 Xiaoyan Wu1,4,5†Wenhui Dong4†Tianyi Zhang4†Haozhen Ren1,2Jinglin Wang1,2Longcheng Shang1Zhengyi Zhu1Wei Zhu3*Xiaolei Shi1,2*
Xiaoyan Wu1,4,5†Wenhui Dong4†Tianyi Zhang4†Haozhen Ren1,2Jinglin Wang1,2Longcheng Shang1Zhengyi Zhu1Wei Zhu3*Xiaolei Shi1,2* Yong Xu4,5
Yong Xu4,5- 1Department of Hepatobiliary Surgery, The Affiliated Nanjing Drum Tower Hospital of Nanjing University Medical School, Nanjing, China
- 2Hepatobiliary Institute, Nanjing University, Nanjing, China
- 3Department of Anesthesiology, The Affiliated Drum Tower Hospital of Nanjing University Medical School, Nanjing, China
- 4Key Laboratory of Targeted Intervention of Cardiovascular Disease, Collaborative Innovation Center for Cardiovascular Translational Medicine, and Center for Experimental Medicine, Department of Pathophysiology, Nanjing Medical University, Nanjing, China
- 5Institute of Biomedical Research, Liaocheng University, Liaocheng, China
Trans-differentiation of quiescent hepatic stellate cells (HSC) into myofibroblast cells is considered the linchpin of liver fibrosis. A myriad of signaling pathways contribute to HSC activation and consequently liver fibrosis. Epidermal growth factor (EGF) family of cytokines signal through the cognate receptor EGFR to promote HSC activation. In the present study we investigated the transcription regulation of epiregulin (EREG), an EGFR ligand, during HSC activation. We report that EREG expression was significantly up-regulated in activated HSCs compared to quiescent HSCs isolated from mice. In addition, there was an elevation of EREG expression in HSCs undergoing activation in vitro. Of interest, deficiency of myocardin-related transcription factor A (MRTF-A), a well-documented regulator of HSC trans-differentiation, attenuated up-regulation of EREG expression both in vivo and in vitro. Further analysis revealed that MRTF-A interacted with serum response factor (SRF) to bind directly to the EREG promoter and activate EREG transcription. EREG treatment promoted HSC activation in vitro, which was blocked by MRTF-A depletion or inhibition. Mechanistically, EREG stimulated nuclear trans-location of MRTF-A in HSCs. Together, our data portray an EREG-MRTF-A feedforward loop that contributes to HSC activation and suggest that targeting the EREG-MRTF-A axis may yield therapeutic solutions against liver fibrosis.
Introduction
Liver fibrosis is a key pathophysiological process taking place in response to various acute and chronic hepatic injuries (Lee et al., 2015). Whereas spatiotemporally controlled liver fibrosis is instrumental to the amelioration of liver injury and restoration of liver function, excessive and/or prolonged liver fibrosis leads to architectural and functional damages to the liver and precipitates the development of such end-stage liver diseases as cirrhosis and hepatocellular carcinoma (Barry et al., 2020). Liver fibrosis can occur following the challenge of a myriad of injurious stimuli including pathogens, toxins, corrosive chemicals, and metabolites. Regardless of the specific triggering factor, myofibroblasts are considered the major effector cell type for liver fibrosis (Kisseleva, 2017). Myofibroblasts possess the characteristics of both muscle cells and fibroblast cells being able to contract and cover the wound and produce and lay down extracellular matrix proteins. The origins from which myofibroblasts arise during liver fibrosis have been a subject matter receiving extensive investigations. Recently lineage fate-mapping experiments have been determined that an overwhelming majority (>90%) of myofibroblasts in the liver are derived from hepatic stellate cells (HSC) that express lecithin retinol acyltransferase (Lrat), a supposedly HSC lineage-specific marker gene (Mederacke et al., 2013). Under physiological settings, quiescent HSCs primarily function as a deposit site for lipids and vitamin A; upon exposure to a pro-fibrogenic microenvironment, HSCs undergo trans-differentiation and become myofibroblasts. In vitro cultured HSCs can also be educated to switch to a myofibroblast-like phenotype by a host of growth factors including transforming growth factor (TGF-β) and platelet-derived growth factor (PDGF) (Hou and Syn, 2018).
Signaling through epidermal growth factor receptor (EGFR) has been shown to contribute to HSC activation and liver fibrosis. Scheving et al. have demonstrated that genetic deletion of EGFR attenuates CCl4 induced liver fibrosis in mice (Scheving et al., 2016). Consistently, pharmaceutical inhibition of EGFR is associated with amelioration of liver fibrosis in different murine models (Fuchs et al., 2014; Liang et al., 2018). Previously, Perugorria et al. have shown the amphiregulin (AR), a ligand for EGFR, plays critical roles in HSC activation and liver fibrosis: AR treatment robustly promotes HSC activation in vitro whereas AR deletion protects the mice from CCl4-induced liver fibrosis (Perugorria et al., 2008). Epiregulin, encoded by EREG, is an EGFR ligand that shares significant homology with amphiregulin (Riese and Cullum, 2014). Whether EREG can contribute to HSC activation remains undetermined.
Mounting evidence suggests that myocardin-related transcription factor A (MRTF-A) plays a pivotal role promoting the differentiation of myofibroblasts in multiple organs (Small, 2012). MRTF-A was initially characterized as a co-factor for serum response factor (SRF) to activate the transcription of muscle-lineage specific genes (Wang et al., 2002). MRTF-A can shuttle between the cytoplasm and the nucleus depending on cytoskeletal reshuffling (Olson and Nordheim, 2010). Previously we have reported that MRTF-A regulates liver fibrosis by transcriptionally programming HSC activation (Fan et al., 2015; Tian et al., 2015, 2016). Here we report that EREG expression is up-regulated during HSC activation both in vivo and in vitro. MRTF-A interacts with SRF to directly bind to the EREG promoter and activate EREG transcription. Reciprocally, EREG contributes to HSC activation by promoting nuclear trans-location of MRTF-A. Therefore, targeting the EREG-MRTF-A axis may yield therapeutic solutions against liver fibrosis.
Materials and Methods
Animals
All animal protocols were reviewed and approved the intramural Ethics Committee on Humane Treatment of Laboratory Animals of Nanjing Medical University. MRTF-A knockout (KO) mice were originally obtained from Steve Morris at St Jude Hospital (Sun et al., 2006). To induce liver fibrosis, MRTF-A KO mice and wild type (WT) littermates were injected with CCl4 (1.0 mL/kg as 50% vol/vol), or injected with thioacetamide (TAA, 100 mg/kg), or subjected to bile duct ligation (BDL) as previously described (Li et al., 2019a, c; Lu et al., 2019).
Cell Culture, Plasmids, and Transient Transfection
Immortalized human HSC (LX-2) were maintained in DMEM supplemented with 10% FBS as previously described (Kong et al., 2019a, b). Primary HSC were isolated and maintained as previously described (Li et al., 2019b). Briefly, the animals were anesthetized by intraperitoneal injection with ketamine-xylazine. A laparotomy was performed and the portal vein was cut to allow retrograde perfusion with pronase (Sigma Aldrich, St. Louis, MO, United States) and collagenase (Roche, Germany) containing solutions. HSCs were isolated from the non-parenchymal fraction by 9.7% Nycodenz gradient centrifugation. Isolated HSCs were seeded in plastic culture dishes and allowed to undergo spontaneous activation. RNA targeting SRF (GAUGGAGUUCAUCGACAACAA) was purchased from Dharmacon. Recombinant TGF-β (100-21) was purchased from Peprotech. Recombinant EREG (1195-EP-025) was purchased from R&D. CCG-1423 (S7719) was purchased from Selleck. Full-length EREG promoter-luciferase construct (−1345/+118) and MRTF-A expression construct have been previously described (Kyotani et al., 2018; Mao et al., 2020b). Truncated and mutated EREG promoter-luciferase constructs were prepared with the QuikChange mutagenesis kit (Agilent). Conditioned media were harvested as previously described (Li et al., 2020a, b). Briefly, the cells were switched to and incubated with serum-free media overnight. The next day, the media were collected, centrifuged at 4,000 × g for 30 min at 4°C using 3-kDa MW cut-off filter units (Millipore) and sterilized through a 0.4-μm filter. Transient transfections were performed with Lipofectamine 2000. Luciferase activities were assayed 24–48 h after transfection using a luciferase reporter assay system (Promega) as previously described (Yang et al., 2019a, b).
Enzyme-Linked Immunosorbent Assay (ELISA)
Secreted epiregulin levels were measured using a commercially available ELISA (LS-F5753, Lifespan Biosciences) per vendor’s recommendations.
Protein Extraction and Western Blot
Whole cell lysates were obtained by re-suspending cell pellets in RIPA buffer (50 mM Tris pH7.4, 150 mMNaCl, 1% Triton X-100) with freshly added protease inhibitor (Roche) as previously described (Fan et al., 2020). Nuclear proteins were extracted using the NE-PER Kit (Pierce) following manufacturer’s recommendation (Mao et al., 2020a). Western blot analyses were performed with anti-MRTF-A (Santa Cruz, sc-32909), anti-SRF (Cell Signaling Technology, 5147), anti-α-tubulin (Sigma, T6074), anti-Lamin A/C (Proteintech, 10298-1), anti-α-SMA (Abcam, ab5694), and anti-β-actin (Sigma, A1978). For densitometrical quantification, densities of target proteins were normalized to those of β-actin as previously described (Lv et al., 2020; Wu et al., 2020). Data are expressed as relative protein levels compared to the control group which is arbitrarily set as 1.
RNA Isolation and Real-Time PCR
RNA was extracted with the RNeasy RNA isolation kit (Qiagen). Reverse transcriptase reactions were performed using a SuperScript First-strand Synthesis System (Invitrogen) as previously described (Zhao et al., 2019; Dong et al., 2020). Real-time PCR reactions were performed on an ABI Prism 7,500 system with the following primers: human EREG, 5′-ACGTGTGGCTCAAGTGTCAA-3′ and 5′-CACTTCACACC TGCAGTAGTTT-3′; mouse Ereg, 5′-TGCTTTGTCTAGGTT CCCACC-3′ and 5′-GGCGGTACAGTTATCCTCGG-3′; human COL1A2, 5′-GTGGCAGTGATGGAAGTGTG-3′ and 5′-AGGA CCAGCGTTACCAACAG-3′; human ACTA2, 5′-CTATGCC TCTGGACGCACAACT-3′ and 5′-CAGATCCAGACGCAT GATGGCA-3′.Ct values of target genes were normalized to the Ct values of housekeekping control gene (18s, 5′-CGCGGTTCTATTTTGTTGGT-3′ and 5′-TCGTCTTCG AAACTCCGACT-3′ for both human and mouse genes) using the ΔΔCt method and expressed as relative mRNA expression levels compared to the control group which is arbitrarily set as 1.
Chromatin Immunoprecipitation
Chromatin Immunoprecipitation (ChIP) assays were performed essentially as described before (Sun et al., 2020). In brief, chromatin in control and treated cells were cross-linked with 1% formaldehyde. Cells were incubated in lysis buffer (150 mMNaCl, 25 mM Tris pH 7.5, 1% Triton X-100, 0.1% SDS, 0.5% deoxycholate) supplemented with protease inhibitor tablet and PMSF. DNA was fragmented into ∼200 bp pieces using a Branson 250 sonicator. Aliquots of lysates containing 200 μg of protein were used for each immunoprecipitation reaction with anti-MRTF-A (Santa Cruz, sc-32909), anti-SRF (Cell Signaling Technology, 5147), or pre-immune IgG. For re-ChIP, immune complexes were eluted with the elution buffer (1% SDS, 100 mM NaCO3), diluted with the re-ChIP buffer (1% Triton X-100, 2 mM EDTA, 150 mMNaCl, 20 mM Tris pH 8.1), and subjected to immunoprecipitation with a second antibody of interest.
Immunofluorescence Microscopy
Immunofluorescence staining was performed as previously described. The cells were fixed with 4% formaldehyde, permeabilized with TBST (0.25% Triton X-100, 150 mM NaCl, 50 mM Tris pH7.4), blocked with 5% BSA, and incubated with indicated primary antibodies overnight. After several washes with PBS, cells were incubated with FITC-labeled secondary antibodies (Jackson) for 30 min. DAPI (Sigma) was added and incubated with cells for 5 min prior to observation. Immunofluorescence was visualized on a co-focal microscope (LSM 710, Zeiss). For each group, at least 10 fields were counted.
Statistical Analysis
One-way ANOVA with post hoc Scheff’e analyses were performed by SPSS software (IBM SPSS v18.0, Chicago, IL, United States). Unless otherwise specified, values of p < 0.05 were considered statistically significant.
Results
EREG Expression Is Up-Regulated in Activated HSCs
Previously it has been shown that amphiregulin (AR), an EGFR ligand closely related to epiregulin (EREG), is activated during HSC trans-differentiation and contributes to liver fibrosis (Perugorria et al., 2008). We asked whether EREG expression levels might be altered during HSC activation. To this end, C57/BL6 mice were injected with CCl4 to induce liver fibrosis (Figure 1A). Picrosirius red staining showed significant liver fibrosis in the CCl4-injected mice compared to the vehicle-injected mice (Figure 1B). Primary HSCs were isolated from the mice with liver fibrosis and from the control mice receiving injection with corn oil. As shown in Figure 1C, expression of α-SMA (Acta2), a myofibroblast marker, was significantly up-regulated, as measured by qPCR, in the activated HSCs compared to the quiescent HSCs; a similar up-regulation of Ereg expression was detected in the HSCs isolated from the fibrotic livers compared to those isolated from the control livers. ELISA measurements confirmed that EREG protein levels were also up-regulated in the activated HSCs compared to the quiescent HSCs (Figure 1D). Next, liver fibrosis was induced in mice by injection with thioacetamide (TAA, Figure 1E). Picrosirius red staining showed significant liver fibrosis in the TAA-injected mice compared to the vehicle-injected mice (Figure 1F). Again, primary HSCs isolated from the fibrotic livers displayed higher levels of Acta2 and Ereg than those isolated from the control livers (Figure 1G). A similar increase in EREG protein levels was detected by ELISA (Figure 1H). Finally, in a third model of liver fibrosis in which the mice were subjected to the BDL surgery (Figure 1I), picrosirius red staining showed significant liver fibrosis in the BDL mice compared to the sham-operated mice (Figure 1J). qPCR (Figure 1K) and ELISA (Figure 1L) assays showed that EREG expression levels were up-regulated during HSC activation in vivo.
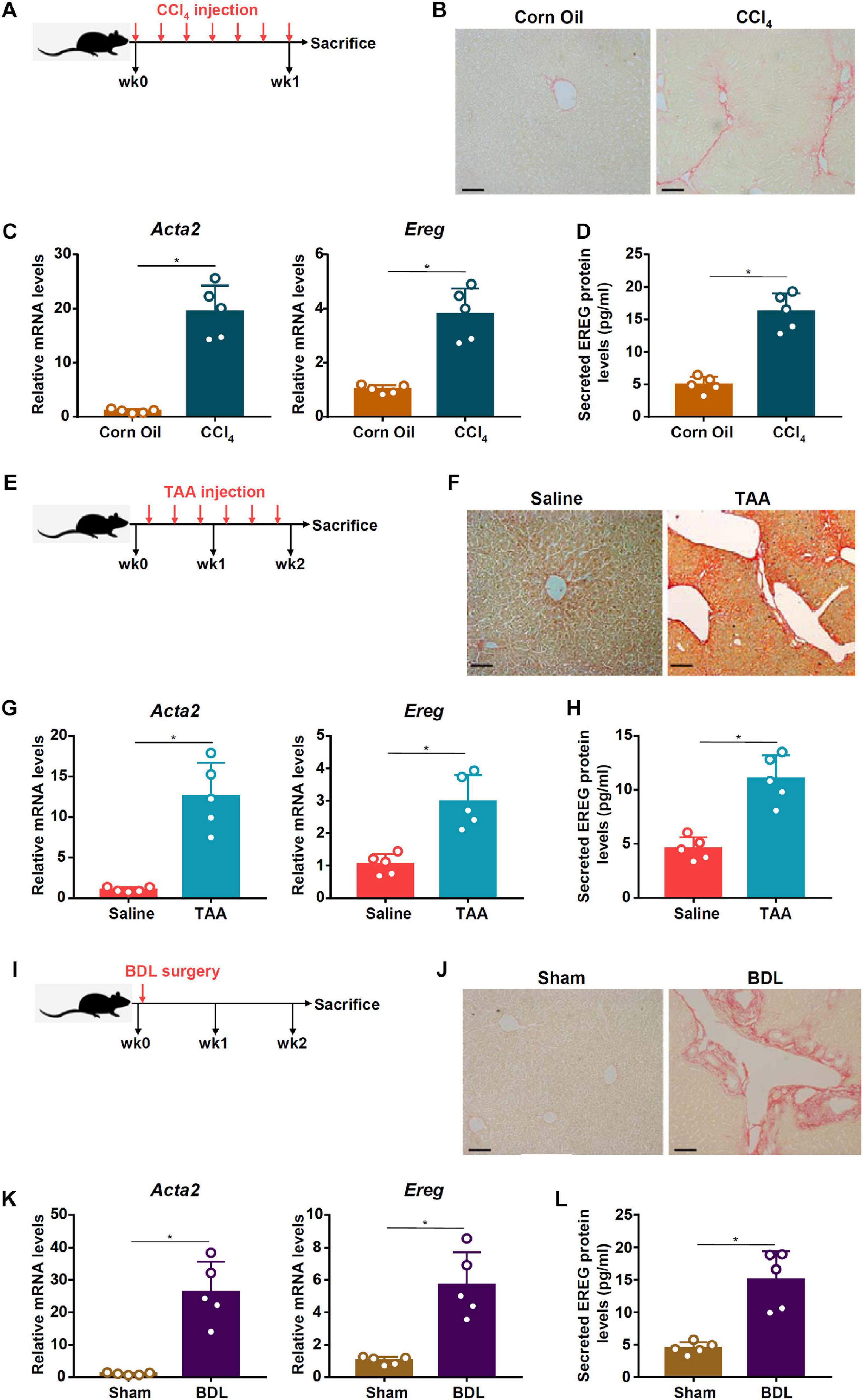
Figure 1. EREG expression is up-regulated in activated HSCs in vivo. (A–D) C57/BL6 mice were injected with CCl4 or corn oil for 7 days. Scheme of protocol (A). Representative images of picrosirius red staining (B). Primary HSCs were isolated from the mice and EREG expression levels were examined by qPCR (C) and ELISA (D). N = 5 mice for each group. (E–H) C57/BL6 mice were injected with TAA or saline for 2 weeks. Scheme of protocol (E). Representative images of picrosirius red staining (F). Primary HSCs were isolated from the mice and EREG expression levels were examined by qPCR (G) and ELISA (H). N = 5 mice for each group. (I–L) C57/BL6 mice were subjected to the BDL procedure or the sham surgery. The mice were sacrificed 2 weeks after the surgery and primary HSCs were isolated. Scheme of protocol (I). Representative images of picrosirius red staining (J). EREG expression levels were examined by qPCR (K) and ELISA (L). N = 5 mice for each group.
We then evaluated the changes in epiregulin expression in cell models of liver fibrosis. In the first model, primary HSCs were isolated from C57/BL mice and allowed to undergo spontaneous activation in vitro. When the cells were harvested at different time points following their isolation, it was observed that Ereg expression was progressively up-regulated mirroring the changes in Acta2 expression (Figures 2A,B). In the second model, LX-2 cells were treated with TGF-β, a well-documented pro-fibrogenic growth factor. Epiregulin expression was significantly up-regulated by TGF-β treatment mirroring the increase in α-SMA expression (Figures 2C,D). Taken together, these data suggest a positive correlation between epiregulin and hepatic stellate cell activation in vivo and in vitro.
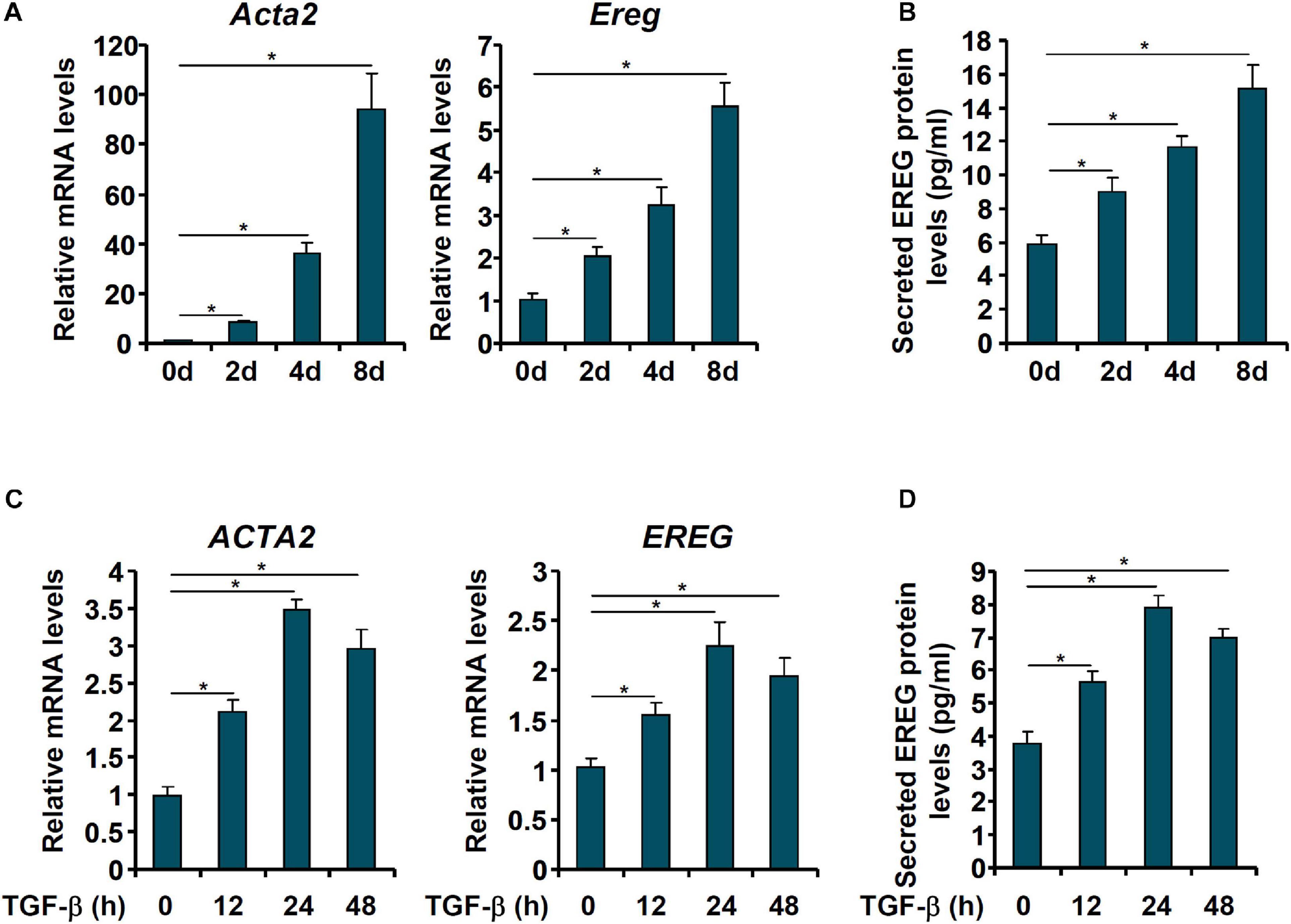
Figure 2. EREG expression is up-regulated in activated HSCs in vitro. (A,B) Primary HSCs were isolated from C57/BL6 mice and underwent spontaneous activation by in vitro culture. The cells were harvested at indicated time points and EREG expression levels were examined by qPCR and ELISA. (C,D) LX-2 cells were treated with or without TGF-β (5 ng/ml) and harvested at indicated time points. EREG expression levels were examined by qPCR and ELISA.
EREG Stimulates Pro-fibrogenic Gene Expression in Hepatic Stellate Cells
Next, we evaluated the effect of epiregulin on pro-fibrogenic gene expression in hepatic stellatecells. Treatment with epiregulin led to a small but appreciable increase in pro-fibrogenic gene expression in LX-2 cells as early as 6 h after the treatment. Induction of pro-fibrogenic genes by epiregulin treatment were detected at 12 and 24 h by qPCR (Figure 3A) and Western blotting (Figure 3B). We also examined the effect of epiregulin treatment on pro-fibrogenic gene expression in spontaneously activated primary HSCs. As shown in Figures 3C,D, the addition of epiregulin augmented the up-regulation of pro-fibrogenic genes as primary HSCs transition from a quiescent state to an activated state. To make a broad point that HSCs can produce and release factors to promote/sustain activation in a feedforward fashion, conditioned media (CM) were harvested from spontaneously activated HSCs to treat quiescent HSCs. Indeed, quiescent HSCs treated with the CM transitioned into an activated state faster than the HSCs cultured in regular media (Supplementary Figure 1). Therefore, it appears that epiregulin may promote the activation of HSCs via an autocrine pathway in vitro.
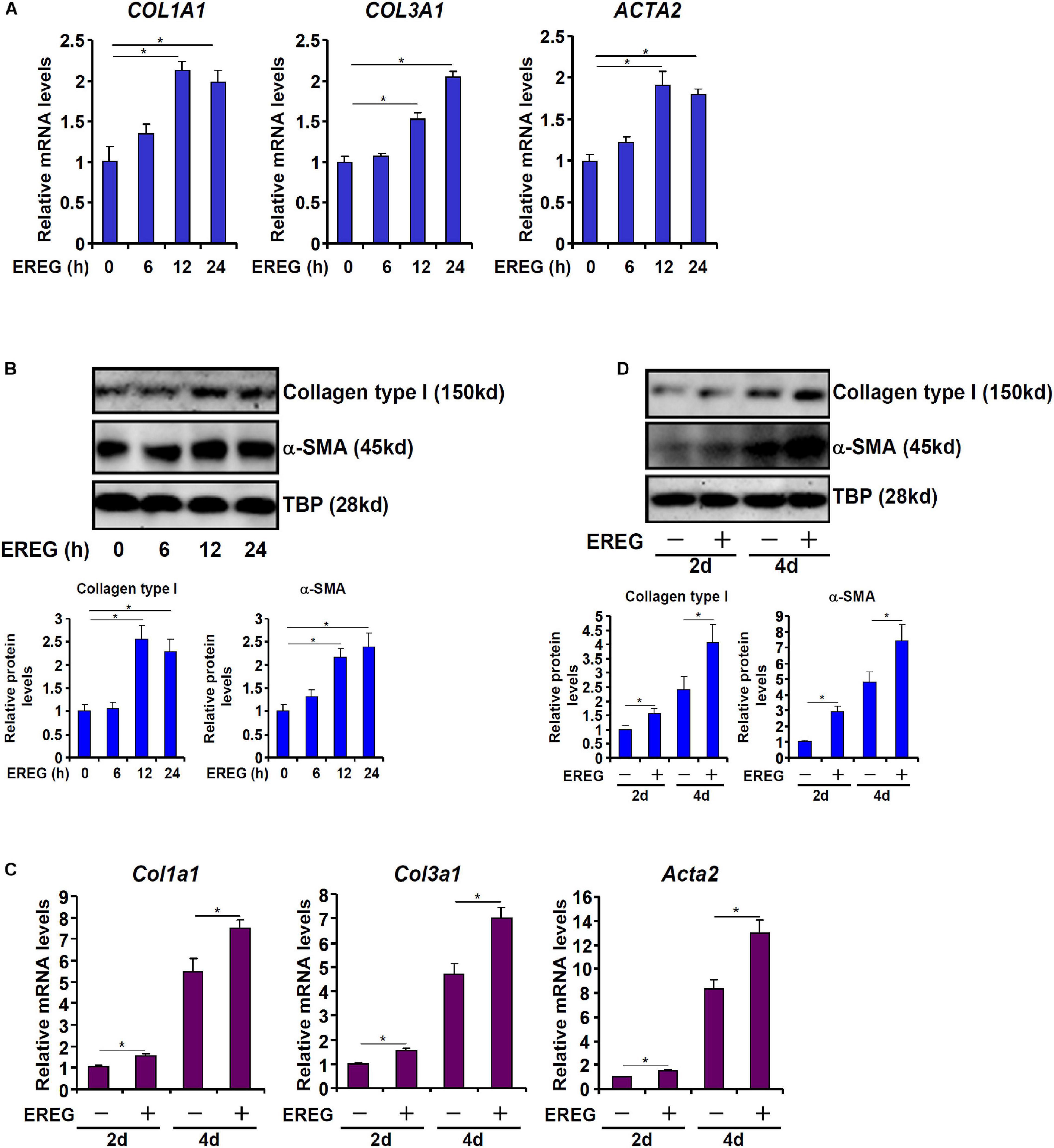
Figure 3. EREG treatment stimulates the expression of fibrogenic genes in HSCs. (A,B) LX-2 cells were treated with or without EREG (5 ng/ml) and harvested at indicated time points. Gene expression levels were examined by qPCR and Western blotting. Quantification was performed with Image Pro based on three independent experiments. (C,D) Primary HSCs were isolated from C57/BL6 mice and underwent spontaneous activation by in vitro culture in the presence or absence of EREG. Gene expression levels were examined by qPCR and Western blotting. Quantification was performed with Image Pro based on three independent experiments.
MRTF-A Deficiency Results in Down-Regulation of EREG Expression in Hepatic Stellate Cells
MRTF-A is key determinant of myofibroblast maturation (Small, 2012). We have previously shown that MRTF-A deletion in mice attenuated liver fibrosis induced by TAA injection (Tian et al., 2016), by CCl4 injection (Tian et al., 2015), or by the BDL procedure (Fan et al., 2015). Consistently, primary HSCs isolated from the MRTF-A KO mice exhibited reduced expression of Acta2 compared to the WT mice following TAA injection (Figure 4A), CCl4 injection (Figure 4C) or the BDL procedure (Figure 4E). Of interest, MRTF-A deficiency comparably decreased epiregulin expression in all three models as measured by qPCR (Figures 4A,C,E) and ELISA (Figures 4B,D,F). In keeping with these observations, induction of epiregulin expression was much more tepid during spontaneous activation of primary HSCs isolated from MRTF-A KO mice than from WT mice (Figures 4G,H). Finally, knockdown of MRTF-A by siRNAs repressed induction of epiregulin expression by TGF-β treatment in LX-2 cells (Figures 4I,J). In contrast, knockdown of MRTF-B, a closely related MRTF-A sibling, did not alter epiregulin expression in either LX-2 cells or primary HSCs (Supplementary Figure 2).
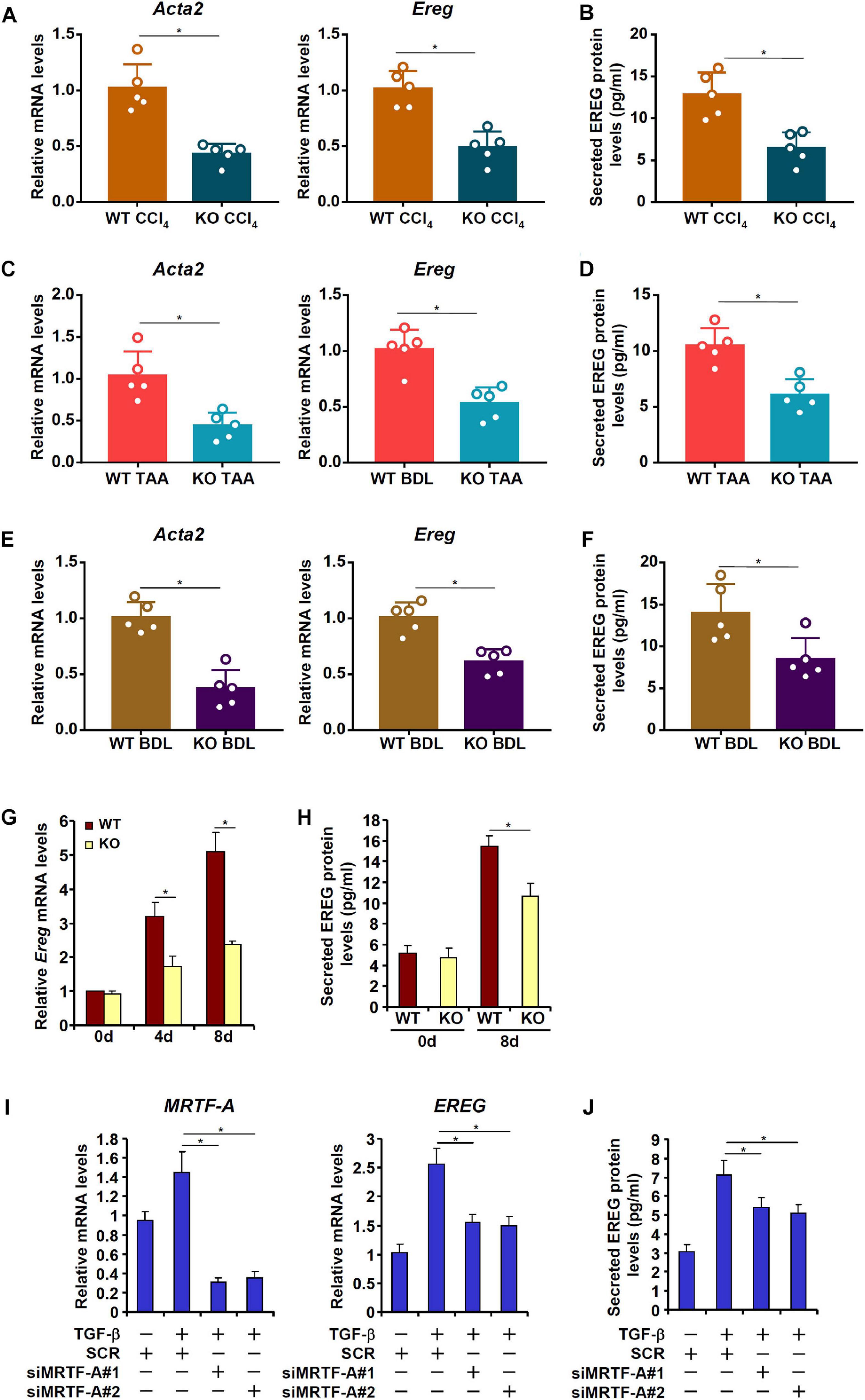
Figure 4. MRTF-A deficiency attenuates EREG activation in HSCs. (A,B) WT and MRTF-A KO mice were injected with CCl4 for 7 days. Primary HSCs were isolated from the mice and EREG expression levels were examined by qPCR and ELISA. N = 5 mice for each group. (C,D) WT and MRTF-A KO mice were injected with TAA for 2 weeks. Primary HSCs were isolated from the mice and EREG expression levels were examined by qPCR and ELISA. N = 5 mice for each group. (E,F) WT and MRTF-A KO mice were subjected to the BDL procedure. The mice were sacrificed 2 weeks after the surgery and primary HSCs were isolated. EREG expression levels were examined by qPCR and ELISA. N = 5 mice for each group. (G,H) Primary HSCs were isolated from WT and MRTF-A KO mice and underwent spontaneous activation for 7 days. EREG expression levels were examined by qPCR and ELISA. (I,J) LX-2 cells were transfected with siRNAs targeting MRTF-A or scrambled siRNAs (SCR) followed by treatment with TGF-β (5 ng/ml) for 24 h. EREG expression levels were examined by qPCR and ELISA.
MRTF-A Activates EREG Transcription by Interacting With SRF
We asked whether MRTF-A might regulate epiregulin expression at the transcriptional level. To test this hypothesis, an EREG promoter-luciferase fusion construct (−1345/ + 118) was transfected into LX-2 cells. Over-expression of MRTF-A dose-dependently up-regulated the EREG promoter activity (Figure 5A). A string of CArG box elements were identified within the EREG promoter (Figure 5A). Inward deletions introduced to the EREG promoter progressively removed the CArG box elements; the removal of the four more distal CArG boxes retained the responsiveness of the EREG promoter to MRTF-A over-expression whereas the removal of the most proximal CArG box rendered the EREG promoter inactive (Figure 5B). Several lines of additional evidence suggest that MRTF-A relies on the proximal CArG box to activate EREG transcription. ChIP assay showed that TGF-β treatment enhanced the association of MRTF-A with the proximal EREG promoter surrounding the innermost CArG box, but not with the intronic region, in LX-2 cells (Figure 5C). Similarly, association of MRTF-A with the Ereg promoter was stronger in activated primary HSCs compared to quiescent primary HSCs (Supplementary Figure 3). Re-ChIP assay confirmed that TGF-β treatment promoted the formation of an SRF-MRTF-A complex on the proximal EREG promoter (Figure 5D). Depletion of SRF with siRNA completely disrupted the binding of MRTF-A to the EREG promoter (Figure 5E). Finally, mutation of the most proximal CArG box abrogated the induction of the EREG promoter by MRTF-A (Figure 5F).
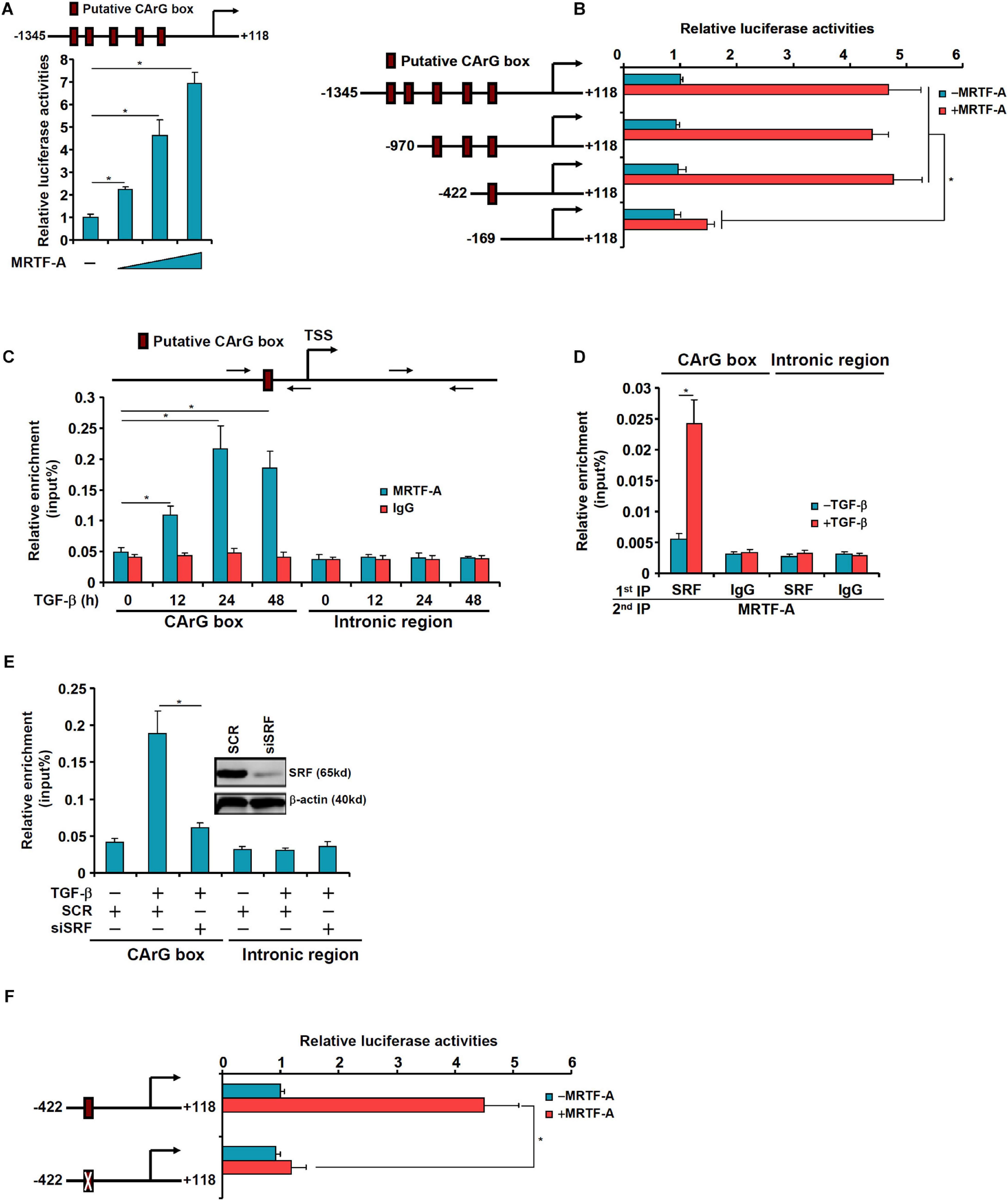
Figure 5. MRTF-A directly activates EREG transcription. (A) A human EREG promoter-luciferase constructs (–1345/ + 118) were transfected into LX-2 cells with or without MRTF-A. Luciferase activities were normalized by protein concentration and GFP fluorescence. (B) Wild type or truncated EREG promoter-luciferase constructs were transfected transfected into LX-2 cells with or without MRTF-A. Luciferase activities were normalized by protein concentration and GFP fluorescence. (C) LX-2 cells were treated with or without TGF-β (5 ng/ml) and harvested at indicated time points. ChIP assays were performed with anti-MRTF-A or IgG. (D) LX-2 cells were treated with or without TGF-β (5 ng/ml) for 24 h. Re-ChIP assay was performed with indicated antibodies. (E) LX-2 cells were transfected with siRNA targeting SRF or scrambled siRNAs (SCR) followed by treatment with TGF-β (5 ng/ml) for 24 h. ChIP assays were performed with anti-MRTF-A. (F) Wild type or CArG mutated EREG promoter-luciferase construct were transfected into LX-2 cells with or without MRTF-A. Luciferase activities were normalized by protein concentration and GFP fluorescence.
EREG Regulates HSC Activation by Promoting Nuclear Trans-Location of MRTF-A
Finally, we asked whether epiregulin could reciprocally influence MRTF-A activity. MRTF-A typically shuttles between the cytoplasm and the nucleus (Olson and Nordheim, 2010). Immunofluorescence staining showed that a large fraction of MRTF-A resides in the cytoplasm with only ∼10% located to the nucleus in LX-2 cells under normal conditions. When exposed to epiregulin treatment, MRTF-A started migrating into the nucleus: at 6 h following epiregulin treatment ∼35% of all MRTF-A proteins whereas at 24 h over 75% of all MRTF-A proteins were detected in the nucleus (Figure 6A). Similar discoveries were made by cell fractionation/Western blotting (Figure 6B). Conversely, induction of pro-fibrogenic genes by epiregulin treatment in LX-2 cells was markedly suppressed by MRTF-A knockdown at both mRNA (Figure 6C) and protein (Figure 6D) levels. Finally, inhibition of MRTF-A activity by CCG-1423 also dampened the up-regulation of pro-fibrogenic genes by epiregulin treatment (Figures 6E,F).
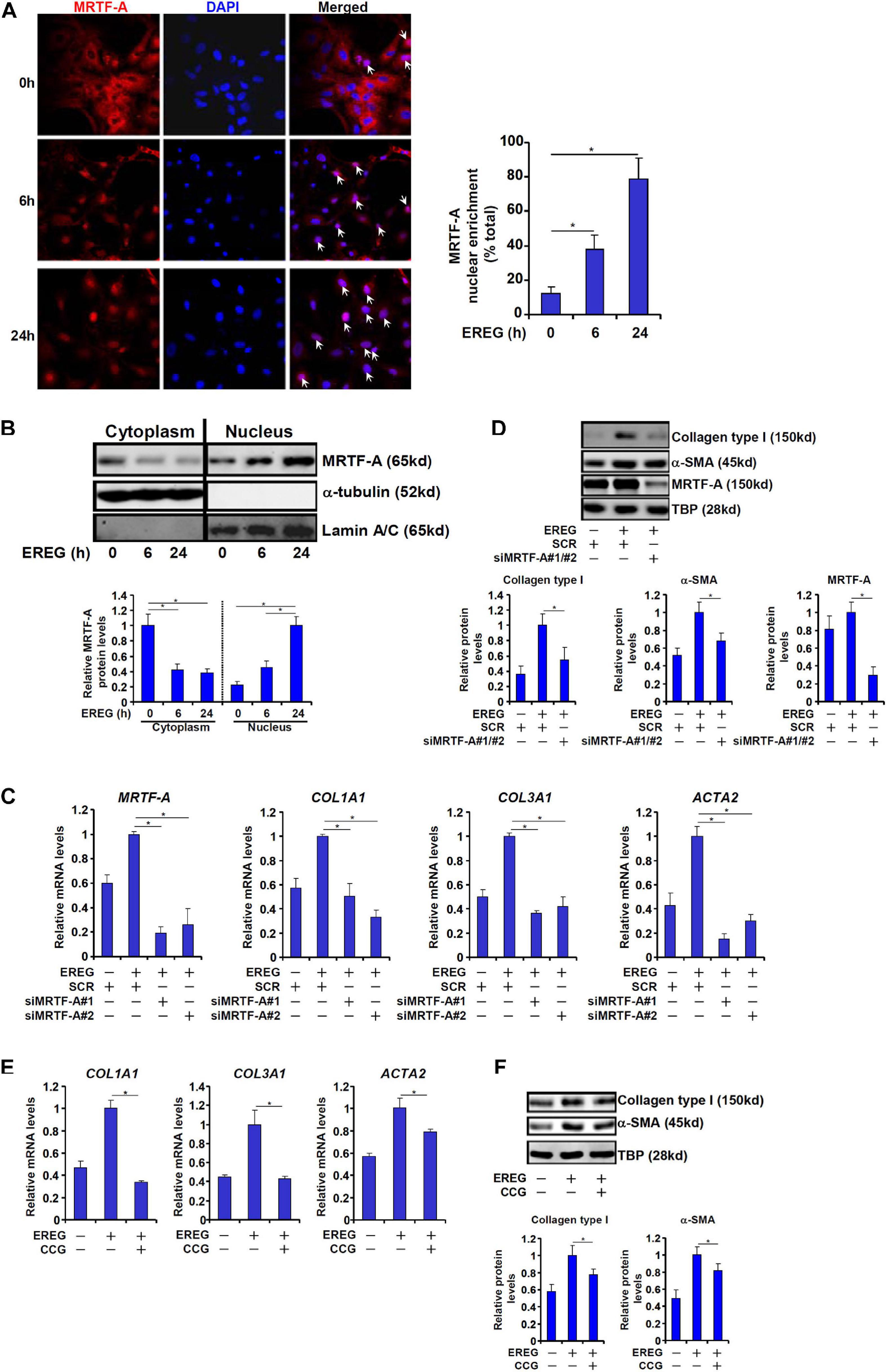
Figure 6. EREG regulates HSC activation through MRTF-A. (A) LX-2 cells were treated with EREG (10 ng/ml) and harvested at indicated time points. MRTF-A localization was examined by immunofluorescence staining. (B) LX-2 cells were treated with EREG (10 ng/ml) and harvested at indicated time points. MRTF-A localization was examined by cell fractionation followed by Western blotting. Quantification was performed with Image Pro based on three independent experiments. (C,D) LX-2 cells were transfected with siRNAs targeting MRTF-A or scrambled siRNA (SCR) treated with recombinant EREG (10 ng/ml) for 24 h. Gene expression was examined by qPCR and Western. Quantification was performed with Image Pro based on three independent experiments. (E,F) LX-2 cells were treated with recombinant EREG (10 ng/ml) in the presence or absence of CCG for 24 h. Gene expression was examined by qPCR and Western. Quantification was performed with Image Pro based on three independent experiments.
Discussion
Activation of HSCs, a hallmark event in liver fibrosis, is programmed by a complex web of signaling cascades that relay the pro-fibrogenic cues to the nucleus, where profound changes in gene expression contribute to the transition of quiescent HSCs to mature myofibroblasts. In the present report, we provide data to show that epiregulin (EREG), belonging to the EGF family of growth factors, is a key regulator of HSC activation. Our data indicate that exposure of cultured HSCs to recombinant epiregulin accelerated the production of pro-fibrogenic genes (Figure 3). This is consistent with previously reported observations that other members of the EGF family including EGF (Lin and Chen, 2008), heparin-bound EGF (HB-EGF) (Takemura et al., 2013), FGF (Wang et al., 2020), and amphiregulin (Perugorria et al., 2008) all contribute to HSC trans-differentiation and liver fibrosis. Of note, all four EGF ligands signal through the same receptor (EGFR) (Riese and Cullum, 2014). Although existing evidence supports the argument that treatment with individual EGF growth factors is sufficient to promote HSC activation at least in vitro, the in vivo requirement/redundancy for each one of these proteins in the development of liver fibrosis is not clear. Deletion of either amphiregulin (AREG) or HB-EGF in mice leads to attenuation of liver fibrosis, suggesting that these growth factors may elicit different signaling cascades and downstream events to promote HSC maturation and fibrogenesis so that the loss of one EGF ligand cannot be fully compensated by other family members. Global EREG-null mice are viable and display no overt gross or liver abnormalities under physiological conditions (Lee et al., 2004). It remains to be ascertained whether these epiregulin deficient mice would phenocopy the AREG–/– mice and the HB-EGF–/– mice in models of liver fibrosis.
We show here that up-regulation of EREG expression during HSC activation in vivo and in vitro is mediated at the transcriptional level by MRTF-A, a well-established pro-fibrogenic molecule (Small, 2012). MRTF-A appears to activate EREG transcription by interacting with SRF and binding to one of the CArG boxes located on the proximal EREG promoter. Of interest, expression of other EGF family members has been shown to be regulated by SRF. For instance, differential regulation of neuregulin 1 (NRG1) in schizophrenia is controlled by several 5′ SNPs that create/abolish binding sites for a string of transcription factors including SRF (Law et al., 2006). On the other hand, SRF can be placed downstream of the EGFR signaling pathway. Augmented SRF activity by EGFR signaling is considered a paradigm in the pathogenesis of multiple cancers (Wee and Wang, 2017). More recently, Stern and colleagues have found that muscular conditional deletion of EGFR, which presumably blocks the signal transduction initiated by EGF, AREG, and EREG, protects the mice from diabetic complications, which is accompanied by changes in gene expression patterns reminiscent of suppressed SRF activity suggesting that EGFR likely regulates muscle cell behavior through stimulating SRF activity (Stern et al., 2020). It would be of great interest to delineate whether a reciprocal regulatory relationship exists between SRF and EREG in the process of HSC activation.
Our data suggest that EREG promotes HSC activation at least in part by inducing MRTF-A nuclear translocation (Figure 6). Sub-cellular localization of MRTF-A is known to be regulated by its post-translational modifications. One of the best characterized modifications of MRTF-A is serine/threonine phosphorylation. The Treisman laboratory has systemically profiled the dynamic alteration of MRTF-A phosphorylation status in response to serum withdrawal/re-addition in fibroblasts uncovering a total of 26 putative S/T residues subjected to phosphorylation (Panayiotou et al., 2016). It is noteworthy that three out of the 26 sites, including S98, T545, and S549, are regulated by EGF stimulation in Hela cells (Olsen et al., 2006). Whereas mutation of T545/S549 that renders the two sites un-phosphorylatable did not impact the cytoplasm-nucleus shuttling of MRTF-A, S98 phosphorylation is critically required for its nuclear accumulation upon serum stimulation (Panayiotou et al., 2016). Thus, it is possible that EREG might promote MRTF-A nuclear translocation by inducing S98 phosphorylation. This hypothesis certainly deserves further investigation.
In summary, we report an epiregulin-MRTF-A feedforward loop that contributes to HSC activation. At least two issues need to be addressed in future studies. First, whether this feedforward loop is relevant in vivo needs to be verified in animal models of liver fibrosis. Second, the mechanism by which epiregulin regulates MRTF-A activity needs to explored in depth. The current data, however, do provide sufficient rationale for designing small-molecule compounds that can blockade this EREG-MRTF-A loop in the intervention of liver fibrosis.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
Ethics Statement
The animal study was reviewed and approved by the Nanjing Medical University Ethics Committee on Humane Treatment of Experimental Animals.
Author Contributions
XS and WZ conceived the project and secured funding and provided the supervision. XW, WD, and TZ designed the experiments. XW, WD, TZ, HR, JW, LS, and ZZ performed experiments and collected the data. YX wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Natural Science Foundation of China (81872359 and 81670566), Jiangsu Province’s Key Provincial Talents Program (ZDRCA2016066), the Nanjing Medical Science and Technique Development Foundation (QRX17129), the Nanjing Health Science and Technology Development Project for Distinguished Young Scholars (JQX19002), the Nanjing Science and Technology Project (201911039), and Innovation and the Entrepreneurship Education Incubation Project of Nanjing University.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2020.591246/full#supplementary-material
References
Barry, A. E., Baldeosingh, R., Lamm, R., Patel, K., Zhang, K., Dominguez, D. A., et al. (2020). Hepatic Stellate Cells and Hepatocarcinogenesis. Front. Cell. Dev. Biol. 8:709. doi: 10.3389/fcell.2020.00709
Dong, W., Kong, M., Zhu, Y., Shao, Y., Wu, D., Lu, J., et al. (2020). Activation of TWIST Transcription by Chromatin Remodeling Protein BRG1 Contributes to Liver Fibrosis in Mice. Front. Cell. Dev. Biol. 8:340. doi: 10.3389/fcell.2020.00340
Fan, Z., Hao, C., Li, M., Dai, X., Qin, H., Li, J., et al. (2015). MKL1 is an epigenetic modulator of TGF-beta induced fibrogenesis. Biochim. Biophys. Acta Gene. Regul. Mech. 1849, 1219–1228. doi: 10.1016/j.bbagrm.2015.07.013
Fan, Z., Kong, M., Li, M., Hong, W., Fan, X., and Xu, Y. (2020). Brahma Related Gene 1 (Brg1) Regulates Cellular Cholesterol Synthesis by Acting as a Co-factor for SREBP2. Front. Cell. Dev. Biol. 8:259. doi: 10.3389/fcell.2020.00259
Fuchs, B. C., Hoshida, Y., Fujii, T., Wei, L., Yamada, S., Lauwers, G. Y., et al. (2014). Epidermal growth factor receptor inhibition attenuates liver fibrosis and development of hepatocellular carcinoma. Hepatology 59, 1577–1590. doi: 10.1002/hep.26898
Hou, W., and Syn, W. K. (2018). Role of Metabolism in Hepatic Stellate Cell Activation and Fibrogenesis. Front. Cell. Dev. Biol. 6:150. doi: 10.3389/fcell.2018.00150
Kisseleva, T. (2017). The origin of fibrogenic myofibroblasts in fibrotic liver. Hepatology 65, 1039–1043. doi: 10.1002/hep.28948
Kong, M., Chen, X., Lv, F., Ren, H., Fan, Z., Qin, H., et al. (2019a). Serum response factor (SRF) promotes ROS generation and hepatic stellate cell activation by epigenetically stimulating NCF1/2 transcription. Redox Biol. 26:101302. doi: 10.1016/j.redox.2019.101302
Kong, M., Hong, W., Shao, Y., Lv, F., Fan, Z., Li, P., et al. (2019b). Ablation of serum response factor in hepatic stellate cells attenuates liver fibrosis. J. Mol. Med. 97, 1521–1533. doi: 10.1007/s00109-019-01831-8
Kyotani, Y., Itaya-Hironaka, A., Yamauchi, A., Sakuramoto-Tsuchida, S., Makino, M., Takasawa, S., et al. (2018). Intermittent hypoxia-induced epiregulin expression by IL-6 production in human coronary artery smooth muscle cells. FEBS Open Bio. 8, 868–876. doi: 10.1002/2211-5463.12430
Law, A. J., Lipska, B. K., Weickert, C. S., Hyde, T. M., Straub, R. E., Hashimoto, R., et al. (2006). Neuregulin 1 transcripts are differentially expressed in schizophrenia and regulated by 5’. SNPs associated with the disease. Proc. Natl. Acad. Sci. U S A. 103, 6747–6752. doi: 10.1073/pnas.0602002103
Lee, D., Pearsall, R. S., Das, S., Dey, S. K., Godfrey, V. L., and Threadgill, D. W. (2004). Epiregulin is not essential for development of intestinal tumors but is required for protection from intestinal damage. Mol. Cell. Biol. 24, 8907–8916. doi: 10.1128/mcb.24.20.8907-8916.2004
Lee, Y. A., Wallace, M. C., and Friedman, S. L. (2015). Pathobiology of liver fibrosis: a translational success story. Gut 64, 830–841. doi: 10.1136/gutjnl-2014-306842
Li, N., Liu, S., Zhang, Y., Yu, L., Hu, Y., Wu, T., et al. (2020a). Transcriptional activation of matricellular protein Spondin2 (SPON2) by BRG1 in vascular endothelial cells promotes macrophage chemotaxis. Front. Cell Dev. Biol. 8:794. doi: 10.3389/fcell.2020.00794
Li, Z., Chen, B., Dong, W., Kong, M., Fan, Z., Yu, L., et al. (2019a). MKL1 promotes endothelial-to-mesenchymal transition and liver fibrosis by activating TWIST1 transcription. Cell Death. Dis. 10:899.
Li, Z., Li, P., Lu, Y., Sun, D., Zhang, X., and Xu, Y. (2019b). A non-autonomous role of MKL1 in the activation of hepatic stellate cells. Biochim. Biophys. Acta Gene. Regul. Mech. 1862, 609–618. doi: 10.1016/j.bbagrm.2019.03.001
Li, Z., Lv, F., Dai, C., Wang, Q., Jiang, C., Fang, M., et al. (2019c). Activation of galectin-3 (LGALS3) transcription by injurious stimuli in the liver is commonly mediated by BRG1. Front. Cell. Dev. Biol. 7:310. doi: 10.3389/fcell.2019.00310
Li, Z., Zhang, Y., Yu, L., Xiao, B., Li, T., Kong, X., et al. (2020b). BRG1 Stimulates Endothelial Derived Alarmin MRP8 to Promote Macrophage Infiltration in an Animal Model of Cardiac Hypertrophy. Front. Cell. Dev. Biol. 8:569. doi: 10.3389/fcell.2020.00569
Liang, D., Chen, H., Zhao, L., Zhang, W., Hu, J., Liu, Z., et al. (2018). Inhibition of EGFR attenuates fibrosis and stellate cell activation in diet-induced model of nonalcoholic fatty liver disease. Biochim. Biophys. Acta Mol. Basis Dis. 1864, 133–142. doi: 10.1016/j.bbadis.2017.10.016
Lin, J., and Chen, A. (2008). Activation of peroxisome proliferator-activated receptor-gamma by curcumin blocks the signaling pathways for PDGF and EGF in hepatic stellate cells. Lab. Invest. 88, 529–540. doi: 10.1038/labinvest.2008.20
Lu, Y., Lv, F., Kong, M., Chen, X., Duan, Y., Sun, D., et al. (2019). A cAbl-MRTF-A Feedback Loop Contributes to Hepatic Stellate Cell Activation. Front. Cell Dev. Biol. 7:243. doi: 10.3389/fcell.2019.00243
Lv, F., Li, N., Kong, M., Wu, J., Miao, D., Ye, Q., et al. (2020). CDKN2a/p16 antagonizes hepatic stellate cell activation and liver fibrosis by modulating ROS levels. Front. Cell. Dev. Biol. 8:176. doi: 10.3389/fcell.2020.00176
Mao, L., Liu, L., Zhang, T., Qin, H., Wu, X., and Xu, Y. (2020a). Histone Deacetylase 11 Contributes to Renal Fibrosis by Repressing KLF15 Transcription. Front. Cell. Dev. Biol. 8:235. doi: 10.3389/fcell.2020.00235
Mao, L., Liu, L., Zhang, T., Wu, X., and Xu, Y. (2020b). MKL1 mediates TGF-beta-induced CTGF transcription to promote renal fibrosis. J. Cell. Physiol. 235, 4790–4803. doi: 10.1002/jcp.29356
Mederacke, I., Hsu, C. C., Troeger, J. S., Huebener, P., Mu, X., Dapito, D. H., et al. (2013). Fate tracing reveals hepatic stellate cells as dominant contributors to liver fibrosis independent of its aetiology. Nat. Commun. 4:2823.
Olsen, J. V., Blagoev, B., Gnad, F., Macek, B., Kumar, C., Mortensen, P., et al. (2006). Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell 127, 635–648. doi: 10.1016/j.cell.2006.09.026
Olson, E. N., and Nordheim, A. (2010). Linking actin dynamics and gene transcription to drive cellular motile functions. Nat. Rev. Mol. Cell Biol. 11, 353–365. doi: 10.1038/nrm2890
Panayiotou, R., Miralles, F., Pawlowski, R., Diring, J., Flynn, H. R., Skehel, M., et al. (2016). Phosphorylation acts positively and negatively to regulate MRTF-A subcellular localisation and activity. Elife 5:e15460.
Perugorria, M. J., Latasa, M. U., Nicou, A., Cartagena-Lirola, H., Castillo, J., Goni, S., et al. (2008). The epidermal growth factor receptor ligand amphiregulin participates in the development of mouse liver fibrosis. Hepatology 48, 1251–1261. doi: 10.1002/hep.22437
Riese, D. J. II, and Cullum, R. L. (2014). Epiregulin: roles in normal physiology and cancer. Semin. Cell Dev. Biol. 28, 49–56. doi: 10.1016/j.semcdb.2014.03.005
Scheving, L. A., Zhang, X., Threadgill, D. W., and Russell, W. E. (2016). Hepatocyte ERBB3 and EGFR are required for maximal CCl4-induced liver fibrosis. Am. J. Physiol. Gastrointest Liver Physiol. 311, G807–G816.
Small, E. M. (2012). The actin-MRTF-SRF gene regulatory axis and myofibroblast differentiation. J. Cardiovasc. Transl. Res. 5, 794–804. doi: 10.1007/s12265-012-9397-0
Stern, C., Schreier, B., Nolze, A., Rabe, S., Mildenberger, S., and Gekle, M. (2020). Knockout of vascular smooth muscle EGF receptor in a mouse model prevents obesity-induced vascular dysfunction and renal damage in vivo. Diabetologia 63, 2218–2234. doi: 10.1007/s00125-020-05187-4
Sun, L., Chen, B., Wu, J., Jiang, C., Fan, Z., Feng, Y., et al. (2020). Epigenetic regulation of a disintegrin and metalloproteinase (ADAM) promotes colorectal cancer cell migration and invasion. Front. Cell Dev. Biol. 8:581692. doi: 10.3389/fcell.2020.581692
Sun, Y., Boyd, K., Xu, W., Ma, J., Jackson, C. W., Fu, A., et al. (2006). Acute myeloid leukemia-associated Mkl1 (Mrtf-a) is a key regulator of mammary gland function. Mol. Cell. Biol. 26, 5809–5826. doi: 10.1128/mcb.00024-06
Takemura, T., Yoshida, Y., Kiso, S., Kizu, T., Furuta, K., Ezaki, H., et al. (2013). Conditional loss of heparin-binding EGF-like growth factor results in enhanced liver fibrosis after bile duct ligation in mice. Biochem. Biophys. Res. Commun. 437, 185–191. doi: 10.1016/j.bbrc.2013.05.097
Tian, W., Fan, Z., Li, J., Hao, C., Li, M., Xu, H., et al. (2016). Myocardin-related transcription factor A (MRTF-A) plays an essential role in hepatic stellate cell activation by epigenetically modulating TGF-beta signaling. Int. J. Biochem. Cell Biol. 71, 35–43. doi: 10.1016/j.biocel.2015.12.005
Tian, W., Hao, C., Fan, Z., Weng, X., Qin, H., Wu, X., et al. (2015). Myocardin related transcription factor A programs epigenetic activation of hepatic stellate cells. J. Hepatol. 62, 165–174. doi: 10.1016/j.jhep.2014.07.029
Wang, C., Li, Y., Li, H., Zhang, Y., Ying, Z., Wang, X., et al. (2020). Disruption of FGF Signaling Ameliorates Inflammatory Response in Hepatic Stellate Cells. Front. Cell. Dev. Biol. 8:601. doi: 10.3389/fcell.2020.00601
Wang, D. Z., Li, S., Hockemeyer, D., Sutherland, L., Wang, Z., Schratt, G., et al. (2002). Potentiation of serum response factor activity by a family of myocardin-related transcription factors. Proc. Natl. Acad. Sci. U S A. 99, 14855–14860. doi: 10.1073/pnas.222561499
Wee, P., and Wang, Z. (2017). Epidermal Growth Factor Receptor Cell Proliferation Signaling Pathways. Cancers 9:52. doi: 10.3390/cancers9050052
Wu, T., Wang, H., Xin, X., Yang, J., Hou, Y., Fang, M., et al. (2020). An MRTF-A-Sp1-PDE5 Axis Mediates Angiotensin-II-Induced Cardiomyocyte Hypertrophy. Front. Cell Dev. Biol. 8:839. doi: 10.3389/fcell.2020.00839
Yang, Y., Liu, L., Fang, M., Bai, H., and Xu, Y. (2019a). The chromatin remodeling protein BRM regulates the transcription of tight junction proteins: Implication in breast cancer metastasis. Biochim. Biophys. Acta Gene. Regul. Mech. 1862, 547–556. doi: 10.1016/j.bbagrm.2019.03.002
Yang, Y., Liu, L., Li, M., Cheng, X., Fang, M., Zeng, Q., et al. (2019b). The chromatin remodeling protein BRG1 links ELOVL3 trans-activation to prostate cancer metastasis. Biochim. Biophys. Acta. Gene. Regul. Mech. 1862, 834–845. doi: 10.1016/j.bbagrm.2019.05.005
Keywords: transcription regulation, hepatic stellate cell, liver fibrosis, epiregulin, MRTF-A, SRF
Citation: Wu X, Dong W, Zhang T, Ren H, Wang J, Shang L, Zhu Z, Zhu W, Shi X and Xu Y (2021) Epiregulin (EREG) and Myocardin Related Transcription Factor A (MRTF-A) Form a Feedforward Loop to Drive Hepatic Stellate Cell Activation. Front. Cell Dev. Biol. 8:591246. doi: 10.3389/fcell.2020.591246
Received: 05 August 2020; Accepted: 28 October 2020;
Published: 15 January 2021.
Edited by:
Pedro M. Fernández-Salguero, University of Extremadura, SpainReviewed by:
Manoj B. Menon, Indian Institute of Technology Delhi, IndiaDae-Sik Lim, Korea Advanced Institute of Science and Technology, South Korea
Copyright © 2021 Wu, Dong, Zhang, Ren, Wang, Shang, Zhu, Zhu, Shi and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaolei Shi, c3hsQG5qdS5lZHUuY24=; Wei Zhu, Y2h6aHV3ZWkxMThAcXEuY29t
†These authors have contributed equally to this work