 Sangam Rajak1†
Sangam Rajak1† Liliana F. Iannucci2,3†
Liliana F. Iannucci2,3† Jin Zhou2
Jin Zhou2 B. Anjum1
B. Anjum1 Nelson George1
Nelson George1 Brijesh K. Singh2
Brijesh K. Singh2 Sujoy Ghosh2
Sujoy Ghosh2 Paul M. Yen2*
Paul M. Yen2* Rohit A. Sinha1*
Rohit A. Sinha1*- 1Department of Endocrinology, Sanjay Gandhi Postgraduate Institute of Medical Sciences, Lucknow, India
- 2Program of Cardiovascular and Metabolic Disorders, Duke-NUS Medical School, Singapore, Singapore
- 3Department of Biology, University of Padua, Padua, Italy
The hepatic mevalonate (MVA) pathway, responsible for cholesterol biosynthesis, is a therapeutically important metabolic pathway in clinical medicine. Using an unbiased transcriptomics approach, we uncover a novel role of Unc-51 like autophagy activating kinase 1 (ULK1) in regulating the expression of the hepatic de novo cholesterol biosynthesis/MVA pathway genes. Genetic silencing of ULK1 in non-starved mouse (AML-12) and human (HepG2) hepatic cells as well as in mouse liver followed by transcriptome and pathway analysis revealed that the loss of ULK1 expression led to significant down-regulation of genes involved in the MVA/cholesterol biosynthesis pathway. At a mechanistic level, loss of ULK1 led to decreased expression of SREBF2/SREBP2 (sterol regulatory element binding factor 2) via its effects on AKT-FOXO3a signaling and repression of SREBF2 target genes in the MVA pathway. Our findings, therefore, discover ULK1 as a novel regulator of cholesterol biosynthesis and a possible druggable target for controlling cholesterol-associated pathologies.
Introduction
Cholesterol, at the physiological level, serves as a major constituent of cellular membranes and a precursor for bile and steroid hormone synthesis (Tabas, 2002). However, the pathological increase in cholesterol levels has been attributed as a leading cause of several cardiovascular and metabolic disorders (Tabas, 2002). Cholesterol can be synthesized de novo via the mevalonate (MVA) pathway in liver, and may also be obtained from food sources (Ye and Debose-Boyd, 2011). Interestingly, it is the endogenous synthesis of cholesterol within liver, via the MVA pathway, which has been more closely linked to the development of cardiovascular complications (Ye and Debose-Boyd, 2011). Not surprisingly, statins, which target the endogenous cholesterol/MVA biosynthesis pathways, have been very successful in countering hypercholesterolemia. However, despite the successful use of statin therapy in most of the patients, either alone or in combination with other agents, some individuals may not respond positively to statins (Reiner, 2014). This crisis warrants initiatives to discover novel alternatives and adjunct therapies to counter hypercholesterolemia. While it has been studied mostly in relation to its role in cholesterol synthesis and cardiovascular diseases, the MVA pathway has become a potential therapeutic target for several cancers, autoimmune disorders, atherosclerosis, fatty liver, and Alzheimer’s disease (Buhaescu and Izzedine, 2007).
Unc-51 like autophagy activating kinase 1 (ULK1) is a proximal protein involved in mammalian autophagic signaling (Lin and Hurley, 2016). It is the only autophagy protein, which possess a kinase activity and triggers autophagy in response to both energy and nutrient depletion (Lin and Hurley, 2016). ULK1 phosphorylates several other autophagy proteins such as ATG13 and Beclin to initiate cellular autophagy (Lin and Hurley, 2016). Besides general autophagy, ULK1 also regulates selective autophagic processes such as mitophagy (Sinha et al., 2015). Interestingly, several non-autophagic roles of ULK1 have been recently uncovered, which include involvement in interferon signaling, ER-to–Golgi cargo transport, glycolysis, and immune response (Konno et al., 2013; Joo et al., 2016; Joshi et al., 2016; Li et al., 2016). In this line of evidence, we had recently demonstrated an autophagy-independent action of ULK1 in regulating the transcriptional activity of nuclear receptors involved in hepatic lipid metabolism (Sinha et al., 2017). Owing to its kinase activity and druggability, new inhibitors for ULK1 have been recently developed (Egan et al., 2015), however, their use in metabolic diseases has not been tested.
In this study, we examined the signaling pathways that are regulated by ULK1 using transcriptomics, followed by pathway analysis during the non-starved (basal) state.
Our results uncover a novel role of ULK1 in regulating the expression of genes involved in hepatic MVA/cholesterol biosynthesis pathway via the AKT-FOXO3a (forkhead box O3a) mediated regulation of sterol regulatory element binding factor 2 (SREBF2).
Results
Genetic and Pharmacological Inhibition of ULK1 Inhibits the Expression of MVA/Cholesterogenic Genes in Mouse AML-12 Cells
To identify the signaling pathways affected by ULK1 during nutrient-rich conditions, we silenced Ulk1 gene in mouse hepatocyte cell line AML-12 using siRNA under non-starved conditions. After confirming knockdown (KD) efficiency of Ulk1 siRNA (Figure 1A) we performed a microarray analysis of the samples followed by pathway analysis (Supplementary Table S1). Our results showed significant down-regulation of the cholesterol biosynthesis pathway upon loss of ULK1 using the Reactome pathway database (Figure 1B and Supplementary Table S1). Besides the regulation of cholesterol biosynthesis, other closely related pathways such as steroid, terpenoid and triglyceride biosynthesis pathways were also downregulated upon Ulk1 silencing (Supplementary Table S1).
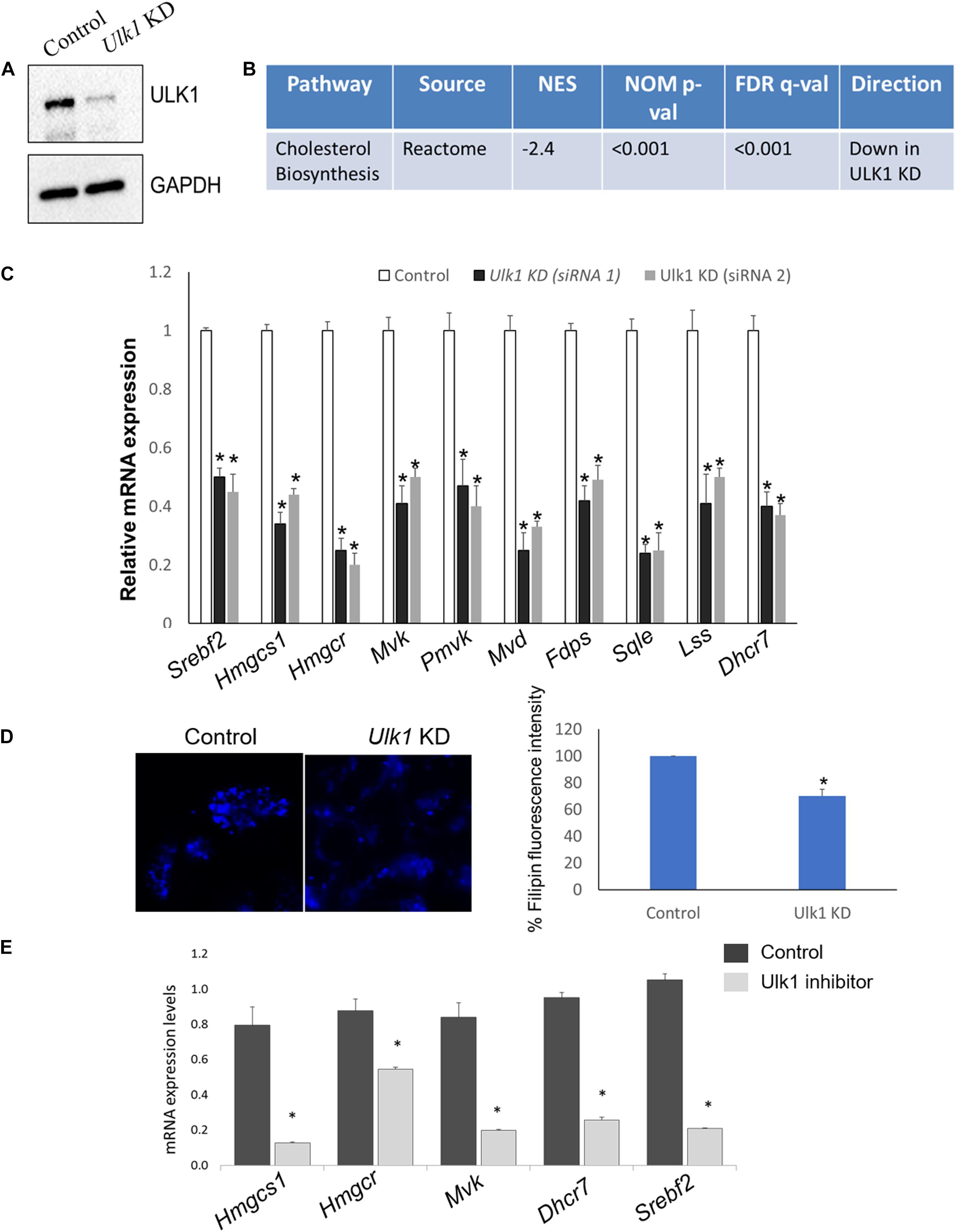
Figure 1. ULK1 regulates mevalonate/cholesterol biosynthesis pathway gene expression in mouse hepatocyte AML12 cells. (A) Representative immunoblot showing knockdown (KD) efficiency of ULK1 in AML12 cells treated with ± Ulk1 siRNA (Thermo Fisher Scientific, s75753 siRNA #1; 10 nM/72 h) or a negative siRNA (denoted as Control). (B) Microarray derived transcriptomic pathway analysis in AML12 cells and mouse liver upon Ulk1 KD as described above. (C) qRT-PCR validation of mevalonate/cholesterol biosynthesis pathway genes ± Ulk1 siRNA’s 1 (Thermo Fisher Scientific, s75753 siRNA #1) and 2 (Thermo Fisher Scientific, s75751 siRNA #2) at 10 nM concentration for 72 h in AML12 cells. Values are means ± SD (n = 4), *p < 0.05. (D) Filipin staining and quantitation showing intracellular cholesterol levels in AML12 cells treated with ± Ulk1 siRNA (Thermo Fisher Scientific, s75753 siRNA #1; 10 nM/72 h) or a negative siRNA (denoted as Control). AML12 cells upon Ulk1 KD (10 nM/72 h) were washed with PBS, fixed, and stained with filipin. All photographs were taken with the same exposure time and 20× magnification. (E) qRT-PCR validation of cholesterol biosynthesis genes enriched in the pathway analysis treated with SBI-0206965 (ULK1 inhibitor) at a dose of 10 uM for 48 h in AML12 cells. Values are means ± SD (n = 3,∗p < 0.05).
Of note, the genes involved in the Reactome cholesterol biosynthesis dataset belong to the broader MVA pathway, which is involved in cholesterol biosynthesis within the hepatocytes (Buhaescu and Izzedine, 2007; Supplementary Table S2). Next, we validated the microarray gene using qRT-PCR, showing that the expression of several rate limiting enzymes of the MVA/cholesterol biosynthesis pathway including hydroxymethylglutaryl-CoA synthase 1 (Hmgcs1), 3-hydroxy-3-methylglutaryl-CoA reductase (Hmgcr) were indeed significantly decreased in the Ulk1 silenced cells in vitro (using two different siRNA) (Figure 1C). Although not present in the gene enrichment dataset, a similar decrease in the expression of the master transcriptional regulator of sterol synthesis, Srebf2 (Madison, 2016) was also observed upon Ulk1 silencing (Figure 1C). Furthermore, repression of the cholesterogenic genes upon Ulk1 KD corroborated with reduced intra-cellular cholesterol in cells (Figure 1D). Additionally, the pharmacological inhibition of ULK1 by a kinase specific inhibitor, SBI-0206965 in AML-12 cells, also exhibited a similar suppression of genes involved in the MVA pathway (Figure 1E) suggesting a role of ULK1 kinase domain in mediating these effects on the MVA pathway genes.
Genetic Inhibition of ULK1 Inhibits the Expression of MVA/Cholesterogenic Genes in Mouse Liver
We next validated our in vitro data in vivo, by silencing Ulk1 gene in mouse liver using siRNA. In consonance with our in vitro results, we observed a similar down-regulation of MVA/cholesterol biosynthesis gene expression upon Ulk1 silencing in mouse liver using microarray, qRT-PCR and western blotting (Figures 2A–C, Supplementary Figure S1 and Supplementary Tables S3, S4). Additionally, the intra-hepatic cholesterol levels were also reduced marginally yet significantly upon Ulk1 silencing (Figure 2D). The reduction in serum cholesterol levels, however, did not reach a significant level (Supplementary Figure S2). Therefore, these results confirmed that ULK1 positively regulates MVA/cholesterol biosynthesis genes in mouse liver in vivo.
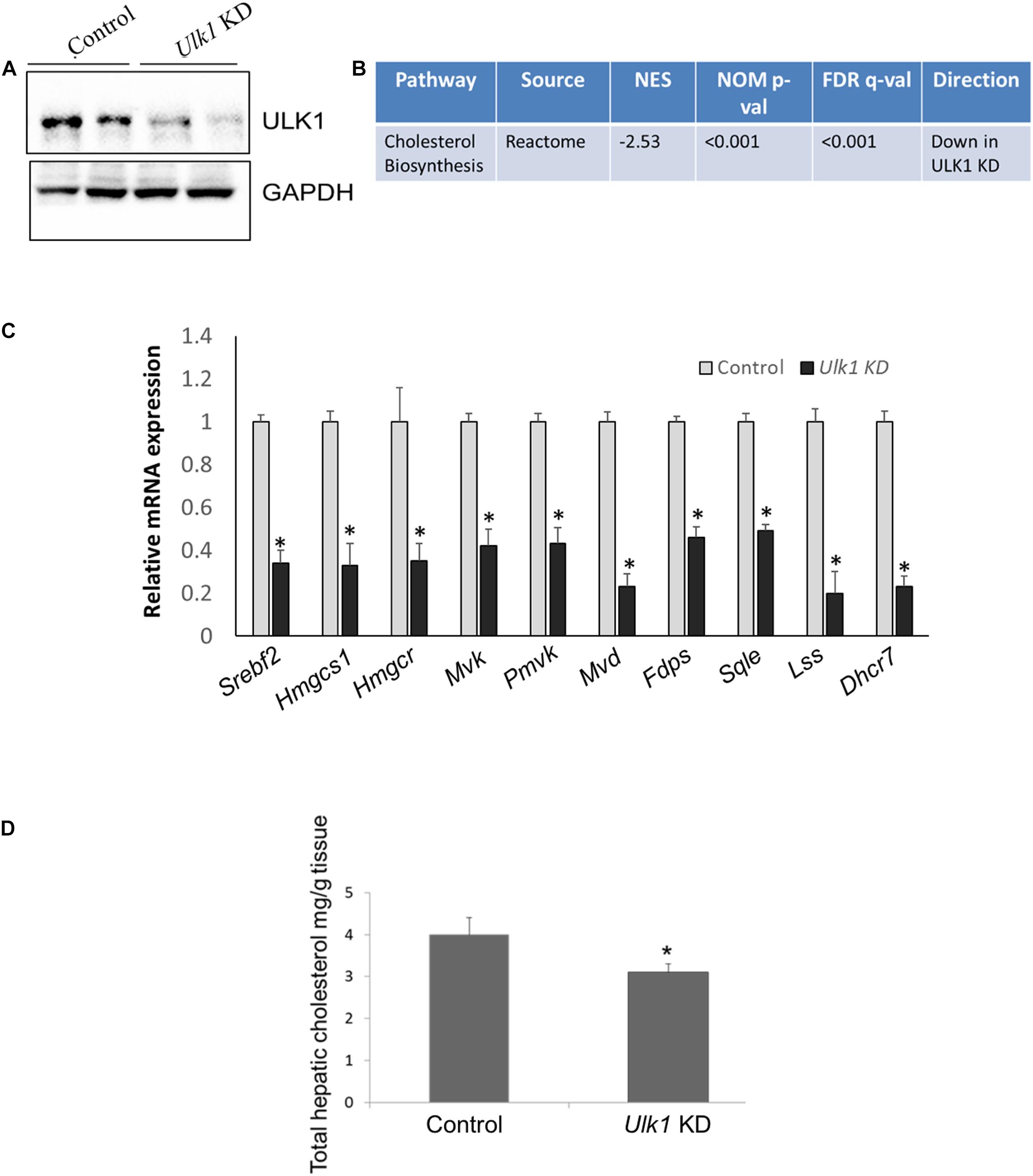
Figure 2. ULK1 regulates mevalonate/cholesterol biosynthesis pathway gene expression in mouse liver. (A) Representative immunoblot showing silencing efficiency of ULK1 in mouse liver treated with hydrodynamic tail vein delivered ± Ulk1 siRNA (40 ug/72 h). (B) Microarray derived transcriptomic pathway analysis in mouse liver upon Ulk1 KD as described above. (C) qRT-PCR validation of mevalonate/cholesterol biosynthesis pathway genes ± Ulk1 siRNA (40 ug/72 h) in AML12 mouse liver. Values are means ± SD (n = 4), ∗p < 0.05. (D) Total cholesterol content in hepatic lipid extracts in mouse liver ± Ulk1 siRNA (40 ug/72 h). Values are means ± SD (n = 4, *p < 0.05).
Loss of Atg5 and Becn1 Does Not Alter Cholesterogenic Gene Expression in AML-12 Cells
Next, to clarify the role of autophagy in this process, we observed the effect of siRNA KD of two additional ATG genes, Atg5 and Beclin1/Becn1, on the expression of key MVA pathway genes in mouse hepatocyte cells, AML-12. Interestingly, neither Atg5 nor Becn1 had any suppressive effect on MVA pathway genes (Supplementary Figure S3) thereby suggesting a probable autophagy-independent action of ULK1 in regulating the expression of these genes.
Genetic Silencing of ULK1 Inhibits the Expression of MVA/Cholesterogenic Genes in Human Hepatic Cells via AKT-FOXO3a Pathway
We also verified our results obtained from the mouse hepatic cells, in human hepatocytes using HepG2 cells. Using RNA-seq and Real-Time PCR validation analysis, we observed a similar down-regulation of signaling pathways related to human cholesterol biosynthesis and SREBF2 in cells that underwent ULK1 KD (Figures 3A–C and Supplementary Tables S5–S7). Thus, ULK1 regulated the MVA pathway across species in the mammalian hepatic cells.
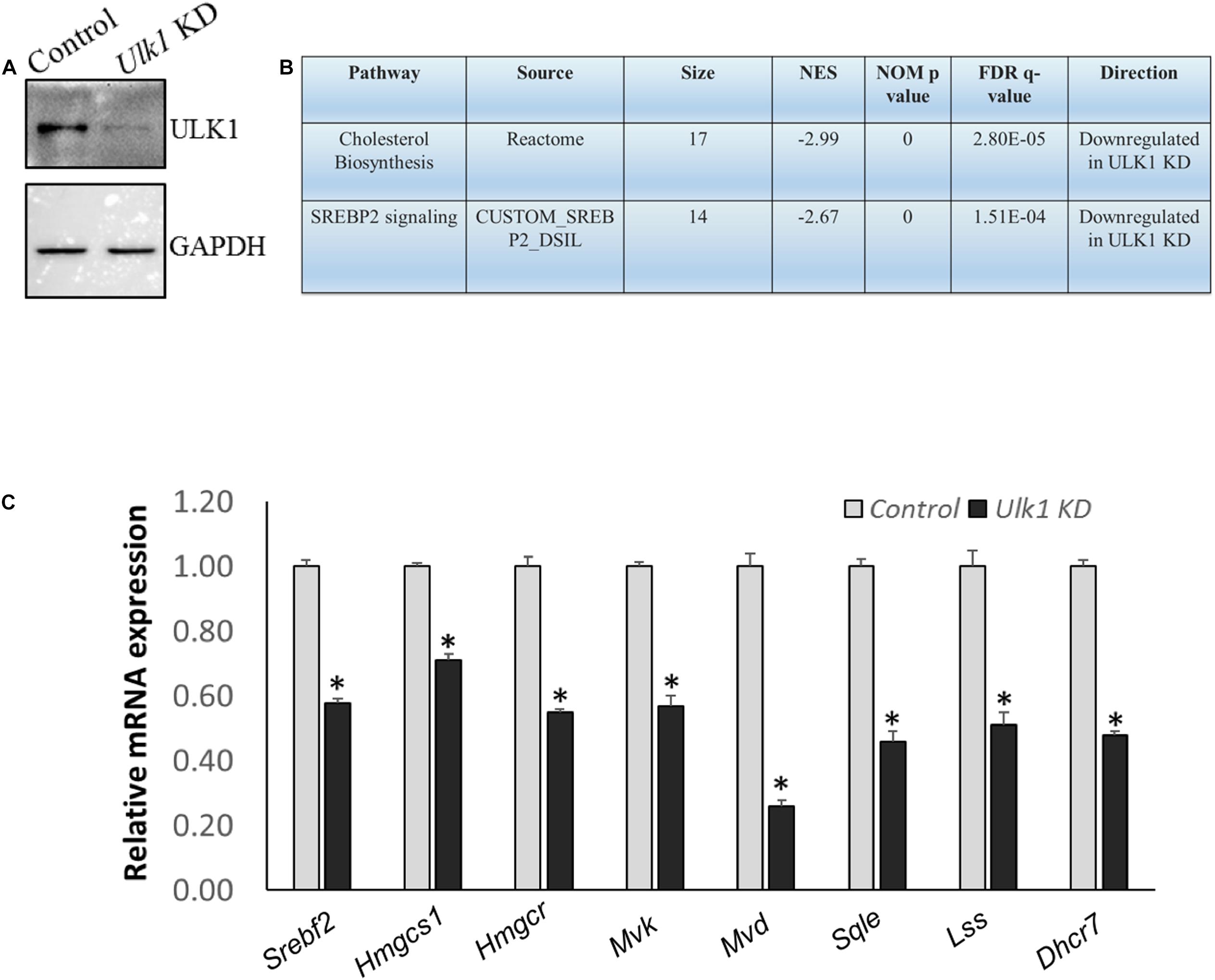
Figure 3. ULK1 loss suppresses the expression of human cholesterogenic genes (A,B) Representative immunoblot showing KD efficiency of ULK1 and RNA-seq derived transcriptomic pathway analysis in HepG2 cells treated with ± Ulk1 siRNA (Thermo Fisher Scientific, s15965; 10 nM/72 h). (C) qRT-PCR validation of mevalonate/cholesterol biosynthesis pathway genes ± Ulk1 siRNA (10 nM/72 h) in HepG2 cells. Values are means ± SD (n = 4), ∗p < 0.05.
As SREBP2/SREBF2 is the master transcriptional regulator of genes involved in the MVA/cholesterol biosynthesis pathway (Madison, 2016), we further investigated its regulation by ULK1 in HepG2 cells. Anabolic signaling by AKT has been shown to upregulate SREBF2 levels in hepatic cells (Luu et al., 2012). Additionally, AKT regulation of Srebf2 gene transcription is mediated through AKT induced inhibitory phosphorylation and nuclear exclusion of FOXO3, a transcription factor (Tzivion et al., 2011) that is a transcriptional repressor of Srebf2 gene (Tao et al., 2013). Therefore, we next investigated whether AKT-FOXO3a signaling was perturbed in the absence of ULK1 in hepatic cells. Interestingly, our results showed that both basal AKT and its substrate FOXO3a phosphorylation was suppressed in hepatic cells in which ULK1 expression was silenced by siRNA (Figures 4A,B). Similar effects were also observed in vivo (Supplementary Figures S4A,B). These results strongly suggest that the activation of FOXO3a is a possible mechanism for the Srebf2 suppression observed in ULK1 KD cells. Furthermore, we also observed increased localization of FOXO3a in the nuclear fraction isolated from ULK1 KD HepG2 cells suggesting increased FOXO3a activity (Supplementary Figures S4C,D). Therefore, to test if FOXO3a activation is indeed involved in the down-regulation of SREBF2 levels in ULK1 KD cells, we performed a rescue experiment in which both ULK1 and FOXO3a were silenced. Our results showed that the loss of FOXO3a in cells, significantly rescued the suppressive effect of ULK1 KD on SREBF2 and its target transcriptional target HMGCR (Figures 4C,D). Intriguingly, as the rescue of HMGCR expression by co-silencing of FOXO3a and ULK1 was not complete, it is possible that other ULK1 regulated FOXO (Forkhead box) proteins such as FOXO1 (Koo et al., 2018) may also be involved in SREBF2 modulation and needs further investigation. The effects of ULK1 KD on AKT activation along with SREBF2 and HMGCR levels were also corroborated using a pharmacological inhibitor of ULK1, SBI-0206965, in HepG2 cells (Figure 4E). Therefore, these results establish a mechanistic link between ULK1 and SREBF2 via AKT-FOXO3a pathway (Figure 4F). However, the molecular basis of AKT regulation by ULK1 remains to be investigated.
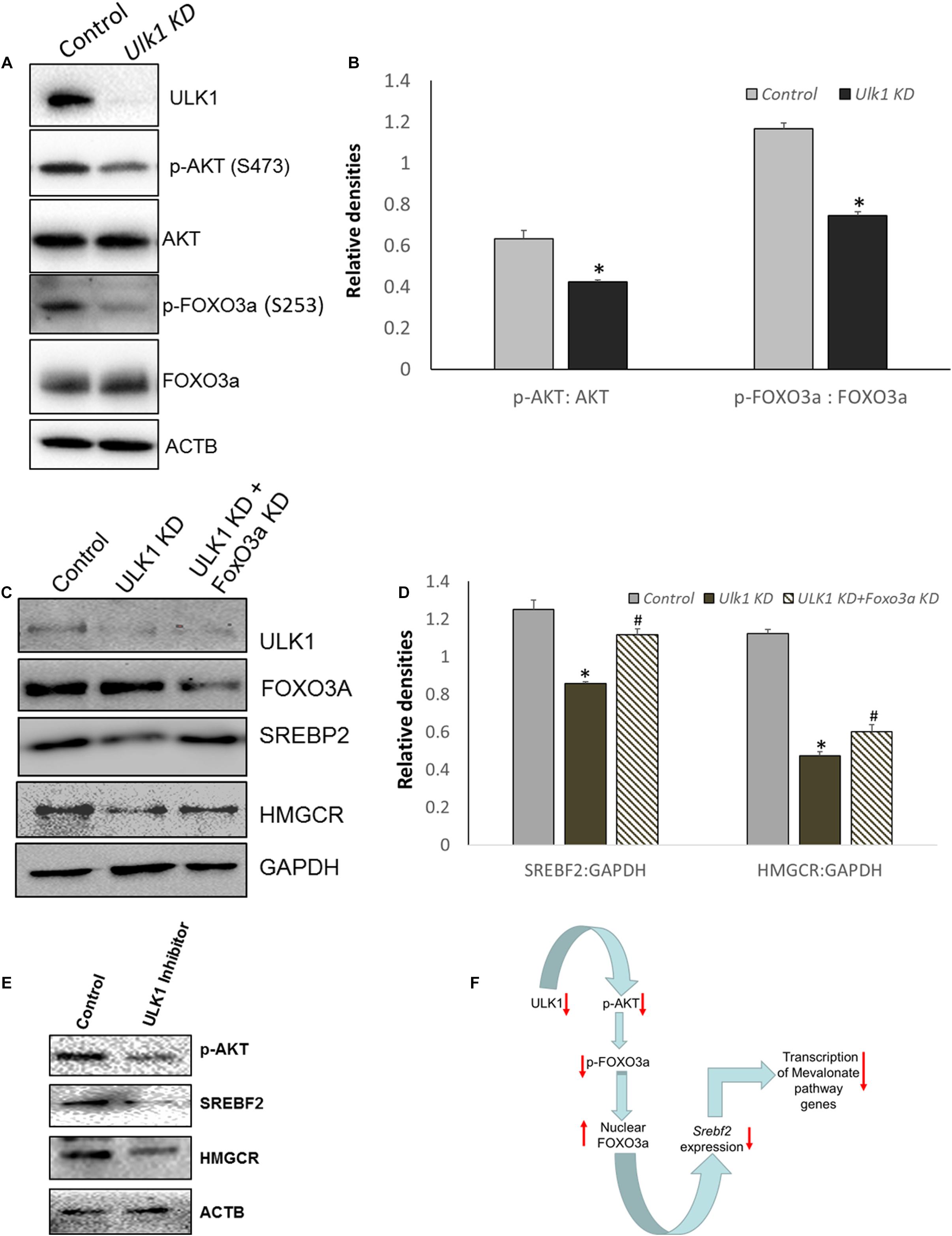
Figure 4. ULK1 loss suppress human cholestrogenic genes via effects on AKT-FOXO3a pathway. (A,B) Representative immunoblot and densitometry showing cellular levels of phosphorylated and total AKT and FoxO3a proteins ± Ulk1 siRNA (10 nM/48 h) in HepG2 cells. Values are means ± SD (n = 3),∗p < 0.05. (C,D) Representative Immunoblot and densitometry showing protein levels of SREBF2 and HMGCR in HepG2 ± Ulk1 alone and with FoxO3a (Cell Signaling Technology, #6302) double KD (10 nM) for 48 h of treatment. Values are means ± SD (n = 3), ∗p < 0.05 between Ulk1 KD vs. Control and #p < 0.05 between Ulk1 KD + FoxO3a KD vs. Ulk1 KD alone. (E) Representative immunoblot of HepG2 cells treated with SBI-0206965 (ULK1 inhibitor) at a dose of 10 uM for 48 h. (F) Schematic diagram showing the regulation of Mevalonate/cholesterol biosynthesis pathway upon loss of ULK1.
Discussion
The MVA pathway is the major metabolic pathway involved in sterol synthesis and has been extensively investigated for its role(s) in cardiovascular diseases. Statins are a very effective class of drugs that target HMGCR, a key rate limiting enzyme in cholesterol biosynthesis (Stancu and Sima, 2001) and are used to treat hypercholesterolemia. However, despite their effectiveness, some patients develop resistance to statins (Reiner, 2014). Given these issues, there have been considerable efforts to develop novel strategies which target cholesterol biosynthesis (Bou Malham and Goldberg, 2016; Sinha et al., 2019). Of note, the inhibition of MVA signaling has shown promising results in limiting cancer progression (Mullen et al., 2016) including liver cancers (Ogura et al., 2018). Interestingly, ULK1 inhibitors have also shown their efficacy in limiting cancer growth (Egan et al., 2015); thus, it is possible that some of the anti-tumor effects of ULK1 inhibitors are mediated through their effect on the MVA pathway.
Our results uncover a novel autophagy independent signaling by ULK1, which involves the regulation of anabolic AKT signaling and nuclear activity of FOXO transcription factor. Although the autophagy-lysosomal pathway is known to regulate nuclear receptor action (Siong Tan et al., 2019; Sinha et al., 2020), the direct effect of ULK1 on nuclear receptor/transcription factor activity remains less explored. As FOXO3a and related FOXO family proteins are involved in a myriad of cellular processes and human diseases (Lee and Dong, 2017), the modulation of ULK1 opens an exciting possibility of targeting FOXO related disorders.
Concerning the effect of ULK1 silencing on systemic cholesterol levels in mouse, we did not observe a significant reduction in serum total cholesterol albeit it showed a downward trend. We believe that this effect could be due to the transient nature of the knockdown experiments performed in this study. Perhaps, a chronic suppression using a liver-specific Ulk1 knockout model or pharmacological inhibitors would be needed to resolve it better. However, considering the functional redundancy of different ULK isoforms, the role of ULK2 in the regulation of cholesterogenic genes remains to be investigated. Although beyond the scope of the present study, further work using ULK1 inhibitors in animal models of hypercholesterolemia or carcinogenesis may be useful to test the translational value of ULK1 inhibitors, either alone or as an adjunct with statins, for the protection or treatment of diseases associated with deranged cholesterol synthesis.
Materials and Methods
Cell Culture
Mouse hepatocytes AML12 (CRL-2254) cells were maintained at 37°C in DMEM-F12 1:1 containing 10% fetal bovine serum (FBS), 1× ITS (Thermo Fisher Scientific, 41400), 10 nM dexamethasone and 1× penicillin/streptomycin, in a 5% CO2 humidified incubator. HepG2 cells were maintained at 37°C in DMEM containing 10% FBS. For siRNA transfection, cells were transfected using RNAiMAX (Thermo Fisher Scientific, 13778150) with Ulk1 (Thermo Fisher Scientific, s75753), Atg5 (Thermo Fisher Scientific, s62452), Becn1 (Thermo Fisher Scientific, s80166), FoxO3a (Cell Signaling Technology, #6302) siRNA (10 nM) for 48 or 72 h, followed by harvesting cells for RNA or protein extraction. ULK1 inhibitor SBI-0206965 (Selleckchem, Catalog No. S7885) was added at a concentration of 10 μM for 48 h.
In vivo siRNA Administration to Liver
C57BL/6N male mice (4–6 weeks old) were administered 40 μg of in vivo-ready Ulk1 (Thermo Fisher Scientific, s75753) or negative siRNA (Thermo Fisher Scientific, 4390843) every 24 h for 3 days via hydrodynamic tail-vein injection protocol using Mirus Bio TransIT-QR Delivery Solution as per manufacturer’s guidelines. All experiments were performed according to institutional animal ethics guidelines at Duke-NUS Medical School, Singapore.
MicroArray and RNA-Seq Analysis
Gene expression microarray profiling was performed using GeneChip Mouse Gene 2.0 ST Array (Affymetrix) on pool of four samples. The cDNA generation, labeling and hybridization were performed at Duke-NUS Genome Biology Facility, Duke-NUS Graduate Medical School, Singapore. Gene expression signals were quantile normalized, and differentially expressed genes were identified via ANOVA models, using treatment specific contrasts (Partek Genomics Suite software, version 6.6). Statistical significance of differentially expressed genes was ascertained via the false discovery rate (FDR). RNA-seq analysis was performed on a pool of four samples per group. RNA-Seq Library Construction and Library QC, c-bot Cluster Generation and Hi-Seq3000 sequencing performed at Duke-NUS Genome Biology Facility, Duke-NUS Graduate Medical School, Singapore. Pathway enrichment analysis was conducted via the Gene Set Enrichment Analysis tool using a list of KEGG and Reactome pathways extracted from the Molecular Signatures Database1. Significance of pathway enrichment was ascertained by permutation testing of gene sets and calculation of FDR. The microarray and RNA-Seq data have been deposited to public databases with accession numbers GSE147579 and PRJNA625896, respectively.
RNA Isolation and qRT-PCR
Total RNA was isolated and qRT-PCR was performed using the QuantiTect SYBR Green PCR Kit (Qiagen, 204141) according to manufacturer’s instructions. Gapdh gene was used for normalization. KiCqStart® SYBR® Green Primers were purchased from Sigma-Aldrich, United States. Primer ID were: Mouse Srebf2 (M_Srebf2_1), Human Srebf2 (H_SREPF2_1), Mouse Hmgcs1 (M_Hmgcs1_1), Human Hmgcs1 (H_HMGCS1_1), Mouse Hmgcr (M_Hmgcr_1), Human Hmgcr (H_HMGCR_1), Mouse Mvk (M_Mvk_1), Human Mvk (H_MVK_1), Mouse Pmvk (M_Pmvk_1), Mouse Mvd (M_Mvd_1), Human Mvd (H_MVD_1), Mouse Fdps (M_Fdps_1), Mouse Sqle (M_Sqle_1), Human Sqle (H_SQLE_1), Mouse Lss (M_Lss_1), Human Lss (H_LSS_1), Mouse Dhcr7 (M_Dhcr7_1), Human Dhcr7 (H_DHCR7_1), Mouse Gapdh (M_Gapdh_1), Human Gapdh (H_GAPDH_1).
Cholesterol Measurement
Intracellular cholesterol levels in AML-12 cells were visualized using Filipin staining and quantitation was performed using Image J software. Four equal sized areas with 20 cells each were chosen in each image and the fluorescence intensity of each area was determined. These values were then used to calculate the average fluorescence of each image and then of control (set to 100%) and Ulk1 silenced group. Mouse liver total cholesterol was measured using commercial kit (MAK043, SIGMA-ALDRICH, United States).
Western Blotting
Cells or tissue samples were lysed using CelLyticTM M Cell Lysis Reagent (Sigma, C2978) and immunoblotting was performed as per manufacturer’s guidelines (Bio-Rad Laboratories, United States). Nuclear fractionation was done using NE-PERTM Nuclear and Cytoplasmic Extraction Reagents (Thermo Fisher Scientific, 78833). Image acquisition was done using ChemiDoc (Bio-Rad ChemiDocTM MP System, 1708280). Densitometry analysis was performed using ImageJ software (NIH, Bethesda, MD, United States). Antibodies used were anti-ULK1 (Cell Signaling Technology, #8054), anti-GAPDH (Cell Signaling Technology, #5174), anti-phospho AKT (Cell Signaling Technology, #4060), anti-AKT (Cell Signaling Technology, #4685), anti-FoxO3a Ser253 (Cell Signaling Technology, #12829), anti-phospho FoxO3a (Cell Signaling Technology, #9466), anti-Beclin (Cell Signaling Technology, #3495), anti-ATG5 (Cell Signaling Technology, #12994), anti-SREBP2 (ABCAM, #ab30682), anti-HMGCR (ABCAM, #ab174830).
Statistics
Results are expressed as mean ± SD. The statistical significance of differences was assessed by unpaired student t-test or one-way ANOVA when comparing different groups.
Data Availability Statement
The datasets generated for this study can be found in the GSEA (GSE147579) and SRA (PRJNA625896).
Ethics Statement
The animal study was reviewed and approved by IACUC, NUS.
Author Contributions
RS conceived the experiments, researched the data and revised, wrote, and approved the manuscript. SG and PY researched the data and revised and approved the manuscript. SR, LI, JZ, NG, BA, and BS performed the experiments. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the SERB (SRG/2019/000398), ICMR (59/05/2019/ONLINE/BMS/TRM), and Wellcome Trust/DBT India Alliance Fellowship (IA/I/16/2/502691) awarded to RS.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2020.523550/full#supplementary-material
Abbreviations
ULK1, Unc-51 like autophagy activating kinase 1; SREBF2, sterol regulatory element binding factor 2; Hmgcs1, hydroxymethylglutaryl-CoA synthase 1; Hmgcr, 3-hydroxy-3-methylglutaryl-CoA reductase; Mvk, mevalonate kinase; Pmvk, phosphomevalonate kinase; Mvd, mevalonate diphosphate decarboxylase; Lss, lanosterol synthase; Sqle, squalene monooxygenase; Dhcr7, 7-dehydrocholesterol reductase; Fdps, farnesyl diphosphate synthase; FOXO3a, forkhead box O3a; NES, normalized enrichment score; NOM p-val, nominal p value.
Footnotes
References
Bou Malham, S., and Goldberg, A. C. (2016). Cardiovascular risk reduction: the future of cholesterol lowering drugs. Curr. Opin. Pharmacol. 27, 62–69. doi: 10.1016/j.coph.2016.01.007
Buhaescu, I., and Izzedine, H. (2007). Mevalonate pathway: a review of clinical and therapeutical implications. Clin. Biochem. 40, 575–584. doi: 10.1016/j.clinbiochem.2007.03.016
Egan, D. F., Chun, M. G., Vamos, M., Zou, H., Rong, J., Miller, C. J., et al. (2015). Small molecule inhibition of the autophagy kinase ULK1 and identification of ULK1 substrates. Mol. Cell 59, 285–297. doi: 10.1016/j.molcel.2015.05.031
Joo, J. H., Wang, B., Frankel, E., Ge, L., Xu, L., Iyengar, R., et al. (2016). The noncanonical role of ULK/ATG1 in ER-to-Golgi trafficking is essential for cellular homeostasis. Mol. Cell 62, 491–506. doi: 10.1016/j.molcel.2016.04.020
Joshi, A., Iyengar, R., Joo, J. H., Li-Harms, X. J., Wright, C., Marino, R., et al. (2016). Nuclear ULK1 promotes cell death in response to oxidative stress through PARP1. Cell Death Differ. 23, 216–230. doi: 10.1038/cdd.2015.88
Konno, H., Konno, K., and Barber, G. N. (2013). Cyclic dinucleotides trigger ULK1 (ATG1) phosphorylation of STING to prevent sustained innate immune signaling. Cell 155, 688–698. doi: 10.1016/j.cell.2013.09.049
Koo, Y., Garneau, M. P., Zhang, Q., and Dale Abel, E. (2018). Regulation of hepatic insulin sensitivity and hepatic steatosis by ULK1. Diabetes 67:1863. doi: 10.2337/db18-1863-p
Lee, S., and Dong, H. H. (2017). FoxO integration of insulin signaling with glucose and lipid metabolism. J. Endocrinol. 233, R67–R79.
Li, T. Y., Sun, Y., Liang, Y., Liu, Q., Shi, Y., Zhang, C. S., et al. (2016). ULK1/2 constitute a bifurcate node controlling glucose metabolic fluxes in addition to autophagy. Mol. Cell 62, 359–370. doi: 10.1016/j.molcel.2016.04.009
Lin, M. G., and Hurley, J. H. (2016). Structure and function of the ULK1 complex in autophagy. Curr. Opin. Cell Biol. 39, 61–68. doi: 10.1016/j.ceb.2016.02.010
Luu, W., Sharpe, L. J., Stevenson, J., and Brown, A. J. (2012). Akt acutely activates the cholesterogenic transcription factor SREBP-2. Biochim. Biophys. Acta 1823, 458–464. doi: 10.1016/j.bbamcr.2011.09.017
Madison, B. B. (2016). Srebp2: a master regulator of sterol and fatty acid synthesis. J. Lipid Res. 57, 333–335. doi: 10.1194/jlr.c066712
Mullen, P. J., Yu, R., Longo, J., Archer, M. C., and Penn, L. Z. (2016). The interplay between cell signalling and the mevalonate pathway in cancer. Nat. Rev. Cancer 16, 718–731. doi: 10.1038/nrc.2016.76
Ogura, S., Yoshida, Y., Kurahashi, T., Egawa, M., Furuta, K., Kiso, S., et al. (2018). Targeting the mevalonate pathway is a novel therapeutic approach to inhibit oncogenic FoxM1 transcription factor in human hepatocellular carcinoma. Oncotarget 9, 21022–21035. doi: 10.18632/oncotarget.24781
Reiner, Z. (2014). Resistance and intolerance to statins. Nutr. Metab. Cardiovasc. Dis. 24, 1057–1066. doi: 10.1016/j.numecd.2014.05.009
Sinha, R. A., Bruinstroop, E., Singh, B. K., and Yen, P. M. (2019). Nonalcoholic fatty liver disease and hypercholesterolemia: roles of thyroid hormones, metabolites, and agonists. Thyroid 29, 1173–1191. doi: 10.1089/thy.2018.0664
Sinha, R. A., Rajak, S., Singh, B. K., and Yen, P. M. (2020). Hepatic lipid catabolism via PPARalpha-lysosomal crosstalk. Int. J. Mol. Sci. 21:2391. doi: 10.3390/ijms21072391
Sinha, R. A., Singh, B. K., Zhou, J., Wu, Y., Farah, B. L., Ohba, K., et al. (2015). Thyroid hormone induction of mitochondrial activity is coupled to mitophagy via ROS-AMPK-ULK1 signaling. Autophagy 11, 1341–1357. doi: 10.1080/15548627.2015.1061849
Sinha, R. A., Singh, B. K., Zhou, J., Xie, S., Farah, B. L., Lesmana, R., et al. (2017). Loss of ULK1 increases RPS6KB1-NCOR1 repression of NR1H/LXR-mediated Scd1 transcription and augments lipotoxicity in hepatic cells. Autophagy 13, 169–186. doi: 10.1080/15548627.2016.1235123
Siong Tan, H. W., Anjum, B., Shen, H. M., Ghosh, S., Yen, P. M., and Sinha, R. A. (2019). Lysosomal inhibition attenuates peroxisomal gene transcription via suppression of PPARA and PPARGC1A levels. Autophagy 15, 1455–1459. doi: 10.1080/15548627.2019.1609847
Stancu, C., and Sima, A. (2001). Statins: mechanism of action and effects. J. Cell. Mol. Med. 5, 378–387. doi: 10.1111/j.1582-4934.2001.tb00172.x
Tao, R., Xiong, X., Depinho, R. A., Deng, C. X., and Dong, X. C. (2013). Hepatic SREBP-2 and cholesterol biosynthesis are regulated by FoxO3 and Sirt6. J. Lipid Res. 54, 2745–2753. doi: 10.1194/jlr.m039339
Tzivion, G., Dobson, M., and Ramakrishnan, G. (2011). FoxO transcription factors; regulation by AKT and 14-3-3 proteins. Biochim. Biophys. Acta 1813, 1938–1945.
Keywords: ULK1, mevalonate pathway, cholesterogenesis, SREBF2, FOXO3A transcription factor
Citation: Rajak S, Iannucci LF, Zhou J, Anjum B, George N, Singh BK, Ghosh S, Yen PM and Sinha RA (2020) Loss of ULK1 Attenuates Cholesterogenic Gene Expression in Mammalian Hepatic Cells. Front. Cell Dev. Biol. 8:523550. doi: 10.3389/fcell.2020.523550
Received: 30 December 2019; Accepted: 11 September 2020;
Published: 30 September 2020.
Edited by:
Prasanna K. Santhekadur, JSS Academy of Higher Education and Research, IndiaReviewed by:
Vladimir Stanislavovich Shavva, Karolinska Institutet (KI), SwedenGopi Sundaram, Central Food Technological Research Institute (CSIR), India
Anjali Devi, JSS Academy of Higher Education and Research, India
Copyright © 2020 Rajak, Iannucci, Zhou, Anjum, George, Singh, Ghosh, Yen and Sinha. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rohit A. Sinha, YW50aG9ueS5yb2hpdEBnbWFpbC5jb20=; Paul M. Yen, cGF1bC55ZW5AZHVrZS1udXMuZWR1LnNn
†These authors have contributed equally to this work