 Elda Pereira Noronha1†
Elda Pereira Noronha1† Luísa Vieira Codeço Marques1†
Luísa Vieira Codeço Marques1† Francianne Gomes Andrade1
Francianne Gomes Andrade1 Luiz Claudio Santos Thuler2
Luiz Claudio Santos Thuler2 Eugênia Terra-Granado1
Eugênia Terra-Granado1 Maria S. Pombo-de-Oliveira1*† and the Brazilian Collaborative Study Group of Acute Leukemia‡
Maria S. Pombo-de-Oliveira1*† and the Brazilian Collaborative Study Group of Acute Leukemia‡- 1Pediatric Hematology-Oncology Program, Research Center, Instituto Nacional de Câncer, Rio de Janeiro, Brazil
- 2Clinical Research Division, Research Center, Instituto Nacional de Câncer, Rio de Janeiro, Brazil
T-cell acute lymphoblastic leukemia (T-ALL) is a biologically heterogeneous malignancy, which reflects distinctive stages of T-cell differentiation arrest. We have revisited a cohort of pediatric T-ALL, in order to test if immunophenotypes associated with molecular alterations would predict the patient's outcome. Genetic mutations, translocations and copy number alterations were identified through Sanger sequencing, RT-PCR, FISH and multiplex ligation-dependent probe amplification (MLPA). We defined 8 immunophenotypic T-ALL subtypes through multiparametric flow cytometry: early T-cell precursor (ETP, n = 27), immature (n = 38), early cortical (n = 15), cortical (n = 50), late cortical (n = 53), CD4/CD8 double negative mature (n = 31), double positive mature (n = 35) and simple positive mature (n = 31) T-ALL. Deletions (del) or amplifications (amp) in at least one gene were observed in 87% of cases. The most frequent gene alterations were CDKN2A/Bdel (71.4%), NOTCH1mut (47.6%) and FBXW7mut (17%). ETP-ALL had frequent FLT3mut (22.2%) and SUZ12del (16.7%) (p < 0.001), while CDKN2A/Bdel were rarely found in this subtype (p < 0.001). The early cortical T-ALL subtype had high frequencies of NOTCH1mut and IL7Rmut (71%, 28.6%, respectively), whereas, mature T-ALL with double positive CD4/CD8 had the highest frequencies of STIL-TAL1 (36.7%), LEF1del (27.3%) and CASP8AP2del (22.7%). The co-existence of two groups of T-ALL with NOTCH1mut/IL7Rmut, and with TLX3/SUZ12del/NF1del/IL7Rmut, were characterized with statistical significance (p < 0.05) but only STIL-TAL1 (pOS 47.5%) and NOTCH1WT/FBXW7WT (pOS 55.3%) are predictors of poor T-ALL outcomes. In conclusion, we have observed that 8 T-ALL subgroups are characterized by distinct molecular profiles. The mutations in NOTCH1/FBXW7 and STIL-TAL1 rearrangement had a prognostic impact, independent of immunophenotype.
Introduction
T-cell acute lymphoblastic leukemia (T-ALL) is a biologically heterogeneous malignancy that reflects the stage of T-cell differentiation arrest. The frequency of T-ALL is about 15% of pediatric ALL cases, being characterized clinically as a predictive high-risk group for long-term outcome (1). The understanding of T-cell biology has substantially improved, and nowadays the 5-years survival rate is about 65–70% in some T-ALL settings (2). Unlike B-cell precursor ALL, in which the immunophenotype-genotype association profile is well established as outcome predictors, T-ALL still needs further investigations. The classification of T-ALL into the four immunophenotypic sub-groups, pro-T, pre-T, cortical, and mature (3), has been under scrutiny for clinical-therapeutical decisions. For instance, CD1a phenotype and CD56 were first associated with outcome (1, 4), but in further studies a null effect has been found (5, 6). The intensification of treatment through consolidation, maintenance regimen and re-intensification has led to considerable benefits to pediatric T-ALL remission rates (7). In 2009, the early T-cell precursor acute lymphoblastic leukemia subtype (ETP-ALL) was characterized by presence of lymphoblasts expressing cytoplasmic CD3 and CD7, in addition to stem cell and myeloid antigens, and the gene mutational spectrum is similar to that of poor differentiated myeloid neoplasm (8, 9). Although this entity was initially described as a predictive high-risk group, recent studies demonstrated no consensus regarding ETP-ALL diagnosis and outcome (2, 10, 11).
Recently, genomic landscape data have shown that the gene lesions frequently found in T-ALL are associated with maturational subtypes of T-ALL (12, 13). Liu et al. (12) have described 10 altered pathways according to T-ALL maturational stage. For instance, NRAS/FLT3 mutations were associated with immature T-ALL (12). It is expected that the genomic landscape provides a logical framework for new therapeutic approaches, therefore, it is very important to establish an algorithm of tests that could quickly predict genetic lesions at diagnosis. Even though T-ALL outcomes have significantly improved in patients that received intensive treatment protocol, unveiling the associations between immunophenotypic profile and genetic abnormalities in T-ALL can be of great importance to establish subsets with distinct prognostic relevance, providing means to avoid late toxicity through risk adaptation of treatment with target therapy. Here, we have revisited a series of pediatric T-ALL and tested if immunophenotypic profiles are associated with distinct molecular alterations and if these associations would predict patient's outcome.
Materials and Methods
Patient Samples
Three hundred forty-one T-ALL cases (age <19 years), sent to the Pediatric Hematology-Oncology Program, Research Center, Instituto Nacional de Câncer, Rio de Janeiro, Brazil, for diagnostic tests (2005–2017), were reviewed in this study. The study design with selection criteria is shown in Supplementary Figure 1. Viable frozen cells were available to complete the immunophenotyping panel according to ETP-ALL criteria from Inukai et al. (11) and to perform additional molecular tests. Cases that had the diagnosis of lymphoblastic lymphoma with <20% of bone marrow infiltration (n = 12) were excluded from this study. Myeloid/T-cell mixed phenotype acute leukemia (n = 10), samples with low cell viability (n = 11) and samples not tested for cytoplasmic CD3, membrane CD3 and/or CD1a (n = 26) were also excluded.
The referring physicians provided information about the demographic and clinical follow up data for the patients. Patients were not formally enrolled in treatment protocols, but were treated according to either the Brazilian Group for Treatment of Childhood Leukemia (GBTLI-ALL99) or the Berlin-Frankfurt-Munster ALL (BFM–ALL) backbone protocols (14, 15).
The treatment outlines were similar, as in the induction phase, all patients received a pre-phase of prednisone (7 days) and intrathecal dose of Methotrexate (MTX). The induction phase lasted 4 weeks, and included prednisone, Vincristine, doxorubicin, L-asparaginase, and intrathecal MTX, Cytarabine and dexamethasone treatment. All patients received adequate treatment for prevention of CNS relapse with chemotherapy. Children with CNS infiltration at the diagnosis received cranial radiotherapy.
Ethics
This study was carried out in accordance with the recommendations of Instituto Nacional de Câncer Research and Ethics Committee. Written informed consent from the parents or legal guardians was obtained from children and adolescents. All young subjects gave written informed consent in accordance with the Declaration of Helsinki. The protocol was approved by the Instituto Nacional de Câncer Research and Ethics Committee under the registry number CEP/INCA#117/12; CONEP: PB #888.2772.3.
T-ALL Immunophenotypic Characterization
In all cases, the immunophenotyping by multiparametric flow cytometry was performed utilizing the panel of monoclonal antibodies in Supplementary Table 1. FACS Calibur and FACS Canto II flow cytometers (Becton, Dickinson, and Company, CA, USA) were used for the sample acquisition and all the immunophenotypic analyses were performed in the Infinicyt™ program version 1.8 (Cytognos—Salamanca—Spain), according to previously published procedures (16, 17). A sample was considered positive for a marker when at least 20% of lymphoblasts in a CD45low/intermediate gate had its expression.
The immunophenotypic classification of T-ALL subtypes was applied using previously published criteria (3, 8, 11). Immunophenotyping was performed by 6 color combination of monoclonal antibodies, applying the Coustan-Smith et al. (8) criteria with the addition of the score system from Inukai et al. (11) to identify ETP-ALL cases.
T-ALL Molecular Characterization
Diagnostic samples from T-ALL were first subjected to total RNA extraction using TRIzol (Life Technologies, Grand Island, NY, USA) according to the manufacturer's instructions. Complementary DNA (cDNA) was synthesized using 2 μg of total RNA with the First-Strand cDNA Synthesis Kit™ (Amersham Pharmacia Biotech, Amersham Biosciences UK Limited, Little Chalfont/UK); cDNA integrity was examined by amplifying a fragment of the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) constitutive gene, accordingly. The STIL-TAL1 and TX3 rearrangement were investigated by Reverse Transcriptase-PCR, as previously published (18). Partial clinical, molecular and prognostic data on Brazilian samples diagnosed from 2005-2012 have been previously published (19, 20).
Genomic DNA from the same leukemic cell samples was also obtained using a QIAamp® DNA Blood Mini Kit (Qiagen GmbH, Hilden, Germany), as recommended by the manufacturer. Mutations were investigated in the hotspot regions of the following genes: NOTCH1, FBXW7, IL7R, RAS (NRAS and KRAS), and FLT3. NOTCH1 mutations were analyzed by screening of the heterodimerization (HD) (exon 26 and 27) and polypeptide enriched in proline, glutamate, serine and threonine (PEST) (exon 34) domains (19, 21). To evaluate the mutational status of FBXW7, we screened exons 9 and 10 (22); mutations in IL7R were investigated in exon 6 (23); mutations in FLT3 were investigated at the tyrosine kinase domain (TKD) in codon 835 and the juxtamembrane domain in exons 11/12 as internal tandem duplications (ITD) and N/KRAS status was determined by searching mutations in exon 1 (codons 12/13) (24). All PCR products were purified, and Sanger sequencing was performed using a BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, Carlsbad, CA) in a 3,500 Genetic Analyzer (Applied Biosystems). The analyses were performed with BioEdit 7.0.9 software, comparing electropherograms with the reference sequences accessed from the National Center for Biotechnology Information (NCBI): NOTCH1 (NG_007458.1; NM_017617.3), FBXW7 (NM_1013415.1; NG_029466.1), IL7R (NG_95671.1; NM_2184.3), KRAS (NG_7524.1; NM_004985.4) and NRAS (NM_002524.4; NG_007572.1).
The multiplex ligation-dependent probe amplification (MLPA) was performed using the SALSA MLPA probe mix P383-A1 TALL (MRC Holland, Amsterdam, The Netherlands) to identify copy number alterations (CNA). This kit is able to assess alterations (deletions or amplifications) in transcription factors (LEF1 and MYB); in signal transduction (PTEN, NF1 and PTPN2); in cell cycling (CDKN2A, CDKN2B and CASP8AP2) and in epigenetic regulator genes (EZH2, SUZ12 and PHF6) and identify the STIL-TAL1 and NUP214-ABL1 fusion genes. The procedure and analyses of data were done according to the manufacturer's instructions. Analyzes was done in Coffalyser software v.140721.1958 (MRC-Holland, Amsterdam, The Netherlands).
Statistical Analyses
To compare the distribution of demography, clinical variables, cellular and molecular alterations between T-ALL subtypes, we have used the χ2 test. Univariate p values were calculated using Pearson's chi-square test or Fisher exact test. Two-sided p-values with a significance limit of <0.05 were considered throughout the study. The frequency of genetic aberration and analysis of concomitances were illustrated by Circos plots (25).
The probability of overall survival (pOS) in 60 months was determined using the Kaplan–Meier method in months from the diagnosis to the outcome (death, survival or last follow-up). Patients who lost to follow-up were censored at their date of last known information. The differences between T-ALL survival distributions were compared by the log-rank test. The multivariate Cox proportional hazard regression method was used to determine the independent prognostic factors influencing pOS. Multivariate Cox analysis was performed with variables associated with a p < 0.2 in univariate analysis. To compare the immunophenotypic profiles regarding demographic, clinical and molecular characteristics, and OS each subtype vs. the remaining subtypes together were considered.
SPSS (Statistical Product and Services Solutions, version 18.0, SPSS Inc, Chicago, IL, USA) software was used for data analyses.
Results
The T-ALL cases previously characterized according to EGIL classification [T-I (n = 7), T-II (n = 73), T-III (n = 103), and T-IV (n = 99)] were revisited, and with the addition of CD117, CD11b, and CD15 analysis, they were re-classified according to Inukai et al. (11) score system (Supplementary Figure 2). Twenty-seven cases previously classified as T-I (n = 7) and T-II (n = 20) were re-classified as ETP-ALL, representing 9.7% of the total T-ALL cohort. The remaining cases were subdivided as follows: (1) immature T-ALL (CD2pos and/or CD5pos and/or CD8pos or CD4pos; n = 38); (2) early cortical (CD4pos and CD8pos, CD1aneg, mCD3neg; n = 15); cortical (CD1apos and mCD3neg; n = 50); late cortical (CD1apos and mCD3pos; n = 53); and (3) mature T-ALL, (CD1aneg and mCD3pos; n = 99). The latter was subdivided in CD4/CD8 double positive (DP) (n = 35), single positive (SP) either CD4neg/CD8pos or CD4pos/CD8neg (n = 31) and CD4/CD8 double negative (DN) (n = 31). All T-ALL cases were cCD3 and CD7 positive. The demographic and clinical features were analyzed according to reviewed T-ALL subtypes and shown in Table 1. ETP-ALL subtype was associated with patients older than 10 years at the diagnosis and circulating white blood cell count (WBC) of <50 × 109/L (p = 0.02 and 0.042, respectively). No significant differences were found in demographic and/or clinical features according to immature, early and late cortical and/or mature T-ALL subtypes (Supplementary Table 2).
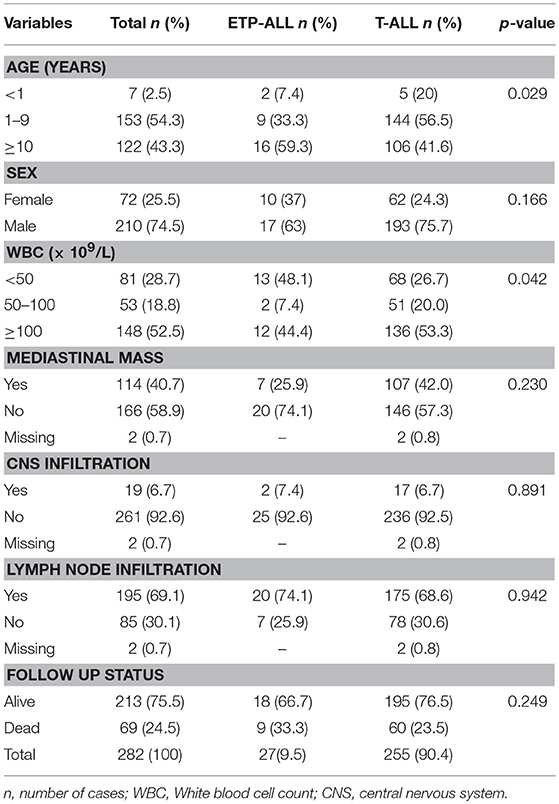
Table 1. Demographic, clinical-laboratorial and molecular features of pediatric T-ALL in Brazil, 2005–2017.
The frequencies of molecular alterations in overall cases and according to revisited T-cell subtypes are shown in Table 2. NOTCH1 mutations were found in 110 out of 231 (47.6%) cases; among these NOTCH1mut cases, 62 (56.4%) had solely HD domain mutations, 27 (24.5%) in the PEST/TAD domain, and 21 (19.1%) had mutations in both HD and PEST domains (data not shown). Thirty-eight cases had FBXW7mut (17%), 19 N/KRASmut (8.2%), 16 IL7Rmut (7.1%), and 9 cases FLT3mut (4.1%). The STIL-TAL1 fusion gene was found in 49 cases (21.2%) and 25 cases were positive for TLX3 (10.2%), which were mutually exclusive. The co-occurrence of both NOTCH1 and FBXW7 mutations were found in 129 cases (57.6%). A heterogeneous distribution of frequencies of these molecular alterations was observed among the T-ALL subtypes, although few specific molecular alterations were associated with the immunophenotyping. The frequencies of NOTCH1mut varied from 37% to 71.4% across the T-ALL subtypes. The early cortical T-ALL presented the highest frequencies of NOTCH1mut and IL7Rmut (71.4% and 28.6%, respectively; p = 0.01). FLT3mut was most frequent in the ETP-ALL subgroup (p < 0.001), whereas IL7Rmut in early cortical T-ALL and N/KRASmut in mature DN T-ALL (p < 0.05). IL7Rmut was absent in ETP-ALL, and N/KRASmut in late cortical and DP mature T-ALL (Table 2). Some molecular alterations were age-associated: 16 out 19 (84.2%) of T-ALL with N/KRASmut were found in children <10 years old (p = 0.037).

Table 2. Molecular alterations according to pediatric T-cell Acute Lymphoblastic Leukemia subtypes, Brazil 2015–2017.
One hundred and sixty-eight T-ALL cases had a good quality of available DNA to perform MLPA tests. To test the possible selection bias, we have compared the demographic and clinical characteristics, T-ALL subsets and molecular aberrations of these 168 cases tested (Supplementary Table 3). There were equal frequency distributions of all variables among the selected cases tested, but there was a decreased number of cases with WBC lower than 100 × 109/L among non MLPA tested T-ALLs (p < 0.01).
The overall frequencies and the concomitance of gene alterations in 168 T-ALL cases are shown in Figures 1A,B. Deletions (del)/amplifications (amp) in at least one gene was identified in 87% of cases. The most frequent genomic aberration was CDKN2A/Bdel found in 71.4% of the cases, with a predominant biallelic status (78.3%). LEF1del (13%), PTENdel (11.3%) and CASP8AP2del (9.5%) were also recurrent. The most frequent gene amplifications were found in MYB (9.5%), and NUP214-ABL1 fusion gene was identified in 6% of cases. The IL7Rmut (n = 16) was found associated with NOTCH1mut (n = 13) and TLX3pos (n = 5) (p < 0.01). Likewise, NF1del and SUZ12del were found concomitantly in three out of eleven TLX3pos cases (p < 0.01). On the other hand, co-occurrence of NF1del and SUZ12del with CDKN2A/Bdel was rarely observed (p < 0.05). In summary, the molecular concomitance reflects the T-ALL heterogeneity, and two groups were well defined: one with NOTCH1mut/IL7Rmut, and another with TLX3pos/SUZ12del/NF1del/IL7Rmut (Figure 1; p < 0.05).
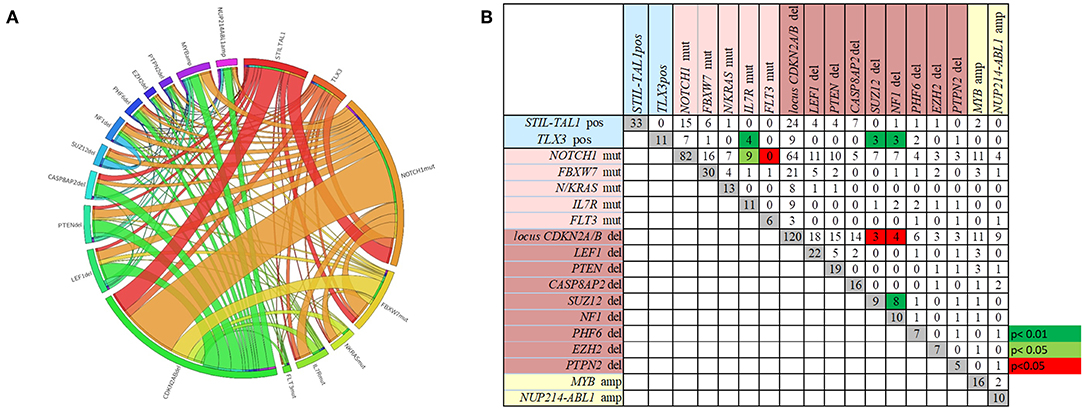
Figure 1. The frequencies and the concomitance of molecular alterations in 168 pediatric T-ALL cases. (A) Circos plot showing the overall co-occurrence of mutations, rearrangements, and CNA assessed through MLPA. Outer segments proportionally represent the alterations found in T-ALL. Interior lines connecting the outer segments proportionally demonstrate the concomitances among genetic alterations. (B) Number of cases with each molecular alteration in gray squares, and number of cases with concomitances between two alterations in white, green and red squares. Green squares represent positive while the red squares negative associations. Alterations ordered by type of abnormality. Rearrangements in blue, mutations in pink, CNA deletions in brown and amplifications in yellow.
The profile of T-ALL subtypes and the coexistence of molecular alterations, according to genes with functions as transcription factors, cell cycling regulator, cell signaling pathways and epigenetic regulators, are better visualized in the Figure 2. We have observed that ETP and immature T-ALL subtypes were enriched with alterations in epigenetic regulators EZH2, SUZ12, and PHF6 (p < 0.001 and p = 0.02, respectively), whereas these alterations were less frequently found in the late cortical and mature T-ALL subtypes (p = 0.013). ETP-ALL subtype has the highest frequencies of SUZ12del (16.7%; p = 0.025), while CDKN2A/Bdel (p < 0.001), CASP8AP2del, LEF1del, PTENdel, and IL7Rmut, were not found in this subtype. CDKN2A/Bdel was present in all early-cortical T-ALL subtype, cortical (96%) and mature DP subtype (91%), (p < 0.01; p = 0.040, respectively). Mature DP T-ALL subtypes have the highest frequencies of LEF1del and CASP8AP2del (27.3 and 22.7%, respectively, p = 0.04; p = 0.046).
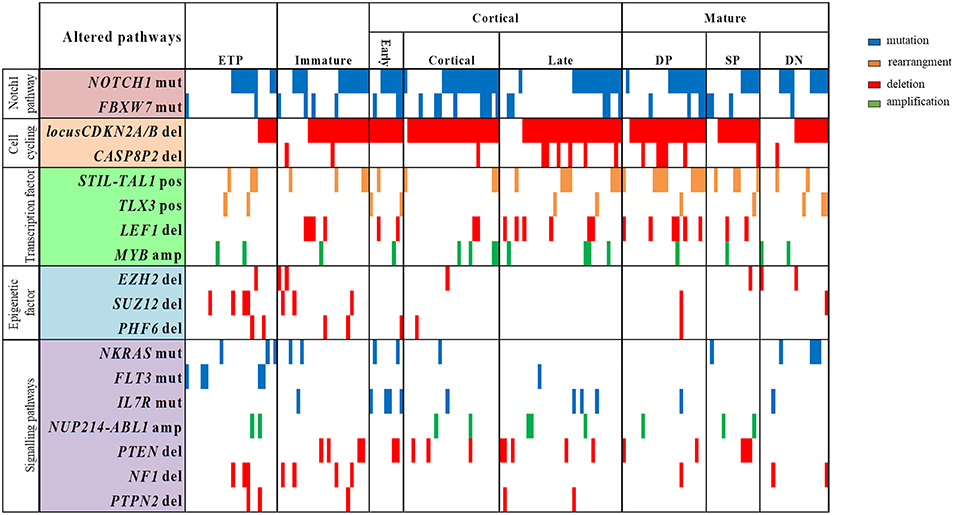
Figure 2. Overview of molecular alterations identified in 168 samples according to T-ALL subtypes and grouped into recurrently altered pathways. ETP, early T cell precursror; DP, douple positive for CD4/CD8; SP, single positive for CD4/CD8; DN, double negative for CD4/CD8; mut, mutation; del, deletion; pos, positive; amp, amplification. Vertical lines represent each patient.
The results of the univariate analysis for pOS of T-ALL cases, regarding clinical, immunophenotypic subtypes and molecular alterations are shown in Supplementary Table 4. The median time of OS of the whole cohort was 42.7 months (95%CI: 39.5-46.2; 5-years pOS 62% ± 0.04). No significant difference in pOS was found for the ETP-ALL when compared to other T-ALL subtypes, WBC level, sex and central nervous system (CNS) infiltration. However, patients with an age <12 months-old had a lower pOS (22.2%; p = 0.036) and the presence of lymph node infiltration and mediastinal mass had higher pOS (68.8%,) compared to those without (49.2%; p = 0.008 and 0.04, respectively), and infiltration in CNS was 57.9%. Regarding molecular alterations, the pOS rates were not significant for TLX3pos, IL7Rmut, RASmut and CNAs. The variables with a p < 0.2 according to univariate analysis are demonstrated in Table 3.
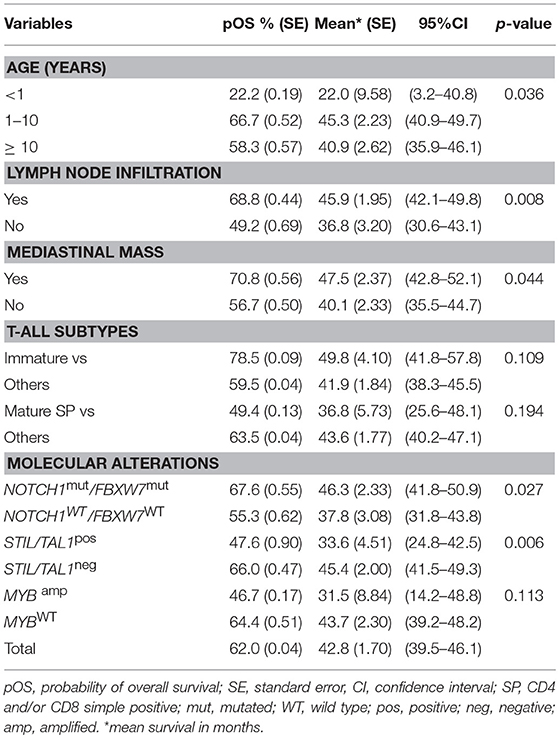
Table 3. Pediatric T-cell acute lymphoblastic leukemia and overall survival in univariate analysis, 2005-2017, Brazil.
In the multivariate analysis model shown in Table 4, the absence of lymph node infiltrations, NOTCH1/FBXW7WT and STIL-TAL1pos were independent risk factors to predict low pOS. Cases with NOTCH1 and/or FBXW7mut were associated with better pOS than NOTCH1WT and FBXW7WT cases. The presence of STIL-TAL1 fusion genes was predictive of worse outcome as shown in Figure 3.
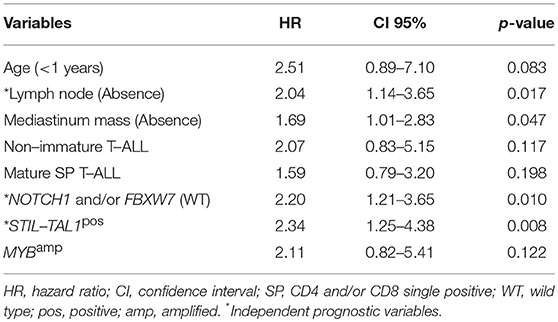
Table 4. Cox regression model of the overall survival variables of pediatric T-cell acute lymphoblastic leukemia, Brazil, 2005–2017.
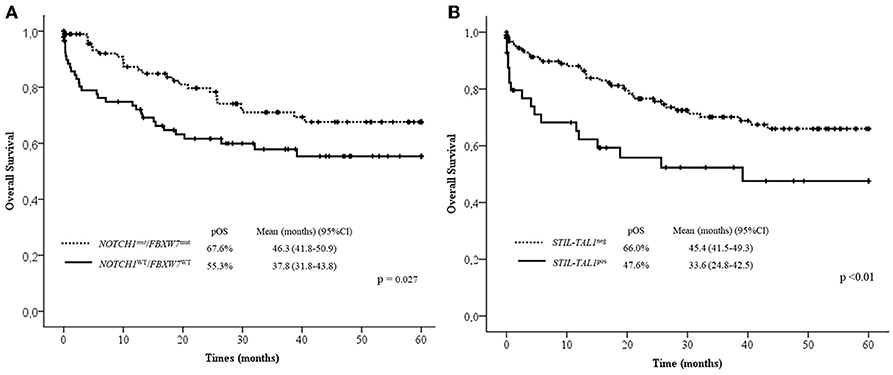
Figure 3. Overall survival curves in 60 months according to NOTCH1/FBXW7 (A) and STIL-TAL1 (B) status. Mut, Mutated; WT, Wild Type; pOS, probability of overall survival; CI, confidential interval; pos, positive.
Discussion
Since T-ALL can be arrested at a variety of T-cell development stages, with common phenotype aberrations, we can conclude that, although important, the current EGIL definition of T-ALL is no longer able to predict genotype abnormalities or patient outcome (5, 12, 13). We have revisited the immunophenotyping of a large cohort of pediatric T-ALL to compare the subtypes with distinctive genomic alterations, implementing a new subdivision based on key differentiation markers, to validate the Inukai et al. (11) score system and to distinguish ETP-ALL among immature T-ALL (8, 11, 12, 26). ETP-ALL has emerged from genomic approaches and was correlated with immunophenotypic profile that has myeloid markers as a differentiating category, in contrast to other T-ALL subtypes. The revisited immunophenotyping demonstrated the value of CD1a, cytoplasmatic CD3, CD5 and the inclusion of myeloid markers (CD117, CD11b, CD15), not previously included in other studies, in the identification of ETP-ALL cases. All cases previously classified as pro-T (T-I) in the EGIL criteria and some of pre-T (T-II) cases were now categorized as ETP-ALL.
In the normal T-cell development process, the commitment to T-cell lineage is characterized by a gradual decrease of CD34, CD44, acquisition of CD1a and, as a later T-cell developmental event, the loss of CD1a (27). Recently, we have demonstrated the aberrant antigenic pattern of CD44 in T-ALL according to T-ALL cell subtypes. CD44 expression in mature subtypes seems to be influenced by genomic alterations in NOTCH1 signaling pathway validating the studies performed in an animal model and/or in co-cultures of human cell lines (16, 27). In the cortical stage, double positivity for CD4/CD8 was found in the majority of T-ALL cases, while the mature T-ALL subtypes were carefully subdivided according to CD4 and CD8 expression (DP, SP and DN) (28, 29). This approach allowed us to identify, by flow cytometry, the immature and mature subtypes of T-ALL with significant association with genomic abnormalities. Overall, 87% of the patients tested by MLPA for CNA have harbored genetic aberrations (either deletions or amplifications) in different frequency rates, according to the level of maturity of the T-cell leukemia. The frequencies of recurrent genetic mutations and rearrangements in our whole T-ALL cohort were in agreement with other clinical data described in the literature, with CDKN2A/B deletions, NOTCH1 mutations and STIL-TAL1 gene fusion being the more prevalent alterations (12, 13, 30). The frequencies of NOTCH1 mutations in our series (47.6%) were similar to frequencies described in Asian patients (31, 32) and slightly lower than other studies in European and North American cohorts (52.2 to 56%), (21, 33, 34). It is important to highlight that these studies, such as ours, screened NOTCH1 mutations in hot spot exons (26, 27 and 34). High frequencies, such as 79% described by Liu et al. (12), can be found when whole-exome sequencing is used. Additionally, the frequencies of MYB and NUP214-ABL1 amplifications were also in accordance with previous studies (35, 36).
The molecular profile of ETP-ALL cases was associated with FLT3 mutations, and enriched with alterations in epigenetic regulators (EZH2, SUZ12, and PHF6). Inactivation-associated epigenetic alterations in hematopoietic progenitors were established as arresting T-cell development and directly leading to aberrant upregulation of early hematopoietic programs transcription in ETP-ALL mouse model (37, 38). For instance, EZH2 and RUNX1 deletions were shown to increase RAS pathway-associated transcription (38), possibly creating a transcriptional environment susceptible to additional hits, such as FLT3-ITD and RAS pathway mutations, which further increases proliferation.
Recently, the role for PHF6 was described in hematopoietic stem cell homeostasis and T-ALL leukemia initiating cell self-renewal, characterizing PHF6 mutations as early events and drivers of leukemia stem cell activity in the pathogenesis of T-ALL (39). Although alterations in PHF6 are often seen in ETP-ALL (9), they can also occur in more mature T-ALL subtypes (40), as observed in our cohort. In addition, the presence of deletions in CDKN2A/B, PTEN and LEF1 were rarely observed in the ETP-ALL cases. On the other hand, mature T-ALL with CD4/CD8 DP has presented a high frequency of deletions in LEF1, CASP8AP2, CDKN2A/B genes and the presence of STIL-TAL1 gene fusion. The mature T-ALL SP to CD4 or CD8 had low NOTCH1 mutations and DN mature T-ALL was associated with an increased frequency of N/KRAS mutations. These differences highlight that these subtypes can represent distinct biological subsets and should not be classified as a single subtype (as only mature), without considering the expression level of CD4 and CD8 antigens.
The molecular aberration profile of ETP-ALL in our cases is partially in accordance with previous publications of T-ALL genomic landscapes (9, 12). However, in this subtype, IL7Rmut was not observed and the frequency of NOTCH1mut did not differ significantly from other subtypes. The great majority of T-cell development research has been performed in animal model or in co-cultures of human cell lines. For instance, in murine thymocyte progenitors, the altered expression of LMO2 and/or BCL11B were shown to cooperate with IL7R mutations before the CD4/CD8 double-negative stage (DN2) and led to the development of ETP-ALL (41). In another murine model, the presence of IL7R mutation, together with an intracellular active form of NOTCH1 (ICN1), led to the development of an aggressive T-ALL with CD4/CD8 DP profile (42, 43). The progression from DN to DP CD4/CD8 stage is characterized by thymocyte expansion, regulated by IL-7R and NOTCH1 signaling as well as LEF1, the transcription factor that is indispensable for thymocyte maturation (44). These in vivo studies might reflect our findings, since most T-ALL cases characterized as CD4/CD8 DP had IL7Rmut and NOTCH1mut (81.2%), while CDKN2A/B and LEF1 deletions by contrast, were rarely found in our ETP-ALL cohort.
One of our aims was to test if any molecular alterations were associated with T-ALL maturation arrest and could predict the outcome. We have first analyzed the impact of the classic prognostic factors such as age strata, sex, increased WBC and CNS infiltration. Two clinical factors presented significantly poor outcome: children with an age <1 year at the diagnosis of T-ALL and the absence of adenomegaly. We have previously tested the association of molecular alterations with both maturational subtype (ETP-ALL vs. more mature T-ALL) and the outcome. Among the subtypes identified in our cohort, the immature subtype had a reasonably good 5-years pOS (75.4%), although not statistically significant. In contrast, the ETP-ALL had a 5-year pOS of 56.7%. Recently we demonstrated that ETP-ALL with NOTCH1mut was associated with significantly better pOS (90%) than NOTCH1wt (pOS 37%; p = 0.017) (45). Interestingly, although the early cortical subtype had higher frequencies of NOTCH1mut, it presented a relatively low pOS (48.6%). In addition, this subtype was associated with high frequencies of IL7Rmut and CDKN2A/Bdel. In a large cohort of children with T-ALL, Schrappe et al. (46) reported that MRD status classified as standard, intermediate or high, allowed for discrimination of prognostic subgroups associated with T-ALL maturational stage. The cortical T-ALL corresponded to the MRD standard patients, having a better event free-survival, which led to the affirmation that the outcome differed by maturational stage (46). The genetic basis of these results could be a specific pathways genetic mutation underlying the T-ALL MRD status and outcome, for instance, CDKN2A/Bdel and NOTCH1mut.
We found a high frequency of CDKN2A/B deletions in all T-ALL subtypes, except ETP-ALL, although this genetic lesion had a null effect in the pOS. Two groups of co-occurring aberrations, such as NOTCH1mut/IL7Rmut and TLX3pos/SUZ12del/NF1del/IL7Rmut were characterized with statistical significance. These genes are implicated in the pathogenesis of T-ALL in cortical and in immature subtypes. The screening of these alterations would be important to drive the selection of target therapy.
The multivariate analysis shows that NOTCH1WT and FBXW7WT, STIL-TAL1pos and the absence of lymph node enlargement are the most relevant indicators of inferior pOS, despite age, T-ALL subtypes and WBC.
Despite the lack of unique protocol applied, all patients have received adequate treatment for prevention of CNS relapse with poly-chemotherapy, and children with high WBC and CNS infiltration at the initial diagnosis received cranial radiotherapy (15). Therefore, our cohort study has confirmed, that the presence of STIL-TAL1 is predictive of poor outcome, while NOTCH1/FBXW7 mutations have the opposite effect, reinforcing the idea that the prognostic impact of NOTCH1 and FBXW7 seems not to be dependent on the treatment protocol applied, despite contrary studies.
Our data support the premises that genetic lesions are associated with T-ALL immunophenotypic profiles and that identifying molecular aberrations is relevant as it would allow patients to receive novel treatment agents, such as target therapies, as the front line treatment.
Conclusion
In conclusion, our results show 8 T-ALL subgroups identified by flow cytometry. These subsets are characterized by distinct molecular profiles, as ETP-ALL and mature T-ALL subtypes, classified according to CD4 and CD8 expression. Nevertheless, immunophenotypic subtypes, classified based on T-cell differentiation, was not predictive for outcome. Of the molecular alterations, only mutations in NOTCH1/FBXW7 and STIL-TAL1 rearrangement had a prognostic impact independent of immunophenotype.
Ethics Statement
This study was carried out in accordance with the recommendations of Instituto Nacional de Câncer Research and Ethics Committee. Written informed consent from the parents or legal guardians were obtained from children and adolescent. All young subjects gave written informed consent in accordance with the Declaration of Helsinki. The protocol was approved by the Instituto Nacional de Câncer Research and Ethics Committee under the registry number CEP/INCA#117/12; CONEP: PB #888.2772.3.
Author Contributions
EN conducted, supervised, and analyzed all the experiments in this study, and wrote the manuscript. LM performed mutation tests and MLPA analyses and wrote the manuscript. FA performed gene mutation tests and collected follow-up information ET-G conducted flow cytometry diagnosis. LT performed OS analyses. MP-d-O designed and supervised the study, and wrote the final version of the manuscript. All authors critically reviewed and approved the final draft of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to all clinicians that provided samples to this study. EN was supported by the Ministry of Health (INCA-Brazil)-Scholarship Program; MP-d-O is supported by Brazilian National Council of Technological and Scientific Development-CNPq (PQ-#301594/2015-5). Partial support was obtained from TUCCA-Associação para Crianças e Adolescentes com Câncer, São Paulo, Brazil and SwissBridge Fundation- Fundação do Câncer, Rio de Janeiro,Brazil (# L0J SWB 2014 SUB-PROJECT 1.A). This study was financed in part by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior - Brasil (CAPES) - Finance Code 001.
Brazilian Collaborative Study Group of Acute Leukemia
Carolina da Paz Zampier, Thayana da Conceição Barbosa, Paulo Chagas Neto, Gisele Dallapicola Brisson, Filipe Vicente dos Santos Bueno, Ingrid Cezar Sardou, Bruno Gonçalves Aguiar, Anna Carolina Silva Dias, Patrícia Carneiro de Brito, Geraldo Pedral Sampaio, Raimundo Antônio Gomes Oliveira, Claudia Teresa de Oliveira, César Casagranda, Geni Ramos Vera, Gustavo Ribeiro Neves, Isis Maria Quezado Magalhães, José Carlos Córdoba, Juliana Teixeira Costa, Rebeca Ferreira Marques, Renata Pereira de Souza Barros, Renata Sarkis Alves, Renato Guedes, Sidnei Epelman.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2019.00316/full#supplementary-material
References
1. Chiaretti S, Foa R. T-cell acute lymphoblastic leukemia. Haematologica. (2009) 94:160–2. doi: 10.3324/haematol.2008.004150
2. Patrick K, Wade R, Goulden N, Mitchell C, Moorman AV, Rowntree C, et al. Outcome for children and young people with Early T-cell precursor acute lymphoblastic leukaemia treated on a contemporary protocol, UKALL 2003. Br J Haematol. (2014) 166:421–4. doi: 10.1111/bjh.12882
3. Bene MC, Castoldi G, Knapp W, Ludwig WD, Matutes E, Orfao A, et al. Proposals for the immunological classification of acute leukemias. European Group for the Immunological Characterization of Leukemias (EGIL). Leukemia. (1995) 9:1783–6.
4. Fischer L, Gokbuget N, Schwartz S, Burmeister T, Rieder H, Bruggemann M, et al. CD56 expression in T-cell acute lymphoblastic leukemia is associated with non-thymic phenotype and resistance to induction therapy but no inferior survival after risk-adapted therapy. Haematologica. (2009) 94:224–9. doi: 10.3324/haematol.13543
5. van Grotel M, Meijerink JP, van Wering ER, Langerak AW, Beverloo HB, Buijs-Gladdines JG, et al. Prognostic significance of molecular-cytogenetic abnormalities in pediatric T-ALL is not explained by immunophenotypic differences. Leukemia. (2008) 22:124–31. doi: 10.1038/sj.leu.2404957
6. Meijerink JP. Genetic rearrangements in relation to immunophenotype and outcome in T-cell acute lymphoblastic leukaemia. Best Pract Res Clin Haematol. (2010) 23:307–18. doi: 10.1016/j.beha.2010.08.002
7. Schrappe M, Moricke A, Reiter A, Henze G, Welte K, Gadner H, et al. Key treatment questions in childhood acute lymphoblastic leukemia: results in 5 consecutive trials performed by the ALL-BFM study group from 1981 to 2000. Klinische Padiatrie. (2013) 225 (Suppl. 1):S62–72. doi: 10.1055/s-0033-1337966
8. Coustan-Smith E, Mullighan CG, Onciu M, Behm FG, Raimondi SC, Pei D, et al. Early T-cell precursor leukaemia: a subtype of very high-risk acute lymphoblastic leukaemia. Lancet Oncol. (2009) 10:147–56. doi: 10.1016/s1470-2045(08)70314-0
9. Zhang J, Ding L, Holmfeldt L, Wu G, Heatley SL, Payne-Turner D, et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature. (2012) 481:157–63. doi: 10.1038/nature10725
10. Zuurbier L, Gutierrez A, Mullighan CG, Cante-Barrett K, Gevaert AO, de Rooi J, et al. Immature MEF2C-dysregulated T-cell leukemia patients have an early T-cell precursor acute lymphoblastic leukemia gene signature and typically have non-rearranged T-cell receptors. Haematologica. (2014) 99:94–102. doi: 10.3324/haematol.2013.090233
11. Inukai T, Kiyokawa N, Campana D, Coustan-Smith E, Kikuchi A, Kobayashi M, et al. Clinical significance of early T-cell precursor acute lymphoblastic leukaemia: results of the Tokyo Children's Cancer Study Group Study L99-15. Br J Haematol. (2012) 156:358–65. doi: 10.1111/j.1365-2141.2011.08955
12. Liu Y, Easton J, Shao Y, Maciaszek J, Wang Z, Wilkinson MR, et al. The genomic landscape of pediatric and young adult T-lineage acute lymphoblastic leukemia. Nat Genet. (2017) 49:1211–8. doi: 10.1038/ng.3909
13. Spinella JF, Cassart P, Richer C, Saillour V, Ouimet M, Langlois S, et al. Genomic characterization of pediatric T-cell acute lymphoblastic leukemia reveals novel recurrent driver mutations. Oncotarget. (2016) 7:65485–503. doi: 10.18632/oncotarget.11796
14. Brandalise SR, Viana MB, Pinheiro VR, Mendonca N, Lopes LF, Pereira WV, et al. Shorter maintenance therapy in childhood acute lymphoblastic leukemia: the experience of the prospective, randomized brazilian GBTLI ALL-93 protocol. Front Pediatr. (2016) 4:110. doi: 10.3389/fped.2016.00110
15. Stary J, Zimmermann M, Campbell M, Castillo L, Dibar E, Donska S, et al. Intensive chemotherapy for childhood acute lymphoblastic leukemia: results of the randomized intercontinental trial ALL IC-BFM 2002. J Clin Oncol. (2014) 32:174–84. doi: 10.1200/jco.2013.48.6522
16. Marques LVC, Noronha EP, Andrade FG, Dos Santos-Bueno FV, Mansur MB, Terra-Granado E, et al. CD44 expression profile varies according to maturational subtypes and molecular profiles of pediatric T-cell lymphoblastic leukemia. Front Oncol. (2018) 8:488. doi: 10.3389/fonc.2018.00488
17. Noronha EP, Andrade FG, Zampier C, de Andrade CF, Terra-Granado E, Pombo-de-Oliveira MS. Immunophenotyping with CD135 and CD117 predicts the FLT3, IL-7R and TLX3 gene mutations in childhood T-cell acute leukemia. Blood Cells Mol Dis. (2016) 57:74–80. doi: 10.1016/j.bcmd.2015.12.003
18. Mansur MB, Emerenciano M, Brewer L, Sant'Ana M, Mendonca N, Thuler LC, et al. SIL-TAL1 fusion gene negative impact in T-cell acute lymphoblastic leukemia outcome. Leukem Lymphoma. (2009) 50:1318–25. doi: 10.1080/10428190903040014
19. Mansur MB, Hassan R, Barbosa TC, Splendore A, Jotta PY, Yunes JA, et al. Impact of complex NOTCH1 mutations on survival in paediatric T-cell leukaemia. BMC Cancer. (2012) 12:9. doi: 10.1186/1471-2407-12-9
20. Mansur MB, van Delft FW, Colman SM, Furness CL, Gibson J, Emerenciano M, et al. Distinctive genotypes in infants with T-cell acute lymphoblastic leukaemia. Br J Haematol. (2015) 171:574–84. doi: 10.1111/bjh.13613
21. Weng AP, Ferrando AA, Lee W, Morris JPt, Silverman LB, Sanchez-Irizarry C, et al. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science. (2004) 306:269–71. doi: 10.1126/science.1102160
22. Kox C, Zimmermann M, Stanulla M, Leible S, Schrappe M, Ludwig WD, et al. The favorable effect of activating NOTCH1 receptor mutations on long-term outcome in T-ALL patients treated on the ALL-BFM 2000 protocol can be separated from FBXW7 loss of function. Leukemia. (2010) 24:2005–13. doi: 10.1038/leu.2010.203
23. Zenatti PP, Ribeiro D, Li W, Zuurbier L, Silva MC, Paganin M, et al. Oncogenic IL7R gain-of-function mutations in childhood T-cell acute lymphoblastic leukemia. Nature genetics. (2011) 43:932–9. doi: 10.1038/ng.924
24. Andrade FG, Noronha EP, Brisson GD, Dos Santos Vicente Bueno F, Cezar IS, Terra-Granado E, et al. Molecular characterization of pediatric acute myeloid leukemia: results of a multicentric study in Brazil. Arch Med Res. (2016) 47:656–67. doi: 10.1016/j.arcmed.2016.11.015
25. Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, et al. Circos: an information aesthetic for comparative genomics. Genome Res. (2009) 19:1639–45. doi: 10.1101/gr.092759.109
26. Ferrando AA, Neuberg DS, Staunton J, Loh ML, Huard C, Raimondi SC, et al. Gene expression signatures define novel oncogenic pathways in T cell acute lymphoblastic leukemia. Cancer Cell. (2002) 1:75–87. doi: 10.1016/s1535-6108(02)00018-1
27. Cante-Barrett K, Mendes RD, Li Y, Vroegindeweij E, Pike-Overzet K, Wabeke T, et al. Loss of CD44dim expression from early progenitor cells marks T-cell lineage commitment in the human thymus. Front Immunol. (2017) 8:32. doi: 10.3389/fimmu.2017.00032
28. Asnafi V, Radford-Weiss I, Dastugue N, Bayle C, Leboeuf D, Charrin C, et al. CALM-AF10 is a common fusion transcript in T-ALL and is specific to the TCRgammadelta lineage. Blood. (2003) 102:1000–6. doi: 10.1182/blood-2002-09-2913
29. Van Coppernolle S, Vanhee S, Verstichel G, Snauwaert S, van der Spek A, Velghe I, et al. Notch induces human T-cell receptor gammadelta+ thymocytes to differentiate along a parallel, highly proliferative and bipotent CD4 CD8 double-positive pathway. Leukemia. (2012) 26:127–38. doi: 10.1038/leu.2011.324
30. Vicente C, Schwab C, Broux M, Geerdens E, Degryse S, Demeyer S, et al. Targeted sequencing identifies associations between IL7R-JAK mutations and epigenetic modulators in T-cell acute lymphoblastic leukemia. Haematologica. (2015) 100:1301–10. doi: 10.3324/haematol.2015.130179
31. Yuan L, Lu L, Yang Y, Sun H, Chen X, Huang Y, et al. Genetic mutational profiling analysis of T cell acute lymphoblastic leukemia reveal mutant FBXW7 as a prognostic indicator for inferior survival. Ann Hematol. (2015) 94:1817–28. doi: 10.1007/s00277-015-2474-0
32. Yeh TC, Liang DC, Liu HC, Jaing TH, Chen SH, Hou JY, et al. Clinical and biological relevance of genetic alterations in pediatric T-cell acute lymphoblastic leukemia in Taiwan. Pediatr Blood Cancer. (2019) 66:e27496. doi: 10.1002/pbc.27496
33. Breit S, Stanulla M, Flohr T, Schrappe M, Ludwig WD, Tolle G, et al. Activating NOTCH1 mutations predict favorable early treatment response and long-term outcome in childhood precursor T-cell lymphoblastic leukemia. Blood. (2006) 108:1151–7. doi: 10.1182/blood-2005-12-4956
34. Clappier E, Collette S, Grardel N, Girard S, Suarez L, Brunie G, et al. NOTCH1 and FBXW7 mutations have a favorable impact on early response to treatment, but not on outcome, in children with T-cell acute lymphoblastic leukemia (T-ALL) treated on EORTC trials 58881 and 58951. Leukemia. (2010) 24:2023–31. doi: 10.1038/leu.2010.205
35. Graux C, Cools J, Melotte C, Quentmeier H, Ferrando A, Levine R, et al. Fusion of NUP214 to ABL1 on amplified episomes in T-cell acute lymphoblastic leukemia. Nat Genet. (2004) 36:1084–9. doi: 10.1038/ng1425
36. Lahortiga I, De Keersmaecker K, Van Vlierberghe P, Graux C, Cauwelier B, Lambert F, et al. Duplication of the MYB oncogene in T cell acute lymphoblastic leukemia. Nat Genet. (2007) 39:593–5. doi: 10.1038/ng2025
37. Danis E, Yamauchi T, Echanique K, Zhang X, Haladyna JN, Riedel SS, et al. Ezh2 Controls an early hematopoietic program and growth and survival signaling in early T cell precursor acute lymphoblastic leukemia. Cell Rep. (2016) 14:1953–65. doi: 10.1016/j.celrep.2016.01.064
38. Booth CAG, Barkas N, Neo WH, Boukarabila H, Soilleux EJ, Giotopoulos G, et al. Ezh2 and Runx1 mutations collaborate to initiate lympho-myeloid leukemia in early thymic progenitors. Cancer Cell. (2018) 33:274–91 e8. doi: 10.1016/j.ccell.2018.01.006
39. Wendorff AA, Quinn SA, Rashkovan M, Madubata CJ, Ambesi-Impiombato A, Litzow MR, et al. Phf6 loss enhances HSC self-renewal driving tumor initiation and leukemia stem cell activity in T-ALL. Cancer Discov. (2018) 9:436–51. doi: 10.1158/2159-8290.cd-18-1005
40. Van Vlierberghe P, Palomero T, Khiabanian H, Van der Meulen J, Castillo M, Van Roy N, et al. PHF6 mutations in T-cell acute lymphoblastic leukemia. Nat Genet. (2010) 42:338–42. doi: 10.1038/ng.542
41. Treanor LM, Zhou S, Janke L, Churchman ML, Ma Z, Lu T, et al. Interleukin-7 receptor mutants initiate early T cell precursor leukemia in murine thymocyte progenitors with multipotent potential. J Exp Med. (2014) 211:701–13. doi: 10.1084/jem.20122727
42. Yokoyama K, Yokoyama N, Izawa K, Kotani A, Harashima A, Hozumi K, et al. In vivo leukemogenic potential of an interleukin 7 receptor alpha chain mutant in hematopoietic stem and progenitor cells. Blood. (2013) 122:4259–63. doi: 10.1182/blood-2012-08-451278
43. Gonzalez-Garcia S, Garcia-Peydro M, Alcain J, Toribio ML. Notch1 and IL-7 receptor signalling in early T-cell development and leukaemia. Curr Topics Microbiol Immunol. (2012) 360:47–73. doi: 10.1007/82_2012_231
44. Yu S, Zhou X, Steinke FC, Liu C, Chen SC, Zagorodna O, et al. The TCF-1 and LEF-1 transcription factors have cooperative and opposing roles in T cell development and malignancy. Immunity. (2012) 37:813–26. doi: 10.1016/j.immuni.2012.08.009
45. Noronha EP, Marques LVC, Andrade FG, Sardou-Cezar I, Dos Santos-Bueno FV, Zampier CP, et al. T-lymphoid/myeloid mixed phenotype acute leukemia and early T-cell precursor lymphoblastic leukemia similarities with NOTCH1 mutation as a good prognostic factor. Cancer Manag Res. (in press).
Keywords: T-cell acute lymphoblastic leukemia, childhood, immunophenotypic subtypes, molecular alterations, early T-cell precursor acute lymphoblastic leukemia, overall survival
Citation: Noronha EP, Marques LVC, Andrade FG, Thuler LCS, Terra-Granado E, Pombo-de-Oliveira MS and the Brazilian Collaborative Study Group of Acute Leukemia (2019) The Profile of Immunophenotype and Genotype Aberrations in Subsets of Pediatric T-Cell Acute Lymphoblastic Leukemia. Front. Oncol. 9:316. doi: 10.3389/fonc.2019.00316
Received: 29 January 2019; Accepted: 08 April 2019;
Published: 30 April 2019.
Edited by:
Naval Daver, University of Texas MD Anderson Cancer Center, United StatesReviewed by:
Belinda Pinto Simoes, University of São Paulo, BrazilRehan Khan, Mayo Clinic, United States
Copyright © 2019 Noronha, Marques, Andrade, Thuler, Terra-Granado, Pombo-de-Oliveira and the Brazilian Collaborative Study Group of Acute Leukemia. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maria S. Pombo-de-Oliveira, bXBvbWJvQGluY2EuZ292LmJy
†These authors have contributed equally to this work
‡The members of the Brazilian Collaborative Study Group of Acute Leukemia are listed at the end of the article