 Sara Galimberti1*
Sara Galimberti1* Mario Petrini1
Mario Petrini1 Claudia Baratè2
Claudia Baratè2 Federica Ricci1Serena Balducci1
Federica Ricci1Serena Balducci1 Susanna Grassi1
Susanna Grassi1 Francesca Guerrini1Elena Ciabatti1Sandra Mechelli2
Francesca Guerrini1Elena Ciabatti1Sandra Mechelli2 Antonello Di Paolo1Chiara Baldini1
Antonello Di Paolo1Chiara Baldini1 Laura Baglietto1
Laura Baglietto1 Lisa Macera3Pietro Giorgio Spezia3Fabrizio Maggi3
Lisa Macera3Pietro Giorgio Spezia3Fabrizio Maggi3- 1Department of Clinical and Experimental Medicine, University of Pisa, Pisa, Italy
- 2Hematology, AOUP, Pisa, Italy
- 3Department of Translational Research and New Technologies in Medicine and Surgery, University of Pisa, Pisa, Italy
SARS-CoV-2 is the viral agent responsible for the pandemic that in the first months of 2020 caused about 400,000 deaths. Among compounds proposed to fight the SARS-CoV-2-related disease (COVID-19), tyrosine kinase inhibitors (TKIs), already effective in Philadelphia-positive acute lymphoblastic leukemia (Ph+ ALL) and chronic myeloid leukemia (CML), have been proposed on the basis of their antiviral action already demonstrated against SARS-CoV-1. Very few cases of COVID-19 have been reported in Ph+ ALL and in CML Italian cohorts; authors suggested that this low rate of infections might depend on the use of TKIs, but the biological causes of this phenomenon remain unknown. In this study, the CML model was used to test if TKIs would sustain or not the viral replication and if they could damage patient immunity. Firstly, the infection and replication rate of torquetenovirus (TTV), whose load is inversely proportional to the host immunological control, have been measured in CML patients receiving nilotinib. A very low percentage of subjects were infected at baseline, and TTV did not replicate or at least showed a low replication rate during the follow-up, with a mean load comparable to the measured one in healthy subjects. Then, after gene expression profiling experiments, we found that several “antiviral” genes, such as CD28 and IFN gamma, were upregulated, while genes with “proviral” action, such as ARG-1, CEACAM1, and FUT4, were less expressed during treatment with imatinib, thus demonstrating that TKIs are not detrimental from the immunological point of view. To sum up, our data could offer some biological explanations to the low COVID-19 occurrence in Ph+ ALL and CML patients and sustain the use of TKIs in COVID-19, as already proposed by several international ongoing studies.
Introduction
In March 2020, the pandemic sustained by the Severe Acute Respiratory Syndrome-Coronavirus 2 (SARS-CoV-2) was officially declared, involving the entire world, after which a new Coronavirus was isolated for the first time in China, in the Hubei province, in December 2019. On June 7, 2020, 6,750,521 cases (USA: 1,886,794, Europe: 2,271,919, and China: 84,629) and 395,779 deaths (USA: 100,038, Europe: 182,790, and China: 4,645) had been recorded (https://www.cdc.gov/coronavirus/2019-nCoV/index.html), an enormous number that well-reflects the gravity of the worldwide situation.
The disease caused by SARS-CoV-2, now known as Coronavirus Disease 19 (COVID-19), is characterized by different clinical manifestations and severity, ranging from cough and cold across fever and mild symptoms to capillary leak, respiratory distress, thrombotic events, and renal, hepatic, and coagulation failure (1). Recently, many efforts have been made in order to detect, isolate, and characterize the virus; understand the COVID-19 pathogenesis; and, especially, find effective treatments.
The SARS-CoV-2 structure was rapidly characterized: it is an RNA virus, with a nucleotide identity higher than 80% with the genome of SARS-CoV-1, the virus responsible for the Asian outbreak of 2002/2003 (2). One of the viral proteins, the surface spike (S) protein, is fundamental for virus attachment, fusion, and entry into the human host cells through the angiotensin-converting enzyme 2 (ACE2) receptor (3). More recently, the dipeptidyl peptidase 4 (DPP4) (also known as CD26) (4) has been characterized as a further virus receptor. DPP4 axis is used by virus for blocking autophagy, for impairing host immune response and sustaining the hyper-inflammatory status (5). Interestingly, both angiotensin and CD26 are involved in the senescence phenomenon, which, in addition to being responsible for further virus dissemination, might explain why COVID-19 is more severe in older people and why azithromycin seems to be effective (6). Coronavirus enters the human cells via a pH- and receptor-dependent way (7); once it has entered the host cells, the virus uses the proteasome ubiquitin system to destroy the host antiviral proteins and increase the production of those proteins that are necessary for its replication (8).
The COVID-19 pathogenesis has been ascribed to the “cytokine storm” that is characterized by increased levels of IL1, IL6, IFN gamma, TNF alpha, IL12, and IL15, in a way similar to the one observed in the macrophage activation syndrome (9), in the cytokine release disease following CAR-T infusion (10), and in the acute graft-versus-host disease (aGVHD), a complication of allogeneic stem cell transplantation (11).
Among the available therapeutic approaches against COVID-19, some of them belong to the rheumatological and hematological armamentarium (12). The efficacy of chloroquine and hydroxychloroquine, which act as immunomodulating agents by modifying the pH of endocytic vesicles (so blocking the first infection phases), is a matter of debate. Indeed, several studies focused their attention on the increased risk of prolongation of QTc and possible onset of arrhythmias that these compounds, especially in combination with antibiotics, seem to induce (13). On the other hand, a meta-analysis conducted on nine different studies sustained the efficacy of these drugs (14). Recently, Lancet retracted a paper published 13 days before where the authors neglected the efficacy of chloroquine and hydroxychloroquine, used alone or in combination with macrolides, in a series of 14,888 patients and 81,144 untreated controls (15), once again making this issue a matter of debate. Other drugs borrowed by the “rheumatological world” have been used in COVID-19 because of their anti-inflammatory power: tocilizumab, an anti-IL6 antibody; anakinra, an anti-IL1 antibody; and baricitinib, an anti-JAK1 compound (16).
Moreover, other possible effective drugs have been derived from the “hematological world,” such as ruxolitinib, a powerful JAK1/2 inhibitor, already effective also in aGVHD (17, 18); begelomab, an anti-CD26 monoclonal antibody that can block the viral occupation of CD26 receptors, restore autophagy, and reduce the hyper-inflammatory status (5); and proteasome inhibitors that block the synthesis of the proteins necessary for the production of new virions (8).
In March 31 2020, a randomized clinical trial, “COUNTER-COVID—Oral imatinib to prevent pulmonary vascular leak in COVID-19,” was registered in Netherlands (https://www.clinicaltrialsregister.eu/ctr-search/trial/2020-001236-10/NL). The primary endpoint of this study was to test whether imatinib might reduce the aggressiveness of COVID-19 pneumonitis. Other clinical trials involving imatinib, alone (NCT04357613) or in combination with baricitinib, lopinavir/ritonavir (NCT04346147), or hydroxychloroquine (NCT04356495), were registered on the “ClinicalTrials.gov” website and are now ongoing.
The basis for the possible efficacy of imatinib in COVID-19 can be determined from previous studies conducted in the SARS-CoV-1 model, where imatinib reduced the number of infected cells by preventing syncytia formation induced by the viral S protein. Considering that Abl1 kinase controls the cytoskeletal rearrangement, the authors suggested that imatinib, inhibiting Abl1, could interfere with the actin dynamics required for virus entry and cell fusion (19, 20). Two recently published Italian surveys, one focused on patients affected by Philadelphia-positive acute lymphoblastic leukemia (Ph+ ALL) and the other one on chronic myeloid leukemia (CML) cases can be the real proof that these observations done in vitro are true also in vivo. Indeed, among 267 adults with Ph+ ALL currently managed in participating centers (including 128 subjects coming from the four Italian regions most affected by the new coronavirus), only one patient has proven to be COVID-19 positive, with a quite rapid resolution of pneumonitis and cellulitis (21). The second article (22) reports that in 6,883 CML patients observed at 51 centers, only 12 cases were SARS-CoV-2 positive. The authors of these articles hypothesized that this low percentage of infections and absence of COVID-19-related severe symptoms might be related to a “protective” action exerted by tyrosine kinase inhibitors (TKIs).
With these assumptions, we conducted two different types of experiments, with the aim of trying to explain why TKIs would protect Ph+ ALL and CML patients from COVID-19.
Firstly, as a “viral” model, we employed the torquetenovirus (TTV), a DNA-virus assigned to the family of Anelloviridae (23), detectable in more than 60% of the asymptomatic healthy subjects (24). It has been reported that TTV load significantly increases after immunosuppressive therapy and solid organ or stem cell transplantation. Translated in the COVID-19 context, if TKIs would sustain the coronavirus infection or replication, we might expect to observe a significant increase of TTV load during treatment of our patients with nilotinib.
Then, we analyzed the immune profile of five CML patients during imatinib treatment in order to test if and how much several hundreds of immunity-related genes were modulated by this TKI. Indeed, we previously reported about the anti-inflammatory power of imatinib (25), which, when translated in the COVID-19 scenario, might represent an important strong point. Nevertheless, we could assume that this anti-inflammatory action could be accompanied by an immune control level reduction, which might be detrimental for the final fight of the virus. With these premises, in the second part of the study, we evaluated the middle-term activity of imatinib on the immune system of our CML patients.
Patients and Methods
Patients
Nilotinib Cohort
TTV load was measured by quantitative real-time PCR in 60 peripheral blood samples from 10 CML patients in the chronic phase receiving nilotinib as a first-line therapy. The clinical features and outcomes of these subjects are reported in Table 1. In particular, six patients were male and four female; their median age was 46 years; the Sokal risk score was high in three cases, intermediate in six, and low in one. All except one achieved an early molecular response (BCR-ABL1/ABL1 ratio ≤ 10% IS after 3 months of treatment), and 9/10 achieved MR3 (BCR-ABL1/ABL1 ratio ≤ 0.1% IS) at 12 months.

Table 1. Features of patients treated with nilotinib as first-line treatment tested for TTV load.
Imatinib Cohort
A possible deregulation of 770 mRNAs caused by imatinib was measured by NanoString technology in peripheral blood samples of five CML patients in the chronic phase after 6 months of 400 mg/day of this TKI with respect to the diagnosis. Clinical features and outcomes of these subjects are reported in Table 2. In particular, three patients were male and two female; their median age was 55 years; the Sokal risk score was high in one case, intermediate in three, and low in another one. In this cohort, two patients were resistant to imatinib, while the other three became optimal responders, according to the ELN 2013 guidelines (26). Before participating in the study, all patients signed an informed consent approved by the Ethical Committee of the AOUP where they declared to donate leftover samples used for diagnostics for further scientific non-profit purposes.

Table 2. Features of patients treated with imatinib as first-line treatment enrolled in the gene expression profile assays.
Methods
TTV Load Measure
Viral DNA was extracted from 200 μl whole peripheral blood anticoagulated with EDTA using QIAamp DNA minikit (Qiagen, Chatsworth, CA, USA) and stored at −20°C. Presence and load of TTV genome were determined by single-step quantitative real-time PCR, as described elsewhere (27). The method showed high sensitivity, being able to identify as positive samples those containing ≥10 viral genomes per milliliter; the specificity had been previously demonstrated, since this technique does not detect other human anelloviruses.
BCR-ABL1/ABL1 Ratio IS Detection
The BCR-ABL1/ABL1 ratio was measured by quantitative PCR on the concomitantly harvested peripheral blood, according to the standardized operative procedures of the Italian cooperative group GIMEMA LabNet (www.gimema.it/labnet-cml/). A minimum of 20,000 ABL1 copies was necessary for considering a sample as “evaluable”; 32,000 ABL1 copies were necessary for defining MR4.5 or 100,000 ABL1 copies for MR5, according to the ELN guidelines (28).
NanoString Assays
NanoString technology (NanoString, Seattle, USA) has been employed for analyzing the immune transcriptome profile of five CML patients after 6 months of treatment with imatinib in comparison with diagnosis. The “Human nCounter Myeloid Innate Immunity panel” that measures the expression of 770 genes involved in 19 different pathways, fundamental for the innate immune response, has been adopted.
Statistical Analysis
Results from the NanoString were analyzed by the nCounter Advanced Analysis 2.0 software that allows us to identify genes significantly upregulated or downregulated, design volcano plots, and perform principal component analysis. For the remaining data, SPSS software version 22 (IBM, Bologna, Italy) was used. Viral load variable was used after transformation of TTV load in log format. Fisher's exact test has been applied to the contingency tables. Differences between distributions were calculated by a non-parametric Mann–Whitney U test. The association among variables was evaluated by the Kruskal–Wallis test. Correlations between variables were assessed using the Spearman r correlation coefficient and Student's t-test. Regression analyses were conducted to evaluate the association between the dependent variable TTV viremia and BCR-ABL1/ABL1 ratio. All p-values presented are based on two-tailed tests, and p ≤ 0.05 was considered as statistically significant.
Results
TTV Infection and Replication Rates in CML Patients
TTV load was measured by quantitative real-time PCR in 60 peripheral blood samples from 10 CML patients in the chronic phase receiving nilotinib 600 mg/day as a first-line therapy. At diagnosis, only two patients showed detectable TTV genome; both TTV-positive patients were male, aged 40 and 53 years; no correlation with any of their clinical features and TTV presence was found. Because a correlation between age and sex with TTV replication has been previously reported in healthy subjects (29), we tested if this observation would be reproducible also in our series. Even if our cohort was very small, no correlation between age or sex and TTV infection rate was found.
Then, we analyzed the TTV load at different time points in order to test if in the TTV-negative cases, the virus started to replicate during treatment with nilotinib and if and how TTV load eventually would change during follow-up. Eleven patients were tested after 3, 6, and 9 months; 9 cases were also tested at 12 months, 15 at 18 months, and 3 patients also after 21 months of therapy.
Overall, during follow-up, 41 samples (68%) were TTV negative, while 19 (32%) presented a TTV quantifiable load. The mean TTV load in positive cases was 2.8 log copies per milliliter (95% confidence interval: 2.5–3.1 log copies per milliliter). It is worth remembering that in different series of immunocompromised patients, the mean TTV load was 4.1 log copies per milliliter (95% confidence interval: 3.9–4.3 log copies per milliliter) (25, 26, 29–32) and that the mean TTV load in healthy blood donors was 2.3 log copies per milliliter (95% confidence interval: 1.7–2.9 log copies per milliliter) (24).
When TTV load was measured at the last time points of follow-up, 9 of the 10 treated CML patients showed either unchanged or slightly increased values (<0.4 log copies per milliliter of variation) relative to baseline. In particular, in one of the two initially TTV-positive patients, TTV did not replicate already at the first time point (after 3 months of therapy), while in the second case, the mean value of TTV load measured at six different time points was the same as that measured baseline (2.7 log copies per milliliter). In the subgroup of cases who were initially TTV negative, four out of eight remained TTV negative during the whole follow-up; in the other four, TTV load was detectable in half of the occasions, but with a maximum increase value of 0.4 log copies per milliliter. When we analyzed TTV load with respect to molecular response assessed by the BCR-ABL1/ABL1 ratio measured by real-time PCR at the same time point, a statistically significant correlation between genome TTV detection and absence of optimal response was found. Indeed, according to the ELN 2013 classification (28), we counted seven failing time points: in all these occasions, the TTV genome was detectable. On the other hand, we had 53 time points where molecular response was optimal or a warning: in 33 of these occasions, TTV was not detectable (p = 0.02).
In conclusion, this first part of the study showed that nilotinib does not sustain the viral replication, even with a medium-term follow-up.
Deregulation of Immunity-Related Genes in CML Patients
In the second phase of our study, we employed the NanoString technology for analyzing the expression of 770 inflammation- and immunity-related genes in five CML patients before and after 6 months of treatment with imatinib, with the aim of testing the impact of this TKI on the possible immunological control of viral infection.
Overall, 58 genes were deregulated by imatinib, with 18 genes being upregulated and 40 downregulated. We previously reported that some genes involved in several different autoimmune/inflammatory conditions were downregulated by imatinib (25), which might have a positive impact on COVID-19. Interestingly, 20 out of these 58 deregulated genes were strictly correlated with immune or antiviral response, so representing the focus of the present study (see Supplemental File 1 for the original raw data).
Among these 20 genes, 11 appeared upregulated and 9 downregulated during treatment with imatinib (see Table 3). In more detail, we found a reduced expression of Arginase 1 (ARG-1) (44), Complement C3a Receptor 1 (C3AR1) (46), Carcinoembryonic antigen-related cell adhesion molecule-1 (CEACAM1) (45), Gelsolin (GSN) (49), Nectin 1 (50), and Fucosyltransferase 4 (FUT4) (48), all genes that usually play a “proviral” role. Indeed, ARG-1 displays a negative effect on immunity in several contexts, including hematological neoplasms: by depleting the microenvironment of arginine, which is essential for T-lymphocyte function, arginase makes them anergic. In CML, ARG-1 has been shown to be highly expressed at diagnosis, when myeloid-derived suppressor cells are very active against T-cell activity (53). CEACAM1 is an important regulator of virus-specific CD8+ T-cell functions. In an in vitro model, treatment with anti-CEACAM1 antibody prevented CD8+ T-cell exhaustion, thus improving the control of viral infections (54). FUT4 is a gene strictly involved in the PD1 axis, whose overexpression has been associated with a shorter survival in lung adenocarcinoma (48). Consequently, FUT4 downregulation could contribute to the inhibition of the PD1–PDL1 axis, with consequent recovery of the immune disease control.
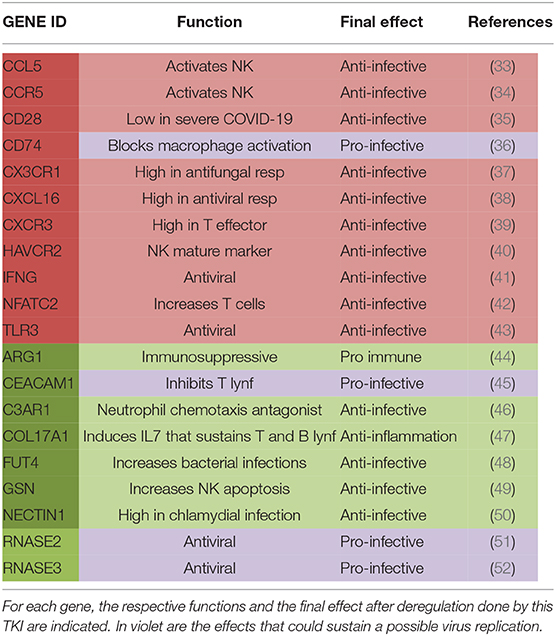
Table 3. In the table are listed genes that were upregulated (in red) or downregulated (in green) in CML patients during treatment with imatinib.
On the other hand, imatinib upregulated some genes that could finally exert an “antiviral” action by sustaining the host immunological control of the viral attack, such as CD28 (35), Interferon gamma (IFN gamma) (41), C-C Motif Chemokine Ligand 5 (CCL5) (33), C-C chemokine receptor type 5 (CCR5) (34), and Toll-like receptor 3 (TLR3) (43). Particularly interesting is CD28, a co-stimulatory molecule also used for CAR-T production (55). CD28 is located immediately downstream of PD1, thus participating in the PD1-derived T-cell suppression (56). The role of CD28 in COVID-19 has been recently investigated: its expression on CD8+ T cells seems to be lower in patients with severe relative to those with mild COVID-19 (57); consequently, its overexpression induced by imatinib might be not detrimental. CCL5 is another interesting chemokine with antiviral action: indeed, in a murine model, CCL5-overexpressing NK cells were hyperactivated and made animals resistant to the viral infection (33). The pro-immune action of IFN gamma is well-known (41); in CML, interferon has a long history of successes, and it seems to be able to delete ABL1 mutations when added to TKIs, hence making patients sensitive again to treatment (58). Finally, also, the increased expression of TLR3 might be positively translated into the COVID-19 context: indeed, it has been previously reported that its overexpression protects neutropenic mice from meningoencephalitis (59).
In conclusion, even if performed on a small series of cases, results from our experiments might support the idea that imatinib could sustain the immunological control of infected subjects (Figure 1). These findings, translated in the coronavirus scenario, could help to explain why very few patients affected by Ph+ ALL and CML developed COVID-19 symptoms.
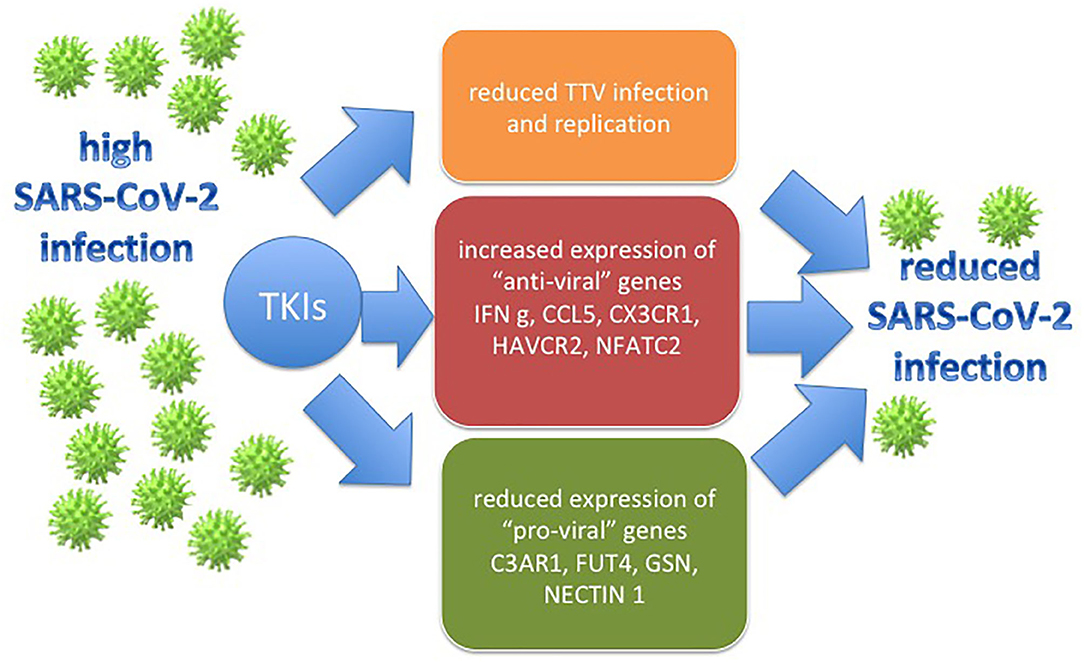
Figure 1. The figure represents how the effects that TKIs have on the CML models employed in the study (the TTV replication and immune gene deregulation) might be translated in the SARS-CoV-2 context, with a possible final antiviral action.
Discussion
The SARS-CoV-2 outbreak and its related disease, COVID-19, represent a new world war that in just a few months produced about 400,000 deaths. Consequently, all of the scientific community is now working to find effective diagnostic tests and therapeutic approaches. Recently, a Dutch group started to treat COVID-19 pneumonitis with imatinib, a TKI already employed in CML (60), Ph+ ALL (61), and gastrointestinal stromal tumor (GIST) (62). The hypothesis that imatinib could be effective also in COVID-19 was based on some previous experimental observations done in SARS and MERS models where imatinib was able to block the virus entrance into human host cells (20). Recently, very few cases of COVID-19 have been reported in the Italian cohort of Ph+ ALL and CML patients (21). Considering that it has been proven that at diagnosis, the immunity of these patients is severely impaired (63), the low infection rate observed during the 2020 pandemic could prove that TKIs play an antiviral role or, at least, could not impair the host response against the new coronavirus.
In our study, we tested if nilotinib could favor or not the viral replication by using the TTV model. We found that in 50% of patients, TTV did not replicate at all evaluated time points and that, in subjects where the TTV load was measurable, its replication rate was low. Indeed, the mean TTV load in positive cases was 2.8 log copies per milliliter, lower than that observed in immunocompromised patients (where the mean TTV load was 4.1 log copies per milliliter) (25, 26, 29–32) and comparable with that of healthy individuals (where the mean TTV load was 2.3 log copies per milliliter) (24). In our opinion, these results might be considered as a good proof that nilotinib does not favor the viral replication.
Moreover, in our series, we observed that only 20% of patients presented a measurable TTV load baseline, a result that mimics the very low rate of infection by the new coronavirus in Ph+ ALL and CML patients. The mechanisms that TTV uses for entering the host cells are still unknown, and we might hypothesize that TTV receptors on CML cells could be lower or absent. This may not be relevant in the coronavirus scenario, because we know that CD26, highly expressed in CML leukemic cells (64), is one of the SARS-CoV-2 receptors. Consequently, in our opinion, the very low percentage of CML patients found to be SARS-CoV-2 positive could be not explained by the inability of the virus to attack the host cells. On the contrary, we can consider that, as already described for SARS-CoV-1, Bcr-Abl1 tyrosine kinase, expressed at very high levels at diagnosis, could change the conformational status of the host cells, making them “impenetrable” by the virus. Indeed, Bcr-Abl1 tyrosine kinase modifies the actin structure and cytoskeleton function (65), probably also by actin adaptors, such as FAK, whose codifying gene is expressed at a very low level in CML, especially in advanced phases of the disease (66). In conclusion, we cannot exclude that Bcr-Abl1 might make Ph+ cells more impenetrable for viruses compared with normal cells. Moreover, about the low rate of TTV replication during treatment with nilotinib, we can hypothesize that some “antiviral” genes could be more expressed. In our previous study, we observed that imatinib upregulated two “antiviral” genes: Interferon-stimulated gene 15 (ISG15) and Mov10 RISC Complex RNA Helicase (MOV10) (67). ISG15 has been reported to exert a direct negative effect on viral replication, probably by restoring autophagy and sustaining NK proliferation and dendritic cell maturation (68). Moreover, MOV10, in a model of porcine respiratory syndrome, blocked virus replication by affecting its nuclear import (69).
In conclusion, the TTV model seems to offer interesting explanations to the low COVID-19 percentages in Ph+ patients, showing that these subjects are probably less infected than healthy individuals and that TKIs seem to not favor viral replication.
On the other hand, the results from the second part of our study could help to reduce the doubt that TKIs might damage the immunological control of infection. Indeed, we observed the upregulation of some “pro-immune” genes, such as CCL5, CD28, and IFNg, and a reduced expression of some “anti-immune” genes, such as ARG-1 and FUT4. We measured the expression of more than 700 genes involved in inflammation and immunity after 6 months of treatment with imatinib, a very long time with respect to that of a viral infection. This might not be a limitation of the study, because the absence of immunological damage after a medium-length treatment might further sustain the idea that TKIs would be not detrimental in shorter time intervals.
Certainly, we are aware that this study has some limitations, especially the low number of patients enrolled and the fact that the first time point of observation was, according to clinical practice, after 3 months of therapy. However, we think that our study might present some valid findings that could help to explain why very few Ph+ patients have been spared by SARS-CoV-2 or did not present severe forms of COVID-19. Obviously, we have to still wait for results of the ongoing large clinical trials before definitively establishing a possible effective role of TKIs in the coronavirus pandemic.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
Ethics Statement
Ethical approval was not provided for this study on human participants because at the first access to the Hematology Division, Pisa, all patients signed an informed consent approved by the Ethical Committee of the AOUP where they declared to donate leftover samples for further scientific non-profit purposes and for authorized researchers to use the clinical data. The patients/participants provided their written informed consent to participate in this study.
Author Contributions
SG, MP, CBar, CBal, LB, and AD designed the study. CBal, FR, SB, and SM enrolled and managed patients. FG, SG, and EC conducted experiments and analyses. SG and FM wrote the first draft of the manuscript and prepared figures and tables. All authors reviewed the draft before submission.
Funding
This work was supported by the University of Pisa, Italy, with the PRA 2018 grant, MP.
Conflict of Interest
SG, MP, and CBar participated to advisory boards organized by Novartis and were speakers during meeting supported by Novartis.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank all our CML patients, our nurse C. Baroni for her daily precious help, and our English lecturer I. Di Vita.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2020.01428/full#supplementary-material
References
1. Harcourt J, Tamin A, Lu X, Kamili S, Sakthivel SK, Murray J, et al. Severe acute respiratory syndrome coronavirus 2 from patient with 2019. Novel Coronavirus Disease, United States. Emerg Infect Dis. (2020) 26:1266–73. doi: 10.3201/eid2606.200516
2. Chan JF, Kok KH, Zhu Z, Chu H, To KK, Yuan S, et al. Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg Microbes Infect. (2020) 9:221–36. doi: 10.1080/22221751.2020.1719902
3. Tai W, He L, Zhang X, Pu J, Voronin D, Jiang S, et al. Characterization of the receptor-binding domain (RBD) of 2019 novel coronavirus: implication for development of RBD protein as a viral attachment inhibitor and vaccine. Cell Mol Immunol. (2020) 17:613–20. doi: 10.1038/s41423-020-0400-4
4. Vankadari N, Wilce JA. Emerging WuHan (COVID-19) coronavirus: glycan shield and structure prediction of spike glycoprotein and its interaction with human CD26. Emerg Microbes Infect. (2020) 9:601–4. doi: 10.1080/22221751.2020.1739565
5. Galimberti S, Morabito F, Gentile M, Baratè C, Benedetti E, Buda G, Petrini M. Dipeptidyl-peptidase 4 (Cd26): a possible therapeutic target in Covid-19. LOJ Phar Clin Res. (2020) 2. doi: 10.32474/LOJPCR.2020.02.000128
6. Malavolta M, Giacconi R, Brunetti D, Provinciali M, Maggi F. Exploring the relevance of senotherapeutics for the current SARS-CoV-2 emergency and similar future global health threats. Cells. (2020) 9:909. doi: 10.3390/cells9040909
7. Yang N, Shen HM. Targeting the endocytic pathway and autophagy process as a novel therapeutic strategy in COVID-19. Int J Biol Sci. (2020) 16:1724–31. doi: 10.7150/ijbs.45498
8. Longhitano L, Tibullo D, Giallongo C, Lazzarino G, Tartaglia N, Galimberti S. et al. Proteasome inhibitors as a possible therapy for SARS-CoV-2. Int J Mol Sci. (2020) 21:E3622. doi: 10.3390/ijms21103622
9. Schulert GS, Grom AA. Pathogenesis of macrophage activation syndrome and potential for cytokine- directed therapies. Annu Rev Med. (2015) 66:145–59. doi: 10.1146/annurev-med-061813-012806
10. Thakar MS, Kearl TJ, Malarkannan S. Controlling cytokine release syndrome to harness the full potential of CAR-based cellular therapy. Front Oncol. (2020) 9:1529. doi: 10.3389/fonc.2019.01529
11. Nassereddine S, Rafei H, Elbahesh E, Tabbara I. Acute graft versus host disease: a comprehensive review. Anticancer Res. (2017) 37:1547–55. doi: 10.21873/anticanres.11483
12. Ferro F, Elefante E, Puxeddu I, Baldini C, Bartoloni E, Baratè C. et al. Covid-19: the new challenge for rheumatologists. first update. Clin Exp Rheumatol. (2020) 38:373–82.
13. Gopinathannair R, Merchant FM, Lakkireddy DR, Etheridge SP, Feigofsky S, Han JK, et al. COVID-19 and cardiac arrhythmias: a global perspective on arrhythmia characteristics and management strategies [published online ahead of print, 2020. Jun 3. J Interv Card Electrophysiol. (2020). doi: 10.1007/s10840-020-00789-9. [Epub ahead of print].
14. Meo SA, Klonoff DC, Akram J. Efficacy of chloroquine and hydroxychloroquine in the treatment of COVID-19. Eur Rev Med Pharmacol Sci. (2020) 24:4539–47. doi: 10.26355/eurrev_202004_21038
15. Mehra MR, Desai SS, Ruschitzka F, Patel AN. RETRACTED: hydroxychloroquine or chloroquine with or without a macrolide for treatment of COVID-19: a multinational registry analysis [published online ahead of print, 2020. Lancet. (2020) doi: 10.1016/S0140-6736(20)31180-6
16. Sarzi-Puttini P, Giorgi V, Sirotti S, Marotto D, Ardizzone S, Rizzardini G, et al. COVID-19, cytokines and immunosuppression: what can we lea rn from severe acute respiratory syndrome? Clin Exp Rheumatol. (2020) 38:337–42.
17. Jagasia M, Perales MA, Schroeder MA, Ali H, Shah NN, Chen YB, et al. Ruxolitinib for the treatment of steroid-refractory acute GVHD (REACH1): a multicenter, open-label, phase 2 trial. Blood. (2020) 135:1739–49. doi: 10.1182/blood.2020004823
18. Stebbing J, Phelan A, Griffin I, Tucker C, Oechsle O, Smith D, et al. COVID-19: combining antiviral and anti-inflammatory treatments. Lancet Infect Dis. (2020) 20:400–2. doi: 10.1016/S1473-3099(20)30132-8
19. Coleman CM, Sisk JM, Mingo RM, Nelson EA, White JM, Frieman MB. Abelson kinase inhibitors are potent inhibitors of severe acute respiratory syndrome coronavirus and middle east respiratory syndrome coronavirus fusion. J Virol. (2016) 90:8924–33. doi: 10.1128/JVI.01429-16
20. Sisk JM, Frieman MB, Machamer CE. Coronavirus S protein-induced fusion is blocked prior to hemifusion by Abl kinase inhibitors. J Gen Virol. (2018) 99:619–30. doi: 10.1099/jgv.0.001047
21. Foà R, Bonifacio M, Chiaretti S, Curti A, Candoni A, Fava C. et al. Ph+ acute lymphoblastic leukaemia in Italy during the Covid-19 pandemic. a campus all study [published online ahead of print, 2020 May 5. Br J Haematol. (2020) 190:e3–5. doi: 10.1111/bjh.16758
22. Breccia M, Abruzzese E, Bocchia M, Bonifacio M, Castagnetti F, Fava C, et al. Chronic myeloid leukemia management at the time of the COVID-19 pandemic in Italy. A campus CML survey. Leukemia. (2020) 34:2260–61. doi: 10.1038/s41375-020-0904-z
23. Bendinelli M, Pistello M, Maggi F, Fornai C, Freer G, Vatteroni ML. Molecular properties, biology, and clinical implications of TT virus, a recently identified widespread infectious agent of humans. Clin Microbiol Rev. (2001) 14:98–113. doi: 10.1128/CMR.14.1.98-113.2001
24. Focosi D, Spezia PG, Macera L, Salvadori S, Navarro D, Lanza M, et al. Assessment of prevalence and load of torquetenovirus viraemia in a large cohort of healthy blood donors. Clin Microbiol Infect. (2020). doi: 10.1016/j.cmi.2020.01.011
25. Galimberti S, Baldini C, Baratè C, Ricci F, Balducci S, Grassi S. et al. The CoV-2 outbreak: how hematologists could help to fight Covid-19. Pharmacol Res. (2020) 157:104866. doi: 10.1016/j.phrs.2020.104866
26. Baccarani M, Deininger MW, Rosti G, Hochhaus A, Soverini S, Apperley JF, et al. European LeukemiaNet recommendations for the management of chronic myeloid leukemia: 2013. Blood. (2013) 122:872–84. doi: 10.1182/blood-2013-05-501569
27. Maggi F, Pistello M, Vatteroni M, Presciuttini S, Marchi S, Isola P, et al. Dynamics of persistent TT virus infection, as determined in patients treated with alpha interferon for concomitant hepatitis C virus infection. J Virol. (2001) 75:11999–2004. doi: 10.1128/JVI.75.24.11999-12004.2001
28. Cross NC, Hochhaus A, Müller MC. Molecular monitoring of chronic myeloid leukemia: principles and interlaboratory standardization. Ann Hematol. (2015) 94 (Suppl. 2):S219–25. doi: 10.1007/s00277-015-2315-1
29. Focosi D, Maggi F, Albani M, Macera L, Ricci V, Gragnani S, et al. Torque Teno Virus viremia kinetics after autologous stem cell transplantation are predictable and may serve as a surrogate marker of functional immune reconstitution. J Clin Virol. (2010) 47:189–92. doi: 10.1016/j.jcv.2009.11.027
30. Burra P, Masier A, Boldrin C, Calistri A, Andreoli E, Senzolo M, et al. Torque Teno Virus: any pathological role in liver transplanted patients? Transpl Int. (2008) 21:972–9. doi: 10.1111/j.1432-2277.2008.00714.x
31. Maggi F, Pifferi M, Tempestini E, Fornai C, Lanini L, Andreoli E, et al. TT virus loads and lymphocyte subpopulations in children with acute respiratory diseases. J Virol. (2003) 77:9081–3. doi: 10.1128/JVI.77.16.9081-9083.2003
32. Maggi F, Focosi D, Statzu M, Bianco G, Costa C, Macera L, et al. Early post-transplant torquetenovirus viremia predicts cytomegalovirus reactivations in solid organ transplant recipients. Sci Rep. (2018) 8:15490. doi: 10.1038/s41598-018-33909-7
33. Kong Q, Wu Y, Gu Y, Lv Q, Qi F, Gong S, et al. Analysis of the molecular mechanism of Pudilan (PDL) treatment for Covid-19 by network pharmacology tools published online ahead of print, 2020 May 30. Biomed Pharmacother. (2020) 128:110316. doi: 10.1016/j.biopha.2020.110316
34. Li F, Sheng Y, Hou W, et al. CCL5-armed oncolytic virus augments CCR5-engineered NK cell infiltration and antitumor efficiency. J Immunother Cancer. (2020) 8:e000131. doi: 10.1136/jitc-2019-000131
35. Hui E, Cheung J, Zhu J, Su X, Taylor MJ, Wallweber HA, et al. T cell costimulatory receptor CD28 is a primary target for PD-1-mediated inhibition. Science. (2017) 355:1428–33. doi: 10.1126/science.aaf1292
36. Wang ZQ, Milne K, Webb JR, Watson PH. CD74 and intratumoral immune response in breast cancer. Oncotarget. (2017) 8:12664–74. doi: 10.18632/oncotarget.8610
37. Choi JY, Kim JH, Hossain FMA, Uyangaa E, Park SO, Kim B, et al. Indispensable role of CX3CR1+ dendritic cells in regulation of virus-induced neuroinflammation through rapid development of antiviral immunity in peripheral lymphoid tissues. Front Immunol. (2019) 10:1467. doi: 10.3389/fimmu.2019.01467
38. Touzelet O, Broadbent L, Armstrong SD, Aljabr W, Cloutman-Green E, Power UF, et al. The secretome profiling of a pediatric airway epithelium infected with hRSV identified aberrant apical/basolateral trafficking and novel immune modulating (CXCL6, CXCL16, CSF3) and antiviral (CEACAM1) proteins. Mol Cell Proteomics. (2020) 19:793–807. doi: 10.1074/mcp.RA119.001546
39. Groom JR, Luster AD. CXCR3 in T cell function. Exp Cell Res. (2011) 317:620–31. doi: 10.1016/j.yexcr.2010.12.017
40. Liong S, Lim R, Barker G, Lappas M. Hepatitis A virus cellular receptor 2 (HAVCR2) is decreased with viral infection and regulates pro-labour mediators OA. Am J Reprod Immunol. (2017) 78. doi: 10.1111/aji.12696
42. Klein-Hessling S, Muhammad K, Klein M, Pusch T, Rudolf R, Flöter J, et al. NFATc1 controls the cytotoxicity of CD8+ T cells. Nat Commun. (2017) 8:511. doi: 10.1038/s41467-017-00612-6
43. Liu H, Zhou RH, Liu Y, Guo L, Wang X, Hu WH, et al. HIV infection suppresses TLR3 activation-mediated antiviral immunity in microglia and macrophages. Immunology. (2020) 160:269–79. doi: 10.1111/imm.13181
44. Arlauckas SP, Garren SB, Garris CS, Kohler RH, Oh J, Pittet MJ, et al. Arg1 expression defines immunosuppressive subsets of tumor-associated macrophages. Theranostics. (2018) 8:5842–54. doi: 10.7150/thno.26888
45. Nagaishi T, Pao L, Lin SH, Iijima H, Kaser A, Qiao SW, et al. SHP1 phosphatase-dependent T cell inhibition by CEACAM1 adhesion molecule isoforms. Immunity. (2006) 25:769–81. doi: 10.1016/j.immuni.2006.08.026
46. Brennan FH, Jogia T, Gillespie ER, Blomster LV, Li XX, Nowlan B, et al. Complement receptor C3aR1 controls neutrophil mobilization following spinal cord injury through physiological antagonism of CXCR2. JCI Insight. (2019) 4:98254. doi: 10.1172/jci.insight.98254
47. Krześniak M, Zajkowicz A, Gdowicz-Kłosok A, Głowala-Kosinska M, Łasut-Szyszka B, Rusin M. Synergistic activation of p53 by actinomycin D and nutlin-3a is associated with the upregulation of crucial regulators and effectors of innate immunity. Cell Signal. (2020) 69:109552. doi: 10.1016/j.cellsig.2020.109552
48. Liu C, Li Z, Wang S, Fan Y, Zhang S, Yang X, et al. FUT4 is involved in PD-1-related immunosuppression and leads to worse survival in patients with operable lung adenocarcinoma. J Cancer Res Clin Oncol. (2019) 145:65–76. doi: 10.1007/s00432-018-2761-y
49. Watek M, Wnorowska U, Wollny T, Durnaś B, Wolak P, Kościołek-Zgódka S, et al. Hypogelsolinemia in patients diagnosed with acute myeloid leukemia at initial stage of sepsis. Med Sci Monit. (2019) 25:1452–8. doi: 10.12659/MSM.911904
50. Slade JA, Hall JV, Kintner J, Phillips-Campbell R, Schoborg RV. Host nectin-1 promotes chlamydial infection in the female mouse genital tract, but is not required for infection in a novel male murine rectal infection model. PLoS ONE. (2016) 11:e0160511. doi: 10.1371/journal.pone.0160511
51. Rosenberg HF. Eosinophil-Derived Neurotoxin (EDN/RNase 2) and the mouse eosinophil-associated RNases (mEars): expanding roles in promoting host defense. Int J Mol Sci. (2015) 16:15442–55. doi: 10.3390/ijms160715442
52. Salazar VA, Arranz-Trullén J, Navarro S, Blanco JA, Sánchez D, Moussaoui M, et al. Exploring the mechanisms of action of human secretory RNase 3 and RNase 7 against Candida albicans. Microbiologyopen. (2016) 5:830–45. doi: 10.1002/mbo3.373
53. Giallongo C, Parrinello N, Tibullo D, La Cava P, Romano A, Chiarenza A, et al. Myeloid derived suppressor cells (MDSCs) are increased and exert immunosuppressive activity together with polymorphonuclear leukocytes (PMNs) in chronic myeloid leukemia patients. PLoS ONE. (2014) 9:e101848. doi: 10.1371/journal.pone.0101848
54. Khairnar V, Duhan V, Patil AM, Zhou F, Bhat H, Thoens C, et al. CEACAM1 promotes CD8+ T cell responses and improves control of a chronic viral infection. Nat Commun. (2018) 9:2561. doi: 10.1038/s41467-018-04832-2
55. Savoldo B, Ramos CA, Liu E, Mims MP, Keating MJ, Carrum G, et al. CD28 costimulation improves expansion and persistence of chimeric antigen receptor-modified T cells in lymphoma patients. J Clin Invest. (2011) 121:1822–6. doi: 10.1172/JCI46110
56. O'Donnell JS, Smyth MJ, Teng MWL. PD1 functions by inhibiting CD28-mediated co-stimulation. Clin Transl Immunol. (2017) 6:e138. doi: 10.1038/cti.2017.15
57. Wang F, Hou H, Luo Y, Tang G, Wu S, Huang M. et al. The laboratory tests and host immunity of Covid-19 patients with different severity of illness. JCI Insight. (2020) 5:e137799. doi: 10.1172/jci.insight.137799
58. Talpaz M, Mercer J, Hehlmann R. The interferon-alpha revival in CML. Ann Hematol. (2015) 94 (Suppl. 2):S195–207. doi: 10.1007/s00277-015-2326-y
59. Ribes S, Arcilla C, Ott M, Schütze S, Hanisch UK, Nessler S, et al. Pre-treatment with the viral Toll-like receptor 3 agonist poly(I:C) modulates innate immunity and protects neutropenic mice infected intracerebrally with Escherichia coli. J Neuroinflammation. (2020) 17:24. doi: 10.1186/s12974-020-1700-4
60. Massimino M, Stella S, Tirrò E, Pennisi MS, Vitale SR, Puma A, et al. ABL1-directed inhibitors for CML: efficacy, resistance and future perspectives. Anticancer Res. (2020) 40:2457–65. doi: 10.21873/anticanres.14215
61. Abou Dalle I, Jabbour E, Short NJ, Ravandi F. Treatment of philadelphia chromosome-positive acute lymphoblastic leukemia. Curr Treat Options Oncol. (2019) 20:4. doi: 10.1007/s11864-019-0603-z
62. Farag S, Smith MJ, Fotiadis N, Constantinidou A, Jones RL. Revolutions in treatment options in gastrointestinal stromal tumours (GISTs): the latest updates. Curr Treat Options Oncol. (2020) 21:55. doi: 10.1007/s11864-020-00754-8
63. Hughes A, Yong ASM. Immune effector recovery in chronic myeloid leukemia and treatment-free remission. Front Immunol. (2017) 8:469. doi: 10.3389/fimmu.2017.00469
64. Galimberti S, Grassi S, Baratè C, Guerrini F, Ciabatti E, Perutelli F. et al. The polycomb BMI1 protein is co-expressed with CD26+ in leukemic stem cells of chronic myeloid leukemia. Front Oncol. (2018) 8:555. doi: 10.3389/fonc.2018.00555
65. Chang X, Liu XL, Du QF, Li R, Feng R, Chen Q, et al. Study of adhesion-related molecule beta1-integrin and focal adhesion kinase in chronic myeloid leukemia. Di Yi Jun Yi Da Xue Xue Bao. (2003) 23:1047–9.
66. Chapman NM, Houtman JC. Functions of the FAK family kinases in T cells: beyond actin cytoskeletal rearrangement. Immunol Res. (2014) 59:23–34. doi: 10.1007/s12026-014-8527-y
67. Grassi S, Palumbo S, Mariotti V, Liberati D, Guerrini F, Ciabatti E, et al. The WNT pathway is relevant for the BCR-ABL1-independent resistance in chronic myeloid leukemia. Front Oncol. (2019) 9:532. doi: 10.3389/fonc.2019.00532
68. Perng YC, Lenschow DJ. ISG15 in antiviral immunity and beyond. Nat Rev Microbiol. (2018) 16:423–39. doi: 10.1038/s41579-018-0020-5
Keywords: CML, TKIs, imatinib, nilotinib, TTV, immunity, NanoString, COVID-19
Citation: Galimberti S, Petrini M, Baratè C, Ricci F, Balducci S, Grassi S, Guerrini F, Ciabatti E, Mechelli S, Di Paolo A, Baldini C, Baglietto L, Macera L, Spezia PG and Maggi F (2020) Tyrosine Kinase Inhibitors Play an Antiviral Action in Patients Affected by Chronic Myeloid Leukemia: A Possible Model Supporting Their Use in the Fight Against SARS-CoV-2. Front. Oncol. 10:1428. doi: 10.3389/fonc.2020.01428
Received: 13 May 2020; Accepted: 06 July 2020;
Published: 02 September 2020.
Edited by:
Pierluigi Porcu, Sidney Kimmel Cancer Center, United StatesReviewed by:
Gina Keiffer, Sidney Kimmel Cancer Center, United StatesMargaret Kasner, Thomas Jefferson University, United States
Copyright © 2020 Galimberti, Petrini, Baratè, Ricci, Balducci, Grassi, Guerrini, Ciabatti, Mechelli, Di Paolo, Baldini, Baglietto, Macera, Spezia and Maggi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sara Galimberti, c2FyYS5nYWxpbWJlcnRpQG1lZC51bmlwaS5pdA==