 Laura J. Smyth1
Laura J. Smyth1 Christopher C. Patterson1
Christopher C. Patterson1 Elizabeth J. Swan1
Elizabeth J. Swan1 Alexander P. Maxwell1,2
Alexander P. Maxwell1,2 Amy Jayne McKnight1*
Amy Jayne McKnight1*- 1Centre for Public Health, Queen’s University Belfast, Belfast, United Kingdom
- 2Regional Nephrology Unit, Belfast City Hospital, Belfast, United Kingdom
A subset of individuals with type 1 diabetes will develop diabetic kidney disease (DKD). DKD is heritable and large-scale genome-wide association studies have begun to identify genetic factors that influence DKD. Complementary to genetic factors, we know that a person’s epigenetic profile is also altered with DKD. This study reports analysis of DNA methylation, a major epigenetic feature, evaluating methylome-wide loci for association with DKD. Unique features (n = 485,577; 482,421 CpG probes) were evaluated in blood-derived DNA from carefully phenotyped White European individuals diagnosed with type 1 diabetes with (cases) or without (controls) DKD (n = 677 samples). Explicitly, 150 cases were compared to 100 controls using the 450K array, with subsequent analysis using data previously generated for a further 96 cases and 96 controls on the 27K array, and de novo methylation data generated for replication in 139 cases and 96 controls. Following stringent quality control, raw data were quantile normalized and beta values calculated to reflect the methylation status at each site. The difference in methylation status was evaluated between cases and controls; resultant P-values for array-based data were adjusted for multiple testing. Genes with significantly increased (hypermethylated) and/or decreased (hypomethylated) levels of DNA methylation were considered for biological relevance by functional enrichment analysis using KEGG pathways. Twenty-two loci demonstrated statistically significant fold changes associated with DKD and additional support for these associated loci was sought using independent samples derived from patients recruited with similar inclusion criteria. Markers associated with CCNL1 and ZNF187 genes are supported as differentially regulated loci (P < 10–8), with evidence also presented for AFF3, which has been identified from a meta-analysis and subsequent replication of genome-wide association studies. Further supporting evidence for differential gene expression in CCNL1 and ZNF187 is presented from kidney biopsy and blood-derived RNA in people with and without kidney disease from NephroSeq. Evidence confirming that methylation sites influence the development of DKD may aid risk prediction tools and stimulate research to identify epigenomic therapies which might be clinically useful for this disease.
Introduction
Diabetes and associated complications are major personal and public health concerns, with diabetic kidney disease (DKD) contributing a substantial financial burden to healthcare providers (Franciosi et al., 2013; Campbell et al., 2017; Arredondo et al., 2018; Disease et al., 2018; Kawaguchi et al., 2020). DKD develops in approximately one-third of individuals with diabetes and remains the most common primary diagnosis of chronic kidney disease (CKD) leading to end-stage kidney disease (ESKD) worldwide (Hill et al., 2014; US Renal and Data System., 2018; Lassalle et al., 2019; UK Renal Registry, 2019). Current treatments are based on modification of risk factors and include the reduction in elevated blood pressure, hyperglycemia and hyperlipidemia. Epidemiological evidence confirms that heritable factors play a major role in the development and progression of DKD, but despite the identification of several genetic loci associated with DKD most of the inherited risk factors remain unknown (Sandholm et al., 2012, 2013, 2014, 2017; Canadas-Garre et al., 2018, 2019; van Zuydam et al., 2018; Fu et al., 2019; Salem et al., 2019).
Emerging evidence for epigenetic phenomena has transformed investigations of heritable influences on disease and, complementary to genome-wide association studies (GWAS), it is now cost-effective to perform population-based studies of the epigenome (Rakyan et al., 2011b; Canadas-Garre et al., 2018; Kato and Natarajan, 2019; Kerr et al., 2020). Epigenetic modifications modulate gene expression without changing the DNA sequence; these may be either stably inherited or dynamic epigenetic marks. Methylation is a key epigenetic feature that plays an important role in chromosomal integrity and regulation of gene expression with different methylation profiles now being associated with many complex diseases (Murphy and Mill, 2014; Greenberg and Bourc’his, 2019; Bayoumi et al., 2020; Hoang et al., 2020). Epigenome-wide association studies (EWAS) have revealed methylation features associated with type 1 diabetes (Rakyan et al., 2011a; Stefan et al., 2014; Elboudwarej et al., 2016), gestational diabetes (Haertle et al., 2017; Dias et al., 2019; Howe et al., 2020), and type 2 diabetes (Florath et al., 2016; Walaszczyk et al., 2018; Ochoa-Rosales et al., 2020). EWAS have also identified methylation features associated with chronic kidney disease (Smyth et al., 2014a,b, 2018; Chu et al., 2017; Qiu et al., 2018).
Differential DNA methylation has been associated with “metabolic memory” of glycemic control and with a higher risk of developing DKD (Swan et al., 2015; Keating et al., 2018; Aranyi and Susztak, 2019; Gluck et al., 2019; Gu, 2019; Jia et al., 2019; Park et al., 2019). As DKD is not clinically detectable until significant organ damage has developed (albuminuria and/or reduced eGFR), more effective diagnostic tools and treatments, guided by a better understanding of pathophysiology, are urgently required. The identification of novel epigenetic risk markers and biological pathways influencing DKD would contribute more to the understanding of this serious diabetic complication. This manuscript describes an EWAS from carefully phenotyped individuals who were specifically recruited to investigate molecular risk factors for DKD in people with type 1 diabetes, followed by in silico and de novo replication. Our overarching aim is to identify differentially methylated CpG sites associated with DKD.
Materials and Methods
Participants
All recruited individuals provided written, informed consent and this study was approved by a United Kingdom Multicentre Research Ethics Committee (MREC/98/6/71). The discovery group comprised a subset of individuals (150 cases compared to 100 controls) selected from an established United Kingdom case-control collection that was recruited specifically to investigate risk factors for DKD (McKnight et al., 2010). One-third of the case group had end stage renal disease (ESRD). All individuals were White, from the United Kingdom and were diagnosed with type 1 diabetes prior to the age of 31 years. Participants in the case group had persistent proteinuria (>0.5 g protein/24 h) at least 10 years after diagnosis of diabetes, hypertension (BP > 135/85 mmHg or treatment with antihypertensive agents) and diabetic retinopathy. Individuals in the control group had at least 15 years duration of type 1 diabetes with normal renal function and were not receiving antihypertensive treatment. Cases and controls in the discovery group were matched for age, gender and duration of diabetes. The in silico replication groups comprised independent samples from the remainder of this collection and had similar characteristics to those involved in the discovery group. Similarly, all individuals included in the de novo typing phase were selected using blood-derived DNA from the larger collection (Table 1).
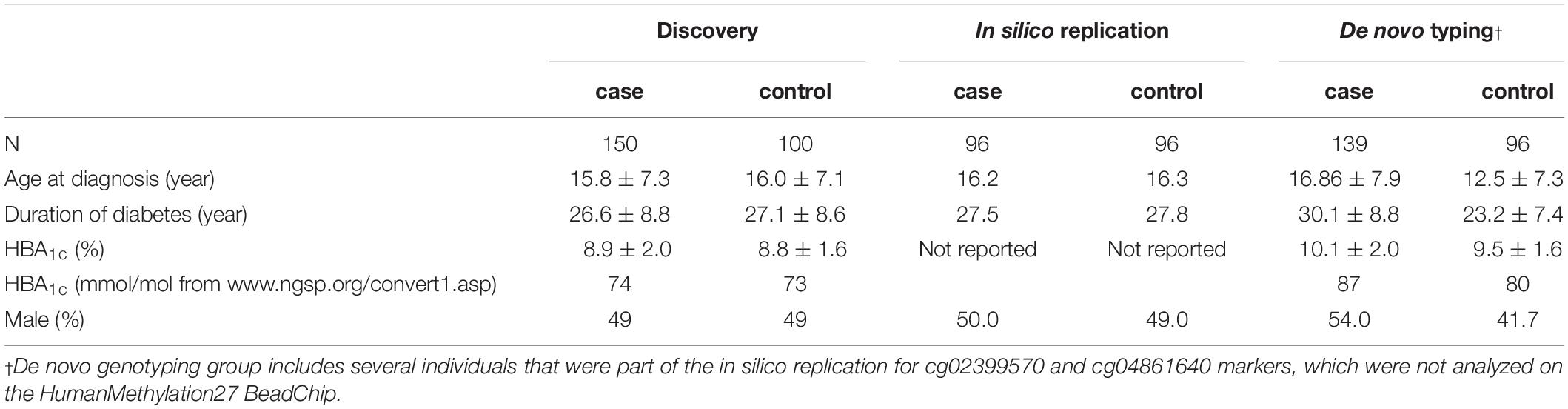
Table 1. Clinical characteristics of individuals participating in this study.
Methylation Typing
482,421 unique CpG features were evaluated in the discovery group. Existing blood-derived DNA [extracted using the salting-out method as previously described (Bell et al., 2010)] was accurately quantitated using PicoGreen®, normalized, and bisulfite treated using the EZ-96 DNA Methylation-GoldTM Kit (Zymo Research, Irvine, CA, United States) with case and control samples randomly distributed across plates. The Infinium Human Methylation 450K BeadChip (Sandoval et al., 2011) (Illumina Inc., San Diego, CA, United States) was employed according to manufacturer’s instructions. Raw data were adjusted for dye bias and quantile normalized at the probe level with data derived from sites using Infinium I or Infinium II assay chemistry considered separately. This high-throughput platform enables quantitative evaluation of methylation levels with single nucleotide resolution, generating a methylation score per individual (a β value ranging from 0 for unmethylated to 1 representing complete methylation) for each CpG site.
In silico support for candidate loci was sought using normalized data available at the Gene Expression Omnibus accession GSE20067 (Bell et al., 2010). We previously analyzed 27,578 CpGs based on data from the Illumina Infinium® HumanMethylation27 BeadChip in 192 individuals diagnosed as having type 1 diabetes with and without nephropathy using time to event (duration of diabetes until nephropathy) analysis (Bell et al., 2010).
De novo replication was performed using Sequenom Epityper assays (Sequenom Inc., Hamburg, Germany). Sequenom facilitates the quantitative analysis of DNA methylation using matrix-assisted laser desorption ionization time-of-flight mass spectrometry. Assays were designed using the default settings (except mass window range changed to 1500–8000) at www.epidesigner.com. Amplicons were carefully designed to cover target sites, results were generated following the manufacturer’s protocol and data analyzed using Epityper viewer 1.2 (Sequenom). An additional 235 independent individuals were typed as part of the replication population for de novo confirmation with the threshold for significance set at P < 0.05 without adjustment for multiple testing (Table 1).
Analysis
For the discovery cohort, stringent quality control included evaluation of bisulfite conversion efficiency, staining, hybridization, target removal, extension, dye specificity and 600 integral negative controls. Samples were excluded where more than 10% of probes did not generate useful data and all sites with poor detection P-values (detection P ≥ 0.05) were set to “missing.” Known non-CpG targeting probes (n = 3,091) were excluded from all results (Chen et al., 2013; Zhou et al., 2017). Probes on autosomes were evaluated for association with DKD adjusted for sex, duration of diabetes, age at diagnosis, and HBA1c with subgroup analysis performed for cases with ESRD. Sex-specific analysis was performed for probes on chromosome X. Converting intensity levels to beta values and initial preprocessing were performed using the default settings within GenomeStudio’s methylation module v1.9 (Illumina) (Smyth et al., 2014b). Principal component analysis and multi-dimensional scaling were employed and potential outliers from gender, non-White ethnicity or experimental batch effects were excluded from further investigation. Proportional white cell counts from whole blood (B cells, granulocytes, monocytes, NK cells, and T cells subsets) were estimated using Houseman’s and Reinius’ approaches (Houseman et al., 2012; Reinius et al., 2012). Microarray quality control metrics reports were generated using the arrayQualityMetrics package with the recommended parameters in Bioconductor (Kauffmann and Huber, 2010); arrays that did not pass the default quality control thresholds were excluded from further analysis. After correction for dye bias, raw data were normalized using quantile normalization using methylumi1. The Bioconductor package Limma (Wettenhall and Smyth, 2004; Ritchie et al., 2015) was used to generate association results. Significance values for the 450K array-based analysis were adjusted for multiple testing using the Benjamini & Hochberg, “fdr” adjustment, implemented in limma and reported as adjusted p-values.
For in silico replication, previously generated association data from the Illumina Infinium® HumanMethylation27 BeadChip for 192 individuals diagnosed as having type 1 diabetes with and without kidney disease (Bell et al., 2010) was “looked up.”
For de novo replication, new genotype data was generated using Sequenom’s MassARRAY® System. Sequenom-based data were analyzed by the large samples z test statistic with regression analysis to adjust for sex, duration of diabetes, age at diagnosis, and HBA1c. The area under the receiver operating characteristic (ROC) curve was generated using SPSS (version 15) to assess the ability of a CpG to distinguish between cases and controls.
In silico functional support for replicated loci was sought from datasets within NephroSeq (last accessed 14th September 2020)2 for publicly available gene expression data using a p-value threshold of 0.05 and a fold change of at least 1.5.
To investigate if SNPs from our GWAS for DKD were located near differentially methylated CpG probes, SNPs in key chromosomal locations (5 kb flanking top ranked markers or their associated genes) were evaluated for association with DKD using publicly available data from our genome-wide association study (dbGAP phs000389.v1.p1); for SNP-based association analysis of this GWAS data, P-values were corrected by genomic control and adjusted for age at diagnosis, duration of diabetes, gender, biochemistry center and the first ten components of the study specific principal component analysis (Sandholm et al., 2012).
Provisionally significant genes (adjusted P < 0.0001) were analyzed for enrichment of KEGG pathway membership; enrichment was assessed for genes that showed increased or decreased methylation values separately. Additionally, DAVID Bioinformatics Resources 6.7 was interrogated for top-ranked gene results in the genetic association database, Online Mendelian Inheritance in Man, Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways and articles recorded in PubMed (Huang da et al., 2009).
Results
Greater than 99% concordance was observed between duplicate samples and experimentally defined genders matched each individual submitted for analysis based on Y-chromosome specific loci. Experimental controls generated expected results, but 5 arrays were identified as outliers and were thus removed from all subsequent analyses. There was no significant difference in the estimated proportional white cell counts between case and control groups (Supplementary Table 1). In silico adjustment for white cell composition does not alter the top-ranked genes associated with DKD in this study. As the discovery collection included 250 individuals, only CpGs with a sizable, significant difference in DNA methylation (Δβ) were considered for subsequent analysis (Figure 1). Twenty-two unique sites were identified with Δβ ≥ 0.2 and P < 10–8 where significance values were adjusted for multiple testing using the Benjamini & Hochberg method for controlling the false discovery rate (FDR). These sites are primarily “promoter associated” and affect 22 genes (Table 2); all 22 sites were taken forward for in silico and wet-lab replication. Subgroup analysis comparing those individuals who received a kidney transplant for DKD with controls show that 20 of these 22 sites were also highlighted in the ESRD focused dataset (Table 2). Sex-specific analyses for cg26399113 on the X chromosome was significant from female only (P = 1 × 10–12) and male only (P = 8.9 × 10–11) analyses. The discovery group generated results for 15 CpGs in the CCNL1 gene, of which 12 showed an increase in methylation between cases and controls. Similarly for ZNF187, nine CpGs were examined of which eight showed differential methylation and all were in the same direction as the index marker.
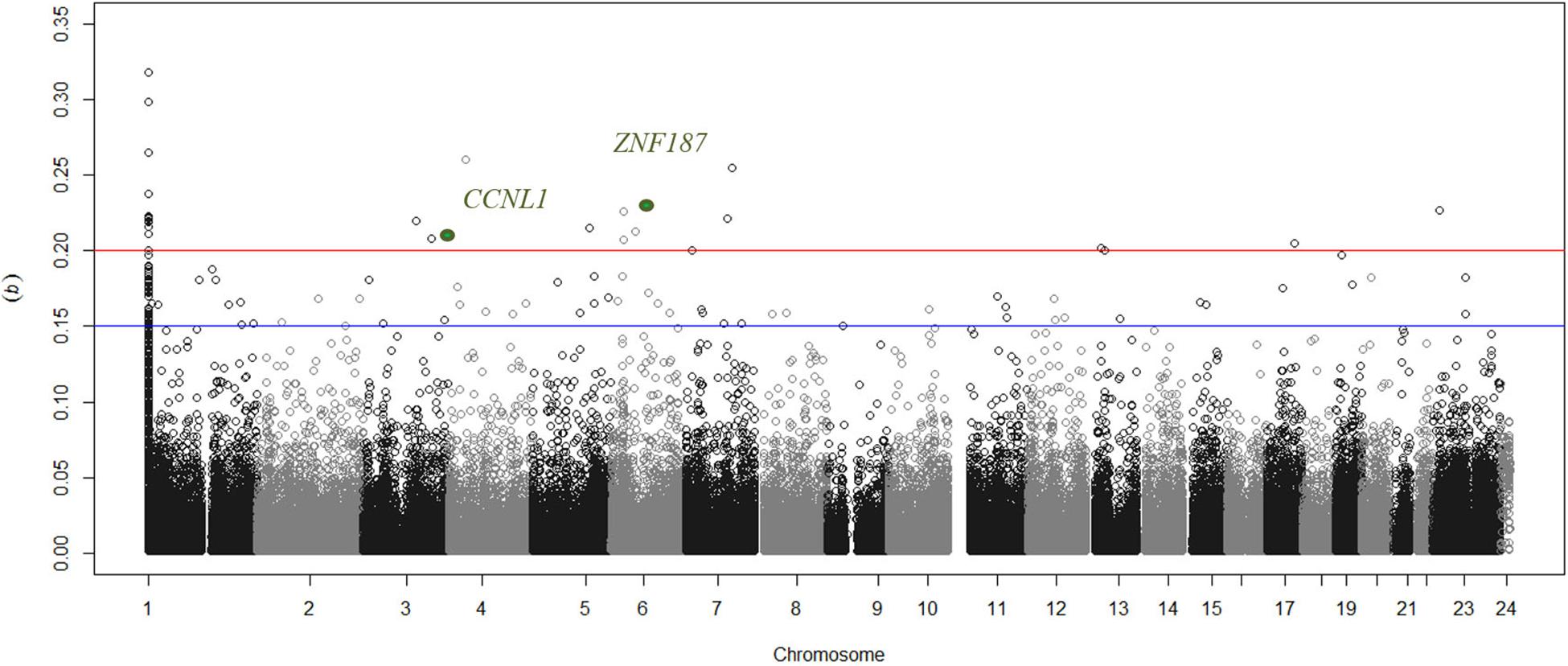
Figure 1. Manhattan plot showing distribution of delta beta values for uniquely mapped sites across all chromosomes. The blue line discriminates sites that have suggestive differences in methylation between cases with nephropathy compared to non-nephropathic individuals in the control group. Circles above the red line are loci where a substantial difference in beta values (>0.2) were observed and the two markers supported in the replication group are highlighted in green. Markers that are not uniquely mapped to a chromosome position based on Illumina’s updated bead pool manifest are assigned to chromosome 1 in this figure.
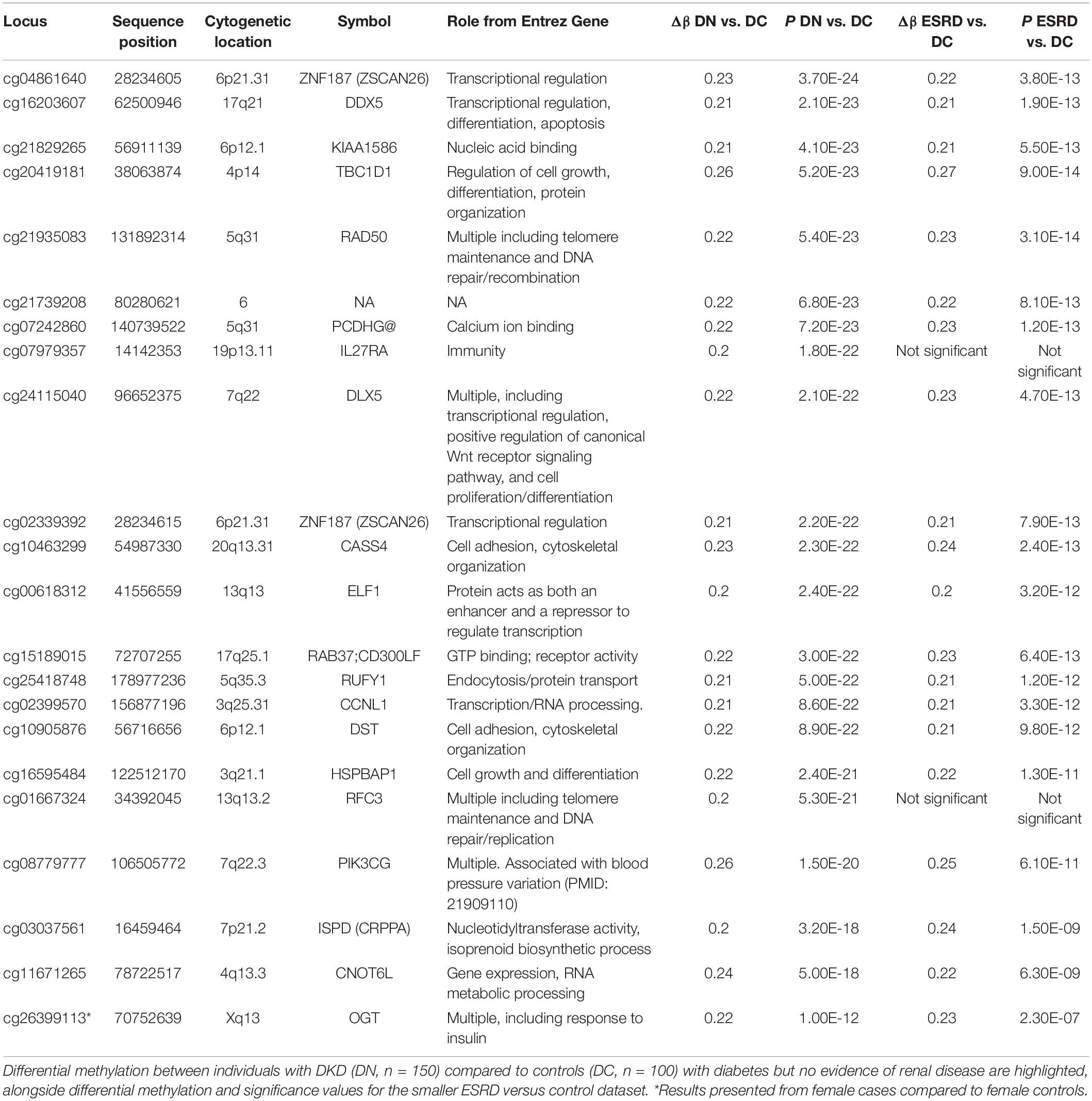
Table 2. Details for markers showing significant (FDR adjusted P-value), substantial differences in mean methylation (Δβ) between case and control groups in the discovery phase.
Functional enrichment analysis for differentially regulated loci (adjusted P < 0.0001) in the discovery group revealed both hypermethylation and hypomethylation across several pathways. Top ranked pathways enriched with significant probes include genes in Wnt signaling pathway (04310, enrichment P = 3.9 × 10–5), focal adhesion (04510, enrichment P = 7.9 × 10–5), MAPK signaling (04010, enrichment P = 1.0 × 10–10) pathways. Annotation from DAVID Bioinformatics Resources 6.7 for top-ranked genes is presented in Supplementary Table 2.
Several SNPs from our British - Irish genome-wide association study revealed nominal significance for SNPs in the region of interest from the 450K discovery analysis. The SNP demonstrating most evidence for association with DKD within 5 kb flanking top-ranked methylation probes are presented in Supplementary Table 3. Rs16888186 showed the most evidence for association in the DST gene with P = 0.0008.
In silico replication data were available for six probes on the 27K array (cg25418748, cg07979357, cg21935083, cg21829265, cg24115040, cg26399113), but none of these sites confirmed differential methylation levels between cases and controls. De novo replication gave z test P-values supporting the association of two loci: cg02399570 on 3q25.31 in the CCNL1 gene, P = 7.2 × 10–14 and cg04861640 on 6p21.31 in the ZNF187 gene, P = 1.5 × 10–15 (Figure 2). Selecting a β level of 25% (as standard for unmethylated loci), the area under the ROC curve was 0.74 for cg02399570 and 0.81 for cg04861640 (Figure 2).
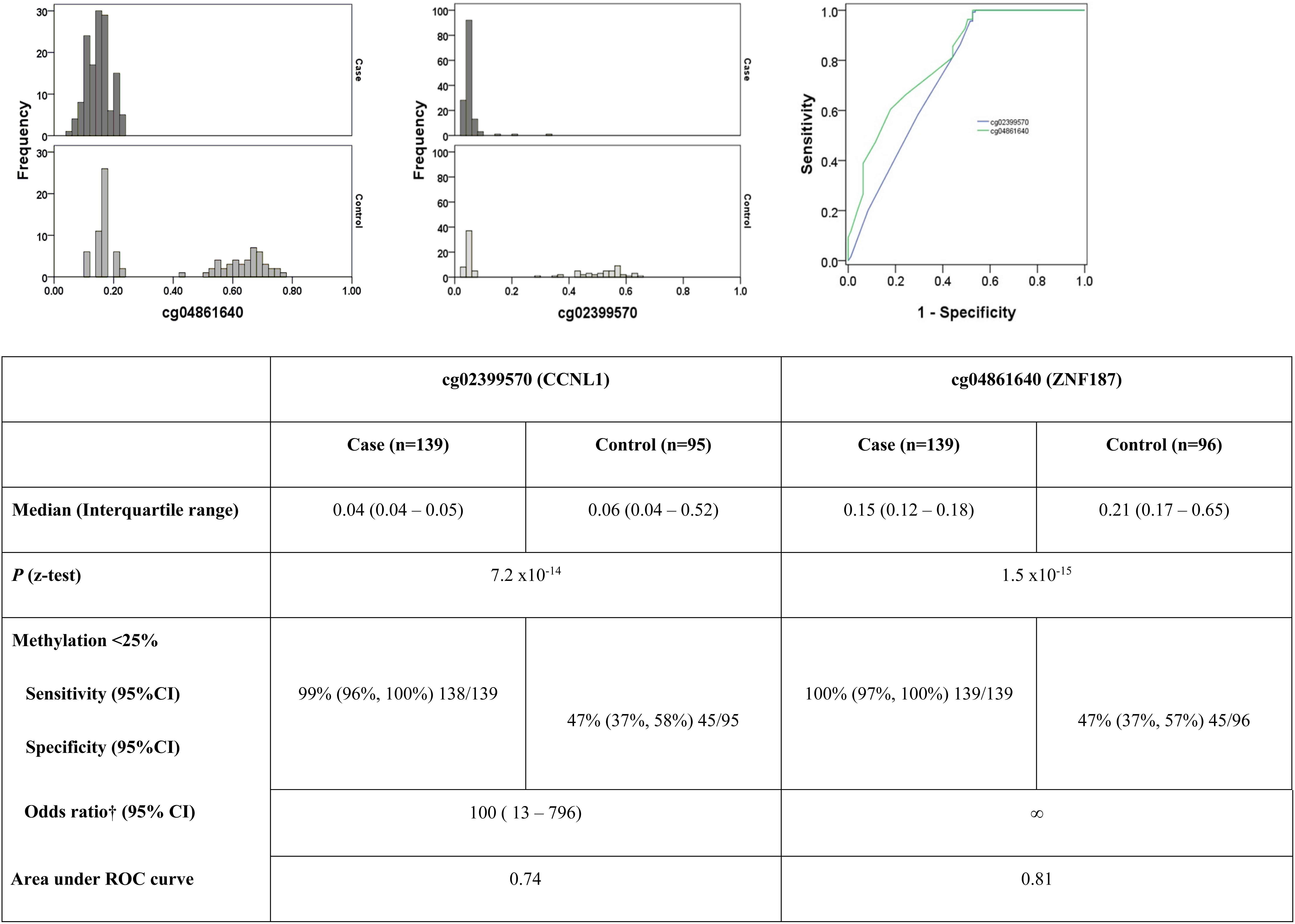
Figure 2. Histograms highlighting differences in the pattern of methylation between cases and controls for the two top ranked markers in the replication population. ROC curve demonstrating sensitivity and specificity of each marker. †Adjusted for age, HbA1c and gender. Neither age at diagnosis, nor duration of diabetes significantly influenced the model.
Multiple significant analyses for CCNL1 gene expression associated with kidney function were returned from NephroSeq (Supplementary Figure 1). Higgins et al. (2004) observed that CCNL1 is more highly expressed in renal glomeruli than other renal tissues. Downregulation of CCNL1 gene expression was reported in multiple datasets comparing people with kidney disease versus controls without kidney disease [P = 3.55E-09 for thin basement membrane disease, based on 199 micro-dissected glomerular samples from CKD patients and living donors analyzed on Affymetrix Human U133 Plus 2.0 and Affymetrix Human U133A (altCDF v10) platforms, and P = 1.84E-05 for 53 kidney biopsies from people with chronic kidney disease tubular damage compared to healthy controls analyzed on Agilent Whole Human Genome Microarrays (Nakagawa et al., 2015)]. Gunther et al. (2014) reported a slight increase in blood-derived CCNL1 gene expression (P = 0.006, fold change 1.6) for kidney transplant recipients with acute rejection compared to kidney transplant recipients with no rejection based on analysis of RNA from 40 samples on the Affymetrix Human Genome U133 Plus 2.0 array. Cox et al. (2015) explored blood-derived gene expression changes associated with glomerular filtration rate in eight patients with IgA nephropathy compared to nine healthy control participants, observing only a small change in expression for CCNL1 (P = 0.02).
Multiple significant analyses were returned from NephroSeq ZNF187 kidney gene expression data from dissected renal lobes of five adult human kidneys using cDNA microarrays representing ∼30,000 different human genes (Higgins et al., 2004). ZNF187 is more highly expressed in renal glomeruli than other renal tissues (Higgins et al., 2004; Lindenmeyer et al., 2010). The most significant result returned from searching NephroSeq for ZNF187 was for tissue type [P = 1.7 × 10–5 comparing tubulointerstitium to glomerular tissue in six transplant living donors (Lindenmeyer et al., 2010)] and acute rejection following kidney transplantation from 48 patients [P = 2.8 × 10–5 (Sarwal et al., 2003)]. Gene expression changes were associated with glomerular filtration rate in kidney biopsy samples from people with IgA nephropathy (P = 6.27 × 10–4) (Reich et al., 2010), diabetes (P = 0.008) (Woroniecka et al., 2011), and blood derived gene expression from healthy individuals who had no evidence of kidney disease (P = 0.018) (Flechner et al., 2004). Focusing in on available data from 22 racially diverse microdissected human kidney samples with type 2 DKD measured on the Affymetrix U133A 2.0 array, visualizes decreased renal function associated with a decrease in ZNF187 gene expression (P = 0.008) (Woroniecka et al., 2011).
Discussion
Using the Infinium Human Methylation 450K BeadChip (Sandoval et al., 2011) we identified differentially methylated CpG sites associated with DKD. To minimize false positive associations in our discovery cohort, we applied stringent quality control and adjusted association analyses; we used both a genome-wide significance threshold and a clear absolute methylation difference (Δβ ≥ 0.2) to minimize artifactual associations and ensure the selection of differentially methylated CpG probes (Dedeurwaerder et al., 2014). No SNPs are reported to affect methylation probes for ZNF187, however, one SNP resides 40 bases from the 3′ end of the probe for the CCNL1 gene. This SNP (rs75624594, NP_064703.1:p.His112 = His, synonymous coding) has a reported minor allele frequency of 50% in African individuals, but only 4% in the US NHLBI Exome Sequencing Project (dbSNP ss342150967)3 so is unlikely to account for the differential methylation observed in this study.
The majority of probes evaluated using the 27K array were also present on the 450K array (n = 25,978), and demonstrate good correlation for many, but not all, CpG probes. For this reasons we sought supporting data from a previous study exploring DKD using the 27K array. It is clearly only possible to seek support for top-ranked markers that were present across both the 27K and 450K arrays and unfortunately data was only available for seven probes, none of which supported an association. Acknowledging the limitations of the 27K array to support top-ranked markers in this study (primarily that the majority of our top-ranked markers were not present on that much smaller array) and in the absence of a replication cohort with 450K data available for a population with similar phenotype characteristics, we sought independent replication. Pragmatically, independent replication was sought using all available samples with high molecular weight DNA (139 cases and 96 controls) for the 22 loci that were significantly associated with DKD from the 450K array-based discovery analysis using Δβ ≥ 0.2 and FDR adjusted P < 10–8 thresholds. Importantly, the replication population was recruited with similar phenotypic characteristics to that of the discovery cohort and we used a completely different wet-lab approach (mass spectrometry) to validate the microarray-based data, which minimizes artifacts due to the microarray analysis. Only two of these CpG sites were supported by the new methylation data generated in our replication population – this is not unusual in genome-wide studies and may be due to false positives in the original EWAS or due to the fact that our replication population was not sufficiently powered to identify significant associations with all loci. Both discovery and de novo replication using a different experimental platform (mass spectrometry by Sequenom analyzed using EpiTyper software) to generate new laboratory data and an independent population strongly support the association of CCNL1 and ZNF187 genes with methylome-wide significance from 450K array-based (microarray by Illumina’s iScan) discovery association results. RNA-based gene expression data also supports a functional influence of CCNL1 and ZNF187 in kidney disease.
Methylation patterns for CCNL1 and ZNF187 genes showed a striking higher methylation level for controls compared to individuals in the case group (Figure 2). It should be noted that DNA samples available for replication were not age and gender matched in this study, rather they were pragmatically selected as all available samples with high quality DNA and careful phenotyping. Cases were older, diagnosed with type 1 diabetes later, and had higher HBA1c values with a longer duration of diabetes than individuals in the control group. Differential methylation between cases and controls was significant in both the CCNL1 and ZNF187 genes following adjustment for age, duration of diabetes, HBA1c and gender.
The CCNL1 gene encodes cyclin L1, which is localized in nuclear speckles (splicing factor storage compartment) (Herrmann et al., 2007), is functionally related to the spliceosome, and is involved in pre-mRNA splicing activities (Chen et al., 2007; Tannukit et al., 2008). The transcription start site for CCNL1 is located 89 bp upstream of the initiation codon and the first two exons overlap the CpG island (Dickinson et al., 2002; Figure 2). CCNL1 was consistently hypomethylated in cases compared to controls. Overexpression (usually associated with hypomethylation) of CCNL1 has been associated with cancer (Sticht et al., 2005; Mitra et al., 2010; Peng et al., 2011). The discovery group generated results for 15 CpGs in the CCNL1 gene, of which 12 showed an increase in methylation between cases and controls. Replication using independent samples and a different wet-lab experimental approach supported the association of CCNL1 with DKD. Based on published gene expression data from the Affymetrix® GeneChip® Whole Transcript Expression Arrays, CCNL1 was one of four genes differentially expressed in patients with kidney stones compared to controls (2.6 fold change, downregulated, P = 6.58E-05) (Liang et al., 2019). Based on gene expression data within NephroSeq, differential gene expression was observed for CCNL1 in kidney biopsy tissues from people with kidney disease compared to controls (Gunther et al., 2014; Ju et al., 2015; Nakagawa et al., 2015); no adjustment was made for cell heterogeneity in the disease compared to control collections for these gene expression datasets. CCNL1 resides in chromosome band 3q25, which has been previously suggested to harbor risk loci for DKD (McKnight et al., 2009). Of particular interest, the CCNL1 gene was ranked 4th from a meta-analysis for association with severe diabetic retinopathy (P = 7.1 × 10–7), but was no longer top-ranked for association with diabetic retinopathy when individuals with nephropathy were removed from the case group (Grassi et al., 2011). Subsequent studies have highlighted CCNL1 SNPs associated with retinopathy and measures of renal function (Lin et al., 2016). Genetic variation near the CCNL1 gene is robustly associated with low birth weight in European individuals (Freathy et al., 2010; Yaghootkar and Freathy, 2012; Horikoshi et al., 2013), specifically with growth restriction from early pregnancy onward (Mook-Kanamori et al., 2011). Another study suggests that individuals who carry a risk allele for rs900400 (near CCNL1) are more vulnerable to stress impacting on birth weight (Ali Khan et al., 2012). The relationship between birth weight and kidney disease has been debated with some groups suggesting that low birth weight is a risk factor for DKD (Rossing et al., 1995) while others report that low birth weight does not increase the risk of DKD (Fagerudd et al., 2006). The birth weight lowering effect rs900400 C allele has also been associated with increased insulin response following oral glucose stimulation in a meta-analysis based on Danish and Finnish non-diabetic individuals (Andersson et al., 2011). Published literature suggests that CCNL1 may affect an individual’s inherited and dynamic responses to their environment, perhaps reflecting both genetic and epigenetic contributions. While the molecular mechanism for CCNL1 influencing DKD remains to be resolved, this is clearly a candidate gene that warrants further investigation having demonstrated genetic (SNPs), epigenetic (methylation) and transcriptomic (gene expression) associations with kidney disease across multiple collections.
In the discovery cohort, nine CpGs were examined for ZNF187; eight of these showed differential methylation and all were in the same direction as the index marker. Replication demonstrated strong support for association of ZNF187 with DKD. Analysis within NephroSeq revealed gene expression changes associated with glomerular filtration rate in kidney biopsy and blood-derived samples from people with IgA nephropathy (Reich et al., 2010), diabetes (Woroniecka et al., 2011), and renal function in healthy individuals (Flechner et al., 2004; Supplementary Figure 1). ZNF187 is involved with transcriptional regulation, but there are few publications describing this gene. The ZNF187 gene is was located at 6p21.31 and encodes the zinc finger protein 187. Gene ontology suggests that ZNF187 is involved with transcriptional regulation, but there are no specific publications for this gene (or any aliases) in PubMed (accessed 12/05/20)4. The protein coding ZNF187 gene has been renamed as ZSCAN26 (zinc finger and SCAN domain containing 26), but we have retained the ZNF187 nomenclature throughout this manuscript to keep the methylation array information consistent and facilitate easier searching and replication within methylation and gene expression datasets.
Both CCNL1 and ZNF187 were more highly expressed in renal glomeruli than other renal tissues, which may be consistent with DKD in people with type 1 diabetes primarily affecting the glomerulus (Higgins et al., 2004). The results for both significantly replicated genes in our methylation dataset are hypomethylated in cases compared to controls, while publicly available gene expression evidence for both genes suggests there is less RNA product in samples from people with kidney disease. This may be because less gene is expressed, or because the mRNA measured on arrays is not present for long, or because different isoforms are not captured by the gene expression array. cg02399570 is in the body of CCNL1 while cg04861640 is within the transcription start site of ZNF187. Most promoters and CpG sites in gene bodies that are hypomethylated are basically expressed, but gene regulation is complex and genes may be hypomethylated and “overexpressed” in disease states (Haney et al., 2016; Li et al., 2017). Significant further research is required in populations that have blood derived, kidney biopsy derived and in vitro models with SNP, CpG, and gene expression data available for the same individuals to tease out the molecular signatures of these genes (CCNL1 and ZNF187) for kidney disease.
Pathway analysis revealed significant gene enrichment in the focal adhesion, Wnt and MAPK signaling pathways. These pathways have been previously top-ranked as differentially regulated in renal tubuli of individuals with DKD compared to healthy tissue from living kidney donors (Woroniecka et al., 2011). The focal adhesion pathway also demonstrates enrichment in the glomeruli of individuals with DKD (Woroniecka et al., 2011) and is a key molecular pathway in the formation and progression of the cardiorenal system (Muhlberger et al., 2012). The Wnt signally pathway has been shown to influence survival of glomerular mesangial cells exposed to high glucose (Lin et al., 2006) (41) and dysregulation of the Wnt pathway may represent and important pathogenic mechanism of DKD (Kavanagh et al., 2011; Zhou et al., 2012).
We incorporated existing GWAS data (Sandholm et al., 2012) with this novel methylation data to provide exploratory analysis seeking a provisional assessment of functionality. i.e., are SNPs demonstrating suggestive association with DKD from GWAS near CpG probes that are differentially methylated using the same case-control study population. As SNPs may have a functional role for each gene at a considerable genetic distance, we pragmatically selected 5 kb flanking each probe for this analysis. No strong associations were identified for SNPs near top-ranked differentially methylated genes. We also considered a similar analysis by looking up CpG sites for association near top-ranked GWAS SNPs (Sandholm et al., 2012). Meta-analysis of genome-wide association studies for DKD revealed novel association (P = 1.2 × 10–8) with SNPs in the AFF3 gene, a transcriptional activator that influences renal fibrosis through the TGFβ1 pathway, in individuals who had progressed to end stage renal disease (Sandholm et al., 2012). Additionally, the top-ranked marker associated with DKD was in the ERBB4 gene (P = 2.1 × 10–7) (Sandholm et al., 2012). Significant differential methylation was observed at both of these loci when comparing cases and controls on the 450K array (Table 3) suggesting that a combined genetic-epigenetic factor may influence the risk of DKD.

Table 3. Methylation probes of interest associated with genes identified from recent meta-analysis of genome-wide association studies for DKD.
The role of epigenetics in common, complex diseases is beginning to be unraveled at a population level using relatively high throughput tools. Evidence is increasing that inter-individual epigenetic variation, in particular DNA methylation, may help explain some of the “missing heritability” that has not been identified through genome-wide association and resequencing approaches. Illumina’s 450K BeadChip was proposed as the method of choice for cost-effective, high throughput epigenome-wide association studies with single-base resolution (Rakyan et al., 2011b). The content of this array (485,764 sites distributed across all chromosomes) was selected based on input from 22 methylation experts across the world. Included are unique markers that cover 99% of RefSeq genes with an average of 17 CpG sites per gene region distributed across the promoter, 5′ untranslated region, first exon, gene body, and 3′ untranslated region. This array also includes dedicated content for CpG sites outside CpG islands and microRNA promoter regions. Illumina have released a higher density EPIC array, the Infinium MethylationEPIC BeadChip, which facilitates evaluation of 862,927 sites at significantly increased financial cost. More comprehensive analysis of the methylome may be conducted through whole-methylome-sequencing, but this is financially prohibitive for most researchers using cohorts of more than 200 participants. However, technological and analytical advances now offer the potential for targeted, high throughput bisulfite sequencing with deep coverage as an attractive option for technical validation and replication. Using high density methylation arrays is currently the most cost-effective approach for EWAS using population-based study designs. Stringent quality control, strong significance values, and independent replication are essential to minimize false positive findings when investigating sequence changes to elucidate the genetic architecture of multifactorial disease.
Although this epigenetic study does not include the large sample numbers traditionally associated with genome-wide association studies, we have applied rigorous analysis approaches throughout and gained support using independent samples by a different technology – important to minimize technical artifacts. A sample size of only 65 heavy smokers and 56 non-smokers was sufficient to identify differential patterns of methylation (P = 2.68–31) associated with smoking using the Illumina Human Methylation 27K BeadChip (Breitling et al., 2011). Similarly, a type 1 diabetes-methylation variation position signature was detected by assaying a relatively modest number of samples (n = 15 monozygotic twin pairs discordant for type 1 diabetes) on the 27K array (Bell et al., 2010; Rakyan et al., 2011a). Associations using the 450K array have been reported from 11 cell lines for rheumatoid arthritis (Nakano et al., 2013), 48 individuals for irritable bowel disease (Harris et al., 2012), and 165 females for alcohol use (Philibert et al., 2012), although study sizes are now increasing. Previous studies exploring blood-derived DNA methylation used an EWAS approach for DKD focused on the 27K array (Sapienza et al., 2011), the mitochondrial genome (Swan et al., 2015) or renal function decline in 181 Pima Indians with diabetes (Qiu et al., 2018). Targeted DNA methylation studies have been conducted using blood-derived DNA (Aldemir et al., 2017; Smyth et al., 2018), cell models of DKD (Brennan et al., 2010; Li et al., 2019) and an EWAS reported on kidney biopsy samples from 91 individuals of whom 45% had diabetes (Gluck et al., 2019) and 11 individuals with diabetes (Ko et al., 2013). The case and control population employed in this study has >60% power to detect a true positive (defined as detected CpGs with a meaningful difference in mean blood derived DNA methylation ±0.2 with a false discovery rate P ≤ 0.05) association using pwrEWAS (Graw et al., 2019). More comprehensive whole genome bisulfite sequencing has been reported for kidney biopsy samples from five individuals with DKD compared to one person with diabetes without kidney disease, and four people with neither diabetes nor kidney disease (Park et al., 2019).
Careful phenotyping is critically important for methylation studies, as is the consistent extraction and storage of DNA. We and others have previously demonstrated that differences in DNA extraction approaches and storage methods significantly alter methylation profiles. Importantly for this study, all DNA was extracted using the same approach in the same laboratory (by two persons) with extracted DNA stored at −80°C in multiple aliquots with only one freeze-thaw cycle. Individuals were carefully phenotyped by consultant nephrologists using internationally agreed phenotype criteria. We restricted analysis to individuals with type 1 diabetes and known kidney function to minimize phenotypic heterogeneity and used a matched design for the discovery population.
Many cell types have unique methylation profiles so where possible it is important to adjust for cell heterogeneity in all studies using blood-derived or kidney-derived DNA. In our study, there was no significant difference in proportional white cell counts from whole blood (B cells, granulocytes, monocytes, NK cells, and T cells subsets) and adjusting for cell composition does not change the top-ranked association results for this study. While adjusting for white cell subpopulations is critically important for cancer studies, immune-mediated responses, and case-control approaches not matched for age and gender, this is a result that we and others have reported previously for carefully phenotyped populations with stringent wet-lab protocols from blood sampling through to array scanning. The proportion of these cell types may also reflect changing disease pathology (Lappalainen and Greally, 2017; Johnson et al., 2020). Epigenetic signatures may display tissue specificity linked to disease mechanisms, however, obtaining kidney biopsy material is invasive and is not performed as part of routine clinical practice in people with DKD and T1D in the United Kingdom. Peripheral blood-based methylation biomarkers have shown promise in several clinical fields (Moore et al., 2014; Agha et al., 2019; Cardenas et al., 2019; DiTroia et al., 2019; Henderson-Smith et al., 2019; Kerr et al., 2019a,b, 2020; Ladd-Acosta and Fallin, 2019; Zhou et al., 2019) including kidney disease (Smyth et al., 2014b, 2018; Swan et al., 2015; Aranyi and Susztak, 2019; Gluck et al., 2019; Kato and Natarajan, 2019; Kerr et al., 2019b; Park et al., 2019). We have previously demonstrated that blood-derived differential methylation is also reflected in kidney-derived differential methylation for CKD (Smyth et al., 2014b). Blood-derived DNA methylation offers clinical utility in biomarker development, incorporating a minimally invasive approach that could be cost-effectively implemented in a routine clinical setting. Indeed, ROC curve analysis suggests that 25% methylation for the two key CCNL1 and ZNF187 markers is reasonably good at differentiating individuals in case and control groups. This is particularly critical for translation in DKD given the difficulty obtaining serial renal biopsies to establish diagnosis, track progression, and monitor response (Kim et al., 2018). Array-based approaches using blood-derived DNA have previously identified risk factors and biomarkers associated with complex phenotypes (Sandholm et al., 2013, 2017; Keating et al., 2018; Canadas-Garre et al., 2019; Gu, 2019; Park et al., 2019).
Interpreting epigenetic factors as disease-causing or consequences of disease processes, alongside genetic and/or environmental heterogeneity, are a significant problem for complex disease. Nevertheless, this study demonstrates that using high density methylation arrays are an appropriate, cost-effective tool to identify differential methylation profiles that may deliver minimally invasive biomarkers that are relevant for diabetic complications. We have identified CCNL1 and ZNF187 as differentially methylated genes associated with DKD in multiple cohorts. Larger EWAS exploring more markers with larger sample sizes will deliver the same gains identifying molecular biomarkers as has been observed for GWAS in recent years. Using longitudinal cohort designs will allow researchers to observe how DNA methylation changes over time. More complex analytical tools are being developed for DNA methylation such as MethylNet (Levy et al., 2020), which offers further opportunities for novel discoveries and improved understanding. The integration of multi-omic profiling will lead to a better understanding of inherited susceptibility to DKD and biomarkers for this common disease.
Data Availability Statement
The data supporting the conclusion of this article will be made available by the authors, without undue reservation.
Ethics Statement
The studies involving human participants were reviewed and approved by the South and West Multicentre Research Ethics Committee (MREC/98/6/71). The patients/participants provided their written informed consent to participate in this study.
Author Contributions
AMcK had full access to the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis. LS, AM, and AMcK conceived and designed the study. LS and ES performed sample analysis. CP performed the statistical analyses in the replication group. All authors provided important intellectual content and agreed the final version of this manuscript.
Funding
Funding was provided by the Science Foundation Ireland and the Department for the Economy, Northern Ireland (15/IA/3152), the Medical Research Council (MC_PC_15025), and the NI Public Health Agency R&D Division as part of a US Ireland award (STL/4760/13). LS is the recipient of a Northern Ireland Kidney Research Fund Fellowship. ES is the recipient of Diabetes UK Ph.D. Studentship BDA 10/0004154.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We appreciate the support provided by Fraser Morton at Fios Genomics who conducted independent analysis of the 450K array data, which was generated by Hologic Gen-Probe Inc. The Warren3/United Kingdom GoKinD Study Group was jointly funded by Diabetes UK and the Juvenile Diabetes Research Foundation and includes the following individuals: Prof. AM, Prof. AMcK, Dr. D. A. Savage (Belfast), Dr. J. Walker (Edinburgh), Dr. S. Thomas, Prof. G. C. Viberti (London), Prof. A. J. M. Boulton (Manchester), Prof. S. Marshall (Newcastle), Prof. A. G. Demaine, Dr. B. A. Millward (Plymouth), and Prof. S. C. Bain (Swansea).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2020.561907/full#supplementary-material
Footnotes
- ^ http://tinyurl.com/methylumi
- ^ www.nephroseq.org
- ^ https://esp.gs.washington.edu/drupal/
- ^ www.ncbi.nlm.nih.gov/pubmed
References
Agha, G., Mendelson, M. M., Ward-Caviness, C. K., Joehanes, R., Huan, T., Gondalia, R., et al. (2019). Blood leukocyte DNA methylation predicts risk of future myocardial infarction and coronary heart disease. Circulation 140, 645–657.
Aldemir, O., Turgut, F., and Gokce, C. (2017). The association between methylation levels of targeted genes and albuminuria in patients with early diabetic kidney disease. Ren. Fail. 39, 597–601. doi: 10.1080/0886022x.2017.1358180
Ali Khan, A., Rodriguez, A., Sebert, S., Kaakinen, M., Cauchi, S., Froguel, P., et al. (2012). The interplay of variants near LEKR and CCNL1 and social stress in relation to birth size. PLoS One 7:e38216. doi: 10.1371/journal.pone.0038216
Andersson, E. A., Harder, M. N., Pilgaard, K., Pisinger, C., Stancakova, A., Kuusisto, J., et al. (2011). The birth weight lowering C-allele of rs900400 near LEKR1 and CCNL1 associates with elevated insulin release following an oral glucose challenge. PLoS One 6:e27096. doi: 10.1371/journal.pone.0027096
Aranyi, T., and Susztak, K. (2019). Cytosine methylation studies in patients with diabetic kidney disease. Curr. Diab. Rep. 19:91.
Arredondo, A., Orozco, E., Duarte, M. B., Cuadra, M., Recaman, A. L., and Azar, A. (2018). Trends and challenges in diabetes for middle-income countries: evidence from Mexico. Glob. Public Health 14, 227–240. doi: 10.1080/17441692.2018.1498115
Bayoumi, A., Gronbaek, H., George, J., and Eslam, M. (2020). The epigenetic drug discovery landscape for metabolic-associated fatty liver disease. Trends Genet. 36, 429–441. doi: 10.1016/j.tig.2020.03.003
Bell, C. G., Teschendorff, A. E., Rakyan, V. K., Maxwell, A. P., Beck, S., and Savage, D. A. (2010). Genome-wide DNA methylation analysis for diabetic nephropathy in type 1 diabetes mellitus. BMC Med. Genom. 3:33. doi: 10.1186/1755-8794-3-33
Breitling, L. P., Yang, R., Korn, B., Burwinkel, B., and Brenner, H. (2011). Tobacco-smoking-related differential DNA methylation: 27K discovery and replication. Am. J. Hum. Genet. 88, 450–457. doi: 10.1016/j.ajhg.2011.03.003
Brennan, E. P., Ehrich, M., O’Donovan, H., Brazil, D. P., Crean, J. K., Murphy, M., et al. (2010). DNA methylation profiling in cell models of diabetic nephropathy. Epigenetics 5, 396–401. doi: 10.4161/epi.5.5.12077
Campbell, J. A., Bishu, K. G., Walker, R. J., and Egede, L. E. (2017). Trends of medical expenditures and quality of life in US adults with diabetes: the medical expenditure panel survey, 2002-2011. Health Qual. Life Outcomes 15:70.
Canadas-Garre, M., Anderson, K., Cappa, R., Skelly, R., Smyth, L. J., McKnight, A. J., et al. (2019). Genetic susceptibility to chronic kidney disease - some more pieces for the heritability puzzle. Front. Genet. 10:453. doi: 10.3389/fgene.2019.00453
Canadas-Garre, M., Anderson, K., McGoldrick, J., Maxwell, A. P., and McKnight, A. J. (2018). Genomic approaches in the search for molecular biomarkers in chronic kidney disease. J. Transl. Med. 16:292.
Cardenas, A., Lutz, S. M., Everson, T. M., Perron, P., Bouchard, L., and Hivert, M. F. (2019). Placental DNA methylation mediates the association of prenatal maternal smoking on birth weight. Am. J. Epidemiol. 188, 1878–1886. doi: 10.1093/aje/kwz184
Chen, H. H., Wong, Y. H., Geneviere, A. M., and Fann, M. J. (2007). CDK13/CDC2L5 interacts with L-type cyclins and regulates alternative splicing. Biochem. Biophys. Res. Commun. 354, 735–740. doi: 10.1016/j.bbrc.2007.01.049
Chen, Y. A., Lemire, M., Choufani, S., Butcher, D. T., Grafodatskaya, D., Zanke, B. W., et al. (2013). Discovery of cross-reactive probes and polymorphic CpGs in the Illumina Infinium HumanMethylation450 microarray. Epigenetics 8, 203–209. doi: 10.4161/epi.23470
Chu, A. Y., Tin, A., Schlosser, P., Ko, Y. A., Qiu, C., Yao, C., et al. (2017). Epigenome-wide association studies identify DNA methylation associated with kidney function. Nat. Commun. 8:1286.
Cox, S. N., Serino, G., Sallustio, F., Blasi, A., Rossini, M., Pesce, F., et al. (2015). Altered monocyte expression and expansion of non-classical monocyte subset in IgA nephropathy patients. Nephrol. Dial. Transplant. 30, 1122–1232. doi: 10.1093/ndt/gfv017
Dedeurwaerder, S., Defrance, M., Bizet, M., Calonne, E., Bontempi, G., and Fuks, F. (2014). A comprehensive overview of infinium humanmethylation450 data processing. Brief Bioinform. 15, 929–941. doi: 10.1093/bib/bbt054
Dias, S., Adam, S., Rheeder, P., Louw, J., and Pheiffer, C. (2019). Altered genome-wide DNA methylation in peripheral blood of south african women with gestational diabetes mellitus. Int. J. Mol. Sci. 20:5828. doi: 10.3390/ijms20235828
Dickinson, L. A., Edgar, A. J., Ehley, J., and Gottesfeld, J. M. (2002). Cyclin L is an RS domain protein involved in pre-mRNA splicing. J. Biol. Chem. 277, 25465–25473. doi: 10.1074/jbc.m202266200
Disease, G. B. D., Injury, I., and Prevalence, C. (2018). Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990-2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet 392, 1789–1858.
DiTroia, S. P., Percharde, M., Guerquin, M. J., Wall, E., Collignon, E., Ebata, K. T., et al. (2019). Maternal vitamin C regulates reprogramming of DNA methylation and germline development. Nature 573, 271–275. doi: 10.1038/s41586-019-1536-1
Elboudwarej, E., Cole, M., Briggs, F. B., Fouts, A., Fain, R., Quach, H., et al. (2016). Hypomethylation within gene promoter regions and type 1 diabetes in discordant monozygotic twins. J. Autoimmun. 68, 23–29. doi: 10.1016/j.jaut.2015.12.003
Fagerudd, J., Forsblom, C., Pettersson-Fernholm, K., Saraheimo, M., Waden, J., Ronnback, M., et al. (2006). Low birth weight does not increase the risk of nephropathy in Finnish type 1 diabetic patients. Nephrol. Dial. Transplant. 21, 2159–2165. doi: 10.1093/ndt/gfl217
Flechner, S. M., Kurian, S. M., Head, S. R., Sharp, S. M., Whisenant, T. C., Zhang, J., et al. (2004). Kidney transplant rejection and tissue injury by gene profiling of biopsies and peripheral blood lymphocytes. Am. J. Transplant. 4, 1475–1489. doi: 10.1111/j.1600-6143.2004.00526.x
Florath, I., Butterbach, K., Heiss, J., Bewerunge-Hudler, M., Zhang, Y., Schottker, B., et al. (2016). Type 2 diabetes and leucocyte DNA methylation: an epigenome-wide association study in over 1,500 older adults. Diabetologia 59, 130–138. doi: 10.1007/s00125-015-3773-7
Franciosi, M., Lucisano, G., Amoretti, R., Capani, F., and Bruttomesso, D. Di Bartolo. (2013). Nicolucci, A., Costs of treatment and complications of adult type 1 diabetes. Nutr. Metab Cardiovasc. Dis. 23, 606–611. doi: 10.1016/j.numecd.2012.03.002
Freathy, R. M., Mook-Kanamori, D. O., Sovio, U., Prokopenko, I., Timpson, N. J., Berry, D. J., et al. (2010). Variants in ADCY5 and near CCNL1 are associated with fetal growth and birth weight. Nat. Genet. 42, 430–435.
Fu, H., Liu, S., Bastacky, S. I., Wang, X., Tian, X. J., and Zhou, D. (2019). Diabetic kidney diseases revisited: a new perspective for a new era. Mol. Metab 30, 250–263. doi: 10.1016/j.molmet.2019.10.005
Gluck, C., Qiu, C., Han, S. Y., Palmer, M., Park, J., Ko, Y. A., et al. (2019). Kidney cytosine methylation changes improve renal function decline estimation in patients with diabetic kidney disease. Nat. Commun. 10:2461.
Grassi, M. A., Tikhomirov, A., Ramalingam, S., Below, J. E., Cox, N. J., and Nicolae, D. L. (2011). Genome-wide meta-analysis for severe diabetic retinopathy. Hum. Mol. Genet. 20, 2472–2481. doi: 10.1093/hmg/ddr121
Graw, S., Henn, R., Thompson, J. A., and Koestler, D. C. (2019). pwrEWAS: a user-friendly tool for comprehensive power estimation for epigenome wide association studies (EWAS). BMC Bioinform. 20:218. doi: 10.1186/s12859-019-2804-7
Greenberg, M. V. C., and Bourc’his, D. (2019). The diverse roles of DNA methylation in mammalian development and disease. Nat. Rev. Mol. Cell Biol. 20, 590–607. doi: 10.1038/s41580-019-0159-6
Gu, H. F. (2019). Genetic and epigenetic studies in diabetic kidney disease. Front. Genet. 10:507. doi: 10.3389/fgene.2019.00507
Gunther, O. P., Shin, H., Ng, R. T., McMaster, W. R., McManus, B. M., Keown, A., et al. (2014). Novel multivariate methods for integration of genomics and proteomics data: applications in a kidney transplant rejection study. OMICS 18, 682–695. doi: 10.1089/omi.2014.0062
Haertle, L., El Hajj, N., Dittrich, M., Muller, T., Nanda, I., Lehnen, H., et al. (2017). Epigenetic signatures of gestational diabetes mellitus on cord blood methylation. Clin. Epigenet. 9:28.
Haney, S. L., Upchurch, G. M., Opavska, J., Klinkebiel, D., Hlady, R. A., Suresh, A., et al. (2016). Promoter hypomethylation and expression is conserved in mouse chronic lymphocytic leukemia induced by decreased or inactivated Dnmt3a. Cell Rep. 15, 1190–1201. doi: 10.1016/j.celrep.2016.04.004
Harris, R. A., Nagy-Szakal, D., Pedersen, N., Opekun, A., Bronsky, J., Munkholm, P., et al. (2012). Genome-wide peripheral blood leukocyte DNA methylation microarrays identified a single association with inflammatory bowel diseases. Inflamm. Bowel. Dis. 18, 2334–2341. doi: 10.1002/ibd.22956
Henderson-Smith, A., Fisch, K. M., Hua, J., Liu, G., Ricciardelli, E., Jepsen, K., et al. (2019). DNA methylation changes associated with Parkinson’s disease progression: outcomes from the first longitudinal genome-wide methylation analysis in blood. Epigenetics 14, 365–382. doi: 10.1080/15592294.2019.1588682
Herrmann, A., Fleischer, K., Czajkowska, H., Muller-Newen, G., and Becker, W. (2007). Characterization of cyclin L1 as an immobile component of the splicing factor compartment. FASEB J. 21, 3142–3152. doi: 10.1096/fj.07-8377com
Higgins, J. P., Wang, L., Kambham, N., Montgomery, K., Mason, V., Vogelmann, S. U., et al. (2004). Gene expression in the normal adult human kidney assessed by complementary DNA microarray. Mol. Biol. Cell 15, 649–656. doi: 10.1091/mbc.e03-06-0432
Hill, C. J., Cardwell, C. R., Patterson, C. C., Maxwell, A. P., Magee, G. M., Young, R. J., et al. (2014). Chronic kidney disease and diabetes in the national health service: a cross-sectional survey of the U.K. national diabetes audit. Diabet. Med. 31, 448–454. doi: 10.1111/dme.12312
Hoang, T. T., Sikdar, S., Xu, C. J., Lee, M. K., Cardwell, J., Forno, E., et al. (2020). Epigenome-Wide association study of DNA methylation and adult asthma in the agricultural lung health study. Eur. Respir. J. 56:2000217.
Horikoshi, M., Yaghootkar, H., Mook-Kanamori, D. O., Sovio, U., Taal, H. R., Hennig, B. J., et al. (2013). New loci associated with birth weight identify genetic links between intrauterine growth and adult height and metabolism. Nat. Genet. 45, 76–82.
Houseman, E. A., Accomando, W. P., Koestler, D. C., Christensen, B. C., Marsit, C. J., Nelson, H. H., et al. (2012). DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinform. 13:86. doi: 10.1186/1471-2105-13-86
Howe, C. G., Cox, B., Fore, R., Jungius, J., Kvist, T., Lent, S., et al. (2020). Maternal gestational diabetes mellitus and newborn DNA methylation: findings from the pregnancy and childhood epigenetics consortium. Diabetes Care 43, 98–105.
Huang, da, W., Sherman, B. T., and Lempicki, R. A. (2009). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57. doi: 10.1038/nprot.2008.211
Jia, Y., Reddy, M. A., Das, S., Oh, H. J., Abdollahi, M., Yuan, H., et al. (2019). Dysregulation of histone H3 lysine 27 trimethylation in transforming growth factor-beta1-induced gene expression in mesangial cells and diabetic kidney. J. Biol. Chem. 294, 12695–12707. doi: 10.1074/jbc.ra119.007575
Johnson, R. K., Vanderlinden, L. A., Dong, F., Carry, M., Seifert, J., Waugh, K., et al. (2020). Longitudinal DNA methylation differences precede type 1 diabetes. Sci. Rep. 10:3721.
Ju, W., Nair, V., Smith, S., Zhu, L., Shedden, K., Song, P., et al. (2015). Tissue transcriptome-driven identification of epidermal growth factor as a chronic kidney disease biomarker. Sci. Transl. Med. 7:316ra193.
Kato, M., and Natarajan, R. (2019). Epigenetics and epigenomics in diabetic kidney disease and metabolic memory. Nat. Rev. Nephrol. 15, 327–345. doi: 10.1038/s41581-019-0135-6
Kauffmann, A., and Huber, W. (2010). Microarray data quality control improves the detection of differentially expressed genes. Genomics 95, 138–142. doi: 10.1016/j.ygeno.2010.01.003
Kavanagh, D. H., Savage, D. A., Patterson, C. C., McKnight, A. J., Crean, J. K., Maxwell, A. P., et al. (2011). Warren 3 Uk GoKin. association analysis of canonical wnt signalling genes in diabetic nephropathy. PLoS One 6:e23904. doi: 10.1371/journal.pone.0023904
Kawaguchi, H., Moriyama, M., and Hashimoto, H. (2020). Does disease management for diabetic nephropathy reduce medical expenditure? evidence from a three-period difference-in-differences analysis. BMC Health Serv. Res. 20:403. doi: 10.1186/s12913-020-05297-0
Keating, S. T., van Diepen, J. A., Riksen, N. P., and El-Osta, A. (2018). Epigenetics in diabetic nephropathy, immunity and metabolism. Diabetologia 61, 6–20. doi: 10.1007/s00125-017-4490-1
Kerr, K., McAneney, A., Smyth, L., Flanagan, C., Silvestri, J., and Nesbitt, M. A. (2019a). A systematic review of differential methylation in rare ophthalmic diseases. BMJ Open Opthalmol. 4:e000342. doi: 10.1136/bmjophth-2019-000342
Kerr, K., McAneney, H., Flanagan, C., Maxwell, A. P., and McKnight, A. J. (2019b). Differential methylation as a diagnostic biomarker of rare renal diseases: a systematic review. BMC Nephrol. 20:320. doi: 10.1186/s12882-019-1517-5
Kerr, K., McAneney, H., Smyth, L. J., Bailie, C., McKee, S., and McKnight, A. J. (2020). A scoping review and proposed workflow for multi-omic rare disease research. Orphanet. J. Rare Dis. 15:107.
Kim, H., Wang, X., and Jin. (2018). Developing DNA methylation-based diagnostic biomarkers. J. Genet. Genom. 45, 87–97. doi: 10.1016/j.jgg.2018.02.003
Ko, Y. A., Mohtat, D., Suzuki, M., Park, A. S., Izquierdo, M. C., Han, S. Y., et al. (2013). Cytosine methylation changes in enhancer regions of core pro-fibrotic genes characterize kidney fibrosis development. Genome Biol. 14:R108.
Ladd-Acosta, C., and Fallin, M. D. (2019). DNA methylation signatures as biomarkers of prior environmental exposures. Curr. Epidemiol. Rep. 6, 1–13. doi: 10.1007/s40471-019-0178-z
Lappalainen, T., and Greally, J. M. (2017). Associating cellular epigenetic models with human phenotypes. Nat. Rev. Genet. 18, 441–451. doi: 10.1038/nrg.2017.32
Lassalle, M., Monnet, E., Ayav, C., Hogan, J., Moranne, O., Couchoud, C., et al. (2019). 2017 Annual report digest of the Renal Epidemiology Information Network (REIN) registry. Transpl. Int. 32, 892–902. doi: 10.1111/tri.13466
Levy, J. J., Titus, A. J., Petersen, C. L., Chen, Y., Salas, L. A., and Christensen, B. C. (2020). MethylNet: an automated and modular deep learning approach for DNA methylation analysis. BMC Bioinform. 21:108. doi: 10.1186/s12859-020-3443-8
Li, W., Sargsyan, D., Wu, R., Li, S., Wang, L., Cheng, D., et al. (2019). DNA Methylome and transcriptome alterations in high glucose-induced diabetic nephropathy cellular model and identification of novel targets for treatment by tanshinone IIA. Chem. Res. Toxicol. 32, 1977–1988. doi: 10.1021/acs.chemrestox.9b00117
Li, Z., Zhuang, X., Zeng, J., and Tzeng, C. M. (2017). Integrated analysis of DNA methylation and mrna expression profiles to identify key genes in severe oligozoospermia. Front. Physiol. 8:261. doi: 10.3389/fphys.2017.00261
Liang, X., Lai, Y., Wu, W., Chen, D., Zhong, F., Huang, J., et al. (2019). LncRNA-miRNA-mRNA expression variation profile in the urine of calcium oxalate stone patients. BMC Med. Genom. 12:57. doi: 10.1186/s12920-019-0502-y
Lin, C. L., Wang, J. Y., Huang, Y. T., Kuo, Y. H., Surendran, K., and Wang, F. S. (2006). Wnt/beta-catenin signaling modulates survival of high glucose-stressed mesangial cells. J. Am. Soc. Nephrol. 17, 2812–2820. doi: 10.1681/asn.2005121355
Lin, X., Wang, J., Yun, L., Jiang, S., Li, L., Chen, X., et al. (2016). Association between LEKR1-CCNL1 and IGSF21-KLHDC7A gene polymorphisms and diabetic retinopathy of type 2 diabetes mellitus in the Chinese Han population. J. Gene Med. 18, 282–287. doi: 10.1002/jgm.2926
Lindenmeyer, M. T., Eichinger, F., Sen, K., Anders, H. J., Edenhofer, I., Mattinzoli, D., et al. (2010). Systematic analysis of a novel human renal glomerulus-enriched gene expression dataset. PLoS One 5:e11545. doi: 10.1371/journal.pone.0011545
McKnight, A. J., O’Donoghue, D., and Maxwell, A. (2009). Annotated chromosome maps for renal disease. Hum. Mutat. 30, 314–320. doi: 10.1002/humu.20885
McKnight, A. J., Patterson, C. C., Pettigrew, K. A., Savage, D. A., Kilner, J., Murphy, M., et al. (2010). A GREM1 gene variant associates with diabetic nephropathy. J. Am. Soc. Nephrol. 21, 773–781. doi: 10.1681/asn.2009070773
Mitra, S., Mazumder Indra, D., Basu, S., Mondal, R. K., Roy, A., Roychoudhury, S., et al. (2010). Amplification of CyclinL1 in uterine cervical carcinoma has prognostic implications. Mol. Carcinog 49, 935–943. doi: 10.1002/mc.20671
Mook-Kanamori, D. O., Marsh, J. A., Warrington, N. M., Taal, H. R., Newnham, J. P., Beilin, L. J., et al. (2011). Variants near CCNL1/LEKR1 and in ADCY5 and fetal growth characteristics in different trimesters. J. Clin. Endocrinol. Metab 96, E810–E815.
Moore, K., McKnight, A. J., Craig, D., and O’Neill, F. (2014). Epigenome-wide association study for Parkinson’s disease. Neuromol. Med. 16, 845–855. doi: 10.1007/s12017-014-8332-8
Muhlberger, I., Monks, K., Fechete, R., Mayer, G., Oberbauer, R., Mayer, B., et al. (2012). Molecular pathways and crosstalk characterizing the cardiorenal syndrome. OMICS 16, 105–112. doi: 10.1089/omi.2011.0121
Murphy, T. M., and Mill, J. (2014). Epigenetics in health and disease: heralding the EWAS era. Lancet 383, 1952–1954. doi: 10.1016/s0140-6736(14)60269-5
Nakagawa, S., Nishihara, K., Miyata, H., Shinke, H., Tomita, E., Kajiwara, M., et al. (2015). Molecular markers of tubulointerstitial fibrosis and tubular cell damage in patients with chronic kidney disease. PLoS One 10:e0136994. doi: 10.1371/journal.pone.0136994
Nakano, K., Whitaker, J. W., Boyle, D. L., Wang, W., and Firestein, G. S. (2013). DNA methylome signature in rheumatoid arthritis. Ann. Rheum. Dis. 72, 110–117. doi: 10.1136/annrheumdis-2012-201526
Ochoa-Rosales, C., Portilla-Fernandez, E., Nano, J., Wilson, R., Lehne, B., Mishra, P., et al. (2020). Epigenetic link between statin therapy and Type 2 diabetes. Diabetes Care 43, 875–884.
Park, J., Guan, Y., Sheng, X., Gluck, C., Seasock, M. J., Hakimi, A. A., et al. (2019). Functional methylome analysis of human diabetic kidney disease. JCI Insight 4:e128886.
Peng, L., Yanjiao, M., Ai-guo, W., Pengtao, G., Jianhua, L., Ju, Y., et al. (2011). A fine balance between CCNL1 and TIMP1 contributes to the development of breast cancer cells. Biochem. Biophys. Res. Commun. 409, 344–349. doi: 10.1016/j.bbrc.2011.05.021
Philibert, R. A., Plume, J. M., Gibbons, F. X., Brody, G. H., and Beach, S. R. (2012). The impact of recent alcohol use on genome wide DNA methylation signatures. Front. Genet. 3:54. doi: 10.3389/fgene.2012.00054
Qiu, C., Hanson, R. L., Fufaa, G., Kobes, S., Gluck, C., Huang, J., et al. (2018). Cytosine methylation predicts renal function decline in American Indians. Kidney Int. 93, 1417–1431. doi: 10.1016/j.kint.2018.01.036
Rakyan, V. K., Beyan, H., Down, T. A., Hawa, M. I., Maslau, S., Aden, D., et al. (2011a). Identification of type 1 diabetes-associated DNA methylation variable positions that precede disease diagnosis. PLoS Genet. 7:e1002300. doi: 10.1371/journal.pgen.1002300
Rakyan, V. K., Down, T. A., Balding, D. J., and Beck, S. (2011b). Epigenome-wide association studies for common human diseases. Nat. Rev. Genet. 12, 529–541. doi: 10.1038/nrg3000
Reich, H. N., Tritchler, D., Cattran, D. C., Herzenberg, A. M., Eichinger, F., Boucherot, A., et al. (2010). A molecular signature of proteinuria in glomerulonephritis. PLoS One 5:e13451. doi: 10.1371/journal.pone.0013451
Reinius, L. E., Acevedo, N., Joerink, M., Pershagen, G., Dahlen, S. E., Greco, D., et al. (2012). Differential DNA methylation in purified human blood cells: implications for cell lineage and studies on disease susceptibility. PLoS One 7:e41361. doi: 10.1371/journal.pone.0041361
Renal, U. S. Data System. (2018). 2018 USRDS Annual Data Report: Epidemiology of Kidney Disease in the United States. Amsterdam: Elsevier.
Ritchie, M. E., Phipson, B., Wu, D., Hu, Y., Law, C. W., Shi, W., et al. (2015). limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43:e47. doi: 10.1093/nar/gkv007
Rossing, P., Tarnow, L., Nielsen, F. S., Hansen, B. V., Brenner, B. M., and Parving, H. H. (1995). Low birth weight. a risk factor for development of diabetic nephropathy? Diabetes 44, 1405–1407. doi: 10.2337/diabetes.44.12.1405
Salem, R. M., Todd, J. N., Sandholm, N., Cole, J. B., Chen, W. M., Andrews, D., et al. (2019). Genome-Wide association study of diabetic kidney disease highlights biology involved in glomerular basement membrane collagen. J. Am. Soc. Nephrol. 30, 2000–2016.
Sandholm, N., Forsblom, C., Makinen, V. P., McKnight, A. J., Osterholm, A. M., He, B., et al. (2014). Genome-wide association study of urinary albumin excretion rate in patients with type 1 diabetes. Diabetologia 57, 1143–1153.
Sandholm, N., McKnight, A. J., Salem, R. M., Brennan, E. P., Forsblom, C., Harjutsalo, V., et al. (2013). Chromosome 2q31.1 associates with ESRD in women with type 1 diabetes. J. Am. Soc. Nephrol. 24, 1537–1543. doi: 10.1681/asn.2012111122
Sandholm, N., Salem, R. M., McKnight, A. J., Brennan, E. P., Forsblom, C., Isakova, T., et al. (2012). New susceptibility loci associated with kidney disease in type 1 diabetes. PLoS Genet. 8:e1002921. doi: 10.1371/journal.pgen.1002921
Sandholm, N., Van Zuydam, N., Ahlqvist, E., Juliusdottir, T., Deshmukh, H. A., Rayner, N. W., et al. (2017). The Genetic Landscape of Renal Complications in Type 1 Diabetes. J. Am. Soc. Nephrol. 28, 557–574.
Sandoval, J., Heyn, H., Moran, S., Serra-Musach, J., Pujana, M. A., Bibikova, M., et al. (2011). Validation of a DNA methylation microarray for 450,000 CpG sites in the human genome. Epigenetics 6, 692–702. doi: 10.4161/epi.6.6.16196
Sapienza, C., Lee, J., Powell, J., Erinle, O., Yafai, F., Reichert, J., et al. (2011). DNA methylation profiling identifies epigenetic differences between diabetes patients with ESRD and diabetes patients without nephropathy. Epigenetics 6, 20–28. doi: 10.4161/epi.6.1.13362
Sarwal, M., Chua, M. S., Kambham, N., Hsieh, S. C., Satterwhite, T., Masek, M., et al. (2003). Molecular heterogeneity in acute renal allograft rejection identified by DNA microarray profiling. N. Engl. J. Med. 349, 125–138. doi: 10.1056/nejmoa035588
Smyth, L. J., Duffy, S., Maxwell, A. P., and McKnight, A. J. (2014a). Genetic and epigenetic factors influencing chronic kidney disease. Am. J. Physiol. Renal. Physiol. 307, F757–F776.
Smyth, L. J., McKay, G. J., Maxwell, A. P., and McKnight, A. J. (2014b). DNA hypermethylation and DNA hypomethylation is present at different loci in chronic kidney disease. Epigenetics 9, 366–376. doi: 10.4161/epi.27161
Smyth, L. J., Maxwell, A. P., Benson, K. A., Kilner, J., McKay, G. J., and McKnight, A. J. (2018). Validation of differentially methylated microRNAs identified from an epigenome-wide association study; sanger and next generation sequencing approaches. BMC Res. Notes 11:767. doi: 10.1186/s13104-018-3872-x
Stefan, M., Zhang, W., Concepcion, E., Yi, Z., and Tomer, Y. (2014). DNA methylation profiles in type 1 diabetes twins point to strong epigenetic effects on etiology. J. Autoimmun. 50, 33–37. doi: 10.1016/j.jaut.2013.10.001
Sticht, C., Hofele, C., Flechtenmacher, C., Bosch, F. X., Freier, K., Lichter, P., et al. (2005). Amplification of Cyclin L1 is associated with lymph node metastases in head and neck squamous cell carcinoma (HNSCC). Br. J. Cancer 92, 770–774. doi: 10.1038/sj.bjc.6602400
Swan, E. J., Maxwell, A. P., and McKnight, A. J. (2015). Distinct methylation patterns in genes that affect mitochondrial function are associated with kidney disease in blood-derived DNA from individuals with Type 1 diabetes. Diabet. Med. 32, 1110–1115. doi: 10.1111/dme.12775
Tannukit, S., Wen, X., Wang, H., and Paine, M. L. (2008). TFIP11, CCNL1 and EWSR1 Protein-protein Interactions, and Their Nuclear Localization. Int. J. Mol. Sci. 9, 1504–1514. doi: 10.3390/ijms9081504
UK Renal Registry (2019). UK Renal Registry 21st Annual Report – Data to 31/12/2017, Bristol, UK. Available online at: https://www.renalreg.org/publications-reports/
van Zuydam, N. R., Ahlqvist, E., Sandholm, N., Deshmukh, H., Rayner, N. W., Abdalla, M., et al. (2018). A genome-wide association study of diabetic kidney disease in subjects with Type 2 diabetes. Diabetes 67, 1414–1427.
Walaszczyk, E., Luijten, M., Spijkerman, A. M. W., Bonder, M. J., Lutgers, H. L., et al. (2018). DNA methylation markers associated with type 2 diabetes, fasting glucose and HbA1c levels: a systematic review and replication in a case-control sample of the lifelines study. Diabetologia 61, 354–368. doi: 10.1007/s00125-017-4497-7
Wettenhall, J. M., and Smyth, G. K. (2004). limmaGUI: a graphical user interface for linear modeling of microarray data. Bioinformatics 20, 3705–3706. doi: 10.1093/bioinformatics/bth449
Woroniecka, K. I., Park, A. S., Mohtat, D., Thomas, D. B., Pullman, J. M., and Susztak, K. (2011). Transcriptome analysis of human diabetic kidney disease. Diabetes 60, 2354–2369. doi: 10.2337/db10-1181
Yaghootkar, H., and Freathy, R. M. (2012). Genetic origins of low birth weight. Curr. Opin. Clin. Nutr. Metab. Care 15, 258–264. doi: 10.1097/mco.0b013e328351f543
Zhou, F., Wang, R., Yuan, P., Ren, Y., Mao, Y., Li, R., et al. (2019). Reconstituting the transcriptome and DNA methylome landscapes of human implantation. Nature 572, 660–664. doi: 10.1038/s41586-019-1500-0
Zhou, T., He, X., Cheng, R., Zhang, B., Zhang, R. R., Chen, Y., et al. (2012). Implication of dysregulation of the canonical wingless-type MMTV integration site (WNT) pathway in diabetic nephropathy. Diabetologia 55, 255–266. doi: 10.1007/s00125-011-2314-2
Keywords: epigenetic, diabetes, kidney, renal, methylation, association
Citation: Smyth LJ, Patterson CC, Swan EJ, Maxwell AP and McKnight AJ (2020) DNA Methylation Associated With Diabetic Kidney Disease in Blood-Derived DNA. Front. Cell Dev. Biol. 8:561907. doi: 10.3389/fcell.2020.561907
Received: 14 May 2020; Accepted: 15 September 2020;
Published: 15 October 2020.
Edited by:
Masaru Katoh, National Cancer Center, JapanReviewed by:
Lucas A. Salas, Dartmouth College, United StatesHarvest F. Gu, China Pharmaceutical University, China
Copyright © 2020 Smyth, Patterson, Swan, Maxwell and McKnight. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Amy Jayne McKnight, YS5qLm1ja25pZ2h0QHF1Yi5hYy51aw==