 Wenhao Jiang
Wenhao Jiang Tricia Z. King
Tricia Z. King Jessica A. Turner
Jessica A. Turner- 1Department of Psychology and the Neuroscience Institute, Georgia State University, Atlanta, GA, United States
- 2Mind Research Network, Albuquerque, NM, United States
Current diagnoses of schizophrenia and related psychiatric disorders are classified by phenomenological principles and clinical descriptions while ruling out other symptoms and conditions. Specific biomarkers are needed to assist the current diagnostic system. However, complicated gene and environment interactions induce great disease heterogeneity. This unclear etiology and heterogeneity raise difficulties in distinguishing schizophrenia-related effects. Simultaneously, the overlap in symptoms, genetic variations, and brain alterations in schizophrenia and related psychiatric disorders raises similar difficulties in determining disease-specific effects. Imaging genetics is a unique methodology to assess the impact of genetic factors on both brain structure and function. More importantly, imaging genetics builds a bridge to understand the behavioral and clinical implications of genetics and neuroimaging. By characterizing and quantifying the brain measures affected in psychiatric disorders, imaging genetics is contributing to identifying potential biomarkers for schizophrenia and related disorders. To date, candidate gene analysis, genome-wide association studies, polygenetic risk score analysis, and large-scale collaborative studies have made contributions to the understanding of schizophrenia with the potential to serve as biomarkers. Despite limitations, imaging genetics remains promising as more aggregative, clustering methods and imaging genetics-compatible clinical assessments are employed in future studies. We review imaging genetics’ contribution to our understanding of the heterogeneity within schizophrenia and the commonalities across schizophrenia and other diagnostic borders, and we will discuss whether imaging genetics is ready to form its own diagnostic system.
Introduction
The current diagnosis of schizophrenia and psychiatric disorders is mainly based on phenomenological observation and clinical descriptions. Although these descriptions are reliable, they are not established on valid pathological bases (1). The heterogeneity of the symptoms, treatment response, and outcomes implies that there are different subtypes within schizophrenia, while phenomenological observation fails to generate precise subgroups revealing etiological and pathological differences (2). Additionally, similar psychotic symptoms aggregate in different disorders and in families. Behind this aggregation, shared biological mechanisms including genetics and neurophysiology are found (1). These findings suggest that the boundaries of psychiatric disorders are merging beyond the traditional categorical diagnostic system. The precise subgroups and disorder boundaries may optimize treatment and prognosis, and research-based biomarkers may help to fulfill this goal. Importantly, efforts have already been made as part of the Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition (DSM-5).
Combining genetics and imaging to assess accumulating genetic variations on brain function and morphometry has become the integrated research method known as imaging genetics (3). Imaging genetics not only serves as a tool to understand the impact of genetic variations on both structural and functional brain, but it also enables researchers to capture the behavioral implication of those genes and associated brain alterations (4). Importantly, imaging genetics characterizes different pathways from genes, imaging, and behavior data. Its quantified findings make it possible to contribute to the currently unknown map of future diagnosis (5). The common technologies in imaging genetic include candidate gene analysis, genome-wide association study (GWAS) using imaging phenotypes, polygenic approaches (polygenic scores, pathway analysis, and multivariate methods), and developing novel approaches (6–8). We focus in this paper on the subset of imaging genetics that focuses on the relationships from gene to brain to behavior, which have generally focused on common variants in the single nucleotide polymorphisms (SNPs).
Various genetically related brain abnormalities have been revealed in SZ. SZ patients generally show smaller brain volume, overall reductions in gray matter in fronto-temporal, thalamo-cortical, and subcortical-limbic circuits and enlargement of ventricles (9). These brain alterations induced in partly by genetic variations (10) are bridging the gap between gene and the phenotype and even clinical symptoms of SZ (see Figure 1) (11). It is encouraging that some shared genetics, imaging, and imaging genetics findings have been recognized across SZ, bipolar disorder (BD), and disorders under other categories. At the same time, imaging and genetics are helping to form subtypes with different mechanisms in SZ.
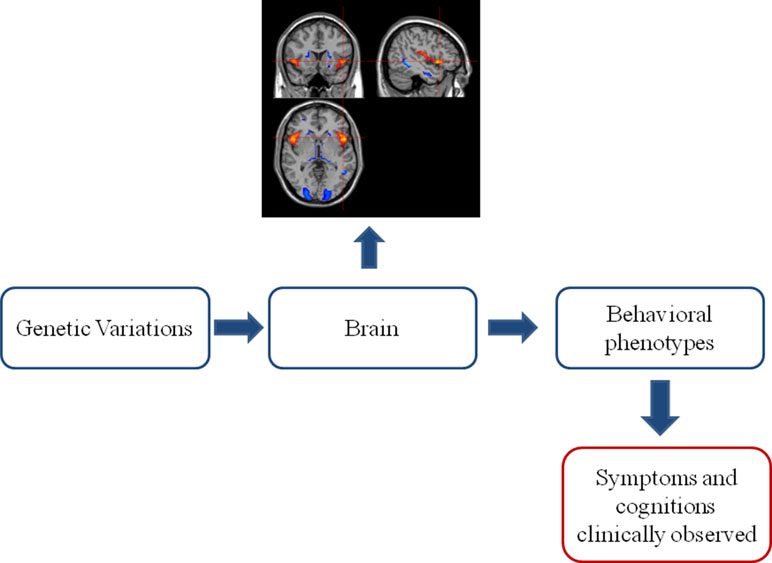
Figure 1 The classic “bottom–up” model in imaging genetics. Genetic variations acclimate their influences on the brain. The brain alterations further develop into behavioral phenotype changes, which can be clinically observed as symptoms and cognitive impairments. This observed clinical profile established the base of current phenomenological diagnostic system of psychiatric disorders.
In this paper, we review the major imaging genetics findings on SZ with closely related psychotic disorders with an eye toward the following questions: 1) to date, what contribution have genetics, neuroimaging, and imaging genetics made to our understanding of the heterogeneity of SZ and the boundaries among psychiatric disorders and 2) whether imaging genetics is ready to form its own diagnostic system.
Traditional and Current Diagnoses of SZ
Feighner and colleagues published the criteria for highly reproducible diagnoses based on behavioral observation in 1972 (12). From this historical view, clinical description, laboratory studies, delineation from other disorders, follow-up studies with retreatment response, and family studies are considered as major theoretical bases for validating a diagnosis (13). However, the follow-up study and treatment response may be questioned for whether they could validate the diagnosis, per se. Antipsychotics are a major choice not only for SZ spectrum but also for depression and BD.
Accumulating new biological understanding does not always agree with the criteria and classification proposed at Feighner’s time. It is now accepted that family coaggregation implies shared abnormal genetic markers and mechanism in the family line. For example, SZ, BD, and schizoaffective disorder (SAD) are in different diagnostic categories, but observations of diagnoses in families of patients showed significant overlap among them, which is still being studied to explore the genetic background (14). Thus, the traditional methods for determining the diagnostic category boundaries are not sufficient.
Genetics and its Impact on the Diagnostic Problem
Genetic Overlap Among SZ and Other Psychiatric Disorders
Genes contribute greatly to the etiology of SZ, and meta-analysis in SZ twin study shows a heritability around 80% (15). Val158Met single polymorphism (SNP) of catechol-O-methyltransferase (COMT), the Val66Met SNP of brain-derived neurotrophic factor (BDNF), and the Ser704Cys SNP of disrupted-in-SZ 1 (DISC1) is the most well-known gene alteration examined by candidate gene analysis (7). The first few reports of GWAS, in contrast, demonstrated several loci associated with SZ including Zinc finger protein 804A (ZNF804A), neurogranin (NRGN), and the major histocompatibility complex (MHC) region. More recent GWAS studies with increased sample size discovered more SZ related loci (16), and some of these loci are shared by BD and other psychiatric disorders (17–25).
SZ and BD are often studied together to elucidate the genetic overlap and disorder boundaries. A genetic correlation around 0.6 is suggested by family, twin, and adoption study (26). However, applying a hierarchical or nonhierarchical diagnostic system has provided conflicting co-occurrence results at the same time (27). SAD is often included in the study of SZ and BD and that genetic relationship could also be potentially affected (28).
In addition to epidemiological evidence, the GWAS study has brought more insight into the actual genetic overlap. ZNF804A is the first discovered marker that may increase the risk for both SZ and BD, and meta-analysis has supported its role (29). The combined SZ and BD GWAS study from Psychiatric Genomics Consortium (PGC) has identified calcium voltage-gated channel subunit alpha1 C (CACNA1C), ankyrin-3(ANK3) and inter-alpha-trypsin inhibitor heavy chain 3–4 (ITIH3-ITIH4) as risk for both disorders (30, 31). Later by introducing pleiotropy-informed conditional false discovery rate, 14 loci were associated with both disorders, and CACNA1C and ITIH4 were identified again (32). PGC’s diagnostic specificity of five disorders analyses has also shown 5′-nucleotidase, cytosolic II (NT5C2), and coiled-coil domain containing 68 (CCDC68) is associated with both disorders (33). The combined GWAS studies will continue to reveal more important loci, but the functional implications and roles of these distinct genes in SZ and BD will need further investigation.
Another idea is using a polygenetic method to combine and count the accumulating effects of a large number of loci, which may or may not reach the GWAS threshold for significance. Again in the PGC study, the cross-disorder group stated SZ and BD were affected by genetic correlation of 0.68 based on their common SNPs (33). Additional polygenic studies blur the distinction across categories and indicate a broad genetic mechanism for these psychiatric disorders (34–36).
However, there is also genetic evidence showing distinctions between SZ and BD (37). Large and rare copy number variations (CNV) have been identified in SZ and certain developmental disorders, but less consistently in BD. In addition, Sz pathogenic CNV carriers showed reduced subcortical regions including thalamus, putamen, pallidum, hippocampus, and accumbens, which were previously identified in Sz participants (38). This finding is consistent with the diagnosis hierarchy, by which BD is only diagnosed with the absence of SZ and developmental disorders.
Genetics Helps Reveal Heterogeneity and Future Subtypes of SZ
Many researchers have tried to provide genetic explanations for SZ’s heterogeneities. Arnedo and colleagues made a promising attempt trying to uncover the hidden genetic architecture of different subtypes of SZ (39). The basic idea of their research was to measure the complexity of hidden architecture in genotype and phenotype. It was expected that the association between distinct sets of phenotypes and SNPs could be revealed in heterogeneous SZ, and it would represent subtypes of SZ with the respective genetic mechanism.
Arnedo et al. generated phenotypic sets using non-negative matrix factorization from the data of series questionnaire and structured interview results. The factorization divided the SZ patients into distinct subgroups with different disease severity, process, and symptom domain (positive, negative, and disorganized symptoms) regardless of their genetic background. SNP sets were generated by a generalized factorization method combined with non-negative matrix factorization. The overlap of patients and SNPs in these sets ensured to be disjoint, to reflect the heterogeneity of SZ. Finally, the association between phenotypic sets and SNP sets were tested in the molecular genetics of schizophrenia (MGS) study. It was also largely replicated by them in the National Institute of Mental Health Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) project and Portuguese Island family samples.
The results were encouraging: Arnedo et al. found 42 SNP sets had >70% risk for SZ, and these SNP sets were significantly associated with different phenotypic sets. For instance, a phenotypic set indicating a general process of severe deterioration (severe process, with positive and negative symptom; moderate severity of impairment; unable to function since onset) was highly correlated with certain SNP set including polypyrimidine tract binding protein 2 (PTBP2) and several other genes which might play a role in neuron differentiation. This severe deterioration SZ may be a potential clinical valid subtype, and following the track of PTBP2 and its SNP cluster may facilitate the examination of the mechanisms underlying severe deterioration.
Based on their findings, it was believed that SZ could be seen as “syndromes group” in which distinct clinical syndromes are associated with disjoint genotypic networks. The interaction map of disjoint genotype and distinct syndromes have shown a possible way of shaping SZ into biological markers or a networks-based subtype.
Imaging and Its Impact on the Diagnostic Issue
Anatomical changes in fronto-temporal, thalamo-cortical, subcortical-limbic circuits, enlargement of ventricles, and widespread white matter fibers abnormalities have been found in many structural studies of schizophrenia (40–43). With the growing sample size and collaboration through different sites, many large-scale meta-analyses have provided new information. The Enhancing Neuroimaging Genetics through Meta-Analysis (ENIGMA) SZ working group’s meta-analysis of subcortical regions across several thousand subjects reported the consistent findings of smaller hippocampus, amygdala, thalamus, accumbens, and intracranial volumes, but larger pallidum and lateral ventricle volumes (44). The putamen and caudate volume results were not reliable across different populations and studies even with this sample size, indicating the possibility of clinical heterogeneity affecting those regions (44). The development of these differences prior to, with, and after disease onset and diagnosis is also important for understanding the disease (45), and comparing the course of the morphometric reductions and increases across diagnoses will be informative. Functional imaging studies have also discovered various abnormal brain regions and connections in SZ. Partially overlapped with structural findings, functional alterations including the prefrontal cortex, superior temporal gyrus, thalamus, frontal lobe, and parietal lobe have been reported in either resting state or task fMRI (46).
Many of the above regions have been identified as structural or functional commonalities among DSM categories (1, 47, 48). Starting from the same point as genetics, there are also imaging research efforts trying to redraw the boundaries between psychotic disorders. One pioneer study is from the Bipolar-SZ Network on Intermediate Phenotypes (B-SNIP) Consortium, Clementz et al. applied neurobiological measures among SZ, BD, and SAD and tried to regroup them into different “biotypes” rather than DSM catalogs (49). A selection of psychotic biomarkers and functional brain activity were collected in this study. Not only patients with psychosis but also their first-degree relatives and healthy controls were included. Clementz et al. then identified three “biotypes,” which were also believed to be more heritable than their original DSM diagnoses. Sensorimotor reactivity and cognitive control distinguish three biotypes: biotype 1 patients showed serious impairment across sensorimotor reactivity and cognitive control; biotype 2 patients show only deficits in cognitive control; and biotype 3 patients seem to be the mildest in cognitive symptoms. The B-SNIP group has also been trying to find the factors that contribute to its biotyping; one attempt is using the flow–frequency fluctuations (ALFF/fALFF) across the SZ, BD, and SAD from the large B-SNIP family study (50). More recently, gray matter density was checked in these three biotypes, and the density loss followed the same order as cognitive decline: biotype 1 showed whole brain gray matter density loss, while type 2 showed largely overlapping results with type 1, and the largest effects were found in fronto-temporal circuits, parietal cortex, and cerebellum. The findings were much more localized and of less magnitude for type 1. Type 3 only showed small reductions in frontal, cingulate, and temporal regions despite their similar DSM diagnoses (51).
Imaging Genetics to Refine the Diagnosis
Imaging Genetics Linking Genetics, Intermediate Imaging, and Cognitive Phenotypes
There are hundreds of papers using imaging genetics method to study SZ in the past 10 years, but here, we will focus on the findings with relatively clear functional implications. First, we selected the genes that have been highlighted in SZ and cognitive functions and if there is more than one report implicating those genes. The details of included genes can be found in Tables 1 and 2 under “Risk SNPs/allele” column. We then searched PubMed database using the terms: [“gene symbols”] (genes we selected) AND [“schizophrenia”] AND [“symptom” OR “cognition” OR “cognitive function”] AND [“MRI”]. Abstracts and main texts were assessed with the following inclusion/exclusion criteria. The inclusion criteria were the following: 1) publications between January 2000 and January 2017, 2) diagnosis of any psychiatric disorders or risk gene, 3) brain structure with volume, concentration, thickness, and surface area, 4) brain function including resting state or task, and 5) including modalities of gene, imaging, and behavior simultaneously. Exclusion criteria were the following: 1) publications including letters, short reports, and brief communication; 2) MRI scanning sequences other than T1, T2, or BOLD; 3) in functional studies, the association between genes, images, and behavior were not directly assessed; and 4) in structural studies, symptoms, cognition, or behavior was not evaluated and collected at the same time window as images were acquired. After excluding 7 studies, 24 studies remained in Table 1 for functional studies. Table 1 lists the selected functional papers, and we highlight findings below by symptom/cognitive domains and possible intermediate imaging phenotype. However, most of these imaging genetic studies were done in healthy risk allele carriers.
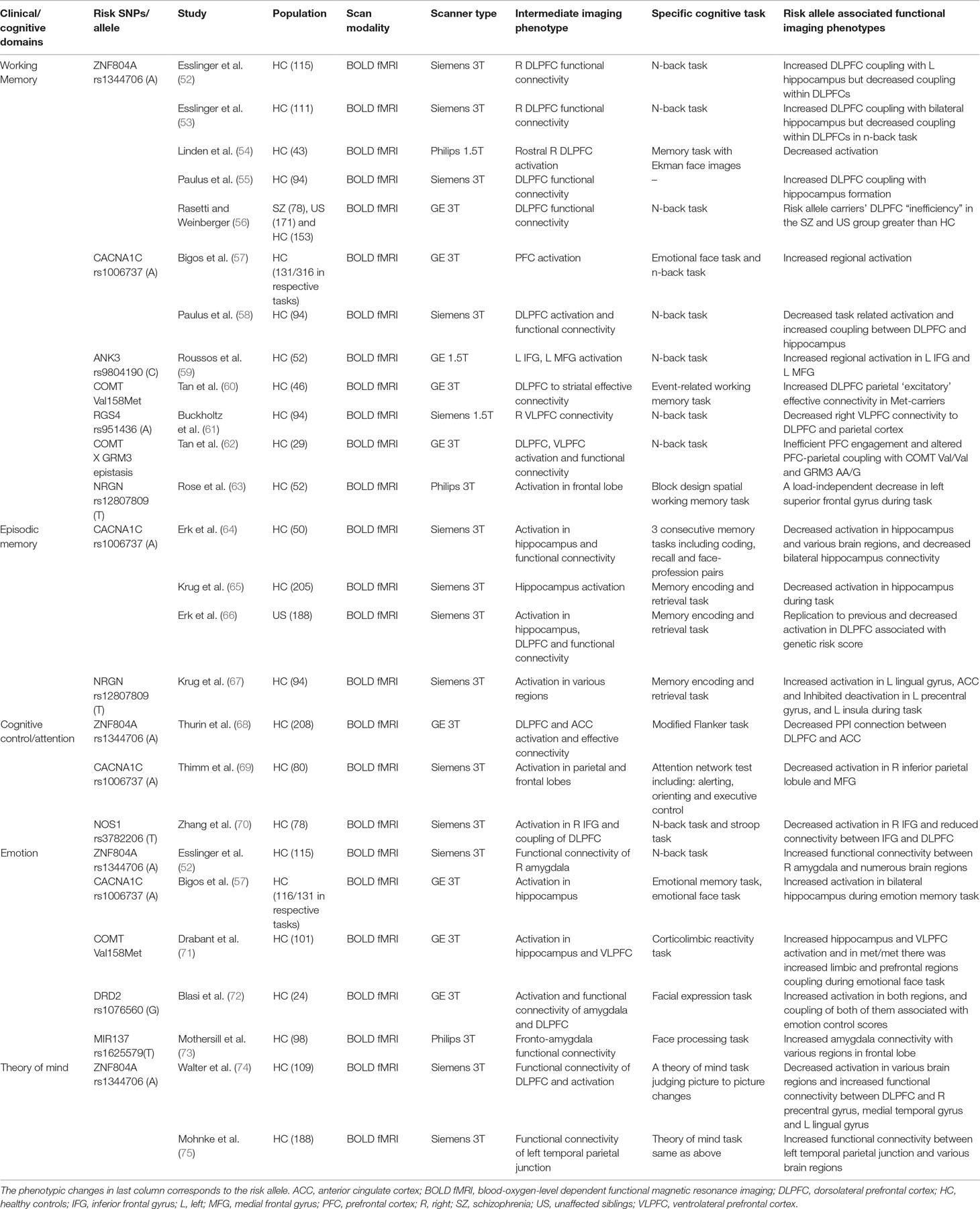
Table 1 Clinical/cognitive domain specific imaging genetic evidence and potential intermediate functional imaging phenotypes.
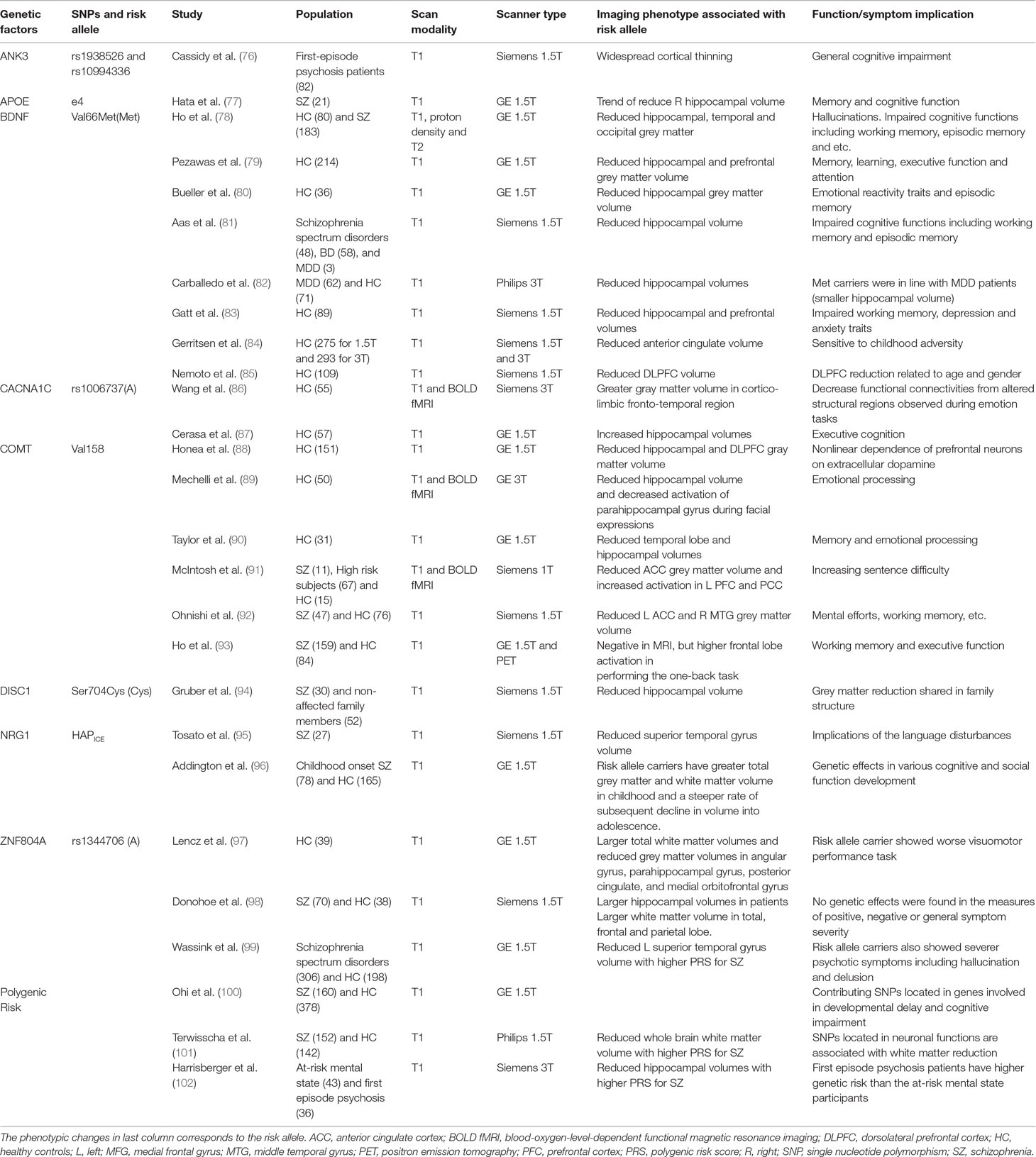
Table 2 Potential structural imaging genetic phenotypes and possible function association.
Working memory deficit is fundamental and critical in SZ. The most well-studied possible intermediate imaging phenotype was the connection abnormalities between dorsolateral prefrontal cortex and hippocampus (DLPFC-HC). ZNF804A (52, 55, 56) and CACNA1C (58) were associated with DLPFC-HC connection alteration. In the healthy controls, risk allele of ZNF804A was associated with the increased DLPFC-HC connection. COMT (60), regulator of G protein signaling 4 (RGS4) (61), and COMT X glutamate metabotropic receptor 3 (GRM3) epistasis were connected to prefrontal cortex-parietal coupling.
Episodic memory or long-term memory was also often disturbed in SZ. Possible intermediate imaging phenotypes included decreased coupling of the hippocampus–parietal cortex, hippocampus and ventrolateral prefrontal cortex (VLPFC), and bilateral hippocampus. However, the genetic association within this thread is elusive (46). In healthy controls, CACNA1C risk allele carriers showed the decreased activation during recall in decreased coupling between bilateral hippocampus (64). The NRGN rs12807809 was found with increased activation in the left lingual gyrus and decreased deactivation in the left precentral gyrus, cingulate, and left insula during the different stages of memory retrieval (67).
SZ patients often show attention or cognitive control deficits. Disturbances in PFC and DLPFC and connection alterations were the most important issue regarding this deficit. NOS1 risk allele carriers showed reduced inferior frontal gyrus and DLPFC connection associated with attention performance (103). For other risk genes, CACNA1C risk allele carriers showed decreased activation in the right inferior parietal lobule and medial frontal gyrus during an attention task (69). Again, ZNF804A showed association with the anterior cingulate cortex (ACC) and DLPFC coupling during attention and cognitive control (68). During emotional memory, SZ CACNA1C risk allele carriers showed increased activation in the bilateral hippocampus, which was in line with finding in BD (57).
Emotion processing is another important disruption common in SZ. ZNF804A (52) and DRD2 (72) have shown correlation with the amygdala and ACC/medial prefrontal cortex (mPFC) within emotion processing. Increased connectivity between amygdala and VLPFC, which was considered as another intermediate imaging phenotype for emotion processing, has been found in healthy risk allele carriers of the COMT (71) and MIR137 (73).
As part of social cognition that is often impaired in SZ, the theory of mind capabilities tends to help people understand mental states of themselves and others. ZNF804A risk alleles correlated with the PFC and various cortical regions in social information processes (74). Decreased activation in bilateral dorsal medial PFC, the left temporoparietal cortex, left inferior parietal cortex, posterior cingulate, and the left lateral PFC was found while investigating ZNF804A (74). There was also a trend for increased functional connectivity of the left temporal parietal junction with several regions (75).
Rather than a localized abnormality, most findings are notably in line with a “disconnection disorder” (104). Additionally, as noted above, these genes are not specific to risk for schizophrenia but show risk as well for other psychiatric disorders; the common functional impairments showing the genetic relationship in SZ and BD tend to be closely associated with connection disturbances and involve multiple brain regions (46, 105, 106).
Structural Brain Imaging Genetics Findings in SZ
It is more difficult for researchers to relate risk gene factors, brain structural alteration, and symptoms or cognitive impairments; large numbers of these structural brain imaging genetic studies have conflicting results (10). We reviewed structural brain imaging studies with the relatively clear and consistent symptom or cognitive implications following the criteria we described above. Note that only research involving genetics, structural brain, symptoms, or cognitions and the analysis between them were included. After excluding 8 studies, 27 structural studies remained (see Table 2).
Some genes like BDNF are engaged in many cognitive domains that are commonly impaired in SZ, although their associations with SZ per se may not be strong. BDNF is essential in nervous system development and prevention of cell loss in various brain regions including the hippocampus, striatum, and more. The Val66Met has been found to be related with reduced hippocampal (107), temporal (78), and frontal volume (80), which may affect various cognitive functions including working memory, episodic memory, executive function, and hallucinations. Its interaction with early life abuse may also result in reduced hippocampal volume in SZ, BD, and MDD (81, 82). As the disease progresses, BDNF is found to be connected with reduced frontal volume and impaired executive function (85, 108).
Other genes may have a closer relationship with SZ, but their imaging genetic findings with brain regions and clinical phenotypes are less consistent. COMT may be involved not only in reduced hippocampal volume but also in reduced cingulate and DLPFC volume, which may potentially affect memory, attention, and executive function (87, 91). Risk allele carriers with rs1006737(A) in CACNA1C show greater gray matter volume in a cortico-limbic and fronto-temporal region but generally in BD (86). The neuregulin 1 gene (NRG1) and its risk haplotype may also contribute to the hippocampal and temporal volume (95, 109). Other critical genes including ANK3 (76), Apoe (77), DISC 1 (110), and ZNF804A (97–99) and more have been found connected to reduced brain volume in hippocampus, cingulate, frontal, temporal, and various brain region volume, suggesting their role in SZ-related cognitive impairment and symptoms.
Polygenic risk score studies also provide imaging genetic evidence for SZ imaging genetic. Temporal volume (100), whole brain white matter volume (101), and hippocampal volume abnormality (102) have been suggested through these approaches.
Overall, the genetic influence on brain structure are widely spread, and their functional or clinical implications are complex. At the moment, the gene to the brain and behavior/symptom links are extensive, affecting many cognitive domains when tested in nonaffected individuals. The specificity of genetic effects on SZ need to be carefully examined, and uncovering better methods to form a link from imaging genetics to clinical phenotypes is important to contribute to the diagnostic issue.
Limitations and Future Directions
Imaging genetics has contributed greatly to our understanding of the biological mechanism behind psychiatric disorders by revealing the potential association between genetics and imaging phenotypes. The merging boundaries between disorders and subtypes within SZ revealed by imaging genetics will continue to shape the future diagnostic approach of psychiatric disorders. However, it also has inevitable limitations, and the pathways linking genetics, neuroimaging intermediate phenotypes, and clinically assessable phenotypes remain far from clear. A diagnostic system built on imaging genetics requires further research efforts.
Limitations
Typically, the effect size of candidate gene analyses is rather small and explains limited brain structural or functional variations (111). Alternatively, large sample imaging genetics research often report encouraging findings supporting vast common variations influence on the human brain (112).
However, these findings and even the logic behind imaging genetics has been questioned. Franke et al. mega-analyzed the largest GWAS data for SZ to date from PGC (33,636 cases and 43,008 controls) and eight structural MRI brain measures from ENIGMA (11,840 individuals) to evaluate the relationship between the common variations and SZ-associated subcortical brain regions (113). For instance, the hippocampal volume deficit was thought fundamental in SZ (114). The hippocampus deficits in SZ are one of the most reliable findings of volumetric deficits (44). The ENIGMA analysis identified common genetic variations related to hippocampal volume without regard to disorder (112); the PGC identified common genetic variations highlighted by 108 loci from GWAS, which were thought to play important roles in the etiology of SZ without regard to hippocampal volume (16). Franke et al. did several analyses to investigate the correlation between these genetic and imaging findings. They used linkage disequilibrium score regression to estimate the SNP-based heritability of volumetric measures, computed and compared genetic predisposition scores to volumes, and quantified rank–rank hypergeometric overlap test and listed genetic variants influencing the brain volume. Unfortunately, all these analyses reported no significant results. They also analyzed the 128 index SNPs from PGC and their association with brain volume including the hippocampus, meta-analyses, conjunction analysis and compare the genetic effect sizes for SZ and volumes. Again, these analyses resulted in nonsignificant findings.
Although Franke et al. emphasized that there were several limitations that may result in this null finding, it strongly reminded us to think carefully about the logic of imaging genetics. Brain measures or structural brain deficits believed to be important pathological alterations of SZ may not be induced by those primary genetic causes of SZ as a diagnostic category. They may be reflecting prenatal and later development environmental effects that correlate with but are not specific to SZ, or the diagnostic category of SZ may not be uniformly organized so the large-scale studies of disease risk may have introduced too many heterogeneities.
Instead, the field must consider whether brain volume is a good bridge to look into the genetic influence on disorders. The idea of “intermediate phenotype” succeeded the idea of endophenotype, which was first used by Gottesman and Shields (115). Either structural or functional imaging was believed to be good intermediate phenotypes, as they provide a large amount of data that can show the effect of genes. Although many imaging genetics studies used the concept of intermediate phenotypes to conduct the hypothesis and research flow, they did not fully meet the criteria of intermediate phenotype. To fulfill the criteria, the phenotype must have the following: good psychometric properties, disorder and symptoms related in general population, stable over time, increased expression in unaffected relatives, cosegregation in families, and common genetic influences shown in the disorder. We have to verify whether a chosen brain measure meets each of these criteria.
Hippocampal volumes, in particular, did seem to fulfill these criteria, in that the volumes were more similar in unaffected siblings (116, 117), seemed to decrease with younger disease onset (118), and the smaller volumes were a strong effect in comparing SZ and controls (119). The other brain regions especially caudate and putamen, which showed a small effect size in ENIGMA, would also need to pass these criteria if they are to be used as intermediate phenotypes. However, these brain volume alterations may not be specific to SZ. As for hippocampal volume among psychiatric disorders, it is also affected in MDD (120, 121), obsessive–compulsive disorder (122), and attention deficit hyperactivity disorder (123). Other than psychiatric disorders, cardiovascular disease, diabetes, hypertension, obesity, physical activity, and various somatic factors may also play a role in modifying hippocampal volume to different extents (124, 125). The hippocampus is vulnerable to various environmental factors from the prenatal stage throughout the lifetime, which makes the hippocampal structure sensitive to neurodisruption but not necessarily specific to SZ (126). The specificity of these altered brain volume will need careful examination before being considered as part of SZ’s pathology in complicated clinical situations.
Another approach is to reconsider other imaging intermediate phenotypes bridging genetics and SZ. For example, there are various anatomic measures other than volumes that should be assessed for genetic effects (127, 128). Gray/white matter density, cortical thickness, cortical folding, cortical surface area, and white matter integrity are potential useful intermediate phenotypes from which to choose. However, although it may also be difficult to fully grasp, the functional implication of these brain measures and their compatibility with genetics will need further investigation (129). As for these other brain volumes and functional measurements, their stability, situation in unaffected relatives, families, and general population will need to be further investigated to answer the criteria question as well as their specificity to the diagnosis or clinical subgrouping.
It may be helpful to expand the genetic modality of imaging genetic study. More heritability could be captured by involving rare variance and chromosome structural variations like CNVs (38). Both options will need better imaging genetic analysis methods and models.
Future Directions
The current review summarized genetics, imaging, and imaging genetics in schizophrenia to date. Imaging genetics may continue to shape the future conceptualization of SZ and psychotic disorders in both clinical and research field.
One future direction is collecting a large number of genetic effects. The method applied by Arnedo et al. is promising in coupling both genetic and phenotypic clusters, but it may need to establish its association with imaging data or physiological measures. The clustering method shows great complexity, while its compatibility with neuroimaging is unknown. The other polygenic method like polygenic risk score is also promising. However, it will call for more common variations and the combination with other data (e.g., the B-SNIP biotype study).
Parallel independent component analysis (pICA) may be another useful tool in this field. This method allows independent components from two modalities to be identified simultaneously, and the association between these two modalities is optimized. pICA is designed to be totally theoretically blind and data-driven, but pICA with reference allows a priori knowledge as the reference to improve robustness. For instance, a set of genes from the same pathway can be used as a reference to highlight their effect on certain brain components as well as behavioral data (130). Chen et al. used pICA and reported that the gray matter density of frontal, precuneus, and cingulate regions might potentially be affected by various genes participating in synaptic plasticity, axon guidance, and molecular signal transduction (131).
Another possible direction is refining the clinical assessment tools to better complement imaging genetics. As raised in the B-SNIP study, a series of symptom rating scales including the Global Assessment of Functioning scale, the Positive and Negative Syndrome Scale, the Young Mania Rating Scale, the Montgomery–Åsberg Depression Rating Scale, the Schizo-Bipolar Scale, and the Birchwood Social Functioning Scale were obtained from the participants. These measures were not able to distinguish SZ, BD, and SAD significantly or contribute much in the building of biotypes (132). Imaging genetic compatible comprehensive symptom scales are needed. These scales are not aiming at distinguishing traditional diagnostic groups or a certain diagnostic group usage. However, they would provide comprehensive clinical profiles “scanning” the symptom domains (2). Some scales like the Symptom Checklist-90 (SCL-90) and its revised version (133) might be worth trying (134). More detailed multidimensional symptoms reflecting scale need to be developed to fit the need of imaging genetics and clarify the path linking genotypic variation, intermediate brain imaging, and clinical phenotypes.
Finally, future research will need to be enhanced by improving power and replicability. Studies with small number of subjects (below 100 participants) will be able to show moderate power with effect size of 0.5. However, it is critical to replicate them independently with same genetic variants, imaging, and behavioral measurements, and direction of the effects by Carter et al. (135). It is also argued, in such studies, null results or conflicting associations with failed replication should still be considered for publications as potentially informative or innovative studies (6, 136). In this case, meta-analytic studies addressing the conflicted results and the issues of publication bias will help to avoid the misleading information potentially generated from small sample research results (6, 137).
Author Contributions
All authors listed have made substantial, direct, and intellectual contribution to the work and approved it for publication.
Funding
WJ and JT were supported in this work by a grant from the National Institute of Mental Health (R01 MH094524).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Lawrie SM, O’Donovan MC, Saks E, Burns T, Lieberman JA. Towards diagnostic markers for the psychoses. Lancet Psychiatry (2016) 3(4):375–85. doi: 10.1016/S2215-0366(16)00021-3
2. Bhati MT. Defining psychosis: the evolution of DSM-5 schizophrenia spectrum disorders. Curr Psychiatry Rep (2013) 15(11):409. doi: 10.1007/s11920-013-0409-9
3. Turner JA, Smyth P, Macciardi F, Fallon JH, Kennedy JL, Potkin SG. Imaging phenotypes and genotypes in schizophrenia. Neuroinformatics (2006) 4(1):21–49. doi: 10.1385/NI:4:1:21
4. Bigos KL, Weinberger DR. Imaging genetics—days of future past. NeuroImage (2010) 53(3):804–9. doi: 10.1016/j.neuroimage.2010.01.035
5. Lawrie SM, O’Donovan MC, Saks E, Burns T, Lieberman JA. Improving classification of psychoses. Lancet Psychiatry (2016) 3(4):367–74. doi: 10.1016/S2215-0366(15)00577-5
6. Bogdan R, Salmeron BJ, Carey CE, Agrawal A, Calhoun VD, Garavan H, et al. Imaging genetics and genomics in psychiatry: a critical review of progress and potential. Biol Psychiatry (2017) 82(3):165–75. doi: 10.1016/j.biopsych.2016.12.030
7. Hashimoto R, Ohi K, Yamamori H, Yasuda Y, Fujimoto M, Umeda-Yano S, et al. Imaging genetics and psychiatric disorders. Curr Mol Med (2015) 15(2):168–75. doi: 10.2174/1566524015666150303104159
8. Fan CC, Smeland OB, Schork AJ, Chen CH, Holland D, Lo MT, et al. Beyond heritability: improving discoverability in imaging genetics. Hum Mol Genet (2018) 27(R1):R22–R28. doi: 10.1093/hmg/ddy082
9. Palaniyappan L, Maayan N, Bergman H, Davenport C, Adams CE, Soares-Weiser K. Voxel-based morphometry for separation of schizophrenia from other types of psychosis in first episode psychosis. Cochrane Database Syst Rev (2015) (8), CD011021. doi: 10.1002/14651858.CD011021.pub2
10. van Haren NE, Bakker SC, Kahn RS. Genes and structural brain imaging in schizophrenia. Curr Opin Psychiatry (2008) 21(2):161–7. doi: 10.1097/YCO.0b013e3282f4f25b
11. Nenadic I, Gaser C, Sauer H. Heterogeneity of brain structural variation and the structural imaging endophenotypes in schizophrenia. Neuropsychobiology (2012) 66(1):44–9. doi: 10.1159/000338547
12. Feighner JP, Robins E, Guze SB, Woodruff RA Jr., Winokur G, Munoz R. Diagnostic criteria for use in psychiatric research. Arch Gen Psychiatry (1972) 26(1):57–63. doi: 10.1001/archpsyc.1972.01750190059011
13. Gershon ES, Grennan KS. Genetic and genomic analyses as a basis for new diagnostic nosologies. Dialog Clin Neurosci (2015) 17(1):69–78.
14. Cross-Disorder Group of the Psychiatric Genomics C, Lee SH, Ripke S, Neale BM, Faraone SV, Purcell SM, et al. Genetic relationship between five psychiatric disorders estimated from genome-wide SNPs. Nat Genet (2013) 45(9):984–94. doi: 10.1038/ng.2711
15. Sullivan PF, Kendler KS, Neale MC. Schizophrenia as a complex trait: evidence from a meta-analysis of twin studies. Arch Gen Psychiatry (2003) 60(12):1187–92. doi: 10.1001/archpsyc.60.12.1187
16. Schizophrenia Working Group of the Psychiatric Genomics C. Biological insights from 108 schizophrenia-associated genetic loci. Nature (2014) 511(7510):421–7. doi: 10.1038/nature13595
17. Schwab SG, Wildenauer DB. Genetics of psychiatric disorders in the GWAS era: an update on schizophrenia. Eur Arch Psychiatry Clin Neurosci (2013) 263 Suppl 2:S147–154. doi: 10.1007/s00406-013-0450-z
18. O’Donovan MC, Craddock N, Norton N, Williams H, Peirce T, Moskvina V, et al. Identification of loci associated with schizophrenia by genome-wide association and follow-up. Nat Genet (2008) 40(9):1053–5.
19. Guan J, Cai JJ, Ji G, Sham PC. Commonality in dysregulated expression of gene sets in cortical brains of individuals with autism, schizophrenia, and bipolar disorder. Transl Psychiatry (2019) 9(1):152. doi: 10.1038/s41398-019-0488-4
20. Musliner KL, Mortensen PB, McGrath JJ, Suppli NP, Hougaard DM, Bybjerg-Grauholm J, et al. Association of polygenic liabilities for major depression, bipolar disorder, and schizophrenia with risk for depression in the danish population. JAMA Psychiatry (2019) 76(5):516–25. doi: 10.1001/jamapsychiatry.2018.4166
21. Smeland OB, Bahrami S, Frei O, et al. Genome-wide analysis reveals extensive genetic overlap between schizophrenia, bipolar disorder, and intelligence. Mol Psychiatry (2019) . doi: 10.1038/s41380-018-0332-x
22. Markota M, Coombes BJ, Larrabee BR, McElroy SL, Bond DJ, Veldic M, et al. Association of schizophrenia polygenic risk score with manic and depressive psychosis in bipolar disorder. Transl Psychiatry (2018) 8(1):188. doi: 10.1038/s41398-018-0242-3
23. Gandal MJ, Haney JR, Parikshak NN, Leppa V, Ramaswami G, Hartl C, et al. Shared molecular neuropathology across major psychiatric disorders parallels polygenic overlap. Science (2018) 359(6376):693–7. doi: 10.1126/science.aad6469
24. Khanzada NS, Butler MG, Manzardo AM. GeneAnalytics pathway analysis and genetic overlap among autism spectrum disorder, bipolar disorder and schizophrenia. Int J Mol Sci (2017) 18(3):527. doi: 10.3390/ijms18030527
25. Forstner AJ, Hecker J, Hofmann A, Maaser A, Reinbold CS, Muhleisen TW, et al. Identification of shared risk loci and pathways for bipolar disorder and schizophrenia. PloS one (2017) 12(2):e0171595.
26. Lichtenstein P, Yip BH, Björk C, Pawitan Y, Cannon TD, Sullivan PF, et al. Common genetic determinants of schizophrenia and bipolar disorder in Swedish families: a population-based study. Lancet (2009) 373(9659):234–9. doi: 10.1016/S0140-6736(09)60072-6
27. Cardno AG, Owen MJ. Genetic relationships between schizophrenia, bipolar disorder, and schizoaffective disorder. Schizophr Bull (2014) 40(3):504–15. doi: 10.1093/schbul/sbu016
28. Kotov R, Leong SH, Mojtabai R, Erlanger AC, Fochtmann LJ, Constantino E, et al. Boundaries of schizoaffective disorder: revisiting Kraepelin. JAMA Psychiatry (2013) 70(12):1276–86. doi: 10.1001/jamapsychiatry.2013.2350
29. Williams HJ, Norton N, Dwyer S, Moskvina V, Nikolov I, Carroll L, et al. Fine mapping of ZNF804A and genome-wide significant evidence for its involvement in schizophrenia and bipolar disorder. Mol Psychiatry (2011) 16(4):429–41. doi: 10.1038/mp.2011.21
30. Schizophrenia Psychiatric Genome-Wide Association Study C. Genome-wide association study identifies five new schizophrenia loci. Nat Genet (2011) 43(10):969–76. doi: 10.1038/ng.940
31. Sullivan PF, Daly MJ, O’Donovan M. Genetic architectures of psychiatric disorders: the emerging picture and its implications. Nat Rev Genet (2012) 13(8):537–51. doi: 10.1038/nrg3240
32. Andreassen OA, Thompson WK, Schork AJ, Ripke S, Mattingsdal M, Kelsoe JR, et al. Improved detection of common variants associated with schizophrenia and bipolar disorder using pleiotropy-informed conditional false discovery rate. PLoS Genet (2013) 9(4):e1003455.
33. Cross-Disorder Group of the Psychiatric Genomics C. Identification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysis. Lancet (2013) 381(9875):1371–9. doi: 10.1016/S0140-6736(12)62129-1
34. Duncan LE, Ratanatharathorn A, Aiello AE, Almli LM, Amstadter AB, Ashley-Koch AE, et al. Largest GWAS of PTSD (N = 20 070) yields genetic overlap with schizophrenia and sex differences in heritability. Mol Psychiatry (2018) 23(3):666–73.
35. St Pourcain B, Robinson EB, Anttila V, Sullivan BB, Maller J, Golding J, et al. ASD and schizophrenia show distinct developmental profiles in common genetic overlap with population-based social communication difficulties. Mol Psychiatry (2018) 23(2):263–70. doi: 10.1038/mp.2016.198
36. Maier R, Moser G, Chen GB, Ripke S, Cross-Disorder Working Group of the Psychiatric Genomics C, Coryell W, et al. Joint analysis of psychiatric disorders increases accuracy of risk prediction for schizophrenia, bipolar disorder, and major depressive disorder. Am J Human Genet (2015) 96(2):283–94.
37. Lee KW, Woon PS, Teo YY, Sim K. Genome wide association studies (GWAS) and copy number variation (CNV) studies of the major psychoses: what have we learnt? Neurosci Biobehav Rev (2012) 36(1):556–71. doi: 10.1016/j.neubiorev.2011.09.001
38. Warland A, Kendall KM, Rees E, Kirov G, Caseras X. Schizophrenia-associated genomic copy number variants and subcortical brain volumes in the UK Biobank. Mol Psychiatry (2019). doi: 10.1038/s41380-019-0355-y
39. Arnedo J, Svrakic DM, Del Val C, Romero-Zaliz R, Hernandez-Cuervo H, Molecular Genetics of Schizophrenia C, et al. Uncovering the hidden risk architecture of the schizophrenias: confirmation in three independent genome-wide association studies. Am J Psychiatry (2015) 172(2):139–53. doi: 10.1176/appi.ajp.2014.14040435
40. Karlsgodt KH, Sun D, Cannon TD. Structural and functional brain abnormalities in schizophrenia. Curr Dir Psychol Sci (2010) 19(4):226–31. doi: 10.1177/0963721410377601
41. Dazzan P, Arango C, Fleischacker W, Galderisi S, Glenthoj B, Leucht S, et al. Magnetic resonance imaging and the prediction of outcome in first-episode schizophrenia: a review of current evidence and directions for future research. Schizophr Bull (2015) 41(3):574–83. doi: 10.1093/schbul/sbv024
42. Pearlson GD, Marsh L. Structural brain imaging in schizophrenia: a selective review. Biol Psychiatry (1999) 46(5):627–49. doi: 10.1016/S0006-3223(99)00071-2
43. Dietsche B, Kircher T, Falkenberg I. Structural brain changes in schizophrenia at different stages of the illness: a selective review of longitudinal magnetic resonance imaging studies. Aust N Z J Psychiatry (2017) 51(5):500–8. doi: 10.1177/0004867417699473
44. van Erp TG, Hibar DP, Rasmussen JM, Glahn DC, Pearlson GD, Andreassen OA, et al. Subcortical brain volume abnormalities in 2028 individuals with schizophrenia and 2540 healthy controls via the ENIGMA consortium. Mol Psychiatry (2016) 21(4):547–53. doi: 10.1038/mp.2015.63
45. Shenton ME, Dickey CC, Frumin M, McCarley RW. A review of MRI findings in schizophrenia. Schizophr Res (2001) 49(1–2):1–52. doi: 10.1016/S0920-9964(01)00163-3
46. Cao H, Dixson L, Meyer-Lindenberg A, Tost H. Functional connectivity measures as schizophrenia intermediate phenotypes: advances, limitations, and future directions. Curr Opin Neurobiol (2016) 36:7–14. doi: 10.1016/j.conb.2015.07.008
47. Isobe M, Miyata J, Hazama M, Fukuyama H, Murai T, Takahashi H. Multimodal neuroimaging as a window into the pathological physiology of schizophrenia: current trends and issues. Neurosci Res (2016) 102:29–38. doi: 10.1016/j.neures.2015.07.009
48. Schwarz E, Tost H, Meyer-Lindenberg A. Working memory genetics in schizophrenia and related disorders: an RDoC perspective. Am J Med GenetPart B Neuropsychiatr Genet (2016) 171B(1):121–31. doi: 10.1002/ajmg.b.32353
49. Clementz BA, Sweeney JA, Hamm JP, Ivleva EI, Ethridge LE, Pearlson GD, et al. Identification of distinct psychosis biotypes using brain-based biomarkers. Am J Psychiatry (2016) 173(4):373–84. doi: 10.1176/appi.ajp.2015.14091200
50. Meda SA, Wang Z, Ivleva EI, Poudyal G, Keshavan MS, Tamminga CA, et al. Frequency-specific neural signatures of spontaneous low-frequency resting state fluctuations in psychosis: evidence from Bipolar-Schizophrenia Network on Intermediate Phenotypes (B-SNIP) Consortium. Schizophr Bull (2015) 41(6):1336–48. doi: 10.1093/schbul/sbv064
51. Ivleva EI, Clementz BA, Dutcher AM, Arnold SJM, Jeon-Slaughter H, Aslan S, et al. Brain structure biomarkers in the psychosis biotypes: findings from the bipolar-schizophrenia network for intermediate phenotypes. Biol Psychiatry (2017) 82(1):26–39.
52. Rasetti R, Sambataro F, Chen Q, Callicott JH, Mattay VS, Weinberger DR. Neural mechanisms of a genome-wide supported psychosis variant. Science (2009) 324(5927):605. doi: 10.1126/science.1167768
53. Esslinger C, Kirsch P, Haddad L, Mier D, Sauer C, Erk S, et al. Cognitive state and connectivity effects of the genome-wide significant psychosis variant in ZNF804A. NeuroImage (2011) 54(3):2514–23.
54. Linden DE, Lancaster TM, Wolf C, Baird A, Jackson MC, Johnston SJ, et al. ZNF804A genotype modulates neural activity during working memory for faces. Neuropsychobiology (2013) 67(2):84–92.
55. Esslinger C, Walter H, Kirsch P, Erk S, Schnell K, Arnold C, et al. Partial support for ZNF804A genotype-dependent alterations in prefrontal connectivity. Hum Brain Mapp (2013) 34(2):304–13. doi: 10.1002/hbm.21434
56. Rasetti R, Sambataro F, Chen Q, Callicott JH, Mattay VS, Weinberger DR. Altered cortical network dynamics: a potential intermediate phenotype for schizophrenia and association with ZNF804A. Arch Gen Psychiatry (2011) 68(12):1207–17. doi: 10.1001/archgenpsychiatry.2011.103
57. Bigos KL, Mattay VS, Callicott JH, Straub RE, Vakkalanka R, Kolachana B, et al. Genetic variation in CACNA1C affects brain circuitries related to mental illness. Arch Gen Psychiatry (2010) 67(9):939–45. doi: 10.1001/archgenpsychiatry.2010.96
58. Paulus FM, Bedenbender J, Krach S, Pyka M, Krug A, Sommer J, et al. Association of rs1006737 in CACNA1C with alterations in prefrontal activation and fronto-hippocampal connectivity. Hum Brain Mapp (2014) 35(4):1190–200. doi: 10.1002/hbm.22244
59. Roussos P, Katsel P, Davis KL, Bitsios P, Giakoumaki SG, Jogia J, et al. Molecular and genetic evidence for abnormalities in the nodes of Ranvier in schizophrenia. Arch Gen Psychiatry (2012) 69(1):7–15.
60. Tan HY, Chen AG, Kolachana B, Apud JA, Mattay VS, Callicott JH, et al. Effective connectivity of AKT1-mediated dopaminergic working memory networks and pharmacogenetics of anti-dopaminergic treatment. Brain: J Neurol (2012) 135(Pt 5):1436–45. doi: 10.1093/brain/aws068
61. Buckholtz JW, Meyer-Lindenberg A, Honea RA, Straub RE, Pezawas L, Egan MF, et al. Allelic variation in RGS4 impacts functional and structural connectivity in the human brain. J Neurosci (2007) 27(7):1584–93. doi: 10.1523/JNEUROSCI.5112-06.2007
62. Tan HY, Chen Q, Sust S, Buckholtz JW, Meyers JD, Egan MF, et al. Epistasis between catechol-O-methyltransferase and type II metabotropic glutamate receptor 3 genes on working memory brain function. Proc Natl Acad Sci U S A (2007) 104(30):12536–41.
63. Rose EJ, Morris DW, Fahey C, Robertson IH, Greene C, O’Doherty J, et al. The effect of the neurogranin schizophrenia risk variant rs12807809 on brain structure and function. Twin Res Hum Genet (2012) 15(3):296–303.
64. Erk S, Meyer-Lindenberg A, Schnell K, Opitz von Boberfeld C, Esslinger C, Kirsch P, et al. Brain function in carriers of a genome-wide supported bipolar disorder variant. Arch Gen Psychiatry (2010) 67(8):803–11. doi: 10.1001/archgenpsychiatry.2010.94
65. Krug A, Witt SH, Backes H, Dietsche B, Nieratschker V, Shah NJ, et al. A genome-wide supported variant in CACNA1C influences hippocampal activation during episodic memory encoding and retrieval. Eur Arch Psychiatry Clin Neurosci (2014) 264(2):103–10.
66. Erk S, Meyer-Lindenberg A, Schmierer P, Mohnke S, Grimm O, Garbusow M, et al. Hippocampal and frontolimbic function as intermediate phenotype for psychosis: evidence from healthy relatives and a common risk variant in CACNA1C. Biol Psychiatry (2014) 76(6):466–75.
67. Krug A, Krach S, Jansen A, Nieratschker V, Witt SH, Shah NJ, et al. The effect of neurogranin on neural correlates of episodic memory encoding and retrieval. Schizophr Bull (2013) 39(1):141–50. doi: 10.1093/schbul/sbr076
68. Thurin K, Rasetti R, Sambataro F, Safrin M, Chen Q, Callicott JH, et al. Effects of ZNF804A on neurophysiologic measures of cognitive control. Mol Psychiatry (2013) 18(8):852–4. doi: 10.1038/mp.2012.134
69. Thimm M, Kircher T, Kellermann T, Markov V, Krach S, Jansen A, et al. Effects of a CACNA1C genotype on attention networks in healthy individuals. Psychol Med (2011) 41(7):1551–61. doi: 10.1017/S0033291710002217
70. Zhang Z, Chen X, Yu P, Zhang Q, Sun X, Gu H, et al. Evidence for the contribution of NOS1 gene polymorphism (rs3782206) to prefrontal function in schizophrenia patients and healthy controls. Neuropsychopharmacology (2015) 40(6):1383–94.
71. Drabant EM, Hariri AR, Meyer-Lindenberg A, Munoz KE, Mattay VS, Kolachana BS, et al. Catechol O-methyltransferase val158met genotype and neural mechanisms related to affective arousal and regulation. Arch Gen Psychiatry (2006) 63(12):1396–406. doi: 10.1001/archpsyc.63.12.1396
72. Blasi G, Lo Bianco L, Taurisano P, Gelao B, Romano R, Fazio L, et al. Functional variation of the dopamine D2 receptor gene is associated with emotional control as well as brain activity and connectivity during emotion processing in humans. J Neurosci (2009) 29(47):14812–9. doi: 10.1523/JNEUROSCI.3609-09.2009
73. Mothersill O, Morris DW, Kelly S, Rose EJ, Fahey C, O’Brien C, et al. Effects of MIR137 on fronto-amygdala functional connectivity. NeuroImage (2014) 90:189–95. doi: 10.1016/j.neuroimage.2013.12.019
74. Walter H, Schnell K, Erk S, Arnold C, Kirsch P, Esslinger C, et al. Effects of a genome-wide supported psychosis risk variant on neural activation during a theory-of-mind task. Mol Psychiatry (2011) 16(4):462–70. doi: 10.1038/mp.2010.18
75. Mohnke S, Erk S, Schnell K, Schutz C, Romanczuk-Seiferth N, Grimm O, et al. Further evidence for the impact of a genome-wide-supported psychosis risk variant in ZNF804A on the Theory of Mind Network. Neuropsychopharmacology (2014) 39(5):1196–205. doi: 10.1038/npp.2013.321
76. Cassidy C, Buchy L, Bodnar M, Dell’elce J, Choudhry Z, Fathalli F, et al. Association of a risk allele of ANK3 with cognitive performance and cortical thickness in patients with first-episode psychosis. J Psychiatry Neurosci (2014) 39(1):31–9. doi: 10.1503/jpn.120242
77. Hata T, Kunugi H, Nanko S, Fukuda R, Kaminaga T. Possible effect of the APOE epsilon 4 allele on the hippocampal volume and asymmetry in schizophrenia. Am J Med Genet (2002) 114(6):641–2. doi: 10.1002/ajmg.10556
78. Ho BC, Milev P, O’Leary DS, Librant A, Andreasen NC, Wassink TH. Cognitive and magnetic resonance imaging brain morphometric correlates of brain-derived neurotrophic factor Val66Met gene polymorphism in patients with schizophrenia and healthy volunteers. Arch Gen Psychiatry (2006) 63(7):731–40. doi: 10.1001/archpsyc.63.7.731
79. Pezawas L, Verchinski BA, Mattay VS, Callicott JH, Kolachana BS, Straub RE, et al. The brain-derived neurotrophic factor val66met polymorphism and variation in human cortical morphology. J Neurosci (2004) 24(45):10099–102.
80. Bueller JA, Aftab M, Sen S, Gomez-Hassan D, Burmeister M, Zubieta JK. BDNF Val66Met allele is associated with reduced hippocampal volume in healthy subjects. Biol Psychiatry (2006) 59(9):812–5. doi: 10.1016/j.biopsych.2005.09.022
81. Aas M, Haukvik UK, Djurovic S, Bergmann O, Athanasiu L, Tesli MS, et al. BDNF val66met modulates the association between childhood trauma, cognitive and brain abnormalities in psychoses. Prog Neuro-psychopharmacol Biol Psychiatry (2013) 46:181–8. doi: 10.1016/j.pnpbp.2013.07.008
82. Carballedo A, Morris D, Zill P, Fahey C, Reinhold E, Meisenzahl E, et al. Brain-derived neurotrophic factor Val66Met polymorphism and early life adversity affect hippocampal volume. Am J Med GenetPart B Neuropsychiatr Genet (2013) 162B(2):183–90. doi: 10.1002/ajmg.b.32130
83. Gatt JM, Nemeroff CB, Dobson-Stone C, Paul RH, Bryant RA, Schofield PR, et al. Interactions between BDNF Val66Met polymorphism and early life stress predict brain and arousal pathways to syndromal depression and anxiety. Mol Psychiatry (2009) 14(7):681–95.
84. Gerritsen L, Tendolkar I, Franke B, Vasquez AA, Kooijman S, Buitelaar J, et al. BDNF Val66Met genotype modulates the effect of childhood adversity on subgenual anterior cingulate cortex volume in healthy subjects. Mol Psychiatry (2012) 17(6):597–603.
85. Nemoto K, Ohnishi T, Mori T, Moriguchi Y, Hashimoto R, Asada T, et al. The Val66Met polymorphism of the brain-derived neurotrophic factor gene affects age-related brain morphology. Neurosci Lett (2006) 397(1-2):25–9. doi: 10.1016/j.neulet.2005.11.067
86. Wang F, McIntosh AM, He Y, Gelernter J, Blumberg HP. The association of genetic variation in CACNA1C with structure and function of a frontotemporal system. Bipolar Disord (2011) 13(7-8):696–700. doi: 10.1111/j.1399-5618.2011.00963.x
87. Cerasa A, Gioia MC, Labate A, Liguori M, Lanza P, Quattrone A. Impact of catechol-O-methyltransferase Val(108/158) Met genotype on hippocampal and prefrontal gray matter volume. Neuroreport (2008) 19(4):405–8. doi: 10.1097/WNR.0b013e3282f5f784
88. Honea R, Verchinski BA, Pezawas L, Kolachana BS, Callicott JH, Mattay VS, et al. Impact of interacting functional variants in COMT on regional gray matter volume in human brain. NeuroImage (2009) 45(1):44–51.
89. Mechelli A, Tognin S, McGuire PK, Prata D, Sartori G, Fusar-Poli P, et al. Genetic vulnerability to affective psychopathology in childhood: a combined voxel-based morphometry and functional magnetic resonance imaging study. Biol Psychiatry (2009) 66(3):231–7.
90. Taylor WD, Zuchner S, Payne ME, Messer DF, Doty TJ, MacFall JR, et al. The COMT Val158Met polymorphism and temporal lobe morphometry in healthy adults. Psychiatry Res (2007) 155(2):173–7.
91. McIntosh AM, Baig BJ, Hall J, Job D, Whalley HC, Lymer GK, et al. Relationship of catechol-O-methyltransferase variants to brain structure and function in a population at high risk of psychosis. Biol Psychiatry (2007) 61(10):1127–34. doi: 10.1016/j.biopsych.2006.05.020
92. Ohnishi T, Hashimoto R, Mori T, Nemoto K, Moriguchi Y, Iida H, et al. The association between the Val158Met polymorphism of the catechol-O-methyl transferase gene and morphological abnormalities of the brain in chronic schizophrenia. Brain (2006) 129(Pt 2):399–410.
93. Ho BC, Wassink TH, O’Leary DS, Sheffield VC, Andreasen NC. Catechol-O-methyl transferase Val158Met gene polymorphism in schizophrenia: working memory, frontal lobe MRI morphology and frontal cerebral blood flow. Mol Psychiatry (2005) 10(3):229, 287-298.
94. Gruber O, Falkai P, Schneider-Axmann T, Schwab SG, Wagner M, Maier W. Neuregulin-1 haplotype HAP(ICE) is associated with lower hippocampal volumes in schizophrenic patients and in non-affected family members. J Psychiatr Res (2008) 43(1):1–6.
95. Tosato S, Bellani M, Bonetto C, Ruggeri M, Perlini C, Lasalvia A, et al. Is neuregulin 1 involved in determining cerebral volumes in schizophrenia? Preliminary results showing a decrease in superior temporal gyrus volume. Neuropsychobiology (2012) 65(3):119–25. doi: 10.1159/000330584
96. Addington AM, Gornick MC, Shaw P, Seal J, Gogtay N, Greenstein D, et al. Neuregulin 1 (8p12) and childhood-onset schizophrenia: susceptibility haplotypes for diagnosis and brain developmental trajectories. Mol Psychiatry (2007) 12(2):195–205.
97. Lencz T, Szeszko PR, DeRosse P, Burdick KE, Bromet EJ, Bilder RM, et al. A schizophrenia risk gene, ZNF804A, influences neuroanatomical and neurocognitive phenotypes. Neuropsychopharmacology (2010) 35(11):2284–91. doi: 10.1038/npp.2010.102
98. Donohoe G, Rose E, Frodl T, Morris D, Spoletini I, Adriano F, et al. ZNF804A risk allele is associated with relatively intact gray matter volume in patients with schizophrenia. NeuroImage (2011) 54(3):2132–7. doi: 10.1016/j.neuroimage.2010.09.089
99. Wassink TH, Epping EA, Rudd D, Axelsen M, Ziebell S, Fleming FW, et al. Influence of ZNF804a on brain structure volumes and symptom severity in individuals with schizophrenia. Arch Gen Psychiatry (2012) 69(9):885–92. doi: 10.1001/archgenpsychiatry.2011.2116
100. Ohi K, Hashimoto R, Ikeda M, Yamashita F, Fukunaga M, Nemoto K, et al. Genetic risk variants of schizophrenia associated with left superior temporal gyrus volume. Cortex (2014) 58:23–6. doi: 10.1016/j.cortex.2014.05.011
101. Terwisscha van Scheltinga AF, Bakker SC, van Haren NE, Derks EM, Buizer-Voskamp JE, Boos HB, et al. Genetic schizophrenia risk variants jointly modulate total brain and white matter volume. Biol Psychiatry (2013) 73(6):525–31. doi: 10.1016/j.biopsych.2012.08.017
102. Harrisberger F, Smieskova R, Vogler C, Egli T, Schmidt A, Lenz C, et al. Impact of polygenic schizophrenia-related risk and hippocampal volumes on the onset of psychosis. Transl Psychiatry (2016) 6(8):e868. doi: 10.1038/tp.2016.143
103. Zhang Z, Chen X, Yu P, Zhang Q, Sun X, Gu H, et al. Evidence for the contribution of NOS1 gene polymorphism (rs3782206) to prefrontal function in schizophrenia patients and healthy controls. Neuropsychopharmacology (2015) 40(6):1383–94. doi: 10.1038/npp.2014.323
104. Friston KJ, Frith CD. Schizophrenia: a disconnection syndrome? Clin Neurosci (1995) 3(2):89–97.
105. Tost H, Bilek E, Meyer-Lindenberg A. Brain connectivity in psychiatric imaging genetics. NeuroImage (2012) 62(4):2250–60. doi: 10.1016/j.neuroimage.2011.11.007
106. Wheeler AL, Voineskos AN. A review of structural neuroimaging in schizophrenia: from connectivity to connectomics. Front Hum Neurosci (2014) 8:653. doi: 10.3389/fnhum.2014.00653
107. Molendijk ML, Bus BA, Spinhoven P, Kaimatzoglou A, Oude Voshaar RC, Penninx BW, et al. A systematic review and meta-analysis on the association between BDNF val(66)met and hippocampal volume—a genuine effect or a winners curse? Am J Med GenetPart B Neuropsychiatr Genet (2012) 159B(6):731–40. doi: 10.1002/ajmg.b.32078
108. Ho BC, Andreasen NC, Dawson JD, Wassink TH. Association between brain-derived neurotrophic factor Val66Met gene polymorphism and progressive brain volume changes in schizophrenia. Am J Psychiatry (2007) 164(12):1890–9. doi: 10.1176/appi.ajp.2007.05111903
109. Munafo MR, Thiselton DL, Clark TG, Flint J. Association of the NRG1 gene and schizophrenia: a meta-analysis. Mol Psychiatry (2006) 11(6):539–46. doi: 10.1038/sj.mp.4001817
110. Duff BJ, Macritchie KA, Moorhead TW, Lawrie SM, Blackwood DH. Human brain imaging studies of DISC1 in schizophrenia, bipolar disorder and depression: a systematic review. Schizophr Res (2013) 147(1):1–13. doi: 10.1016/j.schres.2013.03.015
111. Hibar DP, Stein JL, Renteria ME, Arias-Vasquez A, Desrivieres S, Jahanshad N, et al. Common genetic variants influence human subcortical brain structures. Nature (2015) 520(7546):224–9.
112. Thompson PM, Stein JL, Medland SE, Hibar DP, Vasquez AA, Renteria ME, et al. The ENIGMA Consortium: large-scale collaborative analyses of neuroimaging and genetic data. Brain Imaging Behav (2014) 8(2):153–82.
113. Franke B, Stein JL, Ripke S, Anttila V, Hibar DP, van Hulzen KJ, et al. Genetic influences on schizophrenia and subcortical brain volumes: large-scale proof of concept. Nat Neurosci (2016) 19(3):420–31. doi: 10.1038/nn.4228
114. Haijma SV, Van Haren N, Cahn W, Koolschijn PC, Hulshoff Pol HE, Kahn RS. Brain volumes in schizophrenia: a meta-analysis in over 18 000 subjects. Schizophr Bull (2013) 39(5):1129–38. doi: 10.1093/schbul/sbs118
115. Gottesman II, Shields J. A polygenic theory of schizophrenia. Proc Nat Acad Sci U S A (1967) 58(1):199–205. doi: 10.1073/pnas.58.1.199
116. Hu M, Li J, Eyler L, Guo X, Wei Q, Tang J, et al. Decreased left middle temporal gyrus volume in antipsychotic drug-naive, first-episode schizophrenia patients and their healthy unaffected siblings. Schizophr Res (2013) 144(1–3):37–42. doi: 10.1016/j.schres.2012.12.018
117. Ordonez AE, Luscher ZI, Gogtay N. Neuroimaging findings from childhood onset schizophrenia patients and their non-psychotic siblings. Schizophr Res (2016) 173(3):124–31. doi: 10.1016/j.schres.2015.03.003
118. Meyer-Lindenberg A, Tost H. Neuroimaging and plasticity in schizophrenia. Restor Neurol Neurosci (2014) 32(1):119–27.
119. Adriano F, Caltagirone C, Spalletta G. Hippocampal volume reduction in first-episode and chronic schizophrenia: a review and meta-analysis. Neuroscientist (2012) 18(2):180–200. doi: 10.1177/1073858410395147
120. Schmaal L, Veltman DJ, van Erp TG, Samann PG, Frodl T, Jahanshad N, et al. Subcortical brain alterations in major depressive disorder: findings from the ENIGMA Major Depressive Disorder working group. Mol Psychiatry (2016) 21(6):806–12.
121. Frodl T, Janowitz D, Schmaal L, Tozzi L, Dobrowolny H, Stein DJ, et al. Childhood adversity impacts on brain subcortical structures relevant to depression. J Psychiatr Res (2017) 86:58–65. doi: 10.1016/j.jpsychires.2016.11.010
122. Boedhoe PS, Schmaal L, Abe Y, Ameis SH, Arnold PD, Batistuzzo MC, et al. Distinct Subcortical volume alterations in pediatric and adult OCD: a worldwide meta- and mega-analysis. Am J Psychiatry (2017) 174(1):60–9.
123. Hoogman M, Bralten J, Hibar DP, Mennes M, Zwiers MP, Schweren LS, et al. Subcortical brain volume differences in participants with attention deficit hyperactivity disorder in children and adults: a cross-sectional mega-analysis. Lancet Psychiatry (2017) 4(4):310–9. doi: 10.1016/S2215-0366(17)30160-8
124. Fotuhi M, Do D, Jack C. Modifiable factors that alter the size of the hippocampus with ageing. Nat Rev Neurol (2012) 8(4):189–202. doi: 10.1038/nrneurol.2012.27
125. Jochem C, Baumeister SE, Wittfeld K, Leitzmann MF, Bahls M, Schminke U, et al. Domains of physical activity and brain volumes: A population-based study. NeuroImage (2017) 156:101–8. doi: 10.1016/j.neuroimage.2017.05.020
126. Schmitt A, Malchow B, Hasan A, Falkai P. The impact of environmental factors in severe psychiatric disorders. Front Neurosci (2014) 8:19. doi: 10.3389/fnins.2014.00019
127. Bois C, Ronan L, Levita L, Whalley HC, Giles S, McIntosh AM, et al. Cortical surface area differentiates familial high risk individuals who go on to develop schizophrenia. Biol Psychiatry (2015) 78(6):413–20. doi: 10.1016/j.biopsych.2014.12.030
128. Brandt CL, Doan NT, Tonnesen S, Agartz I, Hugdahl K, Melle I, et al. Assessing brain structural associations with working-memory related brain patterns in schizophrenia and healthy controls using linked independent component analysis. NeuroImage Clin (2015) 9:253–63. doi: 10.1016/j.nicl.2015.08.010
129. Gurung R, Prata DP. What is the impact of genome-wide supported risk variants for schizophrenia and bipolar disorder on brain structure and function? A systematic review. Psychol Med (2015) 45(12):2461–80. doi: 10.1017/S0033291715000537
130. Chen J, Calhoun VD, Pearlson GD, Perrone-Bizzozero N, Sui J, Turner JA, et al. Guided exploration of genomic risk for gray matter abnormalities in schizophrenia using parallel independent component analysis with reference. NeuroImage (2013) 83:384–96. doi: 10.1016/j.neuroimage.2013.05.073
131. Chen J, Calhoun VD, Arias-Vasquez A, Zwiers MP, van Hulzen K, Fernandez G, et al. G-protein genomic association with normal variation in gray matter density. Hum Brain Mapp (2015) 36(11):4272–86. doi: 10.1002/hbm.22916
132. Pearlson GD, Clementz BA, Sweeney JA, Keshavan MS, Tamminga CA. Does biology transcend the symptom-based boundaries of psychosis? Psychiatr Clin North Am (2016) 39(2):165–74. doi: 10.1016/j.psc.2016.01.001
133. Derogatis LR, Cleary PA. Factorial invariance across gender for the primary symptom dimensions of the SCL-90. Br J Soc Clin Psychol (1977) 16(4):347–56. doi: 10.1111/j.2044-8260.1977.tb00241.x
134. Schmitz N, Hartkamp N, Kiuse J, Franke GH, Reister G, Tress W. The Symptom Check-List-90-R (SCL-90-R): a German validation study. Qual Life Res (2000) 9(2):185–93. doi: 10.1023/A:1008931926181
135. Carter CS, Bearden CE, Bullmore ET, Geschwind DH, Glahn DC, Gur RE, et al. Enhancing the informativeness and replicability of imaging genomics studies. Biol Psychiatry (2017) 82(3):157–64. doi: 10.1016/j.biopsych.2016.08.019
136. Ioannidis JP. Why most published research findings are false. PLoS Med (2005) 2(8):e124. doi: 10.1371/journal.pmed.0020124
Keywords: imaging genetics, diagnostic catalogues, heterogeneity, genetic overlap, brain alterations
Citation: Jiang W, King TZ and Turner JA (2019) Imaging Genetics Towards a Refined Diagnosis of Schizophrenia. Front. Psychiatry 10:494. doi: 10.3389/fpsyt.2019.00494
Received: 26 March 2019; Accepted: 24 June 2019;
Published: 12 July 2019.
Edited by:
Lena K. Palaniyappan, University of Western Ontario, CanadaReviewed by:
Rajeev Krishnadas, NHS Greater Glasgow and Clyde, United KingdomNina Kraguljac, University of Alabama at Birmingham, United States
Copyright © 2019 Jiang, King and Turner. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wenhao Jiang, d2ppYW5nNUBzdHVkZW50LmdzdS5lZHU=