQRoqueJtinez-ColonGJMcKechnieJLet alA single-cell atlas of the peripheral immune response to severe COVID-19.medRxiv. (2020). [Preprint]. 10.1101/2020.04.17.20069930
129.
PintoDParkY-JBeltramelloMWallsACTortoriciMABianchiSet alStructural and functional analysis of a potent sarbecovirus neutralizing antibody.bioRxiv. (2020). [Preprint]. 10.1101/2020.04.07.023903
130.
ChenYFengZDiaoBWangRWangGWangCet alThe novel severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) directly imates human spleens and lymph nodes.medRxiv. (2020). [Preprint]. 10.1101/2020.03.27.20045427.
131.
LavinYMorthaARahmanAMeradM.Regulation of macrophage development and function in peripheral tissues.Nat Rev Immunol. (2015) 15:731–44. 10.1038/nri3920
132.
MeradMtinJC.Pathological inflammation in patients with COVID-19: a key role for monocytes and macrophages.Nat Rev Immunol [Internet]. (2020) 6:355–62. 10.1038/s41577-020-0331-4
133.
UralBBYeungSTDamani-YokotaPDevlinJCde VriesMVera-LiconaPet alIdentification of a nerve-associated, lung-resident interstitial macrophage subset with distinct localization and immunoregulatory properties.Sci Immunol. (2020) 5:eaax8756. 10.1126/sciimmunol.aax8756
134.
LiaoMLiuYYuanJWenYXuGZhaoJet alThe landscape of lung bronchoalveolar immune cells in COVID-19 revealed by single-cell RNA sequencing.medRxiv. (2020). [Preprint]. 10.1101/2020.02.23.20026690
135.
ZhouJChuHLiCWongBH-YChengZ-SPoonVK-Met alActive replication of middle east respiratory syndrome coronavirus and aberrant induction of inflammatory cytokines and chemokines in human macrophages: implications for pathogenesis.J Infect Dis. (2013) 209:1331–42. 10.1093/infdis/jit504
136.
Giaellos-BourboulisEJNeteaMGRovinaNAkinosoglouKAntoniadouAAntonakosNet alComplex immune dysregulation in COVID-19 patients with severe respiratory failure.Cell Host Microbe. (2020) 27:992–1000.e3.
137.
MenacheryVDSchäferABurnum-JohnsonKEMitchellHDEisfeldAJWaltersKBet alMERS-CoV and H5N1 influenza virus antagonize antigen presentation by altering the epigenetic landscape.Proc Natl Acad Sci USA. (2018) 115:E1012.
138.
GordonDEgGMBouhaddouMXuJObernierKWhiteKMet alA SARS-CoV-2 protein interaction map reveals targets for drug repurposing.Nature [Internet]. (2020) 583:1–13. 10.1038/s41586-020-2286-9
139.
KeskinenPRonniTMatikainenSLehtonenAkunenI.Regulation of HLA class I and II expression by interferons and influenza A virus in human peripheral blood mononuclear cells.Immunology. (1997) 91:421–9. 10.1046/j.1365-2567.1997.00258.x
140.
FangW-FChenY-MLinC-YHuangH-LYehHChangY-Tet alHistone deacetylase 2 (HDAC2) attenuates lipopolysaccharide (LPS)-induced inflammation by regulating PAI-1 expression.J Inflamm. (2018) 15:3.
141.
KongXFangMLiPFangFXuY.HDAC2 deacetylates class II transactivator and suppresses its activity in macrophages and smooth muscle cells.J Mol Cell Cardiol. (2009) 46:292–9. 10.1016/j.yjmcc.2008.10.023
142.
ZhangYZhangJChenYLuoBYuanYHuangFet alThe ORF8 protein of SARS-CoV-2 mediates immune evasion through potently downregulating MHC-I.bioRxiv. (2020). [Preprint]. 10.1101/2020.05.24.111823
143.
DirkBSPawlakENJohnsonALVan NynattenLRJacobRAHeitBet alHIV-1 Nef sequesters MHC-I intracellularly by targeting early stages of endocytosis and recycling.Sci Rep. (2016) 6:37021.
144.
DirkBSHeitBDikeakosJD.Visualizing interactions between HIV-1 nef and host cellular proteins using ground-state depletion microscopy.AIDS Res Hum Retroviruses. (2015) 31:671–2. 10.1089/aid.2014.0333
145.
CendrowskiJMamiñskaAMiaczynskaM.Endocytic regulation of cytokine receptor signaling.Cytokine Growth Factor Rev. (2016) 32:63–73. 10.1016/j.cytogfr.2016.07.002
146.
McCormickPJtinaJABonifacinoJS.Involvement of clathrin and AP-2 in the trafficking of MHC class II molecules to antigen-processing compartments.Proc Natl Acad Sci USA. (2005) 102:7910. 10.1073/pnas.0502206102
147.
RaoVUSArakeriGSubashARaoJJadhavSSuhail SayeedMet alCOVID-19: Loss of bridging between innate and adaptive immunity?Med Hypotheses. (2020) 144:109861. 10.1016/j.mehy.2020.109861
148.
LawHKWCheungCYNgHYSiaSFChanYOLukWet alChemokine up-regulation in SARS-coronavirus-infected, monocyte-derived human dendritic cells.Blood. (2005) 106:2366–74. 10.1182/blood-2004-10-4166
149.
TsengC-TKPerroneLAZhuHMakinoSPetersCJ.Severe acute respiratory syndrome and the innate immune responses: modulation of effector cell function without productive infection.J Immunol. (2005) 174:7977. 10.4049/jimmunol.174.12.7977
150.
Cervantes-BarraganLZüstRWeberFSpiegelMLangKSAkiraSet alControl of coronavirus infection through plasmacytoid dendritic-cell–derived type I interferon.Blood. (2006) 109:1131–7. 10.1182/blood-2006-05-023770
151.
ScheupleinVASeifriedJMalczykAHMillerLHöckerLVergara-AlertJet alHigh secretion of interferons by human plasmacytoid dendritic cells upon recognition of Middle East respiratory syndrome coronavirus.J Virol. (2015) 89:3859–69. 10.1128/jvi.03607-14
152.
ZhouRToKK-WWongY-CLiuLZhouBLiXet alAcute SARS-CoV-2 infection impairs dendritic cell and T cell responses.Immunity. (2020) 32:132.
153.
ZhangZWangFZhaoMLiuJXuDJinLet al[Characterization of peripheral dendritic cell subsets and its implication in patients infected with severe acute respiratory syndrome].Zhonghua Yi Xue Za Zhi. (2004) 84:22–6.
154.
KolodnyOBergerMFeldmanMWRamY.A new perspective for mitigation of SARS-CoV-2 infection: priming the innate immune system for viral attack.Open Biol. (2020) 10:200138. 10.1098/rsob.200138
155.
PfeifferJKVirginHW.Transkingdom control of viral infection and immunity in the mammalian intestine.Science. (2016) 351:aad5872. 10.1126/science.aad5872
156.
TuvimMJEvansSEClementCGDickeyBFGilbertBE.Augmented lung inflammation protects against influenza A pneumonia.PLoS One. (2009) 4:e4176. 10.1371/journal.pone.0004176
157.
AabyPSambBSimondonFSeckAMKnudsenKWhittleH.Non-specific beneficial effect of measles immunisation: analysis of mortality studies from developing countries.BMJ. (1995) 311:481–5. 10.1136/bmj.311.7003.481
158.
Van’t WoutJWPoellRVan FurthR.The role of BCG/PPD-activated macrophages in resistance against systemic candidiasis in mice.Scand J Immunol. (1992) 36:713–20. 10.1111/j.1365-3083.1992.tb03132.x
159.
Biering-SørensenSAabyPLundNMonteiroIJensenKJEriksenHBet alEarly BCG-denk and neonatal mortality among infants weighing <2500 g: A randomized controlled trial.Clin Infect Dis Off Publ Infect Dis Soc Am. (2017) 65:1183–90. 10.1093/cid/cix525
160.
HoangAChorathKMoreiraAEvansMBurmeister-MortonFBurmeisterFet alCOVID-19 in 7780 pediatric patients: a systematic review.EClinicalMedicine [Internet]. (2020) 24:100433. 10.1016/j.eclinm.2020.100433
161.
ZanettiniCOMDinalankaraWImadaELColantuoniEParmigianiGet alInfluenza Vaccination and COVID19 Mortality in the USA.medRxiv. (2020). [Preprint]. 10.1101/2020.06.24.20129817
162.
MillerAReandelarMJFasciglioneKRoumeaVLiYOtazuGH.Correlation between universal BCG vaccination policy and reduced morbidity and mortality for COVID-19: an epidemiological study.medRxiv. (2020). [Preprint]. 10.1101/2020.03.24.20042937
163.
FidelPLNoverrMC.Could an unrelated live attenuated vaccine serve as a preventive measure to dampen tic inflammation associated with COVID-19 infection?mBio. (2020) 11:e907–20.
164.
KlingerDBlassIRappoportNLinialM.Significantly improved COVID-19 outcomes in countries with Higher BCG Vaccination coverage: a multivariable analysis.medRxiv. (2020). [Preprint]. 10.20944/preprints202006.0073.v1
165.
CurtisNSparrowAGhebreyesusTANeteaMG.Considering BCG vaccination to reduce the impact of COVID-19.Lancet. (2020) 395:1545–6. 10.1016/s0140-6736(20)31025-4
166.
O’NeillLAJNeteaMG.BCG-induced trained immunity: can it offer protection against COVID-19?Nat Rev Immunol. (2020) 20:335–7. 10.1038/s41577-020-0337-y
167.
KeatingSTGrohLvan der HeijdenCDCCRodriguezHdos SantosJCFanucchiSet alThe Set7 lysine methyltransferase regulates plasticity in oxidative phosphorylation necessary for trained immunity induced by β-glucan.Cell Rep. (2020) 31:107548. 10.1016/j.celrep.2020.107548
168.
JesenakMMajtanJRennerovaZKyselovicJBacinPHrubiskoM.Immunomodulatory effect of pleuran (β-glucan from Pleurotus ostreatus) in children with recurrent respiratory tract infections.Int Immunopharmacol. (2013) 15:395–9. 10.1016/j.intimp.2012.11.020
169.
VetvickaVVetvickovaJ.Glucan supplementation enhances the immune response against an influenza challenge in mice.Ann Transl Med. (2015) 3:22–22.
170.
LiBCaiYQiCHansenRDingCMitchellTCet alOrally administered particulate β-Glucan modulates tumor-capturing dendritic cells and improves antitumor T-cell responses in cancer.Clin Cancer Res. (2010) 16:5153. 10.1158/1078-0432.ccr-10-0820
171.
Hassanzadeh-KiabiNYáñezADangItinsGAUnderhillDMGoodridgeHS.Autocrine type I IFN signaling in dendritic cells stimulated with fungal β-glucans or lipopolysaccharide promotes CD8 T cell activation.J Immunol. (2017) 198:375. 10.4049/jimmunol.1601143
172.
ChaungH-CHuangT-CYuJ-HWuM-LChungW-B.Immunomodulatory effects of β-glucans on porcine alveolar macrophages and bone row haematopoietic cell-derived dendritic cells.Vet Immunol Immunopathol. (2009) 131:147–57. 10.1016/j.vetimm.2009.04.004
173.
ChanASQiuXJonasABPatchenMLBoseN.Imprime PGG, a yeast β-glucan immunomodulator, has the potential to repolarize human monocyte-derived M2 macrophages to M1 phenotype.J Immunother Cancer. (2014) 2:191.
Summary
Keywords
SARS-CoV-2, macropahge, innnate immune, MHC trafficking, cytokine - immunological terms
Citation
Taefehshokr N, Taefehshokr S, Hemmat N and Heit B (2020) Covid-19: Perspectives on Innate Immune Evasion. Front. Immunol. 11:580641. doi: 10.3389/fimmu.2020.580641
Received
06 July 2020
Accepted
11 September 2020
Published
30 September 2020
Volume
11 - 2020
Edited by
Charlotte Odendall, King’s College London, United Kingdom
Reviewed by
Claire Kerridge, King’s College London, United Kingdom; Kushagra Bansal, Harvard Medical School, United States
Updates
Copyright
© 2020 Taefehshokr, Taefehshokr, Hemmat and Heit.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bryan Heit, bheit@uwo.ca
This article was submitted to Molecular Innate Immunity, a section of the journal Frontiers in Immunology
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.