 Yunyun Fang
Yunyun Fang Linlin Ji1†
Linlin Ji1† Jingjing Zhang
Jingjing Zhang Li Wei
Li Wei- 1Shanghai Key Laboratory of Diabetes Mellitus, Department of Endocrinology and Metabolism, Shanghai Diabetes Institute, Shanghai Clinical Center for Diabetes, Shanghai Key Clinical Center for Metabolic Disease, Shanghai Jiao Tong University Affiliated Sixth People’s Hospital, Shanghai, China
- 2National Demonstration Center for Experimental Fisheries Science Education, Shanghai Ocean University, Shanghai, China
- 3Department of Endocrinology and Metabolism, Shanghai Eighth People’s Hospital, Shanghai, China
Liraglutide, a glucagon-like peptide-1 receptor agonist (GLP-1RA), has been demonstrated to alleviate non-alcoholic fatty liver disease (NAFLD). However, the underlying mechanism has not been fully elucidated. Increasing evidence suggests that autophagy is involved in the pathogenesis of hepatic steatosis. In this study, we examined whether liraglutide could alleviate hepatic steatosis through autophagy-dependent lipid degradation and investigated the underlying mechanisms. Herein, the effects of liraglutide on NAFLD were evaluated in a high-fat diet (HFD)-induced mouse model of NAFLD as well as in mouse primary and HepG2 hepatocytes exposed to palmitic acid (PA). The expression of the GLP-1 receptor (GLP-1R) was measured in vivo and in vitro. Oil red O staining was performed to detect lipid accumulation in hepatocytes. Electron microscopy was used to observe the morphology of autophagic vesicles and autolysosomes. Autophagic flux activity was measured by infecting HepG2 cells with mRFP-GFP-LC3 adenovirus. The roles of GLP-1R and transcription factor EB (TFEB) in autophagy-lysosomal activation were explored using small interfering RNA. Liraglutide treatment alleviated hepatic steatosis in vivo and in vitro. In models of hepatic steatosis, microtubule-associated protein 1B light chain-3-II (LC3-II) and SQSTM1/P62 levels were elevated in parallel to blockade of autophagic flux. Liraglutide treatment restored autophagic activity by improving lysosomal function. Furthermore, treatment with autophagy inhibitor chloroquine weakened liraglutide-induced autophagy activation and lipid degradation. TFEB has been identified as a key regulator of lysosome biogenesis and autophagy. The protein levels of nuclear TFEB and its downstream targets CTSB and LAMP1 were decreased in hepatocytes treated with PA, and these decreases were reversed by liraglutide treatment. Knockdown of TFEB expression compromised the effects of liraglutide on lysosome biogenesis and hepatic lipid accumulation. Mechanistically, GLP-1R expression was decreased in HFD mouse livers as well as PA-stimulated hepatocytes, and liraglutide treatment reversed the downregulation of GLP-1R expression in vivo and in vitro. Moreover, GLP-1R inhibition could mimic the effect of the TFEB downregulation-mediated decrease in lysosome biogenesis. Thus, our findings suggest that liraglutide attenuated hepatic steatosis via restoring autophagic flux, specifically the GLP-1R-TFEB-mediated autophagy-lysosomal pathway.
Introduction
Non-alcoholic fatty liver disease (NAFLD), characterized by excessive triglyceride (TG) accumulation in the liver, is commonly associated with insulin resistance, obesity, diabetes, and cardiovascular disease (Arab et al., 2018). Currently, the prevalence of NAFLD is estimated to be approximately 10–35% in the general population, making it the most common liver disease worldwide (Vernon et al., 2011). Thus, there is an urgent need to develop new preventive and therapeutic strategies to alleviate NAFLD.
Liraglutide, a glucagon-like peptide-1 receptor agonist (GLP-1RA), exhibits beneficial effects on weight loss, cardiovascular function, and NAFLD in addition to its glucose-lowering effect, especially in patients with obesity (Drucker, 2018; Muller et al., 2019). Previous studies suggested that liraglutide could alleviate high-fat diet (HFD)−induced hepatic lipid accumulation in a weight loss-independent manner (Lyu et al., 2020). However, the mechanism underlying the effects of liraglutide on NAFLD remains unclear.
Autophagy is an important lysosome-mediated process for the degradation of cellular components, including damaged organelles and misfolded proteins, for the maintenance of intracellular energy homeostasis (Yang and Klionsky, 2010). Increasing evidence has demonstrated that autophagy is necessary for regulating lipid metabolism because lipid droplets are degraded through lipophagy (Singh et al., 2009; Cui et al., 2020). Disruption of autophagy might therefore contribute to the aggravation of hepatic lipid accumulation (Wu et al., 2020). Recent studies revealed that liraglutide could alleviate hepatic steatosis, reduce oxidative stress, and activate autophagy (Tong et al., 2016). However, the exact mechanisms underlying these effects have not been fully elucidated.
Lysosomes are organelles involved in the process of intracellular substrate degradation through autophagosome-lysosome fusion and play a critical role in regulating autophagic flux and lipid droplet clearance. Normally, lysosomal lipid degradation capacity depends on a sufficient number of lysosomes and the activation of acid hydrolase enzymes (Sinha et al., 2014; Pi et al., 2019). Transcription factor EB (TFEB), a key member of the microphthalmia family (MiT/TFE), has been identified as a key regulator of lysosome biogenesis and autophagy (Medina et al., 2011). Under normal conditions, TFEB is inactive and in the cytosol, whereas fasting and stress conditions induce its dephosphorylation-mediated activation and subsequent nuclear translocation. TFEB activation upon nuclear translocation may promote lysosomal biogenesis and substrate degradation (Zhang et al., 2018). Recently, TFEB has emerged as a key regulator of the lysosomal degradation of lipid droplets (Su et al., 2018). Moreover, deficient lysosomal clearance, resulting from reduced lysosome quantity and hydrolytic enzyme activity, was implicated in the development of NAFLD (Wang et al., 2019). These findings highlight a potential role of TFEB in the treatment of NAFLD via the promotion of autophagy-dependent lipid degradation. However, whether TFEB mediates the beneficial effect of liraglutide on hepatic autophagic flux and lipid degradation remains unclear.
In this study, we assessed the effects of liraglutide on hepatic steatosis and investigated the underlying molecular mechanisms. Our results revealed that autophagy activation mediated the effect of liraglutide in decreasing hepatic steatosis. Moreover, liraglutide activated autophagic flux and lipid degradation via the GLP-1R-TFEB-mediated autophagy-lysosomal pathway.
Materials and Methods
Animal Treatment
Eight-week-old C57BL/6J mice weighting 23–28 g were purchased from the Model Animal Research Center of Nanjing University. All animals were maintained in a specific pathogen-free barrier facility under a 12 h light/dark cycle. After a week of adaptation, the mice were randomly divided into two groups (n = 10). They were fed a normal chow diet (D12450J, Research Diets) or HFD (D12492, Research Diets) for 18 weeks. The mice were then intraperitoneally administered either liraglutide (400 μg/kg/day, Novo Nordisk, Denmark) or equivoluminal 0.9% saline for another 4 weeks. Body weight was measured weekly, and food intake was recorded every 3 days. All animal procedures were conducted according to the guidelines for Experimental Animal Research.
Glucose Tolerance Test and Insulin Tolerance Test
The intraperitoneal glucose tolerance test (IPGTT) and insulin tolerance test (ITT) were performed to evaluate glucose tolerance and insulin sensitivity after 4 weeks liraglutide treatment. For IPGTT, the mice were intraperitoneally injected with glucose (2 g/kg body weight) after overnight fasting. For ITT, the mice were fasted for 6 h and injected with insulin (Humulin R, Eli Lilly and Company) at a concentration of 1 U/kg body weight. Blood glucose was monitored with a glucometer (Roche, Switzerland) at regular intervals (0, 15, 30, 60, 90, and 120 min).
Biochemical and Lipid accumulation Measurements
The mice were sacrificed after an overnight fast. Serum triglyceride (TG), total cholesterol (TC), low-density lipoprotein cholesterol (LDL-C), alanine aminotransferase (ALT), and aspartic acid transaminase (AST) levels were examined using an automatic biochemistry analyzer (Rayto Technologies, China). The TG and TC of liver tissues and hepatocytes were measured using an Enzymatic Assay Kit (Applygen Technologies, China; Nanjing Jiancheng, China) according to the manufacturer’s protocol.
Histopathological Analysis
Fresh liver specimens were fixed with 4% paraformaldehyde and embedded in paraffin. Paraffin sections were stained with H&E according to a standard procedure (Chang et al., 2010). Images were captured using a light microscope (Leica Microsystems, Germany).
Oil Red O Staining and BODIPY 493/503 Staining
To assess hepatic steatosis, liver tissues were embedded in optimum cutting temperature and immediately frozen in liquid nitrogen. Frozen liver sections were stained with Oil Red O (O0625, Sigma-Aldrich) according to the manufacturer’s instructions. For in vitro studies, the hepatocytes were washed twice with phosphate-buffered saline and fixed in 4% paraformaldehyde for 30 min. Fixed cells were stained with Oil Red O solution for 0.5–1 h and gently washed with 60% isopropanol. Three images per sample were captured using a light microscope (Leica Microsystems, Germany). Lipid droplets were also detected by labeling with the BODIPY 493/503 (D3922, Invitrogen, United States) lipid dye. The images were obtained using a confocal microscope (Carl Zeiss, Germany).
Electron Microscopy
Autophagic vacuoles and autolysosomes were observed using an electron microscope (Olympus, Japan). Fresh liver tissue pieces were fixed with 2.5% glutaraldehyde, dehydrated in a graded ethanol series, and embedded in Epon 812 (Electron Microscopy Sciences, United States).
Cell Culture and Treatments
Human hepatoma HepG2 cells were obtained from the Type Culture Collection of the Chinese Academy of Sciences (Shanghai, China). Murine primary hepatocytes were isolated from male C57BL/6J mice (8 weeks old) using the collagenase IV perfusion technique as previously described (Seglen, 1976; Klaunig et al., 1981). All cells were cultured in DMEM/HIGH GLUCOSE medium containing 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin (Gibco, United States) in a 5% CO2 incubator at 37°C. To establish a lipid-loaded cell model mimicking NAFLD in vitro, palmitic acid (PA, P9767, Sigma-Aldrich) was added to the culture medium at a final concentration of 0.2–0.6 μM. For in vitro experiments, cells were incubated in serum-free medium overnight and were then treated with or without liraglutide (S8256, Selleck Chemicals; 100–500 nM) in serum-free media containing PA or vehicle for 24 h. To examine the effect of liraglutide on hepatic autophagic flux, chloroquine (CQ, C6628, Sigma Aldrich; 50 μM), a classical autophagy inhibitor that blocks the fusion of autophagosomes with lysosomes, was added to the medium for 4–6 h as a negative control. To investigate the TFEB-mediated effects of liraglutide on lysosome biogenesis and autophagy, HepG2 cells were transfected with small interfering RNA (siRNA) targeted against TFEB (Genepharma, China) for 48 h using Lipofectamine 2000 (Thermo Fisher Scientific, United States). The siRNA sequences used are as follows: sense 5′-GACGAAGGUUCAACAUCAATT-3′ and antisense 5′-UUGAUGUUGAACCUUCGUCTT-3′. To further investigate the mechanism of TFEB nuclear translocation, HepG2 cells were transfected with GLP-1R plasmid (Hanbio Biotechnology, China) and small interfering RNA (siRNA) targeted against GLP-1R (Genepharma, China). The siRNA sequences used were as follows: sense 5′-GGACCAGGAACUCCAACAUTT-3′ and antisense 5′-AUGUUGGAGUUCCUGGUCCTT-3′.
Western Blot and Immunofluorescence Analysis
Total protein from liver tissue and hepatocytes was extracted with RIPA Lysis Buffer (P0013B, Beyotime, China) at 4°C. Nuclear and cytoplasmic fractions were separated for analysis of TFEB translocation using a commercial kit (78835, Thermo Fisher Scientific, United States) according to the manufacturer’s instructions. Protein samples (20–30 μg) were separated using SDS-PAGE (Bio-Rad Laboratories, United States) and transferred to PVDF membranes (Millipore Corporation, United States). The membranes were blocked with 5% skimmed milk and incubated with primary antibody for 12 h followed by incubation with horseradish peroxidase-conjugated secondary antibodies. Primary antibodies against LC3A/B (1:1,000, 12741), Beclin1 (1:1,000, 3495), Atg5 (1:1,000, 12994), Atg7 (1:1,000, 8558), GAPDH (1:1,000, 2118s), cathepsin B (1:1,000, 31718), p62 (1:1,000, 5114s), and β-actin (1:1,000, 4970s) were obtained from Cell Signaling Technology (United States). Antibodies against lysosome-associated membrane protein 1 (LAMP1) (1:1,000, sc-20011) and SIRT1 (1:1,000, sc-15404) were from Santa Cruz Biotechnology, and the anti-TFEB (1:2,000, A303-673A) antibody was from Bethyl Laboratories (United States). The antibody against GLP-1R (1:1,000, ab39072) was from Abcam (United Kingdom). Membrane signals were detected using an enhanced chemiluminescence method (Thermo Fisher Scientific, United States). The band intensities were quantitated using ImageJ software. For immunofluorescence, cells were fixed with 4% paraformaldehyde for 15 min at 37°C. After fixation, the cells were permeabilized with 0.25% Triton X-100 for 15 min, blocked with 2% BSA for 30 min, and then incubated with an anti-TFEB antibody (1: 100, MAB9170-100, R&D Systems, United States) overnight at 4°C. The next day, cells were stained with a secondary antibody for 1 h and DAPI for 5 min. Immunofluorescence was visualized using a fluorescent microscope (Leica Microsystems, Germany).
Analysis of Autophagic Flux
For autophagic flux measurement, human hepatoma HepG2 cells were transfected with adenovirus harboring mRFP-GFP-LC3 (Hanbio Biotechnology, China). The cells were then treated with 0.4 μM PA, liraglutide, or CQ for 24 h. After treatment, yellow and red puncta were detected using a confocal microscope (Carl Zeiss, Germany).
Statistical Analysis
The data are expressed as the mean ± standard error of the mean (SEM). Unpaired two-tailed t-tests were used for intergroup comparisons. One-way analysis of variance with post hoc Bonferroni tests was performed for comparisons between multiple groups.
Results
Liraglutide Improves Systemic Glucose and Lipid Homeostasis in HFD-Fed Mice
To explore the beneficial effect of liraglutide on hepatic lipid accumulation, C57BL/6 mice were fed a HFD or chow diet for 18 weeks and were then administered liraglutide (400 μg/kg/day) or saline for another 4 weeks. From the 4th week of intervention, HFD-fed mice exhibited a higher body weight than control mice, which was the case until the end of the experiment (Figure 1A). Liraglutide treatment significantly decreased body weight in HFD-fed mice (Figure 1B[[AQ11]]). Total food intake was decreased in both liraglutide-treated controls and HFD-fed mice (Figure 1D). Further, although liraglutide treatment induced a transient reduction of food intake during the first 2 weeks, no significant differences were found between the HFD groups treated with or without liraglutide during 3–4 weeks (Supplementary Figure S1). In addition, GTT and ITT indicated that liraglutide alleviated glucose intolerance and insulin resistance in HFD-fed mice (Figures 1E,F). However, no significant differences were observed in GTT results between these groups after blood glucose was adjusted as a% of initial glucose (Supplementary Figures S2A,B). Regarding systemic lipid metabolism, as shown in Figures 1G–I, the serum levels of TG, TC, and LDL-C in HFD-fed mice were decreased by liraglutide treatment. Taken together, these results suggest that liraglutide reduced body weight and restored systemic glucose and lipid homeostasis in diet-induced obese mice.
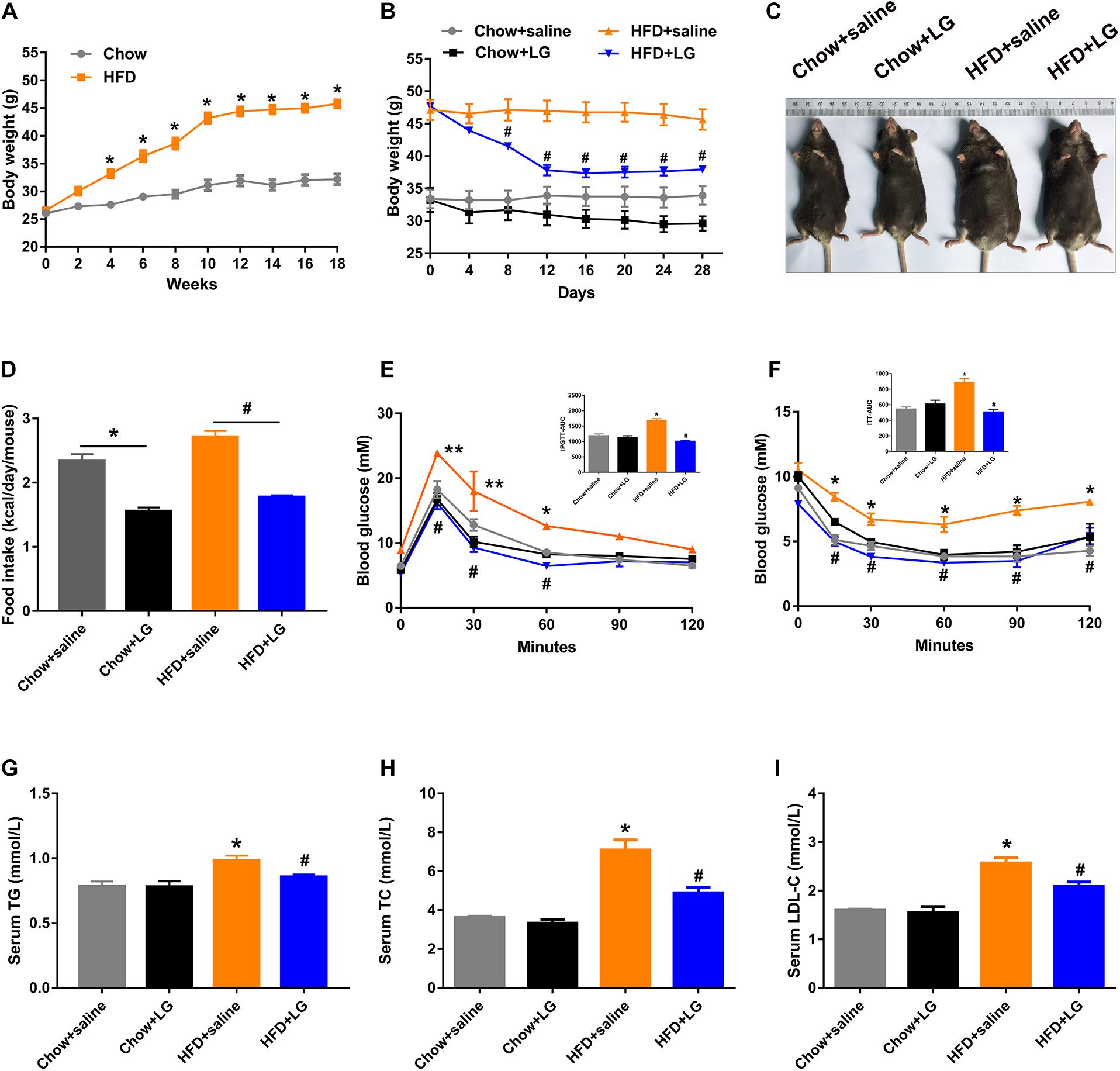
Figure 1. Liraglutide improves systemic glucose and lipid metabolism in HFD mice. (A) Body weight before intervention. (B) Body weight during liraglutide treatment. (C) Representative photos of chow diet- and HFD-fed mice treated with liraglutide (400 μg/kg) or saline daily for 4 weeks. (D) Food intake (kcal/day/mouse). (E,F) IPGTT and ITT results were obtained after liraglutide treatment. AUC: the area under the curve. (G–I) Serum triglycerides (TG), cholesterol (TC), and LDL-C levels. The data are expressed as mean ± SEM; n = 3–5. *P < 0.01, **P < 0.05 vs. Chow + saline group; #P < 0.01 vs. HFD + saline group.
Liraglutide Mitigates HFD-Induced Hepatic Steatosis and Liver Injury
Since liraglutide improved systemic lipid homeostasis, we further investigated its effects on HFD-induced hepatic steatosis. H&E and Oil red O staining revealed that liraglutide treatment visibly attenuated HFD-induced hepatic lipid accumulation, with a reduced number of intracellular lipid droplets and ballooning hepatocytes (Figure 2A). Consistent with these changes, liver weight and hepatic TG content in HFD mice were significantly higher than those in chow diet-fed mice. Liraglutide also decreased liver weight and hepatic TG accumulation in HFD-fed mice. No significant differences were observed in TC content between these groups (Figures 2B–D). Biochemical analysis revealed that elevated serum ALT and AST levels in HFD group mice were clearly decreased by liraglutide (Figures 2E,F). Collectively, these results indicated that liraglutide exerted its hepatoprotective effects by alleviating hepatic steatosis and liver injury.
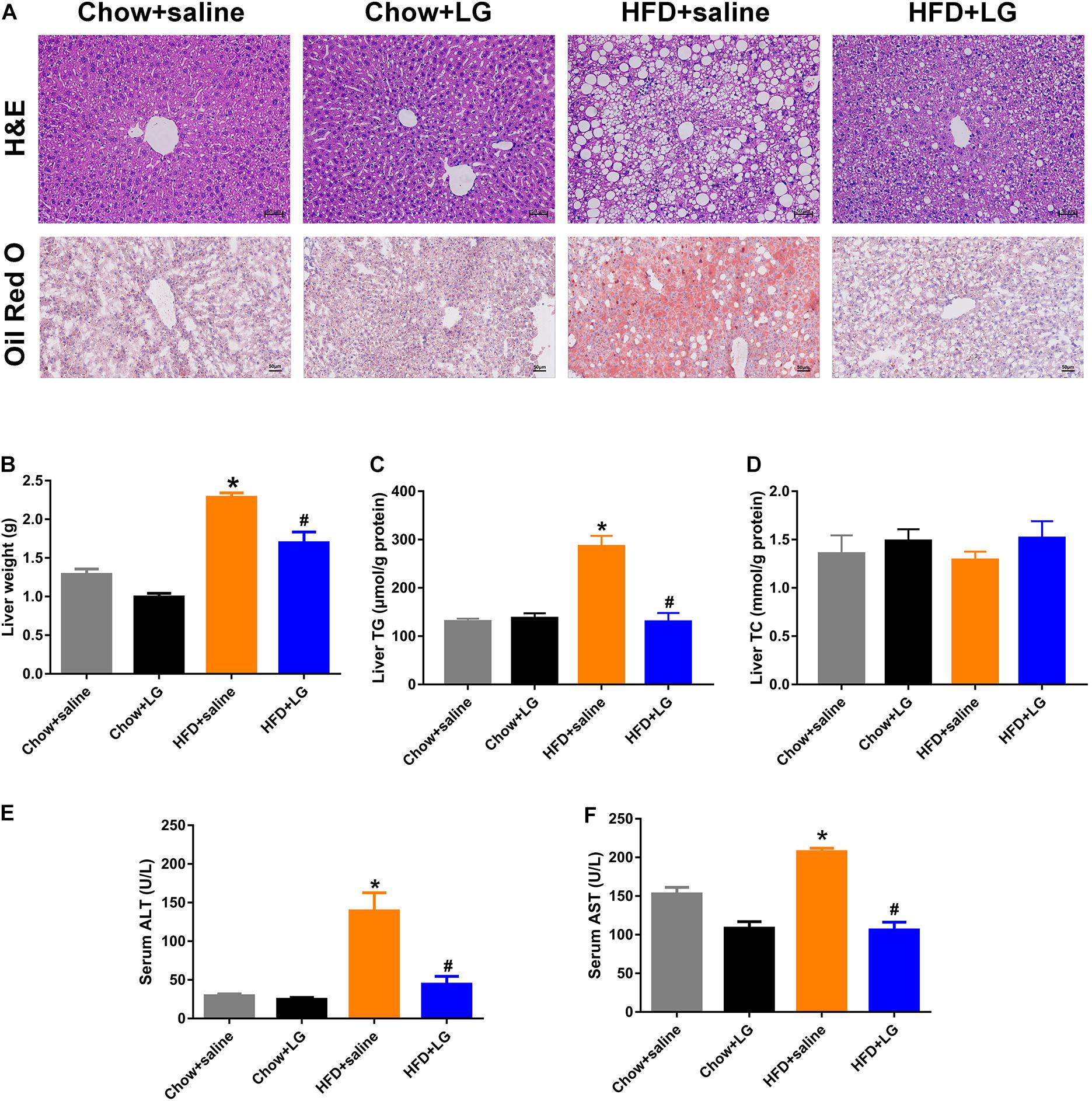
Figure 2. Liraglutide attenuates hepatic steatosis and liver injury in HFD mice. (A) H&E and Oil red O staining of liver tissue (scale bars = 50 μm). (B) Liver weight (g). (C,D) Liver TG and TC levels. (E,F) Serum ALT and AST levels. The data are expressed as mean ± SEM; n = 3–5. *P < 0.01 vs. Chow + saline group; #P < 0.01 vs. HFD + saline group.
Liraglutide Attenuates PA-Induced Lipid Accumulation in vitro
To confirm whether liraglutide could alleviate hepatic lipid accumulation in vitro, primary mouse hepatocytes and HepG2 cells were employed. As shown in Figures 3A,E, lipid accumulation was significantly increased in both PA-stimulated primary mouse hepatocytes and HepG2 cells, an effect which was suppressed by co-incubation with liraglutide. Liraglutide treatment also significantly decreased intracellular TG content in these cell models (Figures 3C,G). However, no significant differences were observed in TC content (Figures 3D,H). Our results revealed that liraglutide directly reduced hepatic steatosis in vitro.
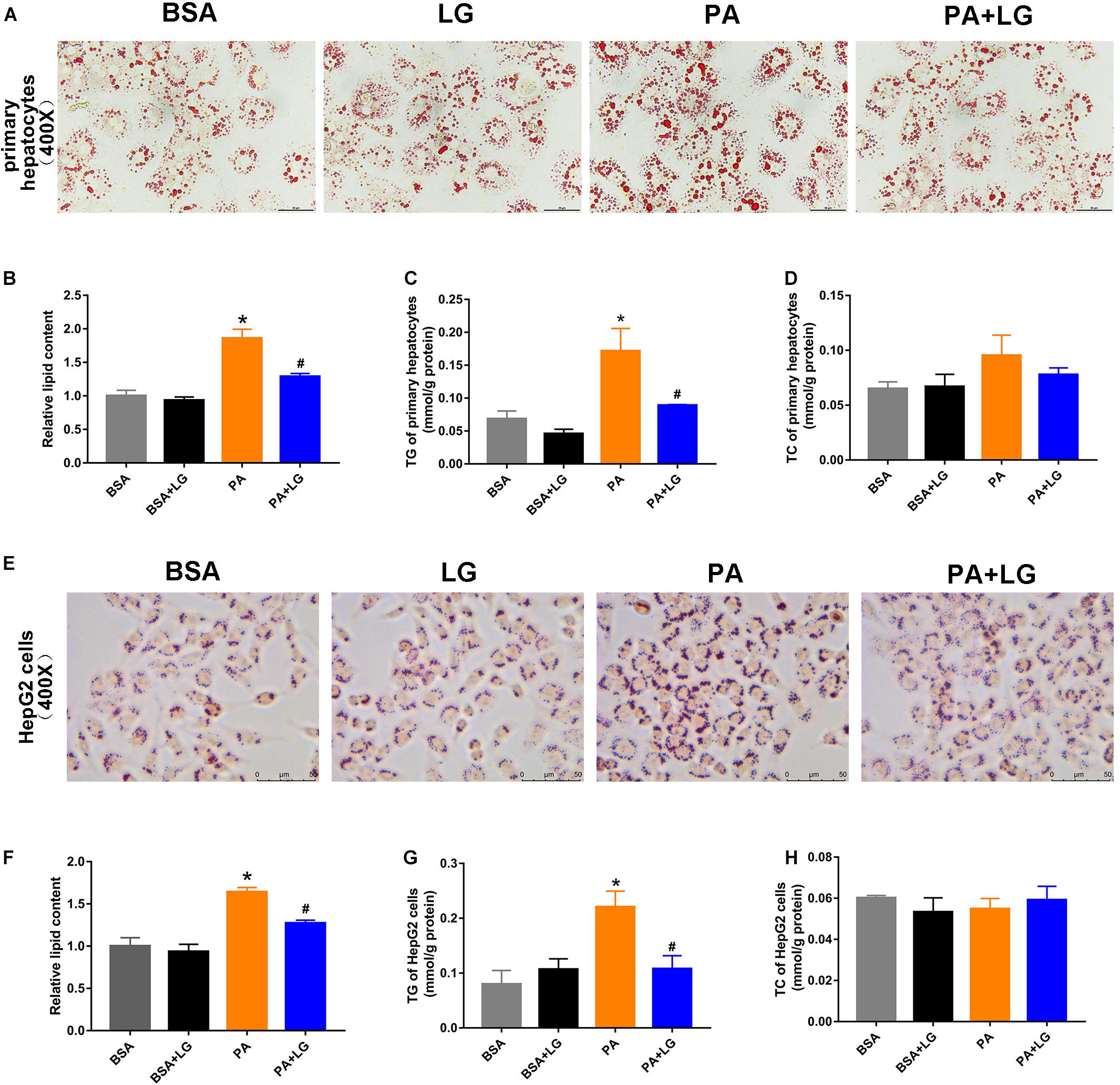
Figure 3. Liraglutide attenuates PA-induced lipid accumulation in primary mouse hepatocytes and HepG2 cells. (A,E) Primary mouse hepatocytes and HepG2 cells were co-incubated with PA and liraglutide for 24 h. Lipid accumulation was assessed by Oil red O staining (scale bars = 50 μm). (B,F) Relative lipid content. (C,G) Hepatocyte TG content. (D,H) Hepatocyte TC content. The data are expressed as mean ± SEM; n = 3. *P < 0.05 vs. BSA group; #P < 0.05 vs. PA group.
Liraglutide Induces Autophagy in HFD Mice
To explore whether liraglutide alleviated hepatic steatosis in HFD mice by activating autophagy, EM was performed. We observed large numbers of lipid droplets and autophagosome-like vesicles in the livers of HFD mice. These vesicles might derive from the sequestration of small lipid droplets or a portion of large lipid droplets, suggestive of lipid-laden autophagosomes. In contrast, virtually no lipid droplets were observed in liver sections from liraglutide-treated HFD mice, as intracellular lipids were engulfed and degraded within the autolysosome compartment (Figures 4A–C). Western blot revealed that the levels of autophagy indicators Atg7 and Beclin1 were lower in HFD mice, while those of LC3-II and autophagic selective substrate SQSTM1/p62 were significantly higher, indicating a blockade of autophagy-lysosomal degradation. Liraglutide treatment restored HFD-inhibited autophagy by increasing the protein expression of Atg7 and Beclin1 (Figures 4D,E) and decreasing the expression of LC3-II and p62 (Figures 4F,G). These findings suggest that hepatic steatosis impaired autophagy degradation, while liraglutide alleviated lipid accumulation by promoting autophagy-lysosomal−dependent lipid degradation.
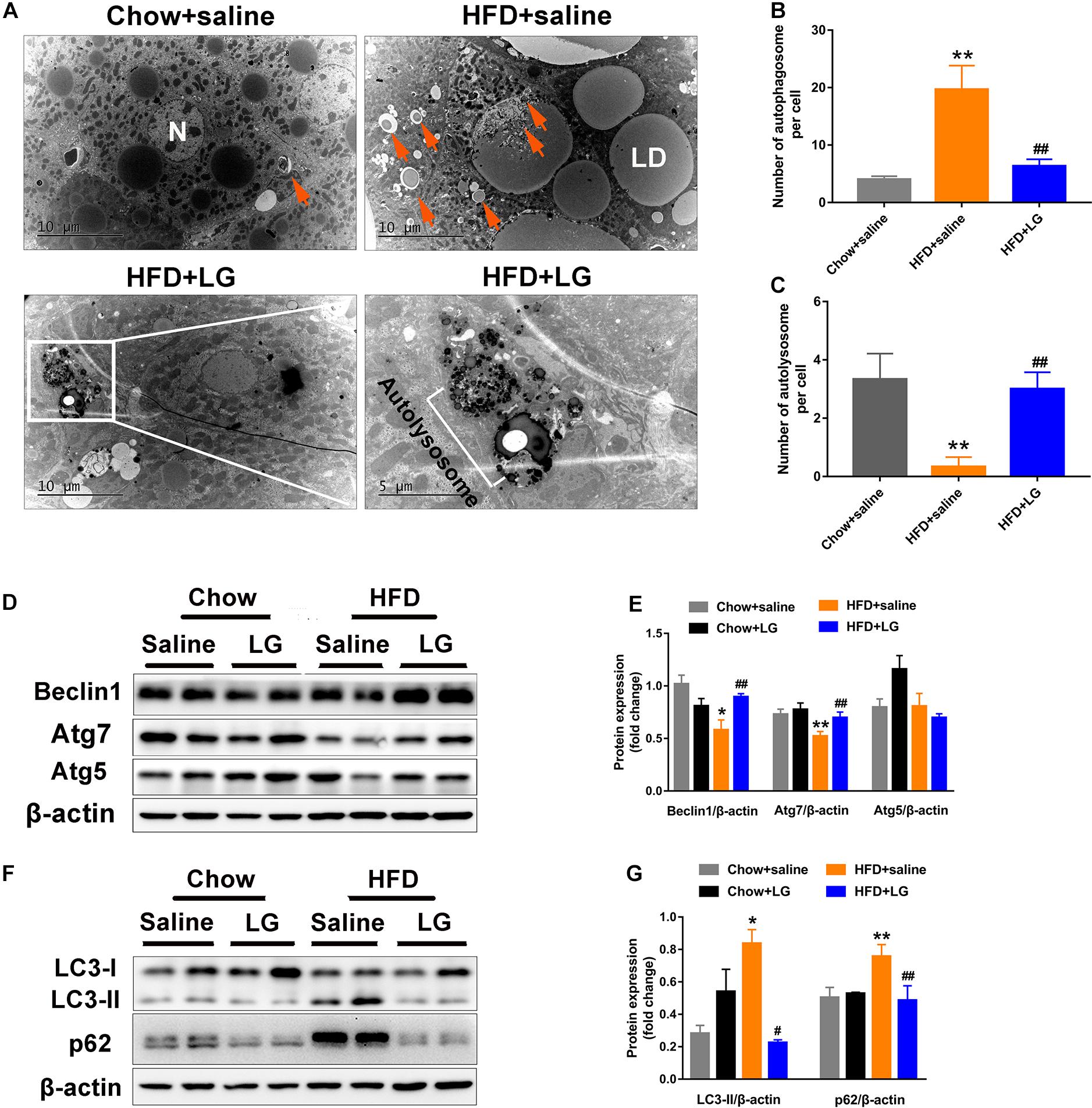
Figure 4. Liraglutide activates autophagy in HFD-fed mice. (A) Representative electron microscopy images of autophagic vacuoles in the liver. Orange arrows indicate autophagic vacuoles (partly containing lipid droplets). Boxed area indicates autolysosome. High magnification of boxed areas in the liraglutide treatment group is presented on the right (scale bars = 10, 5 μm). N, nucleus; LD, lipid droplet; arrows, autophagosome; white box, autolysosome. (B) Quantitative analysis of number of autophagosomes per cell. (C) Quantitative analysis of number of autolysosomes per cell. (D,E) Expression of autophagy-associated proteins Atg5, Atg7, and Beclin1 in livers of HFD-fed mice analyzed by western blot. (F,G) Expression of autophagy-associated proteins LC3-II and autophagic selective substrate p62 in livers of HFD-fed mice analyzed by western blot. The data are expressed as mean ± SEM; n = 3. *P < 0.01, **P < 0.05 vs. Chow + saline group; #P < 0.01 ##P < 0.05 vs. HFD + saline group.
Liraglutide Attenuates PA-Induced Lipid Accumulation by Enhancing Autophagic Flux
To further explore the mechanism underlying the liraglutide-mediated alleviation of hepatic steatosis via autophagic flux activation, primary mouse hepatocytes were isolated and incubated with free fatty acids. An increase in LC3-II and p62 levels was observed in hepatocytes after PA treatment, whereas oleic acid (OA) had no effect on the expression of these proteins (Supplementary Figure S3A). Furthermore, the expression of LC3-II and p62 increased in PA-stimulated primary mouse hepatocytes in a dose-dependent manner (Supplementary Figure S1B). However, the increase was reversed by liraglutide, indicating that liraglutide alleviated the PA−triggered dysfunction of autolysosomal degradation (Figure 5A). Moreover, HepG2 cells were infected with mRFP-GFP-LC3 adenovirus to confirm whether liraglutide could promote autophagic flux (Figures 5B,C). When an Ad-mRFP-GFP-LC3 construct is used, GFP fluorescence only indicates autophagosomes, since it can be easily quenched in the acidic pH of autolysosomes. However, mRFP detects both autophagosomes and autolysosomes, since it is more stable under acidic conditions. Using this approach, we observed that the ratio of mRFP to GFP signals was significantly decreased in PA-stimulated HepG2 cells, suggesting the accumulation of non-acidic autophagosomes. In liraglutide−treated cells, more mRFP signals were observed, indicating that autophagic flux was enhanced without impeding autophagosome-lysosome fusion/or autolysosome function. In addition, blocking fusion by CQ weakened the liraglutide-increased ratio of mRFP to GFP signals.
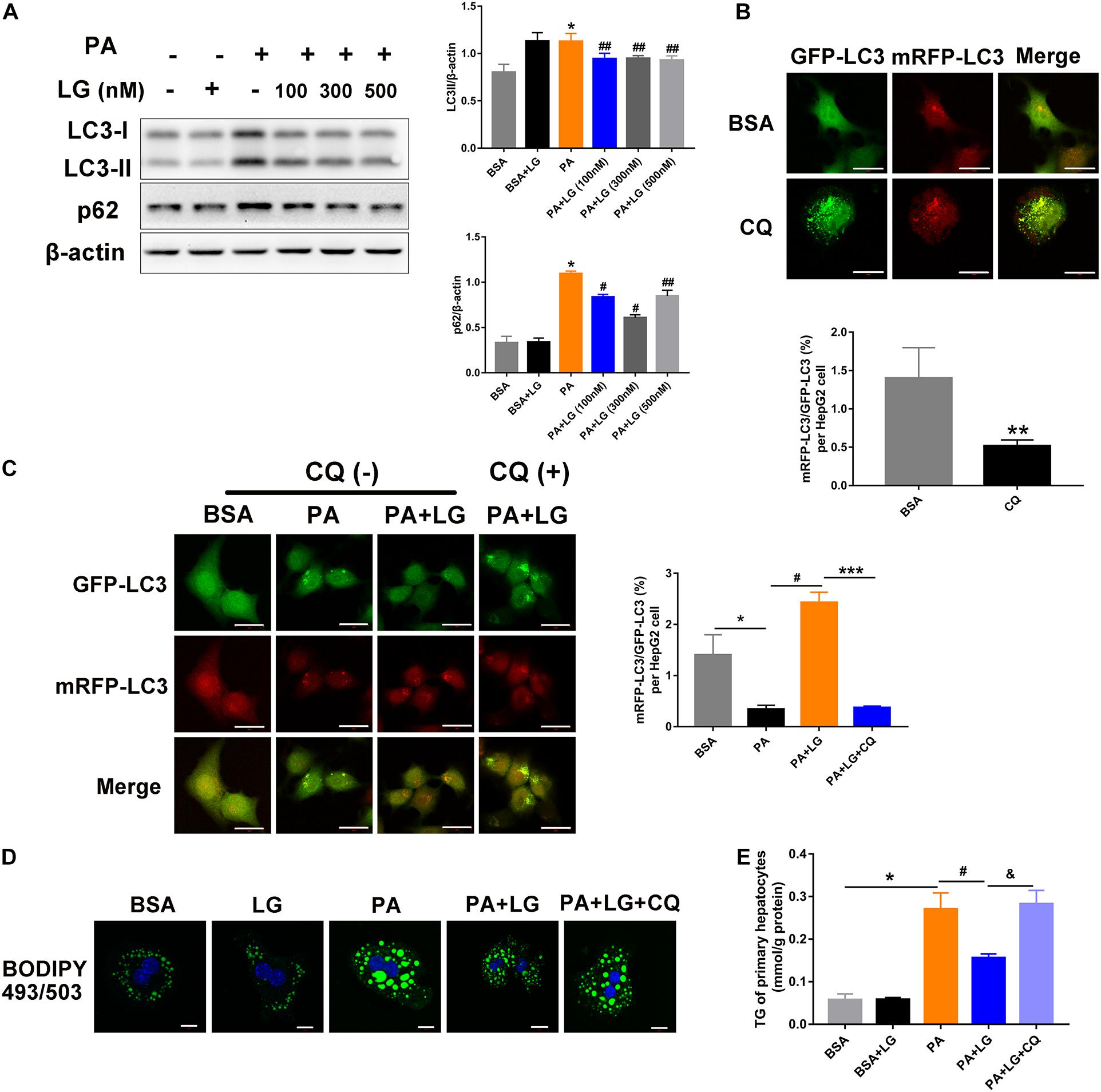
Figure 5. Liraglutide attenuates lipid accumulation by enhancing autophagic flux in vitro. (A). Primary mouse hepatocytes were co-incubated with PA and different concentrations of liraglutide for 24 h. Expression of LC3-II and p62 was analyzed by western blot. (B). Evaluation of autophagic flux in Ad-mRFP-GFP-LC3-infected cells with or without CQ treatment. (C). After Ad-mRFP-GFP-LC3 infection, HepG2 cells were pretreated with PA and liraglutide for 20 h, followed by 4 h of co-treatment with 50 μM of CQ. The ratio of mRFP to GFP per cell (n = 10) was calculated (scale bars = 20 μm). (D) Lipid droplets were detected by labeling with lipid dye BODIPY 493/503 (green) in primary mouse hepatocytes treated with or without liraglutide and CQ. Nuclei were stained with DAPI (blue) (scale bars = 10 μm). (E) Intracellular TG content was measured. The data are expressed as mean ± SEM. *P < 0.01, **P < 0.05 vs. BSA group; ***P < 0.01 vs. PA + LG group; #P < 0.01, ##P < 0.05 vs. PA group; &P < 0.01 vs. PA + LG group.
We then investigated whether liraglutide-induced autophagy was involved in its beneficial effect on lipid accumulation. As shown in Figures 5D,E, liraglutide treatment decreased BODIPY 493/503 fluorescence intensity and intracellular TG content in primary hepatocytes. However, these improvements were significantly diminished with CQ. Together, our results indicate that liraglutide alleviated hepatic steatosis partially by promoting the autophagy-lysosomal pathway.
Liraglutide Stimulates Lysosome Biogenesis by Inducing TFEB Translocation
Given that TFEB plays a critical role in the regulation of autophagy and lysosome biogenesis, we evaluated whether liraglutide alleviated steatosis by promoting TFEB-dependent lysosome biogenesis in vivo and in vitro. Upon its dephosphorylation and nuclear translocation, TFEB activates lysosome-associated gene transcription. Thus, we used subcellular fractionation to confirm its localization. Western blot revealed that nuclear TFEB and its downstream target CTSB were remarkably decreased in the livers of HFD-fed mice, and these decreases were reversed by liraglutide treatment. In contrast, LAMP1 expression was increased, possibly due to a compensatory response for autophagy inhibition (Figure 6A). Consistently, we observed that liraglutide promoted TFEB nuclear translocation and enhanced CTSB and LAMP1 expression in PA−stimulated HepG2 cells (Figures 6C,D). In addition, immunofluorescent results revealed that liraglutide increased the ratio of nuclear to cytosolic TFEB in HepG2 cells (Figure 6B). Overall, these findings suggest that liraglutide stimulated lysosome biogenesis by inducing TFEB nuclear translocation.
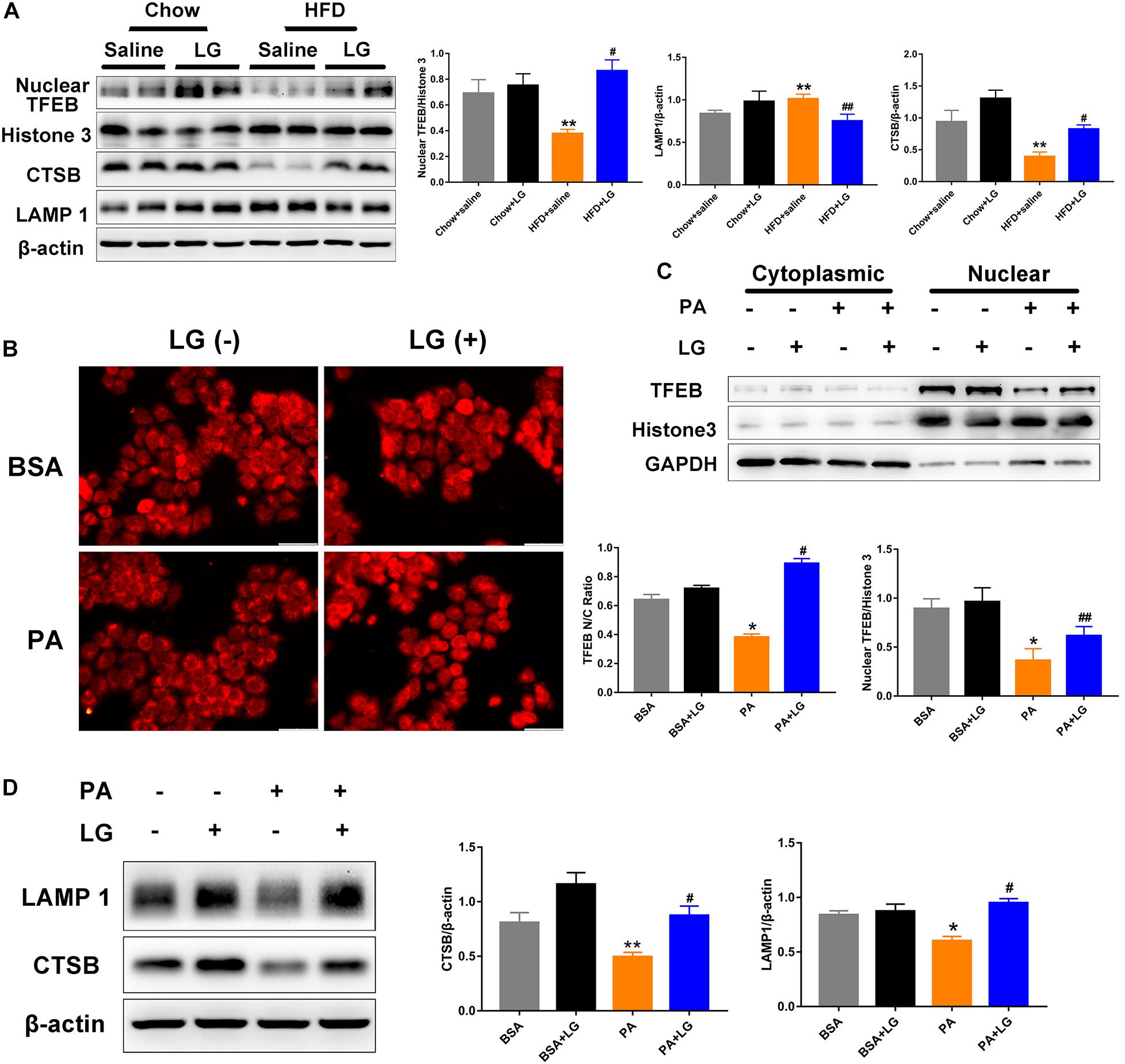
Figure 6. Liraglutide stimulates lysosome biogenesis by inducing TFEB nuclear translocation. (A) Expression of nuclear TFEB, CTSB, and LAMP1 in the livers of HFD-fed mice treated with or without liraglutide. Relative expression levels were normalized to β-actin and Histone 3 levels. (B) HepG2 cells were co-incubated with or without PA and liraglutide for 24 h. After fixation, immunofluorescence staining was performed for TFEB localization (scale bars = 50 μm). (C) Western blot detection of cytoplasm and nuclear TFEB expression in HepG2 cells treated with or without liraglutide. Relative expression levels were normalized to GAPDH and Histone 3 levels, respectively. (D) Western blot detection of CTSB and LAMP1 expression in HepG2 cells. The data are expressed as mean ± SEM; n = 3. *P < 0.01, **P < 0.05 vs. BSA group; #P < 0.01, ##P < 0.05 vs. PA group.
Liraglutide Attenuates PA-Induced Lipid Accumulation by Activating TFEB-Mediated Lysosome Biogenesis
To explore whether TFEB was responsible for the beneficial effects of liraglutide on lysosome biogenesis and lipid accumulation in hepatocytes, a TFEB siRNA (siTFEB) was used to silence TFEB in HepG2 cells. Results showed that TFEB downregulation decreased the expression of CTSB and LAMP1 in liraglutide-treated HepG2 cells (Figure 7A). In addition, decreased BODIPY 493/503 fluorescence intensity and TG content with liraglutide treatment in HepG2 cells were not observed under TFEB knockdown (Figures 7B,C). Our results demonstrate that TFEB plays a critical role in lysosome biogenesis and mediates the beneficial effect of liraglutide on PA-induced intracellular lipid accumulation.
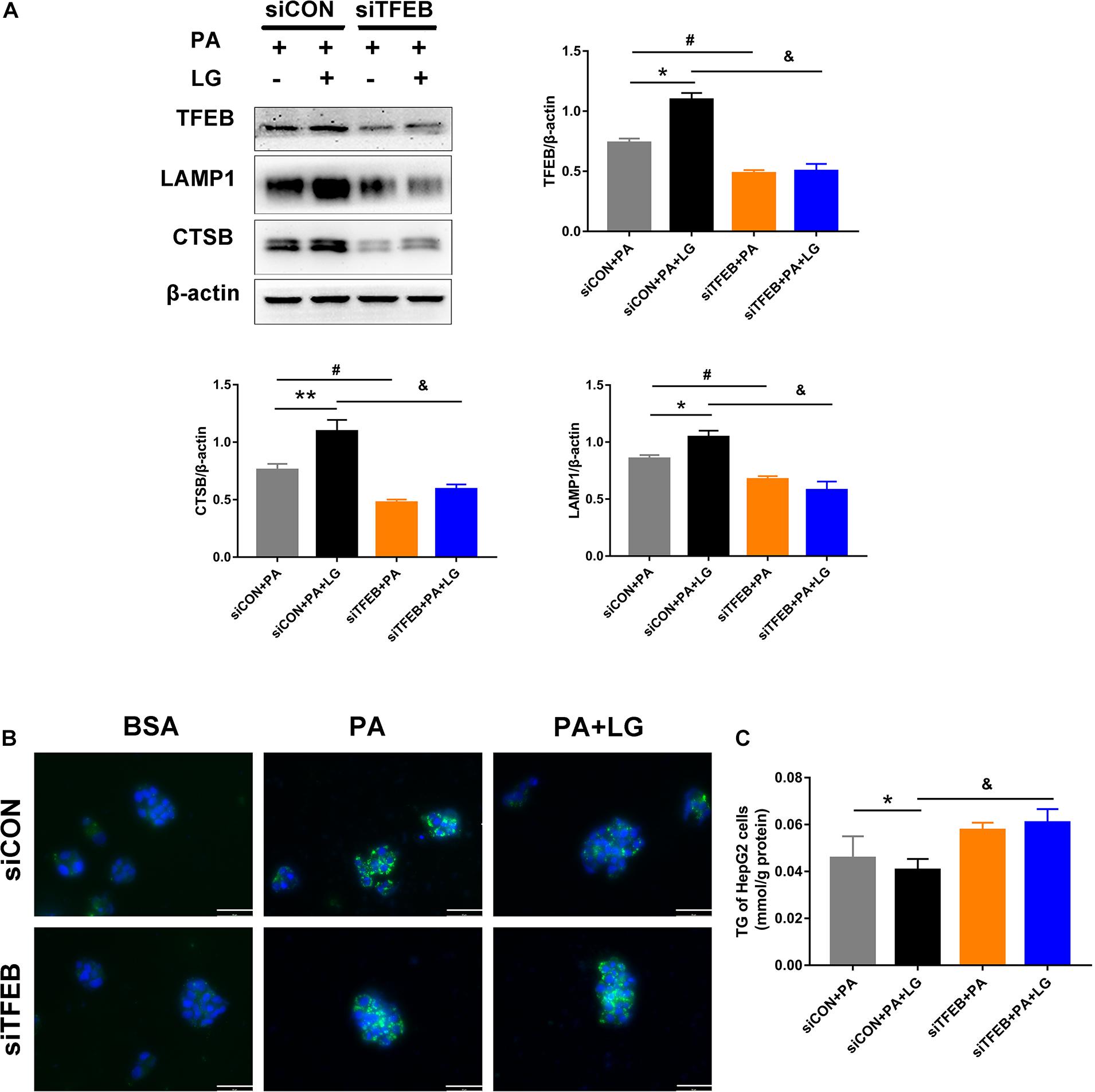
Figure 7. TFEB knockdown weakens the effects of liraglutide on lysosome biogenesis and lipid accumulation. (A) Western blot detection of TFEB and its downstream targets CTSB and LAMP1 in HepG2 cells expressing TFEB-siRNA or control-siRNA. Relative expression levels were normalized to β-actin levels. (B) Lipid droplets in HepG2 cells treated with TFEB-siRNA or control-siRNA were detected via labeling with the lipid dye BODIPY 493/503 (green). Nuclei were stained with DAPI (scale bars = 50 μm). (C) The TG content of HepG2 cells. The data are expressed as the mean ± SEM; n = 3. *P < 0.01, **P < 0.05 and #P < 0.01 vs. siCON + PA group; &P < 0.01 vs. siCON + PA + LG group.
Liraglutide Activates TFEB and Its Downstream Targets Through GLP-1R
To evaluate whether GLP-1R was involved in the action of liraglutide, the GLP-1R protein levels in livers and hepatocytes were detected by western blotting. GLP1-R expression was significantly decreased in HFD mouse livers and PA−stimulated hepatocytes. Further, liraglutide treatment reversed the downregulation of GLP-1R expression in vivo and in vitro (Figures 8A–C). To further investigate whether GLP-1R mediated the effect of liraglutide on TFEB-induced lysosome biogenesis, a GLP-1R siRNA (siGLP-1R) was used to silence GLP-1R in HepG2 cells. Results revealed that the downregulation of GLP-1R decreased the expression of TFEB and its downstream targets CTSB and LAMP1 in liraglutide-treated HepG2 cells (Figure 8D). Simultaneously, a GLP-1R plasmid was used to overexpress GLP-1R in HepG2 cells. GLP-1R overexpression enhanced the TFEB-mediated increase of CTSB and LAMP1 expression in PA-treated HepG2 cells (Figure 8E). Taken together, our results suggest that liraglutide upregulates TFEB and its downstream target proteins through GLP-1R.
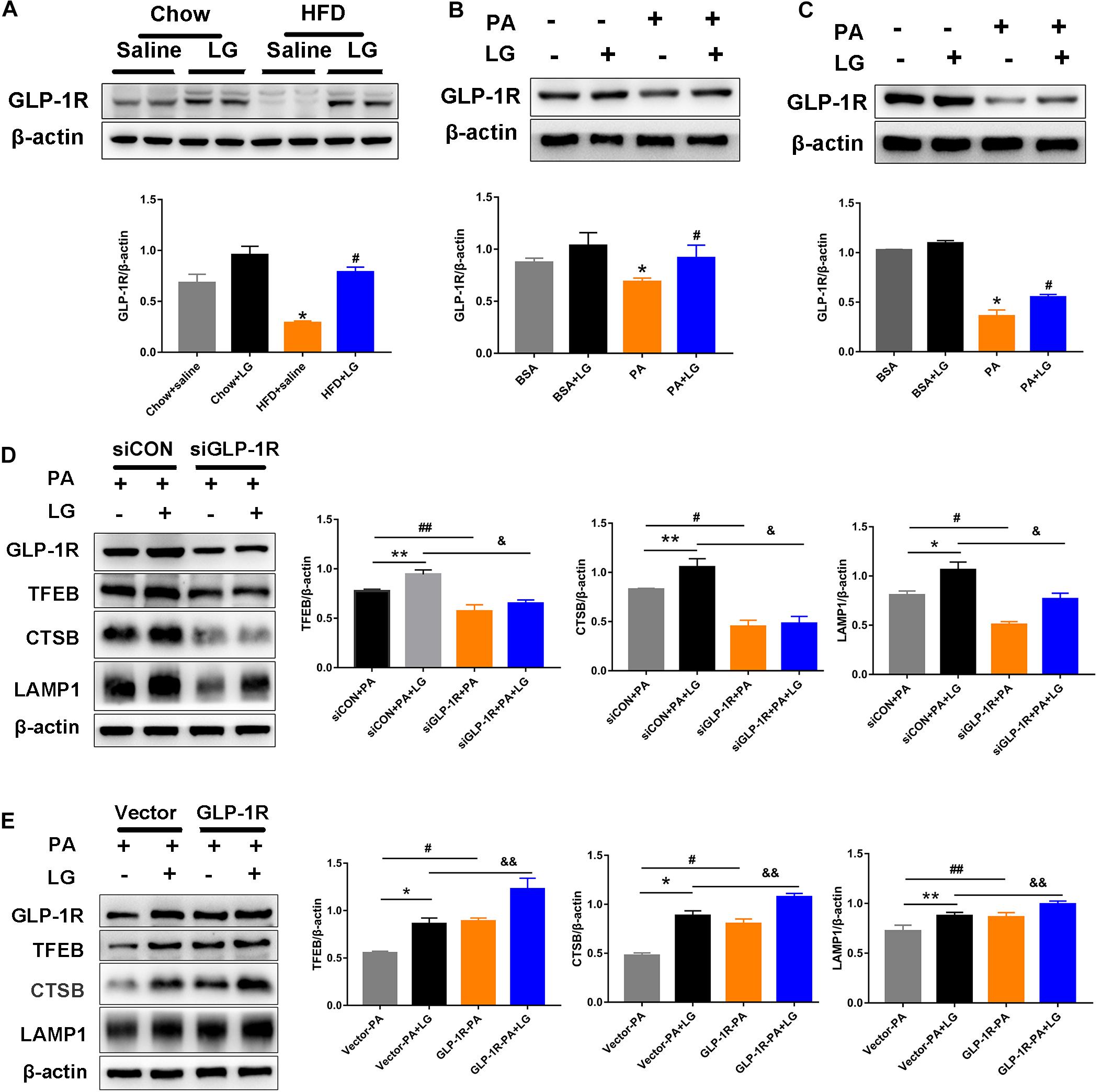
Figure 8. Liraglutide activates TFEB and its downstream targets through GLP-1R. (A) Western blot analysis of GLP-1R protein levels in the liver of HFD-fed mice treated with or without liraglutide. Relative expression levels were normalized to β-actin levels. (B,C) Western blot detection of GLP-1R in primary mouse hepatocytes and HepG2 cells treated with or without liraglutide. (D) Western blot detection of GLP-1R, TFEB, and its downstream targets CTSB and LAMP1 in HepG2 cells transfected with TFEB-siRNA or control-siRNA. (E) Western blot detection of GLP-1R, TFEB, and its downstream targets CTSB and LAMP1 in HepG2 cells with or without GLP-1R overexpression. The data are expressed as the mean ± SEM; n = 3. *P < 0.01, **P < 0.05 vs. Chow + saline group or BSA group or siCON + PA group or Vector-PA group; #P < 0.01, ##P < 0.05 vs. HFD + saline group or PA group or siCON + PA group or Vector-PA group; &P < 0.01, &&P < 0.05 vs. siCON + PA + LG group or Vector-PA + LG group.
Discussion
In this study, we examined the beneficial effect of liraglutide on hepatic steatosis in HFD-fed mice and hepatocytes. Our results demonstrated that liraglutide alleviated hepatic lipid accumulation through activation of the autophagy-lysosomal pathway. Further, we found that, through GLP-1R activation, liraglutide promoted the nuclear translocation of TFEB as well as the expression of its targets CTSB and LAMP1, associated with lysosomal biogenesis and the autophagosome-lysosomal pathway. The increased nuclear translocation of TFEB reduced lipid accumulation in hepatocytes (Figure 9).
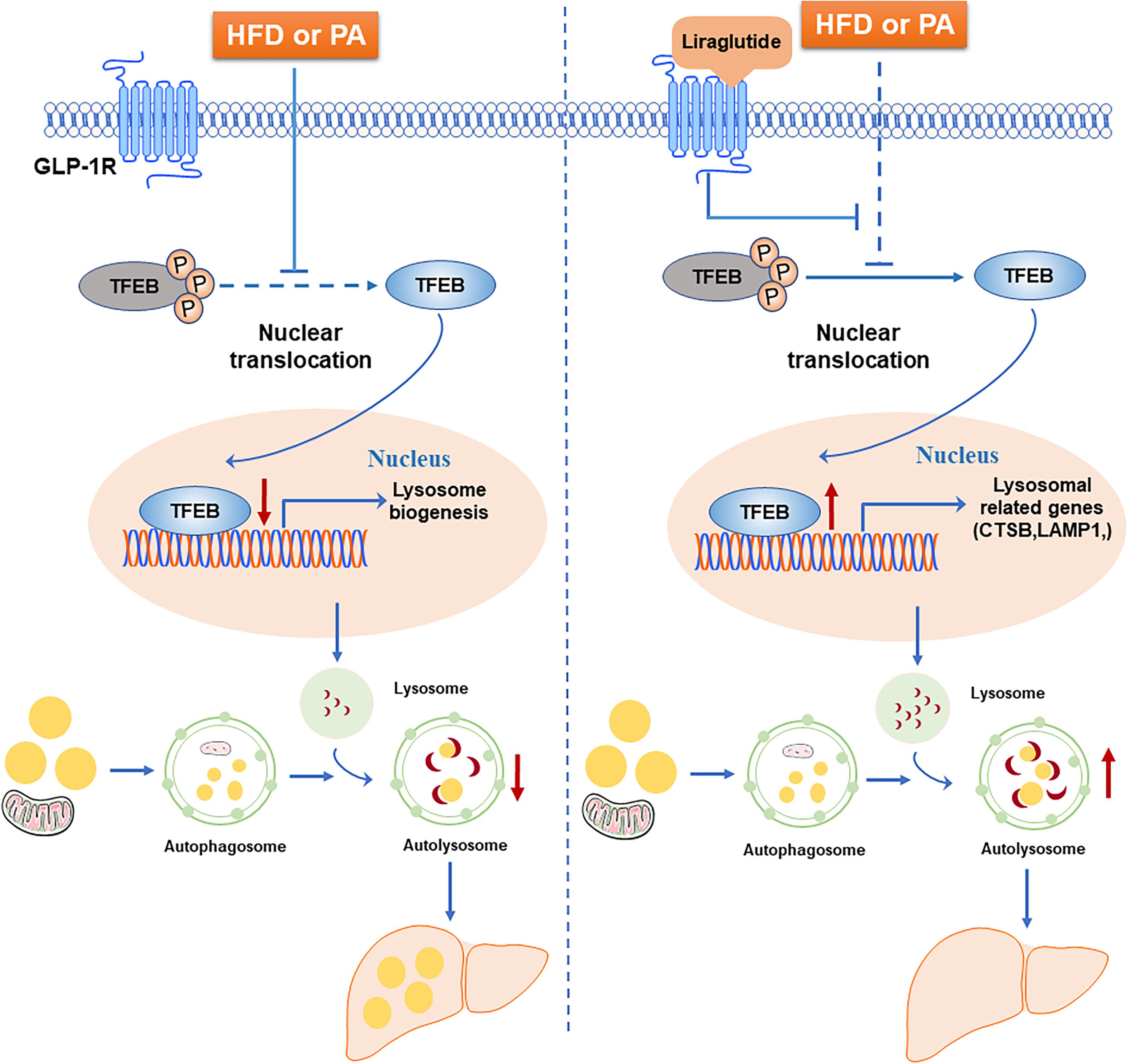
Figure 9. Schematic diagram of the mechanism of liraglutide-mediated alleviation of hepatic steatosis. HFD or PA exposure inhibits TFEB dephosphorylation and nuclear translocation. TFEB regulates lysosomal biogenesis and function. The blocking of TFEB nuclear translocation leads to autophagic flux impairment and subsequently aggravates hepatic steatosis. Liraglutide, a GLP-1R agonist, alleviates hepatic steatosis through enhancing autophagic flux. Mechanistically, liraglutide activates TFEB and its downstream targets through activation of GLP-1R.
As novel potential therapeutic agents for NAFLD, GLP-1RAs were reported to reduce hepatic lipid accumulation (Wu et al., 2019). Further, clinical trial results revealed that liraglutide decreased liver fat content and body weight in patients with type 2 diabetes and NAFLD (Petit et al., 2017). GLP-1RAs also attenuated lipid accumulation in the livers of various animal models (Zheng et al., 2017; Kalavalapalli et al., 2019). These findings were consistent with our results, which indicated that liraglutide significantly decreased liver lipid content as well as plasma TG, ALT, and AST in HFD-fed mice. Another report described a number of effects for GLP-1RAs, including increased postprandial insulin secretion, decreased appetite, and enhanced weight loss (Armstrong et al., 2016). Previous studies suggested that the beneficial effect of GLP-1RAs on weight loss is mainly attributed to the inhibition of food intake, especially for short-term interventions (Kanoski et al., 2011; Wei et al., 2015). A recent research on exendin-4 treatment in mice revealed a decrease in food intake during the first 2 weeks, which disappeared from 2 to 8 weeks (Xu et al., 2016). Consistently, in our study, we observed that the transient reduction in food intake vanished in the last 2 weeks of liraglutide treatment. Thus, we speculated that the weight loss resulting from GLP-1RAs was not entirely dependent on the reduced food intake during long-term administration. Moreover, we investigated the expression of GLP-1R. Interestingly, liraglutide treatment reversed the down-regulation of GLP-1R expression in both in vivo and in vitro NAFLD models. Therefore, the systemic metabolic effects of GLP-1RAs might be attributed to a combination of the above-described mechanisms (Zhou et al., 2019; Mantovani et al., 2020).
Autophagy is important for the maintenance of cellular metabolism and energy homeostasis. Accumulating evidence has indicated that defective autophagy contributes to the aggravation of hepatic steatosis (Miyagawa et al., 2016; Chu et al., 2019). Normally, autophagy is supposed to break down lipid droplets through the lipophagy degradation pathway (Martinez-Lopez and Singh, 2015). A previous study found that GLP-1RA-induced autophagy could remove excess lipid droplets and thus alleviate lipotoxicity (Yu et al., 2019). Moreover, exendin-4 protected hepatocytes and reduced steatosis via autophagy, in turn reducing endoplasmic reticulum stress-related hepatocyte apoptosis (Sharma et al., 2011). However, the mechanism underlying this upregulation of autophagy is unclear. A previous study in mice demonstrated that GLP-1RAs alleviated lipid accumulation through activation of AMPK/mTOR-dependent autophagy, a classic autophagy pathway (He et al., 2016). In the current work, EM examination revealed a large number of lipid droplets surrounded or partly engulfed by autophagosome-like vesicles in the livers of HFD mice. Further, instead of these lipid droplets, lipid-laden autolysosomes were observed following liraglutide treatment. Consistently, the levels of autophagosome formation marker LC3−II and autophagic substrate p62 decreased after treatment with liraglutide. Thus, we inferred that liraglutide restored HFD-inhibited autophagy, perhaps in an autophagy-lysosomal pathway-dependent manner.
Autophagy is a dynamic process. As the terminal stage of autophagic flux, lysosomes fuse with autophagosomes, followed by lysosomal degradation and cellular lipid clearance (Chao et al., 2018a). Defective lysosomal biogenesis and clearance impair autophagic flux, leading to intracellular lipid accumulation during the development of NAFLD (Chao et al., 2018b). Previous studies found that HFD disrupted lysosomal biogenesis in hepatocytes, which was associated with lipid degradation (Pi et al., 2019). We employed lysosomal inhibitor CQ to further investigate autophagy-lysosomal pathway involvement and found that liraglutide stimulated autophagic flux, as demonstrated by a reduction in autophagosome accumulation indicated by mRFP-GFP-LC3. In addition, the liraglutide-mediated attenuation of lipid accumulation in PA-stimulated HepG2 cells was significantly compromised following CQ treatment. Importantly, this is the first work to demonstrate that liraglutide activates the autophagy-lysosomal pathway to alleviate hepatic lipid accumulation.
MiT−TFE subfamily members, including MITF, TFE3, and TFEB, positively regulate lysosomal biogenesis. Among these transcriptional factors, TFEB enhances lipid metabolism by regulating genes that encode lysosome biogenesis- and lipolysis-related factors (Napolitano and Ballabio, 2016). Under nutrient-depleted conditions, TFEB was activated by an autoregulatory feedback loop and subsequently regulated cellular lipid catabolism (Settembre et al., 2013). Recent studies have revealed that TFEB nuclear translocation was promoted by pharmacologically activating the AMPK pathway and eventually alleviated hepatic steatosis in HFD-fed mice (Chen et al., 2019). In addition, TFEB regulated multiple steps of lipid metabolism, such as lipid recruitment and degradation, as well as fatty acid oxidation (Li et al., 2016; Evans et al., 2019). However, the exact influence of liraglutide on TFEB-mediated lysosomal biogenesis had not been studied previously. Our study firstly revealed that liraglutide activated autophagic flux by inducing TFEB nuclear translocation and increasing the expression of its downstream targets CTSB and LAMP1. After silencing TFEB, the liraglutide-mediated promotion of lysosome biogenesis and alleviation of hepatic steatosis were attenuated. Moreover, we found that silencing of GLP-1R could mimic the effects TFEB downregulation in decreasing lysosome biogenesis, whereas its overexpression increased TFEB activation and the expression of its downstream targets CTSB and LAMP1, indicating that GLP-1R mediated the effect of liraglutide in promoting lysosome biogenesis. Therefore, we speculated that liraglutide activates the autophagy-lysosomal pathway to reduce hepatic lipid accumulation through GLP-1R-mediated TFEB activation.
In response to nutrients, GLP-1RAs were also found to alleviate liver steatosis through the upregulation of SIRT1 expression, activating the AMPK pathway and inhibiting SREBP-1c expression (Xu et al., 2014). Moreover, data from recent studies demonstrated that AMPK/SIRT1 pathway activation could upregulate the expression of various autophagy-related genes (Song et al., 2015; Huang et al., 2017). For instance, SIRT1 deacetylation could induce autophagy in an autophagy protein 5-dependent manner (Sun et al., 2018). In the current work, we found that liraglutide decreased SIRT1 expression in PA-treated primary mouse hepatocytes (Supplementary Figure S4C). However, the opposite results were observed in mice, which requires further investigation.
Conclusion
In conclusion, our study reveals that liraglutide attenuates hepatic steatosis via activation of autophagic flux, especially the GLP-1R-TFEB-mediated autophagy-lysosomal pathway. These findings provide a novel mechanism supporting the potential of GLP-1RAs as promising therapeutic agents for NAFLD.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
Ethics Statement
The animal study was reviewed and approved by Animal Ethical and Welfare Committee of Shanghai Sixth People’s Hospital Affiliated to Shanghai Jiao Tong University School.
Author Contributions
YF and LW designed the study. YF and LJ carried out the experimental work. CZ and YX assisted in performing research. JZ and JL collected and analyzed the data. YF interpreted the results and wrote the manuscript. LW and JY contributed to manuscript read and revision. All authors have read and approved the submitted version.
Funding
The authors appreciate the financial support from the National Nature Science Foundation of China (81870603 to LW and 81670790 to JY), the Science and Technology Commission of Shanghai Municipality (20ZR1442500 to LW), and the Shanghai Municipal Commission of Health and Family Planning (20154Y0059 to CZ).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2020.602574/full#supplementary-material
Abbreviations
GLP-1RA, glucagon-like peptide-1 receptor agonist; NAFLD, non-alcoholic fatty liver disease; GLP-1R, glucagon-like peptide-1 receptor; PA, palmitic acid; TFEB, transcription factor EB; siRNA, small interfering RNA; LC3-II, microtubule-associated protein 1B light chain-3-II; HFD, high-fat diet; IGPTT, intraperitoneal glucose tolerance test; ITT, insulin tolerance test; TG, serum triglycerides; TC, total cholesterol; LDL-C, low-density lipoprotein cholesterol; ALT, alanine transferase; AST, aspartic acid transaminase; FBS, fetal bovine serum; OA, oleic acid.
References
Arab, J. P., Arrese, M., and Trauner, M. (2018). Recent insights into the pathogenesis of nonalcoholic fatty liver disease. Annu. Rev. Pathol. 13, 321–350. doi: 10.1146/annurev-pathol-020117-043617
Armstrong, M. J., Gaunt, P., Aithal, G. P., Barton, D., Hull, D., Parker, R., et al. (2016). Liraglutide safety and efficacy in patients with non-alcoholic steatohepatitis (LEAN): a multicentre, double-blind, randomised, placebo-controlled phase 2 study. Lancet 387, 679–690. doi: 10.1016/S0140-6736(15)00803-X
Chang, X., Yan, H., Fei, J., Jiang, M., Zhu, H., Lu, D., et al. (2010). Berberine reduces methylation of the MTTP promoter and alleviates fatty liver induced by a high-fat diet in rats. J. Lipid Res. 51, 2504–2515. doi: 10.1194/jlr.M001958
Chao, X., Ni, H. M., and Ding, W. X. (2018a). Insufficient autophagy: a novel autophagic flux scenario uncovered by impaired liver TFEB-mediated lysosomal biogenesis from chronic alcohol-drinking mice. Autophagy 14, 1646–1648. doi: 10.1080/15548627.2018.1489170
Chao, X., Wang, S., Zhao, K., Li, Y., Williams, J. A., Li, T., et al. (2018b). Impaired TFEB-mediated lysosome biogenesis and autophagy promote chronic ethanol-induced liver injury and steatosis in mice. Gastroenterology 155, 865–879. doi: 10.1053/j.gastro.2018.05.027
Chen, X., Chan, H., Zhang, L., Liu, X., Ho, I., Zhang, X., et al. (2019). The phytochemical polydatin ameliorates non-alcoholic steatohepatitis by restoring lysosomal function and autophagic flux. J. Cell. Mol. Med. 23, 4290–4300. doi: 10.1111/jcmm.14320
Chu, Q., Zhang, S., Chen, M., Han, W., Jia, R., Chen, W., et al. (2019). Cherry Anthocyanins Regulate NAFLD by Promoting Autophagy Pathway. Oxidat. Med. Cell. Longev. 2019, 1–16. doi: 10.1155/2019/4825949
Cui, W., Sathyanarayan, A., Lopresti, M., Aghajan, M., Chen, C., and Mashek, D. G. (2020). Lipophagy-derived fatty acids undergo extracellular efflux via lysosomal exocytosis. Autophagy [Epub ahead of print]. doi: 10.1080/15548627.2020.1728097
Drucker, D. J. (2018). Mechanisms of action and therapeutic application of glucagon-like peptide-1. Cell Metab. 27, 740–756. doi: 10.1016/j.cmet.2018.03.001
Evans, T. D., Zhang, X., Jeong, S. J., He, A., Song, E., Bhattacharya, S., et al. (2019). TFEB drives PGC-1alpha expression in adipocytes to protect against diet-induced metabolic dysfunction. Sci. Signal. 12:eaau2281. doi: 10.1126/scisignal.aau2281
He, Q., Sha, S., Sun, L., Zhang, J., and Dong, M. (2016). GLP-1 analogue improves hepatic lipid accumulation by inducing autophagy via AMPK/mTOR pathway. Biochem. Biophys. Res. Commun. 476, 196–203. doi: 10.1016/j.bbrc.2016.05.086
Huang, Q., Wang, T., Yang, L., and Wang, H. Y. (2017). Ginsenoside rb2 alleviates hepatic lipid accumulation by restoring autophagy via induction of sirt1 and activation of AMPK. Int. J. Mol. Sci. 18:1063. doi: 10.3390/ijms18051063
Kalavalapalli, S., Bril, F., Guingab, J., Vergara, A., Garrett, T. J., Sunny, N. E., et al. (2019). Impact of exenatide on mitochondrial lipid metabolism in mice with nonalcoholic steatohepatitis. J. Endocrinol. 241, 293–305. doi: 10.1530/JOE-19-0007
Kanoski, S. E., Fortin, S. M., Arnold, M., Grill, H. J., and Hayes, M. R. (2011). Peripheral and central GLP-1 receptor populations mediate the anorectic effects of peripherally administered GLP-1 receptor agonists, liraglutide and exendin-4. Endocrinology 152, 3103–3112. doi: 10.1210/en.2011-0174
Klaunig, J. E., Goldblatt, P. J., Hinton, D. E., Lipsky, M. M., Chacko, J., and Trump, B. F. (1981). Mouse liver cell culture. I. Hepatocyte isolation. In Vitro 17, 913–925. doi: 10.1007/BF02618288
Li, X., Zhang, X., Zheng, L., Kou, J., Zhong, Z., Jiang, Y., et al. (2016). Hypericin-mediated sonodynamic therapy induces autophagy and decreases lipids in THP-1 macrophage by promoting ROS-dependent nuclear translocation of TFEB. Cell Death Dis. 7:e2527. doi: 10.1038/cddis.2016.433
Lyu, J., Imachi, H., Fukunaga, K., Sato, S., Kobayashi, T., Dong, T., et al. (2020). Role of ATP-binding cassette transporter A1 in suppressing lipid accumulation by glucagon-like peptide-1 agonist in hepatocytes. Mol. Metab. 34, 16–26. doi: 10.1016/j.molmet.2019.12.015
Mantovani, A., Byrne, C. D., Scorletti, E., Mantzoros, C. S., and Targher, G. (2020). Efficacy and safety of anti-hyperglycaemic drugs in patients with non-alcoholic fatty liver disease with or without diabetes: an updated systematic review of randomized controlled trials. Diabetes Metab. [Epub ahead of print]. doi: 10.1016/j.diabet.2019.12.007
Martinez-Lopez, N., and Singh, R. (2015). Autophagy and lipid droplets in the liver. Annu. Rev. Nutr. 35, 215–237. doi: 10.1146/annurev-nutr-071813-105336
Medina, D. L., Fraldi, A., Bouche, V., Annunziata, F., Mansueto, G., Spampanato, C., et al. (2011). Transcriptional activation of lysosomal exocytosis promotes cellular clearance. Dev. Cell. 21, 421–430. doi: 10.1016/j.devcel.2011.07.016
Miyagawa, K., Oe, S., Honma, Y., Izumi, H., Baba, R., and Harada, M. (2016). Lipid-induced endoplasmic reticulum stress impairs selective autophagy at the step of autophagosome-lysosome fusion in hepatocytes. Am. J. Pathol. 186, 1861–1873. doi: 10.1016/j.ajpath.2016.03.003
Muller, T. D., Finan, B., Bloom, S. R., D’Alessio, D., Drucker, D. J., Flatt, P. R., et al. (2019). Glucagon-like peptide 1 (GLP-1). Mol. Metab. 30, 72–130. doi: 10.1016/j.molmet.2019.09.010
Napolitano, G., and Ballabio, A. (2016). TFEB at a glance. J. Cell. Sci. 129, 2475–2481. doi: 10.1242/jcs.146365
Petit, J. M., Cercueil, J. P., Loffroy, R., Denimal, D., Bouillet, B., Fourmont, C., et al. (2017). Effect of liraglutide therapy on liver fat content in patients with inadequately controlled type 2 diabetes: the Lira-NAFLD study. J. Clin. Endocrinol. Metab. 102, 407–415. doi: 10.1210/jc.2016-2775
Pi, H., Liu, M., Xi, Y., Chen, M., Tian, L., Xie, J., et al. (2019). Long-term exercise prevents hepatic steatosis: a novel role of FABP1 in regulation of autophagy-lysosomal machinery. FASEB J. 33, 11870–11883. doi: 10.1096/fj.201900812R
Seglen, P. O. (1976). Preparation of isolated rat liver cells. Methods Cell Biol. 13, 29–83. doi: 10.1016/s0091-679x(08)61797-5
Settembre, C., De Cegli, R., Mansueto, G., Saha, P. K., Vetrini, F., Visvikis, O., et al. (2013). TFEB controls cellular lipid metabolism through a starvation-induced autoregulatory loop. Nat. Cell. Biol. 15, 647–658. doi: 10.1038/ncb2718
Sharma, S., Mells, J. E., Fu, P. P., Saxena, N. K., and Anania, F. A. (2011). GLP-1 analogs reduce hepatocyte steatosis and improve survival by enhancing the unfolded protein response and promoting macroautophagy. PLoS One 6:e25269. doi: 10.1371/journal.pone.0025269
Singh, R., Kaushik, S., Wang, Y., Xiang, Y., Novak, I., Komatsu, M., et al. (2009). Autophagy regulates lipid metabolism. Nature 458, 1131–1135. doi: 10.1038/nature07976
Sinha, R. A., Farah, B. L., Singh, B. K., Siddique, M. M., Li, Y., Wu, Y., et al. (2014). Caffeine stimulates hepatic lipid metabolism by the autophagy-lysosomal pathway in mice. Hepatology 59, 1366–1380. doi: 10.1002/hep.26667
Song, Y. M., Lee, Y. H., Kim, J. W., Ham, D. S., Kang, E. S., Cha, B. S., et al. (2015). Metformin alleviates hepatosteatosis by restoring SIRT1-mediated autophagy induction via an AMP-activated protein kinase-independent pathway. Autophagy 11, 46–59. doi: 10.4161/15548627.2014.984271
Su, H., Li, Y., Hu, D., Xie, L., Ke, H., Zheng, X., et al. (2018). Procyanidin B2 ameliorates free fatty acids-induced hepatic steatosis through regulating TFEB-mediated lysosomal pathway and redox state. Free Radic. Biol. Med. 126, 269–286. doi: 10.1016/j.freeradbiomed.2018.08.024
Sun, Y., Xia, M., Yan, H., Han, Y., Zhang, F., Hu, Z., et al. (2018). Berberine attenuates hepatic steatosis and enhances energy expenditure in mice by inducing autophagy and fibroblast growth factor 21. Br. J. Pharmacol. 175, 374–387. doi: 10.1111/bph.14079
Tong, W., Ju, L., Qiu, M., Xie, Q., Chen, Y., Shen, W., et al. (2016). Liraglutide ameliorates non-alcoholic fatty liver disease by enhancing mitochondrial architecture and promoting autophagy through the SIRT1/SIRT3-FOXO3a pathway. Hepatol. Res. 46, 933–943. doi: 10.1111/hepr.12634
Vernon, G., Baranova, A., and Younossi, Z. M. (2011). Systematic review: the epidemiology and natural history of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis in adults. Aliment. Pharmacol. Ther. 34, 274–285. doi: 10.1111/j.1365-2036.2011.04724.x
Wang, Y., Zhao, H., Li, X., Wang, Q., Yan, M., Zhang, H., et al. (2019). Formononetin alleviates hepatic steatosis by facilitating TFEB-mediated lysosome biogenesis and lipophagy. J. Nutr. Biochem. 73:108214. doi: 10.1016/j.jnutbio.2019.07.005
Wei, Q., Li, L., Chen, J. A., Wang, S. H., and Sun, Z. L. (2015). Exendin-4 improves thermogenic capacity by regulating fat metabolism on brown adipose tissue in mice with diet-induced obesity. Ann. Clin. Lab. Sci. 45, 158–165.
Wu, K., Zhao, T., Hogstrand, C., Xu, Y., Ling, S., Chen, G., et al. (2020). FXR-mediated inhibition of autophagy contributes to FA-induced TG accumulation and accordingly reduces FA-induced lipotoxicity. Cell Commun. Signal. 18:47. doi: 10.1186/s12964-020-0525-1
Wu, Y. R., Shi, X. Y., Ma, C. Y., Zhang, Y., Xu, R. X., and Li, J. J. (2019). Liraglutide improves lipid metabolism by enhancing cholesterol efflux associated with ABCA1 and ERK1/2 pathway. Cardiovasc. Diabetol. 18:146. doi: 10.1186/s12933-019-0954-6
Xu, F., Li, Z., Zheng, X., Liu, H., Liang, H., Xu, H., et al. (2014). SIRT1 mediates the effect of GLP-1 receptor agonist exenatide on ameliorating hepatic steatosis. Diabetes 63, 3637–3646. doi: 10.2337/db14-0263
Xu, F., Lin, B., Zheng, X., Chen, Z., Cao, H., Xu, H., et al. (2016). GLP-1 receptor agonist promotes brown remodelling in mouse white adipose tissue through SIRT1. Diabetologia 59, 1059–1069. doi: 10.1007/s00125-016-3896-5
Yang, Z., and Klionsky, D. J. (2010). Mammalian autophagy: core molecular machinery and signaling regulation. Curr. Opin. Cell Biol. 22, 124–131. doi: 10.1016/j.ceb.2009.11.014
Yu, X., Hao, M., Liu, Y., Ma, X., Lin, W., Xu, Q., et al. (2019). Liraglutide ameliorates non-alcoholic steatohepatitis by inhibiting NLRP3 inflammasome and pyroptosis activation via mitophagy. Eur. J. Pharmacol. 864:172715. doi: 10.1016/j.ejphar.2019.172715
Zhang, H., Yan, S., Khambu, B., Ma, F., Li, Y., Chen, X., et al. (2018). Dynamic MTORC1-TFEB feedback signaling regulates hepatic autophagy, steatosis and liver injury in long-term nutrient oversupply. Autophagy 14, 1779–1795. doi: 10.1080/15548627.2018.1490850
Zheng, X., Xu, F., Liang, H., Cao, H., Cai, M., Xu, W., et al. (2017). SIRT1/HSF1/HSP pathway is essential for exenatide-alleviated, lipid-induced hepatic endoplasmic reticulum stress. Hepatology 66, 809–824. doi: 10.1002/hep.29238
Keywords: non-alcoholic fatty liver disease, liraglutide, autophagy-lysosome, lysosomal biogenesis, TFEB
Citation: Fang Y, Ji L, Zhu C, Xiao Y, Zhang J, Lu J, Yin J and Wei L (2020) Liraglutide Alleviates Hepatic Steatosis by Activating the TFEB-Regulated Autophagy-Lysosomal Pathway. Front. Cell Dev. Biol. 8:602574. doi: 10.3389/fcell.2020.602574
Received: 03 September 2020; Accepted: 10 November 2020;
Published: 27 November 2020.
Edited by:
Joilson Oliveira Martins, University of São Paulo, BrazilReviewed by:
Angela Castoldi, Federal University of Pernambuco, BrazilMingfeng Xia, Fudan University, China
Copyright © 2020 Fang, Ji, Zhu, Xiao, Zhang, Lu, Yin and Wei. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jun Yin, eWluanVuQHNqdHUuZWR1LmNu; Li Wei, d2VpbGk2M0Bob3RtYWlsLmNvbQ==
†These authors have contributed equally to this work