 Bo-Zong Shao
Bo-Zong Shao Zhe-Qi Xu
Zhe-Qi Xu Bin-Ze Han†
Bin-Ze Han† Ding-Feng Su
Ding-Feng Su Chong Liu
Chong Liu- Department of Pharmacology, Second Military Medical University, Shanghai, China
Inflammasomes are newly recognized, vital players in innate immunity. The best characterized is the NLRP3 inflammasome, so-called because the NLRP3 protein in the complex belongs to the family of nucleotide-binding and oligomerization domain-like receptors (NLRs) and is also known as “pyrin domain-containing protein 3”. The NLRP3 inflammasome is associated with onset and progression of various diseases, including metabolic disorders, multiple sclerosis, inflammatory bowel disease, cryopyrin-associated periodic fever syndrome, as well as other auto-immune and auto-inflammatory diseases. Several NLRP3 inflammasome inhibitors have been described, some of which show promise in the clinic. The present review will describe the structure and mechanisms of activation of the NLRP3 inflammasome, its association with various auto-immune and auto-inflammatory diseases, and the state of research into NLRP3 inflammasome inhibitors.
Introduction
The mammalian immune system defends against internal and external threats using innate immunity and adaptive immunity (Neill et al., 2010). The innate immune response relies on pattern-recognition receptors (PRRs) to target pathogenic microbes and other endogenous or exogenous pathogens. PRRs are expressed mainly in immune and inflammatory cells such as monocytes, macrophages, neutrophils, and dendritic cells (DCs) (Schroder and Tschopp, 2010; Fullard and O’Reilly, 2015). They present antigens to the adaptive immune system to generate long-lasting protection (Alexandre et al., 2014). Pathogen-associated molecular patterns (PAMPs), which are antigens common to a given group of pathogens (Medzhitov, 2009; Abderrazak et al., 2015b), are normally recognized by at least three PRRs: Toll-like receptors (TLRs), C-type lectins (CTLs), and Galectins (Bourgeois and Kuchler, 2012; Dzopalic et al., 2012). The innate immune system is evolutionarily conserved across vertebrates and invertebrates, which means that both human and animal studies can provide valuable insights into innate immunity (Dai et al., 2015).
A newly identified PRR, first described in detail in 2002, is the inflammasome (Martinon et al., 2002; Gentile et al., 2015; Jorgensen and Miao, 2015; Sanders et al., 2015). Numerous inflammasomes have been identified, including NLRP1, NLRP2, NLRP3, double-stranded DNA (dsDNA) sensors absent in melanoma 2 (AIM2) and NLRC4 (Ozaki et al., 2015). The best characterized is the NLRP3 inflammasome, so named because the NLRP3 protein in the complex belongs to the family of nucleotide-binding and oligomerization domain-like receptors (NLRs) and is also known as “pyrin domain-containing protein 3” (Inoue and Shinohara, 2013b; Eigenbrod and Dalpke, 2015). In addition to the NLRP3 protein, the NLRP3 inflammasome also contains adapter protein apoptosis-associated speck-like protein (ASC) and procaspase-1 (Inoue and Shinohara, 2013a; Ito et al., 2015). Interactions among these three proteins tightly regulate inflammasome function in order to ensure immune activity only when appropriate.
In the absence of immune activators, an internal interaction occurs between the NACHT domain and leucine-rich repeats (LRRs), suppressing the interaction between NLRP3 and ASC, thus preventing assembly of the inflammasome (Inoue and Shinohara, 2013a). In the presence of immune activators such as PAMPs, danger-associated molecular patterns (DAMPs), other exogenous invaders or environmental stress, NLRP3 opens up and allows interaction between the pyrin domains (PYDs) in NLRP3 and ASC. Subsequently the caspase recruitment domain (CARD) of ASC binds to the CARD domain on procaspase-1, giving rise to the NLRP3 inflammasome. Formation of this complex triggers procaspase-1 self-cleavage, generating the active caspase-1 p10/p20 tetramer and inducing the conversion of proinflammatory cytokines interleukin (IL)-1β and IL-18 from their immature “pro” forms to active forms that are secreted. Formation of the inflammasome also triggers a process of inflammation-related cell death termed pyroptosis (Willingham et al., 2009; Schroder and Tschopp, 2010; Zhong et al., 2013a; Jorgensen and Miao, 2015).
Activation Of The Nlrp3 Inflammasome
Models of NLRP3 Inflammasome Activation
The NLRP3 inflammasome is present primarily in immune and inflammatory cells following activation by inflammatory stimuli; these cells include macrophages, monocytes, DCs, and splenic neutrophils (Guarda et al., 2011b; Zhong et al., 2013a). Activation of the NLRP3 inflammasome appears to occur in two steps (Zhong et al., 2013a; Sutterwala et al., 2014; Ozaki et al., 2015; Figure 1). The first step involves a priming or initiating signal in which many PAMPs or DAMPs are recognized by TLRs, leading to activation of nuclear factor kappa B (NF-κB)-mediated signaling, which in turn up-regulates transcription of inflammasome-related components, including inactive NLRP3, proIL-1β, and proIL-18 (Bauernfeind et al., 2009; Franchi et al., 2012, 2014). This priming step is often studied in vitro using lipopolysaccharide (LPS; Park et al., 2015). The second step of inflammasome activation is the oligomerization of NLRP3 and subsequent assembly of NLRP3, ASC, and procaspase-1 into a complex. This triggers the transformation of procaspase-1 to caspase-1, as well as the production and secretion of mature IL-1β and IL-18 (Kim et al., 2015; Ozaki et al., 2015; Rabeony et al., 2015).
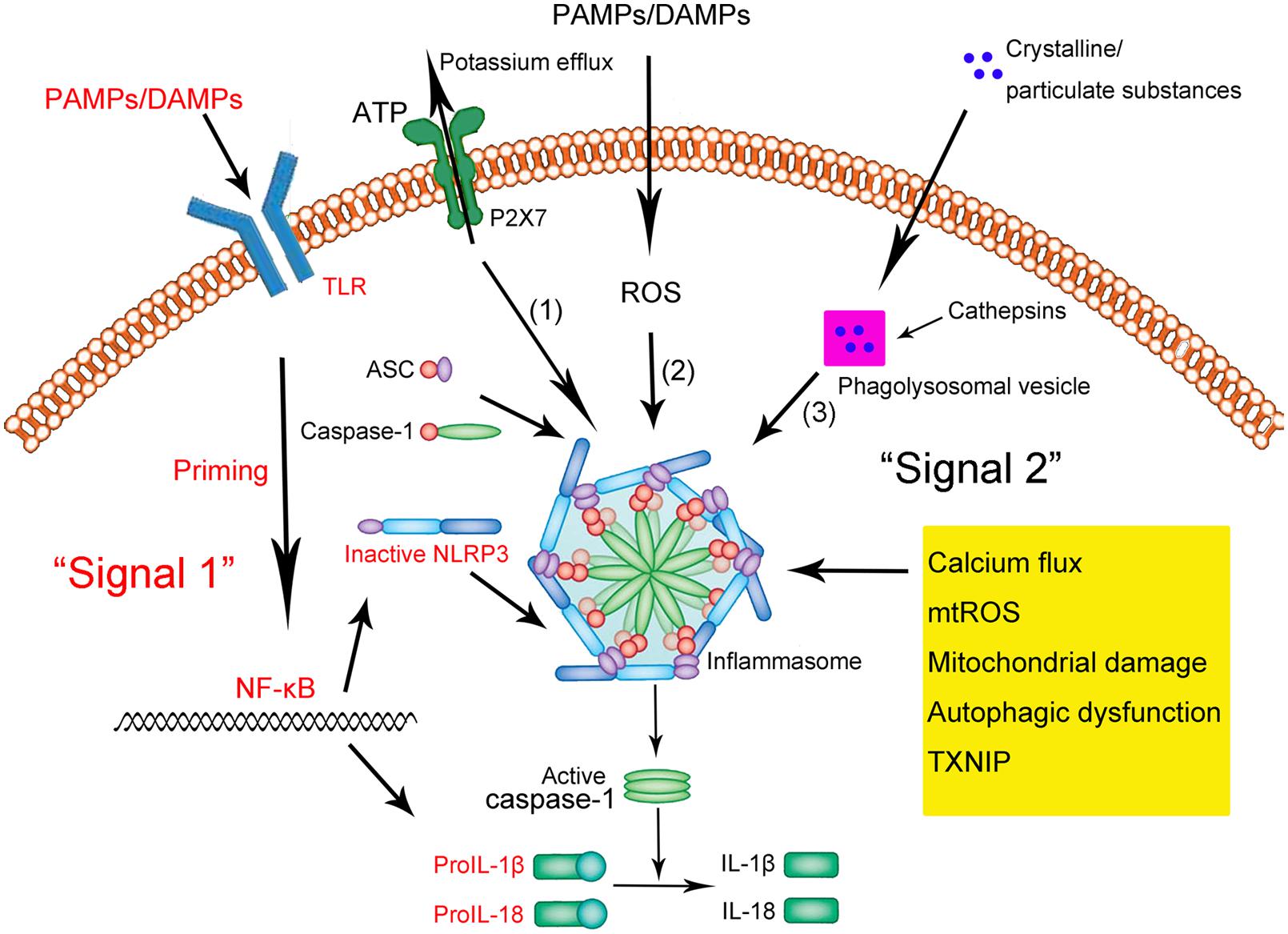
FIGURE 1. Schematic illustration of the NLRP3 inflammasome activation. Upon exposure to pathogen-associated molecular patterns (PAMPs) or danger-associated molecular patterns (DAMPs), Toll-like receptors (TLRs) are phosphorylated and subsequently activate NF-κB. In the nucleus, NF-κB promotes the transcription of NLRP3, proIL-1β, and proIL-18, which, after translation, remain in the cytoplasm in inactive forms. Thus, this signal (depicted in red as “Signal 1”) is a priming event. A subsequent stimulus (shown as “Signal 2” in black) activates the NLRP3 inflammasome by facilitating the oligomerization of inactive NLRP3, apoptosis-associated speck-like protein (ASC), and procaspase-1. This complex, in turn, catalyzes the conversion of procaspase-1 to caspase-1, which contributes to the production and secretion of the mature IL-1β and IL-18. Three models have been proposed to describe the second step of inflammasome activation: (1) Extracellular ATP can induce K+/potassium efflux through a purogenic P2X7-dependent pore, which, leads to the assembly and activation of the NLRP3 inflammasome. Calcium flux is also involved in this process. (2) PAMPs and DAMPs trigger the generation of ROS that promote the assembly and activation of the NLRP3 inflammasome. (3) Phagocytosed environmental irritants form intracellular crystalline or particulate structures leading to lysosomal rupture (magenta box) and release of lysosomal contents like cathepsin B. These induce NLRP3 inflammasome assembly and activation. In addition, other factors and mechanisms have been implicated in the assembly and activation of the NLRP3 inflammasome, including mitochondrial damage, autophagic dysfunction, and thioredoxin-interacting protein (TXNIP).
Three models have been proposed to describe the second step of inflammasome activation, as described in detail by Schroder and Tschopp (2010) (shown in Figure 1). Briefly, all models assume that NLRP3 does not directly interact with exogenous activators, consistent with its ability to sense various pathogens. In the first model, extracellular adenosine triphosphate (ATP), which acts as an NLRP3 agonist, induces K+ efflux through a purogenic P2X7-dependent pore consisting of a pannexin-1 hemichannel. This process leads to NLRP3 inflammasome activation and assembly. Consistent with this model, K+ efflux is a major activator of the NLRP3 inflammasome, while extracellular ATP and pore-forming toxins are the major triggers of IL-1β secretion by the inflammasome (Hari et al., 2014; Liu et al., 2014; Ketelut-Carneiro et al., 2015; Schmid-Burgk et al., 2015). Fluxes of intracellular and endoplasmic reticulum (ER)-related Ca2+ may also activate the NLRP3 inflammasome (Hussen et al., 2012; Zhong et al., 2013b; Shenderov et al., 2014).
In the second model, all known PAMPs and DAMPs, including the activators mentioned above, trigger the generation of reactive oxygen species (ROS), which in turn induce assembly of the NLRP3 inflammasome. For example, damage to NADPH oxidase and other oxidative systems by mitochondrial ROS can activate the inflammasome (van Bruggen et al., 2010; Crane et al., 2014; Lawlor and Vince, 2014; Rajanbabu et al., 2015).
In the third model, assembly and activation of the NLRP3 inflammasome is thought to be triggered by environmental irritants (such as silica, asbestos, amyloid-β, and alum) which form crystalline or particulate structures when engulfed by phagocytes. These aggregates cause lysosomal rupture and release of lysosomal contents via a mechanism mediated by cathepsin B. Consistent with this model, crystalline stimuli such as silica are major triggers of IL-1β secretion by the inflammasome.
Other factors can also activate the NLRP3 inflammasome. These include mitochondrial damage or dysfunction caused by mitochondrial Ca2+ overload (Iyer et al., 2013; Miao et al., 2014; Zhuang et al., 2015), lysosomal disruption (Hornung et al., 2008; Sheedy et al., 2013; Tseng et al., 2013), autophagic dysfunction (Cho et al., 2014; Shao et al., 2014; Jabir et al., 2015) and the activity of thioredoxin-interacting protein (TXNIP; Li et al., 2015; Liu et al., 2015).
The NLRP3 Inflammasome in Disease
While the innate immune response to insults can efficiently protect against disease and death, inappropriate activation of the NLRP3 inflammasome can contribute to the onset and progression of various diseases, particularly age-related diseases such as metabolic disorders and metabolic syndrome (Franceschi et al., 2000; Goldberg and Dixit, 2015). Increased production of IL-1β and IL-18 by the NLRP3 inflammasome contributes to atherosclerotic plaque progression and instability in atherosclerotic patients and animal models (Altaf et al., 2015; Patel et al., 2015; Peng et al., 2015). For example, Patel et al. (2015) showed that genetic ablation of the NLRP3 inflammasome suppressor known as the inhibitor of κB kinase epsilon (IKBKE) enhanced the acute phase response and down-regulated cholesterol metabolism in cultured macrophages and hypercholesterolemic mice. Atherosclerosis and other inflammatory diseases were more severe in animals with the ablation.
Studies in macrophages and animal models have shown that oxidized low-density lipoprotein and cholesterol crystals trigger NLRP3 inflammasome activation (Duewell et al., 2010; Liu et al., 2014). In macrophage and animal models of type II diabetes, hyperglycemia, and free fatty acids trigger inflammasome activation, which harms glucose metabolism and strengthens insulin resistance (Honda et al., 2014; Legrand-Poels et al., 2014; Ruscitti et al., 2015). In macrophage and animal models of uric acid accumulation, monosodium urate crystals activate the NLRP3 inflammasome, causing gout (Hari et al., 2014; Wang et al., 2014; Cleophas et al., 2015). Taken together, these findings suggest that during the progression of many metabolic diseases, the accumulation of abnormal metabolic products activates the NLRP3 inflammasome. Studies in animal models suggest a similar picture in Alzheimer’s disease (Vajjhala et al., 2012; Schnaars et al., 2013; Cho et al., 2014) and obesity induced by a high-fat diet (Haneklaus and O’Neill, 2015; Zhang et al., 2015).
In macrophages and in animal models, studies have also defined a role for the NLRP3 inflammasome in the initiation and development of cerebral and myocardial ischemic diseases, including cerebral ischemia/stroke and myocardial ischemia (Sandanger et al., 2013; Marchetti et al., 2014; Hecker et al., 2015; Ito et al., 2015). Inflammasome activation appears to contribute to post-ischemic inflammation after stroke. For example, Ito et al. (2015) showed that using ibrutinib to inhibit Bruton’s tyrosine kinase (BTK), an essential component of the NLRP3 inflammasome, reduced infarct volume, and neurological damage in a mouse model of cerebral ischemia/reperfusion injury. In addition, it is reported by Hecker et al. (2015) that activation of nicotinic acetylcholine receptors containing subunits α7, α9, and/or α10 inhibited ATP-mediated IL-1β release by human and rat monocytes, helping protect them from collateral damage. NLRP3 inflammasome-related proteins are up-regulated in myocardial fibroblasts following infarction, and this up-regulation may contribute to infarct size in ischemia-reperfusion injury (Sandanger et al., 2013). Consistent with this idea, inhibiting the NLRP3 inflammasome reduces myocardial injury after ischemia-reperfusion in mice (Marchetti et al., 2014).
NLRP3 inflammasome activation has also been linked to various auto-immune and auto-inflammatory diseases. Work from our laboratory and others has demonstrated that NLRP3 inflammasome activation contributes to progression of multiple sclerosis in humans and experimental autoimmune encephalomyelitis (EAE) in animal models (Ming et al., 2002; Jha et al., 2010; Lalor et al., 2011; Inoue et al., 2012a,b; Shao et al., 2014). Severity of multiple sclerosis in patients correlates closely with levels of IL-1β, IL-18, and caspase-1 (Ming et al., 2002; Jha et al., 2010; Lalor et al., 2011); the serum levels of both ILs and of active caspase-1 (p20) are elevated in mice with EAE (Inoue et al., 2012a,b). Studies in macrophages and mouse models of colitis have linked abnormal NLRP3 inflammasome activation with inflammatory bowel disease, including ulcerative colitis and Crohn’s disease (Cheng et al., 2015; Guo et al., 2015; Sun et al., 2015). Polymorphism in the NLRP3 gene is linked to colitis severity and progression in patients (Villani et al., 2009; Lewis et al., 2011), and gain-of-function mutations in the NLRP3 gene that increase production and secretion of IL-1β and IL-18 are associated with cryopyrin-associated periodic fever syndrome (CAPS; Bozkurt et al., 2015; Carta et al., 2015; Zhou et al., 2015). This syndrome comprises several rare hereditary auto-inflammatory diseases in humans and animal models, including familial cold auto-inflammatory syndrome and Muckle–Wells syndrome. Inhibiting IL-1 using specific blocking agents effectively reduces systemic inflammation in CAPS patients (Kuemmerle-Deschner, 2015; Yadlapati and Efthimiou, 2015).
Pharmacological Use Of Nlrp3 Inflammasome Inhibitors
The extensive involvement of the NLRP3 inflammasome in such a range of diseases makes it a highly desirable drug target. Fortunately numerous promising inhibitors of NLRP3 inflammasome activation have been described, several of which are briefly described below together with their pharmacological mechanisms (shown in Table 1).

TABLE 1. Potential mechanisms of several NLRP3 inflammasome inhibitors.
MCC950 and β-Hydroxybutyrate
Two small-molecule inhibitors of the NLRP3 inflammasome were described in groundbreaking reports in Nature Medicine this year (Coll et al., 2015; Youm et al., 2015). Coll et al. (2015) discovered that MCC950, a diarylsulfonylurea-containing compound known to inhibit caspase-1-dependent processing of IL-1β (Perregaux et al., 2001), also inhibits both canonical and non-canonical activation of the NLRP3 inflammasome. MCC950 inhibits secretion of IL-1β and NLRP3-induced ASC oligomerization in mouse and human macrophages. It reduces secretion of IL-1β and IL-18, alleviating the severity of EAE and CAPS in mouse models. Coll et al. (2015) further showed that MCC950 acts specifically on the NLRP3 inflammasome: it does not inhibit the activation of NLRP1, AIM2, or NLRC4 inflammasomes. Baker et al. (2015) have shown that MCC950 inhibits LPS-induced production of IL-1β via a mechanism dependent on the cytoplasmic LPS sensors caspase-4 and caspase-5. Krishnan et al. (2015) demonstrated that hypertension in mice treated with salt and deoxycorticosterone acetate can be reversed by treating them with MCC950, and this reversal depends on the inhibition of inflammasome activation and inflammasome-related IL-1β production.
Youm et al. (2015) discovered that the ketone metabolite β-hydroxybutyrate (BHB), but not acetoacetate or the short-chain fatty acids butyrate and acetate, reduced IL-1β, and IL-18 production by the NLRP3 inflammasome in human monocytes. Like MCC950, BHB appears to block inflammasome activation by inhibiting NLRP3-induced ASC oligomerization. Their in vivo experiments showed that BHB or a ketogenic diet alleviate caspase-1 activation and caspase-1-mediated IL-1β production and secretion, without affecting the activation of NLRC4 or AIM2 inflammasomes. BHB inhibits NLRP3 inflammasome activation independently of AMP-activated protein kinase, ROS, autophagy, or glycolytic inhibition. These studies raise interesting questions about interactions among ketone bodies, metabolic products, and innate immunity. BHB levels increase in response to starvation, caloric restriction, high-intensity exercise, or a low-carbohydrate ketogenic diet (Cotter et al., 2013). Vital organs such as the heart and brain can exploit BHB as an alternative energy source during exercise or caloric deficiency. Future studies should examine how innate immunity, particularly the inflammasome, is influenced by ketones and other alternative metabolic fuels during periods of energy deficiency (Shido et al., 1989; Johnson et al., 2007; McGettrick and O’Neill, 2013; Mercken et al., 2013; Newman and Verdin, 2014).
Although both MCC950 and BHB inhibit NLRP3 inflammasome activation, their mechanisms differ in key respects. BHB inhibits K+ efflux from macrophages, while MCC950 does not. MCC950 inhibits both canonical and non-canonical inflammasome activation, while BHB affects only canonical activation. Nevertheless both inhibitors represent a significant advance toward developing therapies that target IL-1β and IL-18 production by the NLRP3 inflammasome in various diseases (Netea and Joosten, 2015).
Type I Interferon (IFN) and IFN-β
In contrast to these newly described, NLRP3-specific inflammasome inhibitors, type I interferons (IFNs), including IFN-α and IFN-β, have been used for some time to inhibit the NLRP3 and other inflammasomes in various auto-immune and auto-inflammatory diseases. These diseases include multiple sclerosis, systemic-onset juvenile idiopathic arthritis caused by gain-of-function NLRP3 mutations, rheumatic diseases and familial-type Mediterranean fever (Guarda et al., 2011a; Inoue et al., 2012b; Inoue and Shinohara, 2013b; Malhotra et al., 2015; van Kempen et al., 2015). Type I IFNs are produced by specialized immune cells such as macrophages and DCs in response to extracellular stimuli such as bacteria and virus as well as various environmental irritants (Meylan et al., 2006). These IFNs are recognized by the type I IFN receptor (IFNAR), which is a member of the TLR family and is composed of the subunits IFNAR1 and IFNAR2. IFNAR activation involves several proteins, including Janus kinases, tyrosine kinase 2, and several kinds of signal transducers and activators of transcriptions (STATs). However, how type I IFNs affect NLRP3 inflammasome and its production of IL-1β and IL-18 remains unclear (Guarda et al., 2011a), despite numerous studies aimed to improve IFN-based treatments of NLRP3 inflammasome-related diseases. To provide an example of progress in this area, we focus below on studies of IFN therapy against multiple sclerosis in patients and EAE in mice, since type I IFN therapy has been used as a first-line or standard treatment of multiple sclerosis for 15 years (Inoue et al., 2012b).
Malhotra et al. (2015) classified 97 patients with multiple sclerosis into those who responded to IFN-β therapy and those who did not, based on clinico-radiological criteria at 12 and 24 months of treatment. They found that expression of NLRP3 protein and levels of IL-1β were significantly lower among responsive patients who had relapsing-remitting multiple sclerosis than among other patients. Guarda et al. (2011a) found that IL-1β production by primary monocytes was lower in multiple sclerosis patients on IFN-β treatment than in healthy subjects, supporting the value of IFN-β therapy. Studies in mouse bone marrow-derived macrophages by Guarda et al. (2011a) suggest that IFN-β may inhibit IL-1β production through at least two mechanisms. In one pathway, phosphorylation of STAT1 transcription factor leads to repression of NLRP1 and NLRP3 inflammasomes, which in turn inhibits caspase-1-dependent IL-1β maturation. In the second pathway, type I IFNs induce IL-10 production via a STAT-dependent mechanism, and the IL-10 works in an autocrine fashion to reduce levels of pro-IL-1α and pro-IL-1β via a mechanism dependent on STAT3 signaling.
Type I IFN treatment is not effective for all types of multiple sclerosis, and the NLRP3 inflammasome may be a key determinant. Inoue et al. (2012b) conducted studies on mouse primary macrophage cultures as well as EAE mice and concluded that IFN-β therapy is effective only when the NLRP3 inflammasome contributes directly to the disease process. Their studies further showed that IFNAR activation could be inhibited using the suppressor of cytokine signal 1 (SOCS1), which inhibited Rac1 activation and ROS generation, leading in turn to inhibition of NLRP3 inflammasome activity and less severe EAE.
These studies highlight the efficacy of type I IFN therapy and the need for future studies to elucidate the mechanisms of NLRP3 inflammasome inhibition. This work may improve clinical approaches to treating multiple sclerosis and other auto-immune and auto-inflammatory diseases.
Other Kinds of NLRP3 Inflammasome Inhibitors
Several additional ways for inhibiting the NLRP3 inflammasome have opened up in recent years. Autophagy, a self-protective catabolic pathway involving lysosomes, has been shown to inhibit the NLRP3 inflammasome, leading researchers to explore the usefulness of autophagy-inducing treatments (Shao et al., 2014). Chang et al. (2015) showed that the plant polyphenolic compound resveratrol, known to induce autophagy, suppresses mitochondrial damage in macrophages and thereby inhibits NLRP3 inflammasome activation and NLRP3 inflammasome-mediated IL-1β secretion and pyroptosis. Abderrazak et al. (2015a) showed that arglabin inhibits the production and secretion of IL-1β and IL-18 by the NLRP3 inflammasome in a concentration-dependent manner in ApoE-/- mice on a high-fat diet. The reduced IL production translates to less severe atherosclerosis. Those authors reported that arglabin exerts its effects in macrophages by inducing autophagy as well as by reducing inflammation and cholesterol levels.
Cannabinoid receptor 2 (CB2R) is an already demonstrated therapeutic target in inflammation-related diseases (Smoum et al., 2015). Work from our own laboratory (Shao et al., 2014) has shown that autophagy induction may help explain why activation of the anti-inflammatory CB2R leads to inhibition of NLRP3 inflammasome priming and activation in mouse BV2 microglia stimulated with LPS and ATP as well as in a mouse model of EAE. Such CB2R activation reduces the severity of EAE in mice. Thus CB2R agonists similar to the HU-308 used in our work may become an effective therapy for treating NLRP3 inflammasome-related diseases by inducing autophagy.
MicroRNAs may provide another route for inhibiting inflammasomes. These endogenous non-coding RNAs are 20–23 nt long and bind to the 3′ untranslated region (3′ UTR) of protein-coding mRNAs to regulate their translation (Bartel, 2009; Chen and Sun, 2013). MicroRNA-223 binds to a conserved site in the 3′ UTR of the NLRP3 transcript, suppressing protein expression and thereby inhibiting NLRP3 inflammasome priming and IL-1β production (Bauernfeind et al., 2012; Haneklaus et al., 2012; Chen and Sun, 2013). Deficiency in microRNA-223 leads to neutrophilia, spontaneous lung inflammation, and increased susceptibility to endotoxin challenge in mice (Johnnidis et al., 2008; Haneklaus et al., 2013). Several other microRNAs have been reported to be involved in the activation of the NLRP3 inflammasome, including microRNA-155, microRNA-377, and microRNA-133a-1. Reducing the levels of these factors may be useful for treating inflammasome-related disease (Bandyopadhyay et al., 2013; Chen et al., 2015; Wang et al., 2015).
Conclusion
The past decade has witnessed tremendous progress in understanding the structure and activation of the NLRP3 inflammasome, as well as its roles in the initiation and progression of various auto-immune and auto-inflammatory diseases, including metabolic disorders, multiple sclerosis, inflammatory bowel syndrome, and CAPS. Several types of NLRP3 inflammasome inhibitors have been developed and validated in cell culture studies and animal models of NLRP3 inflammasome-related diseases, and type I IFNs have become well established in the clinic. On the other hand, several agents have proven ineffective in clinical settings, and several potential inhibitors require further development, such as autophagy-inducing and microRNA agents. This highlights the need for further research into what pathways activate the NLRP3 inflammasome and can therefore be targeted by appropriate inhibitors. There is still a long way to go toward exploiting NLRP3 inflammasome inhibitors in our fight against diseases.
Author Contributions
B-ZS and Z-QX were in charge of searching all the relative papers and writing this manuscript. B-ZH was in charge of drawing the picture. CL gave her valuable and professional suggestions and guide in organizing and drafting this manuscript. D-FS helped to revise the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgment
This work was supported by a grant from the Shanghai Natural Science Foundation of China (13ZR1448400).
References
Abderrazak, A., Couchie, D., Mahmood, D. F., Elhage, R., Vindis, C., Laffargue, M., et al. (2015a). Anti-inflammatory and antiatherogenic effects of the NLRP3 inflammasome inhibitor arglabin in ApoE2.Ki mice fed a high-fat diet. Circulation 131, 1061–1070. doi: 10.1161/CIRCULATIONAHA.114.013730
Abderrazak, A., Syrovets, T., Couchie, D., El Hadri, K., Friguet, B., Simmet, T., et al. (2015b). NLRP3 inflammasome: from a danger signal sensor to a regulatory node of oxidative stress and inflammatory diseases. Redox Biol. 4, 296–307. doi: 10.1016/j.redox.2015.01.008
Alexandre, Y. O., Cocita, C. D., Ghilas, S., and Dalod, M. (2014). Deciphering the role of DC subsets in MCMV infection to better understand immune protection against viral infections. Front. Microbiol. 5:378. doi: 10.3389/fmicb.2014.00378
Altaf, A., Qu, P., Zhao, Y., Wang, H., Lou, D., and Niu, N. (2015). NLRP3 inflammasome in peripheral blood monocytes of acute coronary syndrome patients and its relationship with statins. Coron. Artery Dis. 26, 409–421. doi: 10.1097/MCA.0000000000000255
Baker, P. J., Boucher, D., Bierschenk, D., Tebartz, C., Whitney, P. G., D’silva, D. B., et al. (2015). NLRP3 inflammasome activation downstream of cytoplasmic LPS recognition by both caspase-4 and caspase-5. Eur. J. Immunol. 45, 2918–2926. doi: 10.1002/eji.201545655
Bandyopadhyay, S., Lane, T., Venugopal, R., Parthasarathy, P. T., Cho, Y., Galam, L., et al. (2013). MicroRNA-133a-1 regulates inflammasome activation through uncoupling protein-2. Biochem. Biophys. Res. Commun. 439, 407–412. doi: 10.1016/j.bbrc.2013.08.056
Bartel, D. P. (2009). MicroRNAs: target recognition and regulatory functions. Cell 136, 215–233. doi: 10.1016/j.cell.2009.01.002
Bauernfeind, F., Rieger, A., Schildberg, F. A., Knolle, P. A., Schmid-Burgk, J. L., and Hornung, V. (2012). NLRP3 inflammasome activity is negatively controlled by miR-223. J. Immunol. 189, 4175–4181. doi: 10.4049/jimmunol.1201516
Bauernfeind, F. G., Horvath, G., Stutz, A., Alnemri, E. S., Macdonald, K., Speert, D., et al. (2009). Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J. Immunol. 183, 787–791. doi: 10.4049/jimmunol.0901363
Bourgeois, C., and Kuchler, K. (2012). Fungal pathogens-a sweet and sour treat for toll-like receptors. Front. Cell. Infect. Microbiol. 2:142. doi: 10.3389/fcimb.2012.00142
Bozkurt, Y., Demir, A., Erman, B., and Gul, A. (2015). Unified modeling of familial mediterranean fever and cryopyrin associated periodic syndromes. Comput. Math. Methods Med. 2015, 893507. doi: 10.1155/2015/893507
Carta, S., Penco, F., Lavieri, R., Martini, A., Dinarello, C. A., Gattorno, M., et al. (2015). Cell stress increases ATP release in NLRP3 inflammasome-mediated autoinflammatory diseases, resulting in cytokine imbalance. Proc. Natl. Acad. Sci. U.S.A. 112, 2835–2840. doi: 10.1073/pnas.1424741112
Chang, Y. P., Ka, S. M., Hsu, W. H., Chen, A., Chao, L. K., Lin, C. C., et al. (2015). Resveratrol inhibits NLRP3 inflammasome activation by preserving mitochondrial integrity and augmenting autophagy. J. Cell. Physiol. 230, 1567–1579. doi: 10.1002/jcp.24903
Chen, S., Smith, B. A., Iype, J., Prestipino, A., Pfeifer, D., Grundmann, S., et al. (2015). MicroRNA-155-deficient dendritic cells cause less severe GVHD through reduced migration and defective inflammasome activation. Blood 126, 103–112. doi: 10.1182/blood-2014-12-617258
Chen, S., and Sun, B. (2013). Negative regulation of NLRP3 inflammasome signaling. Protein Cell 4, 251–258. doi: 10.1007/s13238-013-2128-8
Cheng, Y. L., Song, L. Q., Huang, Y. M., Xiong, Y. W., Zhang, X. A., Sun, H., et al. (2015). Effect of enterohaemorrhagic Escherichia coli O157:H7-specific enterohaemolysin on interleukin-1beta production differs between human and mouse macrophages due to the different sensitivity of NLRP3 activation. Immunology 145, 258–267. doi: 10.1111/imm.12442
Cho, M. H., Cho, K., Kang, H. J., Jeon, E. Y., Kim, H. S., Kwon, H. J., et al. (2014). Autophagy in microglia degrades extracellular beta-amyloid fibrils and regulates the NLRP3 inflammasome. Autophagy 10, 1761–1775. doi: 10.4161/auto.29647
Cleophas, M. C., Crisan, T. O., Lemmers, H., Toenhake-Dijkstra, H., Fossati, G., Jansen, T. L., et al. (2015). Suppression of monosodium urate crystal-induced cytokine production by butyrate is mediated by the inhibition of class I histone deacetylases. Ann. Rheum. Dis. doi: 10.1136/annrheumdis-2014-206258 [Epub ahead of print].
Coll, R. C., Robertson, A. A., Chae, J. J., Higgins, S. C., Munoz-Planillo, R., Inserra, M. C., et al. (2015). A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat. Med. 21, 248–255. doi: 10.1038/nm.3806
Cotter, D. G., Schugar, R. C., and Crawford, P. A. (2013). Ketone body metabolism and cardiovascular disease. Am. J. Physiol. Heart Circ. Physiol. 304, H1060–H1076. doi: 10.1152/ajpheart.00646.2012
Crane, D. D., Bauler, T. J., Wehrly, T. D., and Bosio, C. M. (2014). Mitochondrial ROS potentiates indirect activation of the AIM2 inflammasome. Front. Microbiol. 5:438. doi: 10.3389/fmicb.2014.00438
Dai, L. L., Gao, J. X., Zou, C. G., Ma, Y. C., and Zhang, K. Q. (2015). Mir-233 modulates the unfolded protein response in C. elegans during Pseudomonas aeruginosa infection. PLoS Pathog 11:e1004606. doi: 10.1371/journal.ppat.1004606
Duewell, P., Kono, H., Rayner, K. J., Sirois, C. M., Vladimer, G., Bauernfeind, F. G., et al. (2010). NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 464, 1357–1361. doi: 10.1038/nature08938
Dzopalic, T., Rajkovic, I., Dragicevic, A., and Colic, M. (2012). The response of human dendritic cells to co-ligation of pattern-recognition receptors. Immunol. Res. 52, 20–33. doi: 10.1007/s12026-012-8279-5
Eigenbrod, T., and Dalpke, A. H. (2015). Bacterial RNA: an underestimated stimulus for innate immune responses. J. Immunol. 195, 411–418. doi: 10.4049/jimmunol.1500530
Franceschi, C., Bonafe, M., Valensin, S., Olivieri, F., De Luca, M., Ottaviani, E., et al. (2000). Inflamm-aging. An evolutionary perspective on immunosenescence. Ann. N. Y. Acad. Sci. 908, 244–254. doi: 10.1111/j.1749-6632.2000.tb06651.x
Franchi, L., Eigenbrod, T., Munoz-Planillo, R., Ozkurede, U., Kim, Y. G., Chakrabarti, A., et al. (2014). Cytosolic double-stranded RNA activates the NLRP3 inflammasome via MAVS-induced membrane permeabilization and K+ efflux. J. Immunol. 193, 4214–4222. doi: 10.4049/jimmunol.1400582
Franchi, L., Munoz-Planillo, R., and Nunez, G. (2012). Sensing and reacting to microbes through the inflammasomes. Nat. Immunol. 13, 325–332. doi: 10.1038/ni.2231
Fullard, N., and O’Reilly, S. (2015). Role of innate immune system in systemic sclerosis. Semin. Immunopathol. 37, 511–517. doi: 10.1007/s00281-015-0503-7
Gentile, L. F., Cuenca, A. L., Cuenca, A. G., Nacionales, D. C., Ungaro, R., Efron, P. A., et al. (2015). Improved emergency myelopoiesis and survival in neonatal sepsis by caspase-1/11 ablation. Immunology 145, 300–311. doi: 10.1111/imm.12450
Goldberg, E. L., and Dixit, V. D. (2015). Drivers of age-related inflammation and strategies for healthspan extension. Immunol. Rev. 265, 63–74. doi: 10.1111/imr.12295
Guarda, G., Braun, M., Staehli, F., Tardivel, A., Mattmann, C., Forster, I., et al. (2011a). Type I interferon inhibits interleukin-1 production and inflammasome activation. Immunity 34, 213–223. doi: 10.1016/j.immuni.2011.02.006
Guarda, G., Zenger, M., Yazdi, A. S., Schroder, K., Ferrero, I., Menu, P., et al. (2011b). Differential expression of NLRP3 among hematopoietic cells. J. Immunol. 186, 2529–2534. doi: 10.4049/jimmunol.1002720
Guo, W., Liu, W., Jin, B., Geng, J., Li, J., Ding, H., et al. (2015). Asiatic acid ameliorates dextran sulfate sodium-induced murine experimental colitis via suppressing mitochondria-mediated NLRP3 inflammasome activation. Int. Immunopharmacol. 24, 232–238. doi: 10.1016/j.intimp.2014.12.009
Haneklaus, M., Gerlic, M., Kurowska-Stolarska, M., Rainey, A. A., Pich, D., Mcinnes, I. B., et al. (2012). Cutting edge: miR-223 and EBV miR-BART15 regulate the NLRP3 inflammasome and IL-1beta production. J. Immunol. 189, 3795–3799. doi: 10.4049/jimmunol.1200312
Haneklaus, M., and O’Neill, L. A. (2015). NLRP3 at the interface of metabolism and inflammation. Immunol. Rev. 265, 53–62. doi: 10.1111/imr.12285
Haneklaus, M., O’Neill, L. A., and Coll, R. C. (2013). Modulatory mechanisms controlling the NLRP3 inflammasome in inflammation: recent developments. Curr. Opin. Immunol. 25, 40–45. doi: 10.1016/j.coi.2012.12.004
Hari, A., Zhang, Y., Tu, Z., Detampel, P., Stenner, M., Ganguly, A., et al. (2014). Activation of NLRP3 inflammasome by crystalline structures via cell surface contact. Sci. Rep. 4, 7281. doi: 10.1038/srep07281
Hecker, A., Kullmar, M., Wilker, S., Richter, K., Zakrzewicz, A., Atanasova, S., et al. (2015). Phosphocholine-modified macromolecules and canonical nicotinic agonists inhibit ATP-induced IL-1beta release. J. Immunol. 195, 2325–2334. doi: 10.4049/jimmunol.1400974
Honda, H., Nagai, Y., Matsunaga, T., Okamoto, N., Watanabe, Y., Tsuneyama, K., et al. (2014). Isoliquiritigenin is a potent inhibitor of NLRP3 inflammasome activation and diet-induced adipose tissue inflammation. J. Leukoc. Biol. 96, 1087–1100. doi: 10.1189/jlb.3A0114-005RR
Hornung, V., Bauernfeind, F., Halle, A., Samstad, E. O., Kono, H., Rock, K. L., et al. (2008). Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat. Immunol. 9, 847–856. doi: 10.1038/ni.1631
Hussen, J., Duvel, A., Koy, M., and Schuberth, H. J. (2012). Inflammasome activation in bovine monocytes by extracellular ATP does not require the purinergic receptor P2X7. Dev. Comp. Immunol. 38, 312–320. doi: 10.1016/j.dci.2012.06.004
Inoue, M., and Shinohara, M. L. (2013a). NLRP3 Inflammasome and MS/EAE. Autoimmune Dis. 2013, 859145. doi: 10.1155/2013/859145
Inoue, M., and Shinohara, M. L. (2013b). The role of interferon-beta in the treatment of multiple sclerosis and experimental autoimmune encephalomyelitis – in the perspective of inflammasomes. Immunology 139, 11–18. doi: 10.1111/imm.12081
Inoue, M., Williams, K. L., Gunn, M. D., and Shinohara, M. L. (2012a). NLRP3 inflammasome induces chemotactic immune cell migration to the CNS in experimental autoimmune encephalomyelitis. Proc. Natl. Acad. Sci. U.S.A. 109, 10480–10485. doi: 10.1073/pnas.1201836109
Inoue, M., Williams, K. L., Oliver, T., Vandenabeele, P., Rajan, J. V., Miao, E. A., et al. (2012b). Interferon-beta therapy against EAE is effective only when development of the disease depends on the NLRP3 inflammasome. Sci. Signal. 5, ra38. doi: 10.1126/scisignal.2002767
Ito, M., Shichita, T., Okada, M., Komine, R., Noguchi, Y., Yoshimura, A., et al. (2015). Bruton’s tyrosine kinase is essential for NLRP3 inflammasome activation and contributes to ischaemic brain injury. Nat. Commun. 6, 7360. doi: 10.1038/ncomms8360
Iyer, S. S., He, Q., Janczy, J. R., Elliott, E. I., Zhong, Z., Olivier, A. K., et al. (2013). Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity 39, 311–323. doi: 10.1016/j.immuni.2013.08.001
Jabir, M. S., Hopkins, L., Ritchie, N. D., Ullah, I., Bayes, H. K., Li, D., et al. (2015). Mitochondrial damage contributes to Pseudomonas aeruginosa activation of the inflammasome and is downregulated by autophagy. Autophagy 11, 166–182. doi: 10.4161/15548627.2014.981915
Jha, S., Srivastava, S. Y., Brickey, W. J., Iocca, H., Toews, A., Morrison, J. P., et al. (2010). The inflammasome sensor, NLRP3, regulates CNS inflammation and demyelination via caspase-1 and interleukin-18. J. Neurosci. 30, 15811–15820. doi: 10.1523/JNEUROSCI.4088-10.2010
Johnnidis, J. B., Harris, M. H., Wheeler, R. T., Stehling-Sun, S., Lam, M. H., Kirak, O., et al. (2008). Regulation of progenitor cell proliferation and granulocyte function by microRNA-223. Nature 451, 1125–1129. doi: 10.1038/nature06607
Johnson, J. B., Summer, W., Cutler, R. G., Martin, B., Hyun, D. H., Dixit, V. D., et al. (2007). Alternate day calorie restriction improves clinical findings and reduces markers of oxidative stress and inflammation in overweight adults with moderate asthma. Free Radic. Biol. Med. 42, 665–674. doi: 10.1016/j.freeradbiomed.2006.12.005
Jorgensen, I., and Miao, E. A. (2015). Pyroptotic cell death defends against intracellular pathogens. Immunol. Rev. 265, 130–142. doi: 10.1111/imr.12287
Ketelut-Carneiro, N., Silva, G. K., Rocha, F. A., Milanezi, C. M., Cavalcanti-Neto, F. F., Zamboni, D. S., et al. (2015). IL-18 triggered by the Nlrp3 inflammasome induces host innate resistance in a pulmonary model of fungal infection. J. Immunol. 194, 4507–4517. doi: 10.4049/jimmunol.1402321
Kim, E. H., Park, M. J., Park, S., and Lee, E. S. (2015). Increased expression of the NLRP3 inflammasome components in patients with Behcet’s disease. J. Inflamm. (Lond.) 12, 41. doi: 10.1186/s12950-015-0086-z
Krishnan, S. M., Dowling, J. K., Ling, Y. H., Diep, H., Chan, C. T., Ferens, D., et al. (2015). Inflammasome activity is essential for one kidney/deoxycorticosterone acetate/salt-induced hypertension in mice. Br. J. Pharmacol. doi: 10.1111/bph.13230 [Epub ahead of print].
Kuemmerle-Deschner, J. B. (2015). CAPS – pathogenesis, presentation and treatment of an autoinflammatory disease. Semin. Immunopathol. 37, 377–385. doi: 10.1007/s00281-015-0491-7
Lalor, S. J., Dungan, L. S., Sutton, C. E., Basdeo, S. A., Fletcher, J. M., and Mills, K. H. (2011). Caspase-1-processed cytokines IL-1beta and IL-18 promote IL-17 production by gammadelta and CD4 T cells that mediate autoimmunity. J. Immunol. 186, 5738–5748. doi: 10.4049/jimmunol.1003597
Lawlor, K. E., and Vince, J. E. (2014). Ambiguities in NLRP3 inflammasome regulation: is there a role for mitochondria? Biochim. Biophys. Acta 1840, 1433–1440. doi: 10.1016/j.bbagen.2013.08.014
Legrand-Poels, S., Esser, N., L’homme, L., Scheen, A., Paquot, N., and Piette, J. (2014). Free fatty acids as modulators of the NLRP3 inflammasome in obesity/type 2 diabetes. Biochem. Pharmacol. 92, 131–141. doi: 10.1016/j.bcp.2014.08.013
Lewis, G. J., Massey, D. C., Zhang, H., Bredin, F., Tremelling, M., Lee, J. C., et al. (2011). Genetic association between NLRP3 variants and Crohn’s disease does not replicate in a large UK panel. Inflamm. Bowel Dis. 17, 1387–1391. doi: 10.1002/ibd.21499
Li, Y., Yang, J., Chen, M. H., Wang, Q., Qin, M. J., Zhang, T., et al. (2015). Ilexgenin A inhibits endoplasmic reticulum stress and ameliorates endothelial dysfunction via suppression of TXNIP/NLRP3 inflammasome activation in an AMPK dependent manner. Pharmacol. Res. 99, 101–115. doi: 10.1016/j.phrs.2015.05.012
Liu, W., Gu, J., Qi, J., Zeng, X. N., Ji, J., Chen, Z. Z., et al. (2015). Lentinan exerts synergistic apoptotic effects with paclitaxel in A549 cells via activating ROS-TXNIP-NLRP3 inflammasome. J. Cell. Mol. Med. 19, 1949–1955. doi: 10.1111/jcmm.12570
Liu, W., Yin, Y., Zhou, Z., He, M., and Dai, Y. (2014). OxLDL-induced IL-1 beta secretion promoting foam cells formation was mainly via CD36 mediated ROS production leading to NLRP3 inflammasome activation. Inflamm. Res. 63, 33–43. doi: 10.1007/s00011-013-0667-3
Malhotra, S., Rio, J., Urcelay, E., Nurtdinov, R., Bustamante, M. F., Fernandez, O., et al. (2015). NLRP3 inflammasome is associated with the response to IFN-beta in patients with multiple sclerosis. Brain 138, 644–652. doi: 10.1093/brain/awu388
Marchetti, C., Chojnacki, J., Toldo, S., Mezzaroma, E., Tranchida, N., Rose, S. W., et al. (2014). A novel pharmacologic inhibitor of the NLRP3 inflammasome limits myocardial injury after ischemia-reperfusion in the mouse. J. Cardiovasc. Pharmacol. 63, 316–322. doi: 10.1097/FJC.0000000000000053
Martinon, F., Burns, K., and Tschopp, J. (2002). The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell 10, 417–426. doi: 10.1016/S1097-2765(02)00599-3
McGettrick, A. F., and O’Neill, L. A. (2013). How metabolism generates signals during innate immunity and inflammation. J. Biol. Chem. 288, 22893–22898. doi: 10.1074/jbc.R113.486464
Medzhitov, R. (2009). Approaching the asymptote: 20 years later. Immunity 30, 766–775. doi: 10.1016/j.immuni.2009.06.004
Mercken, E. M., Crosby, S. D., Lamming, D. W., Jebailey, L., Krzysik-Walker, S., Villareal, D. T., et al. (2013). Calorie restriction in humans inhibits the PI3K/AKT pathway and induces a younger transcription profile. Aging Cell 12, 645–651. doi: 10.1111/acel.12088
Meylan, E., Tschopp, J., and Karin, M. (2006). Intracellular pattern recognition receptors in the host response. Nature 442, 39–44. doi: 10.1038/nature04946
Miao, H., Ou, J., Ma, Y., Guo, F., Yang, Z., Wiggins, M., et al. (2014). Macrophage CGI-58 deficiency activates ROS-inflammasome pathway to promote insulin resistance in mice. Cell Rep. 7, 223–235. doi: 10.1016/j.celrep.2014.02.047
Ming, X., Li, W., Maeda, Y., Blumberg, B., Raval, S., Cook, S. D., et al. (2002). Caspase-1 expression in multiple sclerosis plaques and cultured glial cells. J. Neurol. Sci. 197, 9–18. doi: 10.1016/S0022-510X(02)00030-8
Neill, D. R., Wong, S. H., Bellosi, A., Flynn, R. J., Daly, M., Langford, T. K., et al. (2010). Nuocytes represent a new innate effector leukocyte that mediates type-2 immunity. Nature 464, 1367–1370. doi: 10.1038/nature08900
Netea, M. G., and Joosten, L. A. (2015). Inflammasome inhibition: putting out the fire. Cell Metab. 21, 513–514. doi: 10.1016/j.cmet.2015.03.012
Newman, J. C., and Verdin, E. (2014). Ketone bodies as signaling metabolites. Trends Endocrinol. Metab. 25, 42–52. doi: 10.1016/j.tem.2013.09.002
Ozaki, E., Campbell, M., and Doyle, S. L. (2015). Targeting the NLRP3 inflammasome in chronic inflammatory diseases: current perspectives. J. Inflamm. Res. 8, 15–27. doi: 10.2147/JIR.S51250
Park, J. H., Jeong, S. Y., Choi, A. J., and Kim, S. J. (2015). Lipopolysaccharide directly stimulates Th17 differentiation in vitro modulating phosphorylation of RelB and NF-kappaB1. Immunol. Lett. 165, 10–19. doi: 10.1016/j.imlet.2015.03.003
Patel, M. N., Bernard, W. G., Milev, N. B., Cawthorn, W. P., Figg, N., Hart, D., et al. (2015). Hematopoietic IKBKE limits the chronicity of inflammasome priming and metaflammation. Proc. Natl. Acad. Sci. U.S.A. 112, 506–511. doi: 10.1073/pnas.1414536112
Peng, K., Liu, L., Wei, D., Lv, Y., Wang, G., Xiong, W., et al. (2015). P2X7R is involved in the progression of atherosclerosis by promoting NLRP3 inflammasome activation. Int. J. Mol. Med. 35, 1179–1188. doi: 10.3892/ijmm.2015.2129
Perregaux, D. G., Mcniff, P., Laliberte, R., Hawryluk, N., Peurano, H., Stam, E., et al. (2001). Identification and characterization of a novel class of interleukin-1 post-translational processing inhibitors. J. Pharmacol. Exp. Ther. 299, 187–197.
Rabeony, H., Pohin, M., Vasseur, P., Petit-Paris, I., Jegou, J. F., Favot, L., et al. (2015). IMQ-induced skin inflammation in mice is dependent on IL-1R1 and MyD88 signaling but independent of the NLRP3 inflammasome. Eur. J. Immunol. 45, 2847–2857. doi: 10.1002/eji.201445215
Rajanbabu, V., Galam, L., Fukumoto, J., Enciso, J., Tadikonda, P., Lane, T. N., et al. (2015). Genipin suppresses NLRP3 inflammasome activation through uncoupling protein-2. Cell. Immunol. 297, 40–45. doi: 10.1016/j.cellimm.2015.06.002
Ruscitti, P., Cipriani, P., Di Benedetto, P., Liakouli, V., Berardicurti, O., Carubbi, F., et al. (2015). Monocytes from patients with rheumatoid arthritis and type 2 diabetes mellitus display an increased production of interleukin (IL)-1beta via the nucleotide-binding domain and leucine-rich repeat containing family pyrin 3(NLRP3)-inflammasome activation: a possible implication for therapeutic decision in these patients. Clin. Exp. Immunol. 182, 35–44. doi: 10.1111/cei.12667
Sandanger, O., Ranheim, T., Vinge, L. E., Bliksoen, M., Alfsnes, K., Finsen, A. V., et al. (2013). The NLRP3 inflammasome is up-regulated in cardiac fibroblasts and mediates myocardial ischaemia-reperfusion injury. Cardiovasc. Res. 99, 164–174. doi: 10.1093/cvr/cvt091
Sanders, M. G., Parsons, M. J., Howard, A. G., Liu, J., Fassio, S. R., Martinez, J. A., et al. (2015). Single-cell imaging of inflammatory caspase dimerization reveals differential recruitment to inflammasomes. Cell Death Dis. 6, e1813. doi: 10.1038/cddis.2015.186
Schmid-Burgk, J. L., Gaidt, M. M., Schmidt, T., Ebert, T. S., Bartok, E., and Hornung, V. (2015). Caspase-4 mediates non-canonical activation of the NLRP3 inflammasome in human myeloid cells. Eur. J. Immunol. 45, 2911–2917. doi: 10.1002/eji.201545523
Schnaars, M., Beckert, H., and Halle, A. (2013). Assessing beta-amyloid-induced NLRP3 inflammasome activation in primary microglia. Methods Mol. Biol. 1040, 1–8. doi: 10.1007/978-1-62703-523-1_1
Schroder, K., and Tschopp, J. (2010). The inflammasomes. Cell 140, 821–832. doi: 10.1016/j.cell.2010.01.040
Shao, B. Z., Wei, W., Ke, P., Xu, Z. Q., Zhou, J. X., and Liu, C. (2014). Activating cannabinoid receptor 2 alleviates pathogenesis of experimental autoimmune encephalomyelitis via activation of autophagy and inhibiting NLRP3 inflammasome. CNS Neurosci. Ther. 20, 1021–1028. doi: 10.1111/cns.12349
Sheedy, F. J., Grebe, A., Rayner, K. J., Kalantari, P., Ramkhelawon, B., Carpenter, S. B., et al. (2013). CD36 coordinates NLRP3 inflammasome activation by facilitating intracellular nucleation of soluble ligands into particulate ligands in sterile inflammation. Nat. Immunol. 14, 812–820. doi: 10.1038/ni.2639
Shenderov, K., Riteau, N., Yip, R., Mayer-Barber, K. D., Oland, S., Hieny, S., et al. (2014). Cutting edge: endoplasmic reticulum stress licenses macrophages to produce mature IL-1beta in response to TLR4 stimulation through a caspase-8- and TRIF-dependent pathway. J. Immunol. 192, 2029–2033. doi: 10.4049/jimmunol.1302549
Shido, O., Nagasaka, T., and Watanabe, T. (1989). Blunted febrile response to intravenous endotoxin in starved rats. J. Appl. Physiol. 67, 963–969.
Smoum, R., Baraghithy, S., Chourasia, M., Breuer, A., Mussai, N., Attar-Namdar, M., et al. (2015). CB2 cannabinoid receptor agonist enantiomers HU-433 and HU-308: an inverse relationship between binding affinity and biological potency. Proc. Natl. Acad. Sci. U.S.A. 112, 8774–8779. doi: 10.1073/pnas.1503395112
Sun, Y., Zhao, Y., Yao, J., Zhao, L., Wu, Z., Wang, Y., et al. (2015). Wogonoside protects against dextran sulfate sodium-induced experimental colitis in mice by inhibiting NF-kappaB and NLRP3 inflammasome activation. Biochem. Pharmacol. 94, 142–154. doi: 10.1016/j.bcp.2015.02.002
Sutterwala, F. S., Haasken, S., and Cassel, S. L. (2014). Mechanism of NLRP3 inflammasome activation. Ann. N. Y. Acad. Sci. 1319, 82–95. doi: 10.1111/nyas.12458
Tseng, W. A., Thein, T., Kinnunen, K., Lashkari, K., Gregory, M. S., D’amore, P. A., et al. (2013). NLRP3 inflammasome activation in retinal pigment epithelial cells by lysosomal destabilization: implications for age-related macular degeneration. Invest. Ophthalmol. Vis. Sci. 54, 110–120. doi: 10.1167/iovs.12-10655
Vajjhala, P. R., Mirams, R. E., and Hill, J. M. (2012). Multiple binding sites on the pyrin domain of ASC protein allow self-association and interaction with NLRP3 protein. J. Biol. Chem. 287, 41732–41743. doi: 10.1074/jbc.M112.381228
van Bruggen, R., Koker, M. Y., Jansen, M., Van Houdt, M., Roos, D., Kuijpers, T. W., et al. (2010). Human NLRP3 inflammasome activation is Nox1-4 independent. Blood 115, 5398–5400. doi: 10.1182/blood-2009-10-250803
van Kempen, T. S., Wenink, M. H., Leijten, E. F., Radstake, T. R., and Boes, M. (2015). Perception of self: distinguishing autoimmunity from autoinflammation. Nat. Rev. Rheumatol. 11, 483–492. doi: 10.1038/nrrheum.2015.60
Villani, A. C., Lemire, M., Fortin, G., Louis, E., Silverberg, M. S., Collette, C., et al. (2009). Common variants in the NLRP3 region contribute to Crohn’s disease susceptibility. Nat. Genet. 41, 71–76. doi: 10.1038/ng.285
Wang, W., Ding, X. Q., Gu, T. T., Song, L., Li, J. M., Xue, Q. C., et al. (2015). Pterostilbene and allopurinol reduce fructose-induced podocyte oxidative stress and inflammation via microRNA-377. Free Radic. Biol. Med. 83, 214–226. doi: 10.1016/j.freeradbiomed.2015.02.029
Wang, Y., Viollet, B., Terkeltaub, R., and Liu-Bryan, R. (2014). AMP-activated protein kinase suppresses urate crystal-induced inflammation and transduces colchicine effects in macrophages. Ann. Rheum. Dis. doi: 10.1136/annrheumdis-2014-206074 [Epub ahead of print].
Willingham, S. B., Allen, I. C., Bergstralh, D. T., Brickey, W. J., Huang, M. T., Taxman, D. J., et al. (2009). NLRP3 (NALP3, Cryopyrin) facilitates in vivo caspase-1 activation, necrosis, and HMGB1 release via inflammasome-dependent and -independent pathways. J. Immunol. 183, 2008–2015. doi: 10.4049/jimmunol.0900138
Yadlapati, S., and Efthimiou, P. (2015). Impact of IL-1 inhibition on fatigue associated with autoinflammatory syndromes. Mod. Rheumatol. 1–6. doi: 10.3109/14397595.2015.1069459 [Epub ahead of print].
Youm, Y. H., Nguyen, K. Y., Grant, R. W., Goldberg, E. L., Bodogai, M., Kim, D., et al. (2015). The ketone metabolite beta-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease. Nat. Med. 21, 263–269. doi: 10.1038/nm.3804
Zhang, Y., Li, Q., Rao, E., Sun, Y., Grossmann, M. E., Morris, R. J., et al. (2015). Epidermal Fatty Acid binding protein promotes skin inflammation induced by high-fat diet. Immunity 42, 953–964. doi: 10.1016/j.immuni.2015.04.016
Zhong, Y., Kinio, A., and Saleh, M. (2013a). Functions of NOD-like receptors in human diseases. Front. Immunol. 4:333. doi: 10.3389/fimmu.2013.00333
Zhong, Z., Zhai, Y., Liang, S., Mori, Y., Han, R., Sutterwala, F. S., et al. (2013b). TRPM2 links oxidative stress to NLRP3 inflammasome activation. Nat. Commun. 4, 1611. doi: 10.1038/ncomms2608
Zhou, Q., Aksentijevich, I., Wood, G. M., Walts, A. D., Hoffmann, P., Remmers, E. F., et al. (2015). Brief report: cryopyrin-associated periodic syndrome caused by a myeloid-restricted somatic NLRP3 mutation. Arthritis Rheumatol. 67, 2482–2486. doi: 10.1002/art.39190
Keywords: NLRP3 inflammasome, inhibitor, autophagy, MCC950, BHB, interferon
Citation: Shao B-Z, Xu Z-Q, Han B-Z, Su D-F and Liu C (2015) NLRP3 inflammasome and its inhibitors: a review. Front. Pharmacol. 6:262. doi: 10.3389/fphar.2015.00262
Received: 05 September 2015; Accepted: 22 October 2015;
Published: 05 November 2015.
Edited by:
Wenliang Song, Bridgeport Hospital and Yale University, USAReviewed by:
Lydia E. Matesic, University of South Carolina, USAXue Liang, University of Pennsylvania, USA
Copyright © 2015 Shao, Xu, Han, Su and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chong Liu, d2FubGMyMDA0QGFsaXl1bi5jb20=
†These authors have contributed equally to this work.