 Tejas Dharmaraj
Tejas Dharmaraj Youchen Guan
Youchen Guan Julie Liu1
Julie Liu1 Benedicte Gaborit
Benedicte Gaborit Katherine L. Wilson
Katherine L. Wilson- 1Department of Cell Biology, Johns Hopkins University School of Medicine, Baltimore, MD, United States
- 2INSERM, MMG, Aix Marseille Université, Marseille, France
- 3INSERM, INRA, C2VN, Aix Marseille Université, Marseille, France
Emerin (EMD) and barrier to autointegration factor 1 (BANF1) each bind A-type lamins (LMNA) as fundamental components of nuclear lamina structure. Mutations in LMNA, EMD and BANF1 are genetically linked to many tissue-specific disorders including Emery-Dreifuss muscular dystrophy and cardiomyopathy (LMNA, EMD), lipodystrophy, insulin resistance and type 2 diabetes (LMNA) and progeria (LMNA, BANF1). To explore human genetic variation in these genes, we analyzed EMD and BANF1 alleles in the Exome Aggregation Consortium (ExAC) cohort of 60,706 unrelated individuals. We identified 13 rare heterozygous BANF1 missense variants (p.T2S, p.H7Y, p.D9N, p.S22R, p.G25E, p.D55N, p.D57Y, p.L63P, p.N70T, p.K72R, p.R75W, p.R75Q, p.G79R), and one homozygous variant (p.D9H). Several variants are known (p.G25E) or predicted (e.g., p.D9H, p.D9N, p.L63P) to perturb BANF1 and warrant further study. Analysis of EMD revealed two previously identified variants associated with adult-onset cardiomyopathy (p.K37del, p.E35K) and one deemed ‘benign’ in an Emery-Dreifuss patient (p.D149H). Interestingly p.D149H was the most frequent emerin variant in ExAC, identified in 58 individuals (overall allele frequency 0.06645%), of whom 55 were East Asian (allele frequency 0.8297%). Furthermore, p.D149H associated with four ‘healthy’ traits: reduced triglycerides (-0.336; p = 0.0368), reduced waist circumference (-0.321; p = 0.0486), reduced cholesterol (-0.572; p = 0.000346) and reduced LDL cholesterol (-0.599; p = 0.000272). These traits are distinct from LMNA-associated metabolic disorders and provide the first insight that emerin influences metabolism. We also identified one novel in-frame deletion (p.F39del) and 62 novel emerin missense variants, many of which were relatively frequent and potentially disruptive including p.N91S and p.S143F (∼0.041% and ∼0.034% of non-Finnish Europeans, respectively), p.G156S (∼0.39% of Africans), p.R204G (∼0.18% of Latinx), p.R207P (∼0.08% of South Asians) and p.R221L (∼0.15% of Latinx). Many novel BANF1 variants are predicted to disrupt dimerization or binding to DNA, histones, emerin or A-type lamins. Many novel emerin variants are predicted to disrupt emerin filament dynamics or binding to BANF1, HDAC3, A-type lamins or other partners. These new human variants provide a foundational resource for future studies to test the molecular mechanisms of BANF1 and emerin function, and to understand the link between emerin variant p.D149H and a ‘healthy’ lipid profile.
Introduction
Emerin is a conserved ‘LEM-domain’ component of nuclear lamina networks, which have central roles in nuclear structure and function (Simon and Wilson, 2011; Barton et al., 2015; Gruenbaum and Foisner, 2015). Emerin, an integral membrane protein, localizes mainly at the inner membrane of the nuclear envelope (NE) and binds directly to lamins (nuclear intermediate filaments), LINC complexes and an essential chromatin protein named BANF1 (Berk et al., 2013b; Jamin and Wiebe, 2015; Janin et al., 2017). Nuclear lamina proteins have multiple and varied roles from mitosis to tissue-specific signaling (Gerace and Tapia, 2018). For example, during telophase of mitosis, segregating daughter chromosomes are coalesced by BANF1 (Dharmaraj and Wilson, 2017; Samwer et al., 2017). BANF1 then cooperates with emerin, other LEM-domain proteins and lamins to reassemble the nucleus (Liu et al., 2000, 2003; Dechat et al., 2004; Margalit et al., 2005; Barkan et al., 2012; Berk et al., 2013b) and re-establish tissue-specific 3D genome organization (Zullo et al., 2012; Demmerle et al., 2013; Harr et al., 2015; Stevens et al., 2017; van Steensel and Belmont, 2017).
Highlighting these complex roles, missense mutations in lamins and NE membrane proteins cause tissue-specific disorders, termed ‘laminopathies’ (Worman and Schirmer, 2015; Janin et al., 2017). Hundreds of disease-causing variants are reported for the gene encoding A-type lamins (LMNA; Bonne et al., 2004; Dittmer and Misteli, 2011). We previously identified novel LMNA missense alleles at frequencies as high as ∼0.4% in specific ethnic groups (Florwick et al., 2017) among 60,706 unrelated individuals (Lek et al., 2016). This raised questions about potential genetic variation in two key lamin-binding proteins: emerin (EMD) and BANF1 (BANF1).
Emerin is expressed in all tissues and has diverse roles in signaling, cell proliferation, mechanotransduction, transcriptional regulation and 3D genome organization (Demmerle et al., 2013; Le et al., 2016; Iyer et al., 2017; Kirby and Lammerding, 2018). Emerin is specifically important in striated muscle (Barton et al., 2015; Iyer et al., 2017), since loss of emerin causes X-linked recessive EDMD1 (Bonne et al., 2004). EDMD1 is characterized by early contractures of the Achilles, elbow, and neck tendons, progressive muscle weakness and wasting, and dilated cardiomyopathy that can lead to sudden and potentially lethal cardiac arrest. In cardiomyocytes, loss of emerin enhances MAP kinase-dependent gene expression, perturbing heart physiology (Muchir et al., 2007; Huber et al., 2009; Stubenvoll et al., 2015). The same disease, EDMD, is also linked to mutations in proteins that bind emerin including A-type lamins, nesprin-1, nesprin-2, Sun1, Sun2 and LUMA/Tmem43 (Dobrzynska et al., 2016; Pillers and Von Bergen, 2016).
Emerin specifically mediates nuclear responses to mechanical force (Lammerding et al., 2005; Le et al., 2016; Qi et al., 2016; Lele et al., 2018), particularly on stiff substrates (Willer and Carroll, 2017). Emerin is needed to activate mechanoresponsive genes (Lammerding et al., 2005; Le et al., 2016), influences cytoskeletal F-actin networks (Chang et al., 2013) and regulates the SRF-Mkl1 coactivator complex on stiff substrates (Ho et al., 2013; Willer and Carroll, 2017). ‘Pulling’ forces applied to LINC complexes on isolated nuclei cause Tyr-phosphorylation of emerin at unidentified sites, dependent on the integrity of two Tyr residues targeted by Src (emerin Y74 and Y95; Tifft et al., 2009; Guilluy et al., 2014). However, the mechanisms by which emerin, an intrinsically disordered ‘transformer’ protein (Samson et al., 2016), senses force and signals in response to force are open questions. Emerin has at least two modes of self-association with the potential to form networks or filaments (Berk et al., 2014; Herrada et al., 2015; Samson et al., 2017). Other partners include nucleo/cytoskeletal proteins (e.g., F-actin, myosin 1c, tubulin), nuclear membrane proteins (e.g., LAP1, Samp1) and transcriptional regulators such as HDAC3, b-catenin and Lmo7 (Holaska et al., 2006; Berk et al., 2013b; Shin et al., 2013; Barton et al., 2015; Vijayaraghavan et al., 2018).
Additional partners and roles for emerin are emerging in disparate tissues. For example, emerin, BANF1 and lamin A/C associate with the sigma-1 receptor in neurons; association is enhanced by the sigma-1 receptor agonist, cocaine, leading to transcriptional repression of a gene, MAOB1, the product of which degrades dopamine (Tsai et al., 2015). The bacterium Chlamydia psittaci, which causes rapid and potentially lethal pneumonia, targets emerin and emerin-associated nuclear membrane proteins (Mojica et al., 2015). In models of breast and prostate cancer, loss of emerin correlates with increased metastatic potential (Hu et al., 2011; Wozniak et al., 2013; Reis-Sobreiro et al., 2018). Mechanisms and partners for emerin in these contexts are unexplored.
To our knowledge, only one BANF1 variant is reported in humans. This variant, p.A12T, was identified in two homozygous individuals and genetically linked to Nestor-Guillermo progeria syndrome (Puente et al., 2011). This variant perturbs BANF1 binding to dsDNA (Paquet et al., 2014) and weakens BANF1 binding to a progeria-related surface on the Ig-fold domain of lamins A and C (Samson et al., 2018). BANF1 is the conserved partner for all LEM-domain proteins (Laguri et al., 2001), and also binds lamins (Holaska et al., 2003; Samson et al., 2018). Indeed, BANF1 can bind emerin and lamin A simultaneously (Bengtsson and Wilson, 2006; Samson et al., 2018). BANF1 is essential during mitosis: BANF1 collects segregating chromosomes within a single nucleus (Samwer et al., 2017; Dharmaraj and Wilson, 2017) and coordinates with lamins and LEM-domain proteins to reassemble nuclear lamina structure (Haraguchi et al., 2008; Simon and Wilson, 2011). BANF1 is mislocalized in cells that express disease mutations in lamin A (Capanni et al., 2012) and is immobilized by fasting (Bar et al., 2014). BANF1 is an epigenetic regulator (Montes de Oca et al., 2011; Oh et al., 2015) and has roles in chromatin organization (Loi et al., 2015), antiviral defense (Kobayashi and Haraguchi, 2015; Wiebe and Jamin, 2016), suppression of endoreplication (Brayson et al., 2018; Wang et al., 2018) and necrotic pyknosis (Hou et al., 2016). BANF1 regulates the insertion of endogenous mobile elements in eukaryotic genomes; this activity is exploited by retroviruses including HIV-1 (Harris and Engelman, 2000; Wang et al., 2014; Jamin and Wiebe, 2015).
DNA sequencing of human populations has the potential to reveal novel phenotypes or missense variants in specific proteins-of-interest, as sources of insight and information for mechanistic studies. This strategy, applied to the gene encoding A-type lamins (LMNA), revealed unexpectedly high frequencies of disease-causing LMNA alleles in specific ethnic groups, and linked a specific missense variant to type 2 diabetes (Florwick et al., 2017). Other key components of nuclear lamina structure, emerin (EMD) and BANF1 (BANF1), were unexplored. We therefore analyzed EMD and BANF1 variants in the Exome Aggregation Consortium (ExAC) cohort of 60,706 unrelated individuals (Lek et al., 2016), which includes exome sequences from men and women with diverse ancestries (8.6% African, 9.5% Latinx, 7.1% East Asian, 5.4% Finnish, 55% non-Finish European, 13.6% South Asian, 0.7% other). About half of these individuals either have a specific condition (heart disease, type 2 diabetes, schizophrenia or other disorders) or serve as controls for each condition. We anticipated few loss-of-function EMD alleles, since ExAC excludes individuals with severe childhood-onset disorders such as EDMD1. We anticipated few alleles for BANF1, given its conservation and essential roles (Zheng et al., 2000; Montes de Oca et al., 2005; Samwer et al., 2017). Highlighting the value of population diversity represented in ExAC, we identified 14 novel BANF1 missense alleles and many novel EMD alleles, ten of which were relatively frequent in specific ethnic populations. We discovered that emerin variant p.D149H, identified in 0.8% of East Asians, associates with a healthy lipid profile including reduced triglycerides and reduced LDL cholesterol. Our detailed structure-function analysis predicts that many identified BANF1 and emerin variants are potentially disruptive and warrant further study.
Materials and Methods
ExAC Database Searches
Version 0.3.1 ExAC was queried for ‘EMD’ and ‘BANF1’ via the ExAC Browser (Beta), selecting only for variants that passed the quality assurance filter (Lek et al., 2016; ExAC database, 2018). The data was narrowed to include in-frame deletions and missense, synonymous and splice variants, and exported in the .csv file format. To identify variants associated with broadly defined ‘psychiatric disease’ (schizophrenia, bipolar disorder, Tourette syndrome), we compared two variant call files from ExAC version 0.3. The first file (ExAC.r0.3.sites.vep.vcf) contained all ExAC variants; the second file (ExAC.r0.3.nonpsych.sites.vcf) contained all variants not found in the psychiatric cohort. By subtracting the ‘nonpsych’ variants from the full cohort using the UNIX ‘comm’ tool in a shell script, further selecting for emerin and BANF1 alleles, and manually curating the final list to handle the consequences of minimal and multi-allelic representation, we were able to generate a file of emerin and BANF1 variants unique to the psychiatric cohort. To subset against TCGA, we subtracted the variants not reported in the TCGA dataset (ExAC_nonTCGA.r0.3.1.sites.vep.vcf) from the full cohort to generate a list of potentially ‘cancer-unique’ alleles.
Laminopathy Database
To search for previously identified variants, we queried the Universal Mutation Database and the Leiden Muscular Dystrophy Pages (powered by Leiden Open Variation Database v2.0; accessed September 2018) for EMD variants. After curating to exclude all non-missense mutations and non-canonical spliceforms, only three variants were shared with ExAC.
Type 2 Diabetes (T2D) Knowledge Portal
Since we were unable to determine the clinical status of variant carriers in ExAC, we searched the open-access Type 2 Diabetes Knowledge Portal for EMD variants. ‘Associations across all datasets’ for each ExAC EMD variant were obtained using the variant ID or reference SNP ID (rs) as the search term in the T2D portal1. The only protein-coding variant with significant and ‘high-impact’ associations (p.D149H) was identified solely within the 13K Exome Sequence Analysis cohort, which includes samples from 13,007 individuals (half with T2D, half controls) representing five ancestries: European (∼5,000 individuals), African-American, East Asian, South Asian and Hispanic (∼2,000 individuals each). All p.D149H-related statistics come from the analysis reported in the T2D Knowledge Portal.
Statistical Analysis to Query Potential Sex Distribution Bias
To determine if the sex distribution of variants differed from their expected distribution, we calculated expected distributions based on the ExAC sampling of the sex distribution for each ethnic sub-group in which the allele was most concentrated as a first approximation. For example, the allele producing variant p.N91S was found in 19 individuals of non-Finnish European (nFE) ethnicity, so the proportion of males in the nFE sample of ExAC (56.16%) was used to estimate the expected male allele frequency. To compare the expected versus observed sex distributions, we conducted a chi-squared goodness-of-fit test with 1 degree of freedom. No differences in male/female distribution were found to be significant. Even p.R207P, identified only in men, was explained by a very high proportion of males in the South Asian subgroup where this allele was concentrated.
Disorder Prediction
To predict intrinsically disordered regions in ExAC variants, variant emerin sequences were submitted as queries to PONDR-FIT VL-XT2. Raw VL-XT output scores are real numbers from 0 to 1, where 0 is ‘order’ and 1 is ‘disorder,’ and were plotted.
Hydrophobicity and Helical Wheel Plots
Kyte-Doolittle hydrophobicity plots3 were used to assess the impact of variants located in the C-terminal transmembrane domain. Helical wheel projections showing the positions of variants in the emerin transmembrane domain (residues 226–244) were generated using the Emboss pepwheel tool with default settings4. ΔG values for the insertion of transmembrane helices into membranes were calculated using the ΔG prediction server v1.05 (Hessa et al., 2007).
Structural Depictions
PyMOL version 2.2 was used to generate ray-traced images of the solution NMR structure of the emerin LEM domain (RCSB Protein Data Bank ID: 2ODC) and the crystal structure of BANF1 in complex with the emerin LEM domain (RCSB Protein Data Bank ID: 2ODG).
Results
ExAC results for BANF1 are presented first, followed by emerin variants for which we provide allele frequencies, molecular analysis and clinical phenotype.
BANF1 Variants in ExAC
We were frankly surprised to find 14 BANF1 missense alleles, one predicted splice acceptor allele and one frameshift allele (p.F59PfsTer50) in ExAC (Table 1 and Supplementary Table S1). The frameshift (p.F59PfsTer50) and splice acceptor alleles are likely to yield unstable polypeptides, and were not considered further. Missense variant p.S22R was identified in three individuals (all African), for an allele frequency of 0.02905% in Africans. All other BANF1 missense variants were limited to one or two individuals and, surprisingly, one individual was homozygous for the variant identified (p.D9H; Table 1). We also evaluated 3′-UTR alleles of BANF1 (Supplementary Table S1) because this region is functionally important in cervical cancer, where BANF1 mRNA is suppressed by miRNA-203 via binding to 3′-UTR nucleotides 138–159 (Mao et al., 2015). This site was not affected by BANF1 3′-UTR alleles in ExAC, but might be affected by 3′-UTR alleles reported in the dbSNP database: c.∗143T > C (rs1478691828), c.∗144C > T (rs1366306879), c.∗146C > G (rs535232397), c.∗152C > T (rs1442694445). In summary, we identified 14 BANF1 missense variants; all were novel in biology, and most are non-conservative substitutions that might perturb function.
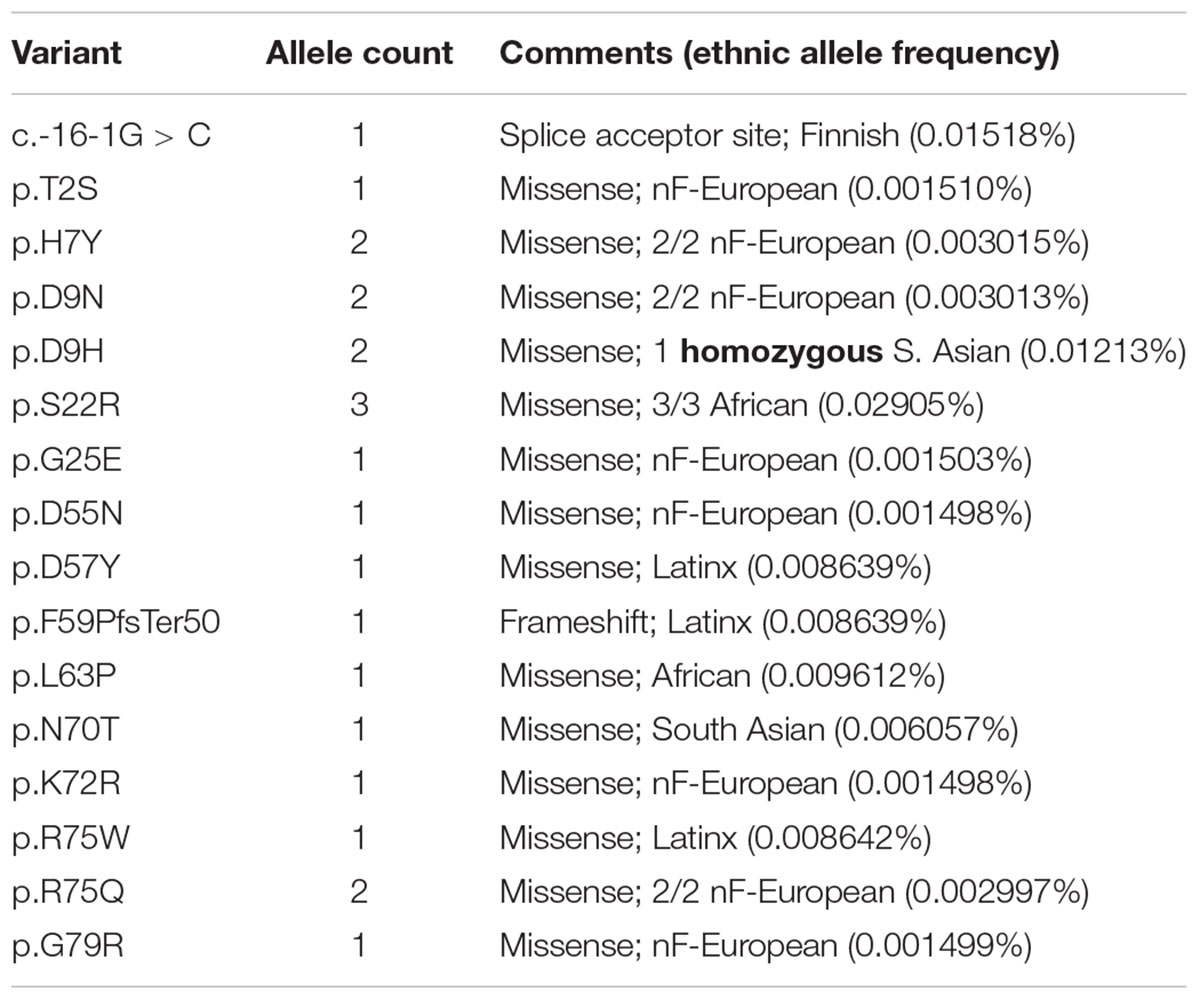
Table 1. BANF1 missense, splice acceptor site and frameshift alleles identified in ExAC.
Two BANF1 missense variants are considered disruptive. Variant p.L63P is likely to disrupt BANF1 folding or dimerization, since WT Leu63 is buried in helix a4 (Figure 1, Ribbon view); whether the resulting protein is degraded or stable is unknown. Interestingly, variant p.G25E was previously created in vitro to disrupt BANF1 dimerization: the resulting protein is stable as a monomer, shows no detectable binding to DNA, histones or emerin (Umland et al., 2000; Montes de Oca et al., 2005), and did not disrupt higher-order chromatin structure in Xenopus egg extracts (Segura-Totten et al., 2002). When over-expressed in HeLa cells, however, GFP-fused BANF1-G25E interfered with the telophase ‘core’ localization and post-mitotic re-assembly of emerin, LAP2β and lamin A (Haraguchi et al., 2001). Whether p.G25E behaves dominantly or recessively when co-expressed at normal levels with WT BANF1 is unknown.
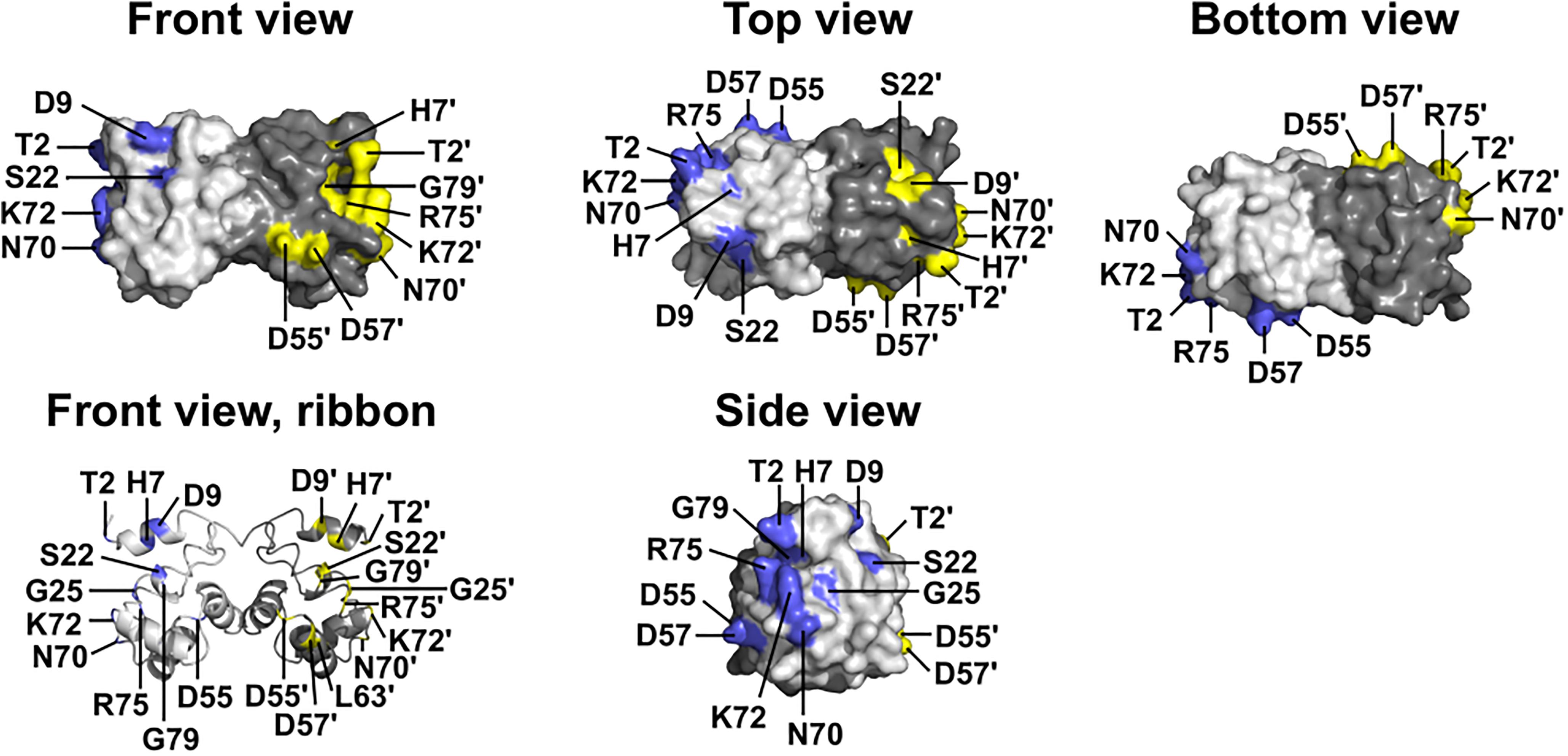
Figure 1. Wildtype BANF1 residues affected by novel ExAC missense variants. Crystal structure (Umland et al., 2000) of the BANF1 dimer showing wildtype residues affected by novel ExAC variants (RCSB Protein Data Bank ID: 1CI4). ‘Top’ and ‘Bottom’ views were obtained by rotating the ‘Front view’ 90° along its long axis out of, and into, the page respectively. ‘Side view’ shows the DNA-binding surface of BAF and was obtained by rotating the Front view 90° to the right. Wildtype residues affected by ExAC variants are colored blue in the pale-gray subunit, and yellow in the dark-gray subunit. Plain and ‘prime’ numbers indicate affected residues in the pale-gray and dark-gray subunits, respectively. Ribbon depiction of the ‘Front view’ reveals residues involved in the dimer interface, and a buried α-helical residue that we predict is disrupted by variant p.L63P.
The other 12 BANF1 missense variants all affect surface-exposed residues (Figure 1), allowing us to evaluate potential molecular impacts. Figure 1 shows ‘front,’ ‘top,’ ‘bottom’ and ‘side’ views of BANF1. WT residues affected by ExAC variants are colored blue in one subunit, yellow in the other subunit (Figure 1). Variants p.D55N and p.D57Y are non-conservative substitutions visible on the ‘front’ and ‘side’ surfaces (Figure 1); the role of the ‘front’ surface is unclear, but may influence binding to histones (Montes de Oca et al., 2005). ExAC variants p.H7Y, p.D9N, p.D9H and p.S22R probably affect the integrity of the ‘top’ surface, but are different from residues (e.g., V11, A12, P14, E83, D86, A87, F88) that contact the Ig-fold of lamins A/C (Samson et al., 2018). Variants p.D9N and p.D9H are potentially more disruptive than an in vitro mutation (p.D9A) that reduced binding to DNA, histones and emerin (Umland et al., 2000; Montes de Oca et al., 2005), but retained the ability to hyper-condense chromatin (Segura-Totten et al., 2002). No variants affected residues on the ‘bottom’ surface (Figure 1), which binds the LEM-domain (via BANF1 residues R37, F39, Q48, V51, L52, L58, E61 and W62; Samson et al., 2018). Interestingly most ExAC variants (e.g., p.H7Y, p.D9N, p.D9H, p.S22R, p.D57Y, p.N70T, p.R75W, p.R75Q, p.G79R) are likely to perturb the ‘side’ surface (Figure 1), responsible for binding to DNA (via BANF1 residues K6, G25, G27, V29 and L30, aided by S4, Q5, A71 and R75 (Bradley et al., 2005); two variants (p.R75W, p.R75Q) affected a residue involved in DNA contact. A previously studied in vitro substitution, p.R75E, reduced binding to DNA and emerin, but enhanced binding to histones (Montes de Oca et al., 2005). Further studies are needed to determine if any of these novel variants affect BANF1 dimerization, binding to histones, BANF1-dependent epigenetic regulation (Montes de Oca et al., 2009, 2011) or other roles (Jamin and Wiebe, 2015).
Emerin Variants in ExAC
Overall, EMD alleles were considered rare (defined as < 1% of the entire ExAC population), comprising 42 synonymous alleles, six nucleotide changes in splice regions with no suggested consequence, 64 missense alleles and two in-frame deletions (Supplementary Table S2). No EMD alleles associated with either cancer or broadly defined psychiatric disease, as determined by subsetting respectively against TCGA and the psychiatric disease cohort in ExAC (see Materials and Methods). Emerin is encoded by an X-linked gene, which allowed us to assess variant distributions in men (hemizygous) versus women (heterozygotes). No EMD alleles were homozygous (Supplementary Table S2). A potential sex-distribution bias of two variants, p.G156S and p.R207P, which appeared to be enriched in women and men, respectively, was an artifact of skewed representation of women versus men in specific ethnic groups (see Materials and Methods).
Further analysis focused on EMD missense and in-frame deletion alleles (henceforth ‘variants’); 39 variants were unique (identified in a single individual; Supplementary Table S2) and 27 were identified in two or more (up to 58) individuals (Table 2). The positions of all 66 variants with respect to the amino acid sequence of emerin are depicted in Figure 2A, with bar heights indicating the number of affected individuals. Residues known to be involved in self-association, filament formation or binding to BANF1, lamin A or nesprins are also depicted (Figures 2B–I).
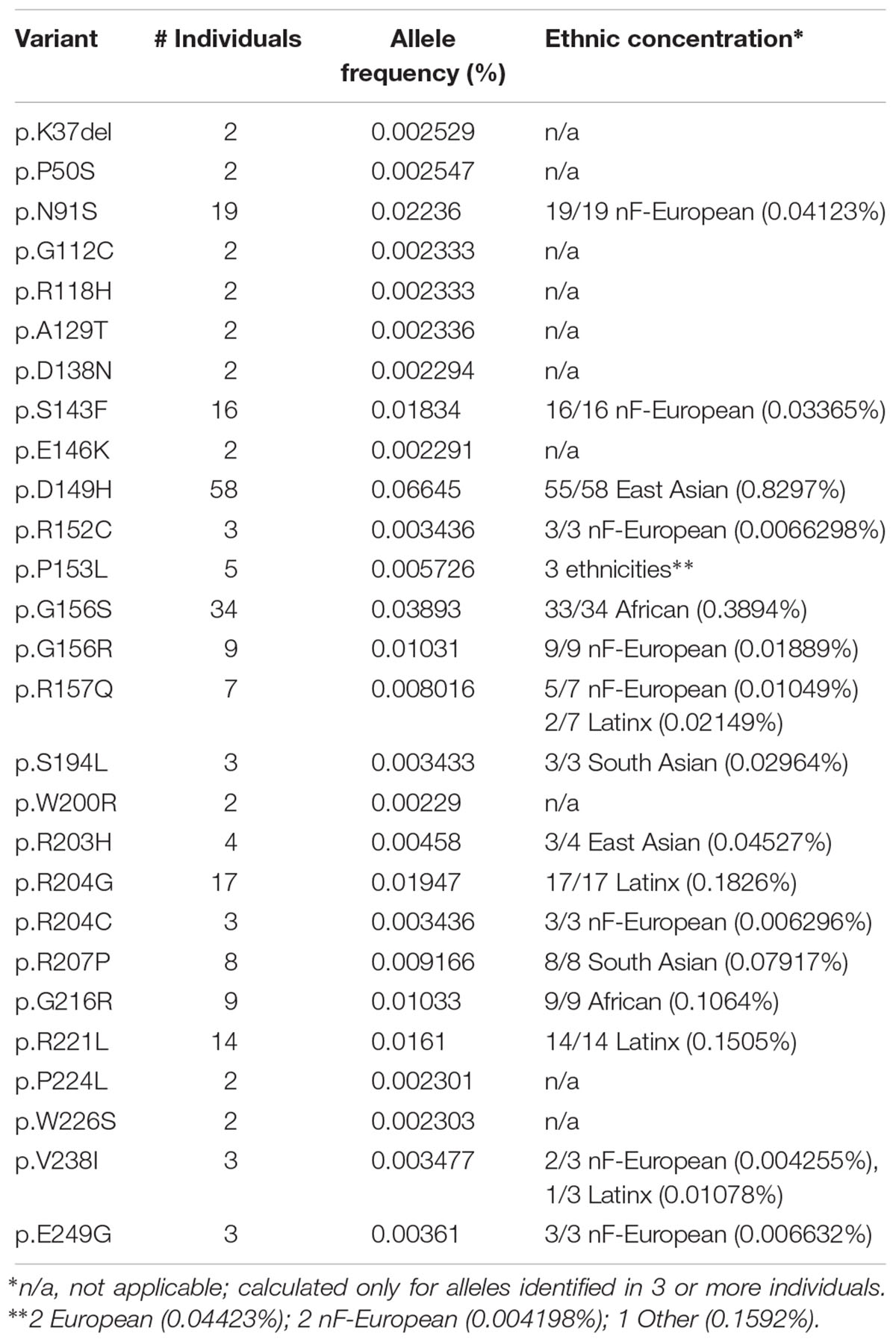
Table 2. EMD missense and in-frame-deletion alleles identified in 2+ individuals in ExAC.
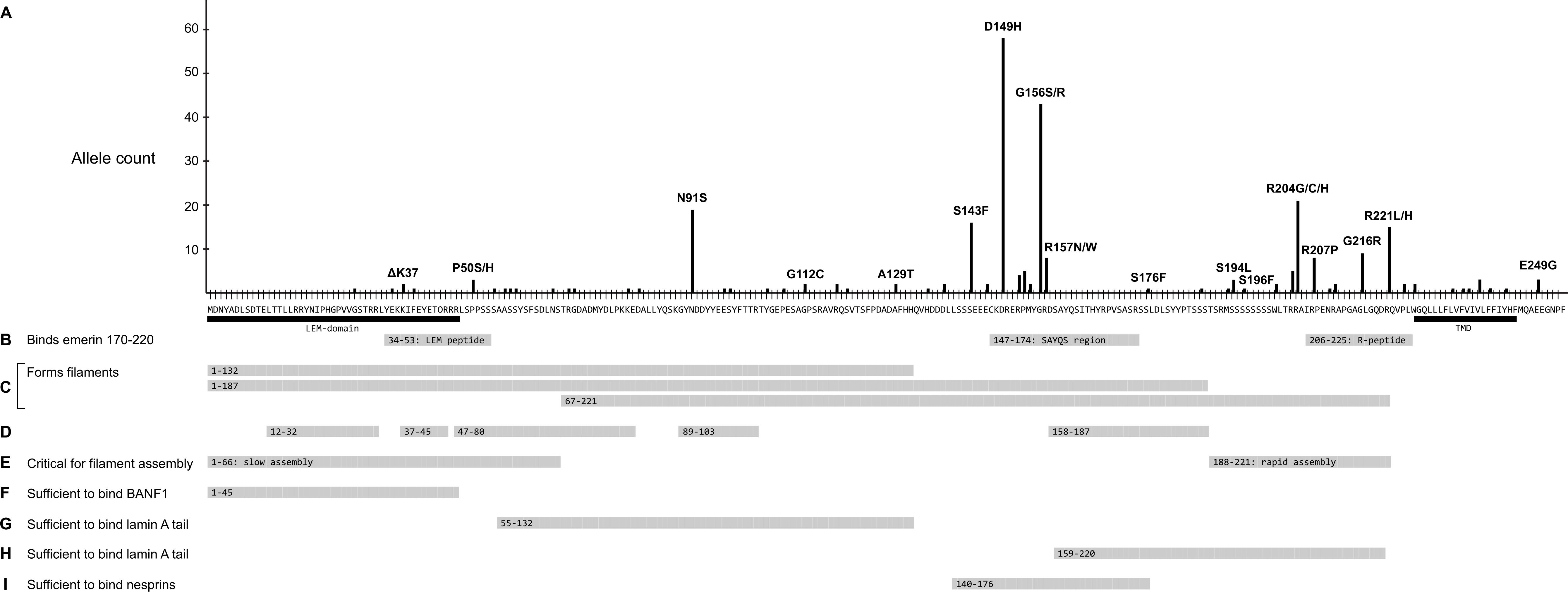
Figure 2. Schematic summary of all identified emerin missense and in-frame deletions in ExAC in relation to functional regions of emerin. (A) Amino acid sequence of wildtype emerin. Y-axis shows the number of variant alleles at each position (‘Allele count’). Each black dot indicates one affected individual. Some variants are named; for a full list see Supplementary Table S2. Black bars indicate the LEM-domain (Cai et al., 2007) and transmembrane domain (‘TMD’). (B) Peptides that mediate intermolecular self-association (Berk et al., 2014). Gray bars indicate three peptides specifically recognized by emerin residues 170–220, named the ‘LEM-peptide’ (residues 34–53), ‘SAYQS region’ (residues 147–174) and ‘R-peptide’ (residues 206–225). (C-E) Regions involved in the assembly of curvilinear filaments in vitro (Herrada et al., 2015; Samson et al., 2018). (C) Gray bars indicate regions that form filaments either slowly (residues 1–132 or 1–187) or rapidly (residues 67–221) in vitro. (D) Gray bars depict residues that resist digestion with protease, thereby identified as backbone (protected) components of emerin filaments. (E) Gray bars depict residues 1–66, critical for (slow) self-assembly of LEM-containing constructs, and residues 188–221, critical for (rapid) self-assembly of residues 67–221. (F) Gray bar indicates residues sufficient to bind BANF1 in vitro. (G) Gray bar indicates residues deduced as sufficient to bind mature lamin A residues 385-646 and, with lower affinity to lamin B1 residues 395–586 (Berk et al., 2014). (H) Gray bar indicates a second region of emerin that independently binds mature lamin A residues 385–646 and, with lower affinity to lamin B1 residues 395–586 (Berk et al., 2014). (I) Gray bar indicates residues sufficient to bind either nesprin-1 or nesprin-2 in vitro (Wheeler et al., 2007). Not depicted: Sun1 and Sun2 bind via deduced emerin residues 134–221 (Haque et al., 2010).
Three Emerin ExAC Variants (p.K37del, p.R203H, p.D149H) Reported in Laminopathy Database
We anticipated little overlap with the laminopathy database, since severe childhood-onset disorders such as EDMD1 are excluded from ExAC (Lek et al., 2016). Indeed, only three emerin ExAC variants were previously reported in the laminopathy database: p.K37del, p.R203H and p.D149H. Variant p.K37del causes the loss-of-function EDMD1 phenotype even though the resulting protein is expressed at normal or near-normal levels (Ellis et al., 1998). Loss of residue K37 destabilizes the LEM-domain (Samson et al., 2017), reduces affinity for BANF1 (Essawy et al., personal communication) and accelerates the rate of emerin self-association as filaments in vitro (Samson et al., 2017). Finding two individuals with p.K37del in ExAC was plausible because this variant is known to cause isolated cardiac disease in adult men (hemizygous) and women (heterozygous; Ben Yaou et al., 2007), and cardiac disease is well-represented in ExAC (Lek et al., 2016). A similar case can be made for variant p.R203H (identified in four ExAC individuals), because the only reported EDMD1 patient with this variant showed cardiac symptoms as an adult (39 years; Funakoshi et al., 1999). These results were encouraging in their suggestion that ExAC populations might also include novel variants that specifically affect the heart.
The third ‘known’ variant, p.D149H, was reported in one EDMD1 patient but nevertheless deemed benign with respect to laminopathy. This was exciting because p.D149H was the most-frequent EMD variant in ExAC and associated with a novel metabolic phenotype, as described below.
Ethnic Concentrations of the Most Frequent Emerin Variants in ExAC
The most frequent emerin variants in ExAC, including nine novel variants, each concentrated in a specific ethnic group (Table 2 and Supplemental Table S2). Topping the list was variant p.D149H, identified predominantly in East Asians (55 of 58 individuals) with an allele frequency of 0.8297% in East Asians; the other individuals were South Asian (one) and Latino (henceforth ‘Latinx’; two). Non-Finnish Europeans (henceforth, ‘Europeans’), the largest ethnic population in ExAC, also had the greatest overall number of emerin variants (Table 2). Three variants were identified only in Europeans: p.N91S (all 19 individuals; allele frequency 0.04123%), p.S143F (all 16 individuals; allele frequency 0.03365%) and p.G156R (all 9 individuals; allele frequency 0.01889%), corresponding to frequencies of 1.8 to 4.1 per 10,000 individuals. Variant p.R157Q was identified predominantly in Europeans (5 of 7 individuals; allele frequency 0.01049%); the other two were Latinx. Variant p.G216R was identified only in Africans (all 9 individuals; allele frequency 0.1064%). Variant p.G156S was identified predominantly in Africans (33 of 34 individuals; allele frequency 0.3894%); the other individual was European. Two variants were identified only in Latinx: p.R204G (all 17 individuals; allele frequency 0.1826%) and p.R221L (all 14 individuals; allele frequency 0.1505%). Variant p.R207P was identified only in South Asians (all 8 individuals; allele frequency 0.07917%). In other words, six variants (p.D149H, p.G156S, p.R204G, p.R207P, p.G216R, p.R221L) were identified at frequencies of 10–83 per 10,000 individuals in specific populations. Note that these frequencies may be skewed by multiple factors including distribution artifacts and potential association with disorders (e.g., cardiomyopathy or type 2 diabetes) that are enriched in ExAC.
Variant p.D149H, Identified in 0.8297% of East Asians, Associates With a ‘Healthy’ Lipid Profile
Variant p.D149H was identified 58 times in ExAC with allele frequencies of 0.0665% overall and 0.8297% in East Asians (Table 2 and Supplementary Table S2). For most disorders, ExAC does not link individuals to clinical phenotypes. However, ExAC includes type 2 diabetes cohorts for whom clinical data is accessible via the T2D Knowledge Portal6. To determine if any variants associated with a metabolic phenotype(s), we searched within 100 kb of EMD (see Materials and Methods). We found no significant associations with body-mass index, diastolic blood pressure, fasting glucose, fasting insulin, glycated hemoglobin (HbA1c), HDL cholesterol, height, hip circumference, systolic blood pressure, type 2 diabetes or waist-hip ratio. By contrast, four traits showed a significant association with emerin variant p.D149H: reduced triglycerides (effect was -0.336; p = 0.0368), reduced waist circumference (effect was -0.321; p = 0.0486), reduced cholesterol (effect was -0.572; p = 0.000346) and reduced LDL cholesterol (effect was -0.599; p = 0.000272). We concluded that emerin variant p.D149H specifically influences metabolism (see Discussion).
‘Top-Ten’ Emerin Variants in Regions That Self-Associate and Bind Lamin A and Nesprins
Variant p.N91S is a disruptive mutation in a core (protease-protected) region of emerin filaments (Figure 2D) that also binds lamin A (Figure 2G). Other ‘top-ten’ variants clustered in two regions: variants p.S143F through p.R157Q affect residues in the ‘SAYQS’ region, while variants p.R203H through p.R221L affect residues in the ‘R-peptide’ region, near the transmembrane domain (TMD; Figure 2B). Both regions mediate emerin self-association, as shown in peptide binding studies (Figure 2B; Berk et al., 2014) and biophysical studies of emerin filaments (Figures 2C,D; Herrada et al., 2015; Samson et al., 2017). Both regions also directly bind lamin A (Figure 2H), nesprins (Figure 2I) and other partners (Berk et al., 2014). The substitutions created by variants p.D149H, p.P153L, p.R204G and p.R207P are predicted to perturb one or more of these functions (see Discussion).
LEM-Domain Variants: One Known (p.K37del) and Three Novel (p.G28A, p.E35K, p.F39del)
Four LEM-domain variants were identified in ExAC: the cardiomyopathy-associated variant p.K37del (discussed above) and three unique novel variants: p.G28A, p.E35K and p.K39del (Supplementary Table S2). WT LEM-domain residues affected by ExAC variants are depicted via surface view (Figure 3A) and ribbon diagram (Figure 3B; PDB code 2ODC; Cai et al., 2007). All four WT residues are surface-exposed (Figure 3A) on the large α-helix (Figure 3B). Residues K35, K37, and F39 each stabilize the α-helix (Figure 3C). Deletion of residue K37 destabilizes the LEM-domain (Samson et al., 2017); we therefore expect the same or worse for variant p.F39del, which is predicted to enhance disorder more than p.K37del (Figure 4). Missense variant p.E35K, which replaces a negative charge (Glu) with a positive charge (Lys), is predicted to increase disorder for residues 21–29 and 35–49 (Figure 4) and thus has the potential to destabilize the LEM-domain. Variant p.G28A is positioned to alter the ‘left’ surface of the LEM-domain (as viewed in Figure 3A). A more-disruptive substitution, p.G28R, is reported for three individuals in the dbSNP database (Praxis fuer Humangenetik Teubingen; ID: rs1064797380). The functions of this surface are unclear, since BANF1 binds the other (‘right’) side (as viewed in Figure 3A) via direct contact with LEM-domain residues Y34 (colored yellow; Figures 3A,B), T13, G24, P25, V27, S29, T30, L33, K36, and K37 (Cai et al., 2007; Samson et al., 2018). Intriguingly, this ‘mystery’ surface of the LEM-domain includes α-helical residues Y34 through R47, which together with disordered residues L48-S53 comprise the ‘LEM-peptide,’ which mediates emerin self-association (Figure 2B) and is largely protease-resistant in the context of emerin filaments (Figure 2D; Samson et al., 2017). Thus, novel ExAC variants p.G28A and p.E35K, along with p.G28R, are predicted to disrupt a conformational change(s) of the LEM-domain that contributes to emerin filament formation, and might also reduce emerin affinity for lamin A, which binds preferentially to emerin filaments (Samson et al., 2018).
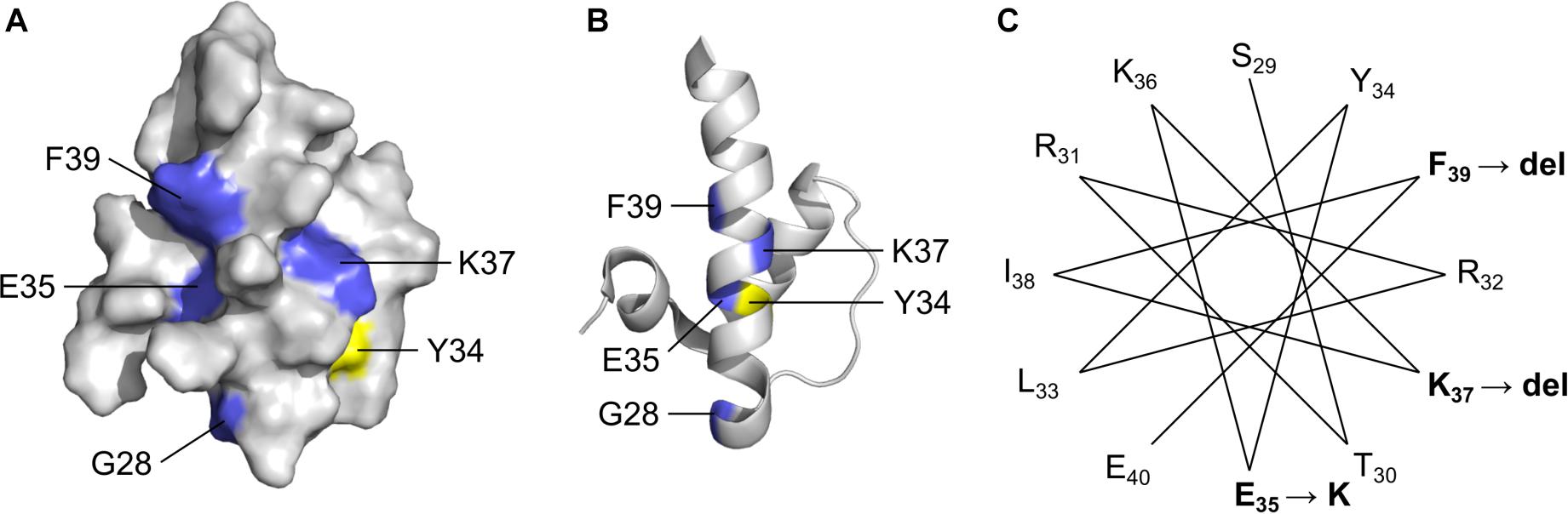
Figure 3. Solution NMR structure (Cai et al., 2007) of the ‘LEM’ domain showing residues affected by ExAC variants (RCSB Protein Data Bank ID: 2ODC). Surface depiction (A) and ribbon depiction (B) of the wildtype LEM domain. Residues affected by ExAC variants p.G28A, p.E35K, p.K37del and p.K39del are shown in blue. Also depicted in yellow for reference is Tyr34, which directly contacts BANF1 (Cai et al., 2007; Samson et al., 2018). (C) A helical projection suggests the longest α-helix, known to be destabilized by p.K37del, is also perturbed by novel variants p.E35K and p.K39del.
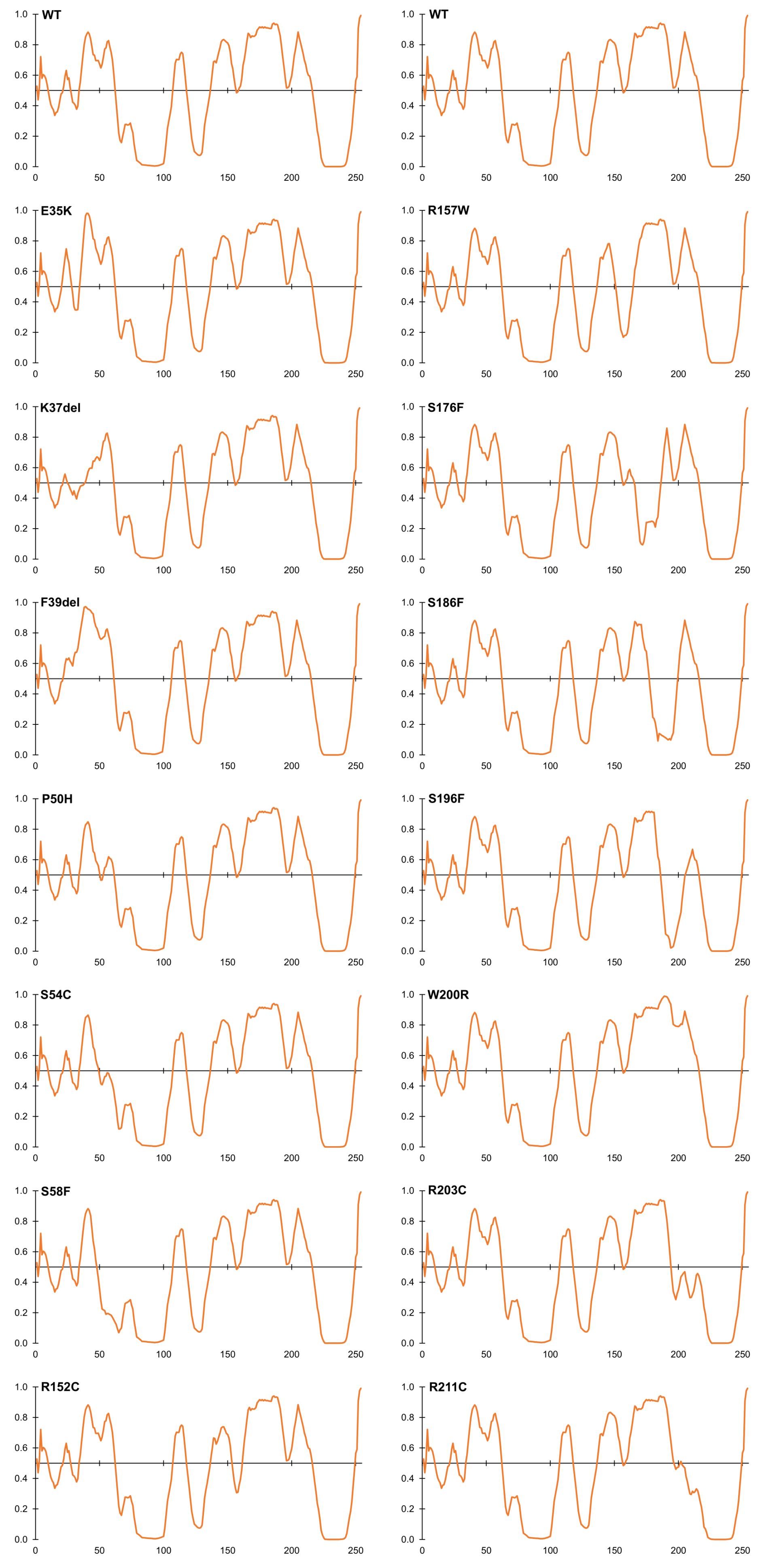
Figure 4. Intrinsic disorder predictions for selected ExAC variants, relative to wildtype emerin (WT). Disorder predictions from PONDR-FIT VL-XT are graphed on the Y-axis: disorder increases above the horizontal line and decreases below the line. The X-axis indicates amino acid positions in emerin.
Variants That Perturb Intrinsic Disorder, a Fundamental Property of Emerin
Beyond the LEM-domain, emerin is dominated by intrinsic disorder, as shown by biophysical analysis of emerin residues 50–132 and residues 67–170 (Samson et al., 2016). Disorder is an important quality that allows such ‘transformer’ proteins to bind specific partners with high affinity, and to undergo regulated conformational changes that create new ‘platforms’ for different sets of partners (Uversky, 2011; Wright and Dyson, 2015; Ruan et al., 2018). About 30% of emerin variants in ExAC were predicted by PONDR VL-XT to increase or decrease disorder (Figure 4). Variants p.P50H, p.S54C, p.S57F, and p.S58F are all predicted to reduce or eliminate disorder near the LEM-domain (Figure 4); interestingly, their proximity to known phosphorylation and O-GlcNAcylation sites (e.g., S55, S56, S60, S62) suggests they might also interfere with posttranslational control of this proposed ‘hinge’ region (Roberts et al., 2006; Berk et al., 2013a,b). Variant p.S54C, identified in one individual in ExAC, is a novel substitution at residue Ser54. A different substitution, p.S54F, disrupts binding to HDAC3 (Demmerle et al., 2012) and other partners (Berk et al., 2013b) and is sufficient to cause EDMD1 (Ellis et al., 1998).
Several novel variants were also predicted to either reduce (p.R152C, p.R152H, p.P153L) or eliminate disorder (p.R157W) in the conserved ‘SAYQS’ region (Figure 4), which mediates emerin self-association (Figures 2B–D) and binding to nesprins (Figure 2I). Seven novel variants (p.S176F, p.S186F, p.S192F, p.S196F, p.R203C, p.R204C, p.R211C) are predicted to eliminate disorder in the distal region (Figure 4) that is both sufficient for binding to lamin A (Figure 2H) and critical for emerin self-association (Figures 2B–E). Most such variants were identified in only one individual each, except p.R152C and p.R204C (three individuals each) and p.P153L (five individuals; Supplementary Table S2). These variants warrant further study (see Discussion).
Emerin Variants in Regions Needed for TRC40-Dependent Membrane Insertion
The hydrophobic C-terminal domain of emerin is recognized by the Transmembrane Recognition Complex 40 (TRC40) pathway for posttranslational insertion into ER membrane (Pfaff et al., 2016). Emerin then diffuses to the NE inner membrane and is retained by binding to A-type lamins (Östlund et al., 2006) and nesprins (Wheeler et al., 2007; Zhang et al., 2007). Several variants including p.L233P, p.F235S and p.Y243S had slightly reduced Kyte-Doolittle hydrophobicity relative to WT emerin (Figure 5A), and less favorable (but still negative) ΔG values for transmembrane helix insertion (Figure 5B). Helical wheel projections of transmembrane residues 226–244 (Figure 5C) showed that variants p.W226S, p.V236M and p.F240L affect conserved residues (Pfaff et al., 2016). Other substitutions (p.L233P, p.F235S, p.Y243S) are likely to disrupt features needed for TRC40-dependent insertion (Figure 5C). These predictions warrant future testing, since most current knowledge about emerin insertion into membranes is based on EDMD-associated frameshifts or deletions that truncate or remove the transmembrane domain (Pfaff et al., 2016). TRC40-dependent insertion is also significantly (∼44%) reduced by EDMD-causing variants p.P183T and p.P183H, located ∼40 residues upstream of the transmembrane domain, through unknown mechanisms (Pfaff et al., 2016). ExAC variants in this upstream region were rare (four unique variants between residues 158–193; Supplementary Table S2). Specific TRC40-dependent targeting to the ER/NE (rather than mitochondria or peroxisomes) requires negatively charged residues on the lumenal C-terminus (Costello et al., 2017; Figueiredo Costa et al., 2018). Emerin has two such residues, one of which is neutralized by variant p.E249G, identified in three individuals in ExAC (Figure 2). We speculate that emerin p.E249G might be inefficiently targeted to the ER/NE, and end up in the wrong organelle.
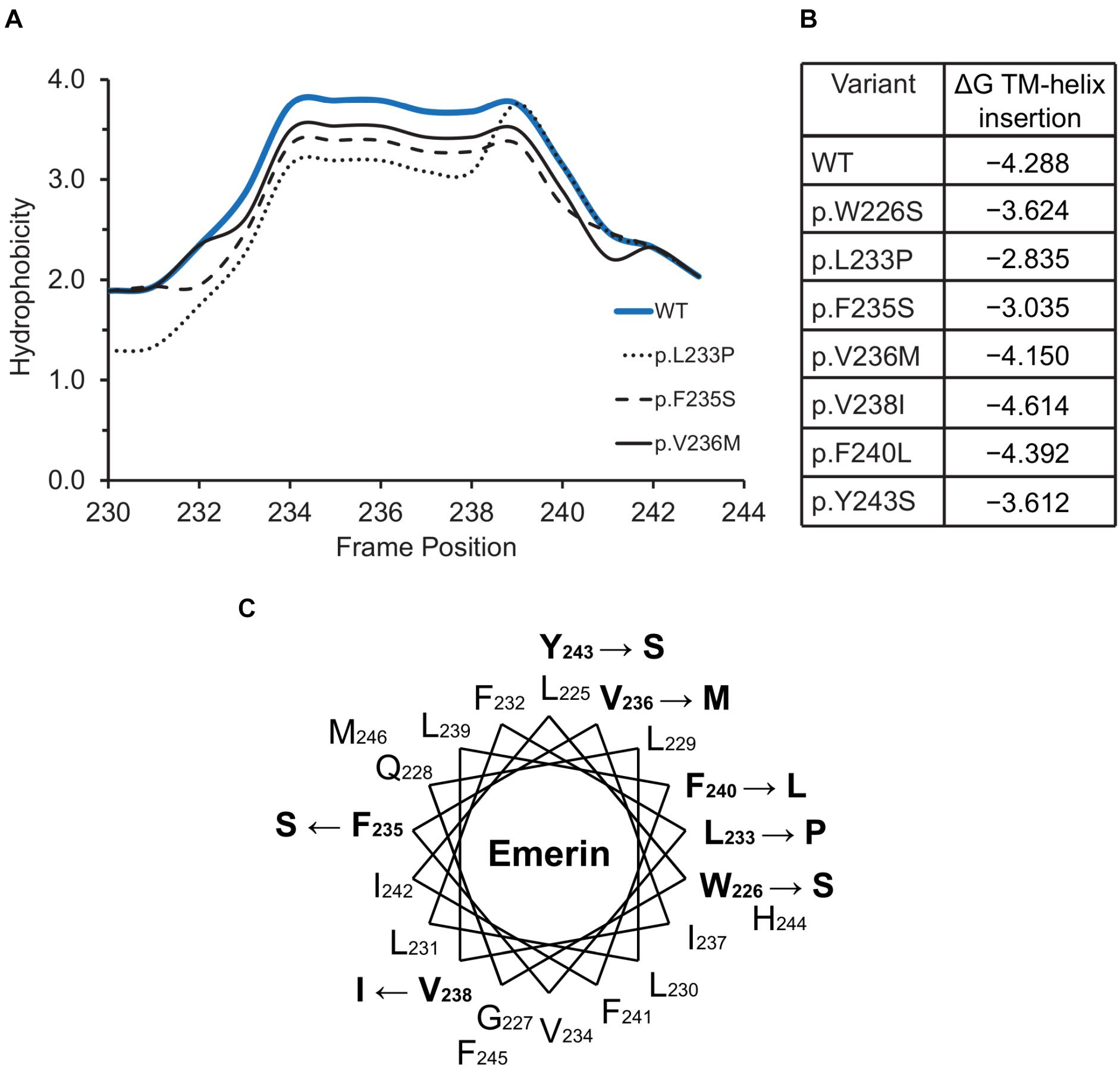
Figure 5. Variants located in or near the emerin C-terminus. (A) Kyte-Doolittle hydrophobicity plots of the transmembrane domains of wildtype (WT) emerin and selected ExAC variants. (B) Predicted ΔG for transmembrane helix insertion of WT emerin and each indicated ExAC variant. (C) Helical wheel projection of the transmembrane domain of emerin (residues 226–244), plotted according to Pfaff and colleagues (Pfaff et al., 2016). WT residues affected by ExAC variants are bold, with an arrow pointing to the substitution.
Discussion
Population diversity in ExAC was key to discovering many novel variants in the genes encoding emerin and BANF1. The first thing to keep in mind is that most or all of the EMD and BANF1 alleles in ExAC could be phenotypically silent, a common theme in human genetics. However as highlighted in this report, certain variants have the potential to perturb emerin or BANF1 at the molecular level. Because EMD is X-linked, men will express the variant emerin exclusively, increasing the possibility of a clinical phenotype. Women express EMD variants exclusively in about 50% of cells, increasing their genetic risk if the variant perturbs emerin function in cardiomyocytes. Novel human variants identified in this study provide new tools to study their molecular roles and potential association with adult-onset heart disease or new physiological roles, as reported here for ‘healthy lipid’ emerin variant p.D149H. We discuss our results for BANF1 first.
Human BANF1 Variants: Novel and Rare
BANF1 was first discovered as a protein essential for retroviral integration into the genome (Lee and Craigie, 1998). Its atomic structure (obligate dimer) and binding sites for dsDNA (two) and the LEM-domain are solved (Bradley et al., 2005; Cai et al., 2007). Residues involved in binding to dsDNA, histone H3, histone H1 or emerin were characterized by in vitro mutagenesis (Umland et al., 2000; Segura-Totten et al., 2002; Montes de Oca et al., 2005). Consistent with its essential roles in mitosis, we found that genetic variation in BANF1 was constrained. BANF1 had a loss-of-function intolerance (pLI) score of 0.74, comparable to that of histone H3 (H3F3A; pLI score 0.69), and more intolerant than LMNA (pLI score 0.99; Florwick et al., 2017).
All 14 identified BANF1 missense variants were novel, and most were non-conservative substitutions with the potential to perturb BANF1 structure or function. The known or predicted molecular impacts of BANF1 variants identified in this study are summarized schematically in Figure 6. Further studies are needed to determine if heterozygous BANF1 variants are recessive or dominant. The only previously reported human BANF variant, p.A12T, causes Nestor-Guillermo progeria syndrome when homozygous (Puente et al., 2011). When expressed ectopically in U2OS cells, variant p.A12T caused a significant increase in the percentage of cells with a misshapen nucleus (Paquet et al., 2014). We therefore speculate that heterozygous variants (e.g., p.G25E, p.L63P, p.D9N) and homozygous variant p.D9H might cause cellular phenotypes. The discovery of rare non-conservative human BANF1 variants is exciting and warrants further study in the contexts of nuclear lamina function, cell cycle control, epigenetics, embryonic stem cells, mechano-transduction or novel roles (Haraguchi et al., 2007; Cox et al., 2011; Graham and Burridge, 2016; Brayson et al., 2018; Wang et al., 2018).
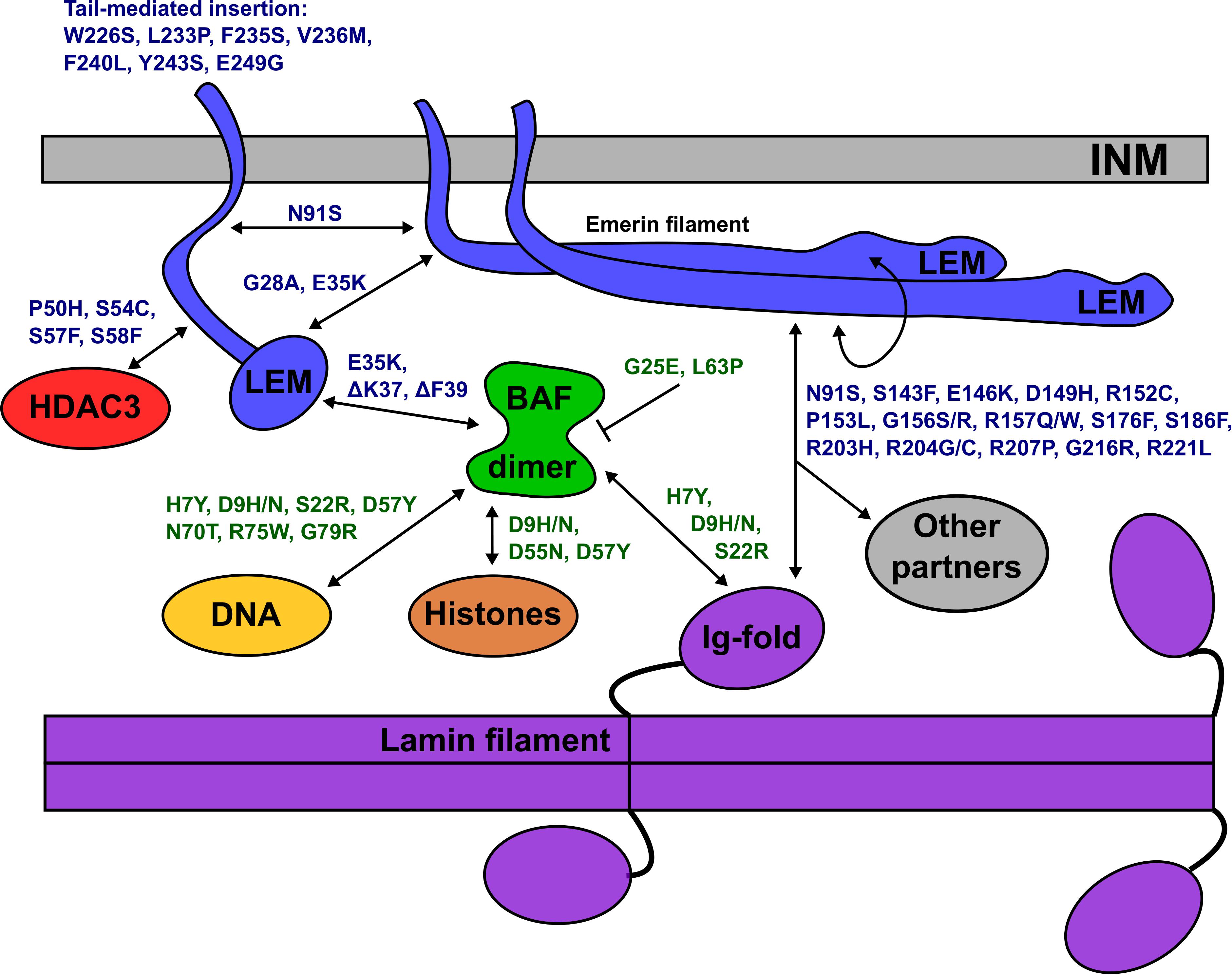
Figure 6. Schematic depiction of the inner nuclear membrane (INM) showing emerin, BANF1, an A-type lamin filament and selected partners (HDAC3, DNA, histones). Emerin polypeptides and variants are shown in blue. BANF1 dimer and variants are shown in green. Lamin filament with four Ig-fold ‘tail’ domains is shown in purple. Double-headed arrows indicate direct association, and each named variant is predicted (or in certain cases, known) to perturb the interaction. Emerin has several modes of self-association and can form filaments (see text). Variant p.G25E inhibits BANF1 dimerization (indicated by the bar) resulting in loss of binding to dsDNA, histones and the LEM-domain; the same is predicted for novel variant p.L63P. ‘Other’ partners not depicted include F-actin, nesprins, β-catenin and other transcription factors (see text). Testing these predictions in future, especially for p.D149H and other ‘high-frequency’ emerin variants, may provide molecular insights into nuclear lamina functions and human disease.
ExAC Variants in or Near the Emerin LEM-Domain
One intriguing question is whether the LEM-domain unfolds or changes conformation in vivo. The rate of emerin filament formation in vitro is accelerated by mutations that destabilize the LEM-domain (Samson et al., 2017), and further accelerated in the absence of the LEM-domain (Figures 2C,E; Samson et al., 2018; Essawy et al., personal communication). As noted earlier, several ExAC variants (e.g., p.P50H, p.S54C, p.S58F) near the LEM-domain have the potential to disrupt a proposed ‘hinge’ region (Figures 2B–D), either by making it inflexible (Figure 4) or by interfering with cellular control (e.g., phosphorylation vs. O-GlcNAcylation at Ser54; Berk et al., 2013a).
Novel Variants-of-Interest in Lamin-Binding Regions
The molecular basis for lamin A binding to emerin is beginning to emerge. Lamin A binds emerin both directly via emerin residues 55–132 (Figure 2G) or 159–220 (Figure 2H), and indirectly through BANF1 (Samson et al., 2018). Furthermore, the Ig-fold domain of lamin A associates with emerin filaments, not emerin monomers (Samson et al., 2018; Essawy et al., personal communication). Thus, we predict that contact with lamin A is perturbed by non-conservative substitutions in peptides required for emerin self-association (e.g., p.P50S, p.P50H, p.D149H, p.G156S, p.R207P, p.G216R, p.R221L), and by variants in regions that bind lamins (e.g., p.N91S, p.R204G/C/H), as illustrated schematically in Figure 6. For example, emerin variants p.R207P and p.G216R are highly disruptive substitutions in the conserved ‘R-peptide’, which is critical for emerin self-association and binding to lamins (Figures 2B,E,H). Several variants (e.g., p.S143F, p.D149H, p.G156R) have the potential to affect binding to nesprins (Figure 2I; ‘Other partners’ in Figure 6).
Variants Near Sites of Force-Stimulated Emerin Tyr-Phosphorylation
‘Pulling’ on LINC complexes of isolated nuclei stiffens the nucleus within seconds (Guilluy et al., 2014). This mechanical response requires Src phosphorylation of emerin, and is significantly reduced by substitutions that block phosphorylation at two sites in emerin: Y74F and Y95F (Tifft et al., 2009; Guilluy et al., 2014). The molecular mechanisms of emerin-dependent stiffening are unknown, and might plausibly involve force-induced and/or phosphorylation-induced changes in emerin conformation, self-association and/or binding to lamin A, BANF1 (chromatin), F-actin or myosin 1c (Holaska and Wilson, 2007; Virtanen and Vartiainen, 2017; Lambert, 2018). Testing these models will be an intriguing challenge, since emerin is flexible and self-association is required to bind many other partners including A-type lamins (Figure 6). We speculate that variants such as p.N91S and p.Y105C might perturb Src-dependent phosphorylation at Y74 and Y95 (Table 2; Tifft et al., 2009). Novel ExAC variants also provide a rich source of human variants to test for potential defects in downstream emerin-dependent transcription regulation (Ho et al., 2013; Willer and Carroll, 2017; Kirby and Lammerding, 2018).
Emerin Variant p.D149H Associated With a ‘Healthy’ (Fasting) Lipid Phenotype
Emerin variant p.D149H, reported as benign with respect to EDMD1, associated significantly with four traits in the Type 2 Diabetes Knowledge Portal: reduced triglycerides and reduced waist circumference, as well as high-impact reductions in LDL cholesterol and cholesterol. To our knowledge, this is the first evidence that emerin directly influences metabolism. The emerin p.D149H phenotype is opposite to that seen in LMNA-associated metabolic disease, especially for triglycerides and waist circumference, both of which are elevated in LMNA-associated lipodystrophy, insulin resistance and metabolic syndrome (Decaudain et al., 2007; Vadrot et al., 2015; Dutour et al., 2011; Brayson and Shanahan, 2017). In contrast to our previous ExAC study, which linked lamin A variant p.G602S with type 2 diabetes (Florwick et al., 2017), we found no association of any emerin variant with type 2 diabetes.
The traits associated with emerin p.D149H, especially the reduction in LDL (‘bad’) cholesterol, are considered healthy. The magnitude of LDL reduction associated with p.D149H is comparable to the expected effects of drugs such as statins, which aim to reduce LDL cholesterol by ≥50% to decrease risk of adverse cardiovascular events (Ridker et al., 2016). The triglyceride phenotype of emerin p.D149H is similar to that caused by loss-of-function mutations in ANGPTL4 (angiopoietin-like protein 4), a secreted cytokine that helps maintain low serum triglyceride levels by inhibiting the enzyme, lipoprotein lipase (Dijk and Kersten, 2014). In mice, ANGPTL4 is expressed mainly in adipose tissue and is upregulated by fasting in both white adipose tissue and liver (Dijk et al., 2018). A missense mutation in ANGPTL4 (p.E40K), detected in ∼3% of European Americans, associates with reduced triglycerides and increased HDL (‘good’) cholesterol (Romeo et al., 2007, 2009), in proportions comparable to the effect of emerin p.D149H. The p.E40K mutation in ANGPTL4 also associates with reduced risk of coronary disease (Stitziel and Myocardial Infarction Genetics and CARDIoGRAM Exome Consortia Investigators, 2016).
The emerin p.D149H lipid phenotype raises important questions. What do these ‘healthy’ lipid traits imply about the normal roles of emerin in striated muscle (Brull et al., 2018), or other tissues? Is this emerin phenotype related to the immobilization of BANF1 at the nuclear lamina in response to fasting in Caenorhabditis elegans intestinal cells (Bar et al., 2014)? Is this ‘healthy lipid’ phenotype seen in both men and women, or is it less pronounced in women, who express WT emerin in ∼50% of cells? We do not know which tissues are affected by emerin p.D149H, although liver and adipose are plausible candidates. Another open question is whether the p.D149H phenotype is related to, or independent of, the metabolic phenotypes caused by dominant LMNA variants. Future exploration of variant p.D149H may provide insight into the protective roles of WT emerin in the heart.
Where to Go From Here
Exome sequence data from human populations is an emerging resource in biology, with the potential to advance personalized medicine for individuals with well-characterized mutations. Our analysis of EMD revealed an unexpected association between emerin variant p.D149H and a ‘healthy lipid’ profile, and showed that nearly 1% (∼0.83%) of East Asians in ExAC carry this variant. Our previous study linked LMNA variant p.G602S to type 2 diabetes and showed that ∼0.3% of African Americans carry this allele, and revealed a dominant lipodystrophy-causing variant (p.I299V) in 0.347% of Latinx individuals in ExAC (Florwick et al., 2017). However, many fundamental aspects of nuclear biology are unexplored or incompletely understood, including the molecular connections between emerin, BANF1 and lamin filaments at the NE. Our analysis of three nuclear lamina genes – LMNA (Florwick et al., 2017), EMD and BANF1 (this study) – in 60,706 unrelated individuals in ExAC has yielded an unexpected trove of novel variants with the potential to perturb specific interactions at the NE, as shown schematically in Figure 6. These variants and predictions await testing.
Author Contributions
KW contributed to the conception and first draft. TD, JL, and YG prepared the figures and tables. TD, CB, BG, and KW analyzed the data. All authors contributed to the intellectual input, manuscript preparation and editing.
Funding
This work was funded by The Johns Hopkins University Claude D. Pepper Older Americans Independence Center National Institutes of Health NIA P30AG021334.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling Editor and reviewer KR declared their involvement as co-editors in the Research Topic, and confirm the absence of any other collaboration.
Acknowledgments
We gratefully acknowledge Nada Essawy, Catherine Coirault, and Sophie Zinn for sharing their unpublished results.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2019.00048/full#supplementary-material
TABLE S1 | List of BANF1 variant alleles in ExAC.
TABLE S2 | List of EMD variant alleles in ExAC.
Abbreviations
BANF1, Barrier to autointegration factor 1; EDMD, Emery-Dreifuss muscular dystrophy; LEM-domain, LAP2-Emerin-MAN1 domain; LINC, linkers of the nucleoskeleton and cytoskeleton; T2D, type 2 diabetes; TCGA, The Cancer Genome Atlas. WT, wildtype.
Footnotes
- ^http://www.type2diabetesgenetics.org/variantInfo/variantInfo/X_153609158_G_C
- ^http://www.pondr.com
- ^https://web.expasy.org/protscale/
- ^http://emboss.bioinformatics.nl/cgi-bin/emboss/pepwheel
- ^http://dgpred.cbr.su.se/index.php?p=TMpred
- ^http://www.type2diabetesgenetics.org/
References
Bar, D. Z., Davidovich, M., Lamm, A. T., Zer, H., Wilson, K. L., and Gruenbaum, Y. (2014). BAF-1 mobility is regulated by environmental stresses. Mol. Biol. Cell 25, 1127–1136. doi: 10.1091/mbc.E13-08-0477
Barkan, R., Zahand, A. J., Sharabi, K., Lamm, A. T., Feinstein, N., Haithcock, E., et al. (2012). Ce-emerin and LEM-2: essential roles in Caenorhabditis elegans development, muscle function, and mitosis. Mol. Biol. Cell 23, 543–552. doi: 10.1091/mbc.E11-06-0505
Barton, L. J., Soshnev, A. A., and Geyer, P. K. (2015). Networking in the nucleus: a spotlight on LEM-domain proteins. Curr. Opin. Cell Biol. 34, 1–8. doi: 10.1016/j.ceb.2015.03.005
Ben Yaou, R., Toutain, A., Arimura, T., Demay, L., Massart, C., Peccate, C., et al. (2007). Multitissular involvement in a family with LMNA and EMD mutations: role of digenic mechanisms? Neurology 68, 1883–1894. doi: 10.1212/01.wnl.0000263138.57257.6a
Bengtsson, L., and Wilson, K. L. (2006). Barrier-to-autointegration factor phosphorylation on Ser-4 regulates emerin binding to lamin A in vitro and emerin localization in vivo. Mol. Biol. Cell 17, 1154–1163. doi: 10.1091/mbc.e05-04-0356
Berk, J. M., Maitra, S., Dawdy, A. W., Shabanowitz, J., Hunt, D. F., and Wilson, K. L. (2013a). O-linked β-N-acetylglucosamine (O-GlcNAc) regulates emerin binding to barrier to autointegration factor (BAF) in a chromatin- and lamin B-enriched “niche”. J. Biol. Chem. 288, 30192–30209. doi: 10.1074/jbc.M113.503060
Berk, J. M., Tifft, K. E., and Wilson, K. L. (2013b). The nuclear envelope LEM-domain protein emerin. Nucleus 4, 298–314. doi: 10.4161/nucl.25751
Berk, J. M., Simon, D. N., Jenkins-Houk, C. R., Westerbeck, J. W., Gronning-Wang, L. M., Carlson, C. R., et al. (2014). The molecular basis of emerin-emerin and emerin-BAF interactions. J. Cell Sci. 127, 3956–3969. doi: 10.1242/jcs.148247
Bonne, G., Leturcq, F., and Ben Yaou, R. (2004). “Emery-Dreifuss muscular dystrophy,” in GeneReviews(® [Internet], eds M. P. Adam, H. H. Ardinger, R. A. Pagon, S. E. Wallace, L. J. H. Bean, K. Stephens, et al. (Seattle, WA: University of Washington).
Bradley, C. M., Ronning, D. R., Ghirlando, R., Craigie, R., and Dyda, F. (2005). Structural basis for DNA bridging by barrier-to-autointegration factor. Nat. Struct. Mol. Biol. 12, 935–936. doi: 10.1038/nsmb989
Brayson, D., Ho, C. Y., and Shanahan, C. M. (2018). Muscle tensions emerge to cause a DNA replication crisis. J. Cell Biol. 217, 1891–1893. doi: 10.1083/jcb.201804041
Brayson, D., and Shanahan, C. M. (2017). Current insights into LMNA cardiomyopathies: existing models and missing LINCs. Nucleus 8, 17–33. doi: 10.1080/19491034.2016.1260798
Brull, A., Morales Rodriguez, B., Bonne, G., Muchir, A., and Bertrand, A. T. (2018). The pathogenesis and therapies of striated muscle laminopathies. Front. Physiol. 9:1533. doi: 10.3389/fphys.2018.01533
Cai, M., Huang, Y., Suh, J., Louis, J. M., Ghirlando, R., Craigie, R., et al. (2007). Solution NMR structure of the barrier-to-autointegration factor-emerin complex. J. Biol. Chem. 282, 14525–14535. doi: 10.1074/jbc.M700576200
Capanni, C., Squarzoni, S., Cenni, V., D’Apice, M. R., Gambineri, A., Novelli, G., et al. (2012). Familial partial lipodystrophy, mandibuloacral dysplasia and restrictive dermopathy feature barrier-to-autointegration factor (BAF) nuclear redistribution. Cell Cycle 11, 3568–3577. doi: 10.4161/cc.21869
Chang, W., Folker, E. S., Worman, H. J., and Gundersen, G. G. (2013). Emerin organizes actin flow for nuclear movement and centrosome orientation in migrating fibroblasts. Mol. Biol. Cell 24, 3869–3880. doi: 10.1091/mbc.E13-06-0307
Costello, J. L., Castro, I. G., Camões, F., Schrader, T. A., McNeall, D., Yang, J., et al. (2017). Predicting the targeting of tail-anchored proteins to subcellular compartments in mammalian cells. J. Cell Sci. 130, 1675–1687. doi: 10.1242/jcs.200204
Cox, J. L., Mallanna, S. K., Ormsbee, B. D., Desler, M., Wiebe, M. S., and Rizzino, A. (2011). Banf1 is required to maintain the self-renewal of both mouse and human embryonic stem cells. J. Cell Sci. 124, 2654–2665. doi: 10.1242/jcs.083238
Decaudain, A., Vantyghem, M. C., Guerci, B., Hécart, A. C., Auclair, M., Reznik, Y., et al. (2007). New metabolic phenotypes in laminopathies: LMNA mutations in patients with severe metabolic syndrome. J. Clin. Endocrinol. Metab. 92, 4835–4844. doi: 10.1210/jc.2007-0654
Dechat, T., Gajewski, A., Korbei, B., Gerlich, D., Daigle, N., Haraguchi, T., et al. (2004). LAP2α and BAF transiently localize to telomeres and specific regions on chromatin during nuclear assembly. J. Cell Sci. 117, 6117–6128. doi: 10.1242/jcs.01529
Demmerle, J., Koch, A. J., and Holaska, J. M. (2012). The nuclear envelope protein emerin binds directly to histone deacetylase 3 (HDAC3) and activates HDAC3 activity. J. Biol. Chem. 287, 22080–22088. doi: 10.1074/jbc.M111.325308
Demmerle, J., Koch, A. J., and Holaska, J. M. (2013). Emerin and histone deacetylase 3 (HDAC3) cooperatively regulate expression and nuclear positions of MyoD, Myf5, and Pax7 genes during myogenesis. Chromosome Res. 21, 765–779. doi: 10.1007/s10577-013-9381-9
Dharmaraj, T., and Wilson, K. L. (2017). How chromosomes unite. Nature 551, 568–569. doi: 10.1038/d41586-017-07439-7
Dijk, W., and Kersten, S. (2014). Regulation of lipoprotein lipase by Angpt14. Trends Endocrinol. Metab. 25, 146–155. doi: 10.1016/j.tem.2013.12.005
Dijk, W., Ruppert, P. M. M., Oost, L. J., and Kersten, S. (2018). Angiopoietin-like 4 promotes the intracellular cleavage of lipoprotein lipase by PCSK3/furin in adipocytes. J. Biol. Chem. 293, 14134–14145. doi: 10.1074/jbc.RA118.002426
Dittmer, T. A., and Misteli, T. (2011). The lamin protein family. Genome Biol. 12:222. doi: 10.1186/gb-2011-12-5-222
Dobrzynska, A., Gonzalo, S., Shanahan, C., and Askjaer, P. (2016). The nuclear lamina in health and disease. Nucleus 7, 233–248. doi: 10.1080/19491034.2016.1183848
Dutour, A., Roll, P., Gaborit, B., Courrier, S., Alessi, M. C., Tregouet, D. A., et al. (2011). High prevalence of laminopathies among patients with metabolic syndrome. Hum. Mol. Genet. 20, 3779–3786. doi: 10.1093/hmg/ddr294
Ellis, J. A., Craxton, M., Yates, J. R., and Kendrick-Jones, J. (1998). Aberrant intracellular targeting and cell cycle-dependent phosphorylation of emerin contribute to the Emery-Dreifuss muscular dystrophy phenotype. J. Cell Sci. 111, 781–792.
ExAC database (2018). Available at: http://exac.broadinstitute.org/ (accessed March 28 and September 28, 2018).
Figueiredo Costa, B., Cassella, P., Colombo, S. F., and Borgese, N. (2018). Discrimination between the endoplasmic reticulum and mitochondria by spontaneously inserting tail-anchored proteins. Traffic 19, 182–197. doi: 10.1111/tra.12550
Florwick, A., Dharmaraj, T., Jurgens, J., Valle, D., and Wilson, K. L. (2017). LMNA sequences of 60,706 unrelated individuals reveal 132 novel missense variants in A-type lamins and suggest a link between variant p.G602S and Type 2 Diabetes. Front. Genet. 8:79. doi: 10.3389/fgene.2017.00079
Funakoshi, M., Tsuchiya, Y., and Arahata, K. (1999). Emerin and cardiomyopathy in Emery-Dreifuss muscular dystrophy. Neuromusc. Disord. 9, 108–114. doi: 10.1016/S0960-8966(98)00097-2
Gerace, L., and Tapia, O. (2018). Messages from the voices within: regulation of signaling by proteins of the nuclear lamina. Curr. Opin. Cell Biol. 52, 14–21. doi: 10.1016/j.ceb.2017.12.009
Graham, D. M., and Burridge, K. (2016). Mechanotransduction and nuclear function. Curr. Opin. Cell Biol. 40, 98–105. doi: 10.1016/j.ceb.2016.03.006
Gruenbaum, Y., and Foisner, R. (2015). Lamins: nuclear intermediate filament proteins with fundamental functions in nuclear mechanics and genome regulation. Annu. Rev. Biochem. 84, 131–164. doi: 10.1146/annurev-biochem-060614-034115
Guilluy, C., Osborne, L. D., Van Landeghem, L., Sharek, L., Superfine, R., Garcia-Mata, R., et al. (2014). Isolated nuclei adapt to force and reveal a mechanotransduction pathway in the nucleus. Nat. Cell Biol. 16, 376–381. doi: 10.1038/ncb2927
Haque, F., Mazzeo, D., Patel, J. T., Smallwood, D. T., Ellis, J. A., Shanahan, C. M., et al. (2010). Mammalian SUN protein interaction networks at the inner nuclear membrane and their role in laminopathy disease processes. J. Biol. Chem. 285, 3487–3498. doi: 10.1074/jbc.M109.071910
Haraguchi, T., Kojidani, T., Koujin, T., Shimi, T., Osakada, H., Mori, C., et al. (2008). Live cell imaging and electron microscopy reveal dynamic processes of BAF-directed nuclear envelope assembly. J. Cell Sci. 121, 2540–2554. doi: 10.1242/jcs.033597
Haraguchi, T., Koujin, T., Osakada, H., Kojidani, T., Mori, C., Masuda, H., et al. (2007). Nuclear localization of barrier-to-autointegration is correlated with progression of S phase in human cells. J. Cell Sci. 120, 1967–1977. doi: 10.1242/jcs.03461
Haraguchi, T., Koujin, T., Segura-Totten, M., Lee, K. K., Matsuoka, Y., Yoneda, Y., et al. (2001). BAF is required for emerin assembly into the reforming nuclear envelope. J. Cell Sci. 114, 4575–4585.
Harr, J. C., Luperchio, T. R., Wong, X., Cohen, E., Wheelan, S. J., and Reddy, K. L. (2015). Directed targeting of chromatin to the nuclear lamina is mediated by chromatin state and A-type lamins. J. Cell Biol. 208, 33–52. doi: 10.1083/jcb.201405110
Harris, D., and Engelman, A. (2000). Both the structure and DNA binding function of the barrier-to-autointegration factor contribute to reconstitution of HIV type 1 integration in vitro. J. Biol. Chem. 275, 39671–39677. doi: 10.1074/jbc.M002626200
Herrada, I., Samson, C., Velours, C., Renault, L., Östlund, C., Chervy, P., et al. (2015). Muscular dystrophy mutations impair the nuclear envelope emerin self-assembly properties. ACS Chem. Biol. 10, 2733–2742. doi: 10.1021/acschembio.5b00648
Hessa, T., Meindl-Beinker, N., Bernsel, A., Kim, J., Sato, Y., Lerch, M., et al. (2007). Molecular code for transmembrane-helix recognition by the Sec61 translocon. Nature 450, 1026–1030. doi: 10.1038/nature06387
Ho, C. Y., Jaalouk, D. E., Vartiainen, M. K., and Lammerding, J. (2013). Lamin A/C and emerin regulate MKL1-SRF activity by modulating actin dynamics. Nature 497, 507–511. doi: 10.1038/nature12105
Holaska, J. M., Lee, K. K., Kowalski, A. K., and Wilson, K. L. (2003). Transcriptional repressor germ cell-less (GCL) and barrier to autointegration factor (BAF) compete for binding to emerin in vitro. J. Biol. Chem. 278, 6969–6975. doi: 10.1074/jbc.M208811200
Holaska, J. M., Rais-Bahrami, S., and Wilson, K. L. (2006). Lmo7 is an emerin-binding protein that regulates the transcription of emerin and many other muscle-relevant genes. Hum. Mol. Genet. 15, 3459–3472. doi: 10.1093/hmg/ddl423
Holaska, J. M., and Wilson, K. L. (2007). An emerin ‘proteome’: purification of distinct emerin-containing complexes from HeLa cells suggests molecular basis for diverse roles including gene regulation, mRNA splicing, signaling, mechanosensing, and nuclear architecture. Biochemistry 46, 8897–8908. doi: 10.1021/bi602636m
Hou, L., Kiu, K., Li, Y. H., Ma, S., Ji, X. M., and Liu, L. (2016). Necrotic pyknosis is a morphologically and biochemically distinct event from apoptotic pyknosis. J. Cell Sci. 129, 3084–3090. doi: 10.1242/jcs.184374
Hu, Q., Guo, C., Li, Y., Aronow, B. J., and Zhang, J. (2011). LMO7 mediates cell-specific activation of the Rho-myocardin-related transcription factor-serum response factor pathway and plays an important role in breast cancer cell migration. Mol. Cell Biol. 31, 3223–3240. doi: 10.1128/MCB.01365-10
Huber, M. D., Guan, T., and Gerace, L. (2009). Overlapping functions of nuclear envelope proteins NEfT25 (Lem2) and emerin in regulation of extracellular signal-regulated kinase signaling in myoblast differentiation. Mol. Cell Biol. 29, 5718–5728. doi: 10.1128/MCB.00270-09
Iyer, A., Koch, A. J., and Holaska, J. M. (2017). Expression profiling of differentiating emerin-null myogenic progenitor identifies molecular pathways implicated in their impaired differentiation. Cells 6:38. doi: 10.3390/cells6040038
Jamin, A., and Wiebe, M. S. (2015). Barrier to autointegration factor (BANF1): interwoven roles in nuclear structure, genome integrity, innate immunity, stress responses and progeria. Curr. Opin. Cell Biol. 34, 61–68. doi: 10.1016/j.ceb.2015.05.006
Janin, A., Bauer, D., Ratti, F., Millat, G., and Méjat, A. (2017). Nuclear envelopathies: a complex LINC between nuclear envelope and pathology. Orphanet. J. Rare Dis. 12:147. doi: 10.1186/s13023-017-0698-x
Kirby, T. J., and Lammerding, J. (2018). Emerging views of the nucleus as a cellular mechanosensor. Nat. Cell Biol. 20, 373–381. doi: 10.1038/s41556-018-0038-y
Kobayashi, S., and Haraguchi, T. (2015). A novel pathway to detect and cope with exogenous dsDNA. Commun. Integr. Biol. 8:e1065361. doi: 10.1080/19420889.2015.1065361
Laguri, C., Gilquin, B., Wolff, N., Romi-Lebrun, R., Courchay, K., Callebaut, I., et al. (2001). Structural characterization of the LEM motif common to three human inner nuclear membrane proteins. Structure 9, 503–511. doi: 10.1016/S0969-2126(01)00611-6
Lambert, M. W. (2018). Spectrin and its interacting partners in nuclear structure and function. Exp. Biol. Med. 243, 507–524. doi: 10.1177/1535370218763563
Lammerding, J., Hsiao, J., Schulze, P. C., Kozlov, S., Stewart, C. L., and Lee, R. T. (2005). Abnormal nuclear shape and impaired mechanotransduction in emerin-deficient cells. J. Cell Biol. 170, 781–791. doi: 10.1083/jcb.200502148
Le, H. Q., Ghatak, S., Yeung, C. Y., Tellkamp, F., Günschmann, C., Dieterich, C., et al. (2016). Mechanical regulation of transcription controls Polycomb-mediated gene silencing during lineage commitment. Nat. Cell Biol. 18, 864–875. doi: 10.1038/ncb3387
Lee, M. S., and Craigie, R. (1998). A previously unidentified host protein protects retroviral DNA from autointegration. Proc. Natl. Acad. Sci. U.S.A. 95, 1528–1533. doi: 10.1073/pnas.95.4.1528
Lek, M., Karczewski, K. J., Minikel, E. V., Samocha, K. E., Banks, E., Fennell, T., et al. (2016). Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291. doi: 10.1038/nature19057
Lele, T. P., Dickinson, R. B., and Gunderson, G. G. (2018). Mechanical principles of nuclear shaping and positioning. J. Cell Biol. 217, 3330–3342. doi: 10.1083/jcb.201804052
Liu, J., Lee, K. K., Segura-Totten, M., Neufeld, E., Wilson, K. L., and Gruenbaum, Y. (2003). MAN1 and emerin have overlapping function(s) essential for chromosome segregation and cell division in Caenorhabditis elegans. Proc. Natl. Acad. Sci. U.S.A. 100, 4598–4603. doi: 10.1073/pnas.0730821100
Liu, J., Rolef Ben-Shahar, T., Riemer, D., Treinin, M., Spann, P., Weber, K., et al. (2000). Essential roles for Caenorhabditis elegans lamin gene in nuclear organization, cell cycle progression, and spatial organization of nuclear pore complexes. Mol. Biol. Cell 11, 3937–3947. doi: 10.1091/mbc.11.11.3937
Loi, M., Cenni, V., Duchi, S., Squarzoni, S., Lopen-Ortin, C., Foisner, R., et al. (2015). Barrier-to-autointegration factor (BAF) involvement in prelamin A-related chromatin organization changes. Oncotarget 7, 15662–15677. doi: 10.18632/oncotarget.6697
Mao, L., Zhang, Y., Mo, W. J., Yu, Y., and Lu, H. (2015). BANF1 is downregulated by IRF1-regulated microRNA-203 in cervical cancer. PLoS One 10:e0117035. doi: 10.1371/journal.pone.0117035
Margalit, A., Segura-Totten, M., Gruenbaum, Y., and Wilson, K. L. (2005). Barrier-to-integration factor is required to segregate and enclose chromosomes within the nuclear envelope and assemble the nuclear lamina. Proc. Natl. Acad. Sci. U.S.A. 102, 3290–3295. doi: 10.1073/pnas.0408364102
Mojica, S. A., Hovis, K. M., Frieman, M. B., Tran, B., Hsia, R. C., Ravel, J., et al. (2015). SINC, a type III secreted protein of Chlamydia psittaci, targets the inner nuclear membrane of infected cells and uninfected neighbors. Mol. Biol. Cell 26, 1918–1934. doi: 10.1091/mbc.E14-11-1530
Montes de Oca, R., Andreassen, P. R., and Wilson, K. L. (2011). Barrier-to-autointegration factor influences specific histone modifications. Nucleus 2, 580–590. doi: 10.4161/nucl.2.6.17960
Montes de Oca, R., Lee, K. K., and Wilson, K. L. (2005). Binding of barrier to autointegration factor (BAF) to histone H3 and selected linker histones including H1.1. J. Biol. Chem. 280, 42252–42262. doi: 10.1074/jbc.M509917200
Montes de Oca, R., Shoemaker, C. J., Gucek, M., Cole, R. N., and Wilson, K. L. (2009). Barrier-to-autointegration factor proteome reveals chromatin-regulatory partners. PLoS One 4:e7050. doi: 10.1371/journal.pone.0007050
Muchir, A., Pavlidis, P., Bonne, G., Hayashi, Y. K., and Worman, H. J. (2007). Activation of MAPK in hearts of EMD null mice: similarities between mouse models of X-linked and autosomal dominant Emery Dreifuss muscular dystrophy. Hum. Mol. Genet. 16, 1884–1895. doi: 10.1093/hmg/ddm137
Oh, H. S., Traktman, P., and Knipe, D. M. (2015). Barrier-to-autointegration factor 1 (BAF/BANF1) promotes association of the SETD1A histone methyltransferase with herpes simplex virus immediate-early gene promoters. mBio 6:e00345–15. doi: 10.1128/mBio.00345-15
Östlund, C., Sullivan, T., Stewart, C. L., and Worman, H. J. (2006). Dependence of diffusional mobility of integral inner nuclear membrane proteins on A-type lamins. Biochemistry 45, 1374–1382. doi: 10.1021/bi052156n
Paquet, N., Box, J. K., Ashton, N. W., Suraweera, A., Croft, L. V., Urquhart, A. J., et al. (2014). Néstor-Guillermo progeria syndrome: a biochemical insight into barrier-to-autointegration factor 1, alanine 12 threonine mutation. BMC Mol. Biol. 15:27. doi: 10.1186/s12867-014-0027-z
Pfaff, J., Monroy, J. R., Jamieson, C., Rajanala, K., Vilardi, F., Schwappach, B., et al. (2016). Emery-Dreifuss muscular dystrophy mutations impair TRC40-dependent targeting of emerin to the inner nuclear membrane. J. Cell Sci. 129, 502–516. doi: 10.1242/jcs.179333
Pillers, D. A., and Von Bergen, N. H. (2016). Emery-Dreifuss muscular dystrophy: a test case for precision medicine. Appl. Clin. Genet. 9, 27–32. doi: 10.2147/TACG.S75028
Puente, X. S., Quesada, V., Osorio, F. G., Cabanillas, R., Cadiñanos, J., Fraile, J. M., et al. (2011). Exome sequencing and functional analysis identifies BANF1 mutation as the cause of a hereditary progeroid syndrome. Am. J. Hum. Genet. 88, 650–656. doi: 10.1016/j.ajhg.2011.04.010
Qi, Y. X., Yao, Q. P., Huang, K., Shi, Q., Zhang, P., Wang, G. L., et al. (2016). Nuclear envelope proteins modulate proliferation of vascular smooth muscle cells during cyclic stretch application. Proc. Natl. Acad. Sci. U.S.A. 113, 5293–5298. doi: 10.1073/pnas.1604569113
Reis-Sobreiro, M., Chen, J. F., Novitskaya, T., You, S., Morley, S., Steadman, K., et al. (2018). Emerin deregulation links nuclear shape instability to metastatic potential. Cancer Res. 78, 6086–6097. doi: 10.1158/0008-5472.CAN-18-0608
Ridker, P. M., Mora, S., Rose, L., and Jupiter Trial Study Group. (2016). Percent reduction in LDL cholesterol following high-intensity statin therapy: potential implication for guidelines and for the prescription of emerging lipid-lowering agents. Eur. Heart J. 37, 1373–1379. doi: 10.1093/eurheartj/ehw046
Roberts, R. C., Sutherland-Smith, A. J., Wheeler, M. A., Jensen, O. N., Emerson, L. J., Spiliotis, I. I., et al. (2006). The Emery-Dreifuss muscular dystrophy associated-protein emerin is phosphorylated on serine 49 by protein kinase A. FEBS J. 273, 4562–4575. doi: 10.1111/j.1742-4658.2006.05464.x
Romeo, S., Pennacchio, L. A., Fu, Y., Boerwinkle, E., Tybjaerg-Hansen, A., Hobbs, H. H., et al. (2007). Population-based resequencing of ANGPTL4 uncovers variations that reduce triglycerides and increase HDL. Nat. Genet. 39, 513–516. doi: 10.1038/ng1984
Romeo, S., Yin, W., Kozlitina, J., Pennacchio, L. A., Boerwinkle, E., Hobbs, H. H., et al. (2009). Rare loss-of-function mutations in ANGPTL family members contribute to plasma triglyceride levels in humans. J. Clin. Invest. 119, 70–79. doi: 10.1172/JCI37118
Ruan, H., Sun, Q., Zhang, W., Liu, Y., and Lai, L. (2018). Targeting intrinsically disordered proteins on the edge of chaos. Drug Discov. Today 24, 217–227. doi: 10.1016/j.drudis.2018.09.017
Samson, C., Celli, F., Hendriks, K., Zinke, M., Essawy, N., Herrada, I., et al. (2017). Emerin self-assembly mechanism: role of the LEM domain. FEBS J. 284, 338–352. doi: 10.1111/febs.13983
Samson, C., Herrada, I., Celli, F., Theillet, F. X., and Zinn-Justin, S. (2016). 1H, 13C and 15N backbone resonance assignment of the intrinsically disordered region of the nuclear envelope protein emerin. Biomol. NMR Assign. 10, 179–182. doi: 10.1007/s12104-015-9662-7
Samson, C., Petitalot, A., Celli, F., Herrada, I., Ropars, V., Le Du, M. H., et al. (2018). Structural analysis of the ternary complex between lamin A/C, BAF and emerin identifies an interface disrupted in autosomal recessive progeroid diseases. Nucleic Acids Res. 46, 10460–10473. doi: 10.1093/nar/gky736
Samwer, M., Schneider, M. W. G., Hoefler, R., Schmalhorst, P. S., Jude, J. G., Zuber, J., et al. (2017). DNA cross-bridging shapes a single nucleus from a set of mitotic chromosomes. Cell 170, 956–972. doi: 10.1016/j.cell.2017.07.038
Segura-Totten, M., Kowalski, A. K., Craigie, R., and Wilson, K. L. (2002). Barrier-to-autointegration factor: major roles in chromatin decondensation and nuclear assembly. J. Cell Biol. 158, 475–485. doi: 10.1083/jcb.200202019
Shin, J. Y., Méndez-Lopez, I., Wang, Y., Hays, A. P., Tanji, K., Lefkowitch, J. H., et al. (2013). Lamina-associated polypeptide-1 interacts with the muscular dystrophy protein emerin and is essential for skeletal muscle maintenance. Dev. Cell 26, 591–603. doi: 10.1016/j.devcel.2013.08.012
Simon, D. N., and Wilson, K. L. (2011). The nucleoskeleton as a genome-associated dynamic ‘network of networks’. Nat. Rev. Mol. Cell Biol. 12, 695–708. doi: 10.1038/nrm3207
Stevens, T. J., Lando, D., Basu, S., Atkinson, L. P., Cao, Y., Lee, S. F., et al. (2017). 3D structures of individual mammalian genomes studied by single-cell Hi-C. Nature 544, 59–64. doi: 10.1038/nature21429
Stitziel and Myocardial Infarction Genetics and CARDIoGRAM Exome Consortia Investigators (2016). Variants in ANGPTL4 and the risk of coronary artery disease. N. Engl. J. Med. 375:2306. doi: 10.1056/NEJMc1607380
Stubenvoll, A., Rice, M., Wietelmann, A., Wheeler, M., and Braun, T. (2015). Attenuation of Wnt/β-catenin activity reverses enhanced generation of cardiomyocytes and cardiac defects caused by loss of emerin. Hum. Mol. Genet. 24, 802–813. doi: 10.1093/hmg/ddu498
Tifft, K. E., Bradbury, K. A., and Wilson, K. L. (2009). Tyrosine phosphorylation of nuclear-membrane protein emerin by Src, Abl and other kinases. J. Cell Sci. 122, 3780–3790. doi: 10.1242/jcs.048397
Tsai, S. Y., Chuang, J. Y., Tsai, M. S., Wang, X. F., Xi, Z. X., Hung, J. J., et al. (2015). Sigma-1 receptor mediates cocaine-induced transcriptional regulation by recruiting chromatin-remodeling factors at the nuclear envelope. Proc. Natl. Acad. Sci. U.S.A. 112, E6562–E6570. doi: 10.1073/pnas.1518894112
Umland, T. C., Wei, S. Q., Craigie, R., and Davies, D. R. (2000). Structural basis of DNA bridging by barrier-to-autointegration factor. Biochemistry 39, 9130–9138. doi: 10.1021/bi000572w
Uversky, V. N. (2011). Intrinsically disordered proteins from A to Z. Int. J. Biochem. Cell Biol. 43, 1090–1103. doi: 10.1016/j.biocel.2011.04.001
Vadrot, N., Duband-Goulet, I., Cabet, E., Attanda, W., Barateau, A., Vicart, P., et al. (2015). The p.R482W substitution in A-type lamins deregulates SREBP1 activity in Dunnigan-type familial partial lipodystrophy. Hum. Mol. Genet. 24, 2096–2109. doi: 10.1093/hmg/ddu728
van Steensel, B., and Belmont, A. S. (2017). Lamina-associated domains: links with chromosome architecture, heterochromatin, and gene repression. Cell 169, 780–791. doi: 10.1016/j.cell.2017.04.022
Vijayaraghavan, B., Figueroa, R. A., Bergqvist, C., Gupta, A. J., Sousa, P., and Hallberg, E. (2018). RanGTPase regulates the interaction between the inner nuclear membrane proteins, Samp1 and emerin. Biochim. Biophys. Acta Biomembr. 1860, 1326–1334. doi: 10.1016/j.bbamem.2018.03.001
Virtanen, J. A., and Vartiainen, M. K. (2017). Diverse functions for different forms of nuclear actin. Curr. Opin. Cell Biol. 46, 33–38. doi: 10.1016/j.ceb.2016.12.004
Wang, S., Stoops, E., Cp, U., Markus, B., Reuveny, A., Ordan, E., et al. (2018). Mechanotransduction via the LINC complex regulates DNA replication in myonuclei. J. Cell Biol. 217, 2005–2018. doi: 10.1083/jcb.201708137
Wang, Y., Wang, J., Devaraj, A., Singh, M., Jimenez Orgaz, A., Chen, J. X., et al. (2014). Suicidal autointegration of sleeping beauty and piggyBac transposons in eukaryotic cells. PLoS Genet. 10:e1004103. doi: 10.1371/journal.pgen.1004103
Wheeler, M. A., Davies, J. D., Zhang, Q., Emerson, L. G., Hunt, J., Shanahan, C. M., et al. (2007). Distinct functional domains in nesprin-1alpha and nesprin-2beta bind directly to emerin and both interactions are disrupted in X-linked Emery-Dreifuss muscular dystrophy. Exp. Cell Res. 313, 2845–2857. doi: 10.1016/j.yexcr.2007.03.025
Wiebe, M. S., and Jamin, A. (2016). The barrier to autointegration factor: interlocking antiviral defense with genome maintenance. J. Virol. 90, 3806–3809. doi: 10.1128/JVI.00178-16
Willer, M. K., and Carroll, C. W. (2017). Substrate stiffness-dependent regulation of the SRF-Mkl1 co-activator complex requires the inner nuclear membrane protein Emerin. J. Cell Sci. 130, 2111–2118. doi: 10.1242/jcs.197517
Worman, H. J., and Schirmer, E. C. (2015). Nuclear membrane diversity: underlying tissue-specific pathologies in disease? Curr. Opin. Cell Biol. 34, 101–112. doi: 10.1016/j.ceb.2015.06.003
Wozniak, M. A., Baker, B. M., Chen, C. S., and Wilson, K. L. (2013). The emerin-binding transcription factor Lmo7 is regulated by association with p130Cas at focal adhesions. PeerJ 1:e134. doi: 10.7717/peerj.134
Wright, P. E., and Dyson, H. J. (2015). Intrinsically disordered proteins in cellular signalling and regulation. Nat. Rev. Mol. Cell Biol. 16, 18–29. doi: 10.1038/nrm3920
Zhang, Q., Bethmann, C., Worth, N. F., Davies, J. D., Wasner, C., Feuer, A., et al. (2007). Nesprin-1 and -2 are involved in the pathogenesis of Emery Dreifuss muscular dystrophy and are critical for nuclear envelope integrity. Hum. Mol. Genet. 16, 2816–2833. doi: 10.1093/hmg/ddm238
Zheng, R., Ghirlando, R., Lee, M. S., Mizuuchi, K., Krause, M., and Craigie, R. (2000). Barrier-to-autointegration factor (BAF) bridges DNA in a discrete, higher-order nucleoprotein complex. Proc. Natl. Acad. Sci. U.S.A. 97, 8997–9002. doi: 10.1073/pnas.150240197
Keywords: barrier to autointegration factor 1, emerin, laminopathy, LDL cholesterol, triglycerides, progeria, mechanotransduction
Citation: Dharmaraj T, Guan Y, Liu J, Badens C, Gaborit B and Wilson KL (2019) Rare BANF1 Alleles and Relatively Frequent EMD Alleles Including ‘Healthy Lipid’ Emerin p.D149H in the ExAC Cohort. Front. Cell Dev. Biol. 7:48. doi: 10.3389/fcell.2019.00048
Received: 17 November 2018; Accepted: 19 March 2019;
Published: 05 April 2019.
Edited by:
Kris Noel Dahl, Carnegie Mellon University, United StatesReviewed by:
David Lutz, Ruhr-Universität Bochum, GermanyKyle J. Roux, Sanford Research, United States
Copyright © 2019 Dharmaraj, Guan, Liu, Badens, Gaborit and Wilson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Katherine L. Wilson, a2x3aWxzb25AamhtaS5lZHU=
†Present address: Tejas Dharmaraj, Department of Medicine, Stanford University School of Medicine, Palo Alto, CA, United States; Youchen Guan, Department of Molecular and Cellular Biology, Baylor College of Medicine, Houston, TX, United States