 Hiroki Yoshioka
Hiroki Yoshioka Sai Shankar Ramakrishnan
Sai Shankar Ramakrishnan Junbo Shim
Junbo Shim Akiko Suzuki
Akiko Suzuki Junichi Iwata
Junichi Iwata- 1Department of Diagnostic and Biomedical Sciences, School of Dentistry, The University of Texas Health Science Center at Houston, Houston, TX, United States
- 2Center for Craniofacial Research, The University of Texas Health Science Center at Houston, Houston, TX, United States
- 3MD Anderson Cancer Center UTHealth Graduate School of Biomedical Sciences, Houston, TX, United States
Cleft palate is the second most common congenital birth defect, and both environmental and genetic factors are involved in the etiology of the disease. However, it remains largely unknown how environmental factors affect palate development. Our previous studies show that several microRNAs (miRs) suppress the expression of genes involved in cleft palate. Here we show that miR-4680-3p plays a crucial role in cleft palate pathogenesis. We found that all-trans retinoic acid (atRA) specifically induces miR-4680-3p in cultured human embryonic palatal mesenchymal (HEPM) cells. Overexpression of miR-4680-3p inhibited cell proliferation in a dose-dependent manner through the suppression of expression of ERBB2 and JADE1, which are known cleft palate-related genes. Importantly, a miR-4680-3p-specific inhibitor normalized cell proliferation and altered expression of ERBB2 and JADE1 in cells treated with atRA. Taken together, our results suggest that upregulation of miR-4680-3p induced by atRA may cause cleft palate through suppression of ERBB2 and JADE1. Thus, miRs may be potential targets for the prevention and diagnosis of cleft palate.
Introduction
The prevalence of cleft lip with or without cleft palate (CL/P) is ~1 in 700 live births worldwide (Ferguson, 1988), and affected individuals require surgical repairs, speech therapy, and dental treatments (Ferguson, 1988). Palate development (which consists of the growth, elevation, and fusion of the palatal shelves) starts at 6–8 weeks of gestation in humans; any failure in these process results in cleft palate (Habib, 1978; Mossey et al., 2009). The etiology of cleft palate is complex and involves environmental and genetic factors and their interactions (Dhulipala et al., 2006; Havasi et al., 2013; Buser and Pohl, 2015; Liu et al., 2018). Maternal use and exposure to tobacco, alcohol, drugs (e.g., retinoic acid and dexamethasone), and chemicals (e.g., dioxin and heavy metals), as well as mutations in genes related to the degradation/metabolism/release of these teratogens, are considered to be risk factors (Prescott et al., 2002; Chevrier et al., 2005; Ramirez et al., 2007). An increasing number of studies suggest that microRNAs (miRs), which are endogenous small non-coding RNAs (~22 nucleotides long) that negatively regulate the expression of their target genes (Bartel, 2004; Obernosterer et al., 2006), play important roles in normal palate development and CL/P in humans and mice (Karsy et al., 2010; Shin et al., 2012; Seelan et al., 2014; Chung et al., 2016; Schoen et al., 2017, 2018; Wang et al., 2017; Mukhopadhyay et al., 2019); however, it remains elusive how and which miRs are crucial roles in CL/P. Our recent studies show that overexpression of either miR-140-5p, miR-133b, miR-374a-5p, miR-381-3p, or miR-4680-3p suppresses cell proliferation in cultured human embryonic palatal mesenchymal (HEPM) cells (Li et al., 2019; Suzuki et al., 2019), suggesting that these miRs may be involved in the pathogenesis of cleft palate.
All-trans retinoic acid (atRA), a derivative of vitamin A, is the most active retinoid that plays important roles in a variety of biological processes, including cell proliferation, differentiation, and extracellular matrix production (Wang and Kirsch, 2002; Lai et al., 2003; Rhinn and Dolle, 2012; Cunningham and Duester, 2015). While atRA is widely used in the treatments of skin diseases and cancers (Karsy et al., 2010; Siddikuzzaman and Berlin Grace, 2011; Mihaly et al., 2012), excessive intake of atRA, a known teratogen, disrupts embryogenesis, causing birth defects (Abbott et al., 1989; Ross et al., 2000; Roberts, 2020) including cleft palate (Abbott et al., 1989; Ross et al., 2000; Wang and Kirsch, 2002; Lai et al., 2003; Yao et al., 2011; Havasi et al., 2013; Hu et al., 2013; Hou et al., 2019; Roberts, 2020). Recent studies show that atRA alters the expression of miRs in human cancer cell lines (Liu et al., 2018, 2019). In this study, we will determine whether and how atRA can alter miR expression, which suppresses the expression of genes related to CL/P.
Methods
Cell Culture
HEPM cells were obtained from American Type Culture Collection (CRL-1486; ATCC) and maintained under Minimum Essential Medium Eagle-alpha modification (αMEM), supplemented with 10% fetal bovine serum (FBS), penicillin/streptomycin, and L-glutamine, at 37°C in a humidified atmosphere with 5% CO2.
Cell Proliferation Assay
The cells were plated onto 96-well cell culture plates at a density of 5,000/well and treated with atRA (R2625, Sigma-Aldrich) at various concentrations (0, 1, 3, 10, and 30 μM), small interfering RNA (siRNA) for either ERBB2 (#103546; Thermo Fisher Scientific) or JADE1 (#109590; Thermo Fisher Scientific), or a negative control (#AM4611; Thermo Fisher Scientific), at 3 pmol in 0.3 μL of transfection reagent (TransIT-X2 system; Mirus Bio LLC) in 0.1 mL αMEM per well for 24, 48, or 72 h. Cell proliferation was measured using Cell Counting Kit 8 (Dojindo Molecular Technologies, Inc.) (n = 6 per group).
Bromodeoxyuridine (BrdU) Incorporation Assay
The cells were plated onto 35-mm dishes at a density of 25,000/dish and treated with 30 μM atRA, or vehicle (dimethyl sulfoxide). After 72 h, the cells were incubated with BrdU for 1 h. Incorporated BrdU was stained with a rat monoclonal antibody against BrdU (ab6326; Abcam, 1:1,000), as previously described (Suzuki et al., 2018a). A total of nine fields, which were randomly selected from three independent experiments, was used for the quantification of BrdU-positive cells.
Terminal 2′-Deoxyuridine, 5′-Triphosphate (dUTP) Nick-End Labeling (TUNEL) Staining
The cells were plated onto 35-mm dishes at a density of 25,000/dish and treated with 30 μM atRA or vehicle for 72 h. The Click-iT Plus TUNEL Assay with Alexa 594 (C10618, Molecular Probes) was used to detect apoptotic cells, as previously described (Suzuki et al., 2018b). A total of four fields, which were randomly selected from two independent experiments, was used for the quantification of TUNEL-positive cells.
Quantitative RT-PCR
The cells were plated onto 60-mm dishes at a density of 50,000/dish and treated with 30 μM atRA or vehicle. After 24 or 72 h, total RNA isolated from HEPM cells (n = 6 per group) was extracted with the QIAshredder and miRNeasy Mini Kit (QIAGEN), according to the manufacturer's instructions. Total RNA (1 μg) from each sample was reverse-transcribed using iScript Reverse Transcription Supermix for quantitative RT-PCR (Bio-Rad), and then the cDNA was amplified with iTaq Universal SYBR Green Supermix (Bio-Rad) using a CFX96 Touch Real-Time PCR Detection system (Bio-Rad). The following PCR primers were used: ERBB2 (NM_004448) sense, 5′-CATTGGGACCGGAGAAACCA-3′, and antisense, 5′-CGCAGCTTCATGTCTGTGC-3′; JADE1 (NM_199320) sense, 5′-AAACGCCAGACCGAGAGTG-3′, and antisense, 5′-AGTTGACAGGCTGCCATTGT-3′; MTHFD1 (NM_005956) sense, 5′-TCCAGTAGTAGTGGCCGTGA-3′, and antisense, 5′-GCTTTGTGTTGAGCTTCGGG-3′; WNT5A (NM_003392) sense, 5′-AAGCAGACGTTTCGGCTACA-3′, and antisense, 5′-GCGCCCAATACGACCAAATC-3′; and GAPDH (NM_002046) sense, 5′-GACAGTCAGCCGCATCTTCT-3′, and antisense, 5′-GCGCCCAATACGACCAAATC-3′. The amount of each quantified target mRNA was normalized by GAPDH. miR expression was measured using the Taqman Fast Advanced Master Mix and Taqman Advanced miR cDNA Synthesis Kit (Thermo Fisher Scientific), according to the manufacturer's instructions. Probes for miR-140-5p (477909_mir), miR-133b (480871_mir), miR-374a-5p (478238_mir), miR381-3p (477816_mir), miR-4680-3p (480701_mir), and miR-26a-5p (477995_mir) were obtained from Thermo Fisher Scientific.
Immunoblotting
The cells were plated onto 60-mm dishes at a density of 50,000/dish and treated with either atRA for 72 h or siRNA for 48 h, as described above. Treated cells were lysed with RIPA buffer (Cell Signaling Technology) containing a protease inhibitor cocktail (Roche). The cells were harvested and centrifuged at 21,130 × g for 10 min at 4°C, and the supernatant of each sample was collected and protein level was determined using the BCA protein kit (Pierce). Protein samples were applied to Mini-PROTEAN TGX Gels (Bio-Rad) and transferred to a polyvinylidene difluoride (PVDF) membrane. Mouse monoclonal antibodies against ERBB2 (MA5-13675, Thermo Fisher Scientific, 1:2,000), JADE1 (MAB6275, R&D, 1:2,000), CDKN1B (3,698, Cell Signaling Technology, 1:1,000), and GAPDH (MAB374, Millipore, 1:6,000), rabbit monoclonal antibodies against CCND1 (2,978, Cell Signaling Technology, 1:1,000), phosphorylated ERK1/2 (4,370, Cell Signaling Technology, 1:1,000), ERK1/2 (4,695, Cell Signaling Technology, 1:1,000), phosphorylated mTOR (5,536, Cell Signaling Technology, 1:1,000), and mTOR (2,983, Cell Signaling Technology, 1:1,000), and a rabbit polyclonal antibody against cleaved caspase 3 (9,661, Cell Signaling Technology, 1:1,000), were used for immunoblotting. Peroxidase-conjugated anti-mouse IgG (7,076, Cell Signaling Technology, 1:100,000) and anti-rabbit IgG (7,074, Cell Signaling Technology, 1:100,000) were used as secondary antibodies. All immunoblotting experiments were performed at least two times to validate the results.
Immunofluorescence Analysis
The cells were plated onto 35-mm glass-bottom dishes at a density of 10,000/dish and treated with 30 μM atRA or vehicle control for 72 h. The immunofluorescence analysis was performed as previously described (Suzuki et al., 2018a), using mouse monoclonal antibodies against ERBB2 (MA5-13675, Thermo Fisher Scientific, 1:200) and JADE1 (MAB6275, R&D, 1:200). Images were taken with a confocal microscope (Ti-E, Nikon).
Knockdown and Overexpression of ERBB2 and JADE1
For the siRNA experiments, the cells were plated onto 35-mm dishes at a density of 20,000/dish. When the cells reached 70% confluency, they were treated with siRNA for either ERBB2, JADE1, or negative control, at 3 pmol in 6 μL of transfection reagent (TransIT-X2 system) in 2 mL of αMEM per dish. Total RNA was isolated after 24 h, and total protein was collected after 48 h. For the overexpression of ERBB2 and JADE1, HEPM cells were plated onto 35-mm dishes at a density of 20,000/dish. When the cells reached 70% confluency, they were treated with plasmid DNA for either ERBB2 [pcDNA3-HER2 (provided by Dr. Mien-Chie Hung through addgene, 16,257)], JADE1 [pCMV-SPORT6-PHF17 (ABIN3826934; genomics-online.com)], or negative controls [pcDNA3.1-RGS-6xHis (provided by Dr. Adam Antebi through addgene, 52,534) or pCMV-HA (provided by Dr. Christopher A Walsh through addgene, 32,530)], at 500 ng in 6 μL of transfection reagent (TransIT-X2 system) in 2 mL of αMEM per dish. Total RNA was isolated after 24 h.
Rescue Experiments
The cells were plated onto 60-mm dishes at a density of 250,000/well and treated with 30 μM atRA or vehicle control. After 24 h, the cells were transfected with either miR-4680-3p inhibitor (3 pmol) or control miR inhibitor (3 pmol; mirVana, Thermo Fisher Scientific), and either ERBB2, JADE1, or control overexpression vector, using the TransIT-X2 system (Mirus Bio LLC), according to the manufacturer's protocol (at 1 μg in 12 μL of transfection reagent in 4 mL of αMEM per dish). The cells were harvested 24 h after transfection and used for further experiments.
Statistical Analysis
All experiments were performed independently at least two times. The statistical significance of the differences between two groups was evaluated using a two-tailed Student t-test. Multiple comparisons were made by one-way analysis of variance with the post-hoc Tukey–Kramer's test. A p < 0.05 was considered to be statistically significant. Data are represented as mean ± standard deviation in the graphs.
Results
atRA Inhibits Cell Proliferation in a Dose-Dependent Manner in HEPM Cells
To determine the dose-dependent effects of atRA on cell proliferation in HEPM cells, we performed cell proliferation assays using HEPM cells treated with atRA at various concentrations (0, 1, 3, 10, and 30 μM). We found that cell proliferation activity was decreased by atRA treatment in a dose-dependent manner (Figure 1A). BrdU incorporation assays confirmed that cell proliferation was significantly decreased in cells treated with 30 μM atRA (Figures 1B,C). While excessive atRA is known to induce apoptosis in several tissues and cells (Okano et al., 2007; Mercader et al., 2008; Nelson et al., 2019; Quan et al., 2019), atRA failed to induce apoptosis in HEPM cells at 30 μM (Supplementary Figure 1). Previous studies show that atRA inhibits cell proliferation through downregulation of cyclin D1 (CCND1) and upregulation of cyclin-dependent kinase inhibitor 1B (CDKN1B; a.k.a. p27, KIP1) in HEPM cells (Dong et al., 2017) as well as in MCF-7 cells, a human breast cancer cell line (Teixeira and Pratt, 1997). We therefore evaluated the expression of CCND1 and CDKN1B by immunoblotting and confirmed that CCND1 expression was downregulated, and CDKN1B expression was upregulated, with atRA treatment (Figure 1D).
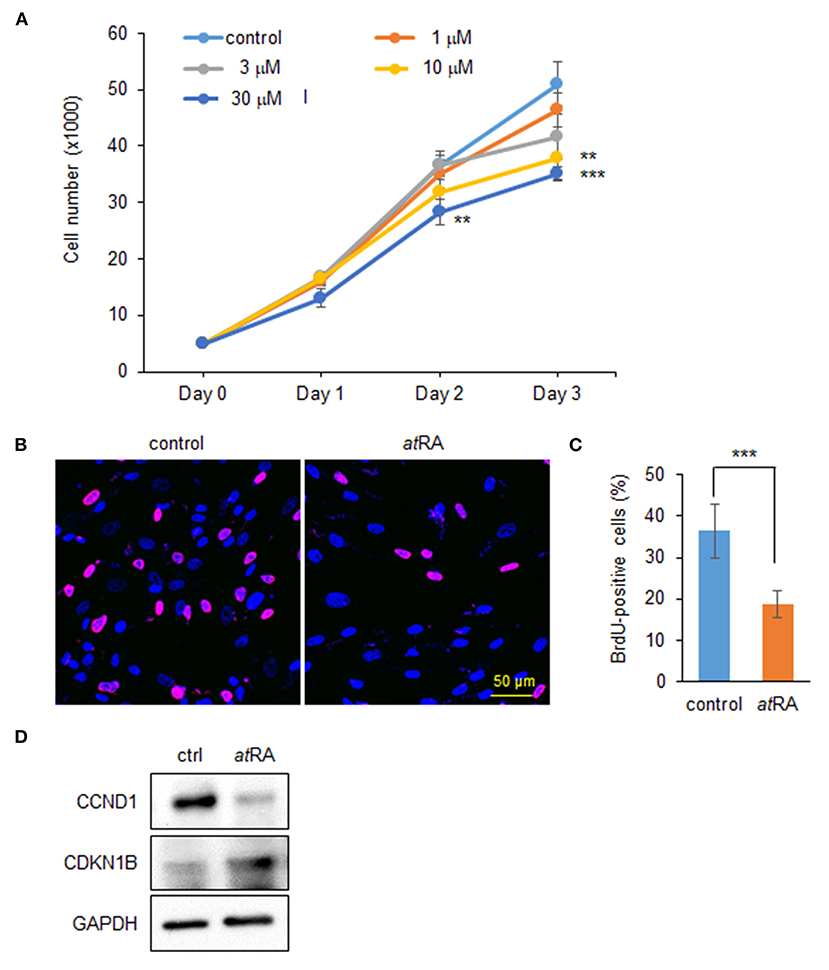
Figure 1. Influence of atRA treatment on proliferation of HEPM cells. (A) Cell proliferation assays in HEPM cells treated with various concentration of atRA for 24, 48, and 72 h. **p < 0.01, ***p < 0.001. Each treatment group was compared with a control vehicle group at each indicated day. (B) BrdU staining (red) in HEPM cells after treatment with 30 μM atRA for 72 h. Nuclei were counterstained with DAPI (blue). Scale bar, 50 μm. (C) Graph shows the quantification of BrdU-positive cells. ***p < 0.001. (D) Immunoblotting for CCND1, CDKN1B, and GAPDH in HEPM cells treated with 30 μM atRA for 72 h. Representative images from two independent experiments are shown.
atRA Suppresses ERBB2 and JADE1 Expression Through Upregulation of miR-4680-3p in HEPM Cells
Our previous studies showed that overexpression of either miR-140-5p, miR-133b, miR-374a-5p, miR-381-3p, or miR-4680-3p inhibits proliferation of HEPM cells through the suppression of genes that are crucial for palate development (Li et al., 2019; Suzuki et al., 2019). We therefore hypothesized that atRA induces the expression of these miRs. To test that hypothesis we analyzed the expression of these miRs after treatment of HEPM cells with atRA for 24 and 72 h, and found that expression of miR-4680-3p, but not miR-140-5p, miR-133b, miR-374a-5p, or miR-381-3p, was specifically and significantly upregulated with atRA treatment (Figure 2A). Next, to identify target genes suppressed by a miR-4680-3p mimic, we performed quantitative RT-PCR (qRT-PCR) analyses for the target genes (ERBB2, JADE1, MTHFD1, and WNT5A), which were predicted through bioinformatic analysis (Suzuki et al., 2019). We found that expression of ERBB2 [a.k.a. HER2, a member of the epidermal growth factor receptor (EGFR) family of transmembrane tyrosine kinase-type receptors (Schechter et al., 1984)] and JADE1 [a.k.a. PHF17, a member of the extended plant homeodomain (PHD) finger protein subfamily (Tzouanacou et al., 2003)] was significantly downregulated in HEPM cells treated with atRA for 24 and 72 h (Figure 2B). This suppression of ERBB2 and JADE1 in atRA-treated cells was confirmed with immunoblotting after 72 h of atRA treatment (Figure 2C).
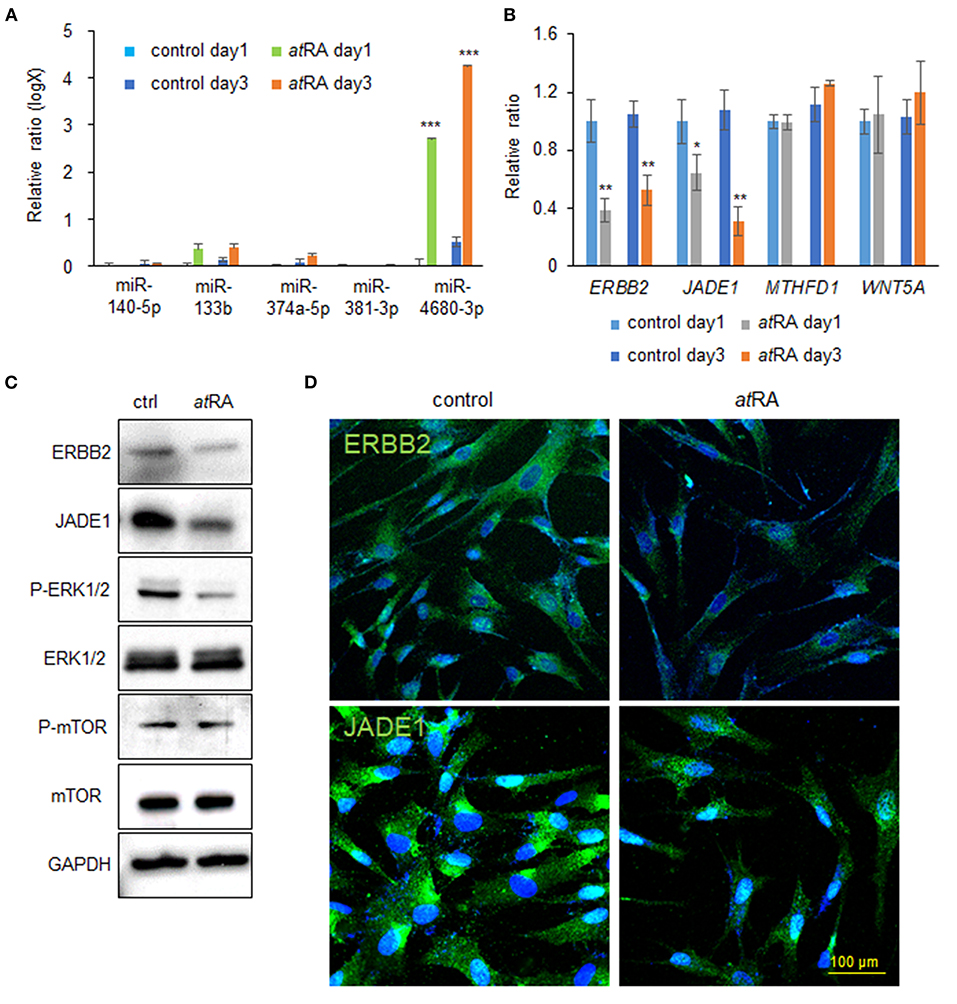
Figure 2. atRA induces miR-4680-3p expression in HEPM cells. (A) Quantitative RT-PCR for the indicated miRs after treatment of HEPM cells with atRA for 1 or 3 days. ***p < 0.001. (B) Quantitative RT-PCR for the indicated genes after treatment of HEPM cells with atRA for 1 or 3 days. *p < 0.05, **p < 0.01. Each treatment group was compared with a control vehicle group at each indicated day. (C) Immunoblotting for ERBB2, JADE1, phosphorylated ERK1/2 (P-ERK1/2), ERK1/2, phosphorylated mTOR (P-mTOR), mTOR, and GAPDH in HEPM cells treated with 30 μM atRA for 72 h. Representative images from two independent experiments are shown. (D) Immunocytochemical analysis of ERBB2 and JADE1 (green) in HEPM cells treated with 30 μM atRA for 72 h. Nuclei were counterstained with DAPI (blue).
Previous studies showed that ERBB2 stimulates several intracellular pathways such as MAPK/ERK and PI3K/AKT/mTOR (Yarden and Pines, 2012; Croessmann et al., 2019). For this reason, we analyzed the ERK1/2 and mTOR pathways in cells treated with atRA and found that ERK1/2 phosphorylation was downregulated, while mTOR phosphorylation was not altered, with atRA treatment (Figure 2C). ERBB2 was detected in the plasma membrane and cytosol in controls, as previously reported (Chung et al., 2016); by contrast, its expression was significantly decreased in atRA-treated cells (Figure 2D). JADE1 was detected in the nucleus and cytosol in controls, as previously reported (Panchenko et al., 2004; Havasi et al., 2013; Siriwardana et al., 2015); however, JADE1 expression was significantly decreased in atRA-treated HEPM cells (Figure 2D). Thus, our results indicate that atRA induces miR-4680-3p expression, leading to the suppression of ERBB2 and JADE1 via ERK1/2 signaling in HEPM cells.
Next, to evaluate the effect of expression of ERBB2 and JADE1 on cell proliferation, we treated HEPM cells with siRNAs for ERBB2 and JADE1. We confirmed that siRNA knockdown of either ERBB2 or JADE1 suppressed their expression at the mRNA and protein levels (Figures 3A–D). Under these conditions, cell proliferation was significantly suppressed by either ERBB2 or JADE1 siRNA knockdown. In addition, additional suppression was observed with a combination of ERBB2 and JADE1 siRNAs (Figure 3E). Furthermore, we confirmed that knockdown of ERBB2 and JADE1 in HEPM cells resulted in downregulated CCND1 and upregulated CDKN1B (Figure 3F). To evaluate the functional significance of ERBB2 and JADE1, we conducted rescue experiments by overexpressing ERBB2 and JADE1 in cells treated with atRA. We first confirmed that expression of ERBB2 and JADE1 was significantly upregulated following overexpression of these genes (Figures 3G,H). Under these conditions, we found that overexpression of ERBB2 and JADE1 partially rescued the cell proliferation inhibited by atRA (Figure 3I). Taken together, our results indicate that atRA inhibits cell proliferation through dysregulation of the ERBB2/JADE1-mediated CCND1/CDKN1B pathway in HEPM cells.
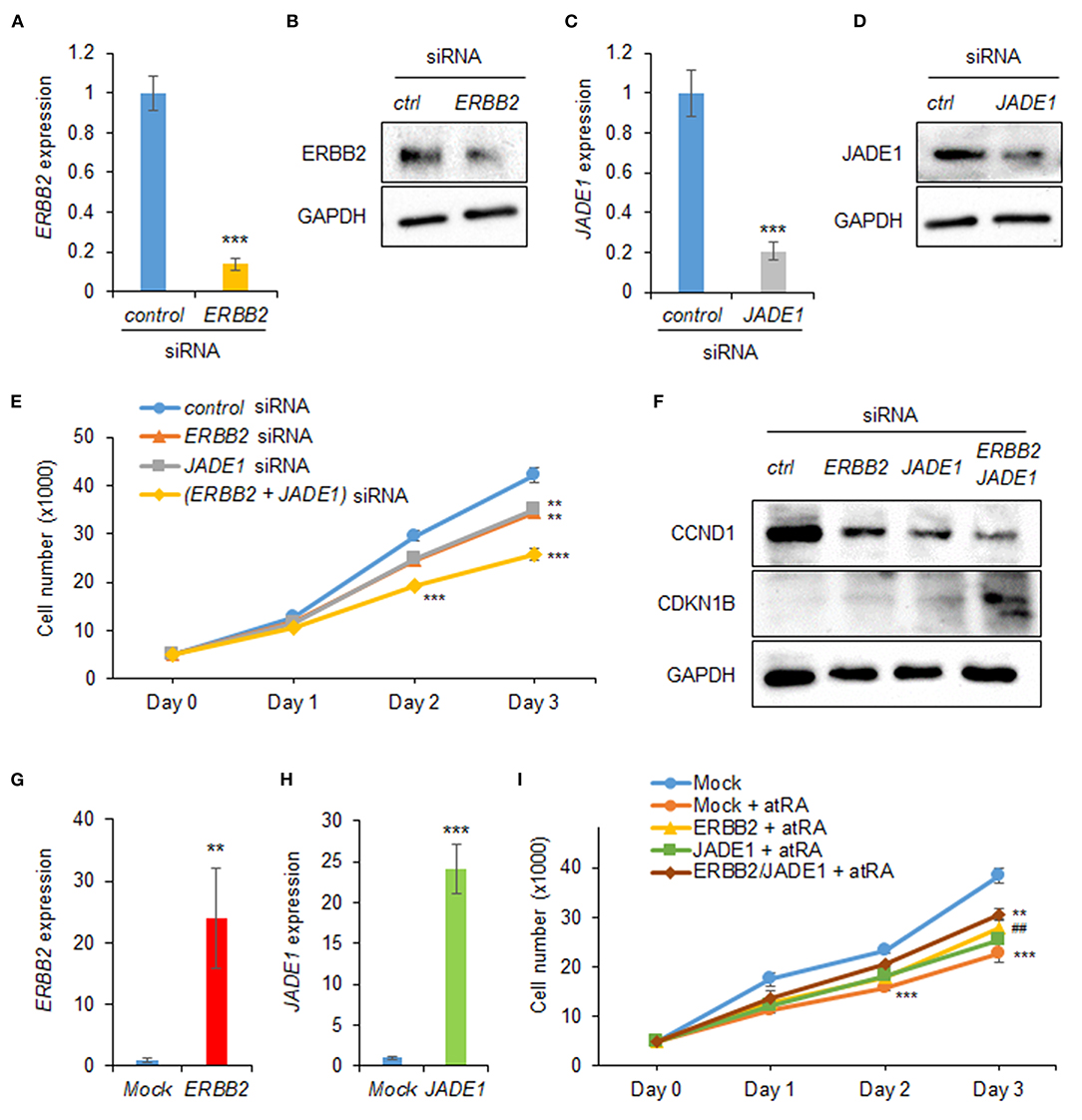
Figure 3. ERBB2 and JADE1 knockdown inhibits cell proliferation in HEPM cells. (A) Quantitative RT-PCR for ERBB2 after treatment with ERBB2 siRNA for 24 h in HEPM cells. ***p < 0.001. (B) Immunoblotting of ERBB2 and GAPDH in HEPM cells treated with an ERBB2 siRNA for 48 h. Representative images from two independent experiments are shown. (C) Quantitative RT-PCR for JADE1 after treatment with JADE1 siRNA for 24 h in HEPM cells. ***p < 0.001. (D) Immunoblotting of JADE1 and GAPDH in HEPM cells treated with JADE1 siRNA for 48 h. Representative images from two independent experiments are shown. (E) Cell proliferation assays in HEPM cells treated with an ERBB2 or JADE1 siRNA for 24, 48, or 72 h. **p < 0.01, ***p < 0.001. Each treatment group was compared with a control siRNA group at each indicated day. (F) Immunoblotting of CCND1, CDKN1B, and GAPDH in HEPM cells treated with siRNA for ERBB2 or JADE1 for 48 h. Representative images from two independent experiments are shown. (G) ERBB2 expression following overexpression in HEPM cells. **p < 0.01. (H) JADE1 expression following overexpression in HEPM cells. ***p < 0.001. (I) Cell proliferation assays in HEPM cells overexpressing ERBB2 and/or JADE1 for 24, 48, or 72 h. **p < 0.01, ***p < 0.001. Each treatment group was compared with control vector (mock) group at each indicated day. ##p < 0.01 vs. mock + atRA at day 3.
Inhibition of miR-4680-3p Can Partially Restore the Decreased Cell Proliferation Induced by atRA
To examine whether normalization of upregulated miR-4680-3p can restore decreased cell proliferation under atRA treatment conditions, we treated HEPM cells with a miR-4680-3p inhibitor, with or without atRA treatment, and found that a miR-4680-3p inhibitor could partially normalize the reduced cell proliferation (Figures 4A–C). As expected, suppression of ERBB2 and JADE1 was normalized by the miR-4680-3p inhibitor under atRA treatment conditions (Figures 4D,E). In addition, we confirmed that phosphorylation of ERK1/2 and expression of CCND1 and CDKN1B were normalized with miR-4680-3p inhibitor (Figure 4E). Taken together, our results indicate that atRA inhibits cell proliferation through miR-4680-3p expression in HEPM cells.
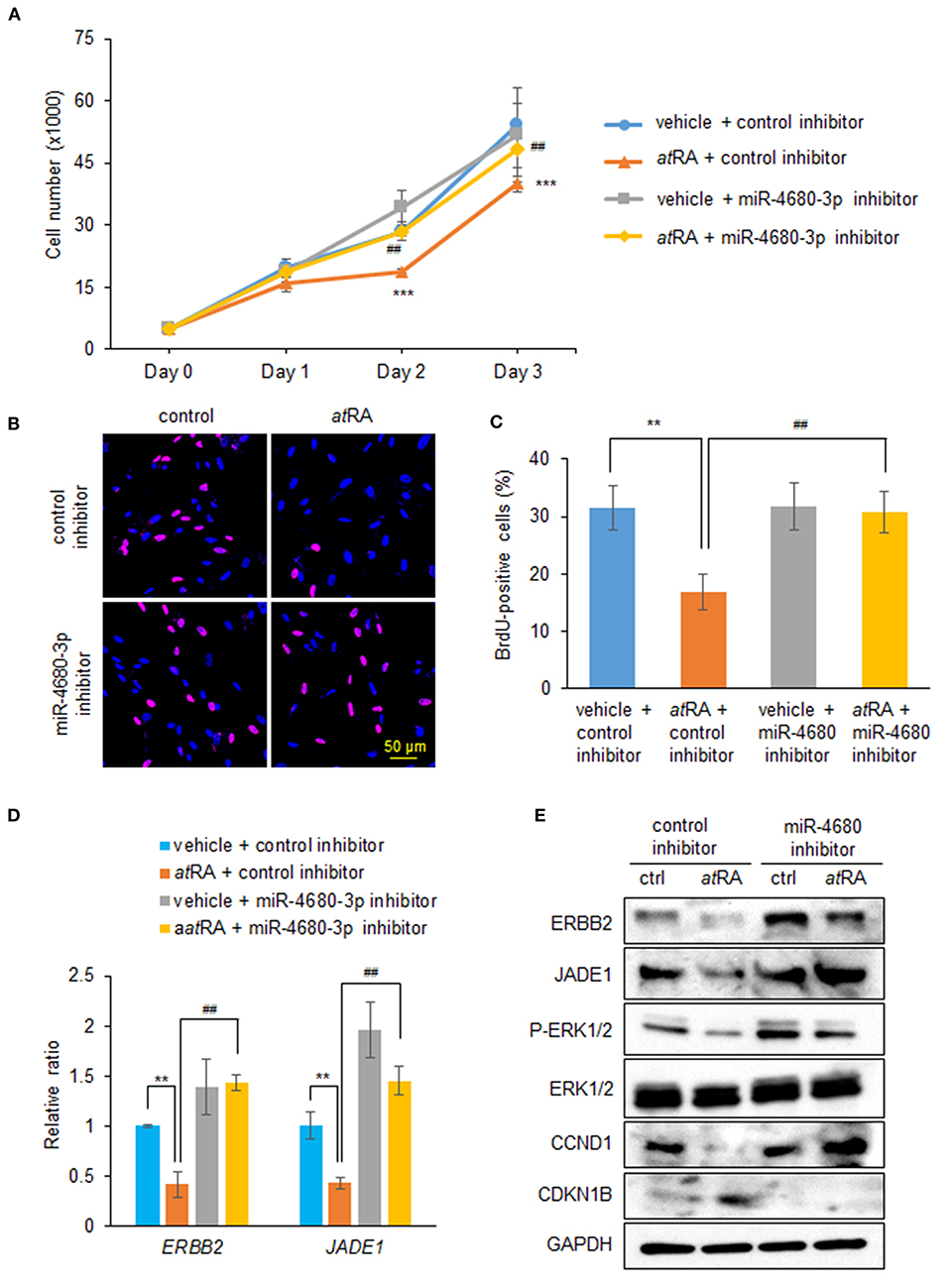
Figure 4. Normalization of miR-4680-3p expression restores atRA-induced cell proliferation suppression in HEPM cells. (A) Cell proliferation assays in HEPM cells treated with 30 μM atRA, with/without miR-4680-3p inhibitor, for 24, 48, or 72 h. ***p < 0.001 vs. control inhibitor + vehicle at each indicated day. ##p < 0.01 vs. control inhibitor + atRA at indicated day. (B) BrdU staining (red) in HEPM cells after treatment with 30 μM atRA, with/without miR-4680-3p inhibitor, for 72 h. Nuclei were counterstained with DAPI (blue). (C) Graph shows the quantification of BrdU-positive cells. **p < 0.01 vs. control inhibitor + vehicle. ##p < 0.01 vs. control inhibitor + atRA. (D) Quantitative RT-PCR for the indicated genes after treatment with atRA, with/without miR-4680-3p inhibitor, for 24 h in HEPM cells. **p < 0.01 vs. control inhibitor + vehicle. ##p < 0.01 vs. control inhibitor + atRA. (E) Immunoblotting of ERBB2, JADE1, phosphorylated ERK1/2 (P-ERK1/2), ERK1/2, CCND1, CDKN1B, and GAPDH in HEPM cells treated with 30 μM atRA, with/without miR-4680-3p inhibitor, for 72 h. Representative images from two independent experiments are shown.
Discussion
Excessive intake of atRA, a teratogenic reagent, induces cleft palate in humans and mice. Previous studies indicate that atRA inhibits cell proliferation and induces apoptosis in HEPM cells and mouse embryonic palatal mesenchymal (MEPM) cells (Yu et al., 2005; Dong et al., 2017). Since miRs are postulated to be essential in various biological processes, such as cell proliferation and apoptosis (Chen et al., 2004; Buser and Pohl, 2015; Shirjang et al., 2019; Akkoc and Gozuacik, 2020), we hypothesized that excessive atRA induces expression of miRs, which suppress genes crucial for palate development, leading to decreased cell proliferation. Our previous studies show that overexpression of miR-140-5p, miR-133b, miR-374a-5p, miR-381-3p, and miR-4680-3p suppresses cell proliferation through downregulation of genes related to cleft palate in HEPM cells (Li et al., 2019; Suzuki et al., 2019). Among them, we found that atRA specifically induced miR-4680-3p expression, which in turn suppressed expression of ERBB2 and JADE1. While expression of some predicted genes targeted by miR-4680-3p was not changed by miR-4680-3p overexpression in HEPM cells, these genes may be regulated by a combination of other miRs or through feedback loops reported in several cancer cell lines and in C2C12 cells, an immortalized mouse myoblast cell line (Hou et al., 2019; Liu et al., 2019; Quan et al., 2019).
ERBB2 is a member of the ERBB receptor tyrosine kinase family including epidermal growth factor receptor (EGFR) (Yarden and Shilo, 2007). The binding of ligands to receptors induces the homo- or hetero-dimerization of receptors and activates the kinase domain that induces downstream signaling cascades such as MAPK/ERK and PI3K/AKT/mTOR pathways, known to be crucial for cell proliferation, migration, and differentiation (Avraham and Yarden, 2011; Arteaga and Engelman, 2014). Our previous bioinformatic study suggests that ERBB signaling pathway may play a substantial role in palate formation (Yan et al., 2020). In fact, the activation of the ERBB2 pathway upregulates CCND1 expression, decreases CDKN1B stability (Lee et al., 2000; Yang et al., 2000; Lenferink et al., 2001), and promotes cell proliferation and angiogenesis in tumors (Le et al., 2005). In addition, inhibition of the ERBB2 pathway with a neutralizing antibody or small-molecule inhibitor for ERBB2 normalizes CDKN1B expression, leading to cell cycle arrest in human breast cancer cells (Le et al., 2003). atRA inhibits cell proliferation through downregulation of CCND1 and upregulation of CDKN1B in HEPM cells (Yu et al., 2005; Dong et al., 2017). Our results show that atRA inhibits cell proliferation through upregulated miR-4680-3p expression, which suppresses ERBB2 expression and its downstream ERK1/2 signaling pathway in HEPM cells. Taken together, our findings for the regulation of miRs by atRA shed light on the link between environmental and genetic factors, and explain how cell proliferation is inhibited by atRA.
JADE1 (a.k.a. PHF17), a transcription factor, contains two variants: JADE1-L (a long form with 842 amino acids) and JADE1-S (a short form without a C-terminal fragment of 333 amino acids) (Borgal et al., 2014; Siriwardana et al., 2015). Although the role of JADE1 remains elusive, the protein exhibits histone acetyltransferase (HAT) activity and acts as a co-factor of the HBO1 complex in histone H4 acetylation during gene regulation, which plays crucial roles in cell cycle regulation (Panchenko et al., 2004; Foy et al., 2008; Panchenko, 2016; Han et al., 2018). The suppression of JADE1 (both JADE1-L and JADE1-S) by siRNA knockdown results in suppression of DNA synthesis in cultured epithelial cell lines and primary fibroblasts (Havasi et al., 2013). Our results indicate that atRA treatment downregulates JADE1 expression, leading to decreased cell proliferation in HEPM cells through downregulated CDKN1 expression and upregulated CDKN1B expression. In future studies, we will characterize JADE1, namely whether and how JADE1 expression is regulated through the ERK1/2 pathway, and whether and how JADE1 regulates expression of CCND1 and CDKN1B. With those data in hand, we will then be able to conclude that JADE1 expression is directly regulated by miR-4680-3p, or regulated through ERBB2–mediated ERK1/2 signaling. In case treatment with both miR-4680-3p mimic and ERK1/2 inhibitor induces additional inhibition of cell proliferation, as well as additional suppression of CCND1 expression, JADE1 expression may be regulated by miR-4680-3p through the ERK1/2 pathway.
In summary, our findings and those of others suggest that excessive intake of retinoic acid during pregnancy leads to reduced cell proliferation through dysregulation of CCND1 and CDKN1B mediated by the miR-4680-3p–ERBB2–ERK1/2–CCND1/CDKN1B cascade in palatal mesenchymal cells, ultimately leading to cleft palate.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
Author Contributions
HY, SR, and JS performed the experiments. HY, AS, and JI wrote the article. All authors reviewed the results and approved the final version of the article.
Funding
This study was supported by grants from the National Institute of Dental and Craniofacial Research, part of the NIH (R03DE026208, R03DE026509, R03DE028340, R01DE029818, and R01DE026767 to JI).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2021.618876/full#supplementary-material
Supplementary Figure 1. atRA does not induces apoptosis in HEPM cells. (A) TUNEL staining (red) in HEPM cells after treatment with 30 μM atRA for 72 h. Nuclei were counterstained with DAPI (blue). Scale bar, 50 μm. (B) Graph shows the quantification of TUNEL-positive cells. N.D., Not Detected. (C) Immunoblotting of cleaved caspase-3 and GAPDH in HEPM cells treated with 30 μM atRA for 72 h. ctl, Control; P.C., Positive Control (mouse small intestine). Representative images from two independent experiments are shown.
References
Abbott, B. D., Harris, M. W., and Birnbaum, L. S. (1989). Etiology of retinoic acid-induced cleft palate varies with the embryonic stage. Teratology 40, 533–553. doi: 10.1002/tera.1420400602
Akkoc, Y., and Gozuacik, D. (2020). MicroRNAs as major regulators of the autophagy pathway. Biochim. Biophys. Acta Mol. Cell Res. 1867:118662. doi: 10.1016/j.bbamcr.2020.118662
Arteaga, C. L., and Engelman, J. A. (2014). ERBB receptors: from oncogene discovery to basic science to mechanism-based cancer therapeutics. Cancer Cell. 25, 282–303. doi: 10.1016/j.ccr.2014.02.025
Avraham, R., and Yarden, Y. (2011). Feedback regulation of EGFR signalling: decision making by early and delayed loops. Nat. Rev. Mol. Cell Biol. 12, 104–117. doi: 10.1038/nrm3048
Bartel, D. P. (2004). MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116, 281–297. doi: 10.1016/S0092-8674(04)00045-5
Borgal, L., Rinschen, M. M., Dafinger, C., Hoff, S., Reinert, M. J., Lamkemeyer, T., et al. (2014). Casein kinase 1 α phosphorylates the Wnt regulator Jade-1 and modulates its activity. J. Biol. Chem. 289, 26344–26356. doi: 10.1074/jbc.M114.562165
Buser, M. C., and Pohl, H. R. (2015). Windows of sensitivity to toxic chemicals in the development of cleft palates. J. Toxicol. Environ. Health B Crit. Rev. 18, 242–257. doi: 10.1080/10937404.2015.1068719
Chen, C. Z., Li, L., Lodish, H. F., and Bartel, D. P. (2004). MicroRNAs modulate hematopoietic lineage differentiation. Science 303, 83–86. doi: 10.1126/science.1091903
Chevrier, C., Perret, C., Bahuau, M., Nelva, A., Herman, C., Francannet, C., et al. (2005). Interaction between the ADH1C polymorphism and maternal alcohol intake in the risk of nonsyndromic oral clefts: an evaluation of the contribution of child and maternal genotypes. Birth Defects Res. A Clin. Mol. Teratol. 73, 114–122. doi: 10.1002/bdra.20103
Chung, I., Reichelt, M., Shao, L., Akita, R. W., Koeppen, H., Rangell, L., et al. (2016). High cell-surface density of HER2 deforms cell membranes. Nat. Commun. 7:12742. doi: 10.1038/ncomms12742
Croessmann, S., Formisano, L., Kinch, L. N., Gonzalez-Ericsson, P. I., Sudhan, D. R., Nagy, R. J., et al. (2019). Combined blockade of activating ERBB2 mutations and ER results in synthetic lethality of ER+/HER2 mutant breast cancer. Clin. Cancer Res. 25, 277–289. doi: 10.1158/1078-0432.CCR-18-1544
Cunningham, T. J., and Duester, G. (2015). Mechanisms of retinoic acid signalling and its roles in organ and limb development. Nat. Rev. Mol. Cell Biol. 16, 110–123. doi: 10.1038/nrm3932
Dhulipala, V. C., Welshons, W. V., and Reddy, C. S. (2006). Cell cycle proteins in normal and chemically induced abnormal secondary palate development: a review. Hum. Exp. Toxicol. 25, 675–682. doi: 10.1177/0960327106070848
Dong, S., Zhang, Y., and Huang, H. (2017). Involvement of RBP4 in alltrans retinoic acid induced cleft palate. Mol. Med. Rep. 16, 5915–5923. doi: 10.3892/mmr.2017.7327
Foy, R. L., Song, I. Y., Chitalia, V. C., Cohen, H. T., Saksouk, N., Cayrou, C., et al. (2008). Role of Jade-1 in the histone acetyltransferase (HAT) HBO1 complex. J. Biol. Chem. 283, 28817–28826. doi: 10.1074/jbc.M801407200
Habib, Z. (1978). Genetic counselling and genetics of cleft lip and cleft palate. Obstet. Gynecol. Surv. 33, 441–447. doi: 10.1097/00006254-197807000-00001
Han, J., Lachance, C., Ricketts, M. D., McCullough, C. E., Gerace, M., Black, B. E., et al. (2018). The scaffolding protein JADE1 physically links the acetyltransferase subunit HBO1 with its histone H3-H4 substrate. J. Biol. Chem. 293, 4498–4509. doi: 10.1074/jbc.RA117.000677
Havasi, A., Haegele, J. A., Gall, J. M., Blackmon, S., Ichimura, T., Bonegio, R. G., et al. (2013). Histone acetyl transferase (HAT) HBO1 and JADE1 in epithelial cell regeneration. Am. J. Pathol. 182, 152–162. doi: 10.1016/j.ajpath.2012.09.017
Hou, L., Zhu, L., Li, H., Jiang, F., Cao, L., Hu, C. Y., et al. (2019). MiR-501-3p forms a feedback loop with FOS, MDFI, and MyoD to regulate C2C12 myogenesis. Cells 8:573. doi: 10.3390/cells8060573
Hu, X., Gao, J., Liao, Y., Tang, S., and Lu, F. (2013). Retinoic acid alters the proliferation and survival of the epithelium and mesenchyme and suppresses Wnt/β-catenin signaling in developing cleft palate. Cell Death Dis. 4:e898. doi: 10.1038/cddis.2013.424
Karsy, M., Albert, L., Tobias, M. E., Murali, R., and Jhanwar-Uniyal, M. (2010). All-trans retinoic acid modulates cancer stem cells of glioblastoma multiforme in an MAPK-dependent manner. Anticancer Res. 30, 4915–4920.
Lai, L., Bohnsack, B. L., Niederreither, K., and Hirschi, K. K. (2003). Retinoic acid regulates endothelial cell proliferation during vasculogenesis. Development 130, 6465–6474. doi: 10.1242/dev.00887
Le, X. F., Claret, F. X., Lammayot, A., Tian, L., Deshpande, D., LaPushin, R., et al. (2003). The role of cyclin-dependent kinase inhibitor p27Kip1 in anti-HER2 antibody-induced G1 cell cycle arrest and tumor growth inhibition. J. Biol. Chem. 278, 23441–23450. doi: 10.1074/jbc.M300848200
Le, X. F., Pruefer, F., and Bast, R. C. Jr. (2005). HER2-targeting antibodies modulate the cyclin-dependent kinase inhibitor p27Kip1 via multiple signaling pathways. Cell Cycle 4, 87–95. doi: 10.4161/cc.4.1.1360
Lee, R. J., Albanese, C., Fu, M., D'Amico, M., Lin, B., Watanabe, G., et al. (2000). Cyclin D1 is required for transformation by activated Neu and is induced through an E2F-dependent signaling pathway. Mol. Cell. Biol. 20, 672–683. doi: 10.1128/MCB.20.2.672-683.2000
Lenferink, A. E., Busse, D., Flanagan, W. M., Yakes, F. M., and Arteaga, C. L. (2001). ErbB2/neu kinase modulates cellular p27(Kip1) and cyclin D1 through multiple signaling pathways. Cancer Res. 61, 6583–6591.
Li, A., Jia, P., Mallik, S., Fei, R., Yoshioka, H., Suzuki, A., et al. (2019). Critical microRNAs and regulatory motifs in cleft palate identified by a conserved miRNA-TF-gene network approach in humans and mice. Brief Bioinform. 21, 1465–1478. doi: 10.1093/bib/bbz082
Liu, D., Zhong, L., Yuan, Z., Yao, J., Zhong, P., Liu, J., et al. (2019). miR-382-5p modulates the ATRA-induced differentiation of acute promyelocytic leukemia by targeting tumor suppressor PTEN. Cell Signal. 54, 1–9. doi: 10.1016/j.cellsig.2018.11.012
Liu, W., Song, Y., Zhang, C., Gao, P., Huang, B., and Yang, J. (2018). The protective role of all-transretinoic acid (ATRA) against colorectal cancer development is achieved via increasing miR-3666 expression and decreasing E2F7 expression. Biomed. Pharmacother. 104, 94–101. doi: 10.1016/j.biopha.2018.05.015
Mercader, J., Granados, N., Bonet, M. L., and Palou, A. (2008). All-trans retinoic acid decreases murine adipose retinol binding protein 4 production. Cell Physiol. Biochem. 22, 363–372. doi: 10.1159/000149815
Mihaly, J., Gericke, J., Aydemir, G., Weiss, K., Carlsen, H., Blomhoff, R., et al. (2012). Reduced retinoid signaling in the skin after systemic retinoid-X receptor ligand treatment in mice with potential relevance for skin disorders. Dermatology 225, 304–311. doi: 10.1159/000345496
Mossey, P. A., Little, J., Munger, R. G., Dixon, M. J., and Shaw, W. C. (2009). Cleft lip and palate. Lancet 374, 1773–1785. doi: 10.1016/S0140-6736(09)60695-4
Mukhopadhyay, P., Smolenkova, I., Warner, D., Pisano, M. M., and Greene, R. M. (2019). Spatio-temporal expression and functional analysis of miR-206 in developing orofacial tissue. Microrna 8, 43–60. doi: 10.2174/2211536607666180801094528
Nelson, S. E., Shoov, E., LaBrie, R. A., and Shaffer, H. J. (2019). Externalizing and self-medicating: heterogeneity among repeat DUI offenders. Drug Alcohol. Depend. 194, 88–96. doi: 10.1016/j.drugalcdep.2018.09.017
Obernosterer, G., Leuschner, P. J., Alenius, M., and Martinez, J. (2006). Post-transcriptional regulation of microRNA expression. RNA 12, 1161–1167. doi: 10.1261/rna.2322506
Okano, J., Suzuki, S., and Shiota, K. (2007). Involvement of apoptotic cell death and cell cycle perturbation in retinoic acid-induced cleft palate in mice. Toxicol. Appl. Pharmacol. 221, 42–56. doi: 10.1016/j.taap.2007.02.019
Panchenko, M. V. (2016). Structure, function and regulation of jade family PHD finger 1 (JADE1. Gene 589, 1–11. doi: 10.1016/j.gene.2016.05.002
Panchenko, M. V., Zhou, M. I., and Cohen, H. T. (2004). von Hippel-Lindau partner Jade-1 is a transcriptional co-activator associated with histone acetyltransferase activity. J. Biol. Chem. 279, 56032–56041. doi: 10.1074/jbc.M410487200
Prescott, N. J., Winter, R. M., and Malcolm, S. (2002). Maternal MTHFR genotype contributes to the risk of non-syndromic cleft lip and palate. J. Med. Genet. 39, 368–369. doi: 10.1136/jmg.39.5.368
Quan, X., Li, X., Yin, Z., Ren, Y., and Zhou, B. (2019). p53/miR-30a-5p/ SOX4 feedback loop mediates cellular proliferation, apoptosis, and migration of non-small-cell lung cancer. J. Cell Physiol. 234, 22884–22895. doi: 10.1002/jcp.28851
Ramirez, D., Lammer, E. J., Iovannisci, D. M., Laurent, C., Finnell, R. H., and Shaw, G. M. (2007). Maternal smoking during early pregnancy, GSTP1 and EPHX1 variants, and risk of isolated orofacial clefts. Cleft Palate Craniofac. J. 44, 366–373. doi: 10.1597/06-011.1
Rhinn, M., and Dolle, P. (2012). Retinoic acid signalling during development. Development 139, 843–858. doi: 10.1242/dev.065938
Roberts, C. (2020). Regulating retinoic acid availability during development and regeneration: the role of the CYP26 enzymes. J. Dev. Biol. 8:6. doi: 10.3390/jdb8010006
Ross, S. A., McCaffery, P. J., Drager, U. C., and De Luca, L. M. (2000). Retinoids in embryonal development. Physiol. Rev. 80, 1021–1054. doi: 10.1152/physrev.2000.80.3.1021
Schechter, A. L., Stern, D. F., Vaidyanathan, L., Decker, S. J., Drebin, J. A., Greene, M. I., et al. (1984). The neu oncogene: an erb-B-related gene encoding a 185,000-Mr tumour antigen. Nature 312, 513–516. doi: 10.1038/312513a0
Schoen, C., Aschrafi, A., Thonissen, M., Poelmans, G., Von den Hoff, J. W., and Carels, C. E. L. (2017). MicroRNAs in palatogenesis and cleft palate. Front. Physiol. 8:165. doi: 10.3389/fphys.2017.00165
Schoen, C., Glennon, J. C., Abghari, S., Bloemen, M., Aschrafi, A., Carels, C. E. L., et al. (2018). Differential microRNA expression in cultured palatal fibroblasts from infants with cleft palate and controls. Eur. J. Orthod. 40, 90–96. doi: 10.1093/ejo/cjx034
Seelan, R. S., Mukhopadhyay, P., Warner, D. R., Appana, S. N., Brock, G. N., Pisano, M. M., et al. (2014). Methylated microRNA genes of the developing murine palate. Microrna 3, 160–173. doi: 10.2174/2211536604666150131125805
Shin, J. O., Lee, J. M., Cho, K. W., Kwak, S., Kwon, H. J., Lee, M. J., et al. (2012). MiR-200b is involved in Tgf-β signaling to regulate mammalian palate development. Histochem. Cell Biol. 137, 67–78. doi: 10.1007/s00418-011-0876-1
Shirjang, S., Mansoori, B., Asghari, S., Duijf, P. H. G., Mohammadi, A., Gjerstorff, M., et al. (2019). MicroRNAs in cancer cell death pathways: apoptosis and necroptosis. Free Radic. Biol. Med. 139, 1–15. doi: 10.1016/j.freeradbiomed.2019.05.017
Siddikuzzaman, Guruvayoorappan, C., and Berlin Grace, V. M. (2011). All trans retinoic acid and cancer. Immunopharmacol. Immunotoxicol. 33, 241–249. doi: 10.3109/08923973.2010.521507
Siriwardana, N. S., Meyer, R. D., and Panchenko, M. V. (2015). The novel function of JADE1S in cytokinesis of epithelial cells. Cell Cycle 14, 2821–2834. doi: 10.1080/15384101.2015.1068476
Suzuki, A., Li, A., Gajera, M., Abdallah, N., Zhang, M., Zhao, Z., et al. (2019). MicroRNA-374a,-4680, and-133b suppress cell proliferation through the regulation of genes associated with human cleft palate in cultured human palate cells. BMC Med. Genomics. 12:93. doi: 10.1186/s12920-019-0546-z
Suzuki, A., Minamide, R., and Iwata, J. (2018a). The role of acetyltransferases for the temporal-specific accessibility of β-catenin to the myogenic gene locus. Sci. Rep. 8:15057. doi: 10.1038/s41598-018-32888-z
Suzuki, A., Minamide, R., and Iwata, J. (2018b). WNT/β-catenin signaling plays a crucial role in myoblast fusion through regulation of nephrin expression during development. Development 145:dev168351. doi: 10.1242/dev.168351
Teixeira, C., and Pratt, M. A. (1997). CDK2 is a target for retinoic acid-mediated growth inhibition in MCF-7 human breast cancer cells. Mol. Endocrinol. 11, 1191–1202. doi: 10.1210/mend.11.9.9977
Tzouanacou, E., Tweedie, S., and Wilson, V. (2003). Identification of Jade1, a gene encoding a PHD zinc finger protein, in a gene trap mutagenesis screen for genes involved in anteroposterior axis development. Mol. Cell. Biol. 23, 8553–8552. doi: 10.1128/MCB.23.23.8553-8562.2003
Wang, S., Sun, C., Meng, Y., Zhang, B., Wang, X., Su, Y., et al. (2017). A pilot study: screening target miRNAs in tissue of nonsyndromic cleft lip with or without cleft palate. Exp. Ther. Med. 13, 2570–2576. doi: 10.3892/etm.2017.4248
Wang, W., and Kirsch, T. (2002). Retinoic acid stimulates annexin-mediated growth plate chondrocyte mineralization. J. Cell. Biol. 157, 1061–1069. doi: 10.1083/jcb.200203014
Yan, F., Dai, Y., Iwata, J., Zhao, Z., and Jia, P. (2020). An integrative, genomic, transcriptomic and network-assisted study to identify genes associated with human cleft lip with or without cleft palate. BMC Med. Genomics. 13:39. doi: 10.1186/s12920-020-0675-4
Yang, H. Y., Zhou, B. P., Hung, M. C., and Lee, M. H. (2000). Oncogenic signals of HER-2/neu in regulating the stability of the cyclin-dependent kinase inhibitor p27. J. Biol. Chem. 275, 24735–24739. doi: 10.1074/jbc.C000147200
Yao, Z., Chen, D., Wang, A., Ding, X., Liu, Z., Ling, L., et al. (2011). Folic acid rescue of ATRA-induced cleft palate by restoring the TGF-β signal and inhibiting apoptosis. J. Oral. Pathol. Med. 40, 433–439. doi: 10.1111/j.1600-0714.2010.00994.x
Yarden, Y., and Pines, G. (2012). The ERBB network: at last, cancer therapy meets systems biology. Nat. Rev. Cancer. 12, 553–563. doi: 10.1038/nrc3309
Yarden, Y., and Shilo, B. Z. (2007). SnapShot: EGFR signaling pathway. Cell. 31:1018. doi: 10.1016/j.cell.2007.11.013
Keywords: all-trans retinoic acid (all-trans RA), cleft palate (CP), microRNA (miR), cell proliferation, environmental factor
Citation: Yoshioka H, Ramakrishnan SS, Shim J, Suzuki A and Iwata J (2021) Excessive All-Trans Retinoic Acid Inhibits Cell Proliferation Through Upregulated MicroRNA-4680-3p in Cultured Human Palate Cells. Front. Cell Dev. Biol. 9:618876. doi: 10.3389/fcell.2021.618876
Received: 18 October 2020; Accepted: 05 January 2021;
Published: 28 January 2021.
Edited by:
Poongodi Geetha-Loganathan, SUNY Oswego, United StatesReviewed by:
Enrico Garattini, Istituto di Ricerche Farmacologiche Mario Negri (IRCCS), ItalyMaddalena Fratelli, Mario Negri Pharmacological Research Institute, Italy
Copyright © 2021 Yoshioka, Ramakrishnan, Shim, Suzuki and Iwata. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Junichi Iwata, anVuaWNoaS5pd2F0YUB1dGgudG1jLmVkdQ==