 Renato Assis Machado1,2*
Renato Assis Machado1,2* Hercílio Martelli-Junior3,4
Hercílio Martelli-Junior3,4 Silvia Regina de Almeida Reis5
Silvia Regina de Almeida Reis5 Erika Calvano Küchler6
Erika Calvano Küchler6 Rafaela Scariot7
Rafaela Scariot7 Lucimara Teixeira das Neves2,8
Lucimara Teixeira das Neves2,8 Ricardo D. Coletta1
Ricardo D. Coletta1- 1Department of Oral Diagnosis, School of Dentistry, University of Campinas (FOP), Piracicaba, Brazil
- 2Hospital for Rehabilitation of Craniofacial Anomalies, University of São Paulo, Bauru, Brazil
- 3Stomatology Clinic, School of Dental, State University of Montes Claros, Montes Claros, Brazil
- 4Center for Rehabilitation of Craniofacial Anomalies, School of Dental, UNIFENAS - Universidade José do Rosario Vellano, Alfenas, Brazil
- 5Department of Basic Science, Bahiana School of Medicine and Public Health, Salvador, Brazil
- 6Department of Orthodontics, University of Regensburg, Regensburg, Germany
- 7Department of Oral and Maxillofacial Surgery, School of Health Science, Federal University of Paraná, Curitiba, Brazil
- 8Department of Biological Sciences, Bauru School of Dentistry, University of São Paulo (FOB), Bauru, Brazil
The identification of genetic risk factors for non-syndromic oral clefts is of great importance for better understanding the biological processes related to this heterogeneous and complex group of diseases. Herein we applied whole-exome sequencing to identify potential variants related to non-syndromic cleft palate only (NSCPO) in the multiethnic Brazilian population. Thirty NSCPO samples and 30 sex- and genetic ancestry-matched healthy controls were pooled (3 pools with 10 samples for each group) and subjected to whole-exome sequencing. After filtering, the functional affects, individually and through interactions, of the selected variants and genes were assessed by bioinformatic analyses. As a group, 399 variants in 216 genes related to palatogenesis/cleft palate, corresponding to 6.43%, were exclusively identified in the NSCPO pools. Among those genes are 99 associated with syndromes displaying cleft palate in their clinical spectrum and 92 previously related to cleft lip palate. The most significantly biological processes and pathways overrepresented in the NSCPO-identified genes were associated with the folic acid metabolism, highlighting the interaction between LDL receptor-related protein 6 (LRP6) and 5-methyltetrahydrofolate-homocysteine methyltransferase (MTR) that interconnect two large networks. This study yields novel data on characterization of specific variants and complex processes and pathways related to NSCPO, including many variants in genes of the folate/homocysteine pathway, and confirms that variants in genes related to syndromic cleft palate and cleft lip-palate may cause NSCPO.
Introduction
Orofacial clefts represent the most common craniofacial malformation in humans with an overall prevalence of approximately 1 per 700 live births, but considerable geographic and ethnic variations exist (Leslie and Marazita, 2013). It may represent the manifestation of a syndrome or as a non-syndromic isolated condition, which corresponds to approximately 70% of all cases (Dixon et al., 2011). Non-syndromic oral clefts (NSOC) comprise two main forms, the cleft lip with or without cleft palate (NSCL ± P) and cleft palate only (NSCPO). Although a multifactorial etiology with a strong genetic component is reported in both NSOC subtypes, many studies have indicated that the genetic factors behind them may be distinct (Mangold et al., 2016; Ludwig et al., 2017). Different genes and chromosomal regions, many identified by genome-wide association studies (GWAS), were described as causal genetic factors for NSCL ± P (Beaty et al., 2016; Yu et al., 2017), but due to lower prevalence, embryological aspects, and recurrence rates, less is known about NSCPO.
To date, five GWAS have been conducted with NSCPO (Beaty et al., 2011; Leslie et al., 2016; Butali et al., 2019; Huang et al., 2019; He et al., 2020). The first studies have identified variants in grainyhead-like transcription factor 3 (GRHL3), a gene underlying van der Woude syndrome, the most common genetic syndrome associated with cleft lip and palate, and other few markers that, in interaction with maternal smoking or multivitamin supplementation (gene–environment interactions), were associated with increased odds to NSCPO development (Beaty et al., 2011; Leslie et al., 2016), whereas the most recent GWAS have identified only single-nucleotide polymorphisms (SNPs) in intergenic regions in association with NSCPO (Butali et al., 2019; Huang et al., 2019; He et al., 2020). The study conducted by Butali et al. (2019) has identified the SNP rs80004662 on chromosome 2, near the catenin alpha 2 (CTNNA2), as a novel marker for NSCPO in the African population, and the study of Huang et al. (2019) has revealed 11 SNPs in nine loci in genome-wide significance with NSCPO in the Han Chinese population. Four SNPs in a novel locus (15q24.3) were reported in the GWAS conducted by He et al. (2020) with a Han Chinese population of patients with NSCPO. These genetic findings suggest that the effects of polymorphic variants in NSCPO are much less frequent or may be caused by rare variants, which are undetectable in GWAS (Hecht et al., 2002; Koillinen et al., 2005; Ishorst et al., 2018). Moreover, it is possible that specific variants in genes associated with syndromic forms of CPO (SCPO) display a causal effect in the non-syndromic clinical phenotype, and the NSCPO may be the result of genetic factors associated with clefts involving both lip and palate (CLP). A previous study supports this hypothesis; out of 350 genes associated with NSCPO, 177 were also identified in patients with CLP and 28 in patients with SCPO (Funato and Nakamura, 2017).
Whole-exome sequencing (WES) has been applied for the genetic characterization of NSCPO (Bureau et al., 2014; Pengelly et al., 2016; Liu et al., 2017; Hoebel et al., 2017; Basha et al., 2018). The study performed by Liu et al. (2017) has identified a rare variant (p.Ser552Pro) in Rho GTPase activating protein 29 (ARHGAP29) in a family spanning NSCPO under the assumption of an autosomal dominant of inheritance. Applying WES in multiple affected NSCPO pedigrees and validation of the potential deleterious variants in a case–control approach, Hoebel et al. (2017) have suggested the participation of acetyl-CoA carboxylase beta (ACACB), CREB-binding protein (CREBBP), GRHL3, MIB E3 ubiquitin protein ligase 1 (MIB1), and protein tyrosine phosphatase receptor type S (PTPRS) in the etiology of NSCPO. In the study conducted by Basha et al. (2018), the c.819_820dupCC and c.3373C > T mutations in tumor protein P63 (TP63) and LDL receptor-related protein 6 (LRP6), respectively, were described in two families with NSCPO. On the other hand, the studies conducted by Bureau et al. (2014); Pengelly et al. (2016) have analyzed multiplex families of patients with NSOC, including NSCPO among those affected, which makes more difficult to understand the exact participation of genes identified in the pathogenesis of NSCPO.
Large-scale genetic studies have confirmed the genetic contribution to the etiology of NSCL ± P but have also demonstrated that these defects can result from variation in multiple genes (Carlson et al., 2019). However, much less is known in terms of genetic etiology of NSCPO. Furthermore, most patients included in the genome-wide studies, including GWAS and genomic sequencing, have been of European or Asian ancestry, and the genetic predisposition to NSOC is ethnicity-dependent, e.g., the association of 8q24 is consistently observed in European patients with NSCL ± P, but much less among patients with Asian ancestry or in Brazilians with high African ancestry (do Rego et al., 2015; Wattanawong et al., 2016). In this context, it is quite important to perform large-scale genetic studies in different populations to define common and ethnic-specific risk genes, but the costs of those assays in a large number of participants are still challenging. To genomic sequencing, pool sequencing (mix DNA samples from several patients prior to sequencing) has proved to be an effective alternative to overcome the problems related to costs (Collins et al., 2016; Popp et al., 2017). In the current study, we applied whole-exome pool sequencing to characterize genetic variants related to NSCPO in the multiethnic Brazilian population. This is the first study to perform WES in Brazilian patients with NSCPO, and our findings revealed a large list of novel variants in previously CPO and CLP-associated genes as well in genes not related to oral clefts.
Materials and Methods
Study Participants
A total of 30 samples from patients with NSCPO and 30 healthy controls were recruited after approval by the Institutional Review Boards at the Center for Rehabilitation of Craniofacial Anomalies, University of José Rosário Vellano, Alfenas, Minas Gerais, and at the Santo Antonio Hospital, Salvador, Bahia. The patients with NSCPO were carefully investigated by specialist teams of the two Centers for the occurrence of associated abnormalities or syndromes, and only unrelated patients with complete NSCPO, without any other congenital malformation or mental disability, were included. The control group consisted of unrelated healthy patients, without congenital malformations, mental disorders, or family history of orofacial clefts, living in the same geographic areas. Cases and controls were unrelated newborn infants or young children. Supplementary Table 1 depicts the main characteristics of the participants of this study, including sex and proportion of genomic ancestry.
Samples and Pooling Strategy
Genomic DNA was isolated from oral mucosa cells, obtained by mouthwash with a 3% sucrose solution or by scraping the buccal mucosa with a swab, using a salting-out protocol (Aidar and Line, 2007). The concentration and quality of DNA samples were assessed by spectrophotometry and agarose gel electrophoresis. Each pool represented 10 samples, and the amount of DNA was adequately balanced to represent each genome equally (Figure 1). After mixing the samples, the pools were quantified using the fluorometric method (Qubit®, Thermo Fisher Scientific, Waltham, MA, United States) and normalized to 10 mM Tris–EDTA buffer pH 8.0 at a final concentration of 5 ng/μl. To ensure greater homogeneity, each pool was composed of patients of the same sex (two pools with females and one pool with males for each group) and from the same geographic location. In addition, the genomic ancestry was assessed in each sample with a 40 biallelic short insertion–deletion polymorphism panel, which was previously validated as ancestry informative of the Brazilian population (Messetti et al., 2017), and the averages of contributions from European, African, and Amerindian ancestries among pools were similar (Supplementary Table 1).
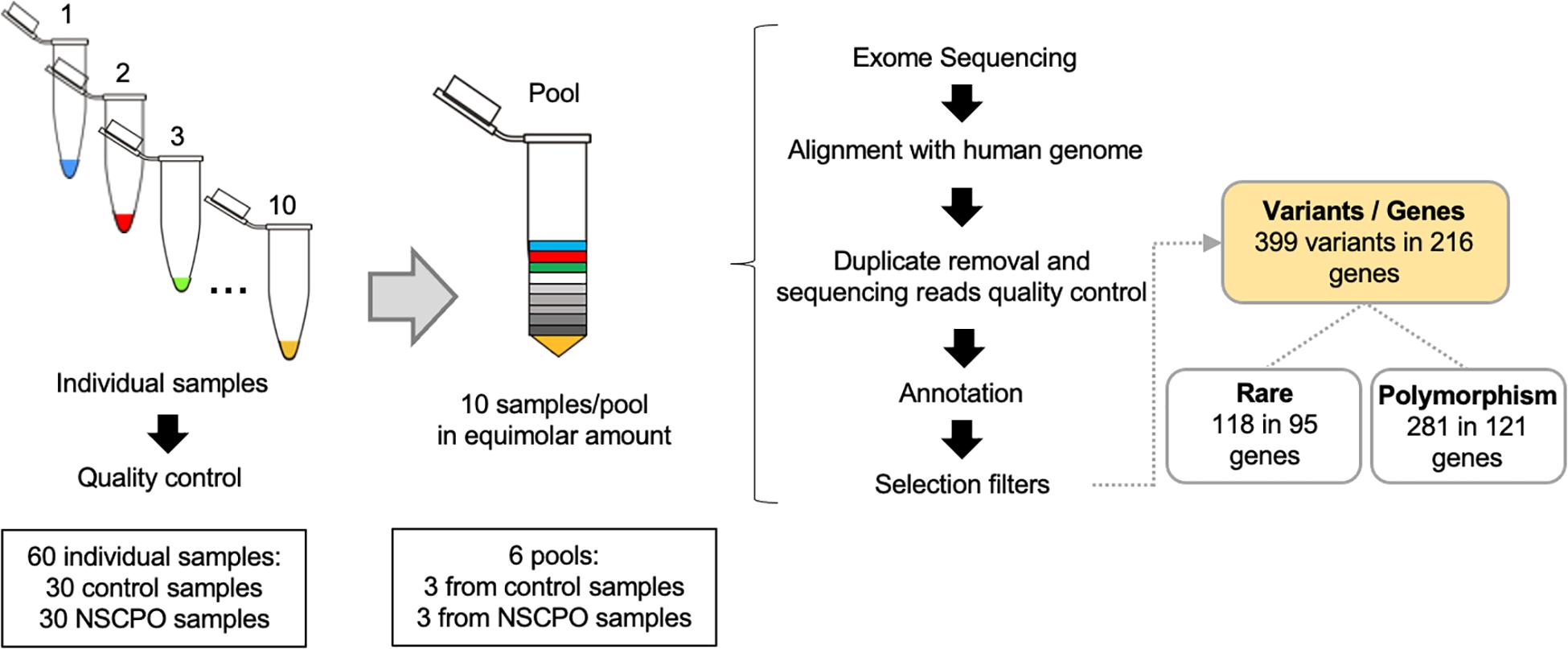
Figure 1. Workflow of whole-exome pool sequencing applied to 30 samples of patients with non-syndromic cleft palate only (NSCPO) and 30 healthy controls. After sequencing and bioinformatic analyses, 399 variants distributed in 216 different genes were found, with 118 rare variants in 95 genes and 281 polymorphisms in 121 genes.
WES and Bioinformatic Analyses
The pools were enriched in libraries with the NexteraTM DNA sample preparation kit (Illumina, San Diego, CA, United States), and each library was quantified by RT-qPCR using the KAPA Library Quantification Kit (KAPA Biosystems Inc., Wilmington, MA, United States). The libraries were diluted to a concentration of 16 pM and pooled by cBOT (Illumina) using the TruSeq PE cluster v3-cBOT-HS Kit (Illumina). Paired sequencing, with a reading length of 100 bp, was performed on the HiSeq 1000 equipment (Illumina), aiming at covering 18 × /pool. Following the sequencing, the sequences of each pool (FASTQ files) were aligned with the human reference genome RCh38/hg19 through the Burrows–Wheeler aligner program [BWA, GNU General Public License version 3.0 (GPLv3), MIT License, Cambridge, MA, United States] (Li and Durbin, 2009). The duplicates were removed, and the quality of the sequencing was verified with the GATK program (Genome Analysis Toolkit). The coverage depth was exported in a BAM file with the Biobambam2 program1, and the variants were identified as VCF files with the Freebayes program2. The BAM and VCF files for each pool were incorporated into the VarSeq® program (version 1.4.8; Golden Helix, Inc., Bozeman, MT, United States) for genetic variant annotation according to the public databases 1000 Genomes, GnomAD Exome, GnomAD Genome, and ExAC. A total of 102,927 variants were identified.
The selection of the variants of interest started with the exclusion of sequences that did not pass the quality control (coverage ≥ 20 and sequencing depth ≥ 100). After applying this filter, 61,398 variants were maintained. Next, the variants present in the control group were excluded, leaving only the variants contained in the pools of patients with NSCPO. The variants that did not show an allele frequency of at least 5% in the pool were also excluded, where in a universe of 20 possible alleles in each pool, at least one allele should be evidenced. The application of these filters reduced the number of variants to 5,129 variants in 3,360 genes. At this point, the GATACA database3, VarElect database4, and Online Mendelian Inheritance in Man (OMIM)5 were used for identification of genes associated with cleft palate, including non-syndromic, syndromic, and CLP. The resulting list of genes was compared to 3,360 genes found in the exome (Figure 1). The polymorphism phenotyping (PolyPhen26) (Adzhubei et al., 2013), sorting intolerant from tolerant (SIFT7) (Kumar et al., 2009), and mutation taster8 were used for biological and functional effect prediction of the variants. STRING, protein–protein interaction networks functional enrichment analysis,9 was used to investigate the functional significance of genes, including biological processes, pathways and interaction network. The p-Values were subjected to false discovery rate to correct multiple tests, and values ≤ 0.05 were considered significant.
Results
Out of 3,360 cleft palate-related genes, 216 (6.43%) displayed at least a variation in the pools of NSCPO. Since phenotypic human gene classification also offers major insights into gene function (Frech and Chen, 2010), the identified genes were categorized on the basis of their previous association with NSCPO (Table 1), SCPO (Supplementary Table 2), and CLP (Supplementary Table 3). Supplementary Table 4 depicts the variants identified in the pools of patients with NSCPO, but not located in the genes previously associated with palatogenesis or oral clefts.
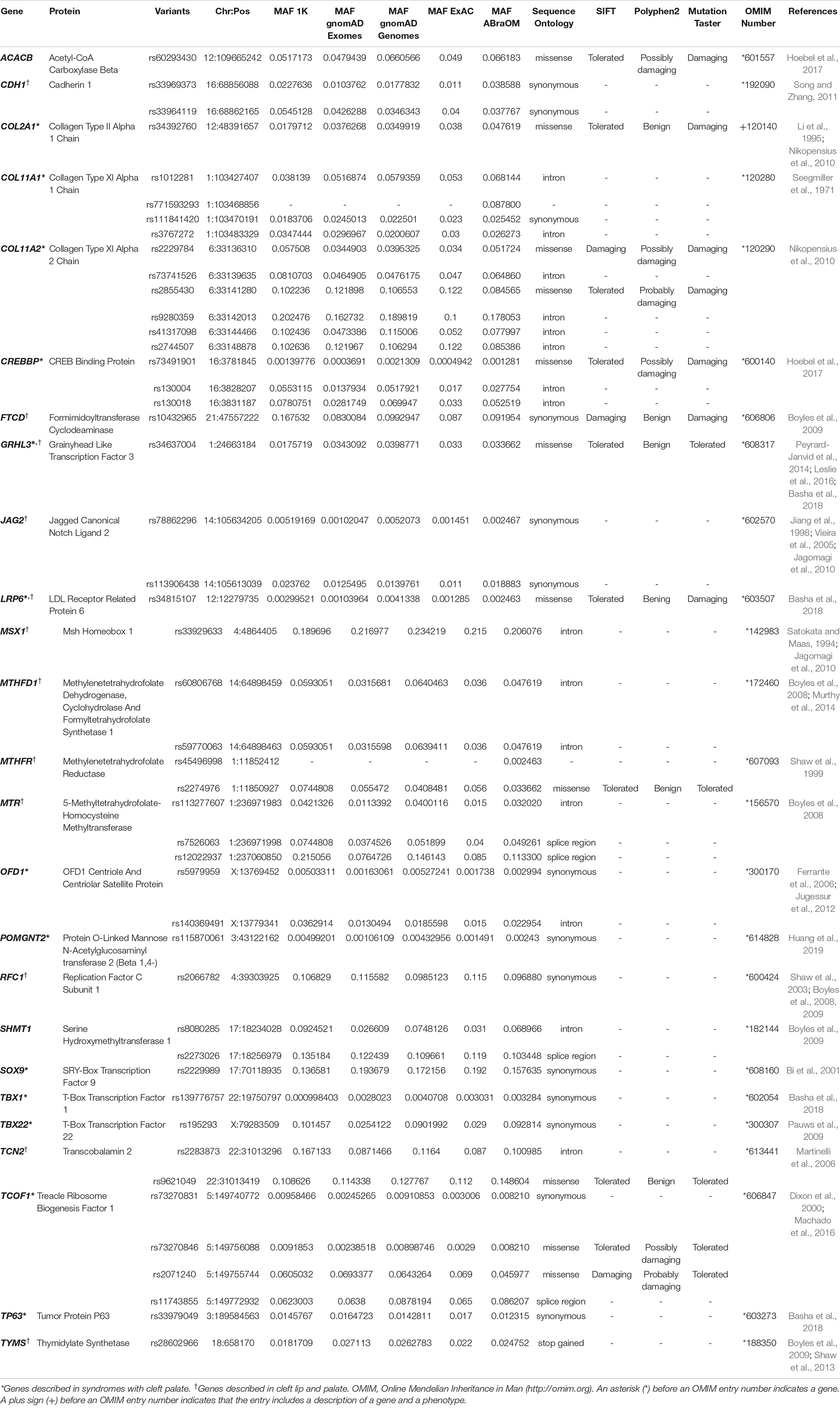
Table 1. Novel variants in genes previously described in association with non-syndromic cleft palate only (NSCPO) found exclusively in the samples of patients with NSCPO.
Among genes previously described in association with NSCPO, 38 novel SNPs (except rs2229989 in SOX9, Jia et al., 2017) and 9 novel rare variants were identified in ACACB, CDH1, COL11A1, COL11A2, COL2A1, CREBBP, FTCD, GRHL3, JAG2, LRP6, MSX1, MTHFD1, MTHFR, MTR, OFD1, POMGNT2, RFC1, SHMT1, SOX9, TBX1, TBX22, TCN2, TCOF1, TP63, and TYMS (Table 1). The biological processes and pathways for these NSCPO-associated genes were generated by STRING (Table 2 and Supplementary Table 5). The most significant biological processes were skeletal system development (GO:0001501, P = 1.96e-10), tetrahydrofolate interconversion (GO:0035999, P = 3.57e-09), and pteridine-containing compound metabolic process (GO:0042558, P = 3.88e-09) (Supplementary Table 5), and the pathways were of one carbon pool by folate (hsa00670, P = 3.63e-11) and antifolate resistance (hsa01523, P = 0.00038) (Table 2). Two main networks interconnected by interactions between LRP6 and MTR were identified in NSCPO-associated genes (Figure 2).
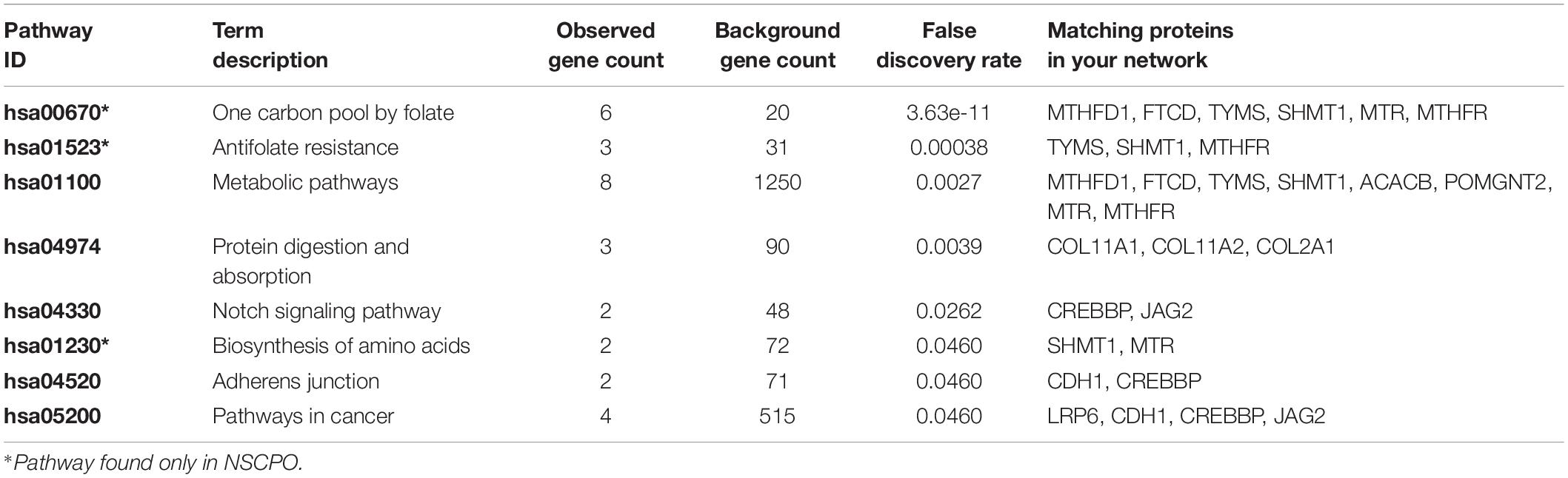
Table 2. List of the overrepresented pathway formatted with the variant-containing-genes identified in patients with non-syndromic cleft palate only (NSCPO).
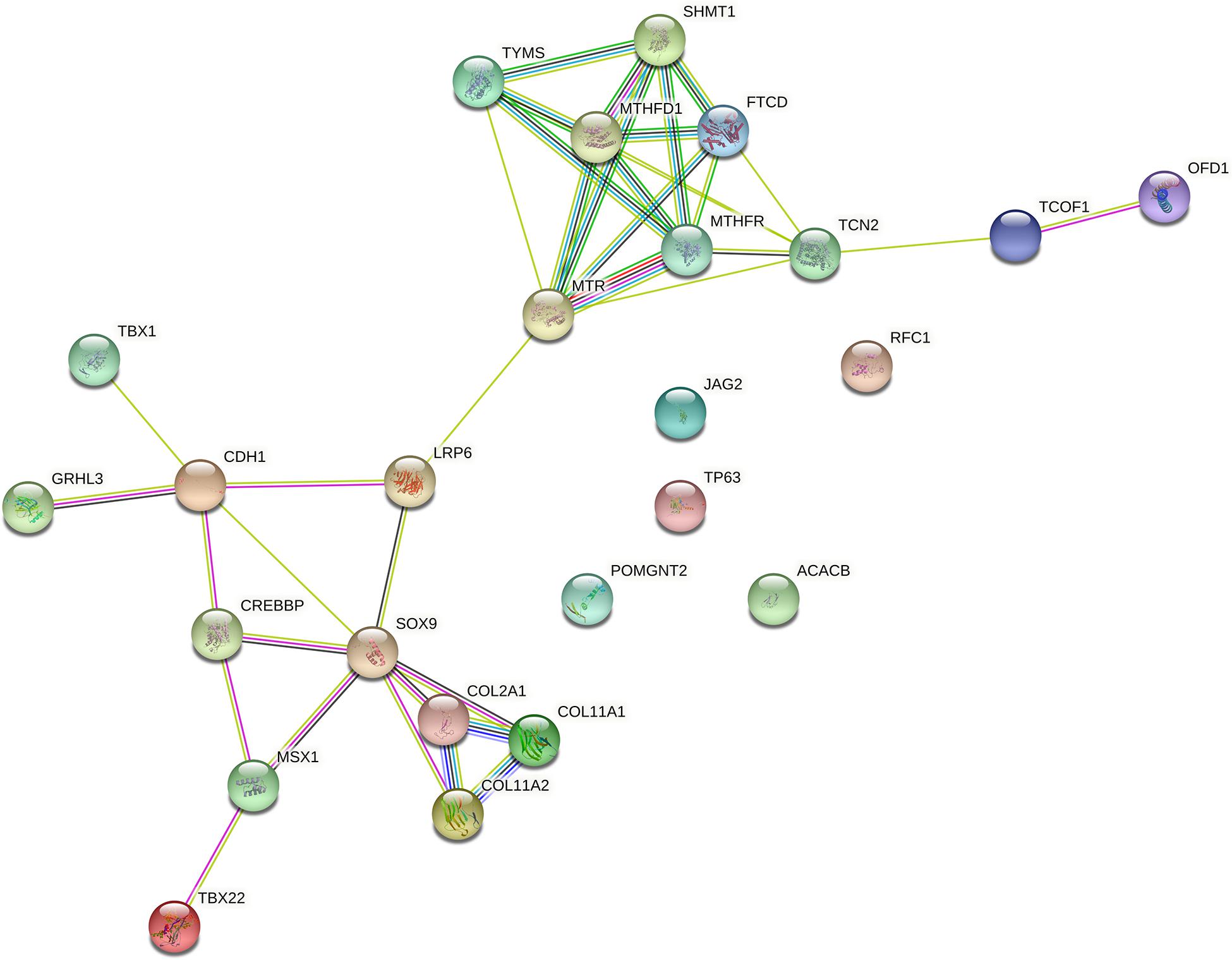
Figure 2. Protein–protein interaction network with the genes associated with non-syndromic cleft palate only (NSCPO). Twenty out of 25 genes formed two nodes interconnected by a validated interaction between LRP6 and MTR. The first encompassed nine genes, including FTCD, MTHFD1, MTHFR, MTR, OFD1, SHMT1, TCN2, TCOF1, and TYMS (P < 1.0e-16), and the second node involved CDH1, COL2A1, COL11A1, COL11A2, CREBBP, GRHL3, LRP6, MSX1, SOX9, TBX1, and TBX22 (P < 1.0e-16). Both nodes were based on curated databases (light blue), experimentally (purple), gene neighborhood (green), gene fusions (red), gene co-occurrence (blue), text mining (light green), co-expression (black), and protein homology (violet). This analysis had an average confidence score of 0.588 (P < 1.0e-16), suggesting a low rate for false-positive interactions.
Variations (n = 192) in 99 SCPO-associated genes were found in NSCPO samples, but not in the control (Supplementary Table 2). These genes participate in 437 biological processes and 31 pathways. The most significantly represented GO terms for biological processes and cellular component ontologies for the encoded proteins were developmental process (GO:0032502, P = 6.42e-12), anatomical structure development (GO:0048856, P = 6.42e-12), and multicellular organism development (GO:0007275, P = 8.60e-12) (Supplementary Table 6), and the pathways were thyroid hormone signaling (hsa04919, P = 0.00047), protein digestion and absorption (hsa04974, P = 0.00077), and proteoglycans in cancer (hsa05205, P = 0.0041) (Supplementary Table 7). The network involved in the SCPO-associated genes included 205 interactions (Supplementary Figure 1).
Since CLP results from failure of fusion of lip and palate, we have also looked into variations of genes related to CLP, and 160 variants in 92 genes were identified (Supplementary Table 3). Together, the CLP-associated genes identified exclusively in the NSCPO pools participate in 873 biological processes and 68 pathways, with the most significant being animal organ development (GO:0048513, P = 2.24e-20) (Supplementary Table 8) and human papillomavirus infection (hsa05165, P = 3.58e-06) (Supplementary Table 9), respectively. A complex network characterized by 244 interactions among these CLP-associated genes identified exclusively in the NSCPO pools was observed (Supplementary Figure 2).
As some genes were exclusive to the NSCPO phenotype and others coincided with the SCPO and CLP phenotypes (Figure 3), we investigated whether biological processes and pathways differ among genes related to NSCPO, SCPO, and CLP, and we compared the biological processes and pathways to identify those ones associated with NSCPO only. Although most genes/pathways are interrelated, 76 biological processes were enriched only for NSCPO-associated genes in the pools of NSCPO, and the most significant were tetrahydrofolate interconversion (GO:0035999, P = 3.57e-09) and pteridine-containing compound metabolic process (GO:0042558, P = 3.88e-09) (processes indicated with an ∗ on Supplementary Table 5). In addition, the pathways of one carbon pool by folate (hsa00670, P = 3.63e-11), antifolate resistance (hsa01523, P = 0.00038), and biosynthesis of amino acids (hsa01230, P = 0.0460), formatted by FTCD, MTHFD1, MTHFR, MTR, SHMT1, and TYMS, were found only in NSCPO-associated genes (pathways indicated with an ∗ on Table 2).
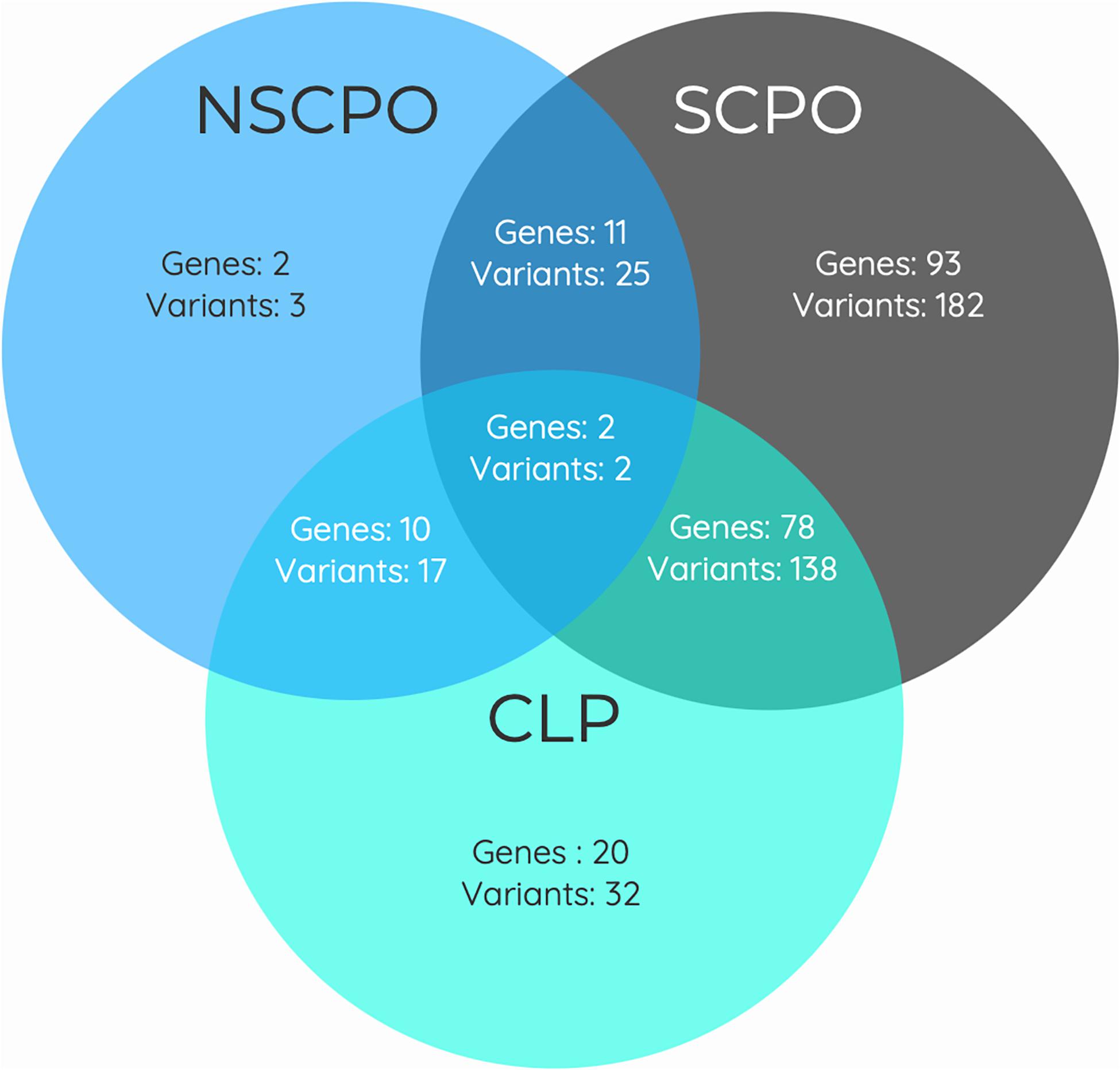
Figure 3. Venn diagram showing the overlap between genes associated with the phenotypes of non-syndromic cleft palate only (NSCPO), syndromic cleft palate only (SCPO), and cleft lip and palate (CLP) found in patients with NSCPO. The numbers in each area represent the gene and variants count for the particular section.
Discussion
Although the palatogenesis is a complex process with involvement of many genes and cellular pathways, genetic variants in association with cleft palate have been identified in only a limited number of these genes. In the present study, after exome sequencing and bioinformatic analyses, we found 399 variants in 216 genes related to palatogenesis/cleft palate exclusively in the DNA pool of patients with NSCPO. Among variations in the 25 NSCPO-associated genes (Table 1), 13 are described in association with syndromes displaying cleft palate in the clinical spectrum (SCPO-associated genes) and 13 were previously associated with CLP. Therefore, variants in ACACB and SHMT1 belonged exclusively to NSCPO. ACACB, encoding the acetyl-coenzyme-A-carboxylase β, an enzyme involved in the oxidation of fatty acids (Ma et al., 2011), was recently reported as a potential candidate for NSCPO by exome sequencing (Hoebel et al., 2017). SHMT1 (serine hydroxymethyltransferase), located at 17p11.2, encodes the cytoplasmic SHMT enzyme consisting of 483 residues (Garrow et al., 1993). Although Shmt1 knockout mice are viable and fertile (MacFarlane et al., 2008), the incidence of neural tube defects among the offspring of Shmt1–/– females is significantly affected by a low-folate diet (Beaudin et al., 2012). Previous studies have also associated SNPs in SMHT1 with NSCPO in a Norwegian population (Boyles et al., 2008), and with NSCL ± P risk in a Chilean population (Salamanca et al., 2020).
Our analyses also revealed novel variants in GHRL3 and LRP6. GRHL3 encodes a transcription factor member of the grainyhead family with significant roles during the embryogenesis of craniofacial structure (Dworkin et al., 2014; Peyrard-Janvid et al., 2014). GRHL3 mutations were reported in patients with VWS (Peyrard-Janvid et al., 2014) and human spina bifida (Lemay et al., 2017), and polymorphisms were associated with both NSCL ± P and NSCPO phenotypes (Leslie et al., 2016; Mangold et al., 2016; Wang Y. et al., 2016; Eshete et al., 2018). In a recent study, we analyzed the previous GRHL3-associated variants (rs10903078, rs41268753, and rs4648975) in a large ancestry-structured case–control sample composed of 1,127 Brazilian participants, and none of the variants withstood the multiple logistic regression analysis with p-Values adjusted to multiple comparisons (Azevedo et al., 2020). As the common GRHL3 variants identified as risk factors for NSCPO in European, Asian, and African populations (Leslie et al., 2016; Mangold et al., 2016; Wang Y. et al., 2016) were not confirmed in our previous study, it is suggested that NSCPO risk associated with particular variants in GRHL3 may differ among ethnic groups and the variant identified in this study (rs34637004) may be the risk variant for the Brazilian population. LRP6, which encodes a central co-receptor in the WNT/β-catenin signaling pathway, has pleiotropic roles in both normal development and complex diseases (Wang et al., 2018). LRP6 controls the canonical WNT/β-catenin pathway promoting development of neural crests and participating in the craniofacial morphogenesis, including lip and palate formation (Tamai et al., 2000; Song et al., 2009; Jin et al., 2011). Variants in LRP6 were associated with congenital neural tube defects (Allache et al., 2014; Lei et al., 2015), tooth agenesis (Massink et al., 2015; Ockeloen et al., 2016), and NSOC (Basha et al., 2018). Although the LRP6-variant rs34815107 (c.4202G > A; p.Arg1401His; MAF < 0.002) was predicted as benign and tolerated and to date no disease was associated with this variant, it is located in the LRP6 cytoplasmic domain, which is essential for Wnt phosphorylation of serine/threonine residues of target proteins (Chen et al., 2014). Further validation in large multiethnic cohort studies and functional analysis are warranted to clarify the association between the novel variants in GRHL3 and LRP6 and NSCPO.
Using ontology analysis, we found 243 biologic processes and 8 pathways formatted by NSCPO-associated genes that interact with each other constituting a large network with two large nodes interconnected by LRP6 and MTR. MTR encodes the methionine synthase protein, which catalyzes the remethylation of homocysteine to form methionine and links of homocysteine cycle to folate metabolism (Leclerc et al., 1996). Strong evidences suggest that the presence of accepted folate/homocysteine levels may not be the only requirement for NSOC prevention, since its absorption, transport, and metabolism, producing active derivatives essentials for DNA methylation and synthesis during embryonic processes, including craniofacial development, are also important to oral cleft predisposing (Desai et al., 2016). Indeed, variants in several genes related to transport and metabolism of folate and homocysteine, including MTR, have been shown to alter the disponibility of the active forms of those molecules and influence cleft risk (Wang W. et al., 2016; Lei et al., 2018). Interestingly, a recent study demonstrated that the methionine-depleted medium inhibits canonical Wnt/β-catenin signaling (Albrecht et al., 2019), and as LRP6 is an indispensable co-receptor for WNT signaling (Gray et al., 2010), our findings reinforce the interlink between MTR and LRP6 and the risk of NSCPO through potential epistatic interactions. Furthermore, as cleft palate can be part of a syndrome or concomitant with cleft lip, indicating that variants in multiple genes may be shared by different conditions with a specific cleft palate phenotype, we applied genetic ontology analysis to identify common and specific pathways. We found 167 biological processes and 5 pathways consisting of ACACB, CDH1, COL2A1, COL11A1, COL11A2, CREBBP, FTCD, LRP6, JAG2, MTHFD1, MTHFR, MTR, POMGNT2, SHMT1, and TYMS enriched in SCPO and CLP. In relation to NSCPO, 76 biological processes and 3 pathways, led by FTCD, MTHFD1, MTHFR, MTR, SHMT1, and TYMS, were found. Thus, these findings suggest that although the genes form distinct biological processes and pathways, similar genes participated in both phenotypes, suggesting common etiologies among them.
The DNA sequencing of pooled patients proved to be an excellent and cost-effective strategy to identify genetic variants related to complex diseases (Collins et al., 2016; Popp et al., 2017; Özdoğan and Kaya, 2020), although it has never been applied to study oral clefts. We detected variants in 6.43% of the investigated genes, which is quite similar to the number of variants identified in the study conducted by Basha et al. (2018), applying individual exome sequencing in 46 multiplex families with NSCPO. Furthermore, we found variants in the same genes identified and validated by Basha et al. (2018) as of risk for NSCPO. However, this technique is associated with problems to be considered when analyzing and interpreting the results. One important problem is to correctly identify rare variants, since sequencing errors can be confused with alleles present at low frequencies, generating false-positive variants. Another potential problem is associated with the estimation of the allelic frequency of the polymorphic variants. The power of genetic analysis depends on the calculation of the allele frequency, and in principle, the pool should provide a robust estimate of the allele frequencies, since it represents several samples, decreasing the general variances of the estimated frequencies. This hypothesis is well supported by mathematical models under the assumption that there are no sequencing errors and each individual contributes an equal amount of DNA to the pools (Anand et al., 2016). Therefore, the lack of validation in a large cohort and the non-characterization of the functional impact of the variants are the limitations of this study. Another limitation that should be noted when interpreting the results is that we did not control for environmental risk factors, although the major source of confounding for genetic studies is population stratification, and our results were adjusted for this.
In summary, our results support the findings of earlier epidemiological studies, indicating that specific variants in genes related to SCPO and CLP may cause NSCPO. Furthermore, gene ontology enrichment analysis identified the interconnection of LRP6 and MTR, reinforcing that variants in the folate/homocysteine pathway may influence risk of NSCPO through potential epistatic interactions. While this study has explored novel variants in previously reported cleft palate genes, we have also identified and described a large group of variants in genes without previous association with NSCPO.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The name of the repository and accession number are as follows: European Nucleotide Archive PRJEB44884 and the samples (SAMEA8730854, SAMEA8730858, SAMEA8730857, SAMEA8730856, SAMEA8730855, SAMEA8730853).
Ethics Statement
The studies involving human participants were reviewed and approved by the Piracicaba Dental School, University of Campinas – 08452819.0.0000.5418. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
Author Contributions
RM contributed to the conception, design, data acquisition, and interpretation, and drafted and critically revised the manuscript. HM-J, SR, EK, RS, and LN contributed to conception, design, data acquisition, and interpretation, and critically revised the manuscript. RC made substantial contributions to the study’s conception and design, and manuscript review and editing. All authors gave their final approval and agreed to be accountable for all aspects of the work.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dra. Ana Cristina Victorino Krepischi (Institute of Biosciences, University of São Paulo, São Paulo, Brazil) for her assistance during exome analysis.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2021.638522/full#supplementary-material
Supplementary Figure 1 | Protein-protein interaction networks constructed with the previously syndromic cleft palate only (SCPO)-associated genes identified in the pools of patients with non-syndromic cleft palate only (NSCPO), but not in the control, using STRING. The networks include 205 edges (interactions) among 98 nodes based on curated databases (light blue), experimentally (purple), gene neighborhood (green), gene fusions (red), gene co-occurrence (blue), textmining (light green), co-expression (black) and protein homology (violet). The overall confidence score of this analysis was 0.446 (P < 1.0e-16), indicating a low change for false-positive interactions.
Supplementary Figure 2 | The protein-protein interaction networks with cleft lip-palate (CLP)-associated targets found in the pools of patients with non-syndromic cleft palate only (NSCPO), but not in the control. Networks include 244 interactions among 91 nodes (overall coefficient score = 0.557; P < 1.0e-16). Different colors represent different levels of evidence of connection between proteins. Light blue represents curated databases, purple experimental evidence, green gene neighborhood, red gene fusions, blue gene co-occurrence, light green evidence from textmining, black co-expression, and violet protein homology.
Footnotes
- ^ https://scfbm.biomedcentral.com/articles/10.1186/1751-0473-9-13
- ^ https://arxiv.org/abs/1207.3907
- ^ https://gataca.cchmc.org/gataca/
- ^ http://varelect.genecards.org
- ^ http://omim.org
- ^ http://genetics.bwh.harvard.edu/pph2/
- ^ http://sift.jcvi.org/
- ^ http://www.mutationtaster.org/
- ^ http:/string-db.org
References
Adzhubei, I., Jordan, D. M., and Sunyaev, S. R. (2013). Predicting functional effect of human missense mutations using PolyPhen-2. Curr. Protoc. Hum. Genet. 7, 7.20.1–7.20.41.
Aidar, M., and Line, S. R. (2007). A simple and cost-effective protocol for DNA isolation from buccal epithelial cells. Braz. Dent. J. 18, 148–152. doi: 10.1590/s0103-64402007000200012
Albrecht, L. V., Bui, M. H., and Robertis, W. M. (2019). Canonical Wnt is inhibited by targeting one-carbon metabolism through methotrexate or methionine deprivation. Proc. Natl. Acad. Sci. U.S.A. 116, 2987–2995. doi: 10.1073/pnas.1820161116
Allache, R., Lachance, S., Guyot, M. C., De Marco, P., Merello, E., Justice, M. J., et al. (2014). Novel mutations in Lrp6 orthologs in mouse and human neural tube defects affect a highly dosage-sensitive Wnt non-canonical planar cell polarity pathway. Hum. Mol. Genet. 23, 1687–1699. doi: 10.1093/hmg/ddt558
Anand, S., Mangano, E., Barizzone, N., Bordoni, R., Sorosina, M., Clarelli, F., et al. (2016). Next generation sequencing of pooled samples: guideline for variants’ Filtering. Sci. Rep. 6:33735.
Azevedo, C. M. S., Machado, R. A., Martelli-Júnior, H., Reis, S. R. A., Persuhn, D. C., Coletta, R. D., et al. (2020). Exploring GRHL3 polymorphisms and SNP-SNP interactions in the risk of non-syndromic oral clefts in the Brazilian population. Oral Dis. 26, 145–151. doi: 10.1111/odi.13204
Basha, M., Demeer, B., Revencu, N., Helaers, R., Theys, S., Saba, S. B., et al. (2018). Whole exome sequencing identifies mutations in 10% of patients with familial non-syndromic cleft lip and/ or palate in genes mutated in well-known syndromes. J. Med. Genet. 55, 449–458. doi: 10.1136/jmedgenet-2017-105110
Beaty, T. H., Marazita, M. L., and Leslie, E. J. (2016). Genetic factors influencing risk to orofacial clefts: today’s challenges and tomorrow’s opportunities. F1000Res. 5:2800. doi: 10.12688/f1000research.9503.1
Beaty, T. H., Ruczinski, I., Murray, J. C., Marazita, M. L., Munger, R. G., Hetmanski, J. B., et al. (2011). Evidence for gene-environment interaction in a genome wide study of nonsyndromic cleft palate. Genet. Epidemiol. 35, 469–478.
Beaudin, A. E., Abarinov, E. V., Malysheva, O., Perry, C. A., Caudill, M., and Stover, P. J. (2012). Dietary folate, but not choline, modifies neural tube defect risk in Shmt1 knockout mice. Am. J. Clin. Nutr. 95, 109–114. doi: 10.3945/ajcn.111.020305
Bi, W., Huang, W., Whitworth, D. J., Deng, J. M., Zhang, Z., Behringer, R. R., et al. (2001). Haploinsufficiency of Sox9 results in defective cartilage primordia and premature skeletal mineralization. Proc. Natl. Acad. Sci. U.S.A. 98, 6698–6703. doi: 10.1073/pnas.111092198
Boyles, A. L., Wilcox, A. J., Taylor, J. A., Meyer, K., Fredriksen, A., Ueland, P. M., et al. (2008). Folate and one-carbon metabolism gene polymorphisms and their associations with oral facial clefts. Am. J. Med. Genet. A 146A, 440–449. doi: 10.1002/ajmg.a.32162
Boyles, A. L., Wilcox, A. J., Taylor, J. A., Shi, M., Weinberg, C. R., Meyer, K., et al. (2009). Oral facial clefts and gene polymorphisms in metabolism of folate/one-carbon and vitamin A: a pathway-wide association study. Genet. Epidemiol. 33, 247–255. doi: 10.1002/gepi.20376
Bureau, A., Parker, M. M., Ruczinski, I., Taub, M. A., Marazita, M. L., Murray, J. C., et al. (2014). Whole exome sequencing of distant relatives in multiplex families implicates rare variants in candidate genes for oral clefts. Genetics 197, 1039–1044. doi: 10.1534/genetics.114.165225
Butali, A., Mossey, P. A., Adeyemo, W. L., Eshete, M. A., Gowans, L. J. J., Busch, T. D., et al. (2019). Genomic analyses in African populations identify novel risk loci for cleft palate. Hum. Mol. Genet. 28, 1038–1051.
Carlson, J. C., Anand, D., Butali, A., Buxo, C. J., Christensen, K., Deleyiannis, F., et al. (2019). A systematic genetic analysis and visualization of phenotypic heterogeneity among orofacial cleft GWAS signals. Genet. Epidemiol. 43, 704–716.
Chen, Q., Su, Y., Wesslowski, J., Hagemann, A., Ramialison, M., Wittbrodt, J., et al. (2014). Tyrosine phosphorylation of LRP6 by Src and Fer inhibits Wnt/β-catenin signalling. EMBO Rep. 15, 1254–1267. doi: 10.15252/embr.201439644
Collins, D. W., Gudiseva, H. V., Trachtman, B., Bowman, A. S., Sagaser, A., Sankar, P., et al. (2016). Association of primary open-angle glaucoma with mitochondrial variants and haplogroups common in African Americans. Mol. Vis. 22, 454–471.
Desai, A., Sequeira, J. M., and Quadros, E. V. (2016). The metabolic basis for developmental disorders due to defective folate transport. Biochimie 126, 31–42. doi: 10.1016/j.biochi.2016.02.012
Dixon, J., Brakebusch, C., Fassler, R., and Dixon, M. J. (2000). Increased levels of apoptosis in the prefusion neural folds underlie the craniofacial disorder, Treacher Collins syndrome. Hum. Mol. Genet. 9, 1473–1480. doi: 10.1093/hmg/9.10.1473
Dixon, M. J., Marazita, M. L., Beaty, T. H., and Murray, J. C. (2011). Cleft lip and palate: understanding genetic and environmental influences. Nat. Rev. Genet. 12, 167–178. doi: 10.1038/nrg2933
do Rego, A. B., Saì, J., Hoshi, R., Viena, C. S., Mariano, L. C., de Castro Veiga, P., et al. (2015). Genetic risk factors for nonsyndromic cleft lip with or without cleft palate in a Brazilian population with high African ancestry. Am. J. Med. Genet. A 167A, 2344–2349. doi: 10.1002/ajmg.a.37181
Dworkin, S., Simkin, J., Darido, C., Partridge, D. D., Georgy, S. R., Caddy, J., et al. (2014). Grainyhead-like 3 regulation of endothelin-1 in the pharyngeal endoderm is critical for growth and development of the craniofacial skeleton. Mech. Dev. 133, 77–90. doi: 10.1016/j.mod.2014.05.005
Eshete, M. A., Liu, H., Li, M., Adeyemo, W. L., Gowans, L. J. J., Mossey, P. A., et al. (2018). Loss-of-function GRHL3 variants detected in African patients with isolated cleft palate. J. Dent. Res. 97, 41–48. doi: 10.1177/0022034517729819
Ferrante, M. I., Zullo, A., Barra, A., Bimonte, S., Messaddq, N., Studer, M., et al. (2006). Oral-facial-digital type I protein is required for primary cilia formation and left-right axis specification. Nat. Genet. 38, 112–117. doi: 10.1038/ng1684
Frech, C., and Chen, N. (2010). Genome-wide comparative gene family classification. PLoS One 5:e13409. doi: 10.1371/journal.pone.0013409
Funato, N., and Nakamura, M. (2017). Identification of shared and unique gene families associated with oral clefts. Int. J. Oral Sci. 9, 104–109. doi: 10.1038/ijos.2016.56
Garrow, T. A., Brenner, A. A., Whitehead, V. M., Chen, X. N., Duncan, R. G., Korenberg, J. R., et al. (1993). Cloning of human cDNAs encoding mitochondrial and cytosolic serine hydroxymethyltransferases and chromosomal localization. J. Biol. Chem. 268, 11910–11916. doi: 10.1016/s0021-9258(19)50286-1
Gray, J. D., Nakouzi, G., Slowinska-Castaldo, B., Dazard, J. E., Rao, J. S., Nadeau, J. H., et al. (2010). Functional interactions between the LRP6 WNT co-receptor and folate supplementation. Hum. Mol. Genet. 19, 4560–4572. doi: 10.1093/hmg/ddq384
He, M., Zuo, X., Liu, H., Wang, W., Zhang, Y., Fu, Y., et al. (2020). Genome-wide analyses identify a novel risk locus for nonsyndromic cleft palate. J. Dent. Res. 99, 1461–1468. (in press) doi: 10.1177/0022034520943867
Hecht, J. T., Mulliken, J. B., and Blanton, S. H. (2002). Evidence for a cleft palate only locus on chromosome 4 near MSX1. Am. J. Med. Genet. 110, 406–407. doi: 10.1002/ajmg.10497
Hoebel, A. K., Drichel, D., van de Vorst, M., Böhmer, A. C., Sivalingam, S., Ishorst, N., et al. (2017). Candidate genes for nonsyndromic cleft palate detected by exome sequencing. J. Dent. Res. 96, 1314–1321.
Huang, L., Jia, Z., Shi, Y., Du, Q., Shi, J., Wang, Z., et al. (2019). Genetic factors define CPO and CLO subtypes of nonsyndromic orofacial cleft. PLoS Genet. 15:e1008357. doi: 10.1371/journal.pgen.1008357
Ishorst, N., Francheschelli, P., Bohmer, A., Khan, M., Heilmann-Heimbach, S., Fricker, N., et al. (2018). Nonsyndromic cleft palate: an association study at GWAS candidate loci in a multiethnic sample. Birth Defects Res. 110, 871–882. doi: 10.1002/bdr2.1213
Jagomagi, T., Nikopensius, T., Krjutskov, K., Tammekivi, V., Viltrop, T., Saag, M., et al. (2010). MTHFR and MSX1 contribute to the risk of nonsyndromic cleft lip/palate. Eur. J. Oral Sci. 118, 213–220. doi: 10.1111/j.1600-0722.2010.00729.x
Jia, Z. L., He, S., Jiang, S. Y., Zhang, B. H., Duan, S. J., Shi, J. Y., et al. (2017). Rs12941170 at SOX9 gene associated with orofacial clefts in Chinese. Arch. Oral Biol. 76, 14–19. doi: 10.1016/j.archoralbio.2016.12.010
Jiang, R., Lan, Y., Chapman, H. D., Norton, C. R., Serreze, D. V., Weinmaster, G., et al. (1998). Defects in limb, craniofacial, and thymic development in Jagged2 mutant mice. Genes Dev. 12, 1046–1057. doi: 10.1101/gad.12.7.1046
Jin, Y. R., Turcotte, T. J., Crocker, A. L., Han, X. H., and Yoon, J. K. (2011). The canonical Wnt signaling activator, R-spondin2, regulates craniofacial patterning and morphogenesis within the branchial arch through ectodermal-mesenchymal interaction. Dev. Biol. 352, 1–13.
Jugessur, A., Skare, O., Lie, R. T., Wilcox, A. J., Christensen, K., Christiansen, L., et al. (2012). X-linked genes and risk of orofacial clefts: evidence from two population-based studies in Scandinavia. PLoS One 7:e39240. doi: 10.1371/journal.pone.0039240
Koillinen, H., Lahermo, P., Rautio, J., Hukki, J., Peyrard-Janvid, M., and Kere, J. (2005). A genome-wide scan of non-syndromic cleft palate only (CPO) in Finnish multiplex families. J. Med. Genet. 42, 177–184. doi: 10.1136/jmg.2004.019646
Kumar, P., Henikoff, S., and Ng, P. C. (2009). Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 4, 1073–1081. doi: 10.1038/nprot.2009.86
Leclerc, D., Campeau, E., Goyette, P., Adjalla, C. E., Christensen, B., Ross, M., et al. (1996). Human methionine synthase: cDNA cloning and identification of mutations in patients of the cblG complementation group of folate/cobalamin disorders. Hum. Mol. Genet. 5, 1867–1874. doi: 10.1093/hmg/5.12.1867
Lei, W., Xia, Y., Wu, Y., Fu, G., and Ren, A. (2018). Associations between MTR A2756G, MTRR A66G, and TCN2 C776G polymorphisms and risk of nonsyndromic cleft lip with or without cleft palate: a meta-analysis. Genet. Test. Mol. Biomarkers 22, 465–473. doi: 10.1089/gtmb.2018.0037
Lei, Y., Fathe, K., McCartney, D., Zhu, H., Yang, W., Ross, M. E., et al. (2015). Rare LRP6 variants identified in spina bifida patients. Hum. Mutat. 36, 342–349. doi: 10.1002/humu.22750
Lemay, P., De Marco, P., Emond, A., Spiegelman, D., Dionne-Laporte, A., Laurent, S., et al. (2017). Rare deleterious variants in GRHL3 are associated with human spina bifida. Hum. Mutat. 38, 716–724. doi: 10.1002/humu.23214
Leslie, E. J., and Marazita, M. L. (2013). Genetics of cleft lip and cleft palate. Am. J. Med. Genet. C Semin. Med. Genet. 163C, 246–258.
Leslie, E. J., Liu, H., Carlson, J. C., Shaffer, J. R., Feingold, E., Wehby, G., et al. (2016). A genome-wide association study of nonsyndromic cleft palate identifies an etiologic missense variant in GRHL3. Am. J. Hum. Genet. 98, 744–754. doi: 10.1016/j.ajhg.2016.02.014
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Li, S. W., Prockop, D. J., Helminen, H., Fassler, R., Lapvetelainen, T., Kiraly, K., et al. (1995). Transgenic mice with targeted inactivation of the Col2 alpha 1 gene for collagen II develop a skeleton with membranous and periosteal bone but no endochondral bone. Genes Dev. 9, 2821–2830. doi: 10.1101/gad.9.22.2821
Liu, H., Busch, T., Eliason, S., Anand, D., Bullard, S., Gowans, L. J., et al. (2017). Exome sequencing provides additional evidence for the involvement of ARHGAP29 in Mendelian orofacial clefting and extends the phenotypic spectrum to isolated cleft palate. Birth Defects Res. 109, 27–37.
Ludwig, K. U., Böhmer, A. C., Bowes, J., Nikolić, M., Ishorst, N., Wyatt, N., et al. (2017). Imputation of orofacial clefting data identifies novel risk loci and sheds light on the genetic background of cleft lip ± cleft palate and cleft palate only. Hum. Mol. Genet. 26, 829–842.
Ma, L., Mondal, A. K., Murea, M., Sharma, N. K., Tönjes, A., Langberg, K. A., et al. (2011). The effect of ACACB cis-variants on gene expression and metabolic traits. PLoS One 6:e23860. doi: 10.1371/journal.pone.0023860
MacFarlane, A. J., Liu, X., Perry, C. A., Flodby, P., Allen, R. H., Stabler, S. P., et al. (2008). Cytoplasmic serine hydroxymethyltransferase regulates the metabolic partitioning of methylenetetrahydrofolate but is not essential in mice. J. Biol. Chem. 283, 25846–25853. doi: 10.1074/jbc.m802671200
Machado, R. A., Messetti, A. C., de Aquino, S. N., Martelli-Junior, H., Swerts, M. S., de Almeida Reis, S. R., et al. (2016). Association between genes involved in craniofacial development and nonsyndromic cleft lip and/or palate in the Brazilian population. Cleft Palate Craniofac. J. 53, 550–556. doi: 10.1597/15-107
Mangold, E., Böhmer, A. C., Ishorst, N., Hoebel, A.-K., Gültepe, P., Schuenke, H., et al. (2016). Sequencing the GRHL3 coding region reveals rare truncating mutations and a common susceptibility variant for nonsyndromic cleft palate. Am. J. Hum. Genet. 98, 755–762. doi: 10.1016/j.ajhg.2016.02.013
Martinelli, M., Scapoli, L., Palmieri, A., Pezzetti, F., Baciliero, U., Padula, E., et al. (2006). Study of four genes belonging to the folate pathway: transcobalamin 2 is involved in the onset of non-syndromic cleft lip with or without cleft palate. Hum. Mutat. 27:294. doi: 10.1002/humu.9411
Massink, M. P., Créton, M. A., Spanevello, F., Fennis, W. M., Cune, M. S., Savelberg, S. M., et al. (2015). Loss-of-function mutations in the WNT co-receptor LRP6 cause autosomal-dominant oligodontia. Am. J. Hum. Genet. 97, 621–626.
Messetti, A. C., Machado, R. A., Oliveira, C. E., Martelli-Júnior, H., Reis, S. R. A., Moreira, H. S. B., et al. (2017). Brazilian multicenter study of association between polymorphisms in CRISPLD2 and JARID2 and non-syndromic oral clefts. J. Oral Pathol. Med. 46, 232–239. doi: 10.1111/jop.12470
Murthy, J., Gurramkonda, V. B., and Lakkakula, B. V. (2014). Significant association of MTHFD1 1958G>A single nucleotide polymorphism with nonsyndromic cleft lip and palate in Indian population. Med. Oral Patol. Oral Cir. Bucal 19, e616–e621.
Nikopensius, T., Jagomagi, T., Krjutskov, K., Tammekivi, V., Saag, M., Prane, I., et al. (2010). Genetic variants in COL2A1, COL11A2, and IRF6 contribute risk to nonsyndromic cleft palate. Birth Defects Res. A Clin. Mol. Teratol. 88, 748–756. doi: 10.1002/bdra.20700
Ockeloen, C. W., Khandelwal, K. D., Dreesen, K., Ludwig, K. U., Sullivan, R., van Rooij, I., et al. (2016). Novel mutations in LRP6 highlight the role of WNT signaling in tooth agenesis. Genet. Med. 18, 1158–1162.
Özdoğan, G. O., and Kaya, H. (2020). Next-Generation sequencing data analysis on pool-seq and low-coverage retinoblastoma data. Interdiscip. Sci. Comput. Life Sci. 12, 302–310. doi: 10.1007/s12539-020-00374-8
Pauws, E., Hoshino, A., Bentley, L., Prajapati, S., Keller, C., Hammond, P., et al. (2009). Tbx22null mice have a submucous cleft palate due to reduced palatal bone formation and also display ankyloglossia and choanal atresia phenotypes. Hum. Mol. Genet. 18, 4171–4179. doi: 10.1093/hmg/ddp368
Pengelly, R. J., Arias, L., Martiìnez, J., Upstill-Goddard, R., Seaby, E. G., Gibson, J., et al. (2016). Deleterious coding variants in multi-case families with non-syndromic cleft lip and/or palate phenotypes. Sci. Rep. 6:30457.
Peyrard-Janvid, M., Leslie, E. J., Kousa, Y. A., Smith, T. L., Dunnwald, M., Magnusson, M., et al. (2014). Dominant mutations in GRHL3 cause Van der Woude syndrome and disrupt oral periderm development. Am. J. Hum. Genet. 94, 23–32. doi: 10.1016/j.ajhg.2013.11.009
Popp, B., Ekici, A. B., Thiel, C. T., Hoyer, J., Wiesener, A., Kraus, C., et al. (2017). Exome Pool-Seq in neurodevelopmental disorders. Eur. J. Hum. Genet. 25, 1364–1376. doi: 10.1038/s41431-017-0022-1
Salamanca, C., González-Hormazábal, P., Recabarren, A. S., Recabarren, P. A., Pantoja, R., Leiva, N., et al. (2020). A SHMT1 variant decreases the risk of nonsyndromic cleft lip with or without cleft palate in Chile. Oral Dis. 26, 159–165. doi: 10.1111/odi.13229
Satokata, I., and Maas, R. (1994). Msx1 deficient mice exhibit cleft palate and abnormalities of craniofacial and tooth development. Nat. Genet. 6, 348–356. doi: 10.1038/ng0494-348
Seegmiller, R., Fraser, F. C., and Sheldon, H. (1971). A new chondrodystrophic mutant in mice. Electron microscopy of normal and abnormal chondrogenesis. J. Cell Biol. 48, 580–593. doi: 10.1083/jcb.48.3.580
Shaw, G. M., Todoroff, K., Finnell, R. H., Rozen, R., and Lammer, E. J. (1999). Maternal vitamin use, infant C677T mutation in MTHFR, and isolated cleft palate risk. Am. J. Med. Genet. 85, 84–85. doi: 10.1002/(sici)1096-8628(19990702)85:1<84::aid-ajmg15>3.0.co;2-v
Shaw, G. M., Yang, W., Perloff, S., Shaw, N. M., Carmichael, S. L., Zhu, H., et al. (2013). Thymidylate synthase polymorphisms and risks of human orofacial clefts. Birth Defects Res. A Clin. Mol. Teratol. 97, 95–100. doi: 10.1002/bdra.23114
Shaw, G. M., Zhu, H., Lammer, E. J., Yang, W., and Finnell, R. H. (2003). Genetic variation of infant reduced folate carrier (A80G) and risk of orofacial and conotruncal heart defects. Am. J. Epidemiol. 158, 747–752. doi: 10.1093/aje/kwg189
Song, L., Li, Y., Wang, K., Wang, Y. Z., Molotkov, A., Gao, L., et al. (2009). Lrp6-mediated canonical Wnt signaling is required for lip formation and fusion. Development 136, 3161–3171. doi: 10.1242/dev.037440
Song, Y., and Zhang, S. (2011). Association of CDH1 promoter polymorphism and the risk of non-syndromic orofacial clefts in a Chinese Han population. Arch. Oral Biol. 56, 68–72. doi: 10.1016/j.archoralbio.2010.08.019
Tamai, K., Semenov, M., Kato, Y., Spokony, R., Liu, C., Katsuyama, Y., et al. (2000). LDL-receptor-related proteins in Wnt signal transduction. Nature 407, 530–535. doi: 10.1038/35035117
Vieira, A. R., Avila, J. R., Daack-Hirsch, S., Dragan, E., Félix, T. M., Rahimov, F., et al. (2005). Medical sequencing of candidate genes for nonsyndromic cleft lip and palate. PLoS Genet. 1:e64. doi: 10.1371/journal.pgen.0010064
Wang, W., Jiao, X. H., Wang, X. P., Sun, X. Y., and Dong, C. M. T. R. (2016). MTRR, and MTHFR gene polymorphisms and susceptibility to nonsyndromic cleft lip with or without cleft palate. Genet. Test. Mol. Biomarkers 20, 297–303. doi: 10.1089/gtmb.2015.0186
Wang, Y., Sun, Y., Huang, Y., Pan, Y., Jia, Z., Ma, L., et al. (2016). Association study between Van der Woude Syndrome causative gene GRHL3 and nonsyndromic cleft lip with or without cleft palate in a Chinese cohort. Gene 588, 69–73. doi: 10.1016/j.gene.2016.04.045
Wang, Z. M., Luo, J. Q., Xu, L. Y., Zhou, H. H., and Zhang, W. (2018). Harnessing low-density lipoprotein receptor protein 6 (LRP6) genetic variation and Wnt signaling for innovative diagnostics in complex diseases. Pharmacogenomics J. 18, 351–358. doi: 10.1038/tpj.2017.28
Wattanawong, K., Rattanasiri, S., McEvoy, M., Attia, J., and Thakkinstian, A. (2016). Association between IRF6 and 8q24 polymorphisms and nonsyndromic cleft lip with or without cleft palate: Systematic review and meta-analysis. Birth Defects Res. A Clin. Mol. Teratol. 106, 773–788. doi: 10.1002/bdra.23540
Keywords: non-syndromic cleft palate only, exome sequence, risk factor, syndrome, oral cleft
Citation: Machado RA, Martelli-Junior H, Reis SRA, Küchler EC, Scariot R, das Neves LT and Coletta RD (2021) Identification of Novel Variants in Cleft Palate-Associated Genes in Brazilian Patients With Non-syndromic Cleft Palate Only. Front. Cell Dev. Biol. 9:638522. doi: 10.3389/fcell.2021.638522
Received: 07 December 2020; Accepted: 30 April 2021;
Published: 08 July 2021.
Edited by:
Mary L. Marazita, University of Pittsburgh, United StatesReviewed by:
Alison L. Reynolds, University College Dublin, IrelandJulian Little, University of Ottawa, Canada
Copyright © 2021 Machado, Martelli-Junior, Reis, Küchler, Scariot, das Neves and Coletta. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Renato Assis Machado, cmVuYXRvYXNzaXNtYWNoYWRvQHlhaG9vLmNvbS5icg==