 Darren Pan
Darren Pan Joshua Richter
Joshua Richter- Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, New York, NY, United States
Multiple myeloma remains an incurable disease despite numerous novel agents being approved in the last decade. Furthermore, disease behavior and susceptibility to current treatments often vary drastically from patient to patient. To date there are no approved therapies in myeloma that are targeted to specific patient populations based on genomic or immunologic findings. Precision medicine, using biomarkers descriptive of a specific tumor’s biology and predictive of response to appropriate agents, may continue to push the field forward by expanding our treatment arsenal while refining our ability to expose patients to only those treatments likely to be efficacious. Extensive research efforts have been carried out in this endeavor including the use of agents targeting Bcl2 and the RAS/MAPK and PI3K/AKT/mTOR pathways. Thus far, clinical trials have yielded occasional successes intermixed with disappointments, reflecting significant hurdles which still remain including the complex crosstalk between oncogenic pathways and the nonlinear genetic development of myeloma, prone to cultivating sub-clones with distinctive mutations. In this review, we explore the landscape of precision therapeutics in multiple myeloma and underscore the degree to which research efforts have produced tangible clinical results.
Introduction
Over the last two decades, the additions of proteasome inhibitors (PIs), immunomodulating agents (IMiDs), and anti-CD38 antibodies to the multiple myeloma treatment arsenal have improved survival materially (1). In recent years, further incremental gains have been made through agents with novel mechanisms, such as panobinostat, selinexor, belantamab mafodotin, and idecabtagene vicleucel. Although treatment approaches can be adjusted to patients’ comorbidities, cytogenetic risk, functional status, and response, the individual drugs that constitute current standard of care are agnostic to molecular characteristics and biomarkers. In spite of the remarkable advances, depth and duration of responses to current agents can vary widely and unpredictably due in large part to extensive intertumor and intratumor genetic variability (2). Drug-refractory relapse remains an inevitability in the vast majority of patients and each successive line of therapy produces shorter responses (3). In this context, the field has seen a renewed push for agents targeted to disease-specific characteristics.
Precision medicine approaches already have become indispensable in several other cancers, from trastuzumab in HER-2-mutated breast cancer to cetuximab in EGFR-mutated non-small cell lung cancer (4). Despite actionable mutations being identified in over three quarters of relapsed multiple myeloma patients, precision medicine has not been incorporated into standard myeloma treatment (5). The advent of genome sequencing technology has facilitated analysis of the tumor genotype with unprecedented resolution. Using these techniques, researchers have been able to compare myeloma samples to normal tissue to identify recurrently mutated pathways that may serve as additional therapeutic targets (2). Furthermore, the use of so-called “umbrella” trials, in which patients with the same cancer but different targetable biomarkers receive different treatments matched to their specific biomarker, and “basket” trials, in which patients with a variety of malignancies sharing a common biomarker are treated with a targeted treatment, has the potential to accelerate discovery of promising precision medicine candidates. The multi-armed MyDRUG protocol is one notable umbrella study in which enrolled patients are screened for targetable mutations and, if identified, are assigned to drug combinations featuring appropriate targeted agents, allowing simultaneous evaluation of multiple targeted approaches that may warrant further exploration (NCT03732703).
Precision medicine approaches hold the promise of finally incorporating this valuable genomic data to target appropriate aspects of each individual’s underlying disease biology. Actionable mutations are now regularly identified in multiple myeloma, with such abnormalities as KRAS mutations, t(11;14), t(4;14), and CDKN2C loss being found in approximately 29%, 20%, 20%, and 15% of cases, respectively (6–9). Here we review progress made in developing agents tailored to disease-specific biology with a focus on dissecting the degree of clinical success observed.
Previous Clinical Pursuits: Targeted Approaches Show Mixed Clinical Results
RAS/MAPK Pathway Inhibition
The mitogen activated protein kinase (MAPK) pathway is commonly mutated in multiple myeloma and is found in over 50% of patients (7). KRAS, NRAS, and BRAF mutations are generally mutually exclusive with rates of up to 29%, 24%, and 12% respectively (7, 10). MAPK pathway mutations can enhance proteasome activity, reduce cellular stress induced by PIs, and can confer resistance to PIs (11, 12). Myeloma cell lines treated with MAPK inhibitors are sensitized to the effects of PIs (12).
The V600E activating BRAF mutation is a poor prognostic sign, with patients harboring the mutation having a propensity for extramedullary disease (13). BRAF-mutated tumors such as melanoma and colorectal cancer have been successfully treated by inhibition of BRAF and the downstream MAPK kinase (MEK) (14, 15). In case reports, heavily pretreated BRAF V600E-mutated multiple myeloma patients experienced durable responses with either the BRAF inhibitor vemurafenib alone and vemurafenib plus the MEK inhibitor cobimetinib and served as proof-of-concept for the efficacy of BRAF inhibition in multiple myeloma (13, 16). Anecdotal activity was also noted with the combination of the BRAF inhibitor dabrafenib and MEK inhibitor trametinib (17, 18). The VE-BASKET study, which treated multiple BRAF V600-mutated tumors with vemurafenib monotherapy, observed a 33% response rate among the 9 multiple myeloma patients included (19). While these early findings show promise, whether vemurafenib efficacy can be improved by means of combination treatment or by introduction as an earlier line of therapy remains to be seen. The phase 2 CAPTUR study will treat a variety of tumor types, including multiple myeloma, according to targetable genetic abnormalities with BRAF V600-mutated disease receiving vemurafenib plus cobimetinib (NCT03297606). See Table 1 for an overview of trials with agents that may favor particular cytogenetic or molecular profile-defined subgroups.
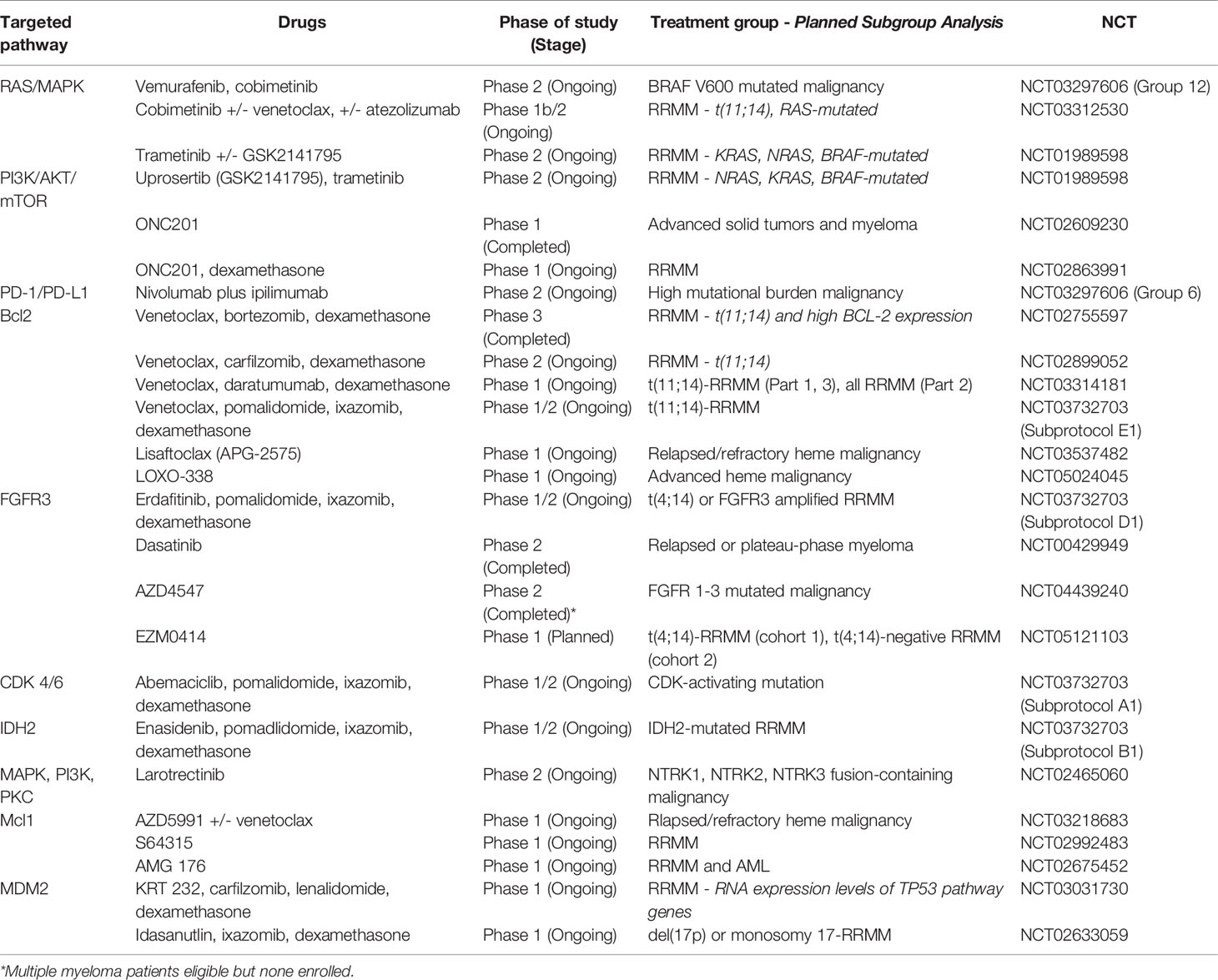
Table 1 Overview of multiple myeloma trials targeting pathways and relevant subgroups.
Cobimetinib is also being investigated independently from vemurafenib. In a preliminary report of a phase Ib/II study of relapsed/refractory multiple myeloma (RRMM), cobimetinib monotherapy demonstrated no efficacy among the 6 treated patients, but showed activity with venetoclax both with and without atezolizumab (20). The authors will be tracking the effects of t(11;14) and RAS mutations on response. Cobimetinib in combination with ixazomib and pomalidomide is also being further investigated in the MYDRUG umbrella protocol in NRAS, KRAS, and BRAF-mutated myeloma.
Single agent trametinib found early success in isolated individuals, including one KRAS-mutated patient with multiply relapsed myeloma and extramedullary disease who attained an impressive response with single agent trametinib (21). Subsequently, a retrospective analysis identified 58 trametinib-treated RRMM cases of which 51 harbored KRAS, NRAS, or BRAF mutations (21). Heterogeneous in both disease characteristics and treatment approach, these real-world patients with a median of 5 lines of prior therapy were treated with trametinib monotherapy, combination therapy, or in some cases monotherapy with additional agents later added. Treatment was well tolerated and produced partial responses or greater in 16 patients, with the 4 occurring on single agent trametinib further confirming its activity in this population. Still, prospective studies investigating trametinib use thus far have been sparse. Due to extensive cross-talk between the MAPK pathway and PI3K/AKT pathways, inhibition of one pathway can activate the other suggesting dual inhibition as a promising therapeutic approach (22, 23). Unfortunately, preliminary results of a phase II trial using trametinib in RRMM with addition of the AKT inhibitor uprosertib (GSK2141795) in non-responders reported an ORR of only 8% in KRAS, NRAS, or BRAF-mutated patients, and no responses among the 12 wild-type patients with trametinib alone (24). Addition of uprosertib increased ORR of study patients to 27%, but in the absence of further published results since this 2016 report, trial-based experience with trametinib remains limited (24).
PI3K/AKT/mTOR Pathway
Multiple myeloma depends heavily on the bone marrow microenvironment for survival and proliferation. Interactions with stromal cells produce cytokines like IL-6, VEGF, and IGF-1 which activate the PI3K/AKT/mTOR pathway in multiple myeloma patients, initiating a signaling cascade which promotes resistance to chemotherapy and cancerous progression (25, 26). Although somatic mutations in the PI3K/AKT pathway are frequently seen in other malignancies, in multiple myeloma no activating mutations in PI3K/AKT genes have been identified (27–29). Similarly, mutations or deletions in the tumor suppressor, PTEN, which can disinhibit the pathway can sensitize tumors to mTOR inhibition are uncommon in multiple myeloma (30, 31). In the absence of targetable mutations, biomarkers that could predict susceptibility pathway inhibition are being actively sought and thus far trials of agents targeting the pathway have yet to adopt a precision approach.
Preclinical studies of PI3K/AKT/mTOR pathway inhibitors in multiple myeloma have long demonstrated therapeutic potential (31–35). However, initial clinical trials, many of which targeted mTOR with rapalogs such as temsirolimus and everolimus, showed muted single agent activity in multiple myeloma patients (36, 37). Everolimus plus lenalidomide, a combination which had demonstrated preclinical synergy, achieved PR or better in 21% of patients in a phase 1 study (38). Notably, a retrospective analysis found that gene expression profiles of responding myeloma patients were characterized by higher baseline expression of mTOR pathway genes (38). These findings suggest that use of microarray to identify patients with favorable gene expression profiles may represent a precision approach for future studies targeting the PI3K pathway. A phase 2 study combined temsirolimus with bortezomib in RRMM with 33% of the 43 patients responding (39). A feedback loop whereby mTOR inhibition increases IGF-1 signaling and activation of AKT may be an important mechanistic explanation for the thus far limited activity of rapalogs (40).
Rapalogs primarily inhibit mTOR’s function as a member of the multiprotein complex, TORC1. Importantly, mTOR also contributes to the function of a second complex, TORC2, whose phosphorylation substrates include AKT (41). Many newer mTOR inhibitors were rationally designed to inhibit both TORC1 and TORC2 to prevent feedback activation of AKT. Disappointingly, a phase 1 study of one such dual TORC1/TORC2 inhibitor, TAK-228, produced only one minimal response out of 26 multiple myeloma patients (42). Similarly, a phase 1 study of CC-223, another dual TORC1/TORC2 inhibitor, produced no responses out of 10 response evaluable myeloma patients, though 2 patients experienced prolonged SD (43).
Inhibition of Akt with perifosine was another approach with efficacy in vitro and in mouse models (34). A phase I study of perifosine, lenalidomide, and dexamethasone in RRMM produced an encouraging 73% ORR and found that responders had higher baseline bone marrow phospho-Akt levels, identifying another potential biomarker for agents targeting the PI3K/AKT pathway (44). A more recent phase III study, however, randomized patients to bortezomib and dexamethasone with or without perifosine but found no signal towards improved response rates or progression-free survival (PFS) at the first interim analysis and was discontinued (45). The pan-Akt inhibitor afuresertib has also been clinically tested in multiple myeloma, with initial monotherapy trials discontinued due to limited single agent activity (46). A basket study of afuresertib with trametinib was discontinued due to intolerable toxicities including grade 3 transaminitis and hypokalemia (47). Although the combination of afuresertib with bortezomib and dexamethasone showed a ORR of 41% in RRMM from preliminary phase 1 data, final results of the since completed study have not been published and a subsequent study of afuresertib and carfilzomib in RRMM has been discontinued (48, 49). Ongoing studies of Akt inhibitors include uprosertib with trametinib, with preliminary ORR of 27% (NCT01951495), and ONC201 as monotherapy or with dexamethasone (NCT02609230, NCT02863991).
PI3K inhibition has also been tested, although the combination of the PI3K inhibitor BYL719 and pan-PIM inhibitor LGH447 in RRMM was poorly tolerated in a phase 1 study, with 26.7% of patients experiencing grade 3 or 4 thrombocytopenia, prompting premature termination (50). With most initial clinical trials hampered by lackluster efficacy or intolerable toxicities, it remains to be seen whether the preclinical promise of the PI3K/AKT/mTOR pathway can truly be translated into tangible clinical results.
Checkpoint Inhibition
Myeloma cells are known to overexpress PD-L1 which contributes to immune evasion (51). While preclinical evidence supported checkpoint inhibitors as a promising treatment approach, particularly when combined with IMiDs, clinical studies have yielded disappointing results (52–54). Not only have nivolumab and pembrolizumab demonstrated minimal single agent activity in myeloma, in some cases the combination of checkpoint inhibitors and IMiDs actually increased mortality (53, 55–57).
It is now clear that a better understanding of myeloma biology is required to guide checkpoint inhibitor therapy in multiple myeloma patients. In solid tumors, the issue of benefit versus immune-mediated risk with checkpoint inhibitors has been particularly favorable in tumors with high mutational burden. Tumors with high mutational burden tend to be associated with higher levels of neoantigens and tumor infiltrating lymphocytes, an environment that can be exploited by checkpoint inhibition (58). In multiple myeloma, apolipoprotein B mRNA editing enzyme (APOBEC) is a source of increased mutational burden and is associated with the MAF t(14;16) translocation (59, 60). While t(14;16) and high mutation and neoantigen burden are indicators of poor prognosis, this rationale supports the potential for these patients to derive benefit from checkpoint inhibitors (61). In the ongoing CAPTUR study, patients with high mutational burden tumors will be assigned to nivolumab plus ipilimumab (NCT03297606).
Standout Success: The Agent Seeing Real-World Use as Targeted Therapy
Venetoclax
Although there are currently no FDA-approved treatments for mutationally defined subsets of multiple myeloma, one agent is backed by a healthy degree of clinical trial data and has begun to see real-world use: venetoclax. Venetoclax, an oral selective inhibitor of anti-apoptotic protein BCL-2, has become a prototypical precision drug in myeloma. Currently approved in CLL and AML, venetoclax in multiple myeloma has demonstrated particular efficacy in patients harboring t(11;14) from preclinical to phase 3 trials (62). The BCL-2 gene is highly expressed in human myeloma cell lines (HMCL) and is thought to play a role in tumorigenesis (63). However, early treatment of HMCLs noted venetoclax sensitivity primarily in the CCND1 subgroup, making the t(11;14) translocation an subgroup of interest (64). Of note, t(11;14) is the most common translocation in newly-diagnosed multiple myeloma (NDMM), seen in about 20% of cases and is associated with increased BCL-2 expression (65, 66). In primary plasma cell leukemia, a particularly aggressive variant of multiple myeloma, t(11;14) incidence is closer to 50% and case reports support venetoclax efficacy among these patients (67–70). BCL-2 expression and dependence is further increased by dexamethasone, offering an attractive combination (71).
As with many myeloma-directed therapies with potential as targeted therapy, early trials applied venetoclax more broadly to assess for biomarker-independent activity. An initial phase 1 study of venetoclax monotherapy in RRMM demonstrated overall response rates of 20% (8). Notably, while 46% of participants harbored t(11;14), 86% of responses were found among these t(11;14) patients (8). Subsequently, the phase 3 BELLINI study randomized 291 RRMM patients to bortezomib and dexamethasone plus either venetoclax or placebo. In all-comers, the venetoclax arm improved median PFS of 22.4 months versus 11.5 months and achieved deeper responses with more patients with MRD negativity. However, patients in the venetoclax group had higher rates of grade ≥3 neutropenia (18% vs. 7% in placebo) and pneumonia (16% vs. 9%). Ultimately, the venetoclax arm experienced poorer overall survival (OS) due in large part to treatment-related deaths from infectious complications, prompting study discontinuation (72). Again, t(11;14) patients benefitted the most from venetoclax addition. Patients harboring t(11;14) or expressing high BCL2 treated with the venetoclax combination attained deeper responses with a median PFS not yet reached compared to 9.9 months with placebo without increased mortality (72). An ongoing phase 2 study of venetoclax with carfilzomib and dexamethasone has also demonstrated safety and efficacy in RRMM, with an ORR of 75% in the 36 patients without t(11;14) and 92% in the 13 patients with t(11;14) (73). Similarly, preliminary phase 1 results in RRMM found that venetoclax, daratumumab, and dexamethasone produced a 96% ORR (all ≥ very good partial response, VGPR) in t(11;14)-only population (n = 24), while the same three agents plus bortezomib produced a 92% ORR (79% ≥ VGPR) in cytogenetically unselected patients (74). The BELLINI study along with other preliminary results have solidified venetoclax’ role as an important combination treatment option in appropriately selected patients.
Based on early efficacy data, venetoclax has begun to see off-label use in the community. One single-center study found that providers most commonly combined venetoclax with dexamethasone alone, followed by triplet regimens with PI and dexamethasone or daratumumab and dexamethasone (75). The 70 patients had a median of 3 prior lines of therapy, the majority were t(11;14) positive (86%), and the median PFS was an encouraging 13 months. Even penta-refractory patients benefitted from a PFS of 7.2 months. A different single-center study found that among 56 patients treated with venetoclax outside of clinical trials, 75% had t(11;14) and the agent was used as monotherapy or with only dexamethasone in 55% of patients while the remainder received triplets or quadruplets (76). Here, venetoclax was introduced later with a median of 6 prior lines and produced a 44% ORR but a shorter 5.8 month PFS. Notably, t(11;14) patients enjoyed longer PFS (9.7 vs. 4.2 months for t(11;14) negative, p = 0.019) and OS (not reached vs. 10.9 months, p = 0.015) (76). Another report found that between 7 Hungarian centers, 33 patients harboring t(11;14) were treated with venetoclax primarily in combination with a PI and dexamethasone either in the relapsed setting (mean 4.8 prior lines) or after suboptimal response to initial pre-transplantation induction (17 patients) (77). The authors noted that an astonishing 96% of patients responded (28% CR, 38% VGPR, 30% PR), particularly impressive when considering the refractoriness of the group. Median PFS was 15.5 months from venetoclax initiation. However, doses utilized varied widely, as only 2 patients received the 800 mg utilized in BELLINI, 1 received 600 mg, and all others received 400mg or less (77). The efficacy observed despite varied doses raises the questions of whether a reduced-intensity venetoclax regimen could represent a viable approach to improving tolerability while preserving efficacy.
A major observation from the BELLINI study was that at 800 mg daily, venetoclax was associated with significant myelosuppression and infectious, often life-threatening complications. A small recent retrospective study specifically assessed low dose venetoclax (≤250 mg/day) in combination with daratumumab, bortezomib, and dexamethasone in RRMM (78). While the 16 patient without t(11;14) had an ORR of only 31%, the 5 patients with the translocation benefited from an 80% ORR. Importantly, despite adding daratumumab to the three agents used in BELLINI, the high rates of infectious complications seen in BELLINI were not observed (no grade ≥3 pneumonia). Randomized clinical trials are needed to clarify the true benefit of reduced-dose venetoclax and whether it could represent an efficacious yet tolerable approach to precision medicine.
While the benefit of venetoclax to date has been most pronounced in t(11;14) or high BCL-2 expressors, multiple ongoing trials continue to investigate the agent in a cytogenetically agnostic fashion. In the relapsed/refractory setting, venetoclax is being studied in combination with carfilzomib and dexamethasone and with daratumumab and dexamethasone with or without bortezomib; aforementioned preliminary data show higher efficacy in t(11;14) patients (NCT02899052, NCT03314181) (73, 74, 79). Venetoclax is also being directly compared to pomalidomide in relapsed/refractory t(11;14) positive patients (NCT03539744) and has also been included in the MyDRUG trial (NCT03732703) (80). Looking forward, a first-in-human phase 1 study of novel BCL-2 inhibitor lisaftoclax is underway in multiple myeloma and other hematologic malignancies with doses up to 1200 mg/day thus far being well-tolerated (NCT03537482) (81). LOXO-338 is another BCL-2 inhibitor being tested in a phase 1 study in advanced hematologic malignancies (NCT05024045).
Looking to the Future: Targeted Approaches Showing Pre-Clinical Promise
FGFR3 Inhibition
In multiple myeloma, the t(4;14)(p16.3;q32) translocation leads to deregulation of FGFR3 and WHSC1/MMSET and is found in up to 20% of newly diagnosed patients (9). Patients harboring this translocation suffer from quicker relapses following chemotherapy and transplant (82, 83). The abnormality remains a poor prognostic marker even in the era of novel agents, and thus identifying effective treatments for this high-risk group remains a significant unmet need (84). FGFR3 inhibition has proved to be an effective strategy among other tumor types such as bladder cancer (85). In multiple myeloma, FGFR3 inhibition has shown preclinical promise as a form of targeted therapy, inducing apoptosis selectively in t(4;14) positive HMCLs and t(4;14) positive multiple myeloma xenografts in mice (86–89). Comparatively, FGFR3 inhibition in the clinical setting has to date been less active.
Clinically, FGFR3 has been targeted by both non-selective multikinase inhibitors and highly selective FGFR inhibitors. In multiple myeloma patients, dovitinib and nintedanib, two multikinase inhibitors which inhibit FGFR, VEGFR, and PDGFR, have demonstrated little more than a tolerable safety profile and the potential to stabilize myelomatous disease (90, 91). Dasatinib, a multikinase inhibitor effective in CML, was investigated in a phase II study of RRMM patients produced only 1 partial response out of 21 treated patients (NCT00429949).
Key to future successes of FGFR3-targeted therapies will be appropriate patient selection. Previously mentioned studies have investigated patients irrespective of mutational status or stratified by t(4;14) status. Importantly, FGFR3 is not upregulated in all cases of t(4;14), and the translocation portends a poor outcome even in patients without altered FGFR3 expression (92). Studies of the novel selective FGFR inhibitors erdafitinib and AZD4547 look to target patients with alterations in the FGFR3 pathway specifically, though there are currently no published results of these agents in multiple myeloma patients. Although the subprotocol of the phase 2 MATCH basket study looked to treat only FGFR3-amplified tumors with AZD4547, no multiple myeloma patients were ultimately enrolled and the subprotocol failed to meet its goal of a 16% response rate among other cancers treated (93). Erdafitinib will be added to ixazomib and pomalidomide in patients with FGFR3 activating mutations in the ongoing MyDRUG protocol (NCT03732703).
CDK4/6 Inhibition
Cyclin-dependent kinases (CDK) 4 and 6 phosphorylate retinoblastoma protein, increasing expression of transcription factors which promote transition from G1 to S phase (94). CDKN2C, located on chromosome 1p, encodes a CDK inhibitor and when deleted in myeloma cells results in increased proliferation (6). Loss of CDKN2C, seen in 15% of myeloma cases, is associated with poorer OS (6, 95). The CDK4/6 inhibitor, abemaciclib, inhibits myeloma cell growth and exhibits cytotoxicity in a dose-dependent manner (96). Abemaciclib also induced regression of tumors in SCID models engrafted with multiple myeloma cells in part by increasing cytokines involved in NK cell recruitment and activation (97). In this model, CDK4/6 inhibition showed synergistic tumor suppression when combined with daratumumab. Patients enrolled in the MyDRUG study with 1p31 (CDKN2C) loss will be treated with abemaciclib in addition to ixazomib and pomalidomide.
Palbociclib is another CDK4/6 inhibitor which shows primarily cytostatic effects in vitro with multiple myeloma cell lines but enhances cytotoxicity when combined with immunomodulatory drugs, bortezomib, and corticosteroids (98–100). Thus far, the agent has only been applied in the clinical setting in a biomarker-independent fashion. A phase 1/2 study of palbociclib with bortezomib and dexamethasone in RRMM attained an ORR in 5 of 25 patients (20%) (101). Though an additional 44% achieved stable disease, the study failed to meet the ORR ≥ 28% needed to proceed to the next stage. A planned trial combining palbociclib with lenalidomide and dexamethasone terminated due to low enrollment (NCT02030483).
Mutant IDH Inhibition
Gain-of-function mutations in isocitrate dehydrogenase (IDH) 1 and 2 results in the metabolism of isocitrate to the oncometabolite 2-hydroxyglutarate rather than α-ketoglutarate. 2-hydroxyglutarate inhibits both histone demethylase and TET2, causing an increase in both histone and DNA methylation which blocks normal cell differentiation (102). Although the use of IDH 1/2 inhibitors, ivosidenib and enasidenib, have found success in AML among the approximately 16% of IDH-mutated patients, in multiple myeloma IDH mutations are only seen in 0.5% (103, 104). Still, as IDH 1/2 mutations represent a relatively recently discovered driver mutation in myeloma, mutant IDH inhibition is now being investigated in humans, with IDH2-mutated patients in the MyDRUG protocol receiving enasidenib-based combinations (105).
SETD2 Methyltransferase Inhibition
SETD2, a histone methyltransferase which catalyzes H3K36 trimethylation, performs a complex array of functions including DNA repair, alternative splicing, and promotion of transcriptional silencing (106). SETD2 is recurrently mutated in a number of tumor types including multiple myeloma, particularly in the relapsed and refractory setting (105, 107). In other diseases, SETD2 is thought to serve as a tumor suppressor, as mutations can hamper DNA repair mechanisms and increase mutation rates (108, 109). However, CRISPR pooled screens have found that myeloma cells are in fact dependent on SETD2 for survival (110). A small molecular inhibitor of SETD2 has demonstrated preclinical efficacy by suppressing proliferation both in myeloma cell lines and in vivo mouse xenografts (111). The authors also state that the agent shows in vitro synergy with general standard of care myeloma therapies. Clinical trials of SETD2 inhibitors are currently being planned.
NTRK Inhibition
Fusions involving the neurotrophin receptor tyrosine kinase genes (NTRK1, NTRK2, and NTRK3) recur in cancers like gliomas, secretory breast cancer, and lung cancer (112). The oncogenic fusion protein retains the TRK kinase domain but contain part of a different gene product leading to ligand-independent constitutive activation of downstream pathways such as MAPK, PI3K, and PKC (113). The NTRK inhibitor larotrectinib has an impressive track record, demonstrating consistent and durable responses across all solid tumors harboring the fusions and has been approved for both adult and pediatric solid tumors with NTRK fusions (114, 115). Unfortunately, NTRK fusions are detected in less than 1% of multiple myeloma cases (116). Still, the MATCH basket trial is looking to enroll patients with both solid and liquid malignancies, including multiple myeloma, harboring NTRK1, NTRK2, or NTRK3 gene fusions into the larotrectinib subprotocol (NCT02465060).
MCL1 Inhibition
Mcl-1 is an antiapoptotic protein known to play an essential role in myeloma cell survival (117, 118). MCL-1 overexpression confers resistance to chemotherapy, with rates of overexpression increasing at relapse (119, 120). The MCL-1 gene is found on chromosome 1q21, along with the gene for the IL-6 receptor, and gains or amplifications of 1q21 seen in approximately 40% of multiple myeloma cases are associated with significantly shorter survival (121). Preclinical data has also demonstrated that myeloma cell lines harboring 1q21 amplifications are particularly sensitive to Mcl-1 inhibition (122). As MCL-1 upregulation is a significant mechanism of venetoclax resistance, whether co-inhibition of Mcl-1 and Bcl-2 can demonstrate clinical synergy is also of interest (66, 123).
Several Mcl-1 inhibitors are being tested in multiple myeloma in various preclinical and early clinical stages. One agent, AMG 176, induced apoptosis in hematologic malignancy cell lines and demonstrating synergy with carfilzomib and dexamethasone (124). A subsequent first-in-human phase 1 trial of AMG 176 in RRMM is ongoing and has shown preliminary tolerability (125). Common treatment-emergent adverse events were cytopenias, nausea, and vomiting. Of note, the combination of AMG 176 and venetoclax showed preclinical synergy in AML, but a dose-finding clinical trial of the combination in AML and lymphoma was suspended over safety concerns (126). AZD5991 also showed potent preclinical efficacy in multiple myeloma and AML, and a phase 1 dose-escalation study of AZD5991 with or without venetoclax is underway in relapse/refractory hematologic malignancies (127).
MDM2 Inhibition
The enzyme mouse double minute 2 (MDM2) controls the ubiquitination of the tumor suppressor p53 and promotes its proteasomal degradation (128, 129). The expression of p53 can be decreased in a number of ways, including MDM2 overexpression, deletion of its chromosome, 17p, and inactivating p53 mutations (seen in ~8% of newly-diagnosed patients) to promote proliferation of myeloma cells (130–132). In multiple myeloma cell lines, the MDM2 inhibitor nutlin increased p53 levels and promoted apoptosis (133). Nutlin also showed preclinical synergy with bortezomib with as both increase p53 levels, but was only effective in myeloma with wild types p53 (134). As such, prolonged treatment with MDM2 inhibitors can select for a resistance line of p53 mutated cells (135, 136). In cases of p53 mutated myeloma, exclusively seen in del(17p) patients, MDM2 inhibition may still have some effect, though at higher drug concentrations (137, 138).
Deletion of 17p presents in about 10% of newly diagnosed multiple myeloma cases and within these patients approximately 37% have a p53 mutation, whereas patients without the deletion generally have wild-type p53 (138). Given the importance of wild-type p53 for the MDM2 mechanism of action, it is not surprising that the MDM2 inhibitor AMG 232, now called KRT-232, was tested in p53 wild-type multiple myeloma and solid tumors and specifically excluded del(17p) multiple myeloma (139). This phase 1 dose finding study demonstrated safety of the agent and found 240mg every 3 weeks to be the highest tolerated dose, limited primarily by cytopenias, though no responses were seen. An ongoing phase 1 study is now testing the safety of KRT-232 in combination with carfilzomib, lenalidomide, and dexamethasone in RRMM (NCT03031730).
Conversely, a phase I/II study of idasanutlin, another MDM2 inhibitor, in combination ixazomib and dexamethasone specifically in RRMM patients harboring del(17p) or monosomy 17 is also underway (NCT02633059).
Discussion
Multiple myeloma treatment continues to evolve rapidly with numerous groundbreaking treatments. In addition to the incorporation of anti-CD38 antibodies into standard of care, CAR-T cell therapy and bispecific T-cell engaging antibodies have emerged as particularly promising treatments poised to define a new wave of myeloma-directed therapeutics. Unfortunately, as of yet, there are no signs these therapies are curative silver bullets; even patients who respond well to the newest immunotherapy approaches ultimately relapse. However, across many tumor types precision medicine approaches are often highly successful both as monotherapy and when targeted agents are combined with therapeutics with broad activity.
As our understanding of multiple myeloma becomes ever more nuanced, precision medicine allows us to provide therapy which reflects the heterogeneity we have long recognized in multiple myeloma with treatments increasingly tailored to each patient’s disease biology. We have transitioned from the original approach of classical chemotherapeutics to novel agents and now to immune-based approaches. We know that a key pathway to maximizing outcomes is to obtain deep responses through eradication of as many sub-clones as possible. As genetic drivers of myeloma are oftentimes sub-clonal, combining immune-based therapies to control bulk disease along with targeted agents to eliminate residual aberrantly driven cancer could be a conceivable pathway to cure!
The ideal precision therapies of the future should be highly efficacious in a subset of myeloma patients, have a readily measurable biomarker predictive of response, and target a mutation or other biomarker with a reasonably high incidence so as to make a significant clinical impact. Venetoclax has seen consistent success in patients with the commonly seen t(11;14) and could represent the first of conceivably many future precision medicine treatments. Agents targeting the RAS/MAPK pathway are well-positioned for clinical investigation given the frequency of NRAS and KRAS mutations in multiple myeloma and early examples of single agent and combination activity. The results of the respective RAS/MAPK-targeting arms of the MyDRUG and CAPTUR studies are eagerly awaited. Conversely, targeting biomarkers like mutant IDH and NTRK fusions, each seen in <1% of myeloma cases, are unlikely to affect the vast majority of patients even if proven useful. However, with almost 200,000 patients in the U.S. living with multiple myeloma in 2022, even low frequency regimens may provide optimal treatment for a meaningful number of patients.
At this time, many additional drugs that hold precision medicine potential are still between preclinical and first-in-human phases and the field as a whole is in its infancy. The future of precision medicine in multiple myeloma does face multiple challenges, foremost among them being myeloma’s extensive intratumoral heterogeneity. The development of this heterogeneity occurs early in myelomagenesis, even preceding clinical symptoms. In addition, this complex genetic makeup is known to change frequently over the disease course (140, 141). The success of future precision approaches will likely depend upon the use of appropriately selected combinations of both targeted and non-targeted agents, the continued use of molecular profiling to identify biomarkers, and an improved understanding of the pathways driving this disease.
Author Contributions
DP researched and wrote the manuscript. JR edited, provided guidance regarding overall direction, and contributed to writing. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. NCI Surveillance E, and End Results Program, Cancer Stat Facts: Myeloma. . Available at: https://seer.cancer.gov/statfacts/html/mulmy.html.
2. Lohr JG, Stojanov P, Carter SL, et al. Widespread Genetic Heterogeneity in Multiple Myeloma: Implications for Targeted Therapy. Cancer Cell (2014) 25:91–101. doi: 10.1016/j.ccr.2013.12.015
3. Yong K, Delforge M, Driessen C, et al. Multiple Myeloma: Patient Outcomes in Real-World Practice. Br J haematology (2016) 175:252–64. doi: 10.1111/bjh.14213
4. A Baudino T. Targeted Cancer Therapy: The Next Generation of Cancer Treatment. Curr Drug Discovery Technol (2015) 12:3–20. doi: 10.2174/1570163812666150602144310
5. Auclair D, Ailawadhi S, Berdeja JG, et al. Utility of Clinical-Grade Sequencing of Relapsed Multiple Myeloma Patients; Interim Analysis of the Multiple Myeloma Research Foundation (MMRF) Molecular Profiling Protocol. Blood (2017) 130:395.
6. Leone PE, Walker BA, Jenner MW, et al. Deletions of CDKN2C in Multiple Myeloma: Biological and Clinical Implications. Clin Cancer Res (2008) 14:6033–41. doi: 10.1158/1078-0432.CCR-08-0347
7. Lionetti M, Barbieri M, Todoerti K, et al. Molecular Spectrum of BRAF, NRAS and KRAS Gene Mutations in Plasma Cell Dyscrasias: Implication for MEK-ERK Pathway Activation. Oncotarget (2015) 6:24205. doi: 10.18632/oncotarget.4434
8. Kumar S, Kaufman JL, Gasparetto C, et al. Efficacy of Venetoclax as Targeted Therapy for Relapsed/Refractory T (11; 14) Multiple Myeloma. Blood J Am Soc Hematol (2017) 130:2401–9.
9. Ronchetti D, Greco A, Compasso S, et al. Deregulated FGFR3 Mutants in Multiple Myeloma Cell Lines With T (4; 14): Comparative Analysis of Y373C, K650E and the Novel G384D Mutations. Oncogene (2001) 20:3553–62. doi: 10.1038/sj.onc.1204465
10. Ryland GL, Jones K, Chin M, et al. Novel Genomic Findings in Multiple Myeloma Identified Through Routine Diagnostic Sequencing. J Clin Pathol (2018) 71:895–9. doi: 10.1136/jclinpath-2018-205195
11. Mulligan G, Lichter DI, Di Bacco A, et al. Mutation of NRAS But Not KRAS Significantly Reduces Myeloma Sensitivity to Single-Agent Bortezomib Therapy. Blood J Am Soc Hematol (2014) 123:632–9. doi: 10.1182/blood-2013-05-504340
12. Shirazi F, Jones RJ, Singh RK, et al. Activating KRAS, NRAS, and BRAF Mutants Enhance Proteasome Capacity and Reduce Endoplasmic Reticulum Stress in Multiple Myeloma. Proc Natl Acad Sci (2020) 117:20004–14. doi: 10.1073/pnas.2005052117
13. Andrulis M, Lehners N, Capper D, et al. Targeting the BRAF V600E Mutation in Multiple Myeloma. Cancer Discov (2013)3:862–9. doi: 10.1158/2159-8290.CD-13-0014
14. Flaherty KT, Infante JR, Daud A, et al. Combined BRAF and MEK Inhibition in Melanoma With BRAF V600 Mutations. New Engl J Med (2012) 367:1694–703. doi: 10.1056/NEJMoa1210093
15. Corcoran RB, Atreya CE, Falchook GS, et al. Combined BRAF and MEK Inhibition With Dabrafenib and Trametinib in BRAF V600–mutant Colorectal Cancer. J Clin Oncol (2015) 33:4023. doi: 10.1200/JCO.2015.63.2471
16. Mey UJ, Renner C, von Moos R. Vemurafenib in Combination With Cobimetinib in Relapsed and Refractory Extramedullary Multiple Myeloma Harboring the BRAF V600E Mutation. Hematological Oncol (2017) 35:890–3. doi: 10.1002/hon.2353
17. Čepulytė R, Žučenka A, Pečeliūnas V. Combination of Dabrafenib and Trametinib for the Treatment of Relapsed and Refractory Multiple Myeloma Harboring BRAF V600E Mutation. Case Rep Hematol (2020) 2020.
18. Salama AK, Li S, Macrae ER, et al. Dabrafenib and Trametinib in Patients With Tumors With BRAFV600E Mutations: Results of the NCI-MATCH Trial Subprotocol H. J Clin Oncol (2020) 38:3895–904. doi: 10.1200/JCO.20.00762
19. Raje N, Chau I, Hyman DM, et al. Vemurafenib in Patients With Relapsed Refractory Multiple Myeloma Harboring BRAFV600 Mutations: A Cohort of the Histology-Independent VE-BASKET Study. JCO Precis Oncol (2018) 2. doi: 10.1200/PO.18.00070
20. Schjesvold F, Ribrag V, Rodriguez-Otero P, et al. Safety and Preliminary Efficacy Results From a Phase Ib/II Study of Cobimetinib as a Single Agent and in Combination With Venetoclax With or Without Atezolizumab in Patients With Relapsed/Refractory Multiple Myeloma. Blood (2020) 136:45–6. doi: 10.1182/blood-2020-135845
21. Heuck C, Jethava Y, Khan R, et al. Inhibiting MEK in MAPK Pathway-Activated Myeloma. Leukemia (2016) 30:976–80. doi: 10.1038/leu.2015.208
22. Aksamitiene E, Kiyatkin A, Kholodenko BN. Cross-Talk Between Mitogenic Ras/MAPK and Survival PI3K/Akt Pathways: A Fine Balance. Biochem Soc Trans (2012) 40:139–46. doi: 10.1042/BST20110609
23. Carracedo A, Ma L, Teruya-Feldstein J, et al. Inhibition of Mtorc1 Leads to MAPK Pathway Activation Through a PI3K-Dependent Feedback Loop in Human Cancer. J Clin Invest (2008) 118:3065–74. doi: 10.1172/JCI34739
24. Trudel S, Bahlis NJ, Venner CP, et al. Biomarker Driven Phase II Clinical Trial of Trametinib in Relapsed/Refractory Multiple Myeloma With Sequential Addition of the AKT Inhibitor, GSK2141795 at Time of Disease Progression to Overcome Treatment Failure: A Trial of the Princess Margaret Phase II Consortium. Blood (2016) 128:4526. doi: 10.1182/blood.V128.22.4526.4526
25. Ramakrishnan V, Kumar S. PI3K/AKT/mTOR Pathway in Multiple Myeloma: From Basic Biology to Clinical Promise. Leukemia Lymphoma (2018) 59:2524–34. doi: 10.1080/10428194.2017.1421760
26. Wang L, Lin N, Li Y. The PI3K/AKT Signaling Pathway Regulates ABCG2 Expression and Confers Resistance to Chemotherapy in Human Multiple Myeloma. Oncol Rep (2019) 41:1678–90. doi: 10.3892/or.2019.6968
27. Maiso P, Liu Y, Morgan B, et al. Defining the Role of TORC1/2 in Multiple Myeloma. Blood J Am Soc Hematol (2011) 118:6860–70. doi: 10.1182/blood-2011-03-342394
28. Mayer IA, Arteaga CL. The PI3K/AKT Pathway as a Target for Cancer Treatment. Annu Rev Med (2016) 67:11–28. doi: 10.1146/annurev-med-062913-051343
29. Ismail SI, Mahmoud IS, Msallam MM, Sughayer MA. Hotspot Mutations of PIK3CA and AKT1 Genes are Absent in Multiple Myeloma. Leukemia Res (2010) 34:824–6. doi: 10.1016/j.leukres.2009.11.018
30. Chang H, Qi XY, Claudio J, Zhuang L, Patterson B, Stewart AK. Analysis of PTEN Deletions and Mutations in Multiple Myeloma. Leukemia Res (2006) 30:262–5. doi: 10.1016/j.leukres.2005.07.008
31. Shi Y, Gera J, Hu L, et al. Enhanced Sensitivity of Multiple Myeloma Cells Containing PTEN Mutations to CCI-779. Cancer Res (2002) 62:5027–34.
32. Raje N, Kumar S, Hideshima T, et al. Combination of the mTOR Inhibitor Rapamycin and CC-5013 has Synergistic Activity in Multiple Myeloma. Blood (2004) 104:4188–93. doi: 10.1182/blood-2004-06-2281
33. Frost P, Moatamed F, Hoang B, et al. In Vivo Antitumor Effects of the mTOR Inhibitor CCI-779 Against Human Multiple Myeloma Cells in a Xenograft Model. Blood (2004) 104:4181–7. doi: 10.1182/blood-2004-03-1153
34. Hideshima T, Catley L, Yasui H, et al. Perifosine, an Oral Bioactive Novel Alkylphospholipid, Inhibits Akt and Induces In Vitro and In Vivo Cytotoxicity in Human Multiple Myeloma Cells. Blood (2006) 107:4053–62. doi: 10.1182/blood-2005-08-3434
35. McMillin DW, Ooi M, Delmore J, et al. Antimyeloma Activity of the Orally Bioavailable Dual Phosphatidylinositol 3-Kinase/Mammalian Target of Rapamycin Inhibitor NVP-Bez235. Cancer Res (2009) 69:5835–42. doi: 10.1158/0008-5472.CAN-08-4285
36. Farag SS, Zhang S, Jansak BS, et al. Phase II Trial of Temsirolimus in Patients With Relapsed or Refractory Multiple Myeloma. Leukemia Res (2009) 33:1475–80. doi: 10.1016/j.leukres.2009.01.039
37. Günther A, Baumann P, Burger R, et al. Activity of Everolimus (RAD001) in Relapsed and/or Refractory Multiple Myeloma: A Phase I Study. Haematologica (2015) 100:541. doi: 10.3324/haematol.2014.116269
38. Yee AJ, Hari P, Marcheselli R, et al. Outcomes in Patients With Relapsed or Refractory Multiple Myeloma in a Phase I Study of Everolimus in Combination With Lenalidomide. Br J Haematology (2014) 166:401–9. doi: 10.1111/bjh.12909
39. Ghobrial IM, Weller E, Vij R, et al. Weekly Bortezomib in Combination With Temsirolimus in Relapsed or Relapsed and Refractory Multiple Myeloma: A Multicentre, Phase 1/2, Open-Label, Dose-Escalation Study. Lancet Oncol (2011) 12:263–72. doi: 10.1016/S1470-2045(11)70028-6
40. Shi Y, Yan H, Frost P, Gera J, Lichtenstein A. Mammalian Target of Rapamycin Inhibitors Activate the AKT Kinase in Multiple Myeloma Cells by Up-Regulating the Insulin-Like Growth Factor Receptor/Insulin Receptor Substrate-1/Phosphatidylinositol 3-Kinase Cascade. Mol Cancer Ther (2005) 4:1533–40. doi: 10.1158/1535-7163.MCT-05-0068
41. Hoang B, Frost P, Shi Y, et al. Targeting TORC2 in Multiple Myeloma With a New mTOR Kinase Inhibitor. Blood J Am Soc Hematol (2010) 116:4560–8. doi: 10.1182/blood-2010-05-285726
42. Ghobrial IM, Siegel DS, Vij R, et al. TAK-228 (Formerly MLN 0128), an Investigational Oral Dual TORC 1/2 Inhibitor: A Phase I Dose Escalation Study in Patients With Relapsed or Refractory Multiple Myeloma, non-Hodgkin Lymphoma, or Waldenström's Macroglobulinemia. Am J Hematol (2016) 91:400–5. doi: 10.1002/ajh.24300
43. Goy A, Ribrag V, Varga A, et al. Phase I Expansion Trial of an Oral TORC1/TORC2 Inhibitor (CC-223) in Diffuse Large B-Cell Lymphoma (DLBCL) and Multiple Myeloma (MM). American Society of Clinical Oncology (2013).
44. Jakubowiak AJ, Richardson PG, Zimmerman T, et al. Perifosine Plus Lenalidomide and Dexamethasone in Relapsed and Relapsed/Refractory Multiple Myeloma: A Phase I Multiple Myeloma Research Consortium Study. Br J Haematology (2012) 158:472–80. doi: 10.1111/j.1365-2141.2012.09173.x
45. Richardson PG, Nagler A, Ben-Yehuda D, et al. Randomized, Placebo-Controlled, Phase 3 Study of Perifosine Combined With Bortezomib and Dexamethasone in Patients With Relapsed, Refractory Multiple Myeloma Previously Treated With Bortezomib. eJHaem (2020) 1:94–102. doi: 10.1002/jha2.4
47. Tolcher AW, Patnaik A, Papadopoulos KP, et al. Phase I Study of the MEK Inhibitor Trametinib in Combination With the AKT Inhibitor Afuresertib in Patients With Solid Tumors and Multiple Myeloma. Cancer Chemotherapy Pharmacol (2015) 75:183–9. doi: 10.1007/s00280-014-2615-5
48. Voorhees PM, Spencer A, Sutherland HJ, et al. Novel AKT Inhibitor Afuresertib in Combination With Bortezomib and Dexamethasone Demonstrates Favorable Safety Profile and Significant Clinical Activity in Patients With Relapsed/Refractory Multiple Myeloma. Washington, DC: American Society of Hematology (2013).
51. Liu J, Hamrouni A, Wolowiec D, et al. Plasma Cells From Multiple Myeloma Patients Express B7-H1 (PD-L1) and Increase Expression After Stimulation With IFN-γ and TLR Ligands via a MyD88-, TRAF6-, and MEK-Dependent Pathway. Blood J Am Soc Hematol (2007) 110:296–304. doi: 10.1182/blood-2006-10-051482
52. Hallett WH, Jing W, Drobyski WR, Johnson BD. Immunosuppressive Effects of Multiple Myeloma are Overcome by PD-L1 Blockade. Biol Blood marrow Transplant (2011) 17:1133–45. doi: 10.1016/j.bbmt.2011.03.011
53. Paul B, Kang S, Zheng Z, Kang Y. The Challenges of Checkpoint Inhibition in the Treatment of Multiple Myeloma. Cell Immunol (2018) 334:87–98. doi: 10.1016/j.cellimm.2018.10.003
54. Görgün G, Samur MK, Cowens KB, et al. Lenalidomide Enhances Immune Checkpoint Blockade-Induced Immune Response in Multiple Myeloma. Clin Cancer Res (2015) 21:4607–18. doi: 10.1158/1078-0432.CCR-15-0200
55. Mateos M-V, Blacklock H, Schjesvold F, et al. A Phase 3 Randomized Study of Pembrolizumab (Pembro) Plus Pomalidomide (Pom) and Dexamethasone (Dex) for Relapsed/Refractory Multiple Myeloma (RRMM): KEYNOTE-183. American Society of Clinical Oncology (2018).
56. Lesokhin AM, Ansell SM, Armand P, et al. Nivolumab in Patients With Relapsed or Refractory Hematologic Malignancy: Preliminary Results of a Phase Ib Study. J Clin Oncol (2016) 34:2698. doi: 10.1200/JCO.2015.65.9789
57. Ribrag V, Avigan DE, Green DJ, et al. Phase 1b Trial of Pembrolizumab Monotherapy for Relapsed/Refractory Multiple Myeloma: KEYNOTE-013. Br J Haematology (2019) 186:e41–4. doi: 10.1111/bjh.15888
58. Goodman AM, Castro A, Pyke RM, et al. MHC-I Genotype and Tumor Mutational Burden Predict Response to Immunotherapy. Genome Med (2020) 12:1–13. doi: 10.1186/s13073-020-00743-4
59. Hoang PH, Cornish AJ, Dobbins SE, Kaiser M, Houlston RS. Mutational Processes Contributing to the Development of Multiple Myeloma. Blood Cancer J (2019) 9:1–11. doi: 10.1038/s41408-019-0221-9
60. Walker BA, Wardell CP, Murison A, et al. APOBEC Family Mutational Signatures are Associated With Poor Prognosis Translocations in Multiple Myeloma. Nat Commun (2015) 6:1–11. doi: 10.1038/ncomms7997
61. Miller A, Asmann Y, Cattaneo L, et al. High Somatic Mutation and Neoantigen Burden are Correlated With Decreased Progression-Free Survival in Multiple Myeloma. Blood Cancer J (2017) 7:e612–2. doi: 10.1038/bcj.2017.94
62. Deeks ED. Venetoclax: First Global Approval. Drugs (2016) 76:979–87. doi: 10.1007/s40265-016-0596-x
63. Pettersson M, Jernberg-Wiklund H, Larsson L, et al. Expression of the Bcl-2 Gene in Human Multiple Myeloma Cell Lines and Normal Plasma Cells1992.
64. Touzeau C, Dousset C, Le Gouill S, et al. The Bcl-2 Specific BH3 Mimetic ABT-199: A Promising Targeted Therapy for T (11; 14) Multiple Myeloma. Leukemia (2014) 28:210–2. doi: 10.1038/leu.2013.216
65. Avet-Loiseau H, Attal M, Moreau P, et al. Genetic Abnormalities and Survival in Multiple Myeloma: The Experience of the Intergroupe Francophone Du Myelome. Blood (2007) 109:3489–95. doi: 10.1182/blood-2006-08-040410
66. Punnoose EA, Leverson JD, Peale F, et al. Expression Profile of BCL-2, BCL-XL, and MCL-1 Predicts Pharmacological Response to the BCL-2 Selective Antagonist Venetoclax in Multiple Myeloma Models. Mol Cancer Ther (2016) 15:1132–44. doi: 10.1158/1535-7163.MCT-15-0730
67. Fernandez de Larrea C, Kyle R, Durie B, et al. International Myeloma Working Group: Plasma Cell Leukemia: Consensus Statement on Diagnostic Requirements, Response Criteria and Treatment Recommendations by the International Myeloma Working Group. Leukemia (2013) 27:780–91. doi: 10.1038/leu.2012.336
68. Touzeau C, Maciag P, Amiot M, Moreau P. Targeting Bcl-2 for the Treatment of Multiple Myeloma. Leukemia (2018) 32:1899–907. doi: 10.1038/s41375-018-0223-9
69. Gonsalves WI, Buadi FK, Kumar SK. Combination Therapy Incorporating Bcl-2 Inhibition With Venetoclax for the Treatment of Refractory Primary Plasma Cell Leukemia With T (11; 14). Eur J Haematology (2018) 100:215–7. doi: 10.1111/ejh.12986
70. Jelinek T, Mihalyova J, Kascak M, et al. Single-Agent Venetoclax Induces MRD-Negative Response in Relapsed Primary Plasma Cell Leukemia With T (11; 14). Am J Hematol (2019) 94:E35–7. doi: 10.1002/ajh.25331
71. Matulis SM, Gupta VA, Nooka AK, et al. Dexamethasone Treatment Promotes Bcl-2 Dependence in Multiple Myeloma Resulting in Sensitivity to Venetoclax. Leukemia (2016) 30:1086–93. doi: 10.1038/leu.2015.350
72. Kumar SK, Harrison SJ, Cavo M, et al. Venetoclax or Placebo in Combination With Bortezomib and Dexamethasone in Patients With Relapsed or Refractory Multiple Myeloma (BELLINI): A Randomised, Double-Blind, Multicentre, Phase 3 Trial. Lancet Oncol (2020) 21:1630–42. doi: 10.1016/S1470-2045(20)30525-8
73. Costa LJ, Davies FE, Monohan GP, et al. Phase 2 Study of Venetoclax Plus Carfilzomib and Dexamethasone in Patients With Relapsed/Refractory Multiple Myeloma. Blood Adv (2021). doi: 10.1182/bloodadvances.2020004146
74. Bahlis NJ, Baz R, Harrison SJ, et al. Phase I Study of Venetoclax Plus Daratumumab and Dexamethasone, With or Without Bortezomib, in Patients With Relapsed or Refractory Multiple Myeloma With and Without T (11; 14). J Clin Oncol (2021), JCO. 21.00443. doi: 10.1200/JCO.21.00443
75. Maples KT, Kaufman JL, Gupta VA, et al. Real-World Outcomes of Venetoclax Refractory Multiple Myeloma Patients. American Society of Clinical Oncology (2020).
76. Sidiqi MH, Al Saleh AS, Kumar SK, et al. Venetoclax for the Treatment of Multiple Myeloma: Outcomes Outside of Clinical Trials. American journal of hematology (2021).
77. Szita VR, Mikala G, Fábián J, et al. Efficacy and Safety of Venetoclax Combinations in T (11; 14) Multiple Myeloma: Real World Data Collected From 7 Hungarian Centers. Blood (2020) 136:32–3. doi: 10.1182/blood-2020-139870
78. Regidor B, Goldwater M-S, Wang J, et al. Low Dose Venetoclax in Combination With Bortezomib, Daratumumab, and Dexamethasone for the Treatment of Relapsed/Refractory Multiple Myeloma Patients—a Single-Center Retrospective Study. Ann Hematol (2021), 1–10.
79. Costa LJ, Stadtmauer EA, Morgan G, et al. Phase 2 Study of Venetoclax Plus Carfilzomib and Dexamethasone in Patients With Relapsed/Refractory Multiple Myeloma. Blood (2018) 132:303. doi: 10.1182/blood-2018-99-117026
80. Mateos M-V, Moreau P, Dimopoulos MA, et al. A Phase III, Randomized, Multicenter, Open-Label Study of Venetoclax or Pomalidomide in Combination With Dexamethasone in Patients With T (11; 14)-Positive Relapsed/Refractory Multiple Myeloma. American Society of Clinical Oncology (2020).
81. Ailawadhi S, Chanan-Khan AAA, Chen Z, et al. First-In-Human Study of Lisaftoclax (APG-2575), a Novel BCL-2 Inhibitor (BCL-2i), in Patients (Pts) With Relapsed/Refractory (R/R) CLL and Other Hematologic Malignancies (HMs). Wolters Kluwer Health (2021).
82. Jaksic W, Trudel S, Chang H, et al. Clinical Outcomes in T (4; 14) Multiple Myeloma: A Chemotherapy-Sensitive Disease Characterized by Rapid Relapse and Alkylating Agent Resistance. J Clin Oncol (2005) 23:7069–73. doi: 10.1200/JCO.2005.17.129
83. Chang H, Sloan S, Li D, et al. The T (4; 14) is Associated With Poor Prognosis in Myeloma Patients Undergoing Autologous Stem Cell Transplant. Br J Haematology (2004) 125:64–8. doi: 10.1111/j.1365-2141.2004.04867.x
84. Chan H, Phillips M, Maganti M, et al. Single-Center Experience in Treating Patients With T (4; 14) Multiple Myeloma With and Without Planned Frontline Autologous Stem Cell Transplantation. Clin Lymphoma Myeloma Leukemia (2018) 18:225–34. doi: 10.1016/j.clml.2017.12.009
85. Markham A. Erdafitinib: First Global Approval. Drugs (2019) 79:1017–21. doi: 10.1007/s40265-019-01142-9
86. Grand E, Chase A, Heath C, Rahemtulla A, Cross N. Targeting FGFR3 in Multiple Myeloma: Inhibition of T (4; 14)-Positive Cells by SU5402 and PD173074. Leukemia (2004) 18:962–6. doi: 10.1038/sj.leu.2403347
87. Trudel S, Stewart AK, Rom E, et al. The Inhibitory Anti-FGFR3 Antibody, PRO-001, is Cytotoxic to T (4; 14) Multiple Myeloma Cells. Blood (2006) 107:4039–46. doi: 10.1182/blood-2005-10-4179
88. Qing J, Du X, Chen Y, et al. Antibody-Based Targeting of FGFR3 in Bladder Carcinoma and T (4; 14)-Positive Multiple Myeloma in Mice. J Clin Invest (2009) 119:1216–29. doi: 10.1172/JCI38017
89. Trudel S, Ely S, Farooqi Y, et al. Inhibition of Fibroblast Growth Factor Receptor 3 Induces Differentiation and Apoptosis in T (4; 14) Myeloma. Blood (2004) 103:3521–8. doi: 10.1182/blood-2003-10-3650
90. Scheid C, Reece D, Beksac M, et al. Phase 2 Study of Dovitinib in Patients With Relapsed or Refractory Multiple Myeloma With or Without T (4; 14) Translocation. Eur J Haematology (2015) 95:316–24. doi: 10.1111/ejh.12491
91. Kropff M, Kienast J, Bisping G, et al. An Open-Label Dose-Escalation Study of BIBF 1120 in Patients With Relapsed or Refractory Multiple Myeloma. Anticancer Res (2009) 29:4233–8.
92. Keats JJ, Reiman T, Maxwell CA, et al. In Multiple Myeloma, T (4; 14)(P16; Q32) is an Adverse Prognostic Factor Irrespective of FGFR3 Expression. Blood J Am Soc Hematol (2003) 101:1520–9.
93. Chae YK, Hong F, Vaklavas C, et al. Phase II Study of AZD4547 in Patients With Tumors Harboring Aberrations in the FGFR Pathway: Results From the NCI-MATCH Trial (EAY131) Subprotocol W. J Clin Oncol (2020) 38:2407. doi: 10.1200/JCO.19.02630
94. McInnes C. Progress in the Evaluation of CDK Inhibitors as Anti-Tumor Agents. Drug Discov Today (2008) 13:875–81. doi: 10.1016/j.drudis.2008.06.012
95. Boyd KD, Ross FM, Walker BA, et al. Mapping of Chromosome 1p Deletions in Myeloma Identifies FAM46C at 1p12 and CDKN2C at 1p32. 3 as Being Genes in Regions Associated With Adverse Survival. Clin Cancer Res (2011) 17:7776–84. doi: 10.1158/1078-0432.CCR-11-1791
96. Iriyama N, Hino H, Moriya S, et al. The Cyclin-Dependent Kinase 4/6 Inhibitor, Abemaciclib, Exerts Dose-Dependent Cytostatic and Cytocidal Effects and Induces Autophagy in Multiple Myeloma Cells. Leukemia Lymphoma (2018) 59:1439–50. doi: 10.1080/10428194.2017.1376741
97. Xu Y, Yao Y, Park WD, et al. Enhancing the Immune Surveillance in Multiple Myeloma Via CDK4/6 Inhibition. Blood (2020) 136:33–4. doi: 10.1182/blood-2020-143327
98. Altenburg JD, Farag SS. The Potential Role of PD0332991 (Palbociclib) in the Treatment of Multiple Myeloma. Expert Opin Investigational Drugs (2015) 24:261–71. doi: 10.1517/13543784.2015.993753
99. Menu E, Garcia J, Huang X, et al. A Novel Therapeutic Combination Using PD 0332991 and Bortezomib: Study in the 5T33MM Myeloma Model. Cancer Res (2008) 68:5519–23. doi: 10.1158/0008-5472.CAN-07-6404
100. Huang X, Jayabalan D, Di Liberto M, et al. Inhibition of CDK4/CDK6 Sensitizes Myeloma to IMiD By Reducing the MEIS2 to Cereblon Ratio That Accelerates IKZF1 and IKZF3 Degradation. Washington, DC: American Society of Hematology (2015).
101. Niesvizky R, Badros AZ, Costa LJ, et al. Phase 1/2 Study of Cyclin-Dependent Kinase (CDK) 4/6 Inhibitor Palbociclib (PD-0332991) With Bortezomib and Dexamethasone in Relapsed/Refractory Multiple Myeloma. Leukemia Lymphoma (2015) 56:3320–8. doi: 10.3109/10428194.2015.1030641
102. Lu C, Ward PS, Kapoor GS, et al. IDH Mutation Impairs Histone Demethylation and Results in a Block to Cell Differentiation. Nature (2012) 483:474–8. doi: 10.1038/nature10860
103. Langer C, Ibañez M, Liebisch P, et al. IDH1 and IDH2 Mutations are Not Frequent in Multiple Myeloma. Blood (2010) 116:4992. doi: 10.1182/blood.V116.21.4992.4992
104. Yen K, Bittinger M, Su S, Fantin V. Cancer-Associated IDH Mutations: Biomarker and Therapeutic Opportunities. Oncogene (2010) 29:6409–17. doi: 10.1038/onc.2010.444
105. Walker BA, Mavrommatis K, Wardell CP, et al. Identification of Novel Mutational Drivers Reveals Oncogene Dependencies in Multiple Myeloma. Blood J Am Soc Hematol (2018) 132:587–97. doi: 10.1182/blood-2018-03-840132
106. Licht JD. SETD2: A Complex Role in Blood Malignancy. Blood J Am Soc Hematol (2017) 130:2576–8. doi: 10.1182/blood-2017-10-811927
107. Vikova V, Jourdan M, Robert N, et al. Comprehensive Characterization of the Mutational Landscape in Multiple Myeloma Cell Lines Reveals Potential Drivers and Pathways Associated With Tumor Progression and Drug Resistance. Theranostics (2019) 9:540. doi: 10.7150/thno.28374
108. Mar BG, Chu SH, Kahn JD, et al. SETD2 Alterations Impair DNA Damage Recognition and Lead to Resistance to Chemotherapy in Leukemia. Blood J Am Soc Hematol (2017) 130:2631–41. doi: 10.1182/blood-2017-03-775569
109. Duns G, van den Berg E, van Duivenbode I, et al. Histone Methyltransferase Gene SETD2 is a Novel Tumor Suppressor Gene in Clear Cell Renal Cell Carcinoma. Cancer Res (2010) 70:4287–91. doi: 10.1158/0008-5472.CAN-10-0120
110. Simoes RDM, Shirasaki R, Tang H, et al. Functional Genomic Landscape of Genes With Recurrent Mutations in Multiple Myeloma. Blood (2018) 132:189. doi: 10.1182/blood-2018-189
111. Jennifer Totman JWL, Riera TV, Tang C, Brach D, Motwani V, Alford JS, et al. Discovery of a Selective Inhibitor of the SETD2 Histone Methyltransferase With Potent In Vitro and In Vivo Activity in Preclinical Models of Multiple Myeloma. Eur Hematol Assoc (2021), Abstract S176.
112. Gatalica Z, Xiu J, Swensen J, Vranic S. Molecular Characterization of Cancers With NTRK Gene Fusions. Modern Pathol (2019) 32:147–53. doi: 10.1038/s41379-018-0118-3
113. Cocco E, Scaltriti M, Drilon A. NTRK Fusion-Positive Cancers and TRK Inhibitor Therapy. Nat Rev Clin Oncol (2018) 15:731–47. doi: 10.1038/s41571-018-0113-0
114. Scott LJ. Larotrectinib: First Global Approval. Drugs (2019) 79:201–6. doi: 10.1007/s40265-018-1044-x
115. Drilon A, Laetsch TW, Kummar S, et al. Efficacy of Larotrectinib in TRK Fusion–Positive Cancers in Adults and Children. New Engl J Med (2018) 378:731–9. doi: 10.1056/NEJMoa1714448
116. Foltz SM, Gao Q, Yoon CJ, et al. Evolution and Structure of Clinically Relevant Gene Fusions in Multiple Myeloma. Nat Commun (2020) 11:1–12. doi: 10.1038/s41467-020-16434-y
117. Slomp A, Peperzak V. Role and Regulation of Pro-Survival BCL-2 Proteins in Multiple Myeloma. Front Oncol (2018) 8:533. doi: 10.3389/fonc.2018.00533
118. Wei AH, Roberts AW, Spencer A, et al. Targeting MCL-1 in Hematologic Malignancies: Rationale and Progress. Blood Rev (2020) 44:100672. doi: 10.1016/j.blre.2020.100672
119. Wuilleme-Toumi S, Robillard N, Gomez P, et al. Mcl-1 is Overexpressed in Multiple Myeloma and Associated With Relapse and Shorter Survival. Leukemia (2005) 19:1248–52. doi: 10.1038/sj.leu.2403784
120. Michels J, Obrist F, Vitale I, et al. MCL-1 Dependency of Cisplatin-Resistant Cancer Cells. Biochem Pharmacol (2014) 92:55–61. doi: 10.1016/j.bcp.2014.07.029
121. Shah V, Sherborne AL, Walker BA, et al. Prediction of Outcome in Newly Diagnosed Myeloma: A Meta-Analysis of the Molecular Profiles of 1905 Trial Patients. Leukemia (2018) 32:102–10. doi: 10.1038/leu.2017.179
122. Slomp A, Moesbergen LM, Gong J-N, et al. Multiple Myeloma With 1q21 Amplification is Highly Sensitive to MCL-1 Targeting. Blood Adv (2019) 3:4202–14. doi: 10.1182/bloodadvances.2019000702
123. Bose P, Gandhi V, Konopleva M. Pathways and Mechanisms of Venetoclax Resistance. Leukemia Lymphoma (2017) 58:2026–39. doi: 10.1080/10428194.2017.1283032
124. Caenepeel S, Brown SP, Belmontes B, et al. AMG 176, a Selective MCL1 Inhibitor, is Effective in Hematologic Cancer Models Alone and in Combination With Established Therapies. Cancer Discov (2018) 8:1582–97.
125. Spencer A, Rosenberg AS, Jakubowiak A, et al. A Phase 1, First-in-Human Study of AMG 176, a Selective MCL-1 Inhibitor, in Patients With Relapsed or Refractory Multiple Myeloma. Clin Lymphoma Myeloma Leukemia (2019) 19:e53–4. doi: 10.1016/j.clml.2019.09.081
127. Tron AE, Belmonte MA, Adam A, et al. Discovery of Mcl-1-Specific Inhibitor AZD5991 and Preclinical Activity in Multiple Myeloma and Acute Myeloid Leukemia. Nat Commun (2018) 9:1–14. doi: 10.1038/s41467-018-07551-w
128. Vassilev LT, Vu BT, Graves B, et al. In Vivo Activation of the P53 Pathway by Small-Molecule Antagonists of MDM2. Science (2004) 303:844–8. doi: 10.1126/science.1092472
129. Michael D, Oren M. The P53–Mdm2 Module and the Ubiquitin System. In: Seminars in Cancer Biology. Elsevier (2003). p. 49–58.
130. Herrero AB, Rojas EA, Misiewicz-Krzeminska I, Krzeminski P, Gutiérrez NC. Molecular Mechanisms of P53 Deregulation in Cancer: An Overview in Multiple Myeloma. Int J Mol Sci (2016) 17:2003. doi: 10.3390/ijms17122003
131. Teoh G, Urashima M, Ogata A, et al. MDM2 Protein Overexpression Promotes Proliferation and Survival of Multiple Myeloma Cells. Blood J Am Soc Hematol (1997) 90:1982–92. doi: 10.1182/blood.V90.5.1982
132. Walker BA, Leone PE, Chiecchio L, et al. A Compendium of Myeloma-Associated Chromosomal Copy Number Abnormalities and Their Prognostic Value. Blood J Am Soc Hematol (2010) 116:e56–65. doi: 10.1182/blood-2010-04-279596
133. Sẗhmer T, Chatterjee M, Hildebrandt M, et al. Nongenotoxic Activation of the P53 Pathway as a Therapeutic Strategy for Multiple Myeloma. Blood (2005) 106:3609–17. doi: 10.1182/blood-2005-04-1489
134. Saha MN, Jiang H, Jayakar J, Reece D, Branch DR, Chang H. MDM2 Antagonist Nutlin Plus Proteasome Inhibitor Velcade Combination Displays a Synergistic Anti-Myeloma Activity. Cancer Biol Ther (2010) 9:936–44. doi: 10.4161/cbt.9.11.11882
135. Skalniak L, Kocik J, Polak J, et al. Prolonged Idasanutlin (RG7388) Treatment Leads to the Generation of P53-Mutated Cells. Cancers (2018) 10:396. doi: 10.3390/cancers10110396
136. Kucab JE, Hollstein M, Arlt VM, Phillips DH. Nutlin-3a Selects for Cells Harbouring TP 53 Mutations. Int J Cancer (2017) 140:877–87. doi: 10.1002/ijc.30504
137. Gu D, Wang S, Kuiatse I, et al. Inhibition of the MDM2 E3 Ligase Induces Apoptosis and Autophagy in Wild-Type and Mutant P53 Models of Multiple Myeloma, and Acts Synergistically With ABT-737. PloS One (2014) 9:e103015. doi: 10.1371/journal.pone.0103015
138. Lodé L, Eveillard M, Trichet V, et al. Mutations in TP53 are Exclusively Associated With Del (17p) in Multiple Myeloma. Haematologica (2010) 95:1973. doi: 10.3324/haematol.2010.023697
139. Gluck WL, Gounder MM, Frank R, et al. Phase 1 Study of the MDM2 Inhibitor AMG 232 in Patients With Advanced P53 Wild-Type Solid Tumors or Multiple Myeloma. Investigational New Drugs (2020) 38:831–43. doi: 10.1007/s10637-019-00840-1
140. Morgan GJ, Walker BA, Davies FE. The Genetic Architecture of Multiple Myeloma. Nat Rev Cancer (2012) 12:335–48. doi: 10.1038/nrc3257
Keywords: precision medicine, targeted therapy, multiple myeloma, novel therapies, RAS/MAPK signaling pathway, PI3K - AKT pathway, Bcl-2 inhibitor, p53
Citation: Pan D and Richter J (2022) Where We Stand With Precision Therapeutics in Myeloma: Prosperity, Promises, and Pipedreams. Front. Oncol. 11:819127. doi: 10.3389/fonc.2021.819127
Received: 20 November 2021; Accepted: 27 December 2021;
Published: 20 January 2022.
Edited by:
Roberto Mina, Università degli Studi di Torino, ItalyReviewed by:
Gregorio Barilà, Ospedale dell’ Angelo, ItalyFrancesca Bonello, University of Turin, Italy
Copyright © 2022 Pan and Richter. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Darren Pan, ZGFycmVuLnBhbkBtb3VudHNpbmFpLm9yZw==