 Kathrin Brockmann
Kathrin Brockmann- 1Center of Neurology, Department of Neurodegeneration and Hertie-Institute for Clinical Brain Research, University of Tübingen, Tübingen, Germany
- 2German Center for Neurodegenerative Disease (DZNE), Bonn, Germany
With disease-modifying compounds targeting alpha-synuclein available in clinical trials, patient stratification according to alpha-synuclein-specific enrichment strategies is a much-needed prerequisite. Such a scenario will be exemplified for GBA, one major genetic risk factor that is specifically associated with the alpha-synucleinopathies: Parkinson’s disease and dementia with Lewy bodies.
Introduction
Due to genetic studies in rare Mendelian cases of Parkinson’s disease (PD) 25 years ago, it became clear that the alpha-synuclein protein is the major component of Lewy bodies and Lewy neurites (Spillantini et al., 1997). It therefore plays a pivotal role in the pathogenesis of alpha-synucleinopathies such as PD and dementia with Lewy bodies (DLB). Subsequently, molecular pathways associated with alpha-synuclein clearance, aggregation, and propagation have been detected. Next to defects in vesicular trafficking, mitochondrial and importantly lysosomal dysfunction represent the most relevant pathways (Jankovic and Tan, 2020). Studying these likely early and initiating events provides “entry points” to develop novel therapeutic targets on an individualized basis.
Notably, histopathology in genetically associated forms of PD can differ. PD patients with LRRK2 mutations show the typical Lewy-body pathology with alpha-synuclein aggregation but also tau aggregation or even nigral degeneration without distinctive histopathology. Nigral degeneration without Lewy body formation is also often seen in PD associated with biallelic mutations in the genes parkin or PINK1 (Schneider and Alcalay, 2017; Henderson et al., 2019). With disease-modifying treatment options targeting alpha-synuclein under way, patient stratification according to alpha-synuclein-specific enrichment strategies as well as knowledge of the disease course and trajectories to disease-related milestones is a much-needed prerequisite to introduce patients to specific therapies. This will be exemplified for the gene glucocerebrosidase (GBA), a major genetic risk factor for PD that is specifically associated with alpha-synuclein pathology.
GBA Mutations Are a Major Genetic Risk Factor for PD
Biallelic mutations in the gene GBA cause Gaucher’s disease (GD), the most common lysosomal storage disorder with tissue accumulation of glucosylceramides due to deficiency of the lysosomal enzyme glucocerebrosidase (GCase). Interestingly, about 25% of GD patients report a first- or second-degree relative to present with typical Parkinsonism (Goker-Alpan et al., 2004; Halperin et al., 2006). This important clinical observation was the hint that heterozygous mutations in the GBA gene might be associated with PD. Subsequently, a large multi-center study across four continents analyzed 5691 PD patients of different ethnic origin compared to 4898 controls and confirmed that with an overall odds ratio (OR) of 5.43, heterozygous mutations in the GBA gene represent a major genetic risk factor for PD (Sidransky et al., 2009). This has now been confirmed across different ethno-racial populations with Caucasian, Asian (Japanese, Chinese, Taiwanese), Hispanic, and African ancestry (Neumann et al., 2009; Lesage et al., 2011; Chen et al., 2014; den Heijer et al., 2020; Mahungu et al., 2020).
Interestingly, some variants that have been reported as non-relevant for Gaucher disease (GD) have been proven to increase the risk for PD, e.g., p.E326K and p.T369M (Zhang et al., 2018; Iwaki et al., 2019). Consequently, GBA-subgroup classification for PD patients is often based on variant severity according to established genotype risks reported for PD.
Moreover, sequencing the GBA gene is very challenging due to the pseudogene. Further, one of the most common severe variants, p.L444P, is not covered well by standard genome-wide arrays such as Neurochip or NeuroXChip. Therefore, interpretation across different studies has to be done carefully, and the most comprehensive analyses are those done by whole gene Sanger sequencing.
GBA Mutations and Parkinson Manifestation
GBA-Associated PD Presents With Non-motor Characteristics
Detailed investigation of the phenotypical spectrum of motor and non-motor symptoms is of utmost importance in order to design studies for disease-modifying therapies. PD patients with GBA mutations (PDGBA) show a younger age at onset with a median onset in the early fifties (Sidransky et al., 2009; Blauwendraat et al., 2019). Of note, this effect is not only attributable to GBA variants in general but is further driven by GBA mutation severity and mutation burden with most severe mutations as well as homozygous and compound heterozygous variants predisposing to the youngest age at onset (Thaler et al., 2017; Malek et al., 2018).
This is of importance as, in general, a younger age and age at onset are typically associated with a more benign disease course, especially in terms of cognitive decline (Forsaa et al., 2010). Keeping this in mind, other important clinical aspects have come to attention in PDGBA. Compared to sporadic PD patients without GBA mutation (PDGBA_wild type), PDGBA present with a higher prevalence of cognitive impairment and more frequently suffer from additional non-motor symptoms including neuropsychiatric disturbances (depression, anxiety, hallucination), autonomic dysfunction, and sleep disturbances such as REM-sleep-behavior disorder (RBD) (Brockmann et al., 2011; Barrett et al., 2014). These findings have been replicated consistently over a the following years in other PD cohorts worldwide, the latest large clinical genome-wide association study in 4093 PD patients (Iwaki et al., 2019). Interestingly, variants that are classified as severe mutations (GBAsevere) have been associated with a more aggressive clinical phenotype suggesting a relevant effect depending on GBA mutation severity (Cilia et al., 2016; Thaler et al., 2018; Petrucci et al., 2020).
Taken together, these clinical findings are of importance for the following reasons:
(1) In addition to demographics (age, age at onset, gender) and co-morbidities, they might offer explanations for the variability of the clinical phenotype in PD.
(2) They might provide defined temporal windows of phenotypical milestones to be addressed in disease-modifying trials beyond pure motor impairment.
GBA-Associated Parkinson’s Disease: More Rapid Progression and Shorter Survival in Prospective Longitudinal Studies
When it comes to clinical trials aiming at disease modification and not just pure symptomatic improvement, the rate of progression is crucial in order to estimate effect sizes and plan study designs (duration, sample sizes, etc.). Following up on these aspects, longitudinally investigated cohorts of PDGBA revealed that this patient group, although younger in age and age at onset, present with an accelerated disease progression in terms of motor impairment, disease staging and cognitive decline. Moreover, survival rates are shorter when compared to PDGBA_wild type (Brockmann et al., 2015b; Stoker et al., 2020).
Shorter and More Prominent Prodromal Phase in GBA-Associated PD
The typical motor manifestation of PD is preceded by a prodromal phase that is characterized by a variety of non-motor and early motor signs (Berg et al., 2015). Non-motor symptoms include amongst others hyposmia, autonomic dysfunction, and neuropsychiatric symptoms, whereas reduced arm swing and bradykinesia indicate early motor signs. However, type, prevalence, time of occurrence, and rate of progression of these prodromal symptoms vary between patients. Given the findings from the manifest disease phase in PDGBA with pronounced non-motor symptoms and a more rapid disease progression, we retrospectively focused on patient’s perception regarding their individual prodromal phase before PD diagnosis. Comparing PDGBA and PDGBA_wild type, we could show that: (1) prevalence and time of occurrence of prodromal symptoms seem more pronounced in PDGBA. They reported a shorter prodromal phase with almost parallel beginning of non-motor and early motor signs before PD diagnosis. Contrarily, PDGBA_wild type showed a long prodromal interval starting with non-motor symptoms long before early motor signs manifested. (2) Patients carrying severe GBA mutations reported the highest total amount of prodromal signs. These findings suggest that clinical trajectories known from the manifest disease might be present already in the prodromal phase (Zimmermann et al., 2018). Indeed, prospective studies found that asymptomatic GBA mutation carriers present with parallel deterioration of non-motor and motor sign when compared to healthy controls without GBA mutation (Beavan et al., 2015; Avenali et al., 2019). There is only one prospective longitudinal study available in patients with RBD that monitored the prodromal phase until phenoconversion to manifest PD. While there were no differences in the severity of prodromal motor and non-motor markers, GBA mutation status was associated with accelerated phenoconversion to PD and/or dementia (Honeycutt et al., 2019).
GBA Mutations and Dementia With Lewy Bodies
The important finding that PDGBA more frequently develop dementia earlier in the disease course than PDGBA_wild type prompted the community to perform a large multicenter analysis across 11 centers evaluating GBA mutations in 721 cases with DLB, which represents a clinico-histopathological continuum to PD. With an even higher OR of 8.28, GBA mutations are also strongly associated with DLB. Similar to PD, GBA mutations also predispose to an earlier age at onset and more pronounced disease severity/progression in DLB (Nalls et al., 2013). This study further supported GBA mutations as a significant genetic risk factor for synucleinopathies and confirmed the overall impression that GBA-related Parkinsonism predisposes to an increased incidence of dementia.
GBA Mutations and Multisystem Atrophy
The higher prevalence of autonomic dysfunction and the link to alpha-synuclein pathology as discussed further on raised the idea that GBA variants might also be associated with increased risk to develop MSA. However, studies in MSA revealed conflicting results, both in clinically diagnosed as well as in autopsy-confirmed cases. One large study investigated 969 MSA patients with Japanese, European, and North American background and found an overall OR of 2.44. Notably, the authors reported a significant association between GBA variants and MSA-C phenotype (Mitsui et al., 2015). Another study with autopsy-confirmed MSA and Alzheimer patients reported a higher frequency of GBA variants in the MSA group compared to the AD group (Sklerov et al., 2017). However, a larger study with autopsy-proven MSA cases could not show an association of GBA variants with MSA (Segarane et al., 2009). Moreover, several studies with clinical diagnosis of MSA also found no clear association (Srulijes et al., 2013; Sun et al., 2013; Asselta et al., 2014).
GBA-Associated Pathomechanism and Histopathology
Evidence from cell models favors the hypothesis that GBA mutations result in disrupted trafficking of GCase from the ER to Golgi and in lower lysosomal GCase enzyme activity which in turn cause a build-up of lysosomal glucosylceramides and impair alpha-synuclein degradation (Mazzulli et al., 2011). Highlighting the role of lysosomal dysfunction in the pathogenesis of PD, results from postmortem brain tissue and IPS cell-derived neurons show that reduced lysosomal GCase activity is paralleled by increased levels of alpha-synuclein, not only in PDGBA but, to a lesser degree, also in cases with PDGBA_wild type (Gegg et al., 2012; Murphy et al., 2014; Schondorf et al., 2014; Moors et al., 2019). Moreover, recent data in human midbrain dopaminergic neurons suggest that conformational changes of alpha-synuclein toward an aggregation-prone pattern can be even induced by the presence of glycosphingolipids irrespective of an underlying mutation in the GBA gene (Zunke et al., 2018). More specifically, it was suggested that lysosomal GCase and alpha-synuclein are linked in a bidirectional pathogenic loop in synucleinopathies as shown in cell cultures and in induced-pluripotent stem (IPS) cell-derived dopaminergic midbrain neurons: (1) Functional loss of GCase activity compromises lysosomal degradation of alpha-synuclein and causes its aggregation due to reduced lysosomal chaperone-mediated autophagy. (2) Alpha-synuclein itself inhibits the activity of GCase (Mazzulli et al., 2011; Schondorf et al., 2014). Consequently, PDGBA fulfill both conditions of this bidirectional loop in parallel leading to a self-reinforcing mechanism. Thereby, alpha-synuclein aggregation and propagation might be enhanced which possibly explains the wide-spread neocortical Lewy body pathology observed in postmortem brain tissue of PDGBA (Neumann et al., 2009; Gundner et al., 2019; Moors et al., 2019). These pathomechanistic aspects in turn offer a reasonable explanation for the more severe and more rapid disease progression seen in the prodromal and in the manifest phase in PDGBA (Figure 1).
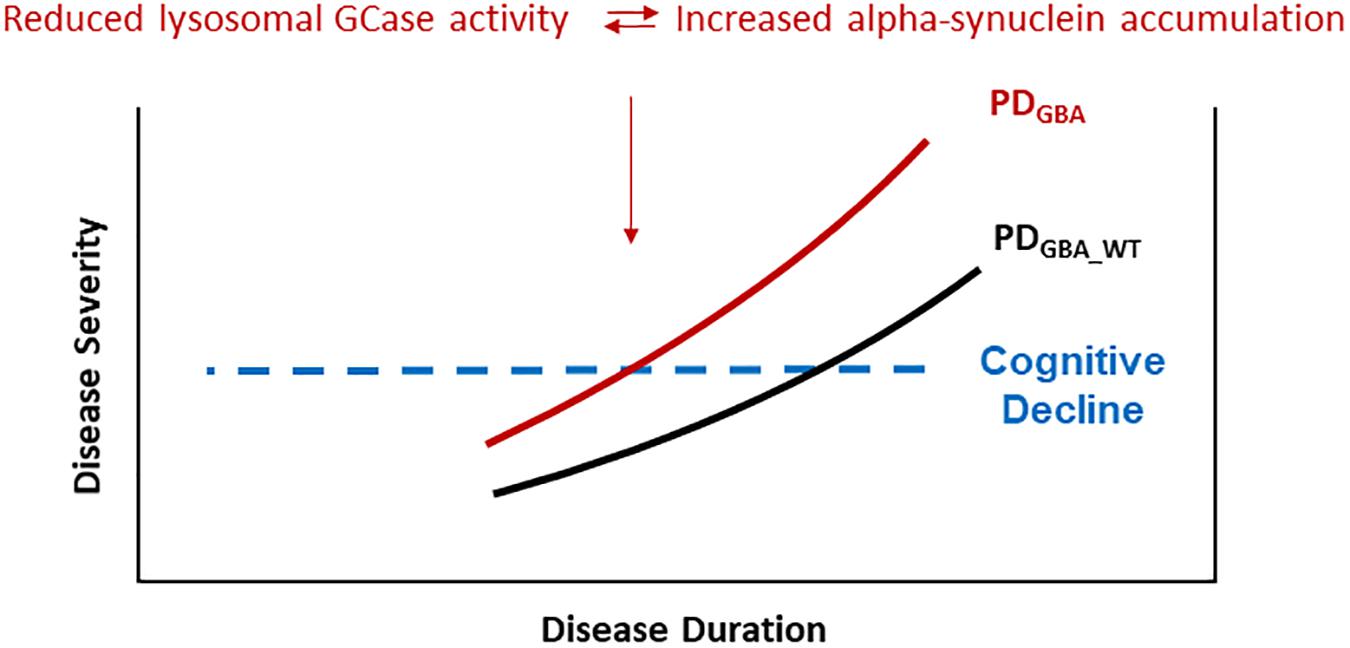
Figure 1. PD patients carrying pathogenic GBA mutations show a more rapid progression of motor symptoms, specifically of cognitive decline. These clinical characteristics seem to be driven by accelerated alpha-synuclein aggregation and propagation due to a self-inforcing bidirectional pathogenic loop between GCase deficiency and alpha-synuclein accumulation. Especially the high risk of cognitive decline in yet cognitively intact PDGBA patients provides a defined window of opportunity for modifying treatment options targeting alpha-synuclein.
GBA-Associated Biomarker Profiles in Patient-Derived Biofluids
Despite this clear experimental evidence, we often fail to translate these findings into clinical research with patient cohorts, and we lack to confirm the impact of genetic mutations on biochemical profiles in patient-derived biomaterial. Moreover, we need to evaluate whether such profiles might be suitable as biochemical readout for target engagement.
Heterozygous variants in the GBA gene are associated with lower levels of lysosomal GCase enzyme activity using a variety of assays in different patient-derived biofluids including blood and CSF (Schondorf et al., 2014; Alcalay et al., 2015; Paciotti et al., 2019). Similar to the findings in brain tissue, GCase activity is also reduced in PD patients without GBA variant, albeit to a lesser degree (Parnetti et al., 2017).
As we have no reliable imaging marker available to assess the cerebral load of alpha-synuclein in vivo, research has focused on CSF. Importantly, it is widely discussed whether CSF profiles of alpha-synuclein species reflect alpha-synuclein pathology in the brain. Analyses in sporadic PD demonstrated CSF levels of total alpha-synuclein to be decreased in PD compared to healthy controls (Malek et al., 2014; Mollenhauer et al., 2019). However, CSF levels of total alpha-synuclein do not correlate with motor-associated disease progression. These findings imply that CSF levels of total alpha-synuclein are not suitable to monitor motor progression in the clinically manifested disease phase and/or that the nature of progression is too slow. Since prodromal PD subjects with hyposmia and/or REM-sleep behavior disorder already show decreased CSF levels of alpha-synuclein (Mollenhauer et al., 2019), one might argue that this phenomenon develops early in the disease and does not parallel with the manifest disease phase.
Given the specific mechanistic link between GBA and alpha-synuclein, it is tempting to speculate whether PDGBA represent a proxy for alpha-synuclein-driven CSF profiles. Indeed, we could show that PDGBA present with lower CSF levels of total alpha-synuclein compared to healthy controls and also compared to PDGBA_wild type. Importantly, PDGBA with severe variants show the lowest mean values (Lerche et al., 2020).
As PDGBA present accelerated cognitive decline, this subgroup of PD patients represent a good model to study biochemical profiles in CSF that might be associated with cognitive impairment. In general, limbic and/or cortical Lewy body pathology is hypothesized to be the main substrate driving cognitive decline in sporadic PD (Aarsland et al., 2005). In more recent years, it became clear that a considerable proportion of sporadic PD patients who developed dementia in their disease course show concomitant amyloid-beta and tau pathology at autopsy in addition to the typical Lewy-body pathology (Halliday et al., 2008; Compta et al., 2011). Correspondingly, reduced CSF levels of amyloid-beta1_42 and/or elevated CSF levels of total-tau and phospho-tau have been reported to be associated with cognitive impairment in sporadic PD (Brockmann et al., 2015a, 2017; Kang et al., 2016; Lerche et al., 2019b). However, this seems not to be the case in PDGBA as CSF levels of Aβ1–42, t-TAU, and p-Tau are similar to those seen in healthy control individuals, whereas levels of alpha-synuclein were lower. These findings suggest that the prominent cognitive impairment in PDGBA is not associated with amyloid-beta or tau pathology but might be driven by alpha-synuclein aggregation.
Based on the genetic link between GBA mutations and DLB, it was tempting to speculate whether findings of CSF alpha-synuclein profiles in PDGBA patients can be also detected in DLB patients carrying a GBA mutation. Similar to PD, GBA mutations are associated with decreased CSF levels of total alpha-synuclein in DLB patients. Again, these findings seem dependent on GBA mutation severity and were most pronounced in DLBGBA patients with severe mutations (Lerche et al., 2019a).
These in vivo data seem to confirm findings from cell models and postmortem analysis: lower GCase activity is associated with prominent CSF profiles of total alpha-synuclein representing a mirror of greater cerebral Lewy pathology in PDGBA and DLBGBA patients (Neumann et al., 2009; Gundner et al., 2018; Moors et al., 2018). Yet, a substantial inter-individual variability and overlap with healthy controls is seen so that CSF levels of total alpha-synuclein are not ideal to be used as a single biomarker. More recently, real−time quaking−induced conversion (RT−QuIC) and protein misfolding cyclic amplification (PMCA) have been successfully implemented to evaluate alpha-synuclein seeding capacities. These assays are based on the conversion of monomeric substrate protein into β−sheet−rich aggregates by seeding with small amounts of protein aggregates (Fairfoul et al., 2016; Shahnawaz et al., 2017). As this method is highly sensitive and specific for alpha-synuclein aggregation, PD and DLB patients with GBA mutations would be prime candidates to be assessed with this assay.
Discussion and Outlook
PD patients carrying pathogenic GBA mutations show a faster motor progression and cognitive decline. Importantly, the higher risk of cognitive decline is not associated with amyloid-β pathology (e.g., CSF Abeta1_42 levels) as shown instead in sporadic PD without GBA mutations. Postmortem studies show that lower levels of lysosomal GCase activity are associated with greater alpha-synuclein pathology in PD, PDD, and PDGBA brains. This has been confirmed by in vivo studies showing that PD and DLB patients carrying pathogenic GBA mutations had reduced levels of lysosomal GCase activity paralleled by lower CSF levels of total alpha-synuclein (possibly mirroring greater Lewy pathology in the brain). Based on these results, high amounts of aggregated alpha-synuclein could play a pivotal role in cognitive decline in PDGBA (Figure 1).
So far, drugs aiming at slowing disease progression in neurodegenerative diseases failed. One reason that could be discussed is that they were administered in manifest disease stages where brain pathology is too advanced. PDGBA patients who are cognitively intact represent a high-risk population to develop PD-associated dementia and thus provide a defined window of opportunity for treatment aiming to delay cognitive decline. Thereby, starting a treatment able to reduce the propagation of aggregated alpha-synuclein in this population would overcome the challenge of starting intervention early enough. As alpha-synuclein aggregation seems to be the main driver of dementia in PDGBA, this specific population represents a role model to study the effect of alpha-synuclein lowering treatment strategies such as monoclonal antibodies targeting aggregated alpha-synuclein.
A more complex picture is now beginning to emerge and points toward a central role for GCase activity not only in GBA-associated PD but also in sporadic as well as other genetic forms. Postmortem brain tissue analyses, patient-derived IPS-cell models, and CSF studies show decreased levels of GCase activity paralleled by alpha-synuclein accumulation in wild-type PD patients and in PD patients with mutations in LRRK2, parkin, and DJ-1. Interestingly, LRRK2 kinase activity seems to regulate GCase activity in an inverse pattern (Parnetti et al., 2017; Burbulla et al., 2019; Ysselstein et al., 2019). These recent findings highlight not only the importance of lysosomal dysfunction in the pathophysiology of PDGBA but the significance of this pathway for PD in general. Thereby, one could imagine that lysosomal-targeted treatment options developed for PDGBA (Mullin et al., 2020) might be also beneficial for other PD subgroups in which lysosomal dysfunction driving alpha-synuclein accumulation plays a major role. However, at this point we lack direct comparisons with the same assay/methodology evaluating the degree of GCase activity reduction, lysosomal dysfunction, and consecutive alpha-synuclein accumulation between wild-type and different genetic forms of PD (GBA, LRKK2, Parkin, PINK, etc.).
Author Contributions
The author confirms being the sole contributor of this work and has approved it for publication.
Funding
KB has received a research grant from the University of Tübingen (Clinician Scientist) and the German Society of Parkinson’s Disease (dPV), funding from the Michael J. Fox Foundation (MJFF) and the German Centre for Neurodegenerative Diseases (DZNE, MIGAP), travel grants from the Movement Disorders Society, and speaker honoraria from Abbvie, Lundbeck, UCB, and Zambon.
Conflict of Interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Aarsland, D., Perry, R., Brown, A., Larsen, J. P., and Ballard, C. (2005). Neuropathology of dementia in Parkinson’s disease: a prospective, community-based study. Ann. Neurol. 58, 773–776. doi: 10.1002/ana.20635
Alcalay, R. N., Levy, O. A., Waters, C. C., Fahn, S., Ford, B., Kuo, S. H., et al. (2015). Glucocerebrosidase activity in Parkinson’s disease with and without GBA mutations. Brain 138, 2648–2658.
Asselta, R., Rimoldi, V., Siri, C., Cilia, R., Guella, I., Tesei, S., et al. (2014). Glucocerebrosidase mutations in primary parkinsonism. Parkinsonism. Relat. Disord. 20, 1215–1220. doi: 10.1016/j.parkreldis.2014.09.003
Avenali, M., Toffoli, M., Mullin, S., Mcneil, A., Hughes, D. A., Mehta, A., et al. (2019). Evolution of prodromal parkinsonian features in a cohort of GBA mutation-positive individuals: a 6-year longitudinal study. J. Neurol. Neurosurg. Psychiatry 90, 1091–1097. doi: 10.1136/jnnp-2019-320394
Barrett, M. J., Shanker, V. L., Severt, W. L., Raymond, D., Gross, S. J., Schreiber-Agus, N., et al. (2014). Cognitive and Antipsychotic Medication Use in Monoallelic GBA-Related Parkinson Disease. JIMD Rep. 16, 31–38. doi: 10.1007/8904_2014_315
Beavan, M., Mcneill, A., Proukakis, C., Hughes, D. A., Mehta, A., and Schapira, A. H. (2015). Evolution of prodromal clinical markers of Parkinson disease in a GBA mutation-positive cohort. JAMA Neurol. 72, 201–208. doi: 10.1001/jamaneurol.2014.2950
Berg, D., Postuma, R. B., Adler, C. H., Bloem, B. R., Chan, P., Dubois, B., et al. (2015). MDS research criteria for prodromal Parkinson’s disease. Mov. Disord. 30, 1600–1611. doi: 10.1002/mds.26431
Blauwendraat, C., Heilbron, K., Vallerga, C. L., Bandres-Ciga, S., Von Coelln, R., Pihlstrom, L., et al. (2019). Parkinson’s disease age at onset genome-wide association study: defining heritability, genetic loci, and alpha-synuclein mechanisms. Mov. Disord. 34, 866–875.
Brockmann, K., Lerche, S., Dilger, S. S., Stirnkorb, J. G., Apel, A., Hauser, A. K., et al. (2017). SNPs in Abeta clearance proteins: lower CSF Abeta1-42 levels and earlier onset of dementia in PD. Neurology 89, 2335–2340. doi: 10.1212/wnl.0000000000004705
Brockmann, K., Schulte, C., Deuschle, C., Hauser, A. K., Heger, T., Gasser, T., et al. (2015a). Neurodegenerative CSF markers in genetic and sporadic PD: classification and prediction in a longitudinal study. Parkinsonism. Relat. Disord. 21, 1427–1434. doi: 10.1016/j.parkreldis.2015.10.008
Brockmann, K., Srulijes, K., Pflederer, S., Hauser, A. K., Schulte, C., Maetzler, W., et al. (2015b). GBA-associated Parkinson’s disease: reduced survival and more rapid progression in a prospective longitudinal study. Mov. Disord. 30, 407–411. doi: 10.1002/mds.26071
Brockmann, K., Srulijes, K., Hauser, A. K., Schulte, C., Csoti, I., Gasser, T., et al. (2011). GBA-associated PD presents with nonmotor characteristics. Neurology 77, 276–280. doi: 10.1212/wnl.0b013e318225ab77
Burbulla, L. F., Jeon, S., Zheng, J., Song, P., Silverman, R. B., and Krainc, D. (2019). A modulator of wild-type glucocerebrosidase improves pathogenic phenotypes in dopaminergic neuronal models of Parkinson’s disease. Sci. Transl. Med. 11:eaau6870. doi: 10.1126/scitranslmed.aau6870
Chen, J., Li, W., Zhang, T., Wang, Y. J., Jiang, X. J., and Xu, Z. Q. (2014). Glucocerebrosidase gene mutations associated with Parkinson’s disease: a meta-analysis in a Chinese population. PLoS One 9:e115747. doi: 10.1371/journal.pone.0115747
Cilia, R., Tunesi, S., Marotta, G., Cereda, E., Siri, C., Tesei, S., et al. (2016). Survival and dementia in GBA-associated Parkinson’s disease: the mutation matters. Ann. Neurol. 80, 662–673. doi: 10.1002/ana.24777
Compta, Y., Parkkinen, L., O’sullivan, S. S., Vandrovcova, J., Holton, J. L., Collins, C., et al. (2011). Lewy- and Alzheimer-type pathologies in Parkinson’s disease dementia: which is more important? Brain 134, 1493–1505. doi: 10.1093/brain/awr031
den Heijer, J. M., Cullen, V. C., Quadri, M., Schmitz, A., Hilt, D. C., Lansbury, P., et al. (2020). A Large-Scale Full GBA1 Gene Screening in Parkinson’s Disease in the Netherlands. Mov Disord. doi: 10.1002/mds.28112 [Epub ahead of print].
Fairfoul, G., Mcguire, L. I., Pal, S., Ironside, J. W., Neumann, J., Christie, S., et al. (2016). Alpha-synuclein RT-QuIC in the CSF of patients with alpha-synucleinopathies. Ann. Clin. Transl. Neurol. 3, 812–818. doi: 10.1002/acn3.338
Forsaa, E. B., Larsen, J. P., Wentzel-Larsen, T., and Alves, G. (2010). What predicts mortality in Parkinson disease: a prospective population-based long-term study. Neurology 75, 1270–1276. doi: 10.1212/wnl.0b013e3181f61311
Gegg, M. E., Burke, D., Heales, S. J., Cooper, J. M., Hardy, J., Wood, N. W., et al. (2012). Glucocerebrosidase deficiency in substantia nigra of parkinson disease brains. Ann. Neurol. 72, 455–463. doi: 10.1002/ana.23614
Goker-Alpan, O., Schiffmann, R., Lamarca, M. E., Nussbaum, R. L., Mcinerney-Leo, A., and Sidransky, E. (2004). Parkinsonism among Gaucher disease carriers. J. Med. Genet. 41, 937–940. doi: 10.1136/jmg.2004.024455
Gundner, A. L., Duran-Pacheco, G., Zimmermann, S., Ruf, I., Moors, T., Baumann, K., et al. (2018). Path mediation analysis reveals GBA impacts Lewy body disease status by increasing alpha-synuclein levels. Neurobiol. Dis. 121, 205–213. doi: 10.1016/j.nbd.2018.09.015
Gundner, A. L., Duran-Pacheco, G., Zimmermann, S., Ruf, I., Moors, T., Baumann, K., et al. (2019). Path mediation analysis reveals GBA impacts Lewy body disease status by increasing alpha-synuclein levels. Neurobiol. Dis. 121, 205–213. doi: 10.1016/j.nbd.2018.09.015
Halliday, G., Hely, M., Reid, W., and Morris, J. (2008). The progression of pathology in longitudinally followed patients with Parkinson’s disease. Acta Neuropathol. 115, 409–415. doi: 10.1007/s00401-008-0344-8
Halperin, A., Elstein, D., and Zimran, A. (2006). Increased incidence of Parkinson disease among relatives of patients with Gaucher disease. Blood Cells Mol. Dis. 36, 426–428. doi: 10.1016/j.bcmd.2006.02.004
Henderson, M. X., Sengupta, M., Trojanowski, J. Q., and Lee, V. M. Y. (2019). Alzheimer’s disease tau is a prominent pathology in LRRK2 Parkinson’s disease. Acta Neuropathol. Commun. 7:183.
Honeycutt, L., Montplaisir, J. Y., Gagnon, J. F., Ruskey, J., Pelletier, A., Gan-Or, Z., et al. (2019). Glucocerebrosidase mutations and phenoconversion of REM sleep behavior disorder to parkinsonism and dementia. Parkinsonism. Relat. Disord. 65, 230–233. doi: 10.1016/j.parkreldis.2019.04.016
Iwaki, H., Blauwendraat, C., Leonard, H. L., Liu, G., Maple-Grodem, J., Corvol, J. C., et al. (2019). Genetic risk of Parkinson disease and progression:: an analysis of 13 longitudinal cohorts. Neurol. Genet. 5:e348.
Jankovic, J., and Tan, E. K. (2020). Parkinson’s disease: etiopathogenesis and treatment. J. Neurol. Neurosurg. Psychiatry 91, 795–808.
Kang, J. H., Mollenhauer, B., Coffey, C. S., Toledo, J. B., Weintraub, D., Galasko, D. R., et al. (2016). CSF biomarkers associated with disease heterogeneity in early Parkinson’s disease: the Parkinson’s Progression Markers Initiative study. Acta Neuropathol. 131, 935–949.
Lerche, S., Machetanz, G., Wurster, I., Roeben, B., Zimmermann, M., Pilotto, A., et al. (2019a). Dementia with lewy bodies: GBA1 mutations are associated with cerebrospinal fluid alpha-synuclein profile. Mov. Disord. 34, 1069–1073.
Lerche, S., Wurster, I., Roben, B., Machetanz, G., Zimmermann, M., Bernhard, F., et al. (2019b). Parkinson’s disease: evolution of cognitive impairment and CSF Abeta1-42 profiles in a prospective longitudinal study. J. Neurol. Neurosurg. Psychiatry 90, 165–170. doi: 10.1136/jnnp-2018-318956
Lerche, S., Wurster, I., Roeben, B., Zimmermann, M., Riebenbauer, B., Deuschle, C., et al. (2020). Parkinson’s disease: Glucocerebrosidase 1 mutation severity is associated with CSF Alpha-Synuclein profiles. Mov. Disord. 35, 495–499. doi: 10.1002/mds.27884
Lesage, S., Anheim, M., Condroyer, C., Pollak, P., Durif, F., Dupuits, C., et al. (2011). Large-scale screening of the Gaucher’s disease-related glucocerebrosidase gene in Europeans with Parkinson’s disease. Hum. Mol. Genet. 20, 202–210.
Mahungu, A. C., Anderson, D. G., Rossouw, A. C., Van Coller, R., Carr, J. A., Ross, O. A., et al. (2020). Screening of the glucocerebrosidase (GBA) gene in South Africans of African ancestry with Parkinson’s disease. Neurobiol. Aging 88, 156.e11–156.e14.
Malek, N., Swallow, D., Grosset, K. A., Anichtchik, O., Spillantini, M., and Grosset, D. G. (2014). Alpha-synuclein in peripheral tissues and body fluids as a biomarker for Parkinson’s disease - a systematic review. Acta Neurol. Scand. 130, 59–72. doi: 10.1111/ane.12247
Malek, N., Weil, R. S., Bresner, C., Lawton, M. A., Grosset, K. A., Tan, M., et al. (2018). Features of GBA-associated Parkinson’s disease at presentation in the UK Tracking Parkinson’s study. J. Neurol. Neurosurg. Psychiatry 89, 702–709.
Mazzulli, J. R., Xu, Y. H., Sun, Y., Knight, A. L., Mclean, P. J., Caldwell, G. A., et al. (2011). Gaucher disease glucocerebrosidase and alpha-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell 146, 37–52. doi: 10.1016/j.cell.2011.06.001
Mitsui, J., Matsukawa, T., Sasaki, H., Yabe, I., Matsushima, M., Durr, A., et al. (2015). Variants associated with Gaucher disease in multiple system atrophy. Ann. Clin. Transl. Neurol. 2, 417–426.
Mollenhauer, B., Caspell-Garcia, C. J., Coffey, C. S., Taylor, P., Singleton, A., Shaw, L. M., et al. (2019). Longitudinal analyses of cerebrospinal fluid alpha-Synuclein in prodromal and early Parkinson’s disease. Mov. Disord. 34, 1354–1364. doi: 10.1002/mds.27806
Moors, T. E., Paciotti, S., Ingrassia, A., Quadri, M., Breedveld, G., Tasegian, A., et al. (2018). Characterization of Brain Lysosomal Activities in GBA-Related and Sporadic Parkinson’s Disease and Dementia with Lewy Bodies. Mol Neurobiol. 56, 1344–1355. doi: 10.1007/s12035-018-1090-0
Moors, T. E., Paciotti, S., Ingrassia, A., Quadri, M., Breedveld, G., Tasegian, A., et al. (2019). Characterization of Brain Lysosomal Activities in GBA-Related and Sporadic Parkinson’s Disease and Dementia with Lewy Bodies. Mol. Neurobiol. 56, 1344–1355. doi: 10.1007/s12035-018-1090-0
Mullin, S., Smith, L., Lee, K., D’souza, G., Woodgate, P., Elflein, J., et al. (2020). Ambroxol for the treatment of patients with Parkinson disease with and without glucocerebrosidase gene mutations: a nonrandomized. Noncontrolled Trial. JAMA Neurol. 77, 427–434. doi: 10.1001/jamaneurol.2019.4611
Murphy, K. E., Gysbers, A. M., Abbott, S. K., Tayebi, N., Kim, W. S., Sidransky, E., et al. (2014). Reduced glucocerebrosidase is associated with increased alpha-synuclein in sporadic Parkinson’s disease. Brain 137, 834–848. doi: 10.1093/brain/awt367
Nalls, M. A., Duran, R., Lopez, G., Kurzawa-Akanbi, M., Mckeith, I. G., Chinnery, P. F., et al. (2013). A multicenter study of glucocerebrosidase mutations in dementia with Lewy bodies. JAMA Neurol. 70, 727–735.
Neumann, J., Bras, J., Deas, E., O’sullivan, S. S., Parkkinen, L., Lachmann, R. H., et al. (2009). Glucocerebrosidase mutations in clinical and pathologically proven Parkinson’s disease. Brain 132, 1783–1794. doi: 10.1093/brain/awp044
Paciotti, S., Gatticchi, L., Beccari, T., and Parnetti, L. (2019). Lysosomal enzyme activities as possible CSF biomarkers of synucleinopathies. Clin. Chim. Acta 495, 13–24. doi: 10.1016/j.cca.2019.03.1627
Parnetti, L., Paciotti, S., Eusebi, P., Dardis, A., Zampieri, S., Chiasserini, D., et al. (2017). Cerebrospinal fluid beta-glucocerebrosidase activity is reduced in parkinson’s disease patients. Mov. Disord. 32, 1423–1431. doi: 10.1002/mds.27136
Petrucci, S., Ginevrino, M., Trezzi, I., Monfrini, E., Ricciardi, L., Albanese, A., et al. (2020). GBA-Related Parkinson’s disease: dissection of genotype-phenotype correlates in a Large Italian Cohort. Mov Disord. doi: 10.1002/mds.28195 [Epub ahead of print].
Schneider, S. A., and Alcalay, R. N. (2017). Neuropathology of genetic synucleinopathies with parkinsonism: review of the literature. Mov. Disord. 32, 1504–1523. doi: 10.1002/mds.27193
Schondorf, D. C., Aureli, M., Mcallister, F. E., Hindley, C. J., Mayer, F., Schmid, B., et al. (2014). iPSC-derived neurons from GBA1-associated Parkinson’s disease patients show autophagic defects and impaired calcium homeostasis. Nat. Commun. 5:4028.
Segarane, B., Li, A., Paudel, R., Scholz, S., Neumann, J., Lees, A., et al. (2009). Glucocerebrosidase mutations in 108 neuropathologically confirmed cases of multiple system atrophy. Neurology 72, 1185–1186. doi: 10.1212/01.wnl.0000345356.40399.eb
Shahnawaz, M., Tokuda, T., Waragai, M., Mendez, N., Ishii, R., Trenkwalder, C., et al. (2017). Development of a Biochemical Diagnosis of Parkinson Disease by Detection of alpha-Synuclein Misfolded Aggregates in Cerebrospinal Fluid. JAMA Neurol. 74, 163–172. doi: 10.1001/jamaneurol.2016.4547
Sidransky, E., Nalls, M. A., Aasly, J. O., Aharon-Peretz, J., Annesi, G., Barbosa, E. R., et al. (2009). Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N. Engl. J. Med. 361, 1651–1661.
Sklerov, M., Kang, U. J., Liong, C., Clark, L., Marder, K., Pauciulo, M., et al. (2017). Frequency of GBA variants in autopsy-proven multiple system atrophy. Mov. Disord. Clin. Pract. 4, 574–581. doi: 10.1002/mdc3.12481
Spillantini, M. G., Schmidt, M. L., Lee, V. M., Trojanowski, J. Q., Jakes, R., and Goedert, M. (1997). Alpha-synuclein in Lewy bodies. Nature 388, 839–840.
Srulijes, K., Hauser, A. K., Guella, I., Asselta, R., Brockmann, K., Schulte, C., et al. (2013). No association of GBA mutations and multiple system atrophy. Eur. J. Neurol. 20, e61–e62.
Stoker, T. B., Camacho, M., Winder-Rhodes, S., Liu, G., Scherzer, C. R., Foltynie, T., et al. (2020). Impact of GBA1 variants on long-term clinical progression and mortality in incident Parkinson’s disease. J Neurol Neurosurg Psychiatry. 91:jnnp-2020-322857.
Sun, Q. Y., Guo, J. F., Han, W. W., Zuo, X., Wang, L., Yao, L. Y., et al. (2013). Genetic association study of glucocerebrosidase gene L444P mutation in essential tremor and multiple system atrophy in mainland China. J. Clin. Neurosci. 20, 217–219. doi: 10.1016/j.jocn.2012.01.055
Thaler, A., Bregman, N., Gurevich, T., Shiner, T., Dror, Y., Zmira, O., et al. (2018). Parkinson’s disease phenotype is influenced by the severity of the mutations in the GBA gene. Parkinsonism. Relat. Disord. 55, 45–49. doi: 10.1016/j.parkreldis.2018.05.009
Thaler, A., Gurevich, T., Bar Shira, A., Gana Weisz, M., Ash, E., Shiner, T., et al. (2017). A “dose” effect of mutations in the GBA gene on Parkinson’s disease phenotype. Parkinsonism. Relat. Disord. 36, 47–51. doi: 10.1016/j.parkreldis.2016.12.014
Ysselstein, D., Nguyen, M., Young, T. J., Severino, A., Schwake, M., Merchant, K., et al. (2019). LRRK2 kinase activity regulates lysosomal glucocerebrosidase in neurons derived from Parkinson’s disease patients. Nat. Commun. 10:5570.
Zhang, Y., Shu, L., Sun, Q., Zhou, X., Pan, H., Guo, J., et al. (2018). Integrated genetic analysis of racial differences of common GBA variants in Parkinson’s disease: a meta-analysis. Front. Mol. Neurosci. 11:43. doi: 10.3389/fnmol.2018.00043
Zimmermann, M., Gaenslen, A., Prahl, K., Srulijes, K., Hauser, A. K., Schulte, C., et al. (2018). Patient’s perception: shorter and more severe prodromal phase in GBA-associated PD. Eur. J. Neurol. 26, 694–698. doi: 10.1111/ene.13776
Keywords: GBA, alpha-synuclein, dementia, GCase, Parkinson
Citation: Brockmann K (2020) GBA-Associated Synucleinopathies: Prime Candidates for Alpha-Synuclein Targeting Compounds. Front. Cell Dev. Biol. 8:562522. doi: 10.3389/fcell.2020.562522
Received: 02 July 2020; Accepted: 09 September 2020;
Published: 25 September 2020.
Edited by:
Friederike Zunke, University of Kiel, GermanyReviewed by:
Hugo Vicente Miranda, New University of Lisbon, PortugalChristos Proukakis, University College London, United Kingdom
Copyright © 2020 Brockmann. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kathrin Brockmann, a2F0aHJpbi5icm9ja21hbm5AdW5pLXR1ZWJpbmdlbi5kZQ==