 Sarah Delbaere1
Sarah Delbaere1 Adelbert De Clercq1
Adelbert De Clercq1 Shuji Mizumoto2
Shuji Mizumoto2 Fredrik Noborn3,4
Fredrik Noborn3,4 Jan Willem Bek1
Jan Willem Bek1 Lien Alluyn1
Lien Alluyn1 Charlotte Gistelinck5
Charlotte Gistelinck5 Delfien Syx1
Delfien Syx1 Phil L. Salmon6
Phil L. Salmon6 Paul J. Coucke1
Paul J. Coucke1 Göran Larson3,4
Göran Larson3,4 Shuhei Yamada2
Shuhei Yamada2 Andy Willaert1†
Andy Willaert1† Fransiska Malfait1*†
Fransiska Malfait1*†- 1Department of Biomolecular Medicine, Center for Medical Genetics Ghent, Ghent University Hospital, Ghent University, Ghent, Belgium
- 2Department of Pathobiochemistry, Faculty of Pharmacy, Meijo University, Nagoya, Japan
- 3Department of Laboratory Medicine, Sahlgrenska Academy at the University of Gothenburg, Gothenburg, Sweden
- 4Laboratory of Clinical Chemistry, Sahlgrenska University Hospital, Gothenburg, Sweden
- 5Department of Orthopaedics and Sports Medicine, University of Washington, Seattle, WA, United States
- 6Bruker MicroCT, Kontich, Belgium
Proteoglycans are structurally and functionally diverse biomacromolecules found abundantly on cell membranes and in the extracellular matrix. They consist of a core protein linked to glycosaminoglycan chains via a tetrasaccharide linkage region. Here, we show that CRISPR/Cas9-mediated b3galt6 knock-out zebrafish, lacking galactosyltransferase II, which adds the third sugar in the linkage region, largely recapitulate the phenotypic abnormalities seen in human β3GalT6-deficiency disorders. These comprise craniofacial dysmorphism, generalized skeletal dysplasia, skin involvement and indications for muscle hypotonia. In-depth TEM analysis revealed disturbed collagen fibril organization as the most consistent ultrastructural characteristic throughout different affected tissues. Strikingly, despite a strong reduction in glycosaminoglycan content, as demonstrated by anion-exchange HPLC, subsequent LC-MS/MS analysis revealed a small amount of proteoglycans containing a unique linkage region consisting of only three sugars. This implies that formation of glycosaminoglycans with an immature linkage region is possible in a pathogenic context. Our study, therefore unveils a novel rescue mechanism for proteoglycan production in the absence of galactosyltransferase II, hereby opening new avenues for therapeutic intervention.
Introduction
Proteoglycans (PGs) comprise a group of complex biomacromolecules, consisting of a core protein and one or multiple glycosaminoglycan (GAG) side chains, that are ubiquitously expressed in the extracellular matrix (ECM) and on cell surfaces and basement membranes in vertebrate and invertebrate species (Prydz and Dalen, 2000; Selleck, 2000; Couchman and Pataki, 2012; Lindahl et al., 2015). Besides their structural role, PGs act as mediators between the ECM and intracellular signaling pathways and contribute to development and tissue homeostasis, influencing a variety of cellular processes including cell fate determination, cell proliferation, migration, adhesion, differentiation, and survival (Malfait et al., 2013).
The PG superfamily is, depending on the composition of the GAG chains, subdivided into heparan sulfate (HS) and chondroitin/dermatan sulfate (CS/DS) PGs. These GAG chains are linear polysaccharides, composed of a repeated disaccharide unit consisting of an uronic acid and amino sugar. HSPGs, such as perlecan, syndecan and glypican, consist of repeating glucuronic acid (GlcA)-N-acetyl-glucosamine (GlcNAc) disaccharides. CSPGs consist of repeating GlcA-N-acetyl-galactosamine (GalNAc) disaccharides. DS is a stereoisomer of CS, in which GlcA is epimerized to iduronic acid (IdoA). In mammalian tissues, CS and DS chains are often present as hybrid CS-DS chains on PGs (e.g. decorin, biglycan) (Sugahara et al., 2003). Tightly controlled modifications, such as epimerization and sulfation reactions, further increase the structural and functional diversity of the HS and CS/DS PGs (Kusche-Gullberg and Kjellen, 2003; Bui et al., 2010). GAG biosynthesis of both HSPGs and CS/DSPGs is initiated in the endoplasmic reticulum and Golgi apparatus by the formation of a common tetrasaccharide linkage region (glucuronic acid-galactose-galactose-xylose-O-) (GlcA-Gal-Gal-Xyl-O-) via O-glycosylation of a serine residue of the core protein (Figure 1A; Prydz and Dalen, 2000; Couchman and Pataki, 2012; Lindahl et al., 2015). The first step in the biosynthesis of this tetrasaccharide linkage region is catalyzed by xylosyltransferases (XylTs) encoded by the paralogs XYLT1 [MIM 608124] and XYLT2 [MIM 608125] (Gotting et al., 2000). Two Gal residues are subsequently added by, respectively, galactosyltransferase I (GalT-I or β4GalT7 encoded by B4GALT7 [MIM 604327]) and galactosyltransferase II (GalT-II or β3GalT6 encoded by B3GALT6 [MIM 615291]) (Almeida et al., 1999; Bai et al., 2001). Further addition of a GlcA by glucuronosyltransferase I (GlcAT-I encoded by B3GAT3) [MIM 606374] completes the formation of the linkage region (Kitagawa et al., 1998).
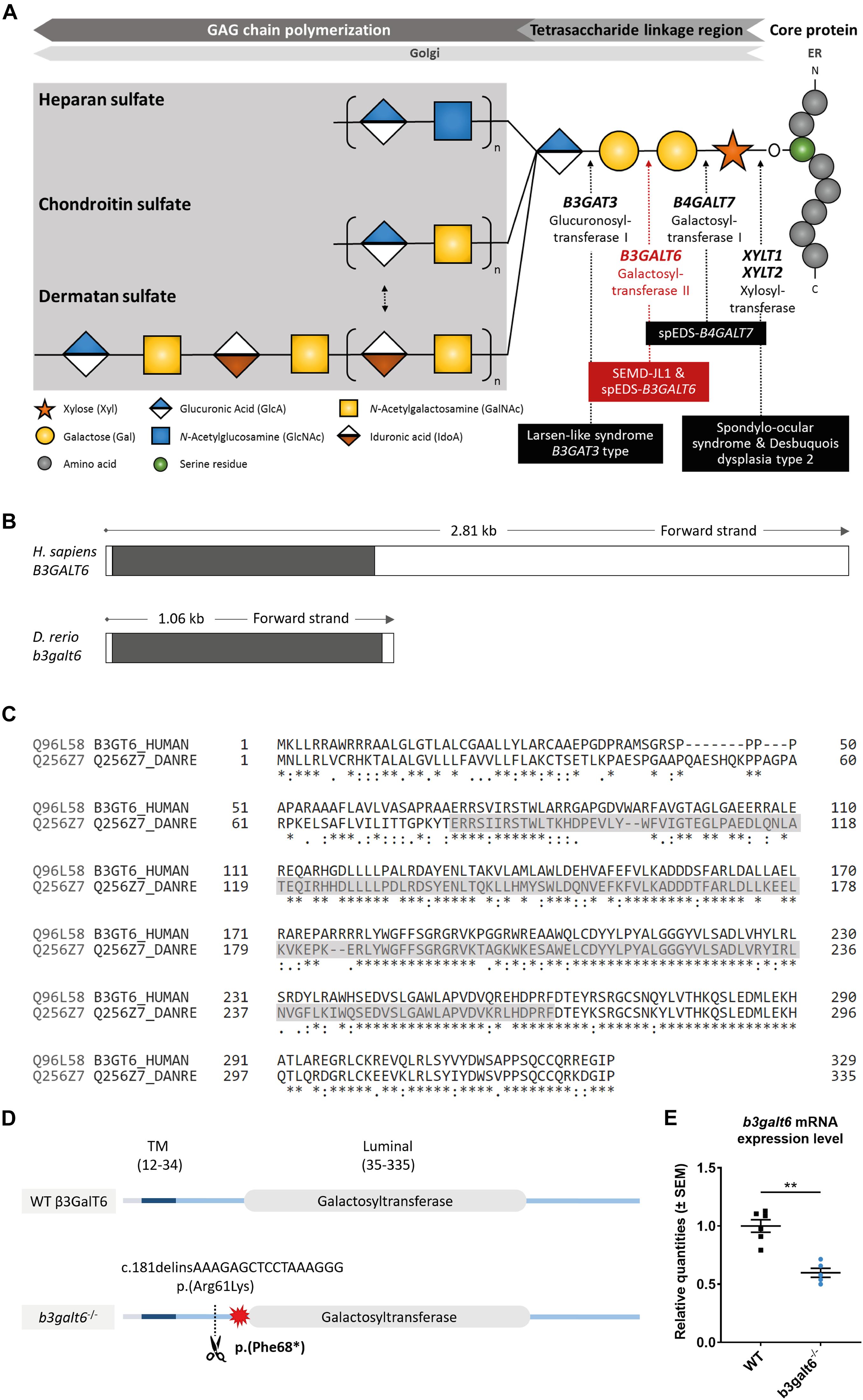
Figure 1. Schematic representation of the GAG biosynthesis and the evolutionary conservation of the zebrafish and human b3galt6/B3GALT6 gene and B3galt6/β3GalT6 protein. (A) Simplified schematic representation of the PG linkage and GAGs biosynthesis. PG core proteins are synthesized in the endoplasmic reticulum and undergo further modification in the Golgi apparatus. First, a tetrasaccharide linkage region is synthesized that originates from the addition of a Xyl onto a serine residue of the core protein. Subsequent addition of two Gals and one GlcA residues completes this process. Depending on the addition of GlcNAc or GalNAc to the terminal GlcA residue of the linkage region, HS or CS/DS PGs will be formed, respectively. The elongation of the GAG chain continues by repeated addition of uronic acid and amino sugar residues. The GAG chain is then further modified by epimerization and sulfation (not shown). The main genes encoding for the enzymes involved in the linkage region biosynthesis are indicated in bold and biallelic mutations in these genes result in different linkeropathies. Disorders resulting from deficiency of the enzymes are indicated in black boxes. The deficient enzyme (β3GalT6) and the related disorder studied here are depicted in red (e.g., spEDS-B3GALT6 and SEMD-JL1). (B) Schematic representation of the genomic structure of the H. sapiens B3GALT6 (NM_080605.4) and D. rerio b3galt6 (NM_001045225.1) genes. The white and black boxes represent the untranslated regions and the single B3GALT6/b3galt6 exon, respectively. The zebrafish b3galt6 gene resides on chromosome 11, spans a region of 1.06 kb and has not been duplicated during teleost evolution. (C) Amino acid (AA) sequence alignment (Clustal Omega) between H. sapiens β3GalT6 (Uniprot: Q96L58) and D. rerio B3galt6 (Uniprot: Q256Z7). The luminal galactosyltransferase domain is highlighted in gray. Conservation of AA sequences is shown below the alignment: “*” residues identical in all sequences in the alignment; “:” conserved substitutions; “.” semi-conserved substitutions; “space” no conservation. (D) Schematic representation of the cmg20 allele in b3galt6 mutants. b3galt6 consists of three parts, a cytosolic domain (AA 1-11), a transmembrane domain (TM, AA 12-34) and a luminal domain (AA 35-335). The location of the CRISPR/Cas9-generated indel variant is indicated by a scissor symbol and the resulting premature termination codon (PTC) is depicted by a red star. (E) RT-qPCR analysis of relative b3galt6 mRNA expression levels in b3galt6–/– adult zebrafish (0.597 ± 0.03919) (n = 5) compared to WT siblings (1 ± 0.05395) (n = 6). Data are expressed as mean ± SEM. A Mann-Whitney U test was used to determine significance, **P < 0.01.
The cellular machinery required for GAG biosynthesis and modification is conserved over a broad range of eukaryotic organisms (Wen et al., 2014). The particular importance of a correct initiation of GAG synthesis is underscored by the identification of a series of severe, overlapping and multisystemic genetic human disorders that result from deficient activity of any of these “linkage-enzymes,” collectively coined as “linkeropathies” (Guo et al., 2013; Nakajima et al., 2013; Jones et al., 2015). The most frequently reported linkeropathy results from galactosyltransferase II deficiency, caused by biallelic pathogenic variants in B3GALT6. This leads to two clinically overlapping conditions, spondylodysplastic Ehlers-Danlos syndrome (EDS) (spEDS-B3GALT6) and spondyloepimetaphyseal dysplasia with joint laxity type I (SEMD-JL1) (Malfait et al., 2013; Nakajima et al., 2013), both of which are characterized by variable degrees of spondyloepimetaphyseal bone dysplasia (with postnatal growth restriction, bowing of long bones, hypo- or dysplasia of iliac and long bones, and vertebral body changes), kyphoscoliosis, bone fragility with spontaneous fractures, joint hypermobility in conjunction with joint contractures, hypotonia with delayed motor development, skin fragility, and craniofacial dysmorphisms, including midfacial hypoplasia and dysplastic teeth (Malfait et al., 2013, 2017; Nakajima et al., 2013). The severe and pleiotropic phenotype suggests a critical role for B3GALT6 in the development and homeostasis of various connective tissues. Pathogenic variants in B3GALT6 either result in mislocalization and/or reduced amounts of active galactosyltransferase II (Ritelli et al., 2015). Previous studies on several biallelic B3GALT6 variants showed that they resulted in a barely detectable in vitro galactosyltransferase II enzymatic activity, but intriguingly HS and CS/DS GAG synthesis was partially preserved in cellulo (Van Damme et al., 2018). While some variants may have a greater deleterious effect on the in vitro than on the in cellulo enzymatic activity, it is currently unknown whether there are rescue mechanisms at play to partially compensate for the galactosyltransferase II activity.
In recent years, zebrafish have emerged as an excellent tool to model disease and to increase our understanding of developmental processes and disease mechanisms. Zebrafish share 70% of their genes with humans (Santoriello and Zon, 2012) and the advantages of their use are reflected by their large breed size, short lifecycle and low husbandry costs (Dooley and Zon, 2000). Zebrafish has proven to be a relevant genetic model for the study of PG biosynthesis since many enzymes involved in this process are strongly conserved between human and zebrafish (Eames et al., 2010), and a number of zebrafish models for defective PG synthesis (including fam20bb1125, xylt1b1189, or b3gat3hi307), provided novel knowledge on PG function in bone and cartilage formation (Amsterdam et al., 2004; Nissen et al., 2006; Eames et al., 2010, 2011).
In this study, we generated b3galt6 knock-out (KO) zebrafish as a model for spEDS-B3GALT6 and SEMD-JL1. These models were used to investigate the structure and amount of different GAG disaccharides and the PG linkage region composition in these disorders. Furthermore, in-depth phenotypic characterization of b3galt6–/– zebrafish was done to reveal to which extent key features of the human disorders are recapitulated and to further elucidate the function of galactosyltransferase II in vertebrate musculoskeletal development.
Results
CRISPR/Cas9-Mediated b3galt6 Knock-Out in Zebrafish Leads to Reduced HS and CS/DS Glycosaminoglycan Concentrations in Bone, Muscle, and Skin
Similar to its human ortholog, zebrafish b3galt6 (NM_001045225.1) is a single-exon gene (Figure 1B). Human and zebrafish galactosyltransferase II (β3GalT6/B3galt6) protein sequence display 82.5% amino acid (AA) similarity, with the highest AA conservation in the luminal domain (Figure 1C).
In order to investigate the consequences of zebrafish B3galt6-deficiency on GAG biosynthesis, we generated two b3galt6 KO zebrafish models using CRISPR/Cas9 gene editing. The first KO model carries the c.181delinsAAAGAGCTCCTAAAGGG indel mutation (cmg20 allele), leading to a premature termination codon (PTC) [p.(Phe68∗)], located upstream to the catalytic domain (Figure 1D). RT-qPCR showed a 40.3% reduction of b3galt6 mRNA expression in homozygous adult b3galt6 KO zebrafish compared to wild-type (WT) siblings (Figure 1E). For the remainder of this manuscript, this model will be referred to as b3galt6–/–. The second KO model carries a four base pair deletion (c.398_401del) (cmg22 allele), predicted to generate a PTC [p.(Asn139∗)] in the catalytic domain (NM_001045225.1). This mutant was solely used for validation purposes (Supplementary Figure S1). Both b3galt6 KO zebrafish were produced at an expected Mendelian ratio and survived into adulthood. Unfortunately, commercial antibodies, which were only available against human β3GalT6, did not work in our hands and precluded the investigation of zebrafish B3galt6 protein levels and localization.
To examine the activity of B3galt6 in different b3galt6–/– tissues, concentrations of CS, DS, and HS disaccharides extracted from bone, muscle and skin from WT and b3galt6–/– zebrafish were quantified using a combination of enzymatic digestion and anion-exchange HPLC (Supplementary Figures S2–S5 and Supplementary Tables S1–S4). Overall, a generalized decrease in HS, CS and DS concentration was detected in all examined tissues of b3galt6–/– zebrafish. In the GAG fractions prepared from bone, the disaccharide concentration of the CS/DS and CS moieties was significantly decreased by, respectively, 55 and 64%, compared to WT zebrafish (Table 1 and Supplementary Tables S1, S2). DS moieties were undetectable in bone of b3galt6–/– zebrafish, whereas their concentration was 50.5 pmol/mg protein in bone of WT zebrafish (Table 1 and Supplementary Table S3). A non-significant decrease in the disaccharide concentration of the HS moiety was observed in b3galt6–/– mutant bone (Table 1 and Supplementary Table S4), compared to WT. In GAG fractions prepared from b3galt6–/– muscle, CS/DS, CS, and DS disaccharide moieties were generally lower than in WT muscle (Table 1 and Supplementary Tables S1–S3). Due to the low concentrations close to the detection limit, a significant reduction could only be detected for the DS moiety (72% reduction) (Table 1 and Supplementary Table S3). In addition, a significant decrease in HS disaccharide moieties by 56% was noted in b3galt6–/– muscle compared to WT muscle (Table 1 and Supplementary Table S4). In the GAG fractions prepared from b3galt6–/– skin, the concentration of all disaccharide moieties was significantly reduced compared to WT (Table 1 and Supplementary Tables S1–S4). The disaccharide concentration of the CS/DS, CS, DS and HS moieties were decreased by, respectively, 77, 73, 55, and 62% compared to WT (Table 1 and Supplementary Tables S1–S4). No altered sulfation patterns were observed in disaccharide moieties when comparing unique GAG chains between WT and b3galt6–/– zebrafish samples (Supplementary Tables S1–S4).
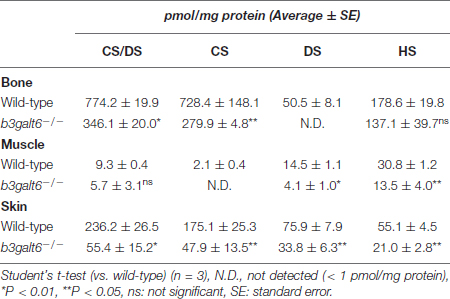
Table 1. Concentrations of GAG disaccharides in wild-type and b3galt6–/– zebrafish tissues.
Despite the strong reduction in disaccharide concentrations observed in b3galt6–/– zebrafish, low amounts of GAGs were still produced in the mutants. Similar results were obtained for the second b3galt6 KO zebrafish (cmg22 allele) (Supplementary Table S5). RT-qPCR revealed no significant upregulation of other galactosyltransferase(s) member(s) from the b3galt family (b3galt1a, b3galt2, b3galt4, b3galnt2) or of other linkage-enzymes (xylt1, xylt2, b4galt7 and b3gat3) (Supplementary Figure S6), suggesting that altered expression of these genes did not compensate for the reduced b3galt6 mRNA levels.
B3galt6-Deficiency Leads to the Biosynthesis of a Non-canonical (GlcA-Gal-Xyl-O-) Trisaccharide Linkage Region in Zebrafish
In order to explain why b3galt6–/– zebrafish can still produce GAG chains on PG core proteins, we investigated the composition of the linkage region. To this purpose, whole zebrafish extracts were enriched for all PGs, their GAG chains depolymerized with bacterial lyases, the core proteins digested with trypsin and the remaining glycopeptides subjected to nano-scale liquid chromatography-tandem mass spectrometry (nLC-MS/MS) using a recently developed glycoproteomic approach (Noborn et al., 2015; Persson et al., 2019). Potential structural differences in PG linkage regions in WT versus b3galt6–/– zebrafish are thus elucidated through the combined identities of peptide backbones and GAG linkage region structures. The glycoproteomic analysis identified two CS-glycopeptides derived from biglycan (UniProt: A8BBH0) in both the WT and b3galt6–/– zebrafish. These CS-glycopeptides displayed different linkage region structures in WT compared with b3galt6–/– zebrafish (Figure 2). While the peptide in the WT zebrafish was modified with a hexasaccharide structure (Figure 2A), the peptide in the b3galt6–/– zebrafish was modified with a pentasaccharide structure (Figure 2B), supporting the hypothesis that b3galt6–/– zebrafish produces a non-canonical trisaccharide linkage region. The precursor ion (m/z 1281.7922; 3+) in the WT zebrafish equated to a mass of a peptide (DQEEGSAVEPYKPEHPTCPFGCR) modified with a hexasaccharide structure, with the peptide containing two carbamidomethyl (CAM) modifications. Furthermore, the hexasaccharide structure was modified with one sulfate group (SO) on the subterminal GalNAc residue [GlcAGalNAc-H2O + SO3 + H]+, and one phosphate group (HPO) on the Xyl residue (Supplementary Figure S7). In contrast, the precursor ion (m/z 1227.7765; 3+) in the b3galt6–/– zebrafish equated to a mass of the same peptide but modified with a pentasaccharide structure, with two phosphate/sulfate groups, with the peptide containing two CAM modifications. The difference in mass between the WT and b3galt6–/– precursor ions corresponds to 162.08 Da, equating to the mass of a hexose (galactose) residue (162 Da). Additional HS- and CS-glycopeptides with the expected tetrasaccharide linkage region were found in the WT. In the b3galt6–/–, no HS-modified structures were identified but additional CS-glycopeptides with the non-canonical trisaccharide linkage region were found. Supplementary Table S6 shows a complete list of all glycopeptides identified in both WT and b3galt6–/– zebrafish. PGs harboring a tetrasaccharide linkage region were never detected in the b3galt6–/– adult zebrafish samples and no trisaccharide linkage region was detected in WT zebrafish samples.
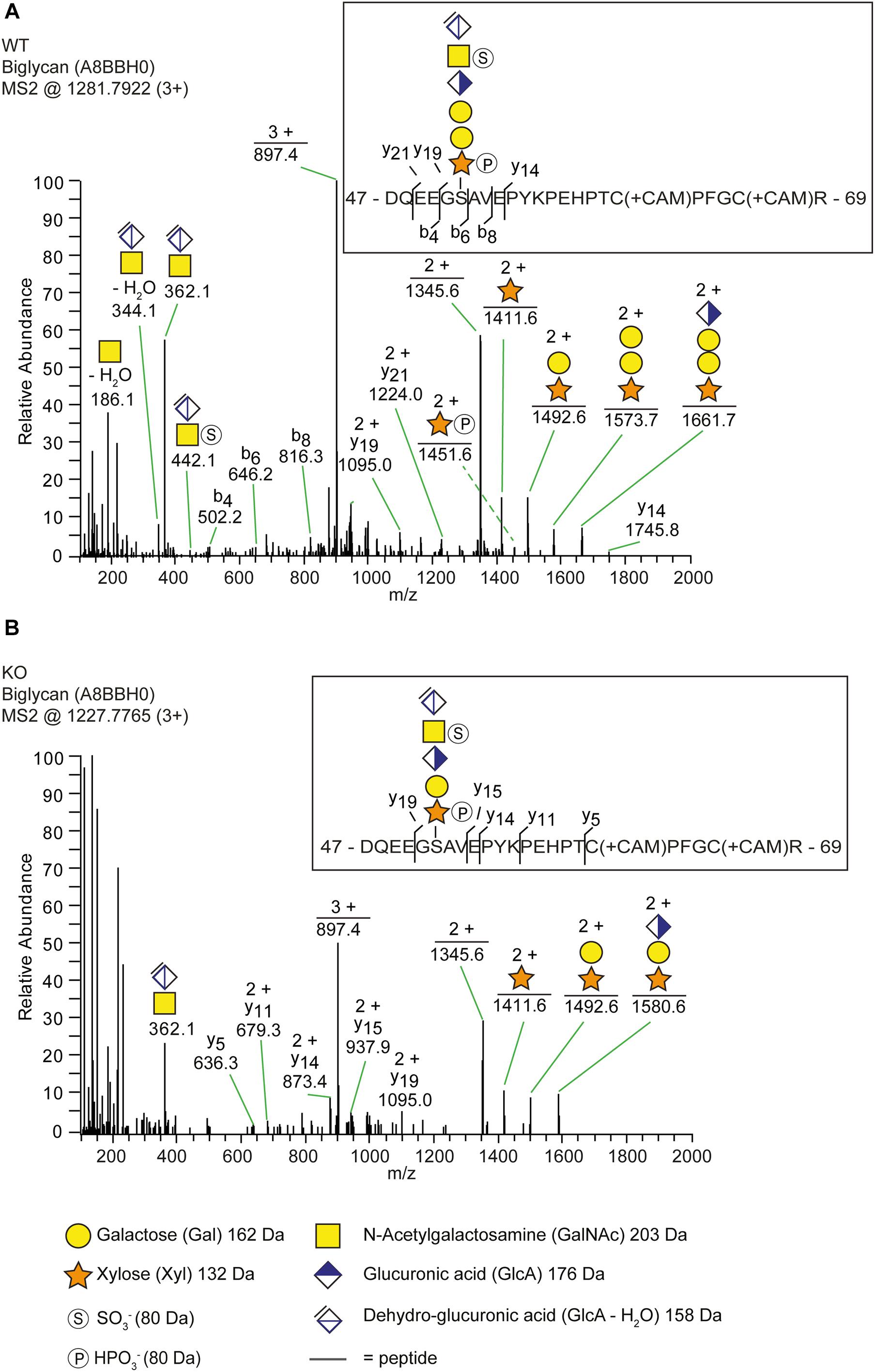
Figure 2. The identification of a trisaccharide linkage region in the b3galt6–/– zebrafish. (A,B) MS2 fragment mass spectra of CS-glycopeptides derived from biglycan (UniProt: A8BBH0) showed different linkage regions in WT compared to the b3galt6–/– zebrafish. (A) The peptide (DQEEGSAVEPYKPEHPTCPFGCR) in WT zebrafish was modified with a residual hexasaccharide structure, corresponding to the canonical tetrasaccharide linkage region (m/z 1281.79; 3+). The hexasaccharide structure was modified with one sulfate group (SO) at the subterminal GalNAc residue and one phosphate group (HPO) at the xylose residue (Supplementary Figure S7). (B) The peptide (DQEEGSAVEPYKPEHPTCPFGCR) in b3galt6–/– zebrafish was modified with a residual pentasaccharide structure, corresponding to a non-canonical trisaccharide linkage region (m/z 1227.78; 3+). The pentasaccharide structure was modified with two phosphate/sulfate groups. Their exact positioning and identity could not be determined due to low intensities of corresponding ion peaks, and the positions are therefore assigned in analogy of the WT structure.
These observations suggest that B3GAT3/GlcAT-I can transfer a GlcA residue not only to Gal-Gal-Xyl, but also to a Gal-Xyl substrate. This was confirmed by an in vitro GlcAT-I assay, which showed that GlcAT-I is indeed capable of adding a GlcA to a Gal-Xyl-p-nitrophynyl, which was chemically synthesized, thus to a substrate ending with a single galactose (Supplementary Figure S8).
b3galt6–/– Zebrafish Show Early Onset, Progressive Morphological Abnormalities and Delay of Cartilage and Bone Formation, Reminiscent of Human spEDS-B3GALT6
One of the clinical hallmarks of human spEDS-B3GALT6 is the pronounced skeletal involvement with the presence of spondyloepimetaphyseal dysplasia with postnatal growth restriction, bowing of long bones, hypoplasia of iliac bones, femoral head dysplasia, vertebral body changes, kyphoscoliosis and bone fragility with spontaneous fractures (Table 2). To evaluate the development of cartilage and bone associated with B3galt6-deficiency in zebrafish, phenotypic characterization of b3galt6–/– zebrafish was performed in juvenile (20 dpf, days post fertilization) and adult stages (4 months).
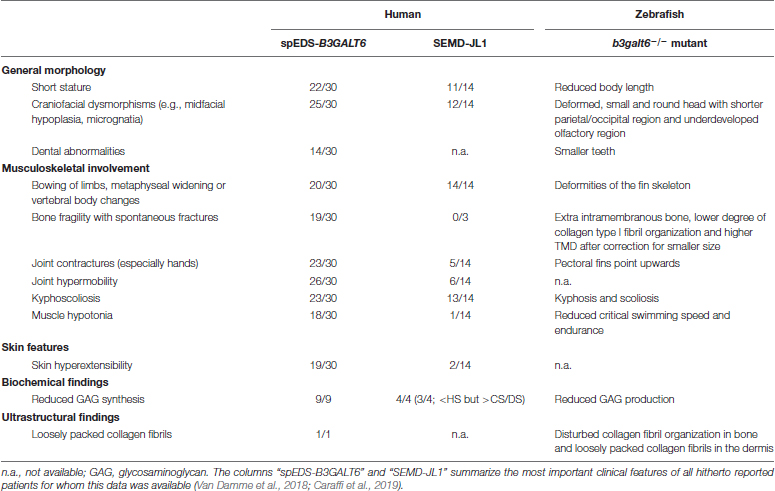
Table 2. Clinical features and ultrastructural and biochemical abnormalities observed in spEDS-B3GALT6 and SEMD-JL1 individuals compared to the b3galt6–/– zebrafish model.
At 20 dpf, the average weight (W), standard length (SL) and Fulton’s condition factor (K), which is used as a measure for the relative proportion of SL to W (Nash et al., 2006), were not significantly different between b3galt6–/– and WT siblings (Figure 3A), although for SL there was a trend toward smaller mutants (P = 0.0640). Several externally visible morphological anomalies could be observed in 20 dpf b3galt6–/– zebrafish as compared to their WT siblings, including a shorter, more compact head, a shorter and rounder frontal region, a shorter parietal/occipital region and a malformed posterior edge of the opercular apparatus (Figure 3B). Alcian blue (AB) cartilage staining showed no morphological abnormalities, but developmental delay of the head and pectoral and caudal fin endoskeleton was noted (Figure 3B and Supplementary Table S7), with cartilage elements being absent or in early development in b3galt6 KO, while these structures were already fully developed in WT sibling zebrafish. Alizarin red (AR) mineral staining showed delayed mineralization in all regions of the head, the pectoral fin girdle and the caudal fin (Figure 3B and Supplementary Table S8), further confirming developmental retardation. In addition, malformations of different mineralized bones, such as the parasphenoid, branchiostegal rays, supracleithrum, and the subopercle bone could be detected in the head of b3galt6–/– zebrafish larvae (Supplementary Figure S9). The vertebral column, i.e., the vertebral centra and their associated elements (neural and haemal arches), did not show delayed mineralization in b3galt6–/– zebrafish larvae, but malformation of the associated elements and fusions of preural vertebrae were observed at a much higher frequency in the b3galt6–/– mutants. Kyphosis, scoliosis and lordosis were also more frequently observed and pronounced in b3galt6–/– larvae compared to WT siblings (Supplementary Figure S10 and Supplementary Table S8). A similar phenotype was observed in the second b3galt6 KO model (cmg22 allele), confirming the specificity of the displayed phenotype (Supplementary Figure S11).
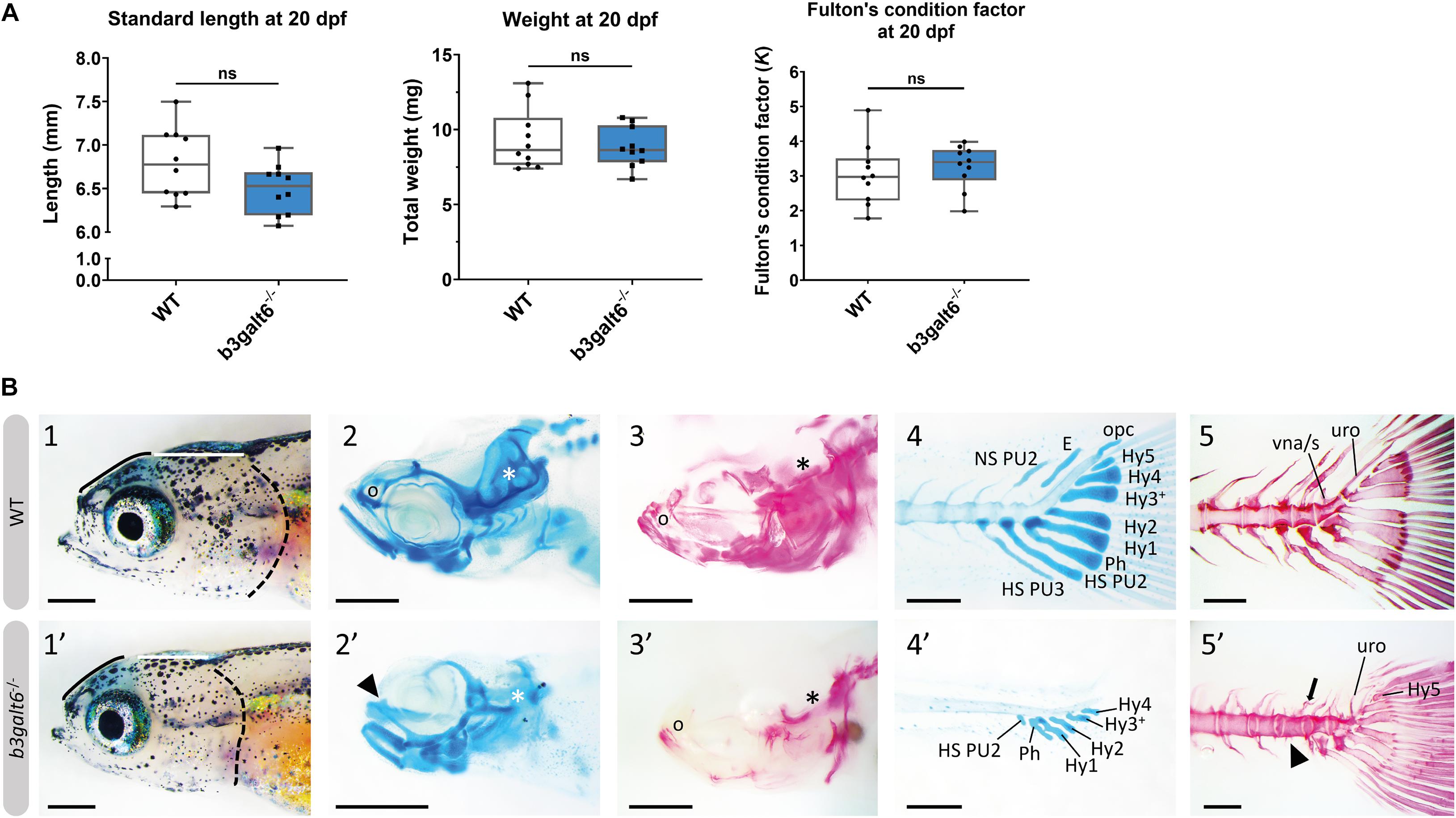
Figure 3. Developmental delay and morphological abnormalities in 20 dpf b3galt6–/– zebrafish. (A) Weight, standard length and Fulton’s condition factor of WT and b3galt6–/– zebrafish larvae at 20 dpf (n = 10). (B) External phenotype of the head of juvenile WT and b3galt6–/– sibling zebrafish. All images are oriented anterior to the left, posterior to the right, dorsal to the top and ventral to the bottom. (1-1’) b3galt6–/– zebrafish display a rounder frontal region (black line), shorter parietal/occipital region (white horizontal line) and a malformed posterior edge of the opercular apparatus (dotted black line), compared to WT control. (2-2’) Representative images of cartilage AB staining of the head. The olfactory region (indicated by “o” in the WT and arrowhead in the b3galt6–/– picture) and the otic region (indicated by white asterisk) show delayed development of cartilage elements in b3galt6–/– zebrafish as compared to WT. (3-3’) Representative images of AR mineral staining of the head. The olfactory region (indicated by “o”) and the otic region (indicated by asterisk) show severely delayed mineralization in b3galt6–/– zebrafish larvae as compared to WT. (4-4’) Representative images of AB staining of the caudal fin. The modified associated elements show developmental delay in b3galt6–/– zebrafish as compared to WT. E, epural; HS PU2, haemal spine of preural 2; HS PU3, haemal spine of preural 3; Hy1–5, hypural 1–5; NS PU2, neural spine preural 2; opc, opistural cartilage; Ph, parhypural (5-5’) Representative images of AR mineral staining of the caudal fin. Caudal vertebrae and their associated elements show no delay in ossification. Malformation or absence of caudal fin associated elements (block arrow indicates a neural arch; arrowhead indicates a missing haemal arch), and malformation of uroneural (uro) and vestigial neural arches and spines (vna/s) are observed in b3galt6–/– zebrafish; hypurals have a bent appearance. Scale bars: images of the head 500 μm, images of the caudal fin 200 μm.
At the adult stage (4 months), b3galt6–/– zebrafish had a significantly reduced average SL (1.834 ± 0.077 cm), and W (0.1196 ± 0.014 g), when compared to their WT siblings (2.371 ± 0.049 cm, and 0.2497 ± 0.016 g, respectively), while the Fulton’s condition factor (K) indicated that SL and W were normally proportioned to each other (Figure 4A). A similar phenotype was observed in the second b3galt6 KO model (cmg22 allele), confirming the specificity of the displayed phenotype (Supplementary Figure S1).
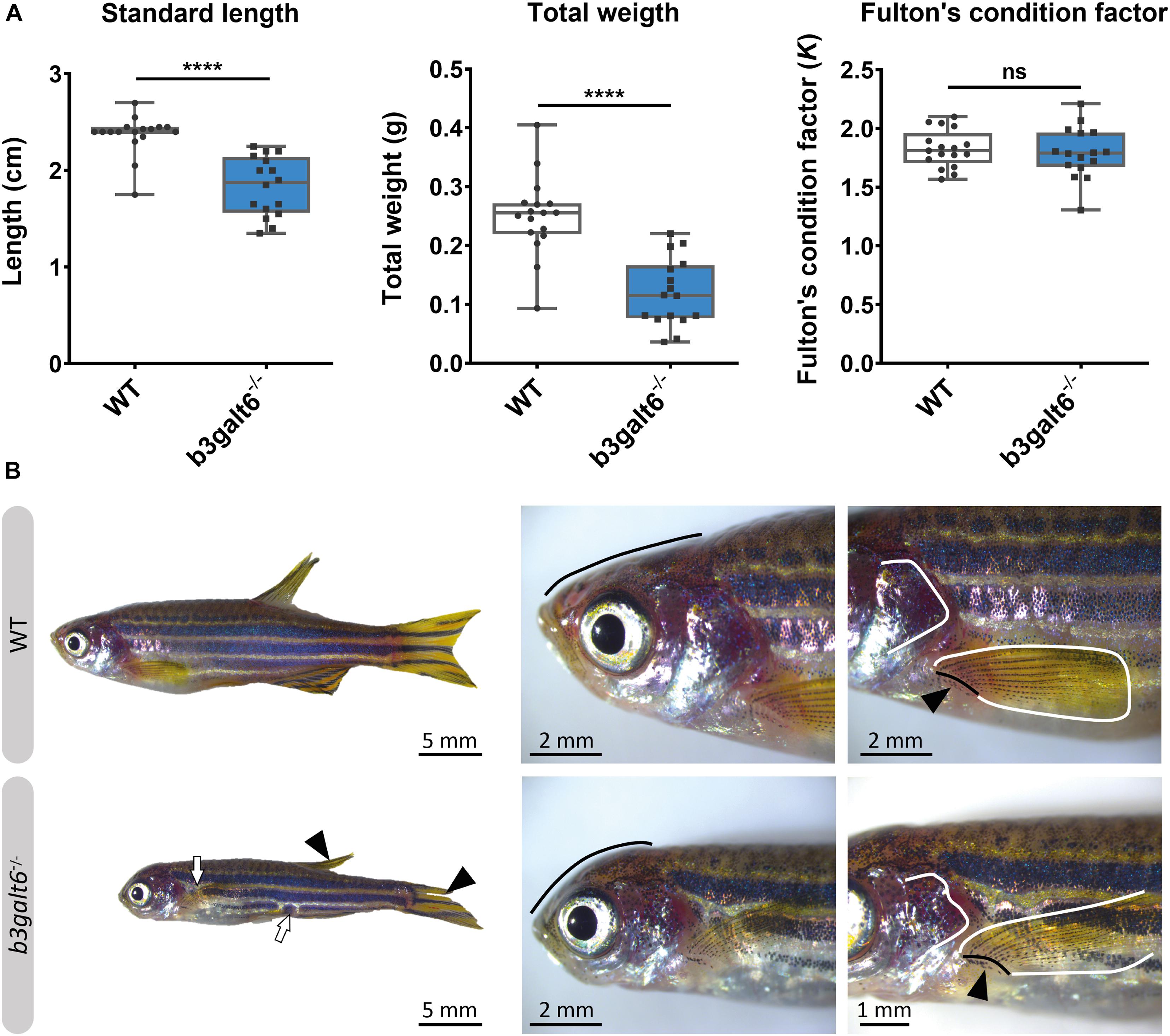
Figure 4. Characterization of the external phenotype of adult b3galt6–/– zebrafish. (A) b3galt6–/– zebrafish (n = 16) had a significantly shorter standard length (SL) compared to their WT siblings (n = 17). The weight (W) was significantly lower in the mutant zebrafish. The Fulton’s condition factor [K = 100*(W/SL3)] was not significantly different between b3galt6–/– zebrafish and WT siblings. Data are expressed as box plots with min-to-max whiskers on which each individual data point is plotted. An unpaired t-test with Welch’s correction was used to determine significance. ****P < 0.0001, ns: not significant. (B) Adult b3galt6–/– zebrafish exhibit morphological abnormalities compared to their WT siblings; b3galt6–/– zebrafish display interruptions of the blue horizontal stripes (white arrows), smaller fins (arrowhead), aberrations of the head shape (black line), of the shape of the opercular apparatus (white line indicates the posterior edge) and of the pectoral fin (black line and arrow indicate the fin rays at the base of the fin, white line shows the outline of the fin).
The head malformations, which were already observed in 20 dpf b3galt6–/– zebrafish, showed further progression, with a more pronounced smaller, rounded head and an abnormal compaction of the opercular apparatus (Figure 4B). In addition, a reduced surface area of the fins, with a crenelated postero-distal fin edge was observed in the mutant animals. The pectoral fins were pointing in an abnormal upwards angle (Figure 4B). In addition, an altered pigmentation pattern was observed, mainly on the ventral part of the body where the dark stripes showed interruptions (Figure 4B).
Further progression of the larval phenotype was also confirmed by AR staining of the mineralized skeleton in adult (4 months) b3galt6–/– zebrafish. The olfactory region was underdeveloped (Figure 5A), the cranial roof bones and sutures were deformed (Supplementary Figure S12) and the opercular apparatus was smaller and compacted in KO zebrafish (Figure 5A and Supplementary Figure S12). The fifth ceratobranchial arch and its attached teeth were smaller in b3galt6–/– zebrafish (Figure 5B). In the pectoral fins of b3galt6–/– zebrafish the radials were smaller, had an irregular shape, and were often fused (Figure 5B). The autocentra, i.e., the inner layer of the vertebral centra in the vertebral column and caudal fin (Arratia and Schultze, 1992) of KO zebrafish had a normal hourglass shape, but the associated elements often showed presence of extra intramembranous bone (Figure 5 and Supplementary Figure S12) and extra bony elements (Figure 5A). Fusions were observed in preural vertebrae (Figure 5A and Supplementary Figures S12B,C). Lordosis and scoliosis was variably observed in the caudal region of the vertebral column of b3galt6–/– mutant zebrafish (Supplementary Figures S12B,C). Extra mineralization was observed at the edge (growth zone) of b3galt6–/– scales (Figure 5B). Notably, variation in the severity of the skeletal malformations in b3galt6–/– zebrafish was observed.
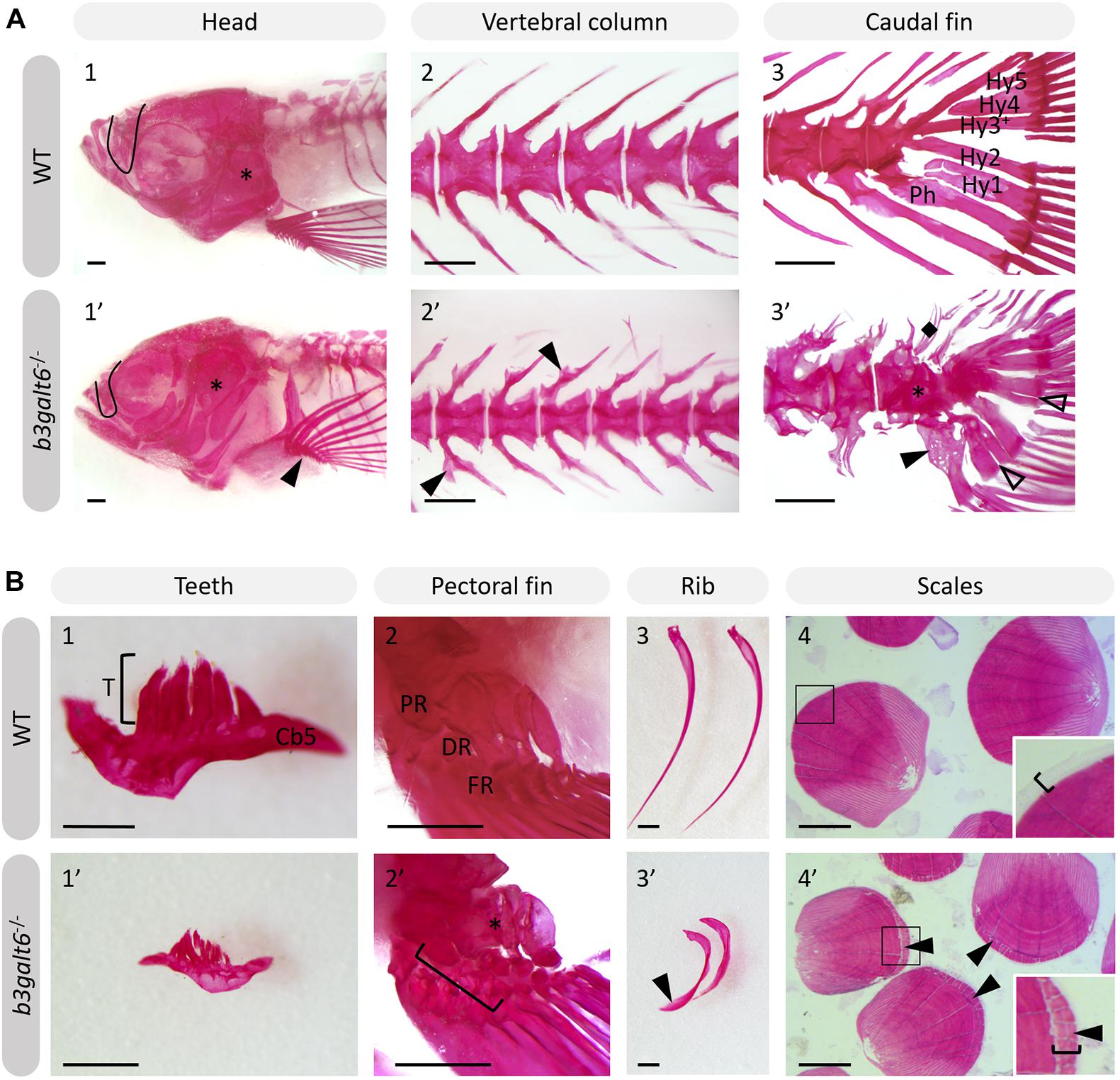
Figure 5. Alizarin red stained mineralized bone in WT and b3galt6–/– adult zebrafish. The mineralized bones in the skeleton were studied in 4 months old WT and mutant zebrafish after AR mineral staining. (A) b3galt6–/– zebrafish display malformation of the cranium, vertebral column and caudal fin. (A1-1’) The cranium shows an underdeveloped olfactory region (black line) and a smaller compacted opercular bone (asterisk). (A2-2’) The vertebral column shows extra intramembranous outgrowths located where the distal tip of the neural and haemal arches fuse to a neural and haemal spine, respectively (arrowheads). (A3-3’) In the caudal fin fusion of preural vertebrae is observed (asterisk). Associated elements show intramembranous outgrowths (arrowhead) and the presence of extra bony elements (diamond). The distal tip of the hypurals is sometimes split (transparent arrowhead). Structures indicated in the WT caudal fin: Hy1-5, hypural 1-5, Ph, parhypural. (B) Details of mineralized structures in b3galt6–/– zebrafish. (B1-1’) The fifth ceratobranchial arch and its teeth are smaller in KO zebrafish. (B2-2’) The proximal (PR) and distal radials (DR) have irregular shapes in mutant zebrafish. Proximal (asterisk) and distal radials are sometimes fused. The bracket indicates a fusion between a proximal and distal radial, and the first fin ray (FR). (B3-3’) Ribs also show extra intramembranous bone (black arrowhead). (B4-4’) More mineralization was observed at the growth zone (bracket) of the scales in mutant zebrafish (arrowhead). Scale bars = 500 μm.
Subsequent quantitative μCT analysis of 4 months old zebrafish revealed multifaceted effects of B3galt6-deficiency on vertebral morphology and mineralization. Most notably, the volume of vertebral centrum, haemal and neural associated elements was significantly reduced throughout the entire vertebral column in b3galt6–/– zebrafish (P < 0.01), with more pronounced differences in the abdominal region (Figure 6). Tissue mineral density (TMD) was not significantly different in the b3galt6–/– vertebral bodies compared to WT siblings. However, TMD in zebrafish is proportionally related to standard length (Parichy et al., 2009; McMenamin et al., 2016), and b3galt6–/– zebrafish exhibited a significantly reduced body size compared to the WT siblings. We therefore normalized the μCT data to standard length using the normalization procedure outlined by Hur et al. (2017). In this procedure, phenotypic data in WT sibling controls was scaled using the mean standard length of b3galt6–/– zebrafish as the reference length. Remarkably, after normalization, a significant increase in TMD of centrum, haemal and neural associated elements was noted in b3galt6–/– zebrafish compared to WT controls (P < 0.001, P < 0.01, and P < 0.01, respectively) (Supplementary Figure S13).
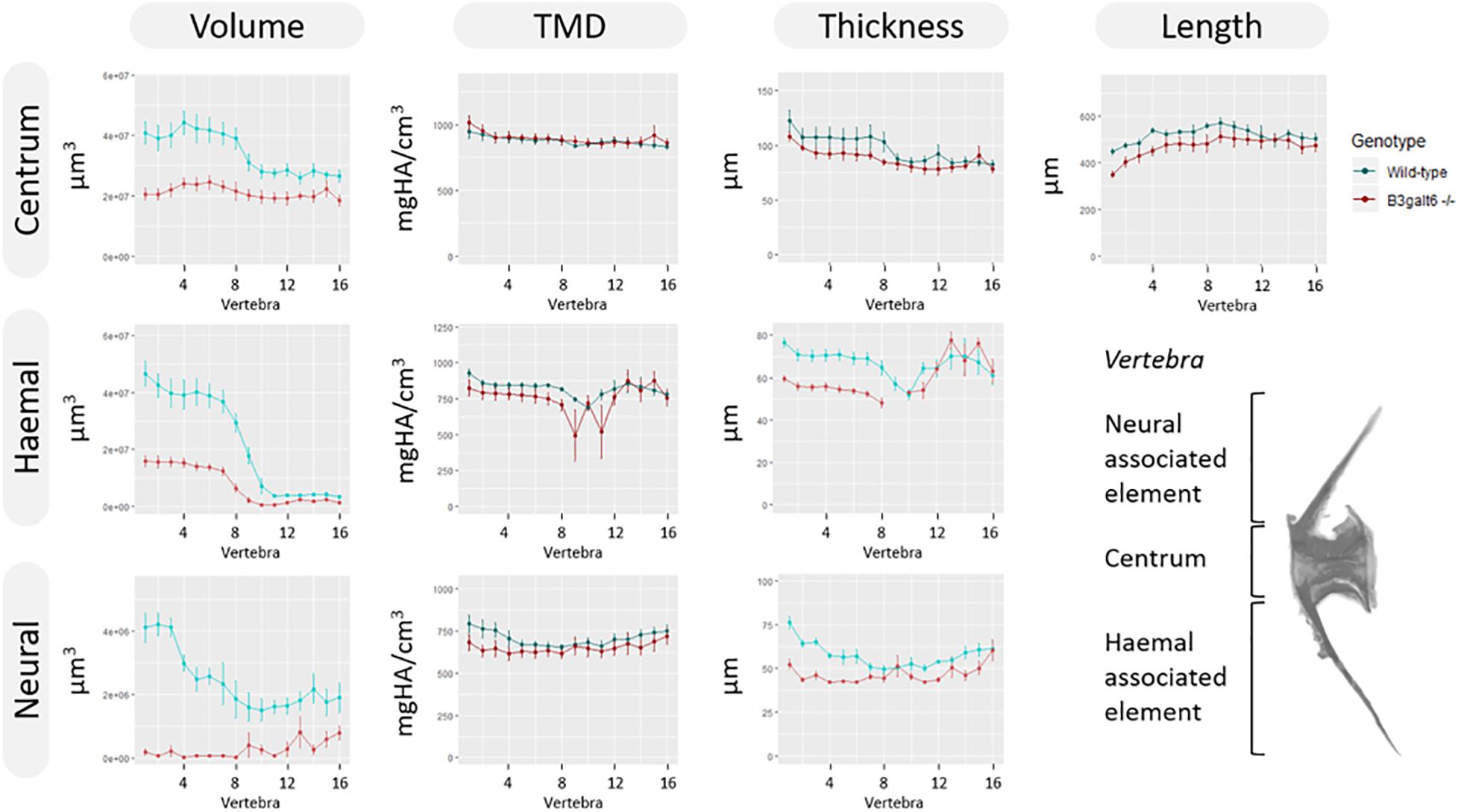
Figure 6. μCT analysis of adult WT and b3galt6–/– zebrafish. Quantitative μCT analysis of the vertebral column in five mutants vs. five WT zebrafish at the age of 4 months. The bone volume, tissue mineral density (TMD) and bone thickness were calculated for three parts of each vertebra: the vertebral centrum, the neural associated element (neural arch and neural spine) and the haemal associated element (haemal arch and haemal spine). Plots associated with a significant difference are colored in a lighter coloring scheme. The X-axis represents each vertebra along the anterior-posterior axis. The volume is significantly reduced for each part of the vertebra in b3galt6–/– zebrafish (P < 0.01, P < 0.001, and P < 0.000001, respectively). The thickness of the haemal (P < 0.01) and neural (P = 0.001) associated element is also significantly reduced in the mutants whereas the centrum thickness is not significantly reduced in b3galt6–/– zebrafish (P = 0.12). TMD is not significantly different between b3galt6–/– zebrafish and WT siblings when not normalized for standard length (P > 0.2). Data are presented as mean ± SEM. TMD, tissue mineral density; HA, hydroxyapatite.
b3galt6 Knock-Out Zebrafish Display a Disturbed Organization of Type I Collagen Fibrils in Skin, Bone, and Intervertebral Space
Transmission electron microscopy (TEM) studies on the dermis of spEDS-B3GALT6 patients revealed an abnormal collagen fibril architecture with loosely packed collagen fibrils of variable size and shape (Malfait et al., 2013). We therefore studied the effect of loss of galactosyltransferase II activity on collagen fibril architecture. TEM sections of skin biopsies of b3galt6–/– zebrafish showed an abnormal dermal collagen fibril architecture characterized by loosely packed collagen fibrils and larger and more interfibrillar spaces compared to WT siblings (Figure 7A). In addition, at the level of the scales a thicker epidermal layer was observed and electron dense structures were present in the interfibrillar spaces (Figure 7A). Next, we examined the intervertebral region and adjacent vertebral centra in the vertebral column of 5 months old zebrafish by means of TEM. In general, the type I collagen fibers in b3galt6–/– zebrafish displayed a lower degree of organization (Figure 7B). This was observed in immature collagen layers, deposited by osteoblasts in the outer edges of the intervertebral space, but also in the mature type I collagen layers of the intervertebral ligament and in the autocentrum, the inner layer of the vertebral bone, where a lack of the typical plywood-like organization was seen. Detailed investigation of TEM sections of the vertebral bone revealed presence of several electron dense spots of unknown origin in knock-outs. These “dark spots” may reflect extrafibrillar hydroxyapatite crystals (Figure 7B).
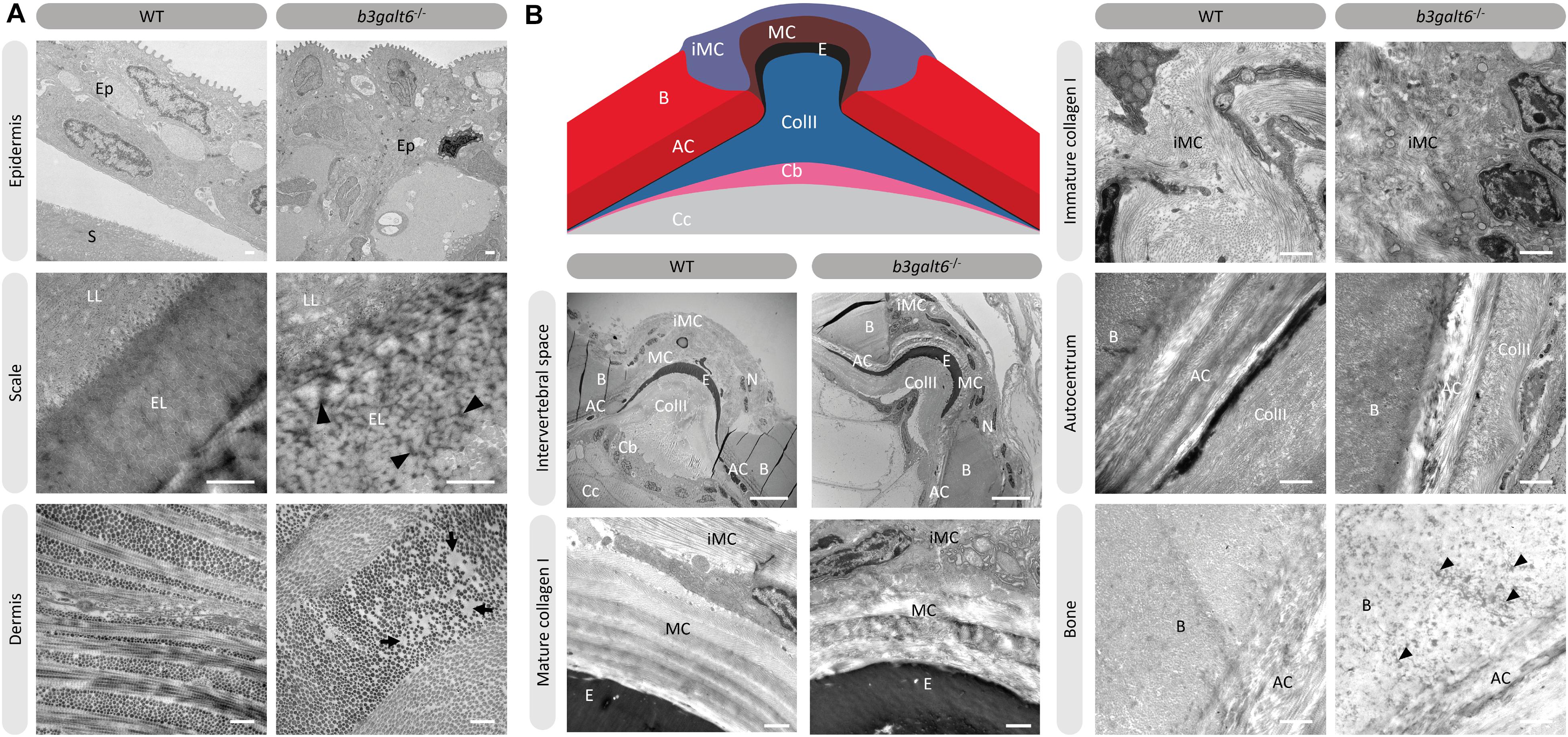
Figure 7. Collagen fibrillar architecture in 5 months old adult zebrafish. (A) Skin from 5 months old WT and b3galt6–/– adult zebrafish. The epidermis (Ep), the outer layer of the skin covering the scales (S) of zebrafish, appears thicker in b3galt6–/– zebrafish compared to the epidermis of WT zebrafish. A mature zebrafish scale consists of several layers. The limiting layer of the scale (LL) is rich in fine granules that in some areas are linearly arranged, forming dense layers. This matrix contrasts with that of the external layer(s) (EL) below. Structures resembling extrafibrillar crystals (arrowhead) are noted in the external layer of the mutant scale, which are rare in WT zebrafish sections. Upon investigation of the dermis of b3galt6–/– mutants, increased interfibrillar spaces are noted (arrows), which are absent in the dermis of WT adults. Scale bars = 500 nm. (B) Schematic representation and TEM images of the zebrafish intervertebral space. Schematic representation of the zebrafish intervertebral space. AC, autocentrum; B, bone; Cb, chordoblasts; Cc, chordocytes; ColII, type II collagen; E, elastin; iMC, immature collagen type I layer; MC, mature collagen type I layer. Starting from the central body axis, several recognizable structures are present in the intervertebral space along a proximo-distal axis: (i) notochord tissue consisting of chordocytes and chordoblasts, (ii) the notochord sheath consisting of a type II collagen layer and an external elastic membrane, (iii) and a connective tissue ligament consistent of a mature and an immature type I collagen layer. The mature type I collagen layer is organized in a typical plywood-like pattern and is continuous with the inner layer of the vertebral bone, i.e., the autocentrum, which also shows a plywood-like organization. The immature type I collagen layers consist of loose collagen fibers connecting the outer layer of vertebral bones. Notice the higher number of nuclei (N) in the b3galt6–/– zebrafish at the intervertebral space. Mature collagen (MC) displays a typical plywood-like organization, in WT zebrafish but not in comparable regions for b3galt6–/– mutant zebrafish. Cross section and longitudinal type I collagen regions are less clearly demarcated and smaller in immature collagen (iMC) from b3galt6–/– zebrafish compared to WT siblings. It is more difficult to pinpoint the different layers of the autocentrum (AC) in the KO compared to WT zebrafish. Bone (B) from the b3galt6–/– zebrafish shows electron dense structures (arrowhead). Scale bars = 10 μm for the intervertebral space and 1 μm for the other images.
Mass spectrometry analysis of type I collagen modification in bone derived from the vertebral column showed no significant difference in hydroxylation status of the α1(I) K87 and C-telopeptide lysine residues (Supplementary Table S9). These lysine residues are conserved in zebrafish and human type I collagen and are crucial for intermolecular crosslinking of collagen fibrils in bone matrix (Gistelinck et al., 2017).
b3galt6 Knock-Out Zebrafish Display Ultrastructural Muscle Abnormalities and Lower Endurance
Human spEDS-B3GALT6 patients often suffer from muscle hypotonia with a delay in motor development. In view of the observed reduction in the concentration of the different GAG moieties in muscle of b3galt6–/– zebrafish, we investigated the function and the ultrastructural appearance of muscle.
First, the critical swimming speed (Ucrit) and endurance of the zebrafish were assessed by means of a custom-made swim tunnel. The Ucrit, defined as the maximum velocity that the tested zebrafish could sustain for at least 5 min (Plaut, 2000), was significantly lower in b3galt6–/– zebrafish (30.1 ± 2.39 cm/s) compared to length-matched WT (41.61 ± 1.942 cm/s) (P < 0.01) (Figure 8A). The stamina of each zebrafish was determined by an effort test where it was tested how long zebrafish could swim at high speed (37 cm/s) after a 3 min warm-up. This effort test showed that the b3galt6–/– zebrafish performed less well compared to length-matched WT zebrafish, indicating a lower endurance (Figure 8B).
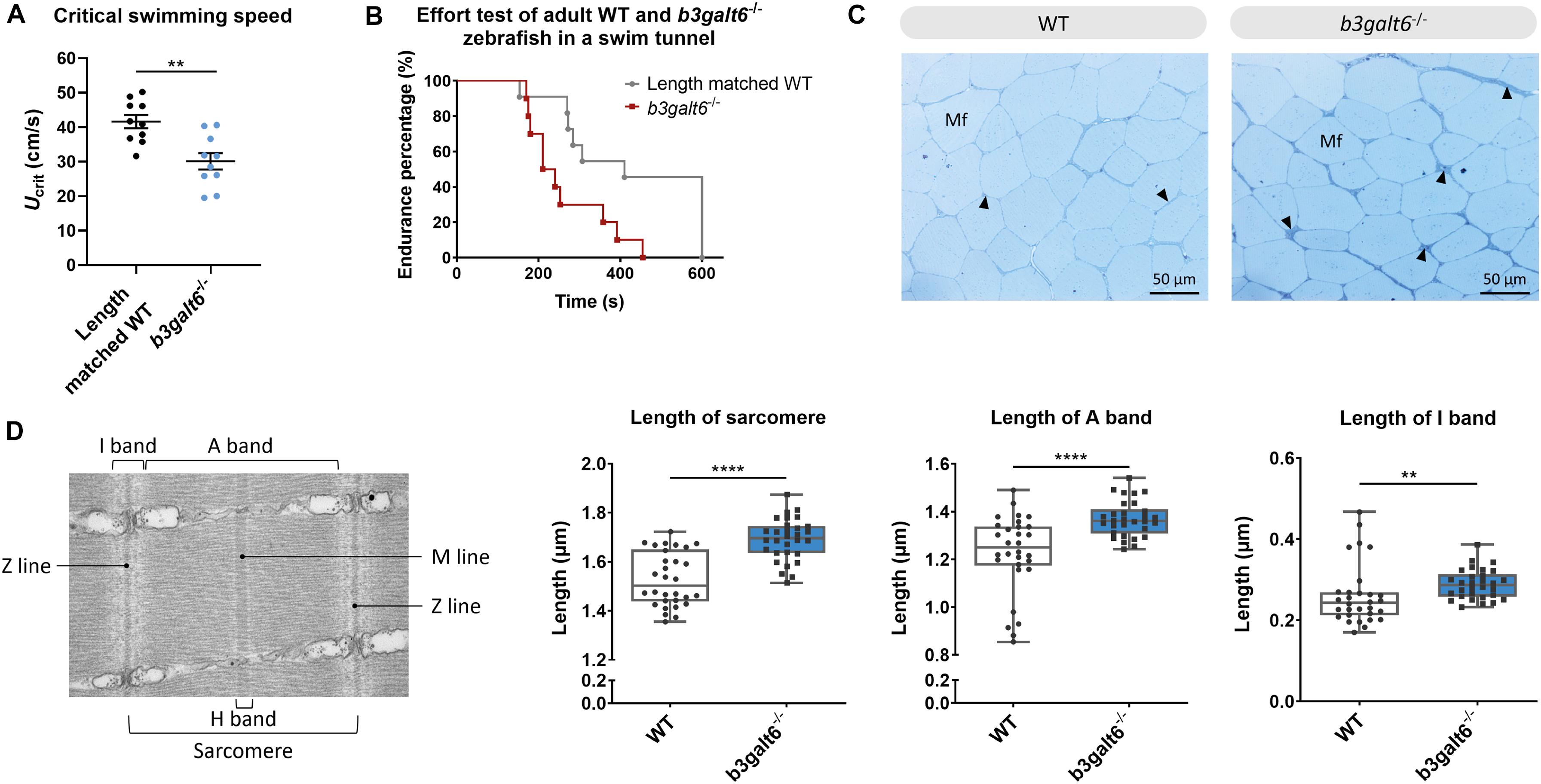
Figure 8. Functional and structural analysis of adult WT and b3galt6–/– zebrafish muscle. (A) The critical swimming speed and (B) the endurance performance was determined for b3galt6–/– and length-matched WT zebrafish. (C) Semi-thin cross sections, stained with toluidine blue, demonstrate the presence of a thicker endomysium (arrowhead), located between the muscle fibers (Mf) at the horizontal myoseptum of b3galt6–/– zebrafish at the age of 5 months. (D) Transmission electron microscope structure of a sarcomere with indications of the Z and M line and A, I, and H band. The sarcomere length was determined at 5 different loci per semi-thin section, for 6 semi-thin sections origination from 3 different zebrafish per genotype (N = 30). A significant increase in sarcomere length for b3galt6–/– is noted. Furthermore, both the A and I band show an increase in length for the mutant zebrafish. Data are expressed as box plots with min-to-max whiskers on which each individual data point is plotted. A Mann-Whitney U test was used to determine significance. ****P < 0.0001, **P < 0.01.
The muscle tissue in the zebrafish trunk was further investigated at the ultrastructural level. Toluidine blue-stained semi-thin cross sections revealed that the endomysium, a layer of connective tissue ensheating the muscle fibers, was enlarged along the horizontal myoseptum of b3galt6–/– zebrafish, although no clear differences in muscle fiber diameter were noted (Figure 8C). Ultra-thin parasaggital sections, investigated by TEM, showed that the length of the sarcomeres, the smallest basic units of striated muscle, as well as the length of the A and I band were significantly increased in the b3galt6–/– zebrafish (Figure 8D).
Discussion
In this study we developed two viable b3galt6 KO zebrafish models using CRISPR/Cas9 mutagenesis, and performed an in-depth characterization of the biochemical, ultrastructural and phenotypical consequences of β3GalT6-deficiency on different tissues, including skin, cartilage, mineralized bone and muscle. These models currently represent the only vertebrate models available for the study of the pathogenetic consequences of β3GalT6-deficiency on tissue development and homeostasis. In both models we detected a significant reduction, yet not a complete absence in b3galt6 mRNA expression (Figure 1E). One possible explanation is the absence of canonical nonsense-mediated decay (NMD) in the single-exon b3galt6 gene, due to a lack of exon junction complexes (EJCs) (Wittkopp et al., 2009). We were unable to examine the effect of the mutations on B3galt6 protein levels and localization due to the unavailability of a suitable antibody. Nevertheless, since the cmg20 and cmg22 alleles are predicted to introduce a PTC, respectively, upstream and in the catalytic domain, residual translation of some mRNA into a truncated protein is not expected to lead to the production of an active enzyme. β3galt6 is a key enzyme in the biosynthesis of the common linkage region of PGs, and reduced levels of active enzyme are thus expected to equally affect the biosynthesis of HS and CS/DS GAG chains. We therefore quantified the disaccharide concentrations in bone, muscle and skin of adult b3galt6–/– zebrafish by means of a combination of enzymatic digestion and anion-exchange HPLC. This revealed a distinct, and relatively proportional decrease in the concentration of CS, DS, hybrid CS/DS and HS moieties in all examined tissues, compared to age-matched WT zebrafish (Table 1). Nonetheless, low amounts of GAGs were still produced in the KO zebrafish in absence of upregulation of other galactosyltransferase member(s) from the b3galt family or by other linkage-enzymes (Supplementary Figure S6). Together with the previous demonstration that the other five β3GalT human enzymes (e.g., β3GalT1, β3GalT2, β3GalT3/β3GalNAcT1, β3GalT4, and β3GalT5) are involved in the biosynthesis of glycoproteins and glycolipids (Bai et al., 2001), compensation for galactosyltransferase II deficiency in zebrafish by other b3galt family members is unlikely. Chinese hamster ovary B3galt6 KO cells were also shown to be capable of producing GAGs, in contrast to Xylt2, B4galt7 or B3gat3 KO cells (Chen et al., 2018). The recent identification of the presence of a non-canonical trisaccharide linkage region, lacking one Gal residue (GlcA-Gal-Xyl-O-), as a minor constituent in the PG bikunin in the urine of healthy human individuals (Persson et al., 2019), prompted us to examine the composition of the GAG linkage region in our b3galt6 KO zebrafish models, by analyzing protein extracts from adult b3galt6–/– and WT zebrafish via LC-MS/MS. This led to the striking finding that the few detected PGs in the b3galt6–/– zebrafish all presented with a trisaccharide PG linkage region, consisting of only one Gal (Figure 2B and Supplementary Table S6). Further in vitro analysis indicated that GlcAT-I/B3GAT3 is capable of adding a GlcA to Gal-Xyl-O- (Supplementary Figure S8). This observation might explain the phenotypic differences observed between GalT-II/B3GALT6 deficiency and deficiencies of the other linkage region enzymes, for which such a rescue mechanism might be absent. This hypothesis is supported by the lack of GAGs in the Chinese hamster ovary Xylt2, B4galt7 or B3gat3 KO cells and by the lethal phenotype observed in the b4galt7 KO zebrafish model (Delbaere et al., 2020b). At present, we have no information on the length of the GAG chains attached to the trisaccharide linkage region or the composition and the possible modifications of these HS and CS/DS chains. As such, the GAG structure, length and modifications in the b3galt6–/– zebrafish might be different from the ones in WT zebrafish.
HSPGs and CS/DSPGs are essential macromolecules for the development, signaling and homeostasis of many tissues. It therefore comes as no surprise that defects in linkage region biosynthesis can affect many organs. Endochondral ossification of cartilage (forming chondral bones) for example is a multi-staged process that must be coordinated in space and time. CS/DSPGs and HSPGS, which have key roles in modulating signaling events during vertebrate skeletal development, are expressed in a spatially and temporally highly controlled manner (Eames et al., 2011), thus regulating the timing of endochondral ossification. Genetic defects disrupting PG synthesis or structure have been shown to disrupt the timing of skeletal development. Two zebrafish models with abnormal PG linkage region biosynthesis, i.e., the fam20b and xylt1-deficient models, revealed partial decrease in CSPGs, which are predominantly present in cartilage, and accelerated endochondral ossification, showing that CSPGs can negatively regulate skeletogenic timing (Eames et al., 2011). On the other hand, a strong reduction in CSPGs and partial decrease in HSPGs was noted in b3gat3 mutant zebrafish, but this led to a delay in endochondral ossification and short and disorganized cartilaginous structures (Holmborn et al., 2012). Our B3galt6-deficient zebrafish models also show a delay in skeletal development at juvenile stages. At 20 dpf, a delay was noticed in the development of cartilage and mineralized bony elements of the craniofacial structures, the pectoral and caudal fin endoskeleton (Figure 3B and Supplementary Tables S7, S8), and these abnormalities had progressed in adult zebrafish. The reduced body length, misshapen cranial bones and smaller teeth observed in the head skeleton, and general skeletal dysplasia with kyphosis and scoliosis seen in the vertebral column of b3galt6–/– adult zebrafish is reminiscent of the skeletal phenotype observed in human β3GalT6-deficient individuals (Table 2).
In addition to skeletal dysplasia, we also observed abnormalities in bone mineralization. Adult mutant zebrafish stained with AR showed the presence of extra intramembranous bone and extra bony elements on the associated elements of the vertebral centra. On the other hand, quantitative μCT analysis of the vertebral column and associated elements revealed a generalized reduction in bone volume and thickness, and a relative increase in TMD after correction for reduced body length of the mutant zebrafish (Figure 6 and Supplementary Figure S13). Although it is not clear so far if the increased fracture risk in spEDS-B3GALT6 patients can be attributed to a lower bone volume combined with a higher TMD, such hypothesis has been put forward for the brittle bone disorder Osteogenesis Imperfecta (OI) based on observations in both patients and animal models (Gistelinck et al., 2016; Fiedler et al., 2018). In the Chihuahua (col1a1achi/+) OI zebrafish model the intrafibrillar mineral platelets were shown to be smaller and more densely packed, leading to an increased mineral to matrix ratio, and consequently a reduced elastic modulus-to-hardness ratio, a measure for fracture toughness (Viguet-Carrin et al., 2006; Bishop, 2016; Fiedler et al., 2018). In addition, in both OI patients and the mouse model oim, reduced collagen matrix organization was shown to lead to the formation of extrafibrillar crystals, which are larger compared to intrafibrillar crystals (Viguet-Carrin et al., 2006). Eventually, additional minerals stabilize the tissue, but they do so at the cost of increased brittleness of the bone (Grabner et al., 2001). In accordance, in b3galt6–/– zebrafish we observed an irregular collagen organization interspersed with large interfibrillar spaces and electron dense spots on ultra-thin sections of skin and bone via TEM. The “dark spots” observed in the vertebral bone could be remnants of extrafibrillar mineral aggregates, contributing to increased mineralization but also increased brittleness of the skeleton.
Abnormalities in collagen fibril organization were observed in many mutant zebrafish tissues. Besides vertebral bone, irregular collagen organization was also observed in the intervertebral space, intervertebral ligament, and the dermis (Figure 7). This strongly resembles the previously reported abnormal dermal collagen fibril architecture in the skin from a β3GalT6-deficient individual (Malfait et al., 2013). This could be attributed to abnormalities in GAG chains of small leucine-rich PGs (SLRPs), such as the DSPGs decorin and biglycan, which are known to be of particular importance for regulating collagen fibril organization in connective tissues through their core protein and GAG chains (Kalamajski and Oldberg, 2010). SLRPs are also thought to influence collagen cross-linking patterns, but the mechanism by which they do so remain largely unknown (Kalamajski and Oldberg, 2010). Although similar hydroxylation levels were observed at K87 and the C-telopeptide of the type I collagen α1 chain in b3galt6–/– and WT adult zebrafish (Supplementary Table S9), we cannot rule out that type I collagen cross-linking might still be affected.
In view of the observation that human β3GalT6-deficient individuals often present quite severe muscle hypotonia with delayed gross motor development and reduced endurance, we also studied the effect of B3galt6-deficiency on GAG synthesis in muscle, and on muscle function and ultrastructure (Figure 8). Similar to the other examined tissues, HS and CS/DS GAG concentrations were severely reduced in muscle tissue. It is known that the ECM in skeletal muscle plays an important role in muscle fiber force transmission, maintenance and repair. Numerous PGs, including the HSPGs syndecans 1–4, glypican-1, perlecan and agrin, and CS/DSPGs, such as decorin and biglycan, are present in muscle ECM. Besides their structural role, many of them are known to bind growth factors (e.g., TGFβ). Those growth factors can be stored and released by the negatively charged GAGs. Certain enzymes in the ECM can cleave GAG chains, thereby releasing associated growth factors, allowing their interaction in cell signaling and mechanotransduction (Gillies and Lieber, 2011). The significant decrease in critical swimming speed (Ucrit) and endurance observed in the b3galt6–/– zebrafish suggest decreased muscle function (Figures 8A,B). Further evidence for an intrinsic muscle dysfunction in the b3galt6–/– model comes from semi-thin muscle sections, showing an enlargement of the connective tissue sheet surrounding the muscle fibers (Figure 8C). Nevertheless, we cannot exclude that the observed skeletal malformations and/or other abnormalities, such as respiratory or neuronal dysfunction, could also influence the outcome of these tests.
Taken together, we generated and characterized the first b3galt6 KO zebrafish models and show that their phenotype largely mimics that of human β3Galt6-deficiency. A strong reduction in HS, CS and DS GAG concentration was observed in b3galt6–/– zebrafish. Yet, we did observe a small amount of GAGs being produced with a unique trisaccharide PG linkage region. Furthermore, we showed that a lack of B3galt6 causes disturbances in collagen fibril organization in skin, ligament and bone, with the latter also showing abnormalities in mineralization, and leads to structural and functional abnormalities of the muscle. These KO zebrafish can serve as useful models to further unravel the underlying pathogenic mechanisms of this complex multisystemic disorder and serve as a tool for the development and evaluation of possible therapeutic interventions, such as the stimulation of intrinsic compensational mechanisms for the production of PGs.
Future studies will focus on verifying the presence of a trisaccharide linkage region in samples (e.g., urine) from human spEDS-B3GALT6 patients. In addition, the hypothesis that B3gat3 is responsible for the remaining GAG synthesis in our b3galt6–/– model should be further explored by inactivating B3gat3 in our b3galt6–/– model via knock-down or knock-out approaches, followed by GAG analysis. Potential therapeutic avenues to explore include stimulation of B3gat3 expression, or enzyme replacement therapy (ERT). For patients with an inborn error of metabolism, such as Gaucher disease and mucopolysaccharidosis, ERT is a well-known therapeutic approach and has already been proven successful (Ferreira and Gahl, 2017). Other (but less straightforward) therapeutic avenues in human are stimulation of residual enzyme via co-enzyme treatment or enzyme enhancement therapy, bone-marrow transplantation or correcting the product deficiency by providing alternate substrate. Nevertheless, we should first investigate the human situation before starting to explore these avenues.
Materials and Methods
Zebrafish Maintenance
Wild-type (AB strain) and b3galt6 knock-out zebrafish lines were reared and maintained by standard protocols (Gistelinck et al., 2018). All animal studies were performed in agreement with EU Directive 2010/63/EU for animals, permit number: ECD 16/18. All zebrafish used in a single experiment were bred at the same density. All efforts were made to minimize pain, distress and discomfort.
Alcian Blue and Alizarin Red Staining
Twenty-day-old larvae (n = 10) and 5 months old WT and b3galt6–/– (n = 5) adult zebrafish were stained as previously described (Delbaere et al., 2020b). See Supplementary Information for details.
μCT Scanning Analysis
Whole-body μCT scans of 4 months old WT (n = 5) and b3galt6–/– (n = 5) siblings were acquired on a SkyScan 1275 (Bruker, Kontich, Belgium) using the following scan parameters: 0.25 mm aluminum filter, 50 kV, 160 μA, 65 ms integration time, 0.5° rotation step, 721 projections/360°, 5 m 57 s scan duration and 21 μm voxel size. DICOM files of individual zebrafish were segmented in MATLAB using custom FishCuT software and data were analyzed and corrected for standard length in the R statistical environment, as previously described (Hur et al., 2017).
Histology and Transmission Electron Microscopy
Zebrafish bone, consisting of the last transitional vertebra and two consecutive caudal vertebrae (Morin-Kensicki et al., 2002; Bird and Mabee, 2003), skin, at the lateral line below the dorsal fin, and muscle samples, located dorsolateral underneath the dorsal fin, dissected from 5 months old WT (n = 5) and b3galt6–/– (n = 5) zebrafish, were fixed and embedded in epon following the procedures outlined by Huysseune and Sire (1992).
Swim Tunnel Experiments
Ten WT (5 months) and ten b3galt6–/– zebrafish (8 months), were selected based on their similar (not significantly different) length and weight. The critical swimming speed (Ucrit) was measured in a swim tunnel for both WT and b3galt6–/– zebrafish as previously described (Brett, 1964). Furthermore, the endurance of each zebrafish was tested by means of an effort test in the swim tunnel. Each zebrafish had a 3 min warm-up and after the third minute, the speed increased until 37 cm/s for maximum 7 min. When the zebrafish was not able to swim anymore, the test was stopped.
Collagen Crosslinking Analysis
The collagen crosslinking was analyzed in the dissected vertebral column (excluding Weberian apparatus and urostyle) from 5 months old wild-type (n = 5) and b3galt6–/– (n = 5) siblings as described before in Gistelinck et al. (2016).
Disaccharide Composition Analysis of CS, DS, and HS Chains
The level of total disaccharides from CS, DS, and HS in bone, muscle and skin tissues from 9 months old zebrafish (n = 5) were determined as described previously (Mizumoto et al., 2009; Mizumoto and Sugahara, 2012).
Glycopeptide Preparation and Analysis
LC-MS/MS Sample Preparation
CS- and HS-glycopeptides were purified from 6 months old WT (n = 3) and b3galt6–/– (n = 3) zebrafish using a combination of extraction, trypsin digestion and anion exchange chromatography, slightly modified (see Supplementary Information) from previously described protocols (Noborn et al., 2015, 2016).
LC-MS/MS Analysis
The samples were analyzed on an Orbitrap Fusion mass spectrometer coupled to an Easy-nLC 1200 liquid chromatography system (Thermo Fisher Scientific., Waltham, MA) (see Supplementary Information). Nanospray Flex ion source was operated in positive ionization mode and three separate higher-energy collision-induced dissociation (HCD) MS/MS spectra were recorded for each precursor ion (Noborn et al., 2015, 2016).
LC-MS/MS Data Analysis
The glycopeptide files were analyzed using both manual interpretation and automated Mascot searches for GAG-glycopeptides. The files were manually interpreted using the Xcalibur software (Thermo Fisher Scientific). Database searches were performed against Danio rerio in the UniProtKB/Swiss-Prot database and NCBI using Mascot Distiller (version 2.6.1.0, Matrix Science, London, United Kingdom) and an in-house Mascot server (version 2.5.1). Glycan structures with very small mass differences were manually evaluated at the MS2-level.
GlcAT-I Assay
GlcA-transferase assay was carried out by the methodology as previously described (Kitagawa et al., 1998; Baasanjav et al., 2011; Job et al., 2016), with slight modifications.
Detailed information regarding the procedures used is provided in Supplementary Material and Methods.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Ethics Statement
The animal study was reviewed and approved by the Ethische Commissie Dierproeven Faculteit Geneeskunde en Gezondheidswetenschappen Universiteit Gent. All animal studies were performed in agreement with EU Directive 2010/63/EU for animals, permit number: ECD 16/18.
Author Contributions
SD, AW, and FM designed the study and wrote the manuscript. SD, AD, SM, FN, JB, LA, and CG performed the research. SM, PS, GL, and SY contributed new reagents and analytic tools. SD, AD, SM, FN, JB, LA, CG, DS, PS, PC, GL, SY, AW, and FM analyzed the data. All authors reviewed the manuscript.
Funding
DS and FM were a postdoctoral fellow and a senior clinical investigator, respectively, of the Research Foundation Flanders (FWO), Belgium. This work was supported by a Methusalem Grant (BOFMET2015000401) from the Ghent University to Prof. Anne De Paepe, by grants from the FWO to DS (12Q5917N) and to FM (1842318N) and by grant (2017-00955) from the Swedish Research Council to FN and GL. CG was supported at the UW by a post-doctoral fellowship of the Belgian American Educational Fund (BAEF). This work was further supported by a Grant-in-Aid for Scientific Research (C) 19K07054 (to SM) from the Japan Society for the Promotion of Science, Japan; by the Takeda Science Foundation (to SM); Grant-in Aid for Research Center for Pathogenesis of Intractable Diseases from the Research Institute of Meijo University (SM and SY).
Conflict of Interest
PS was employed by the company Bruker MicroCT.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank the TEM facility of the Nematology Research Unit, member of the UGent TEM-Expertise Center (life sciences). We are grateful to Hanna De Saffel and Petra Vermassen for their technical assistance on processing the TEM samples. The LC-MS/MS analyses were performed at the BioMS node at the Proteomics Core Facility, Sahlgrenska Academy, University of Gothenburg with support from the Swedish National Infrastructure for Biological Mass Spectrometry (BioMS), funded by the Swedish Research Council. This manuscript has been released as a pre-print at bioRxiv (Delbaere et al., 2020a).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2020.597857/full#supplementary-material
References
Almeida, R., Levery, S. B., Mandel, U., Kresse, H., Schwientek, T., Bennett, E. P., et al. (1999). Cloning and expression of a proteoglycan UDP-galactose:beta-xylose beta1,4-galactosyltransferase I. A seventh member of the human beta4-galactosyltransferase gene family. J. Biol. Chem. 274, 26165–26171. doi: 10.1074/jbc.274.37.26165
Amsterdam, A., Nissen, R. M., Sun, Z., Swindell, E. C., Farrington, S., and Hopkins, N. (2004). Identification of 315 genes essential for early zebrafish development. Proc. Natl. Acad. Sci. U.S.A. 101, 12792–12797. doi: 10.1073/pnas.0403929101
Arratia, G., and Schultze, H. P. (1992). Reevaluation of the caudal skeleton of certain actinopterygian fishes: III. Salmonidae. Homologization of caudal skeletal structures. J. Morphol. 214, 187–249. doi: 10.1002/jmor.1052140209
Baasanjav, S., Al-Gazali, L., Hashiguchi, T., Mizumoto, S., Fischer, B., Horn, D., et al. (2011). Faulty initiation of proteoglycan synthesis causes cardiac and joint defects. Am. J. Hum. Genet. 89, 15–27. doi: 10.1016/j.ajhg.2011.05.021
Bai, X., Zhou, D., Brown, J. R., Crawford, B. E., Hennet, T., and Esko, J. D. (2001). Biosynthesis of the linkage region of glycosaminoglycans: cloning and activity of galactosyltransferase II, the sixth member of the beta 1,3-galactosyltransferase family (beta 3GalT6). J. Biol. Chem. 276, 48189–48195. doi: 10.1074/jbc.m107339200
Bird, N. C., and Mabee, P. M. (2003). Developmental morphology of the axial skeleton of the zebrafish, Danio rerio (Ostariophysi: Cyprinidae). Dev. Dyn. 228, 337–357. doi: 10.1002/dvdy.10387
Bishop, N. (2016). Bone material properties in Osteogenesis imperfecta. J. Bone Miner. Res. 31, 699–708. doi: 10.1002/jbmr.2835
Brett, J. R. (1964). The respiratory metabolism and swimming performance of young sockeye salmon. J. Fish. Res. Board Can. 21, 1183–1226. doi: 10.1139/f64-103
Bui, C., Ouzzine, M., Talhaoui, I., Sharp, S., Prydz, K., Coughtrie, M. W., et al. (2010). Epigenetics: methylation-associated repression of heparan sulfate 3-O-sulfotransferase gene expression contributes to the invasive phenotype of H-EMC-SS chondrosarcoma cells. FASEB J. 24, 436–450. doi: 10.1096/fj.09-136291
Caraffi, S. G., Maini, I., Ivanovski, I., Pollazzon, M., Giangiobbe, S., Valli, M., et al. (2019). severe peripheral joint laxity is a distinctive clinical feature of Spondylodysplastic-Ehlers-danlos syndrome (EDS)-B4GALT7 and spondylodysplastic-EDS-B3GALT6. Genes 10:799. doi: 10.3390/genes10100799
Chen, Y. H., Narimatsu, Y., Clausen, T. M., Gomes, C., Karlsson, R., Steentoft, C., et al. (2018). The GAGOme: a cell-based library of displayed glycosaminoglycans. Nat. Methods 15, 881–888. doi: 10.1038/s41592-018-0086-z
Couchman, J. R., and Pataki, C. A. (2012). An introduction to proteoglycans and their localization. J. Histochem. Cytochem. 60, 885–897. doi: 10.1369/0022155412464638
Delbaere, S., De Clercq, A., Mizumoto, S., Noborn, F., Bek, J. W., Alluyn, L., et al. (2020a). b3galt6 knock-out zebrafish recapitulate β3GalT6-deficiency disorders in human and reveal a trisaccharide proteoglycan linkage region. bioRxiv [Preprint]. doi: 10.1101/2020.06.22.165316
Delbaere, S., Van Damme, T., Syx, D., Symoens, S., Coucke, P., Willaert, A., et al. (2020b). Hypomorphic zebrafish models mimic the musculoskeletal phenotype of beta4GalT7-deficient Ehlers-Danlos syndrome. Matrix Biol. 89, 59–75. doi: 10.1016/j.matbio.2019.12.002
Dooley, K., and Zon, L. I. (2000). Zebrafish: a model system for the study of human disease. Curr. Opin. Genet. Dev. 10, 252–256. doi: 10.1016/s0959-437x(00)00074-5
Eames, B. F., Singer, A., Smith, G. A., Wood, Z. A., Yan, Y. L., He, X., et al. (2010). UDP xylose synthase 1 is required for morphogenesis and histogenesis of the craniofacial skeleton. Dev. Biol. 341, 400–415. doi: 10.1016/j.ydbio.2010.02.035
Eames, B. F., Yan, Y. L., Swartz, M. E., Levic, D. S., Knapik, E. W., Postlethwait, J. H., et al. (2011). Mutations in fam20b and xylt1 reveal that cartilage matrix controls timing of endochondral ossification by inhibiting chondrocyte maturation. PLoS Genet. 7:e1002246. doi: 10.1371/journal.pgen.1002246
Ferreira, C. R., and Gahl, W. A. (2017). Lysosomal storage diseases. Transl. Sci. Rare Dis. 2, 1–71.
Fiedler, I. A. K., Schmidt, F. N., Wolfel, E. M., Plumeyer, C., and Milovanovic, P. (2018). Severely impaired bone material quality in chihuahua zebrafish resembles classical dominant human Osteogenesis imperfecta. J. Bone Miner. Res. 33, 1489–1499. doi: 10.1002/jbmr.3445
Gillies, A. R., and Lieber, R. L. (2011). Structure and function of the skeletal muscle extracellular matrix. Muscle Nerve 44, 318–331. doi: 10.1002/mus.22094
Gistelinck, C., Gioia, R., Gagliardi, A., Tonelli, F., Marchese, L., Bianchi, L., et al. (2017). Zebrafish collagen type I: molecular and biochemical characterization of the major structural protein in bone and skin. Sci. Rep. 6:21540.
Gistelinck, C., Kwon, R. Y., Malfait, F., Symoens, S., Harris, M. P., Henke, K., et al. (2018). Zebrafish type I collagen mutants faithfully recapitulate human type I collagenopathies. Proc. Natl. Acad. Sci. U.S.A. 115, E8037–E8046.
Gistelinck, C., Witten, P. E., Huysseune, A., Symoens, S., Malfait, F., Larionova, D., et al. (2016). Loss of Type I collagen telopeptide Lysyl hydroxylation causes musculoskeletal abnormalities in a zebrafish model of bruck syndrome. J. Bone Miner. Res. 31, 1930–1942. doi: 10.1002/jbmr.2977
Gotting, C., Kuhn, J., Zahn, R., Brinkmann, T., and Kleesiek, K. (2000). Molecular cloning and expression of human UDP-d-Xylose:proteoglycan core protein beta-d-xylosyltransferase and its first isoform XT-II. J. Mol. Biol. 304, 517–528. doi: 10.1006/jmbi.2000.4261
Grabner, B., Landis, W. J., Roschger, P., Rinnerthaler, S., Peterlik, H., Klaushofer, K., et al. (2001). Age- and genotype-dependence of bone material properties in the osteogenesis imperfecta murine model (oim). Bone 29, 453–457. doi: 10.1016/s8756-3282(01)00594-4
Guo, M. H., Stoler, J., Lui, J., Nilsson, O., Bianchi, D. W., Hirschhorn, J. N., et al. (2013). Redefining the progeroid form of Ehlers-Danlos syndrome: report of the fourth patient with B4GALT7 deficiency and review of the literature. Am. J. Med. Genet. A 161A, 2519–2527.
Holmborn, K., Habicher, J., Kasza, Z., Eriksson, A. S., Filipek-Gorniok, B., Gopal, S., et al. (2012). On the roles and regulation of chondroitin sulfate and heparan sulfate in zebrafish pharyngeal cartilage morphogenesis. J. Biol. Chem. 287, 33905–33916. doi: 10.1074/jbc.m112.401646
Hur, M., Gistelinck, C. A., Huber, P., Lee, J., Thompson, M. H., Monstad-Rios, A. T., et al. (2017). MicroCT-based phenomics in the zebrafish skeleton reveals virtues of deep phenotyping in a distributed organ system. eLife 6:e26014.
Huysseune, A., and Sire, J. Y. (1992). Bone and cartilage resorption in relation to tooth development in the anterior part of the mandible in cichlid fish: a light and TEM study. Anat. Rec. 234, 1–14. doi: 10.1002/ar.1092340102
Job, F., Mizumoto, S., Smith, L., Couser, N., Brazil, A., Saal, H., et al. (2016). Functional validation of novel compound heterozygous variants in B3GAT3 resulting in severe osteopenia and fractures: expanding the disease phenotype. BMC Med. Genet. 17:86. doi: 10.1186/s12881-016-0344-9
Jones, K. L., Schwarze, U., Adam, M. P., Byers, P. H., and Mefford, H. C. (2015). A homozygous B3GAT3 mutation causes a severe syndrome with multiple fractures, expanding the phenotype of linkeropathy syndromes. Am. J. Med. Genet. A 167A, 2691–2696. doi: 10.1002/ajmg.a.37209
Kalamajski, S., and Oldberg, A. (2010). The role of small leucine-rich proteoglycans in collagen fibrillogenesis. Matrix Biol. 29, 248–253. doi: 10.1016/j.matbio.2010.01.001
Kitagawa, H., Tone, Y., Tamura, J., Neumann, K. W., Ogawa, T., Oka, S., et al. (1998). Molecular cloning and expression of glucuronyltransferase I involved in the biosynthesis of the glycosaminoglycan-protein linkage region of proteoglycans. J. Biol. Chem. 273, 6615–6618. doi: 10.1074/jbc.273.12.6615
Kusche-Gullberg, M., and Kjellen, L. (2003). Sulfotransferases in glycosaminoglycan biosynthesis. Curr. Opin. Struct. Biol. 13, 605–611. doi: 10.1016/j.sbi.2003.08.002
Lindahl, U., Couchman, J., Kimata, K., and Esko, J. D. (2015). “Proteoglycans and sulfated glycosaminoglycans,” in Essentials of Glycobiology, 3rd Edn, eds A. Varki, R. D. Cummings, J. D. Esko, P. Stanley, G. W. Hart, M. Aebi, et al. (Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press), 207–221.
Malfait, F., Francomano, C., Byers, P., Belmont, J., Berglund, B., Black, J., et al. (2017). The 2017 international classification of the Ehlers-Danlos syndromes. Am. J. Med. Genet. C Semin. Med. Genet. 175, 8–26.
Malfait, F., Kariminejad, A., Van Damme, T., Gauche, C., Syx, D., Merhi-Soussi, F., et al. (2013). Defective initiation of glycosaminoglycan synthesis due to B3GALT6 mutations causes a pleiotropic Ehlers-Danlos-Syndrome-like connective tissue disorder. Am. J. Hum. Genet. 92, 935–945. doi: 10.1016/j.ajhg.2013.04.016
McMenamin, S. K., Chandless, M. N., and Parichy, D. M. (2016). Working with zebrafish at postembryonic stages. Methods Cell Biol. 134, 587–607. doi: 10.1016/bs.mcb.2015.12.001
Mizumoto, S., Mikami, T., Yasunaga, D., Kobayashi, N., Yamauchi, H., Miyake, A., et al. (2009). Chondroitin 4-O-sulfotransferase-1 is required for somitic muscle development and motor axon guidance in zebrafish. Biochem. J. 419, 387–399. doi: 10.1042/bj20081639
Mizumoto, S., and Sugahara, K. (2012). Glycosaminoglycan chain analysis and characterization (glycosylation/epimerization). Methods Mol. Biol. 836, 99–115. doi: 10.1007/978-1-61779-498-8_7
Morin-Kensicki, E. M., Melancon, E., and Eisen, J. S. (2002). Segmental relationship between somites and vertebral column in zebrafish. Development 129, 3851–3860.
Nakajima, M., Mizumoto, S., Miyake, N., Kogawa, R., Iida, A., Ito, H., et al. (2013). Mutations in B3GALT6, which encodes a glycosaminoglycan linker region enzyme, cause a spectrum of skeletal and connective tissue disorders. Am. J. Hum. Genet. 92, 927–934. doi: 10.1016/j.ajhg.2013.04.003
Nash, R., Valencia, A. H., and Geffen, A. J. (2006). The origin of Fulton’s condition factor - Setting the record straight. Fisheries 31, 236–238.
Nissen, R. M., Amsterdam, A., and Hopkins, N. (2006). A zebrafish screen for craniofacial mutants identifies wdr68 as a highly conserved gene required for endothelin-1 expression. BMC Dev. Biol. 6:28. doi: 10.1186/1471-213X-6-28
Noborn, F., Gomez Toledo, A., Green, A., Nasir, W., Sihlbom, C., Nilsson, J., et al. (2016). Site-specific identification of heparan and chondroitin sulfate glycosaminoglycans in hybrid proteoglycans. Sci. Rep. 6:34537. doi: 10.1038/srep34537
Noborn, F., Gomez Toledo, A., Sihlbom, C., Lengqvist, J., Fries, E., Kjellen, L., et al. (2015). Identification of chondroitin sulfate linkage region glycopeptides reveals prohormones as a novel class of proteoglycans. Mol. Cell Proteom. 14, 41–49. doi: 10.1074/mcp.m114.043703
Parichy, D. M., Elizondo, M. R., Mills, M. G., Gordon, T. N., and Engeszer, R. E. (2009). Normal table of postembryonic zebrafish development: staging by externally visible anatomy of the living fish. Dev. Dyn. 238, 2975–3015. doi: 10.1002/dvdy.22113
Persson, A., Nilsson, J., Vorontsov, E., Noborn, F., and Larson, G. (2019). Identification of a non-canonical chondroitin sulfate linkage region trisaccharide. Glycobiology 29, 366–371. doi: 10.1093/glycob/cwz014
Plaut, I. (2000). Effects of fin size on swimming performance, swimming behaviour and routine activity of zebrafish Danio rerio. J. Exp. Biol. 203(Pt 4), 813–820.
Prydz, K., and Dalen, K. T. (2000). Synthesis and sorting of proteoglycans - commentary. J. Cell Sci. 113, 193–205.
Ritelli, M., Chiarelli, N., Zoppi, N., Dordoni, C., Quinzani, S., Traversa, M., et al. (2015). Insights in the etiopathology of galactosyltransferase II (GalT-II) deficiency from transcriptome-wide expression profiling of skin fibroblasts of two sisters with compound heterozygosity for two novel B3GALT6 mutations. Mol. Genet. Metab. Rep. 2, 1–15. doi: 10.1016/j.ymgmr.2014.11.005
Santoriello, C., and Zon, L. I. (2012). Hooked! modeling human disease in zebrafish. J. Clin. Invest. 122, 2337–2343. doi: 10.1172/jci60434
Selleck, S. B. (2000). Proteoglycans and pattern formation: sugar biochemistry meets developmental genetics. Trends Genet. 16, 206–212. doi: 10.1016/s0168-9525(00)01997-1
Sugahara, K., Mikami, T., Uyama, T., Mizuguchi, S., Nomura, K., and Kitagawa, H. (2003). Recent advances in the structural biology of chondroitin sulfate and dermatan sulfate. Curr. Opin. Struct. Biol. 13, 612–620. doi: 10.1016/j.sbi.2003.09.011
Van Damme, T., Pang, X., Guillemyn, B., Gulberti, S., Syx, D., De Rycke, R., et al. (2018). Biallelic B3GALT6 mutations cause spondylodysplastic Ehlers-Danlos syndrome. Hum. Mol. Genet. 27, 3475–3487. doi: 10.1093/hmg/ddy234
Viguet-Carrin, S., Garnero, P., and Delmas, P. D. (2006). The role of collagen in bone strength. Osteoporos. Int. 17, 319–336. doi: 10.1007/s00198-005-2035-9
Wen, J., Xiao, J., Rahdar, M., Choudhury, B. P., Cui, J., Taylor, G. S., et al. (2014). Xylose phosphorylation functions as a molecular switch to regulate proteoglycan biosynthesis. Proc. Natl. Acad. Sci. U.S.A. 111, 15723–15728. doi: 10.1073/pnas.1417993111
Keywords: b3galt6, zebrafish, trisaccharide linkage region, proteoglycans, linkeropathies
Citation: Delbaere S, De Clercq A, Mizumoto S, Noborn F, Bek JW, Alluyn L, Gistelinck C, Syx D, Salmon PL, Coucke PJ, Larson G, Yamada S, Willaert A and Malfait F (2020) b3galt6 Knock-Out Zebrafish Recapitulate β3GalT6-Deficiency Disorders in Human and Reveal a Trisaccharide Proteoglycan Linkage Region. Front. Cell Dev. Biol. 8:597857. doi: 10.3389/fcell.2020.597857
Received: 22 August 2020; Accepted: 17 November 2020;
Published: 10 December 2020.
Edited by:
Mario Antonio Bianchet, Johns Hopkins University, United StatesReviewed by:
Ross F. Collery, Medical College of Wisconsin, United StatesJoseph M. Chambers, Manchester University, United States
Copyright © 2020 Delbaere, De Clercq, Mizumoto, Noborn, Bek, Alluyn, Gistelinck, Syx, Salmon, Coucke, Larson, Yamada, Willaert and Malfait. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fransiska Malfait, RnJhbnNpc2thLk1hbGZhaXRAVUdlbnQuYmU=
†These authors have contributed equally to this work