 Swetha Peesari
Swetha Peesari Jeremy P. McAleer*
Jeremy P. McAleer*- Department of Pharmaceutical Sciences, Marshall University School of Pharmacy, Huntington, WV, United States
Th9 cells are a CD4 T cell subset that produces interleukin-9 (IL-9), a pleiotropic cytokine implicated in allergies, autoimmunity and cancer. Defining the cellular effects of IL-9 and factors regulating its expression are essential for fully understanding its roles in immunity and disease. IL-9 acts on a variety of immune and non-immune cells through a heterodimeric receptor composed of IL-9Rα and the common gamma chain. In CD4 T cells, IL-9 promotes mTOR activation, aerobic glycolysis, proliferation and reinforces its own expression. Additional cellular effects include mast cell activation, B cell antibody production and anti-tumor immunity. These biological activities are complemented by recent studies that expand our understanding of Th9 differentiation beyond canonical cytokine and transcription factor pathways. Notably, glycolytic reprogramming and fatty acid metabolism have emerged as key regulators of IL-9 production, mediated through the activities of mTOR, PPAR-γ and acetyl-CoA carboxylase 1 (ACC1). mTOR-driven aerobic glycolysis is essential for Th9 cell differentiation, supporting survival, proliferation, and IL9 expression through HIF-1α activation. In contrast, ACC1 suppresses IL-9 through fatty acid synthesis, which enhances RARα-mediated transcriptional repression. PPAR-γ appears to have dual functions: it promotes IL-9 production by increasing glucose uptake and activating mTOR, but reduces IL-9 in response to synthetic agonists that may increase fatty acid uptake. Overall, these findings highlight critical roles for metabolic regulators in Th9 responses and suggest that targeting these pathways may offer new therapeutic strategies for IL-9-driven diseases.
Introduction
IL-9 was identified in 1988 as a T helper cell growth factor produced in mitogen-stimulated cultures (1). Originally named P40, IL-9 has 126 amino acids and an unmodified molecular weight of 14 kDa, with post translational glycosylation further increasing the weight to 40kDa (2, 3). Human IL-9 shares 56% amino acid similarity to its murine counterpart (4), with cytokines in both species sharing similar functions. In vitro, IL-9 induces the proliferation and activation of IL-4 responsive CD4+ helper T cells and mast cells (1, 5), although it may also suppress lymphocyte proliferation under some conditions (6). On the other hand, IL-9 was not observed to stimulate cytotoxic CD8+ T cells, B cells or myeloid cells (7). In vivo, several inflammatory conditions induce IL-9, including asthma, atopic and contact dermatitis, food allergies and bacterial infections (8–10). These studies and others stimulated interest in identifying cellular sources of IL-9 and the mechanisms regulating its production.
Cytokine regulation of Th9 cell differentiation
CD4 T cells are the best characterized cell type that produces IL-9, although other cells including mast cells and NKT cells are also capable (11). In 1994, naïve CD4 T cell activation with TGF-β and IL-4 was shown to induce the differentiation of a novel subset later named Th9 (12). Subsequent studies demonstrated indispensable roles for STAT6 and GATA3 in the ability of IL-4 to enhance IL-9 production from T cells treated with TGF-β (13, 14). This was associated with IL-4 suppressing the Treg differentiation factor Foxp3 following TGF-β treatment. On the other hand, IL-10 was expressed at similar levels in murine Th2 and Th9 cells (13). The precise role of GATA3 was not immediately clear: although Th9 cells express negligible levels of the canonical Th2 transcription factor, GATA3 deficiency abrogates IL-9 production. This suggests GATA3 is involved in the early transition of Th2 to Th9 cells prior to its downregulation by TGF-β (13). GATA3 deficiency also increases Foxp3 expression in the presence of TGF-β, highlighting antagonistic functions for these transcription factors. Human Th9 cells share similar differentiation requirements as their murine counterparts, although GATA3 and Foxp3 appear to be expressed at higher levels (15). Other transcription factors involved in Th9 differentiation include PU.1, IRF4, AP1 and NF-kB (7). PU.1 suppresses Th2 cytokines and increases IL-9 production by promoting histone acetylation at the Il9 locus (16). IRF4 contributes to Th9, Th2 and Th17 differentiation (7, 17). Both Th2 and Th17 differentiation conditions can induce IL-9 production from murine CD4 T cells (18). Overall, the coordinated action of multiple transcription factors supports IL-9 production in Th9 cells, which have been classified as a subset of Th2 cells (19). While IL-4 and TGF-β are essential for IL-9 induction, other cytokines have been identified that either increase (e.g. type I IFNs, IL-1β, IL-2, IL-6, IL-9, IL-10, IL-12, IL-21, IL-25) or decrease (e.g. IFN-γ, IL-23, IL-27) IL-9 production from CD4 T cells (7, 14, 15). In addition to Th9 cells, other TGF-β-dependent subsets (e.g. Th17, Tregs) are capable of IL-9 production (18, 20), indicating a degree of functional overlap among these populations.
Cellular effects of IL-9
IL-9 signals through a heterodimeric receptor composed of IL-9 receptor alpha (IL-9Rα) and the common gamma chain (γc), the latter of which is shared with receptors for IL-2, IL-4, IL-7, IL-15 and IL-21. Thus, IL-9 responsiveness is restricted to cells expressing both subunits, including lymphocyte subsets and mast cells. Inflammatory conditions can broaden IL-9 responsiveness in other populations (e.g. antigen presenting cells and epithelial cells) by upregulating IL-9Rα or γc (21). The binding of IL-9 to its receptor complex primarily activates JAK/STAT signaling pathways, with documented roles for JAK1, JAK3, STAT1, STAT3 and STAT5 (22). Additional signaling pathways include Mitogen-Activated Protein Kinase (MAPK) and insulin receptor substrate (IRS)-Phosphatidylinositol-3 Kinase (PI3K). Negative regulation is mediated by receptor ubiquitination and inhibitory molecules such as SOCS proteins, PIAS and SH-PTP2 (22).
Originally identified as a CD4 T cell growth factor (1), IL-9 induces the expression of genes involved in survival and proliferation. While naïve CD4 T cells lack IL-9R and are non-responsive to this cytokine, several effector subsets are regulated by IL-9. For instance, activating murine CD4 T cells with IL-9 upregulates both IL-4 and TGF-β (18), cytokines integral to the differentiation of Th2, Th9, Th17 and regulatory T cell (Treg) subsets. In Th9 cells, IL-9 acts in an autocrine/paracrine manner to promote effector function (14). This positive feedback loop was attributed to the upregulation of lactate transporter MCT1, leading to aerobic glycolysis (23). In Th2 cells, IL-9 augments IL-5 production (24), while in combination with TGF-β promotes Th17 differentiation (18), possibly due to STAT3 activation by IL-9. The same study also found that IL-9 enhances the suppressive function of Tregs. These findings demonstrate that IL-9 can promote pro- or anti-inflammatory responses depending on the cell type.
Beyond T cells, IL-9 influences several hematopoietic and non-hematopoietic populations. In mast cells, IL-9 enhances proliferation, survival, and activation, leading to increased production of cytokines such as IL-1β, IL-5, IL-6 and IL-13 (5, 11). Transgenic overexpression of IL-9 increases the production of antigen-specific antibodies following immunization, presumably due to IL-9-driven B cell expansion (25). In antigen presenting cells, IL-9 induces TGF-β and suppresses IL-12 (26, 27), dampening Th1 responses. Roles for IL-9 in hematopoiesis have been suggested due to its proliferative effects on IL-3-dependent myeloid cell lines (28). Most non-hematopoietic cells remain non-responsive due to their lack of the γc receptor subunit, although inflammatory conditions may lead to its upregulation. For example, IL-4 enhances IL-9R expression on keratinocytes, and treatment with IL-9 stimulates IL-8 production in an ERK-dependent manner (29). This suggests inflammatory environments can transiently render epithelial cells to be IL-9-responsive. Some tumors also directly respond to IL-9, with protective and pathogenic effects described (30). Lymphoma cell lines show increased proliferation and survival against apoptotic-inducing agents in the presence of IL-9 (31). In contrast, melanoma cell lines upregulate anti-proliferative (p21) or pro-apoptotic (TRAIL) molecules in response to IL-9, enhancing apoptosis (32). Currently, the role of tumor-specific IL-9R expression on Th9-mediated anti-tumor immunity in vivo remains unclear. Collectively, the cellular effects of IL-9 support its diverse roles in mucosal host defense, allergic disease, autoimmunity and anti-tumor immunity, as described next (Figure 1).
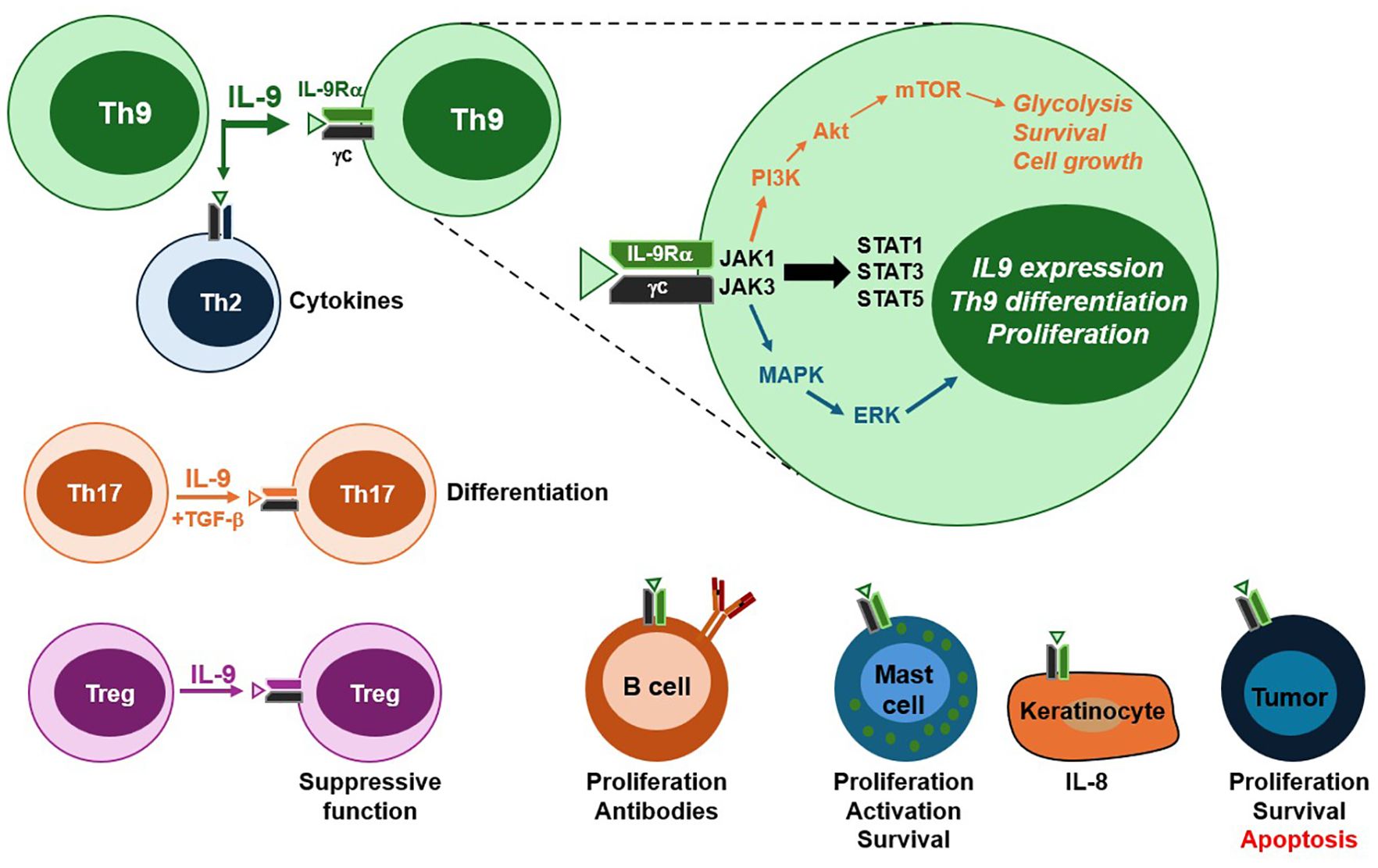
Figure 1. IL-9 receptor signaling and cellular targets. Th9 cells are the primary source of IL-9, although other T cell subsets, including Th17 and Tregs, are also capable of IL-9 production. The IL-9 receptor is a heterodimer composed of IL-9Rα and the common gamma chain (γc). Ligand binding activates three major intracellular signaling pathways. The JAK/STAT pathway promotes Th9 differentiation and IL-9 production. The PI3K/Akt pathway contributes to mTOR activation, resulting in aerobic glycolysis, survival and macromolecule synthesis required for cell growth. The MAP kinase cascade facilitates the transcription of genes involved in effector T cell proliferation. In addition to Th9 cells, IL-9 targets a range of hematopoietic and sometimes non-hematopoietic cells. Among CD4 T cell subsets and B cells, IL-9 enhances proliferation, differentiation and effector function. In mast cells, IL-9 promotes activation, cytokine secretion and survival. Keratinocytes can also respond to IL-9 when they express the receptor complex, leading to pro-inflammatory cytokine production. In cancer, IL-9 plays dual roles through promoting proliferation and survival in lymphomas, while inducing apoptosis in melanoma. The responsiveness of non-hematopoietic populations to IL-9 occurs through the upregulation of IL-9R components in inflammatory settings. Altogether, these pathways and target cell responses contribute to the pleiotropic roles of IL-9 in immunity, inflammation and disease.
Role of IL-9 in diseases
IL-9 contributes to inflammatory reactions in the skin, intestine and lungs, as well as cancer and autoimmunity. While not typically the dominant cytokine, IL-9 modulates disease progression through its diverse effects on immune and non-immune populations (21). For instance, atopic dermatitis (AD) is primarily a Th2-driven disease, although Th9 cells are increasingly recognized due to their preferential skin migration (33), elevated IL9/IL9R expression in lesional tissue (19, 34), correlations between Th9 cell frequencies and disease severity (35), and disease amelioration following IL-9 neutralization (36). Mechanisms through which IL-9 contributes to skin inflammation include promoting IgE production, eosinophil and mast cell infiltration, and VEGF secretion from keratinocytes (37). In asthma, IL-9 was identified as a genetic risk factor in 1997 (38), and subsequent studies confirmed its involvement in airway hyperresponsiveness, eosinophilia, mast cell hyperplasia and tissue pathology (17, 39). Ulcerative colitis is another Th2 disease associated with elevated IL-9 expression. Notably, IL-9 deficiency or neutralization protects mice from experimental colitis (40–42). The IL-9-driven intestinal pathology may result from its direct effects on intestinal epithelial cells, leading to decreased growth, proliferation, wound healing and tight junction protein expression (40, 41, 43). Despite its pathogenic role in colitis, IL-9 protects against intestinal nematode infections by enhancing antibody responses, mast cell function and basophilia (44–46). This suggests IL-9 mediated inflammation on epithelial surfaces may have originally evolved as a host defense mechanism.
In cancer, IL-9 plays dual roles acting as either a tumor suppressor or promoter depending on the tumor type (30). Non-hematopoietic models demonstrate potent anti-tumor activities of IL-9 and Th9 cells. For instance, IL-9 directly induces apoptosis and suppresses proliferation in melanoma, enhances CD8 T cell cytotoxicity, and improves mast cell anti-tumor activity (32, 47–49). Notably, endogenous IL-9 inhibits melanoma growth in pre-clinical models, and adoptively-transferred Th9 cells improve survival (47). The anti-melanoma activity is enhanced by stimulation of the Epidermal Growth Factor Receptor (EGFR) during Th9 differentiation (50). Th9 cells themselves may directly contribute to tumor clearance through expression of granzymes and perforin (51). Additional cytokines involved in the anti-tumor activity of Th9 cells include IL-21 and IL-24 (49, 52, 53). In gastric cancer, high IL-9 expression correlates with improved patient survival, and recombinant IL-9 augments the efficacy anti-PD-1 immunotherapy (54). These findings have led to the investigation of Th9 cells in adoptive transfer approaches for solid tumors (30). Conversely, IL-9 can support tumor growth in IL-9R-expressing malignancies, including lymphomas, lung cancer and pancreatic cancer (30, 55–57). IL-9 may also promote immune evasion by upregulating PD-1 on CD8+ cytotoxic T cells (58), or enhancing Treg-mediated suppression (59). These dual roles underscore the importance of tumor-intrinsic IL-9R expression and the tumor microenvironment in determining whether IL-9 promotes or inhibits tumor growth. Thus, IL-9 is being explored therapeutically for enhancing its activity in cancer immunotherapy or inhibiting its function in IL-9-responsive malignancies.
Interleukin-9 also has complex roles in autoimmune diseases. Rheumatoid arthritis and psoriatic arthritis patients have elevated IL-9 in synovial fluid and tissues (60, 61). Synovial T cell infiltration was associated with high levels of IL-9R expression, and circulating Th9 cells were expanded by citrullinated peptides. Despite this pro-inflammatory profile, IL-9-deficient mice develop worsened antigen-induced-arthritis, including cartilage destruction and impaired Treg function (62). Similarly, systemic lupus erythematosus patients show elevated IL-9 and Th9 cells in circulation (63, 64), although IL-9 neutralization in lupus-prone mice reduces autoantibody production and renal pathology, especially when combined with IL-17 blockade (65). This was attributed to the ability of IL-9 to promote B cell proliferation and antibody production. In multiple sclerosis models, IL-9 and Th9 cells have been shown to contribute to pathology (66, 67), with IL-9R deficiency either exacerbating or suppressing experimental autoimmune encephalomyelitis (68, 69). These results may reflect IL-9 enhancing both Th17 differentiation and Treg suppressive function (18). Collectively, these findings underscore dual roles for IL-9 in autoimmunity, where it promotes pathology through Th9, Th17 or B cells, while protecting against disease through Tregs and dendritic cells.
Metabolic regulation of IL-9 production
As with other CD4 T cell subsets, metabolism plays a central role in the differentiation and function of Th9 cells. Several metabolic products, including lipids, amino acids and TCA cycle intermediates, have been shown to directly affect IL-9 production and/or Th9 differentiation (50, 70–73). Here, we highlight roles of the major metabolic regulators mTOR, PPAR-γ and ACC1, drawing upon what is broadly known from CD4 T cell differentiation, as well as recent studies focused specifically on IL-9. Other reviews have also summarized metabolic control of Th9 cells (74, 75).
mTOR
T cell activation induces profound metabolic changes to support the energy production and biomass accumulation required for clonal expansion and effector cell differentiation. This has been well characterized for glucose metabolism, with Akt and mTOR playing critical roles in driving aerobic glycolysis (76). Among the CD4 T cell subsets, Th9 cells exhibited the highest glycolytic activity, with mTOR required for optimal IL-9 production (77). Accessibility of the IL9 promoter is further enhanced by STAT5-induced histone acetylation, facilitating the binding of transcription factors such as BATF, Foxo1 and HIF-1α (77–80). In the absence of mTOR activation, IL9 transcription is suppressed by Foxp1 and the histone deacetylase SIRT1 (77, 78). Genetic deficiency of SIRT1 increases glycolytic and mTORC1 activity in Th9 cells, enhancing anti-tumor immunity and allergic airway inflammation (77). Importantly, mTOR-driven IL-9 production has been associated with pathology and mast cell hyperplasia in experimental food allergy (81). Both mTORC1 and the rapamycin-insensitive mTORC2 complex contribute to Th9 differentiation, as Rictor deficiency impairs Th9 polarization and allergic airway inflammation (82). Several endogenous molecules can enhance IL-9 production through mTOR. For instance, extracellular ATP induces nitric oxide and mTOR activation, increasing IL-9 production in an HIF-1α-dependent manner (83). Amphiregulin was later shown to activate HIF-1α through the epidermal growth factor receptor (EGFR) (50). The STING ligand 2’3’-cGAMP increases IL-9 in an mTOR-dependent manner, enhancing anti-tumor immunity (84). High glucose concentrations enhance IL-9 production through PPAR-γ mediated aerobic glycolysis (23). This study showed that PPAR-γ suppression decreases mTORC1 phosphorylation, while another demonstrated the microRNA miR-145 suppresses IL-9 through mTOR-HIF1α inhibition (85). Collectively, these studies highlight an indispensable role for mTOR-driven glycolysis in Th9 cell differentiation, and suggest that metabolic interventions targeting this pathway could modulate IL-9-dependent inflammation and immunity.
PPAR-γ
Peroxisome proliferator-activated receptor-γ (PPAR-γ) is a member of a nuclear receptor superfamily of transcription factors activated by fatty acids (86). Ligand binding drives the expression of genes involved in adipogenesis, lipid metabolism, glucose homeostasis and inflammation (86–88). Clinically, PPAR-γ is targeted by thiazolidinediones for type 2 diabetes and 5-aminosalicylates for inflammatory bowel disease. While its role in adipose tissue insulin sensitivity is well established, distinct functions for PPAR-γ in CD4 T cells have been identified. In response to antigen and costimulation, PPAR-γ promotes fatty acid uptake and lipolysis to support T cell proliferation (89). In Th2 cells, IL-4-induced PPARG expression increases lipid metabolism and IL-5 production (90). In vivo, Pparg expression in CD4 T cells contributes to Th2-driven inflammation in the intestine, lungs and skin (91–93). PPAR-γ is also expressed in regulatory T cells (Tregs), where it supports their suppressive function and protects against experimental colitis, psoriasis, graft-versus-host disease and insulin resistance (94–98). Thus, anti-inflammatory effects of PPAR-γ are partially due to the enhancement of Treg-mediated suppression. Interestingly, thiazolidinediones can increase Foxp3 expression and Treg differentiation independently of PPAR-γ (98). Collectively, these studies show that PPAR-γ mediates both pro- and anti-inflammatory functions depending on the T cell subset.
The role of PPAR-γ in Th9 cell differentiation has recently become elucidated. Human Th9 cells were characterized as a subpopulation of Th2 cells expressing high levels of PPAR-γ (19). This population is enriched within memory CD4+ CCR4+ CCR8+ cells in human blood, and the upregulation of IL-9 following T cell activation correlates with a transient decrease in canonical Th2 cytokines IL-4, IL-5 and IL-13. Several genes coordinate the transitioning of Th2 to Th9 cells, including cytokines, growth factors and their receptor signaling pathways (99). PPAR-γ suppression in Th9 cells decreased IL-9 production, as well as the expression of genes promoting T cell activation, glucose metabolism and aerobic glycolysis (19, 23). These pro-glycolytic functions of PPAR-γ occurred under high glucose conditions and were associated with glucose uptake, mTORC1 phosphorylation and proliferation. In line with this, paracrine IL-9 activity increased the expression of genes involved in aerobic glycolysis, including SLC16A1 which encodes for the lactate transporter MCT1, contributing to proliferation (23). The regulatory relationship between PPAR-γ and mTORC1 appears to be bidirectional; however, as another study demonstrated mTORC1 activation prior to PPAR-γ-mediated fatty acid uptake (89). While the endogenous ligand(s) responsible for PPAR-γ activation in Th9 cells remains unknown, the synthetic agonist rosiglitazone suppresses IL-9 in human Th9 cells (100). Combining rosiglitazone with the glucose metabolism inhibitor 2-deoxy-D-glucose decreased IL-9 more potently than either treatment alone, suggesting a glycolysis-independent component of rosiglitazone-mediated suppression. It remains to be determined if this is due to fatty acid uptake or another mechanism, as rosiglitazone exhibits PPAR-γ-dependent and -independent effects on cells (101). Nonetheless, the anti-inflammatory properties of rosiglitazone in vitro align with therapeutic benefits of PPAR-γ agonists in IL-9-associated diseases such as atopic dermatitis and psoriasis (91, 102–104). Collectively, PPAR-γ modulators suppress IL-9 production from Th9 cells through metabolic regulation.
Acetyl-CoA carboxylase 1
T cell activation increases cellular demands for fatty acids, leading to the upregulation of enzymes and transcription factors involved in fatty acid biosynthesis (76, 89). Central to this process is acetyl-CoA Carboxylase 1 (ACC1), which catalyzes the carboxylation of acetyl-CoA to generate malonyl-CoA, the precursor for mid- and long-chain fatty acids (105). In this way, ACC1 supports T cell growth during clonal expansion. Low cellular ATP levels inactivate ACC1 through phosphorylation by AMP-activated protein kinase (AMPK), which diverts metabolism toward fatty acid oxidation to restore ATP levels. Experimentally, ACC1 function can be assessed with pharmacologic inhibitors and/or genetic silencing of its gene ACACA. A mouse model found that diet-induced obesity increases ACC1 expression in memory CD4 T cells, enhancing IL-17 production and Th17-driven pathology (106). Further, ACC1 inhibition reduces IL-17 while increasing Foxp3 expression in Th17 cultures (107), demonstrating its pivotal role in Th17 cell differentiation. Mice lacking ACC1 in CD4 T cells are protected from diseases associated with IL-17, such as asthma, psoriasis and colitis, but show increased susceptibility to infections (108–111). These pro-inflammatory effects of ACC1 are mediated by glycolytic and oxidative metabolic reprogramming (112). Intriguingly, ACC1 inhibition may either enhance or reduce memory CD4 T cell generation depending on the context (113, 114). These studies highlight opposing functions for ACC1 in Th17 and Treg differentiation, raising the question of how this pathway influences other effector subsets such as Th9 cells.
Recent studies identified a suppressive role for ACC1 in Th9 differentiation. Culturing Th9 cells with ACC1 inhibitors or under fatty acid-free conditions substantially increases IL-9 production (100, 115). This increased IL-9 can be restored with exogenous oleic acid or palmitic acid, demonstrating de novo fatty acid synthesis and fatty acid uptake suppress Th9 differentiation. At the chromatin level, ACC1 suppression led to greater histone acetylation at the Il9 and Batf3 promoter regions, consistent with a permissive chromatin landscape (115). This may be due to the accumulation intracellular acetyl-CoA following ACC1 suppression, which is used by histone acetyltransferases to globally increase chromatin histone acetylation (116). In addition, ACC1 suppression enhances the sensitivity of cells to TGF-β by increasing SMAD2/3 phosphorylation and its subsequent binding to Il9 (115). The same study demonstrated that retinoic acid receptor-alpha (RARα) signaling may contribute to the suppressive effects of ACC1 and exogenous oleic acid on IL-9 production. This is consistent with RARα activation suppressing Th9 differentiation (117). Pre-treatment of Th9 cells with an ACC1 inhibitor in vitro enhanced their anti-tumor activity following adoptive transfer to tumor-bearing mice (115), demonstrating robust Th9 immunity. Human Th9 cultures also contain a small population of Foxp3+ IL-9- cells (100), possibly due to exogenous TGF-β or TCR/CD28 stimulation (118, 119). ACC1 suppression increases the ratio of IL-9+:Foxp3- cells, demonstrating a pro-inflammatory shift in vitro (100). This contrasts with Th17 cultures, in which ACC1 suppression decreases the ratio of IL-17+:Foxp3- cells (107). Further studies are necessary to understand differential effects of ACC1 on Foxp3 expression in Th9 versus Th17 cultures. Nonetheless, Foxp3 inhibition did not affect IL-9 production in cultures containing TGF-β (100), suggesting the Foxp3+ cells are not functional Tregs. These findings demonstrate that ACC1 limits IL-9 expression through coordinated metabolic and epigenetic mechanisms. Further elucidating how ACC1 limits IL-9 expression will provide key insights into the lipid-mediated control of inflammatory responses.
Conclusions
Recent insights into the metabolic regulation of Th9 cells have expanded our understanding beyond canonical cytokine signaling and transcription factor pathways (Figure 2). Glycolytic reprogramming and fatty acid metabolism play critical roles in Th9 differentiation and IL-9 production, with important contributions from mTOR, PPAR-γ and ACC1. A more detailed mechanistic understanding will require investigation of additional regulators such as fatty acid synthase (FAS), sterol regulatory element-binding protein 1 (SREBP1) and AMPK. Challenges in using IL-9 targeted therapies may arise from its pleiotropic effects on multiple cell types, including Th9, Th17, Tregs, B cells, epithelial and cancer cells. For instance, the IL-9 neutralizing antibody Medi-528 did not show clinical benefit in an asthma trial (120). In cancer, ACC1 antagonism may facilitate Th9 immunity while also inducing tumor cell apoptosis (100, 115, 121–123). Conversely, ACC1 suppression has been shown to increase Foxp3+ Tregs (107), and induce hypertriglyceridemia (124), possibly limiting its therapeutic value. Similarly, PPAR-γ agonists used clinically for metabolic disorders can suppress IL-9 production (100), but may cause adverse effects including weight gain, fluid retention, and bone loss (86). These challenges underscore the need for cell-specific delivery strategies that target Th9 cells or IL-9R-expressing populations. Moving forward, key priorities should include: (A) defining how metabolic pathways influence IL-9-driven diseases, (B) identifying metabolic signatures that distinguish pathogenic from regulatory IL-9R+ cell populations, and (C) determining how metabolic interventions can fine-tune IL-9-dependent immunity. As Th9 cells gain recognition for their diverse roles in immune regulation, metabolic targeting may become crucial for harnessing their full therapeutic potential.
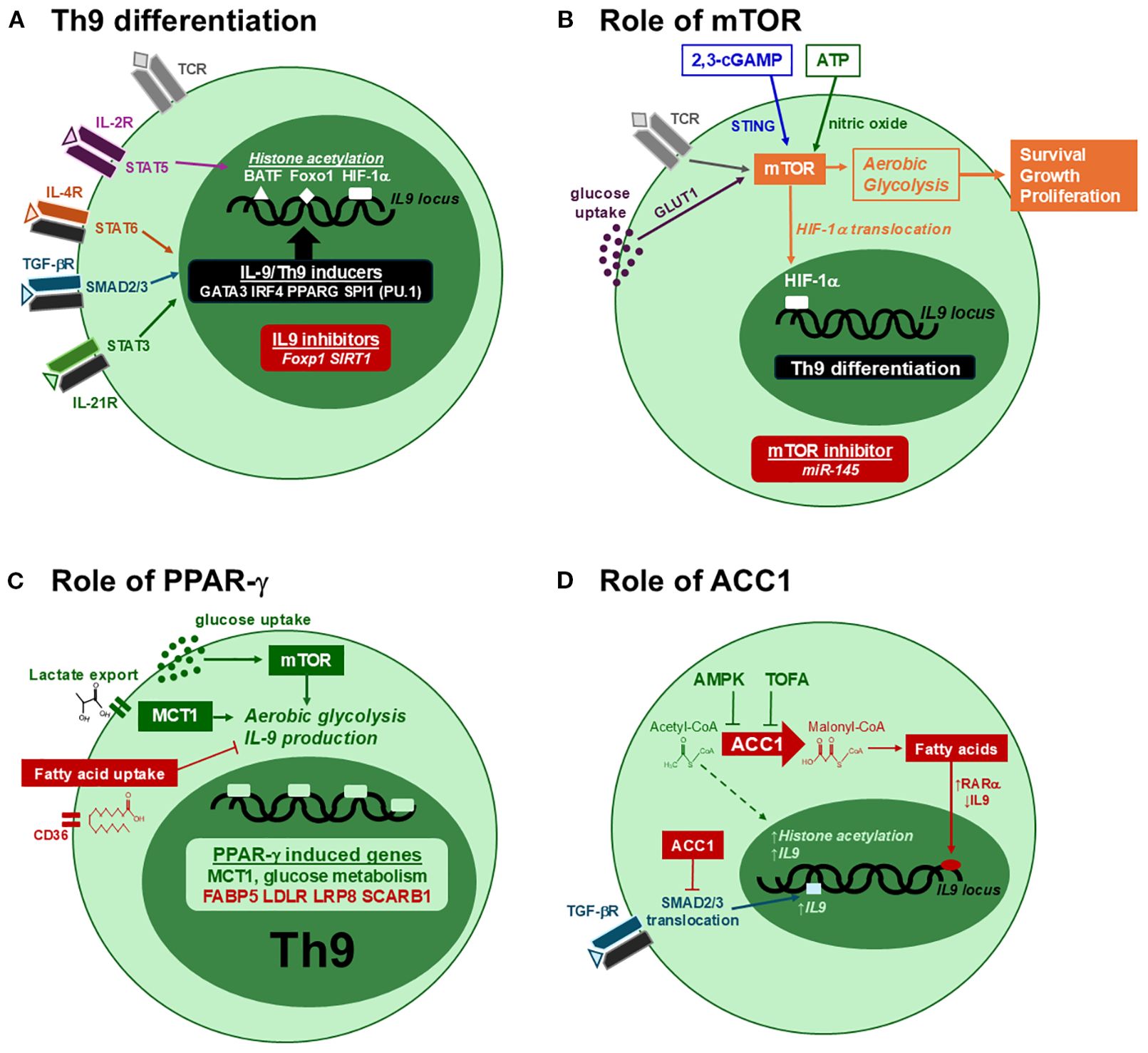
Figure 2. Metabolic regulation of Th9 differentiation. (A) Th9 differentiation is induced through the coordinated activities of STAT and SMAD pathways in CD4 T cells. IL-4, TGF-β and IL-21 induce several transcription factors required Th9 differentiation, while STAT5 promotes histone acetylation to enhance their binding to the IL9 promoter. In the absence of Th9-inducing signals, IL9 expression is repressed by Foxp1 and SIRT1. (B) mTOR promotes Th9 differentiation through aerobic glycolysis, leading to HIF-1α nuclear translocation, IL9 expression, cell growth and proliferation. This activity is enhanced with high extracellular glucose, 2,3-cGAMP and ATP, and is inhibited by miR-145. (C) PPAR-γ has positive and negative effects on IL-9. By enhancing glucose uptake and MCT1-mediated lactate export, PPAR-γ stimulates aerobic glycolysis and IL-9 production. By increasing the expression of genes involved in fatty acid uptake (red), PPAR-γ can also have a negative impact on IL-9. (D) ACC1 suppresses IL-9 production through fatty acid synthesis. The conversion of acetyl-CoA to malonyl-CoA generates the substrate for fatty acids, increasing RARα activity and decreasing IL9 expression. In addition, ACC1 suppresses SMAD2/3 phosphorylation in response to TGF-β, further limiting Th9 differentiation. ACC1 suppression increases IL-9 by suppressing fatty acid synthesis and RARα activity, increasing SMAD2 phosphorylation, and possibly promoting histone acetylation via acetyl-CoA (dotted line).
Author contributions
SP: Writing – original draft, Investigation. JM: Conceptualization, Investigation, Funding acquisition, Writing – review & editing, Writing – original draft, Formal Analysis.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. Costs for this publication were provided by Marshall University School of Pharmacy.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Uyttenhove C, Simpson RJ, and Van Snick J. Functional and structural characterization of P40, a mouse glycoprotein with T-cell growth factor activity. Proc Natl Acad Sci U.S.A. (1988) 85:6934–8. doi: 10.1073/pnas.85.18.6934
2. Simpson RJ, Moritz RL, Rubira MR, Gorman JJ, and Van Snick J. Complete amino acid sequence of a new murine T-cell growth factor P40. Eur J Biochem. (1989) 183:715–22. doi: 10.1111/j.1432-1033.1989.tb21103.x
3. Van Snick J, Goethals A, Renauld JC, Van Roost E, Uyttenhove C, Rubira MR, et al. Cloning and characterization of a cDNA for a new mouse T cell growth factor (P40). J Exp Med. (1989) 169:363–8. doi: 10.1084/jem.169.1.363
4. Yang YC, Ricciardi S, Ciarletta A, Calvetti J, Kelleher K, and Clark SC. Expression cloning of cDNA encoding a novel human hematopoietic growth factor: human homologue of murine T-cell growth factor P40. Blood. (1989) 74:1880–4. doi: 10.1182/blood.V74.6.1880.1880
5. Hultner L, Druez C, Moeller J, Uyttenhove C, Schmitt E, Rude E, et al. Mast cell growth-enhancing activity (MEA) is structurally related and functionally identical to the novel mouse T cell growth factor P40/TCGFIII (interleukin 9). Eur J Immunol. (1990) 20:1413–6. doi: 10.1002/eji.1830200632
6. Schmitt E, Van Brandwijk R, Van Snick J, Siebold B, and Rude E. TCGF III/P40 is produced by naive murine CD4+ T cells but is not a general T cell growth factor. Eur J Immunol. (1989) 19:2167–70. doi: 10.1002/eji.1830191130
7. Goswami R and Kaplan MH. A brief history of IL-9. J Immunol. (2011) 186:3283–8. doi: 10.4049/jimmunol.1003049
8. Angkasekwinai P. Th9 cells in allergic disease. Curr Allergy Asthma Rep. (2019) 19:29. doi: 10.1007/s11882-019-0860-8
9. Gessner A, Blum H, and Rollinghoff M. Differential regulation of IL-9-expression after infection with Leishmania major in susceptible and resistant mice. Immunobiology. (1993) 189:419–35. doi: 10.1016/S0171-2985(11)80414-6
10. Seumois G, Ramirez-Suastegui C, Schmiedel BJ, Liang S, Peters B, Sette A, et al. Single-cell transcriptomic analysis of allergen-specific T cells in allergy and asthma. Sci Immunol. (2020) 5:eaba6087. doi: 10.1126/sciimmunol.aba6087
11. Noelle RJ and Nowak EC. Cellular sources and immune functions of interleukin-9. Nat Rev Immunol. (2010) 10:683–7. doi: 10.1038/nri2848
12. Schmitt E, Germann T, Goedert S, Hoehn P, Huels C, Koelsch S, et al. IL-9 production of naive CD4+ T cells depends on IL-2, is synergistically enhanced by a combination of TGF-beta and IL-4, and is inhibited by IFN-gamma. J Immunol. (1994) 153:3989–96. doi: 10.4049/jimmunol.153.9.3989
13. Dardalhon V, Awasthi A, Kwon H, Galileos G, Gao W, Sobel RA, et al. IL-4 inhibits TGF-beta-induced Foxp3+ T cells and, together with TGF-beta, generates IL-9+ IL-10+ Foxp3(-) effector T cells. Nat Immunol. (2008) 9:1347–55. doi: 10.1038/ni.1677
14. Veldhoen M, Uyttenhove C, van Snick J, Helmby H, Westendorf A, Buer J, et al. Transforming growth factor-beta ‘reprograms’ the differentiation of T helper 2 cells and promotes an interleukin 9-producing subset. Nat Immunol. (2008) 9:1341–6. doi: 10.1038/ni.1659
15. Wong MT, Ye JJ, Alonso MN, Landrigan A, Cheung RK, Engleman E, et al. Regulation of human Th9 differentiation by type I interferons and IL-21. Immunol Cell Biol. (2010) 88:624–31. doi: 10.1038/icb.2010.53
16. Chang HC, Sehra S, Goswami R, Yao W, Yu Q, Stritesky GL, et al. The transcription factor PU.1 is required for the development of IL-9-producing T cells and allergic inflammation. Nat Immunol. (2010) 11:527–34. doi: 10.1038/ni.1867
17. Staudt V, Bothur E, Klein M, Lingnau K, Reuter S, Grebe N, et al. Interferon-regulatory factor 4 is essential for the developmental program of T helper 9 cells. Immunity. (2010) 33:192–202. doi: 10.1016/j.immuni.2010.07.014
18. Elyaman W, Bradshaw EM, Uyttenhove C, Dardalhon V, Awasthi A, Imitola J, et al. IL-9 induces differentiation of TH17 cells and enhances function of FoxP3+ natural regulatory T cells. Proc Natl Acad Sci U.S.A. (2009) 106:12885–90. doi: 10.1073/pnas.0812530106
19. Micosse C, von Meyenn L, Steck O, Kipfer E, Adam C, Simillion C, et al. Human “T(H)9” cells are a subpopulation of PPAR-gamma(+) T(H)2 cells. Sci Immunol. (2019) 4:eaat5943. doi: 10.1126/sciimmunol.aat5943
20. Lu LF, Lind EF, Gondek DC, Bennett KA, Gleeson MW, Pino-Lagos K, et al. Mast cells are essential intermediaries in regulatory T-cell tolerance. Nature. (2006) 442:997–1002. doi: 10.1038/nature05010
21. Pajulas A, Zhang J, and Kaplan MH. The world according to IL-9. J Immunol. (2023) 211:7–14. doi: 10.4049/jimmunol.2300094
22. Chakraborty S, Kubatzky KF, and Mitra DK. An update on interleukin-9: from its cellular source and signal transduction to its role in immunopathogenesis. Int J Mol Sci. (2019) 20:2113. doi: 10.3390/ijms20092113
23. Bertschi NL, Steck O, Luther F, Bazzini C, von Meyenn L, Scharli S, et al. PPAR-gamma regulates the effector function of human T helper 9 cells by promoting glycolysis. Nat Commun. (2023) 14:2471. doi: 10.1038/s41467-023-38233-x
24. Poulin LF, Habran C, Stordeur P, Goldman M, McKenzie A, Van Snick J, et al. Interleukin-9 stimulates the production of interleukin-5 in CD4+ T cells. Eur Cytokine Netw. (2005) 16:233–9.
25. Vink A, Warnier G, Brombacher F, and Renauld JC. Interleukin 9-induced in vivo expansion of the B-1 lymphocyte population. J Exp Med. (1999) 189:1413–23. doi: 10.1084/jem.189.9.1413
26. Pilette C, Ouadrhiri Y, Van Snick J, Renauld JC, Staquet P, Vaerman JP, et al. IL-9 inhibits oxidative burst and TNF-alpha release in lipopolysaccharide-stimulated human monocytes through TGF-beta. J Immunol. (2002) 168:4103–11. doi: 10.4049/jimmunol.168.8.4103
27. Wu B, Huang C, Kato-Maeda M, Hopewell PC, Daley CL, Krensky AM, et al. IL-9 is associated with an impaired Th1 immune response in patients with tuberculosis. Clin Immunol. (2008) 126:202–10. doi: 10.1016/j.clim.2007.09.009
28. Williams DE, Morrissey PJ, Mochizuki DY, de Vries P, Anderson D, Cosman D, et al. T-cell growth factor P40 promotes the proliferation of myeloid cell lines and enhances erythroid burst formation by normal murine bone marrow cells in vitro. Blood. (1990) 76:906–11. doi: 10.1182/blood.V76.5.906.906
29. Hong CH, Chang KL, Wang HJ, Yu HS, and Lee CH. IL-9 induces IL-8 production via STIM1 activation and ERK phosphorylation in epidermal keratinocytes: A plausible mechanism of IL-9R in atopic dermatitis. J Dermatol Sci. (2015) 78:206–14. doi: 10.1016/j.jdermsci.2015.03.004
30. Bick F, Blanchetot C, Lambrecht BN, and Schuijs MJ. A reappraisal of IL-9 in inflammation and cancer. Mucosal Immunol. (2025) 18:1–15. doi: 10.1016/j.mucimm.2024.10.003
31. Lv X, Feng L, Ge X, Lu K, and Wang X. Interleukin-9 promotes cell survival and drug resistance in diffuse large B-cell lymphoma. J Exp Clin Cancer Res. (2016) 35:106. doi: 10.1186/s13046-016-0374-3
32. Fang Y, Chen X, Bai Q, Qin C, Mohamud AO, Zhu Z, et al. IL-9 inhibits HTB-72 melanoma cell growth through upregulation of p21 and TRAIL. J Surg Oncol. (2015) 111:969–74. doi: 10.1002/jso.23930
33. Wong MT, Ong DE, Lim FS, Teng KW, McGovern N, Narayanan S, et al. A high-dimensional atlas of human T cell diversity reveals tissue-specific trafficking and cytokine signatures. Immunity. (2016) 45:442–56. doi: 10.1016/j.immuni.2016.07.007
34. Sismanopoulos N, Delivanis DA, Alysandratos KD, Angelidou A, Vasiadi M, Therianou A, et al. IL-9 induces VEGF secretion from human mast cells and IL-9/IL-9 receptor genes are overexpressed in atopic dermatitis. PloS One. (2012) 7:e33271. doi: 10.1371/journal.pone.0033271
35. Ma L, Xue HB, Guan XH, Shu CM, Zhang JH, and Yu J. Possible pathogenic role of T helper type 9 cells and interleukin (IL)-9 in atopic dermatitis. Clin Exp Immunol. (2014) 175:25–31. doi: 10.1111/cei.12198
36. Makita S, Takatori H, Matsuki A, Kawashima H, Iwata A, Tanaka S, et al. T-bet and STAT6 coordinately suppress the development of IL-9-mediated atopic dermatitis-like skin inflammation in mice. J Invest Dermatol. (2021) 141:1274–1285 e5. doi: 10.1016/j.jid.2020.08.029
37. Clark RA and Schlapbach C. T(H)9 cells in skin disorders. Semin Immunopathol. (2017) 39:47–54. doi: 10.1007/s00281-016-0607-8
38. Nicolaides NC, Holroyd KJ, Ewart SL, Eleff SM, Kiser MB, Dragwa CR, et al. Interleukin 9: a candidate gene for asthma. Proc Natl Acad Sci U.S.A. (1997) 94:13175–80. doi: 10.1073/pnas.94.24.13175
39. Temann UA, Geba GP, Rankin JA, and Flavell RA. Expression of interleukin 9 in the lungs of transgenic mice causes airway inflammation, mast cell hyperplasia, and bronchial hyperresponsiveness. J Exp Med. (1998) 188:1307–20. doi: 10.1084/jem.188.7.1307
40. Gerlach K, Hwang Y, Nikolaev A, Atreya R, Dornhoff H, Steiner S, et al. TH9 cells that express the transcription factor PU.1 drive T cell-mediated colitis via IL-9 receptor signaling in intestinal epithelial cells. Nat Immunol. (2014) 15:676–86. doi: 10.1038/ni.2920
41. Nalleweg N, Chiriac MT, Podstawa E, Lehmann C, Rau TT, Atreya R, et al. IL-9 and its receptor are predominantly involved in the pathogenesis of UC. Gut. (2015) 64:743–55. doi: 10.1136/gutjnl-2013-305947
42. Yuan A, Yang H, Qi H, Cui J, Hua W, Li C, et al. IL-9 antibody injection suppresses the inflammation in colitis mice. Biochem Biophys Res Commun. (2015) 468:921–6. doi: 10.1016/j.bbrc.2015.11.057
43. Gerlach K, McKenzie AN, Neurath MF, and Weigmann B. IL-9 regulates intestinal barrier function in experimental T cell-mediated colitis. Tissue Barriers. (2015) 3:e983777. doi: 10.4161/21688370.2014.983777
44. Faulkner H, Humphreys N, Renauld JC, Van Snick J, and Grencis R. Interleukin-9 is involved in host protective immunity to intestinal nematode infection. Eur J Immunol. (1997) 27:2536–40. doi: 10.1002/eji.1830271011
45. Faulkner H, Renauld JC, Van Snick J, and Grencis RK. Interleukin-9 enhances resistance to the intestinal nematode Trichuris muris. Infect Immun. (1998) 66:3832–40. doi: 10.1128/IAI.66.8.3832-3840.1998
46. Licona-Limon P, Henao-Mejia J, Temann AU, Gagliani N, Licona-Limon I, Ishigame H, et al. Th9 cells drive host immunity against gastrointestinal worm infection. Immunity. (2013) 39:744–57. doi: 10.1016/j.immuni.2013.07.020
47. Purwar R, Schlapbach C, Xiao S, Kang HS, Elyaman W, Jiang X, et al. Robust tumor immunity to melanoma mediated by interleukin-9-producing T cells. Nat Med. (2012) 18:1248–53. doi: 10.1038/nm.2856
48. Wan J, Wu Y, Huang L, Tian Y, Ji X, Abdelaziz MH, et al. ILC2-derived IL-9 inhibits colorectal cancer progression by activating CD8(+) T cells. Cancer Lett. (2021) 502:34–43. doi: 10.1016/j.canlet.2021.01.002
49. You FP, Zhang J, Cui T, Zhu R, Lv CQ, Tang HT, et al. Th9 cells promote antitumor immunity via IL-9 and IL-21 and demonstrate atypical cytokine expression in breast cancer. Int Immunopharmacol. (2017) 52:163–7. doi: 10.1016/j.intimp.2017.08.031
50. Roy S, Rizvi ZA, Clarke AJ, Macdonald F, Pandey A, Zaiss DMW, et al. EGFR-HIF1alpha signaling positively regulates the differentiation of IL-9 producing T helper cells. Nat Commun. (2021) 12:3182. doi: 10.1038/s41467-021-23042-x
51. Lu Y, Wang Q, Xue G, Bi E, Ma X, Wang A, et al. Th9 cells represent a unique subset of CD4(+) T cells endowed with the ability to eradicate advanced tumors. Cancer Cell. (2018) 33:1048–1060 e7. doi: 10.1016/j.ccell.2018.05.004
52. Chen J, Zhang Y, Zhang H, Zhang M, Dong H, Qin T, et al. IL-24 is the key effector of Th9 cell-mediated tumor immunotherapy. iScience. (2023) 26:107531. doi: 10.1016/j.isci.2023.107531
53. Vegran F, Berger H, Boidot R, Mignot G, Bruchard M, Dosset M, et al. The transcription factor IRF1 dictates the IL-21-dependent anticancer functions of TH9 cells. Nat Immunol. (2014) 15:758–66. doi: 10.1038/ni.2925
54. Fang H, Li R, Gu Y, Fei Y, Jin K, Chen Y, et al. Intratumoral interleukin-9 delineates a distinct immunogenic class of gastric cancer patients with better prognosis and adjuvant chemotherapeutic response. Oncoimmunology. (2020) 9:1856468. doi: 10.1080/2162402X.2020.1856468
55. Gruss HJ, Brach MA, Drexler HG, Bross KJ, and Herrmann F. Interleukin 9 is expressed by primary and cultured Hodgkin and Reed-Sternberg cells. Cancer Res. (1992) 52:1026–31.
56. Hu B, Qiu-Lan H, Lei RE, Shi C, Jiang HX, and Qin SY. Interleukin-9 Promotes Pancreatic Cancer Cells Proliferation and Migration via the miR-200a/Beta-Catenin Axis. BioMed Res Int. (2017) 2017:2831056. doi: 10.1155/2017/2831056
57. Ye ZJ, Zhou Q, Yin W, Yuan ML, Yang WB, Xiong XZ, et al. Differentiation and immune regulation of IL-9-producing CD4+ T cells in Malignant pleural effusion. Am J Respir Crit Care Med. (2012) 186:1168–79. doi: 10.1164/rccm.201207-1307OC
58. Boncompagni G, Tatangelo V, Lopresti L, Ulivieri C, Capitani N, Tangredi C, et al. Leukemic cell-secreted interleukin-9 suppresses cytotoxic T cell-mediated killing in chronic lymphocytic leukemia. Cell Death Dis. (2024) 15:144. doi: 10.1038/s41419-024-06528-6
59. Feng LL, Gao JM, Li PP, and Wang X. IL-9 contributes to immunosuppression mediated by regulatory T cells and mast cells in B-cell non-hodgkin’s lymphoma. J Clin Immunol. (2011) 31:1084–94. doi: 10.1007/s10875-011-9584-9
60. Ciccia F, Guggino G, Rizzo A, Manzo A, Vitolo B, La Manna MP, et al. Potential involvement of IL-9 and Th9 cells in the pathogenesis of rheumatoid arthritis. Rheumatol (Oxford). (2015) 54:2264–72. doi: 10.1093/rheumatology/kev252
61. Kundu-Raychaudhuri S, Abria C, and Raychaudhuri SP. IL-9, a local growth factor for synovial T cells in inflammatory arthritis. Cytokine. (2016) 79:45–51. doi: 10.1016/j.cyto.2015.12.020
62. Rauber S, Luber M, Weber S, Maul L, Soare A, Wohlfahrt T, et al. Resolution of inflammation by interleukin-9-producing type 2 innate lymphoid cells. Nat Med. (2017) 23:938–44. doi: 10.1038/nm.4373
63. Dantas AT, Marques CD, da Rocha Junior LF, Cavalcanti MB, Goncalves SM, Cardoso PR, et al. Increased serum interleukin-9 levels in rheumatoid arthritis and systemic lupus erythematosus: pathogenic role or just an epiphenomenon? Dis Markers. (2015) 2015:519638. doi: 10.1155/2015/519638
64. Ouyang H, Shi Y, Liu Z, Feng S, Li L, Su N, et al. Increased interleukin−9 and CD4+IL-9+ T cells in patients with systemic lupus erythematosus. Mol Med Rep. (2013) 7:1031–7. doi: 10.3892/mmr.2013.1258
65. Yang J, Li Q, Yang X, and Li M. Interleukin-9 is associated with elevated anti-double-stranded DNA antibodies in lupus-prone mice. Mol Med. (2015) 21:364–70. doi: 10.2119/molmed.2014.00237
66. Jager A, Dardalhon V, Sobel RA, Bettelli E, and Kuchroo VK. Th1, Th17, and Th9 effector cells induce experimental autoimmune encephalomyelitis with different pathological phenotypes. J Immunol. (2009) 183:7169–77. doi: 10.4049/jimmunol.0901906
67. Li H, Nourbakhsh B, Cullimore M, Zhang GX, and Rostami A. IL-9 is important for T-cell activation and differentiation in autoimmune inflammation of the central nervous system. Eur J Immunol. (2011) 41:2197–206. doi: 10.1002/eji.201041125
68. Nowak EC, Weaver CT, Turner H, Begum-Haque S, Becher B, Schreiner B, et al. IL-9 as a mediator of Th17-driven inflammatory disease. J Exp Med. (2009) 206:1653–60. doi: 10.1084/jem.20090246
69. Yoshimura S, Thome R, Konno S, Mari ER, Rasouli J, Hwang D, et al. IL-9 controls central nervous system autoimmunity by suppressing GM-CSF production. J Immunol. (2020) 204:531–9. doi: 10.4049/jimmunol.1801113
70. Bazzini C, Bertschi NL, Steck O, Luther F, Scharli S, Rolfes ED, et al. Human T helper 9 cells rely on peroxisome proliferator-activated receptor-gamma-mediated cystine uptake to prevent lipid peroxidation and bioenergetic failure. J Invest Dermatol. (2025) 145:1693–1705 e2. doi: 10.1016/j.jid.2024.10.616
71. Ma X, Bi E, Huang C, Lu Y, Xue G, Guo X, et al. Cholesterol negatively regulates IL-9-producing CD8(+) T cell differentiation and antitumor activity. J Exp Med. (2018) 215:1555–69. doi: 10.1084/jem.20171576
72. Rampal R, Awasthi A, and Ahuja V. Retinoic acid-primed human dendritic cells inhibit Th9 cells and induce Th1/Th17 cell differentiation. J Leukoc Biol. (2016) 100:111–20. doi: 10.1189/jlb.3VMA1015-476R
73. Reilly NA, Sonnet F, Dekkers KF, Kwekkeboom JC, Sinke L, Hilt S, et al. Oleic acid triggers metabolic rewiring of T cells poising them for T helper 9 differentiation. iScience. (2024) 27:109496. doi: 10.1016/j.isci.2024.109496
74. Kanno T, Nakano K, and Endo Y. Cellular metabolism in Th9, Th17, and Treg cell differentiation. Int Immunol. (2025) 37:dxaf032. doi: 10.1093/intimm/dxaf032
75. Xu WD, Chen YY, Li YW, Yang J, and Huang AF. Targeting Th9 cells in autoimmune diseases: a narrative review. Front Immunol. (2025) 16:1615611. doi: 10.3389/fimmu.2025.1615611
76. Buck MD, O’Sullivan D, and Pearce EL. T cell metabolism drives immunity. J Exp Med. (2015) 212:1345–60. doi: 10.1084/jem.20151159
77. Wang Y, Bi Y, Chen X, Li C, Li Y, Zhang Z, et al. Histone deacetylase SIRT1 negatively regulates the differentiation of interleukin-9-producing CD4(+) T cells. Immunity. (2016) 44:1337–49. doi: 10.1016/j.immuni.2016.05.009
78. Bi E, Ma X, Lu Y, Yang M, Wang Q, Xue G, et al. Foxo1 and Foxp1 play opposing roles in regulating the differentiation and antitumor activity of T(H)9 cells programmed by IL-7. Sci Signal. (2017) 10:eaak9741. doi: 10.1126/scisignal.aak9741
79. Buttrick TS, Wang W, Yung C, Trieu KG, Patel K, Khoury SJ, et al. Foxo1 promotes th9 cell differentiation and airway allergy. Sci Rep. (2018) 8:818. doi: 10.1038/s41598-018-19315-z
80. Fu Y, Wang J, Panangipalli G, Ulrich BJ, Koh B, Xu C, et al. STAT5 promotes accessibility and is required for BATF-mediated plasticity at the Il9 locus. Nat Commun. (2020) 11:4882. doi: 10.1038/s41467-020-18648-6
81. Yamaki K and Yoshino S. Preventive and therapeutic effects of rapamycin, a mammalian target of rapamycin inhibitor, on food allergy in mice. Allergy. (2012) 67:1259–70. doi: 10.1111/all.12000
82. Chen H, Zhang L, Wang P, Su H, Wang W, Chu Z, et al. mTORC2 controls Th9 polarization and allergic airway inflammation. Allergy. (2017) 72:1510–20. doi: 10.1111/all.13152
83. Roy S and Awasthi A. ATP Triggers Human Th9 Cell Differentiation via Nitric Oxide-Mediated mTOR-HIF1alpha Pathway. Front Immunol. (2019) 101120. doi: 10.3389/fimmu.2019.01120
84. Benoit-Lizon I, Jacquin E, Rivera Vargas T, Richard C, Roussey A, Dal Zuffo L, et al. CD4 T cell-intrinsic STING signaling controls the differentiation and effector functions of T(H)1 and T(H)9 cells. J Immunother Cancer. (2022) 10:e003459. doi: 10.1136/jitc-2021-003459
85. Huang YY, Jiang HX, Shi QY, Qiu X, Wei X, Zhang XL, et al. miR-145 inhibits th9 cell differentiation by suppressing activation of the PI3K/akt/mTOR/p70S6K/HIF-1alpha pathway in Malignant ascites from liver cancer. Onco Targets Ther. (2020) 13:3789–800. doi: 10.2147/OTT.S245346
86. Ahmadian M, Suh JM, Hah N, Liddle C, Atkins AR, Downes M, et al. PPARgamma signaling and metabolism: the good, the bad and the future. Nat Med. (2013) 19:557–66. doi: 10.1038/nm.3159
87. Kliewer SA, Forman BM, Blumberg B, Ong ES, Borgmeyer U, Mangelsdorf DJ, et al. Differential expression and activation of a family of murine peroxisome proliferator-activated receptors. Proc Natl Acad Sci U.S.A. (1994) 91:7355–9. doi: 10.1073/pnas.91.15.7355
88. Mangelsdorf DJ and Evans RM. The RXR heterodimers and orphan receptors. Cell. (1995) 83:841–50. doi: 10.1016/0092-8674(95)90200-7
89. Angela M, Endo Y, Asou HK, Yamamoto T, Tumes DJ, Tokuyama H, et al. Fatty acid metabolic reprogramming via mTOR-mediated inductions of PPARgamma directs early activation of T cells. Nat Commun. (2016) 7:13683. doi: 10.1038/ncomms13683
90. Stark JM, Tibbitt CA, and Coquet JM. The metabolic requirements of th2 cell differentiation. Front Immunol. (2019) 10:2318. doi: 10.3389/fimmu.2019.02318
91. Bapat SP, Whitty C, Mowery CT, Liang Y, Yoo A, Jiang Z, et al. Obesity alters pathology and treatment response in inflammatory disease. Nature. (2022) 604:337–42. doi: 10.1038/s41586-022-04536-0
92. Chen T, Tibbitt CA, Feng X, Stark JM, Rohrbeck L, Rausch L, et al. PPAR-gamma promotes type 2 immune responses in allergy and nematode infection. Sci Immunol. (2017) 2:eaal5196. doi: 10.1126/sciimmunol.aal5196
93. Saubermann LJ, Nakajima A, Wada K, Zhao S, Terauchi Y, Kadowaki T, et al. Peroxisome proliferator-activated receptor gamma agonist ligands stimulate a Th2 cytokine response and prevent acute colitis. Inflammation Bowel Dis. (2002) 8:330–9. doi: 10.1097/00054725-200209000-00004
94. Cipolletta D, Feuerer M, Li A, Kamei N, Lee J, Shoelson SE, et al. PPAR-gamma is a major driver of the accumulation and phenotype of adipose tissue Treg cells. Nature. (2012) 486:549–53. doi: 10.1038/nature11132
95. Guri AJ, Mohapatra SK, Horne WT 2nd, Hontecillas R, and Bassaganya-Riera J. The role of T cell PPAR gamma in mice with experimental inflammatory bowel disease. BMC Gastroenterol. (2010) 10:60. doi: 10.1186/1471-230X-10-60
96. Hontecillas R and Bassaganya-Riera J. Peroxisome proliferator-activated receptor gamma is required for regulatory CD4+ T cell-mediated protection against colitis. J Immunol. (2007) 178:2940–9. doi: 10.4049/jimmunol.178.5.2940
97. Sivasami P, Elkins C, Diaz-Saldana PP, Goss K, Peng A, M.t. Hamersky J, et al. Obesity-induced dysregulation of skin-resident PPARgamma(+) Treg cells promotes IL-17A-mediated psoriatic inflammation. Immunity. (2023) 56:1844–1861 e6. doi: 10.1016/j.immuni.2023.06.021
98. Wohlfert EA, Nichols FC, Nevius E, and Clark RB. Peroxisome proliferator-activated receptor gamma (PPARgamma) and immunoregulation: enhancement of regulatory T cells through PPARgamma-dependent and -independent mechanisms. J Immunol. (2007) 178:4129–35. doi: 10.4049/jimmunol.178.7.4129
99. Khokhar M, Purohit P, Gadwal A, Tomo S, Bajpai NK, and Shukla R. The differentially expressed genes responsible for the development of T helper 9 cells from T helper 2 cells in various disease states: immuno-interactomics study. JMIR Bioinform Biotechnol. (2023) 4:e42421. doi: 10.2196/42421
100. Peesari S and McAleer JP. Regulation of human Th9 cell differentiation by lipid modulators targeting PPAR-gamma and acetyl-CoA-carboxylase 1. Front Immunol. (2024) 15:1509408. doi: 10.3389/fimmu.2024.1509408
101. Dang YF, Jiang XN, Gong FL, and Guo XL. New insights into molecular mechanisms of rosiglitazone in monotherapy or combination therapy against cancers. Chem Biol Interact. (2018) 296:162–70. doi: 10.1016/j.cbi.2018.09.019
102. Ellis CN, Varani J, Fisher GJ, Zeigler ME, Pershadsingh HA, Benson SC, et al. Troglitazone improves psoriasis and normalizes models of proliferative skin disease: ligands for peroxisome proliferator-activated receptor-gamma inhibit keratinocyte proliferation. Arch Dermatol. (2000) 136:609–16. doi: 10.1001/archderm.136.5.609
103. McAleer JP. Obesity and the microbiome in atopic dermatitis: Therapeutic implications for PPAR-gamma agonists. Front Allergy. (2023) 4:1167800. doi: 10.3389/falgy.2023.1167800
104. Tachibana M, Wada K, Katayama K, Kamisaki Y, Maeyama K, Kadowaki T, et al. Activation of peroxisome proliferator-activated receptor gamma suppresses mast cell maturation involved in allergic diseases. Allergy. (2008) 63:1136–47. doi: 10.1111/j.1398-9995.2008.01677.x
105. Wang Y, Yu W, Li S, Guo D, He J, and Wang Y. Acetyl-coA carboxylases and diseases. Front Oncol. (2022) 12:836058. doi: 10.3389/fonc.2022.836058
106. Endo Y, Asou HK, Matsugae N, Hirahara K, Shinoda K, Tumes DJ, et al. Obesity drives th17 cell differentiation by inducing the lipid metabolic kinase, ACC1. Cell Rep. (2015) 12:1042–55. doi: 10.1016/j.celrep.2015.07.014
107. Berod L, Friedrich C, Nandan A, Freitag J, Hagemann S, Harmrolfs K, et al. De novo fatty acid synthesis controls the fate between regulatory T and T helper 17 cells. Nat Med. (2014) 20:1327–33. doi: 10.1038/nm.3704
108. Kao YS, Mamareli P, Dhillon-LaBrooy A, Stuve P, Godoy GJ, Velasquez LN, et al. Targeting ACC1 in T cells ameliorates psoriatic skin inflammation. J Mol Med (Berl). (2023) 101:1153–66. doi: 10.1007/s00109-023-02349-w
109. Koh J, Woo YD, Yoo HJ, Choi JP, Kim SH, Chang YS, et al. De novo fatty-acid synthesis protects invariant NKT cells from cell death, thereby promoting their homeostasis and pathogenic roles in airway hyperresponsiveness. Elife. (2023) 12:RP87536. doi: 10.7554/eLife.87536
110. Mamareli P, Kruse F, Lu CW, Guderian M, Floess S, Rox K, et al. Targeting cellular fatty acid synthesis limits T helper and innate lymphoid cell function during intestinal inflammation and infection. Mucosal Immunol. (2021) 14:164–76. doi: 10.1038/s41385-020-0285-7
111. Stuve P, Minarrieta L, Erdmann H, Arnold-Schrauf C, Swallow M, Guderian M, et al. De novo fatty acid synthesis during mycobacterial infection is a prerequisite for the function of highly proliferative T cells, but not for dendritic cells or macrophages. Front Immunol. (2018) 9:495. doi: 10.3389/fimmu.2018.00495
112. Nakajima T, Kanno T, Yokoyama S, Sasamoto S, Asou HK, Tumes DJ, et al. ACC1-expressing pathogenic T helper 2 cell populations facilitate lung and skin inflammation in mice. J Exp Med. (2021) 218:e20210639. doi: 10.1084/jem.20210639
113. Endo Y, Onodera A, Obata-Ninomiya K, Koyama-Nasu R, Asou HK, Ito T, et al. ACC1 determines memory potential of individual CD4(+) T cells by regulating de novo fatty acid biosynthesis. Nat Metab. (2019) 1:261–75. doi: 10.1038/s42255-018-0025-4
114. Ibitokou SA, Dillon BE, Sinha M, Szczesny B, Delgadillo A, Reda Abdelrahman D, et al. Early inhibition of fatty acid synthesis reduces generation of memory precursor effector T cells in chronic infection. J Immunol. (2018) 200:643–56. doi: 10.4049/jimmunol.1602110
115. Nakajima T, Kanno T, Ueda Y, Miyako K, Endo T, Yoshida S, et al. Fatty acid metabolism constrains Th9 cell differentiation and antitumor immunity via the modulation of retinoic acid receptor signaling. Cell Mol Immunol. (2024) 21:1266–81. doi: 10.1038/s41423-024-01209-y
116. Galdieri L and Vancura A. Acetyl-CoA carboxylase regulates global histone acetylation. J Biol Chem. (2012) 287:23865–76. doi: 10.1074/jbc.M112.380519
117. Schwartz DM, Farley TK, Richoz N, Yao C, Shih HY, Petermann F, et al. Retinoic acid receptor alpha represses a th9 transcriptional and epigenomic program to reduce allergic pathology. Immunity. (2019) 50:106–120.e10. doi: 10.1016/j.immuni.2018.12.014
118. Mantel PY, Ouaked N, Ruckert B, Karagiannidis C, Welz R, Blaser K, et al. Molecular mechanisms underlying FOXP3 induction in human T cells. J Immunol. (2006) 176:3593–602. doi: 10.4049/jimmunol.176.6.3593
119. Tran DQ, Ramsey H, and Shevach EM. Induction of FOXP3 expression in naive human CD4+FOXP3 T cells by T-cell receptor stimulation is transforming growth factor-beta dependent but does not confer a regulatory phenotype. Blood. (2007) 110:2983–90. doi: 10.1182/blood-2007-06-094656
120. Oh CK, Leigh R, McLaurin KK, Kim K, Hultquist M, and Molfino NA. A randomized, controlled trial to evaluate the effect of an anti-interleukin-9 monoclonal antibody in adults with uncontrolled asthma. Respir Res. (2013) 14:93. doi: 10.1186/1465-9921-14-93
121. Guseva NV, Rokhlin OW, Glover RA, and Cohen MB. TOFA (5-tetradecyl-oxy-2-furoic acid) reduces fatty acid synthesis, inhibits expression of AR, neuropilin-1 and Mcl-1 and kills prostate cancer cells independent of p53 status. Cancer Biol Ther. (2011) 12:80–5. doi: 10.4161/cbt.12.1.15721
122. He D, Sun X, Yang H, Li X, and Yang D. TOFA induces cell cycle arrest and apoptosis in ACHN and 786-O cells through inhibiting PI3K/Akt/mTOR pathway. J Cancer. (2018) 9:2734–42. doi: 10.7150/jca.26374
123. Nishi K, Suzuki K, Sawamoto J, Tokizawa Y, Iwase Y, Yumita N, et al. Inhibition of fatty acid synthesis induces apoptosis of human pancreatic cancer cells. Anticancer Res. (2016) 36:4655–60. doi: 10.21873/anticanres.11016
Keywords: Th9 cells, interleukin-9 (IL-9), allergies, autoimmunity, cancer, MTOR activation, PPAR-gamma, ACC1
Citation: Peesari S and McAleer JP (2025) Metabolic regulation of Th9 cell differentiation: insights for IL-9-driven diseases. Front. Immunol. 16:1672072. doi: 10.3389/fimmu.2025.1672072
Received: 23 July 2025; Accepted: 01 September 2025;
Published: 15 September 2025.
Edited by:
Gareth S. D. Purvis, University of Oxford, United KingdomReviewed by:
Suyasha Roy, National Institutes of Health (NIH), United StatesCopyright © 2025 Peesari and McAleer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jeremy P. McAleer, bWNhbGVlckBtYXJzaGFsbC5lZHU=