Abstract
The NLRP3 inflammasome represents a critical inflammatory machinery driving pathology in many acute (e. g., myocardial infarction or stroke) and chronic (Alzheimer's disease, atherosclerosis) human disorders linked to the activity of IL-1 cytokines. Although the therapeutic potential of NLRP3 is undisputed, currently no clinically approved therapies exist to target the NLRP3 inflammasome directly. The recent discovery of BTK as a direct and positive regulator of the NLRP3 inflammasome has, however, raised the intriguing possibility of targeting the NLRP3 inflammasome via existing or future BTK inhibitors. Here, I review the mechanistic basis for this notion and discuss the molecular and cellular role of BTK in the inflammasome process. Specific attention will be given to cell-type dependent characteristics and differences that may be relevant for targeting approaches. Furthermore, I review recent (pre-)clinical evidence for effects of BTK inhibitors on NLRP3 activity and highlight and discuss open questions and future research directions. Collectively, the concept of targeting BTK to target NLRP3-dependent inflammation will be explored comprehensively at the molecular, cellular and therapeutic levels.
Introduction
Within the immune system, Bruton's tyrosine kinase (BTK) appears to be something like a “Swiss Army knife,” a highly versatile molecule that seemingly participates in innumerable processes [reviewed in Weber et al. (2017)]: These range from immune cell development and differentiation in neutrophils and B cells (Khan et al., 1995; Fiedler et al., 2011), to innate functions e.g. Toll-like receptor (TLR), Fc, and growth factor receptor signaling, phagocytosis, and platelet activation (Quek et al., 1998; Horwood et al., 2006; Jongstra-Bilen et al., 2008; Melcher et al., 2008; Singhal et al., 2011; Strijbis et al., 2013), and to adaptive immunity, e.g. BCR signaling (Wilson et al., 2015). To add to this, BTK is not only relevant in normal cells but also in the context of malignancy, most notably as a target for B cell malignancies (Wilson et al., 2015). Recent evidence regarding novel splice variants of BTK in colon (Grassilli et al., 2016) and breast (Eifert et al., 2013) cancer expand its significance from the well-known and critical role of BTK in malignant B cells. It is the latter that has driven intense research and clinical development of pharmacological inhibitors to target the kinase activity of BTK. Ibrutinib was the first inhibitor approved by the FDA for the treatment of chronic lymphocytic leukemia and mantle cell lymphoma (Byrd et al., 2013; Mcnally et al., 2015) and later steroid-resistant chronic Graft-vs.-Host disease (GvHD) (Jaglowski and Blazar, 2018), but multiple other molecules are now in development or clinical testing. So the coming years are not only going to witness better targeting of B cell malignancies but a host of new applications especially in innate immunity await exploration. Especially in innate immunity, one process has recently gained considerable interest as a regulator of inflammation, the NLRP3 inflammasome (Agostini et al., 2004; Swanson et al., 2019; Weber et al., 2020). NLRP3 is a cytoplasmic danger and stress sensor belonging to the Nod-like receptor family of pattern recognition receptors (PRRs) (Agostini et al., 2004; Takeuchi and Akira, 2010), that is activated by homeostasis-perturbing exogenous and endogenous cues. Its expression has been reporter for multiple myeloid cells, ranging from macrophages (Franchi and Nunez, 2008), dendritic cells (Ghiringhelli et al., 2009), platelets (Murthy et al., 2017) and neutrophils (Mankan et al., 2012) to microglia (Freeman et al., 2017), Kupffer cells (Huang et al., 2013) and cardiomyocytes (Yao et al., 2018) [summarized in Guarda et al. (2011) and Weber et al. (2020)]. At least in vitro, NLRP3 activation involves a transcriptional and post-translational priming phase (signal 1), e.g., via TLR signaling, followed by an actual activation step (signal 2), which has been linked to K+ efflux (Munoz-Planillo et al., 2013). The latter leads to conformational changes in NLRP3 (Tapia-Abellan et al., 2019) and the assembly of a large multi-protein complex termed the inflammasome (Figure 1) (Agostini et al., 2004; Swanson et al., 2019). The NLRP3 inflammasome additionally includes an adaptor, ASC, and the enzyme pro-caspase-1 (Swanson et al., 2019). Upon assembly, caspase-1 becomes active and cleaves inactive IL-1 cytokine family members to mature into biologically active and secreted forms that potently trigger inflammation (Swanson et al., 2019). Comprehensive reviews of NLRP3 activation and regulation are found here (Mangan et al., 2018; Swanson et al., 2019; Weber et al., 2020). Inflammation instigated by NLRP3 has been shown to be fundamental to pathophysiological changes in diseases like cryopyrin-associated periodic fever syndrome (CAPS) (Agostini et al., 2004), myocardial infarction (Abbate et al., 2015), stroke (Ito et al., 2015), liver inflammation (Wree et al., 2014), type 2 diabetes (Masters et al., 2010), Alzheimer's disease (Heneka et al., 2013) Parkinson's disease (Gordon et al., 2018) and aging (Swanson et al., 2019; He et al., 2020; Weber et al., 2020). Hence, targeting NLRP3 is one of the most prominent therapeutic goals in innate immunity (Mangan et al., 2018). Unfortunately, there are so far no clinically approved inhibitors for direct targeting of NLRP3 or caspase-1 (Mangan et al., 2018; Swanson et al., 2019). When others and us recently discovered BTK as a novel positive regulator (Ito et al., 2015; Liu et al., 2017) this posed the intriguing possibility to target NLRP3-mediated inflammation using BTK kinase inhibitors (Banoth and Cassel, 2017; Henrickson, 2017; Liu et al., 2017). I here review the mechanistic and clinical basis for this notion, current controversies and open questions.
Figure 1
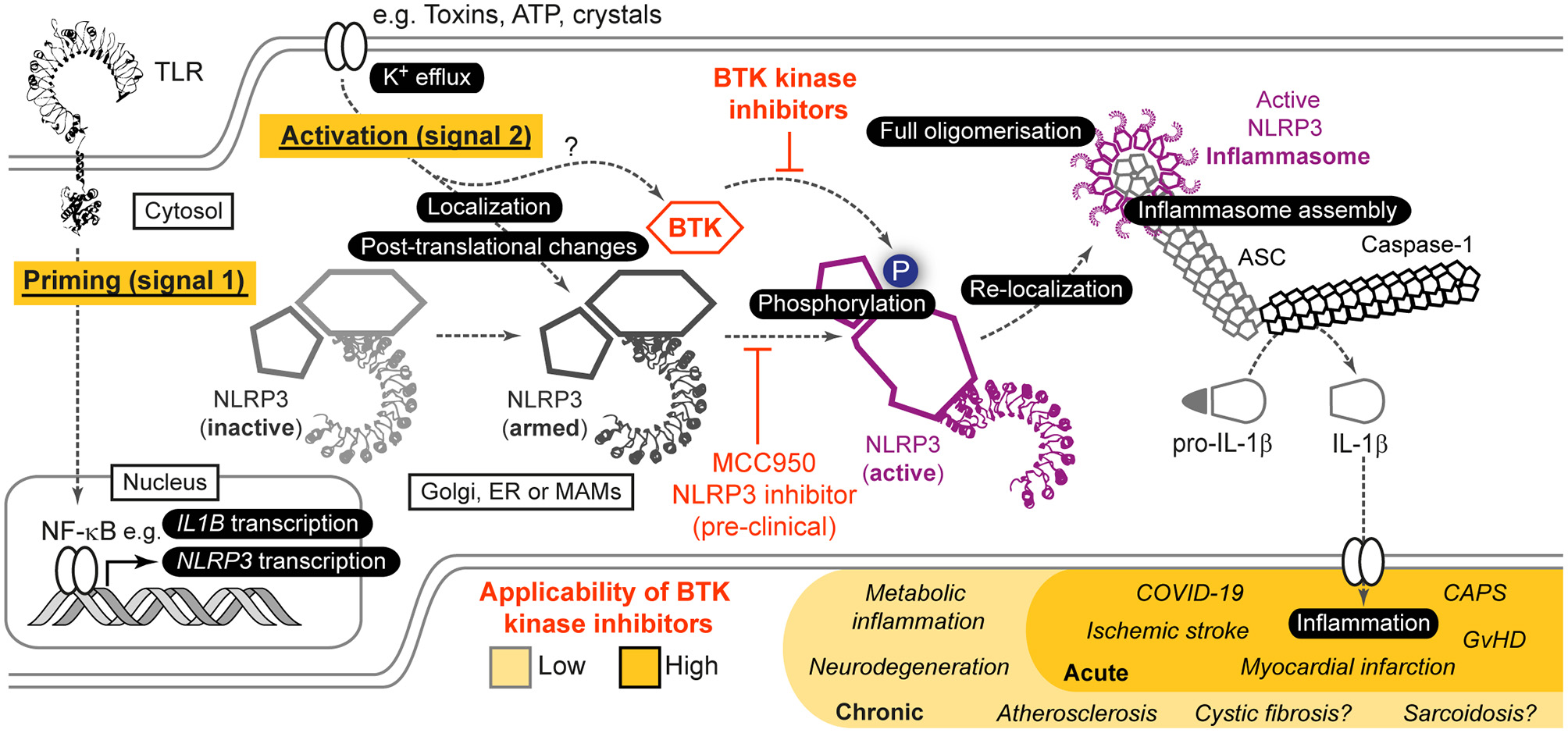
Role of BTK in NLRP3 inflammasome activation and resulting opportunities for indirect targeting of NLRP3 via BTK. Simplified scheme of NLRP3 activation highlighting the role of BTK. For further details see main article and Weber et al. (2020). Based on in vitro, pre-clinical mouse models as well as ex vivo analyses in human samples and patients, indirect targeting of NLRP3 via current BTK inhibitors could be envisaged especially in acute NLRP3/IL-1 axis-mediated inflammatory conditions, whereas in chronic conditions NLRP3-selective targeting would be necessary.
Molecular Contributions of BTK to the NLRP3 Activation Process
BTK was identified as a NLRP3 regulator by two independent groups based on tyrosine kinase inhibitor and phospho-proteomics screens (Ito et al., 2015; Liu et al., 2017): Ito et al. (2015) showed a positive regulatory role for BTK in murine myeloid cells and its targeting reduced NLRP3 inflammasome-dependent IL-1β release. Similar results were obtained in our lab for primary human immune cells (Liu et al., 2017). Both studies also showed a physical interaction of BTK with NLRP3 and its adaptor ASC in overexpression systems (Ito et al., 2015; Liu et al., 2017), hinting to a direct role in the inflammasome process. BTK seemed specific for NLRP3 as other inflammasomes were not affected (Ito et al., 2015). Interestingly, BTK was rapidly phosphorylated upon NLRP3 activation, suggesting that its kinase activity might be relevant for the NLRP3 activation process. We therefore investigated whether BTK also interacted with endogenous NLRP3 in primary immune cells. Indeed, an interaction was detected after LPS stimulation of cells, i.e., after initiation of the priming phase (signal 1) and before the actual activation step (signal 2). Furthermore, we noted that NLRP3 became phospho-tyrosine-modified upon activation of NLRP3 by nigericin, in our hands (Bittner et al., 2020, Preprint) and also observed in Mao et al. (2020). This modification was BTK-dependent as it was reduced in BTK-ablated murine Btk KO BMDM and human XLA PBMC or inhibitor treated cells (Bittner et al., 2020, PrePrint). We subsequently mapped BTK-modified tyrosine residues and found that BTK was able to modify at least four tyrosine residues—three located in a critical localization motif of NLRP3, the so-called polybasic motif (PBM), and one adjacent to this motif (Bittner et al., 2020, PrePrint). The positively charged PBM was shown to direct NLRP3 toward phospho-inositol-4-phosphate (PIP4)-rich membranes (e.g., Golgi and possibly endosomes or mitochondria-associated membranes) via charge interactions (Zhang et al., 2017; Chen and Chen, 2018; Seoane et al., 2020). On these membranes, NLRP3 is thought to oligomerize (Chen and Chen, 2018) but then dissociate and re-locate to the microtubule-organizing center (MTOC) where ASC and NEK7 are engaged (Magupalli et al., 2020). On a peptide level, phospho-modification of tyrosine residues in the PBM altered the charge of this region (Bittner et al., 2020, PrePrint) and therefore may support the translocation or release of NLRP3 from PI4P-rich membranes, which would enable HDAC-6/dynein-dependent transport toward the MTOC where NLPR3 was shown to assemble a macromolecular ASC- and caspase-1 containing inflammasome (Magupalli et al., 2020). This would be similar to the described function of Protein Kinase D to promote membrane dissociation (Zhang et al., 2017), albeit the latter mechanism appears PBM-independent as the modified site, S295, locates outside the PBM, but may also involve charge repulsion upon serine phosphorylation. In line with this notion of BTK-mediated phosphorylation to alter PBM charge, mutation of the BTK-modified tyrosines in overexpressed NLRP3 resulted in lower binding to PIP4 beads (Bittner et al., 2020, Preprint). Furthermore, BTK inhibition or genetic ablation in primary cells coincided with a reduced ability of NLRP3 to form oligomers and to engage ASC (Bittner et al., 2020, Preprint). Furthermore, we observed that a mutant form of NLRP3, in which the modified tyrosines were mutated to phenylalanine failed to induce IL-1 release (Bittner et al., 2020, Preprint), indicating that these tyrosine positions indeed are vital for full NLRP3 activation. Collectively, studies by us (Liu et al., 2017; Bittner et al., 2020, Preprint) and others (Ito et al., 2015) suggest that BTK is a direct and positive regulator of the inflammasome process, raising the intriguing notion of targeting NLRP3-mediated inflammation directly via BTK (Banoth and Cassel, 2017; Henrickson, 2017; Liu et al., 2017).
Targeting NLRP3 Indirectly Via BTK—a Viable Therapeutic Opportunity in the Absence of Clinically Approved Direct NLRP3 Inhibitors
Given its involvement in multiple inflammatory disorders, multiple strategies for targeting NLRP3-mediated inflammation have been proposed. FDA-approved since 2001 is the targeting of IL-1 itself via either recombinant IL-1 receptor antagonist (IL-1RA, tradename Rilonacept) or monoclonal antibodies (tradenames Anakinra and Canakinumab) (Dinarello and Van Der Meer, 2013). These treatments are well-established and show good efficacy; however, they involve regular injections, suffer from resistance mechanisms such as anti-drug antibodies, and do not target NLRP3 directly; rather, they only target one (IL-1) of the several NLRP3-regulated inflammatory alarmins (e.g., IL-18 and HMGB-1) (Wiken et al., 2018) and do not affect pyroptosis. The development of direct NLRP3 inhibitors hence is receiving much attention and industrial efforts, especially since the discovery of MCC950 (also known as CRID3) as a direct NLRP3 inhibitor (Coll et al., 2015). MCC950 binds to the NACHT domain and stabilizes the closed, i.e., inactive, conformation of NLRP3, thus blocking its activity (Coll et al., 2019; Tapia-Abellan et al., 2019). A growing number of pre-clinical in vivo models have since reported efficacy for MCC950, e.g., in models of cryopyrin-associated periodic syndrome [CAPS (Coll et al., 2015)], Alzheimer's disease [APP/PS1 model (Dempsey et al., 2017)], sarcoidosis [trehalose 6,6'-dimycolate-granuloma model (Huppertz et al., 2020)], atherosclerosis [ApoE model (Van Der Heijden et al., 2017)] or cystic fibrosis [Cftr transgenic model (Mcelvaney et al., 2019)].
However, despite several clinical studies, so far no direct NLRP3 inhibitor resembling MCC950 has reached late stage clinical development or approval. Thus, alternative strategies will be of high interest for probably at least the next 5–8 years. Targeting NLRP3 indirectly via BTK could be one of them. It would build upon the fact that BTK is a well-established apharmacological target and that pharmacological inhibitors are either already FDA-approved (ibrutinib, acalabrutinib, and zanubrutinib) or in late-stage clinical development. Compared to anti-IL-1 therapy, inhibiting BTK probably would still target NLRP3 proximally enough to block all known NLRP3-dependent effects, both in terms of alarmins and pyroptosis. Of course similar indirect NLRP3 targeting approaches could be envisaged for other known direct NLRP3 regulators [e.g., protein kinase D (Zhang et al., 2017)], albeit none of the known ones are clinically revant to date.
Evidence From Preclinical IN VIVO Models for the Plausibility of BTK-Focussed NLRP3 Targeting
Insights into the plausibility of the targeting NLRP3 via BTK firstly came from our description of a reduced NLRP3 activity in patients on ibrutinib therapy (Liu et al., 2017). In these patients, who received a daily oral dose of ibrutinib of 420–560 mg, nigericin- and ATP-dependent IL-1 cleavage and release were reduced, whereas TNF release was comparable (Liu et al., 2017). We also observed a moderate effect in an S. aureus in vivo infection model, where ibrutinib administration led to reduced bacterial control (Liu et al., 2017). More prominent are the data by Ito et al. (2015) in an experimental model of ischemic stroke. They reported that ibrutinib suppressed infarct volume growth and neurological damage in line with reduced maturation of IL-1β and caspase-1 activation in infiltrating macrophages and neutrophils in the infarcted area. More recently, cardiac failure in the wake of sepsis was studied in an experimental model under ibrutinib and acalabrutinib administration (O'Riordan et al., 2019). Btk-deficient Xid mice were also protected in a polymicrobial sepsis model, with reduced NLRP3 activation contributing to the phenotype (O'Riordan et al., 2020). Interestingly, BTK inhibition reduced NLRP3 protein levels and IL-1 in the serum. Moreover, Purvis et al. (2020) described that ibrutinib treatment ameliorated the NLRP3 contribution to inflammation upon high-fat-diet in mice, which resulted in improved glycemic control. In stark contrast is a recent study proposing BTK as a negative regulator, based on increased intestinal inflammation in a BTK-inhibited or-deficient context (Mao et al., 2020): In this study, high LPS concentrations used for priming led to increased IL-1 release under conditions of BTK inhibition and in a colitis model, BTK deficiency was associated with greater inflammation. However, the ablation of regulatory B cells by BTK-deficiency (Yanaba et al., 2011; Kondo et al., 2018) or the intestinal involvement of other inflammasomes (e.g., NLRC4, NLRP6, pyrin or AIM2) (Elinav et al., 2011; Romberg et al., 2014; Hu et al., 2015; Sharma et al., 2018), which are not BTK-dependent (Ito et al., 2015), both could explain why IL-1 inhibition in vivo could counteract the seemingly negative regulatory role of BTK proposed (Mao et al., 2020). In humans, effective suppression of the NLRP3/IL-1 axis by BTK inhibitors, and thus a positive regulatory effect of BTK on NLRP3, was instead suggested by a recent off-label trial of acalabrutinib in COVID-19 patients (Roschewski et al., 2020). The improvement of patients under ventilation or oxygenation treatment was attributable to BTK function in macrophages, but not B cells (Roschewski et al., 2020), implicating BTK function in these cells and hence the NLRP3 inflammasome. Although in most of these studies, mechanistic details, e.g., phosphorylation of NLRP3 or interaction or phosphorylation of BTK, need to be clarified, a case for intervening in NLRP3-mediated inflammation at the level of BTK can clearly be made (Figure 1).
Discussion
Given the effect of BTK inhibition on B cell development and function, the applicability of targeting NLRP3 via current BTK inhibitors at this stage appears limited to and most suitable for acute settings like myocardial infarction, stroke or COVID-19 rather than chronic NLRP3-mediated diseases such as Alzheimer's or Parkinson's disease. If strategies could be found that can discriminate between BTK function in B cells and macrophages, e.g., by targeted delivery or inhibitors based on protein-protein interactions, more long-term conditions may also come into view therapeutically. On the back of the promising studies described above, further mechanistic work seems mandatory. Additionally, in pre-clinical in vivo models conditional BTK alleles that circumvent the simultaneous ablation of B cell function characteristic for BTK KO mice will be highly desirable. These should be complemented by ex vivo studies of NLRP3 activity in cancer patients treated with the increasingly specific BTK inhibitors or biomaterial from healthy volunteers and XLA patients. Furthermore, one needs to bear in mind that BTK is only a partial NLRP3 regulator and that some IL-1 can still be released in the absence of BTK, albeit at drastically lower levels (Liu et al., 2017). Evidence from ibrutinib-treated patients (Liu et al., 2017) indicates that sufficient NLRP3 blockade may be clinically achievable via BTK inhibitors at tolerable doses, but prospective off-label clinical studies would be mandatory for the inflammatory setting of interest. Although BTK-NLRP3 interactions have been studied mainly in macrophages, effects of BTK inhibitors on NLRP3 function in platelets, neutrophils, Kupffer cells or microglia will be important to study for capturing the effect of systemic application of BTK inhibitors to block NLRP3-mediated inflammation in the absence of clinically approved NLRP3 inhibitors. Collectively, targeting the NLRP3 inflammasome via BTK represents an attractive yet fully-to-be-explored alternative application of BTK inhibitors to pathological inflammatory states beyond B cell malignancies. In fact, the clinical efficacy of BTK inhibitors in GvHD (Jaglowski and Blazar, 2018) and COVID-19, and the reported role of the NLRP3/IL-1 axis in these disease states (Jankovic et al., 2013; Roschewski et al., 2020) lend support to the notion that BTK-inhibitor therapy for NLRP3 inhibition warrants further preclinical and clinical exploration.
Statements
Author contributions
The author confirms being the sole contributor of this work and has approved it for publication.
Funding
The study was supported by the Deutsche Forschungsgemeinschaft (German Research Foundation, DFG) Grant No. We-4195/15-1 (to AW), University Hospital Tübingen (Fortüne Grant No. 2310-0-0). Infrastructural funding was provided by the University of Tübingen, the University Hospital Tübingen and the DFG Clusters of Excellence iFIT—Image-Guided and Functionally Instructed Tumor Therapies (EXC 2180), CMFI—Controlling Microbes to Fight Infection (EXC 2124). Gefördert durch die Deutsche Forschungsgemeinschaft (DFG) im Rahmen der Exzellenzstrategie des Bundes und der Länder—EXC 2180 (390900677), EXC 2124. Additional research grants funded by IFM Therapeutics and Novartis are gratefully acknowledged.
Acknowledgments
We acknowledge support by Open Access Publishing Fund of University of Tübingen.
Conflict of interest
The author declares that his laboratory received funding from Novartis during the course of the generation of this review. The funder was not involved in the collection, analysis, interpretation of data, the writing of this article or the decision to submit it for publication.
References
1
Abbate A. Kontos M. C. Abouzaki N. A. Melchior R. D. Thomas C. Van Tassell B. W. et al . (2015). Comparative safety of interleukin-1 blockade with anakinra in patients with ST-segment elevation acute myocardial infarction (from the VCU-ART and VCU-ART2 pilot studies). Am. J. Cardiol.115, 288–292. 10.1016/j.amjcard.2014.11.003
2
Agostini L. Martinon F. Burns K. Mcdermott M. F. Hawkins P. N. Tschopp J. (2004). NALP3 forms an IL-1beta-processing inflammasome with increased activity in Muckle-Wells autoinflammatory disorder. Immunity20, 319–325. 10.1016/S1074-7613(04)00046-9
3
Banoth B. Cassel S. L. (2017). Bruton tyrosine kinase inhibition: clinical relevance beyond B cells. J. Allergy Clin. Immunol.140, 985–987. 10.1016/j.jaci.2017.03.041
4
Bittner Z. A. Liu X. Shankar S. Tapia-Abellán A. Kalbacher H. Andreeva L. et al . (2020). BTK operates a phospho-tyrosine switch to regulate NLRP3 inflammasome activity. bioRxiv864702. 10.1101/864702
5
Byrd J. C. Furman R. R. Coutre S. E. Flinn I. W. Burger J. A. Blum K. A. et al . (2013). Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N. Engl. J. Med.369, 32–42. 10.1056/NEJMoa1215637
6
Chen J. Chen Z. J. (2018). PtdIns4P on dispersed trans-Golgi network mediates NLRP3 inflammasome activation. Nature564, 71–76. 10.1038/s41586-018-0761-3
7
Coll R. C. Hill J. R. Day C. J. Zamoshnikova A. Boucher D. Massey N. L. et al . (2019). MCC950 directly targets the NLRP3 ATP-hydrolysis motif for inflammasome inhibition. Nat. Chem. Biol.15, 556–559. 10.1038/s41589-019-0277-7
8
Coll R. C. Robertson A. A. Chae J. J. Higgins S. C. Munoz-Planillo R. Inserra M. C. et al . (2015). A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat. Med.21, 248–255. 10.1038/nm.3806
9
Dempsey C. Rubio Araiz A. Bryson K. J. Finucane O. Larkin C. Mills E. L. et al . (2017). Inhibiting the NLRP3 inflammasome with MCC950 promotes non-phlogistic clearance of amyloid-beta and cognitive function in APP/PS1 mice. Brain Behav. Immun.61, 306–316. 10.1016/j.bbi.2016.12.014
10
Dinarello C. A. Van Der Meer J. W. (2013). Treating inflammation by blocking interleukin-1 in humans. Semin. Immunol.25, 469–484. 10.1016/j.smim.2013.10.008
11
Eifert C. Wang X. Kokabee L. Kourtidis A. Jain R. Gerdes M. J. et al . (2013). A novel isoform of the B cell tyrosine kinase BTK protects breast cancer cells from apoptosis. Genes Chromosomes Cancer52, 961–975. 10.1002/gcc.22091
12
Elinav E. Strowig T. Kau A. L. Henao-Mejia J. Thaiss C. A. Booth C. J. et al . (2011). NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell145, 745–757. 10.1016/j.cell.2011.04.022
13
Fiedler K. Sindrilaru A. Terszowski G. Kokai E. Feyerabend T. B. Bullinger L. et al . (2011). Neutrophil development and function critically depend on Bruton tyrosine kinase in a mouse model of X-linked agammaglobulinemia. Blood117, 1329–1339. 10.1182/blood-2010-04-281170
14
Franchi L. Nunez G. (2008). The Nlrp3 inflammasome is critical for aluminium hydroxide-mediated IL-1beta secretion but dispensable for adjuvant activity. Eur. J. Immunol.38, 2085–2089. 10.1002/eji.200838549
15
Freeman L. Guo H. David C. N. Brickey W. J. Jha S. Ting J. P. (2017). NLR members NLRC4 and NLRP3 mediate sterile inflammasome activation in microglia and astrocytes. J. Exp. Med.214, 1351–1370. 10.1084/jem.20150237
16
Ghiringhelli F. Apetoh L. Tesniere A. Aymeric L. Ma Y. Ortiz C. et al . (2009). Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat. Med.15, 1170–1178. 10.1038/nm.2028
17
Gordon R. Albornoz E. A. Christie D. C. Langley M. R. Kumar V. Mantovani S. et al . (2018). Inflammasome inhibition prevents alpha-synuclein pathology and dopaminergic neurodegeneration in mice. Sci. Transl. Med.10:eaah4066. 10.1126/scitranslmed.aah4066
18
Grassilli E. Pisano F. Cialdella A. Bonomo S. Missaglia C. Cerrito M. G. et al . (2016). A novel oncogenic BTK isoform is overexpressed in colon cancers and required for RAS-mediated transformation. Oncogene35, 4368–4378. 10.1038/onc.2015.504
19
Guarda G. Zenger M. Yazdi A. S. Schroder K. Ferrero I. Menu P. et al . (2011). Differential expression of NLRP3 among hematopoietic cells. J. Immunol.186, 2529–2534. 10.4049/jimmunol.1002720
20
He M. Chiang H. H. Luo H. Zheng Z. Qiao Q. Wang L. et al . (2020). An acetylation switch of the NLRP3 inflammasome regulates aging-associated chronic inflammation and insulin resistance. Cell Metab.31, 580–591.e585. 10.1016/j.cmet.2020.01.009
21
Heneka M. T. Kummer M. P. Stutz A. Delekate A. Schwartz S. Vieira-Saecker A. et al . (2013). NLRP3 is activated in Alzheimer's disease and contributes to pathology in APP/PS1 mice. Nature493, 674–678. 10.1038/nature11729
22
Henrickson S. E. (2017). Teaching an old pathway new tricks: Targeting BTK to block NLRP3. Sci. Immunol.2:eaar2548. 10.1126/sciimmunol.aar2548
23
Horwood N. J. Page T. H. Mcdaid J. P. Palmer C. D. Campbell J. Mahon T. et al . (2006). Bruton's tyrosine kinase is required for TLR2 and TLR4-induced TNF, but not IL-6, production. J. Immunol.176, 3635–3641. 10.4049/jimmunol.176.6.3635
24
Hu S. Peng L. Kwak Y. T. Tekippe E. M. Pasare C. Malter J. S. et al . (2015). The DNA sensor AIM2 maintains intestinal homeostasis via regulation of epithelial antimicrobial host defense. Cell Rep.13, 1922–1936. 10.1016/j.celrep.2015.10.040
25
Huang H. Chen H. W. Evankovich J. Yan W. Rosborough B. R. Nace G. W. et al . (2013). Histones activate the NLRP3 inflammasome in Kupffer cells during sterile inflammatory liver injury. J. Immunol.191, 2665–2679. 10.4049/jimmunol.1202733
26
Huppertz C. Jager B. Wieczorek G. Engelhard P. Oliver S. J. Bauernfeind F. G. et al . (2020). The NLRP3 inflammasome pathway is activated in sarcoidosis and involved in granuloma formation. Eur. Respir. J.55:1900119. 10.1183/13993003.00119-2019
27
Ito M. Shichita T. Okada M. Komine R. Noguchi Y. Yoshimura A. et al . (2015). Bruton's tyrosine kinase is essential for NLRP3 inflammasome activation and contributes to ischaemic brain injury. Nat. Commun.6:7360. 10.1038/ncomms8360
28
Jaglowski S. M. Blazar B. R. (2018). How ibrutinib, a B-cell malignancy drug, became an FDA-approved second-line therapy for steroid-resistant chronic GVHD. Blood Adv.2, 2012–2019. 10.1182/bloodadvances.2018013060
29
Jankovic D. Ganesan J. Bscheider M. Stickel N. Weber F. C. Guarda G. et al . (2013). The Nlrp3 inflammasome regulates acute graft-versus-host disease. J. Exp. Med.210, 1899–1910. 10.1084/jem.20130084
30
Jongstra-Bilen J. Puig Cano A. Hasija M. Xiao H. Smith C. I. Cybulsky M. I. (2008). Dual functions of Bruton's tyrosine kinase and Tec kinase during Fcgamma receptor-induced signaling and phagocytosis. J. Immunol.181, 288–298. 10.4049/jimmunol.181.1.288
31
Khan W. N. Alt F. W. Gerstein R. M. Malynn B. A. Larsson I. Rathbun G. et al . (1995). Defective B cell development and function in Btk-deficient mice. Immunity3, 283–299. 10.1016/1074-7613(95)90114-0
32
Kondo K. Shaim H. Thompson P. A. Burger J. A. Keating M. Estrov Z. et al . (2018). Ibrutinib modulates the immunosuppressive CLL microenvironment through STAT3-mediated suppression of regulatory B-cell function and inhibition of the PD-1/PD-L1 pathway. Leukemia32, 960–970. 10.1038/leu.2017.304
33
Liu X. Pichulik T. Wolz O. O. Dang T. M. Stutz A. Dillen C. et al . (2017). Human NACHT, LRR, and PYD domain-containing protein 3 (NLRP3) inflammasome activity is regulated by and potentially targetable through Bruton tyrosine kinase. J. Allergy Clin. Immunol.140, 1054–1067.e1010. 10.1016/j.jaci.2017.01.017
34
Magupalli V. G. Negro R. Tian Y. Hauenstein A. V. Di Caprio G. Skillern W. et al . (2020). HDAC6 mediates an aggresome-like mechanism for NLRP3 and pyrin inflammasome activation. Science369:eaas8995. 10.1126/science.aas8995
35
Mangan M. S. J. Olhava E. J. Roush W. R. Seidel H. M. Glick G. D. Latz E. (2018). Targeting the NLRP3 inflammasome in inflammatory diseases. Nat. Rev. Drug Discov.17, 588–606. 10.1038/nrd.2018.97
36
Mankan A. K. Dau T. Jenne D. Hornung V. (2012). The NLRP3/ASC/Caspase-1 axis regulates IL-1beta processing in neutrophils. Eur. J. Immunol.42, 710–715. 10.1002/eji.201141921
37
Mao L. Kitani A. Hiejima E. Montgomery-Recht K. Zhou W. Fuss I. et al . (2020). Bruton tyrosine kinase deficiency augments NLRP3 inflammasome activation and causes IL-1beta-mediated colitis. J. Clin. Invest.130, 1793–1807. 10.1172/JCI128322
38
Masters S. L. Dunne A. Subramanian S. L. Hull R. L. Tannahill G. M. Sharp F. A. et al . (2010). Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1beta in type 2 diabetes. Nat. Immunol.11, 897–904. 10.1038/ni.1935
39
Mcelvaney O. J. Zaslona Z. Becker-Flegler K. Palsson-Mcdermott E. M. Boland F. Gunaratnam C. et al . (2019). Specific inhibition of the NLRP3 inflammasome as an antiinflammatory strategy in cystic fibrosis. Am. J. Respir. Crit. Care Med.200, 1381–1391. 10.1164/rccm.201905-1013OC
40
Mcnally G. A. Long J. M. Brophy L. R. Badillo M. R. (2015). Ibrutinib: implications for use in the treatment of mantle cell lymphoma and chronic lymphocytic leukemia. J. Adv. Pract. Oncol.6, 420–431. 10.6004/jadpro.2015.6.5.3
41
Melcher M. Unger B. Schmidt U. Rajantie I. A. Alitalo K. Ellmeier W. (2008). Essential roles for the Tec family kinases Tec and Btk in M-CSF receptor signaling pathways that regulate macrophage survival. J. Immunol.180, 8048–8056. 10.4049/jimmunol.180.12.8048
42
Munoz-Planillo R. Kuffa P. Martinez-Colon G. Smith B. L. Rajendiran T. M. Nunez G. (2013). K(+) efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity38, 1142–1153. 10.1016/j.immuni.2013.05.016
43
Murthy P. Durco F. Miller-Ocuin J. L. Takedai T. Shankar S. Liang X. et al . (2017). The NLRP3 inflammasome and Bruton's tyrosine kinase in platelets co-regulate platelet activation, aggregation, and in vitro thrombus formation. Biochem. Biophys. Res. Commun.483, 230–236. 10.1016/j.bbrc.2016.12.161
44
O'Riordan C. E. Purvis G. S. D. Collotta D. Chiazza F. Wissuwa B. Al Zoubi S. et al . (2019). Bruton's tyrosine kinase inhibition attenuates the cardiac dysfunction caused by cecal ligation and puncture in mice. Front. Immunol.10:2129. 10.3389/fimmu.2019.02129
45
O'Riordan C. E. Purvis G. S. D. Collotta D. Krieg N. Wissuwa B. Sheikh M. H. et al . (2020). X-linked immunodeficient mice with no functional Bruton's tyrosine kinase are protected from sepsis-induced multiple organ failure. Front. Immunol.11:581758. 10.3389/fimmu.2020.581758
46
Purvis G. S. D. Collino M. Aranda-Tavio H. Chiazza F. O'riordan C. E. Zeboudj L. et al . (2020). Inhibition of Bruton's TK regulates macrophage NF-kappaB and NLRP3 inflammasome activation in metabolic inflammation. Br. J. Pharmacol. 177, 4416–4432. 10.1111/bph.15182
47
Quek L. S. Bolen J. Watson S. P. (1998). A role for Bruton's tyrosine kinase (Btk) in platelet activation by collagen. Curr. Biol.8, 1137–1140. 10.1016/S0960-9822(98)70471-3
48
Romberg N. Al Moussawi K. Nelson-Williams C. Stiegler A. L. Loring E. Choi M. et al . (2014). Mutation of NLRC4 causes a syndrome of enterocolitis and autoinflammation. Nat. Genet.46, 1135–1139. 10.1038/ng.3066
49
Roschewski M. Lionakis M. S. Sharman J. P. Roswarski J. Goy A. Monticelli M. A. et al . (2020). Inhibition of Bruton tyrosine kinase in patients with severe COVID-19. Sci. Immunol.5:eabd0110. 10.1126/sciimmunol.abd0110
50
Seoane P. I. Lee B. Hoyle C. Yu S. Lopez-Castejon G. Lowe M. et al . (2020). The NLRP3-inflammasome as a sensor of organelle dysfunction. J. Cell Biol.219:e202006194. 10.1083/jcb.202006194
51
Sharma D. Malik A. Guy C. S. Karki R. Vogel P. Kanneganti T. D. (2018). Pyrin inflammasome regulates tight junction integrity to restrict colitis and tumorigenesis. Gastroenterology154, 948–964.e948. 10.1053/j.gastro.2017.11.276
52
Singhal E. Kumar P. Sen P. (2011). A novel role for Bruton's tyrosine kinase in hepatocyte growth factor-mediated immunoregulation of dendritic cells. J. Biol. Chem.286, 32054–32063. 10.1074/jbc.M111.271247
53
Strijbis K. Tafesse F. G. Fairn G. D. Witte M. D. Dougan S. K. Watson N. et al . (2013). Bruton's tyrosine kinase (BTK) and Vav1 contribute to Dectin1-dependent phagocytosis of Candida albicans in macrophages. PLoS Pathog.9:e1003446. 10.1371/journal.ppat.1003446
54
Swanson K. V. Deng M. Ting J. P. (2019). The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat. Rev. Immunol.19, 477–489. 10.1038/s41577-019-0165-0
55
Takeuchi O. Akira S. (2010). Pattern recognition receptors and inflammation. Cell140, 805–820. 10.1016/j.cell.2010.01.022
56
Tapia-Abellan A. Angosto-Bazarra D. Martinez-Banaclocha H. De Torre-Minguela C. Ceron-Carrasco J. P. Perez-Sanchez H. et al . (2019). MCC950 closes the active conformation of NLRP3 to an inactive state. Nat. Chem. Biol.15, 560–564. 10.1038/s41589-019-0278-6
57
Van Der Heijden T. Kritikou E. Venema W. Van Duijn J. Van Santbrink P. J. Slutter B. et al . (2017). NLRP3 inflammasome inhibition by MCC950 reduces atherosclerotic lesion development in apolipoprotein e-deficient mice-brief report. Arterioscler. Thromb. Vasc. Biol.37, 1457–1461. 10.1161/ATVBAHA.117.309575
58
Weber A. N. Bittner Z. A. Sankar S. Liu X. Chang T. H. Jin T. et al . (2020). Recent insights into the regulatory networks of NLRP3 inflammasome activation. J. Cell Sci. 133:jcs248344. 10.1242/jcs.248344
59
Weber A. N. R. Bittner Z. Liu X. Dang T. M. Radsak M. P. Brunner C. (2017). Bruton's tyrosine kinase: an emerging key player in innate immunity. Front. Immunol.8:1454. 10.3389/fimmu.2017.01454
60
Wiken M. Hallen B. Kullenberg T. Koskinen L. O. (2018). Development and effect of antibodies to anakinra during treatment of severe CAPS: sub-analysis of a long-term safety and efficacy study. Clin. Rheumatol.37, 3381–3386. 10.1007/s10067-018-4196-x
61
Wilson W. H. Young R. M. Schmitz R. Yang Y. Pittaluga S. Wright G. et al . (2015). Targeting B cell receptor signaling with ibrutinib in diffuse large B cell lymphoma. Nat. Med.21, 922–926. 10.1038/nm.3884
62
Wree A. Eguchi A. Mcgeough M. D. Pena C. A. Johnson C. D. Canbay A. et al . (2014). NLRP3 inflammasome activation results in hepatocyte pyroptosis, liver inflammation, and fibrosis in mice. Hepatology59, 898–910. 10.1002/hep.26592
63
Yanaba K. Yoshizaki A. Asano Y. Kadono T. Tedder T. F. Sato S. (2011). IL-10-producing regulatory B10 cells inhibit intestinal injury in a mouse model. Am. J. Pathol.178, 735–743. 10.1016/j.ajpath.2010.10.022
64
Yao C. Veleva T. Scott L. Jr. Cao S. Li L. Chen G. et al . (2018). Enhanced cardiomyocyte NLRP3 inflammasome signaling promotes atrial fibrillation. Circulation138, 2227–2242. 10.1161/CIRCULATIONAHA.118.035202
65
Zhang Z. Meszaros G. He W. T. Xu Y. De Fatima Magliarelli H. Mailly L. et al . (2017). Protein kinase D at the Golgi controls NLRP3 inflammasome activation. J. Exp. Med.214, 2671–2693. 10.1084/jem.20162040
Summary
Keywords
Bruton's tyrosine kinase, NLRP3 inflammasome, inflammation, phosphorylation, Interleukin-1 (IL-1), kinase inhibitor
Citation
Weber ANR (2021) Targeting the NLRP3 Inflammasome via BTK. Front. Cell Dev. Biol. 9:630479. doi: 10.3389/fcell.2021.630479
Received
17 November 2020
Accepted
14 January 2021
Published
25 February 2021
Volume
9 - 2021
Edited by
Cornelia Brunner, Ulm University Medical Center, Germany
Reviewed by
Ankit Malik, University of Chicago, United States; María A. Balboa, Consejo Superior de Investigaciones Científicas, Spain
Updates
Copyright
© 2021 Weber.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alexander N. R. Weber alexander.weber@uni-tuebingen.de
This article was submitted to Signaling, a section of the journal Frontiers in Cell and Developmental Biology
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.