 Laurent Beuret1
Laurent Beuret1 Simon-Pierre Fortier-Beaulieu1
Simon-Pierre Fortier-Beaulieu1 Vincent Rondeau2,3Sophie Roy1Nicolas Houde1
Vincent Rondeau2,3Sophie Roy1Nicolas Houde1 Karl Balabanian2,3Marion Espéli2,3
Karl Balabanian2,3Marion Espéli2,3 Jean Charron1,4*
Jean Charron1,4*- 1Centre de Recherche sur le Cancer de l’Université Laval, Centre de Recherche du CHU de Québec-Université Laval (Oncology), Québec, QC, Canada
- 2Université de Paris, Institut de Recherche Saint Louis, EMiLy, Inserm U1160, Paris, France
- 3OPALE Carnot Institute, The Organization for Partnerships in Leukemia, Hôpital Saint-Louis, Paris, France
- 4Department of Molecular Biology, Medical Biochemistry and Pathology, Université Laval, Québec, QC, Canada
Several studies have established the crucial role of the extracellular signal–regulated kinase (ERK)/mitogen-activated protein kinase pathway in hematopoietic cell proliferation and differentiation. MEK1 and MEK2 phosphorylate and activate ERK1 and ERK2. However, whether MEK1 and MEK2 differentially regulate these processes is unknown. To define the function of Mek genes in the activation of the ERK pathway during hematopoiesis, we generated a mutant mouse line carrying a hematopoietic-specific deletion of the Mek1 gene function in a Mek2 null background. Inactivation of both Mek1 and Mek2 genes resulted in death shortly after birth with a severe anemia revealing the essential role of the ERK pathway in erythropoiesis. Mek1 and Mek2 functional ablation also affected lymphopoiesis and myelopoiesis. In contrast, mice that retained one functional Mek1 (1Mek1) or Mek2 (1Mek2) allele in hematopoietic cells were viable and fertile. 1Mek1 and 1Mek2 mutants showed mild signs of anemia and splenomegaly, but the half-life of their red blood cells and the response to erythropoietic stress were not altered, suggesting a certain level of Mek redundancy for sustaining functional erythropoiesis. However, subtle differences in multipotent progenitor distribution in the bone marrow were observed in 1Mek1 mice, suggesting that the two Mek genes might differentially regulate early hematopoiesis.
Introduction
Mitogen-activated protein kinase (MAPK) signaling pathways consist of sequences of protein kinases that link extracellular stimuli to targets located in different cellular compartments (Wortzel and Seger, 2011). In mammals, the extracellular signal–regulated kinase (ERK) cascade constitutes one of the best-characterized MAPK pathways. It is activated by multiple surface receptors such as growth factor receptors and G protein–coupled receptors to elicit various physiological outputs including fate determination, differentiation, proliferation, survival, and migration (Alberola-Ila and Hernandez-Hoyos, 2003; Fischer et al., 2005; Shaul and Seger, 2007; Geest and Coffer, 2009; Guihard et al., 2010; Wortzel and Seger, 2011; Saulnier et al., 2012; Sun et al., 2015). Following receptor triggering, RAS binds GTP and with the help of scaffolding proteins forms heterodimers with RAF protein kinases that directly phosphorylate and activate MEK1 and MEK2, two dual-specificity kinases that activate ERK1 and ERK2 by phosphorylation (Roskoski, 2012a,b). Although multiple kinases can activate MEK1 and MEK2, they are the only kinases able to phosphorylate ERK1 and ERK2, acting as gatekeepers. As such, MEK proteins have been considered potential therapeutic targets as overactivation of ERK pathway is observed in many cancers (Zhao and Adjei, 2014; Caunt et al., 2015).
The ubiquitously expressed MEK1 and MEK2 proteins share a high level of amino acid sequence homology (Aoidi et al., 2016). Mek1 null mutant mice die during gestation from the underdevelopment of the placenta, whereas Mek2 null mutants are viable and fertile revealing compensatory effects by Mek1 in extraembryonic tissues (Bélanger et al., 2003; Bissonauth et al., 2006; Nadeau et al., 2009; Nadeau and Charron, 2014). However, when the deletion of Mek1 is restricted to the embryo proper, Mek1 null mice are viable and fertile, indicating functional redundancy between Mek1 and Mek2 genes in embryo formation (Bissonauth et al., 2006). Moreover, knock-in of Mek2 cDNA sequence into the Mek1 locus rescues the placental defects in absence of MEK1 revealing MEK redundancy, as well as the importance of the total amount of MEK proteins in placenta development (Aoidi et al., 2016). Functional redundancy between Mek1 and Mek2 is further supported by the lack of phenotypes in animals in which targeted tissues retain either one Mek1 or one Mek2 functional allele (Scholl et al., 2007; Newbern et al., 2008; Yamashita et al., 2011; Li et al., 2012; Shim et al., 2013; Boucherat et al., 2014; Ihermann-Hella et al., 2014; Boucherat et al., 2017). Except for the formation of extraembryonic structures, obvious developmental defects are observed only when all Mek1 and Mek2 alleles are deleted.
Several studies have established the crucial role of the ERK pathway in hematopoietic cell proliferation and differentiation (Alberola-Ila and Hernandez-Hoyos, 2003; Fischer et al., 2005; Geest and Coffer, 2009; Guihard et al., 2010; Saulnier et al., 2012; Kollmann et al., 2017). In vitro studies using differentiation-competent cell lines showed that the pathway is important for erythroid, megakaryocyte, and myeloid cell growth, apoptosis, and differentiation (Matsuzaki et al., 2000; Uchida et al., 2001; Mori et al., 2003; Geest et al., 2009). The ERK signaling pathway was also shown to control early myeloid commitment of hematopoietic stem cells (HSCs) (Hsu et al., 2007). Moreover, in vivo analyses supported a role for the pathway in hematopoiesis. For instance, characterization of B-raf null mutant mice revealed a role for the ERK pathway in the expansion of progenitors but not in their differentiation (Kamata et al., 2005). Erk1 null mutant animals presented accumulation of erythroid progenitors in enlarged spleen indicating that ERK1 may act as a negative regulator of adult steady-state splenic erythropoiesis (Guihard et al., 2010). In bone marrow, Erk1 deficiency caused mild osteopetrosis due to defective myeloid lineage progenitors and subsequent impairment in osteoclastogenesis and osteoclast function (Saulnier et al., 2012). Altogether, in vitro and in vivo studies demonstrated that the integrity of the ERK pathway is critical for both erythroid and myeloid cell fate and differentiation. However, while the implication of the ERK pathway in hematopoiesis is established, the function of each kinase of the pathway remains undefined.
To specifically address the role of the ERK activators, MEK1 and MEK2, in hematopoietic cell differentiation, and to bypass the early embryonic lethality associated with the Mek1 null mutation, we specifically deleted the Mek1 gene in hematopoietic cell lineages using the Vav1-icre deleter mouse line. This conditional mutation was implemented in a Mek2 null background. Mice lacking Mek1 and Mek2 gene functions in the hematopoietic lineages died shortly after birth from anemia. In contrast, mice that retained one functional allele of Mek1 (1Mek1) or of Mek2 (1Mek2) in these lineages were viable and fertile. Both 1Mek1 and 1Mek2 mutant mice showed a splenomegaly with reduction of hematocrit, red blood cell (RBC) counts, and hemoglobin concentration in circulating blood, indicating a trend to anemia. Multipotent hematopoietic progenitors were reduced in 1Mek1 bone marrow but not in spleen. Conversely, multipotent hematopoietic progenitors and erythroid progenitors were significantly increased in 1Mek2 spleen. Together, these data confirmed the importance of the ERK pathway in erythropoiesis. Despite Mek1 and Mek2 gene redundancy in this process, each locus possesses some functional specificity.
Materials and Methods
Mouse Strains, Genotyping, and Tissue Collection
The Mek1flox/flox and Mek2–/– mutant mouse lines and the Sox2-cre and Vav1-icre deleter mouse lines were previously described (Hayashi et al., 2002; Bélanger et al., 2003; de Boer et al., 2003; Bissonauth et al., 2006). Mek1–/– mice (Mek1flox/flox Tg+/Sox2–cre) were obtained by breeding Mek1flox/flox Tg+/Sox2–cre males with Mek1flox/flox females. The 1Mek1 (Mek1+/flox Mek2–/– Tg+/Vav1–iCre) mice were generated by breeding Mek1flox/flox Mek2–/– females with 1Mek1 males, whereas 1Mek2 (Mek1flox/flox Mek2+/– Tg+/Vav1–iCre) mice were produced by mating Mek1flox/flox Mek2–/– females with 1Mek2 males. All mouse lines were maintained in the 129S6/SvEv genetic background. Age of the embryos was estimated by considering the morning of the day of the vaginal plug as E0.5. Genomic DNA from embryonic yolk sac and mouse tail biopsy was extracted, purified, and genotyped by Southern blot and polymerase chain reaction analyses as previously described (de Boer et al., 2003; Nadeau et al., 2009). Experiments were performed according to the guidelines of the Canadian Council on Animal Care and in compliance with the European Union guide for the care and use of laboratory animals and approved by the institutional animal care committee and by an institutional review committee (C2EA-26, Animal Care and Use Committee, Villejuif, France).
Hematologic Parameters
Blood cell counts were analyzed with a UniCel DxC 600 Synchron Clinical Systems (Beckman Coulter, Mississauga, On, Canada). Hematocrit was assessed on a hematocrit centrifuge (IEC Micro MB centrifuge, Saint-Laurent, QC, Canada). Reticulocyte counts were measured as described (Guihard et al., 2010). Briefly, 2 μL of whole blood was incubated with thiazole orange at 10–4 mg/mL in phosphate-buffered saline (PBS) for 1 h. Blood sample diluted in PBS was used as an unstained control. Reticulocyte counts were measured by flow cytometry.
Flow Cytometry
Single-cell suspensions were prepared from mouse hematopoietic tissues by sieving and gentle pipetting through 70-μm nylon mesh. Flow cytometry was used to assess cell populations. Peripheral blood cells and single-cell suspensions (106) were saturated with Fc Block and stained with fluorochrome-conjugated antibodies using standard procedures (Malynn et al., 1995; Bélanger et al., 2003; Espeli et al., 2011). For intracellular staining, cells were fixed 15 min in 2% paraformaldehyde (PFA) at room temperature after labeling with cell surface markers followed by cell permeabilization in ice-cold methanol. Flow cytometry was performed on a BD LSRFortessa cell analyzer. FlowJo software was used for analysis of flow cytometry data using the following mAbs: anti–mouse CD3 [clone 145-2C11, hamster immunoglobulin G1 (IgG1)], CD11b (clone M1/70, rat IgG2b), CD16/32 (clone 93, rat IgG2a), CD34 (clone RAM34, rat IgG2a), CD41 (clone MWReg30, rat IgG1), CD45R/B220 (clone RA3-6B2, rat IgG2a), CD48 (clone HM48-1, Armenian hamster IgG), CD71 (clone RI7217, rat monoclonal), CD117/c-Kit (clone 2B8, rat IgG2b), CD127 (clone A7R34, rat IgG2a), CD135/Flt3 (clone A2F10, rat IgG2a), CD150 (clone TC15-23F12,2, rat IgG2a), Ter119 (clone TER-119, rat IgG2b), Gr-1 (clone RB6-8C5, rat IgG2b), Sca-1 (clone E13-161,7, rat IgG2a), and MEK1/2 (clone D1A5, rabbit IgG). Antibodies were conjugated to biotin, BV650, fluorescein isothiocyanate, phycoerythrin (PE), allophycocyanin (APC), AF700, PE-cyanin (Cy) 5, PE-Cy7, eFluor 450,AF647,APC- eFluor780, peridinin chlorophyll protein PerCP-Cy5,5, or Pacific blue and purchased from BD (Mississauga, ON, Canada), eBioscience (San Diego, CA, United States), or BioLegend (San Diego, CA, United States).
Colony-Forming Assays
Methylcellulose colony-forming assays were performed in MethoCult M3434 complete medium with recombinant cytokines (StemCell Technologies, Vancouver, BC, Canada). In brief, 2 × 104 cells from bone marrow, 1 × 105 cells from spleen, 2 × 104 peripheral blood cells from E12.5 embryos, 2 × 104 fetal liver cells from E14.5 embryos, and the aorta–gonad–mesonephros (AGM) region from E8.5 embryos were plated for colony-forming unit (CFU) assays. Cultures were incubated at 37°C in 5% CO2 for 7 days for the growth of colonies of erythroid burst-forming units (BFU-E); granulocyte, erythrocyte, macrophage, and megakaryocyte progenitors (CFU-GEMM); and granulocytes and macrophages (CFU-GM). All the cultures were done in duplicate. Data as presented are mean ± SEM. Three to eight independent specimens were analyzed.
Proliferation Index
Proliferation index was measured by counting the number of pHH3-immunoreactive cells, divided by the total cell number for each section analyzed. Eight random fields were counted for an average number of 500 cells per field. Four specimens per genotype were tested. The primary antibody used was a rabbit monoclonal antibody against pHH3 (1/200 dilution; Cell Signaling, Danvers, MA, United States). The biotinylated secondary antibody was a goat anti-rabbit (1/250; Vector Laboratories, Burlington, ON, Canada).
Erythrocyte Half-Life
The erythrocyte half-life was evaluated using biotinylation of entire blood cells and monitoring for cell replacement (Gifford et al., 2006). Biotinylation of blood cell was carried out by intravenous injection of 150 μL of 3.25 mg/mL of N-hydroxysulfosuccinimide (NHS) esters (EZ-Link NHS-Biotin reagents; Thermo Fisher Scientific, Saint-Laurent, QC, Canada) for 2 consecutive days. RBCs (3 × 106) obtained from 1 to 5 μL of medial saphenous vein blood were labeled with 1 μg de streptavidin-AlexaFluor 647 (Thermo Fisher Scientific, cat. no. S21374) in 1 mL of PBS. The number of biotinylated cells in circulating blood was monitored at regular intervals by flow cytometry.
Phenylhydrazine Stress Test and Response to Erythropoietin Induction
Two- to four-months-old mice were injected subcutaneously with 80 mg/kg phenylhydrazine (PHZ) hydrochloride solution in PBS at day 0 and 40 mg/kg at day 1. Erythropoietin (EPO) induction was done by subcutaneous injection of 50 U of human recombinant EPO (Eprex) at day 0. Blood was obtained via the medial saphenous vein at regular intervals for hematocrit and reticulocyte count measurements by flow cytometry after thiazole orange staining as described by Siekmeier et al. (2000). Data are presented as mean ± SEM, and they are the result of four to five independent specimens analyzed.
Statistical Analyses
Samples were statistically compared with either the analysis of variance (ANOVA) on linear models with Kruskal–Wallis multiple-comparisons test or mixed-effects analysis with Tukey multiple-comparisons test, when appropriate. In the boxplots, the whiskers go down to the smallest value and up to the largest, whereas the boxes extend from the 25th to 75th percentiles with the median. P < 0.05 was considered statistically significant.
Results
Loss of Mek Function in Hematopoietic Cell Lineages Causes Severe Anemia and Neonatal Death
The specific ablation of Mek1 function in hematopoietic cell lineages was generated with the Vav1-icre deleter mouse line in a Mek2 null background (de Boer et al., 2003). Mek1+/flox Mek2–/– Tg+/Vav1–iCre (thereafter named 1Mek1) males were bred with Mek1flox/flox Mek2–/– females to generate Mek1flox/flox Mek2–/– Tg+/Vav1–icre mutants (Mekhema null). Expected Mendelian ratios were obtained during embryogenesis and up to birth (Figure 1A). At embryonic day (E) 16.5, Mekhema null mutants appeared phenotypically normal with no obvious defect in RBC production and vasculature (Figures 1B,C). At birth (D0), Mekhema null newborns looked similar to their control littermates with no sign of cyanosis (Figure 1D). However, all Mekhema null pups died within the first 36 h of life exhibiting a two-time reduction of hematocrit and a severe decrease in spleen size, indicating anemia and problematic splenic erythropoiesis (Figures 1A,E,F).
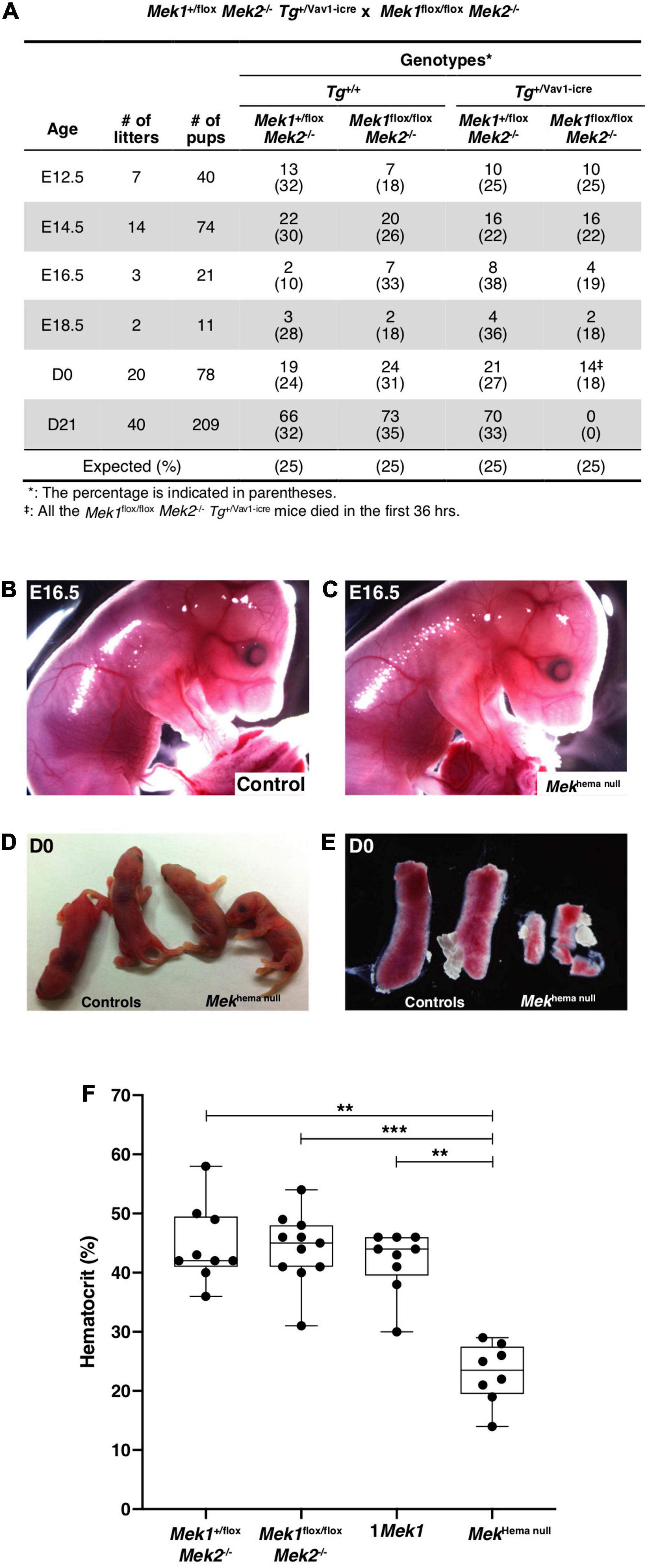
Figure 1. Impact of the conditional Mek1 deletion with the Vav1-icre in the Mek2–/– background. (A) Ratios of embryos and mice genotypes at different embryonic and adult stages obtained from matings between Mek1+/flox Mek2–/– Tg+/Vav1–iCre male with Mek1flox/flox Mek2–/– female. (B–D) Gross morphology of E16.5 (B,C) and D0 (D) control and Mekhema null animals. Normal vascularization and blood circulation were seen in controls and Mekhema null embryos at E16.5. (E) Macroscopic views showing the hypoplastic Mekhema null spleen phenotype at D0 compared with spleen from littermate controls. (F) Hematocrit at birth. No sign of cyanosis was detected at birth in Mekhema null embryos, but they died in the first 36 h with a severe anemia. The results are presented as whiskers-and-boxes graph, and each dot represents an individual mouse. ANOVA with Kruskal–Wallis multiple-comparisons test was performed. **P < 0.01, ***P < 0.001.
Erythropoiesis occurs sequentially at different sites during embryogenesis. It starts in the yolk sac from E7.5 to E10.5, to migrate into the AGM region between E8.5 and E11.5, and then to the placenta from E10.5 to E13.5, and to the fetal liver at E10.5 until birth (Orkin and Zon, 2008). In adult mice, erythropoiesis takes place mainly in bone marrow, but it can also occur in the spleen in response to anemic stress (Paulson et al., 2011). To determine whether anemia and the reduced spleen size seen in Mekhema null pups were due to defective expansion or differentiation of specific hematopoietic progenitors, we measured the number of colonies of BFU-E and CFU-GEMM by performing methylcellulose colony-forming assays with AGM at E10.5 and fetal liver at E14.5 from control (Mek2–/– and 1Mek1) and Mekhema null embryos (Figure 2). No statistical difference was observed in the numbers of BFU-E and CFU-GEMM progenitor populations from AGM of control and Mekhema null mutants. This result was not surprising since the onset of the Vav1-iCre activity occurs around E11.5. However, it precludes the investigation of the role for the ERK pathway at the early stage of hematopoiesis (Figures 2A,C; Bustelo et al., 1993). In contrast, the number of BFU-E colonies from the fetal liver was reduced in Mekhema null mutants, and only few CFU-GEMM colonies were recovered compared to controls (Figures 2B,D). Thus, the lack of ERK signaling in Mekhema null mutants perturbed hematopoiesis by affecting the production or the survival of erythroid progenitors, which most likely contributed to the anemia observed in Mekhema null newborns.
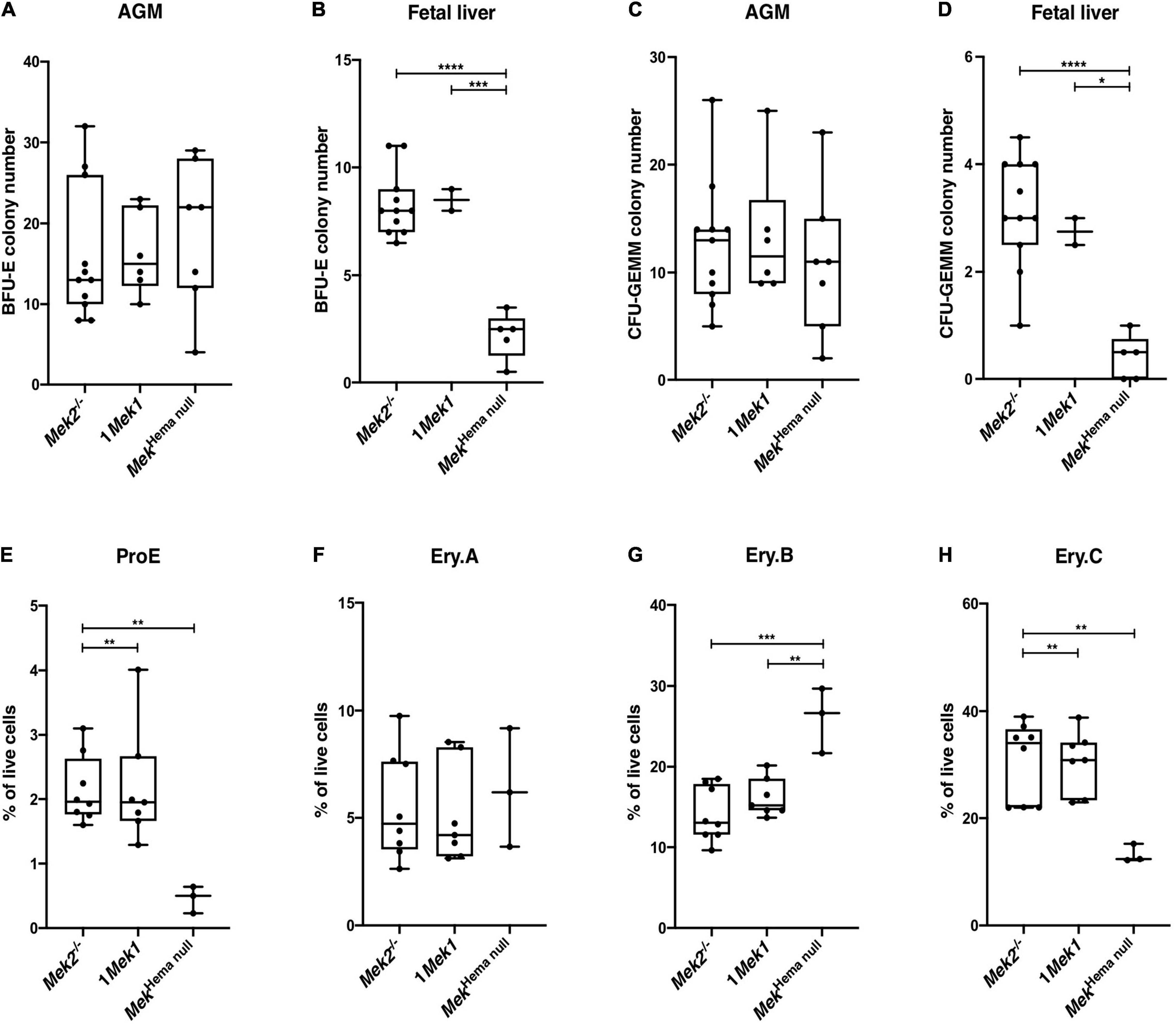
Figure 2. Erythroid progenitors and subsets in Mekhema null mutants. (A–F) Numbers of BFU-E (A–C) and CFU-GEMM (D–F) colonies obtained after in vitro growth of E10.5 AGM region (A,C) and E14.5 fetal liver cells (B,D) from controls and Mekhema null embryos are presented. (E–H) Erythroid subset analysis by flow cytometry analysis in newborn spleen. Frequency of Ter119medCD71highFSChigh (ProE; E), Ter119highCD71highFSChigh (Ery.A; F), Ter119highCD71highFSClow (Ery.B; G), and Ter119highCD71lowFSClow (Ery.C; H) are presented. They revealed a decrease of the ProE and Ery.B populations and an increase of the Ery.C population in Mekhema null samples. The results are presented as whiskers-and-boxes graph, and each dot represents an individual mouse. ANOVA with Kruskal–Wallis multiple-comparisons test was performed. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
To determine if these embryonic defects translate into abnormal erythropoietic cell differentiation at birth, flow cytometry analyses for the expression of Ter119 and CD71 markers and the forward scatter (FSC) parameter were performed in spleen from newborns to monitor the development of the erythropoietic compartment from proerythroblasts (ProE; Ter119medCD71highFSChigh) to successive differentiation stages: basophilic (Ery.A; Ter119highCD71highFSChigh), late basophilic and polychromatic (Ery.B; Ter119high CD71highFSClow), and finally orthochromatic erythroblasts (Ery.C; Ter119highCD71lowFSClow (Guihard et al., 2010; Koulnis et al., 2011). The proportion of committed ProE was reduced by four times in Mekhema null mutants when compared to Mek2–/– and 1Mek1 control littermates (Figure 2E). Moreover, the proportion of differentiated orthochromatic erythroblasts was decreased by half in Mekhema null mutants, whereas the proportion of late basophilic and polychromatic erythroblasts was doubled (Figures 2G,H). No change was observed for the basophilic erythroblasts (Figure 2F). These results, combined with the reduced size of the spleen at birth (Figure 1E), suggested a key role for the ERK pathway in erythroid cell commitment and differentiation. In absence of ERK signaling through the loss of both Mek genes, reduced proerythroblast commitment and a block at the last steps of erythroid cell maturation occurred.
As the lack of MEK function was associated with decreased CFU-GEMM, we also analyzed the myeloid differentiation in splenocytes from newborns by flow cytometry. The proportion of Mac1+ (also known as CD11b) single-positive and Gr1+Mac1+ double-positive myeloid cell populations, as well as CD19+ B cell populations, was significantly reduced in Mekhema null mutants, indicating a diminution in monocytes, macrophages, and B cells (Figures 3A–D). Moreover, no statistical differences in granulocyte and macrophage progenitors (CFU-GM) colony numbers, as assessed by methyl cellulose colony formation assay, were detected in AGM at E10.5 (Figure 3E). In contrast, the number of CFU-GM was reduced in peripheral blood at E12.5 and in fetal liver at E14.5 from Mekhema null embryos when compared to controls (Figures 3F,G). As the size of the spleen in Mekhema null newborn mice was greatly reduced, the decreased numbers of progenitor cell populations might be even more pronounced supporting the importance of the role of MEK in erythroid and myeloid hematopoietic lineage commitment.
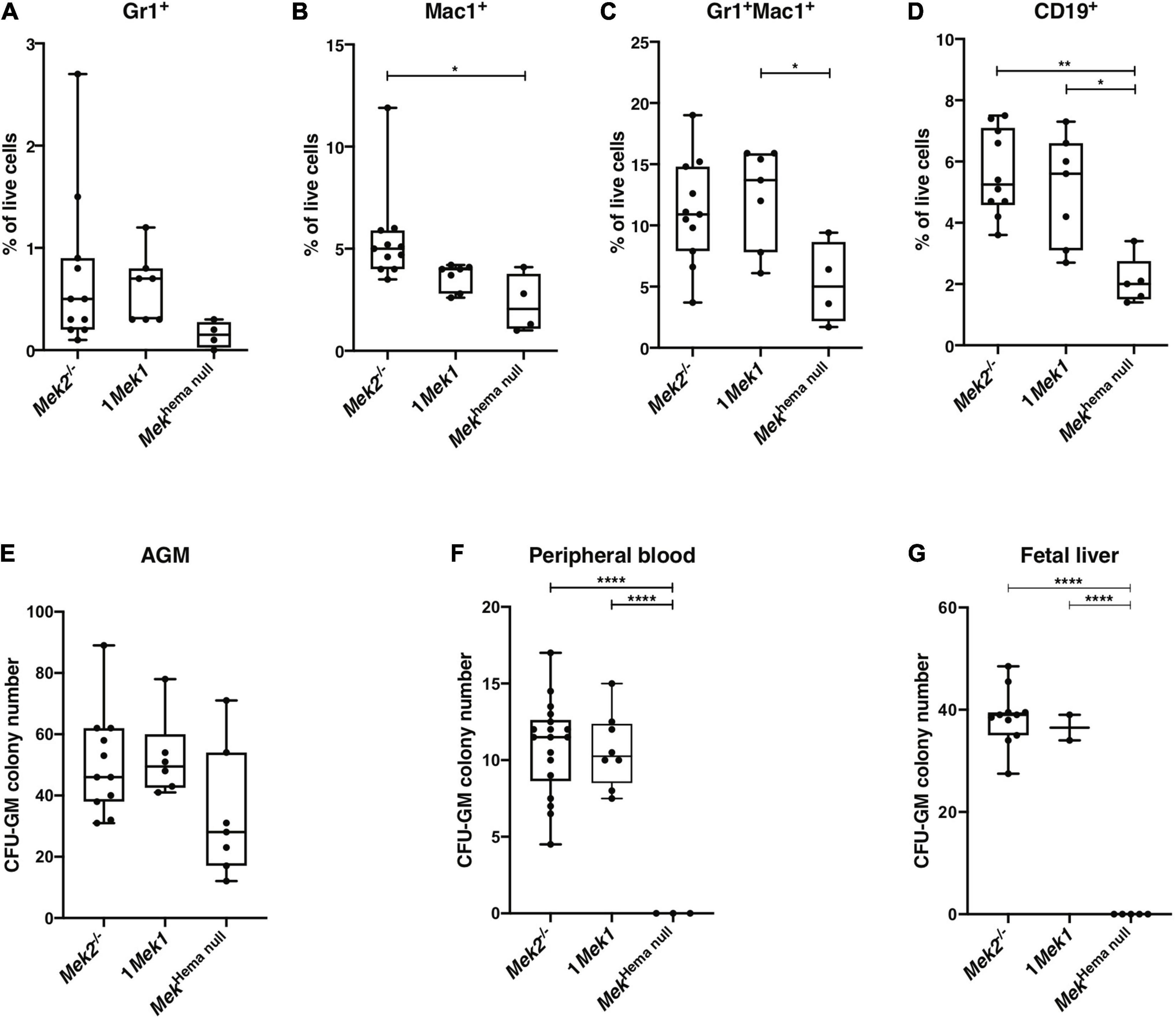
Figure 3. Hematopoiesis cell populations affected in Mekhema null mutants. (A–D) Flow cytometry analyses of newborn spleen revealed a decreased in the proportion of Gr-1+ (A), Mac1+ (B), Mac1+Gr-1+ (C), and CD19+ (D) cells in Mekhema null mutants compared to control animals. (E–G) Numbers of CFU-GM colonies obtained after in vitro growth of E10.5 AGM region (E), E12.5 peripheral blood cells (F), and E14.5 fetal liver cells (G) from controls and Mekhema null embryos are presented. A gradual loss of erythroid and myeloid cell progenitors occurred in Mekhema null embryos during gestation. The results are presented as whiskers-and-boxes graph, and each dot represents an individual mouse. ANOVA with Kruskal–Wallis multiple-comparisons test was performed. *P < 0.05, **P < 0.01, ****P < 0.0001.
Mild Anemia in 1Mek1 and 1Mek2 Mutants
To tear apart the impact of MEK dosage versus MEK isoform on hematopoiesis, we studied mouse models bearing only one allele of Mek1 (Mek1+/flox Mek2–/– Tg+/Vav1–iCre thereafter named 1Mek1) or Mek2 (Mek1flox/flox Mek2+/– Tg+/Vav1–iCre, 1Mek2) in the hematopoietic cell lineages. Both mutant mice were viable, and their hematopoietic parameters were studied from 2 to 4 months of age and compared to samples from wt and single Mek null mutants [Mek1flox/flox Tg+/Sox2–cre (Mek1–/–) and Mek2–/– mice]. While wt and Mek2–/– mutants presented no difference in hematocrit, hemoglobin concentration, and RBC counts, Mek1–/– mutants display significantly reduced hemoglobin concentration and RBC numbers indicating that Mek1 or Mek2 genes are not totally interchangeable for these processes (Figures 4A–C). Moreover, reducing Mek genes to one allele led to reduction in hematocrit levels, hemoglobin concentration, and RBC numbers in both 1Mek1 and 1Mek2 mutants. Of note, the number of circulating white blood cells was similar to wt counts in all Mek mutants, suggesting that one allele of Mek is sufficient to sustain normal leukopoiesis (Figure 4D). Surprisingly, the spleen weight/body weight ratio was two and three times higher in 1Mek1 and 1Mek2 mutants, respectively, when compared to controls (Figure 4E).
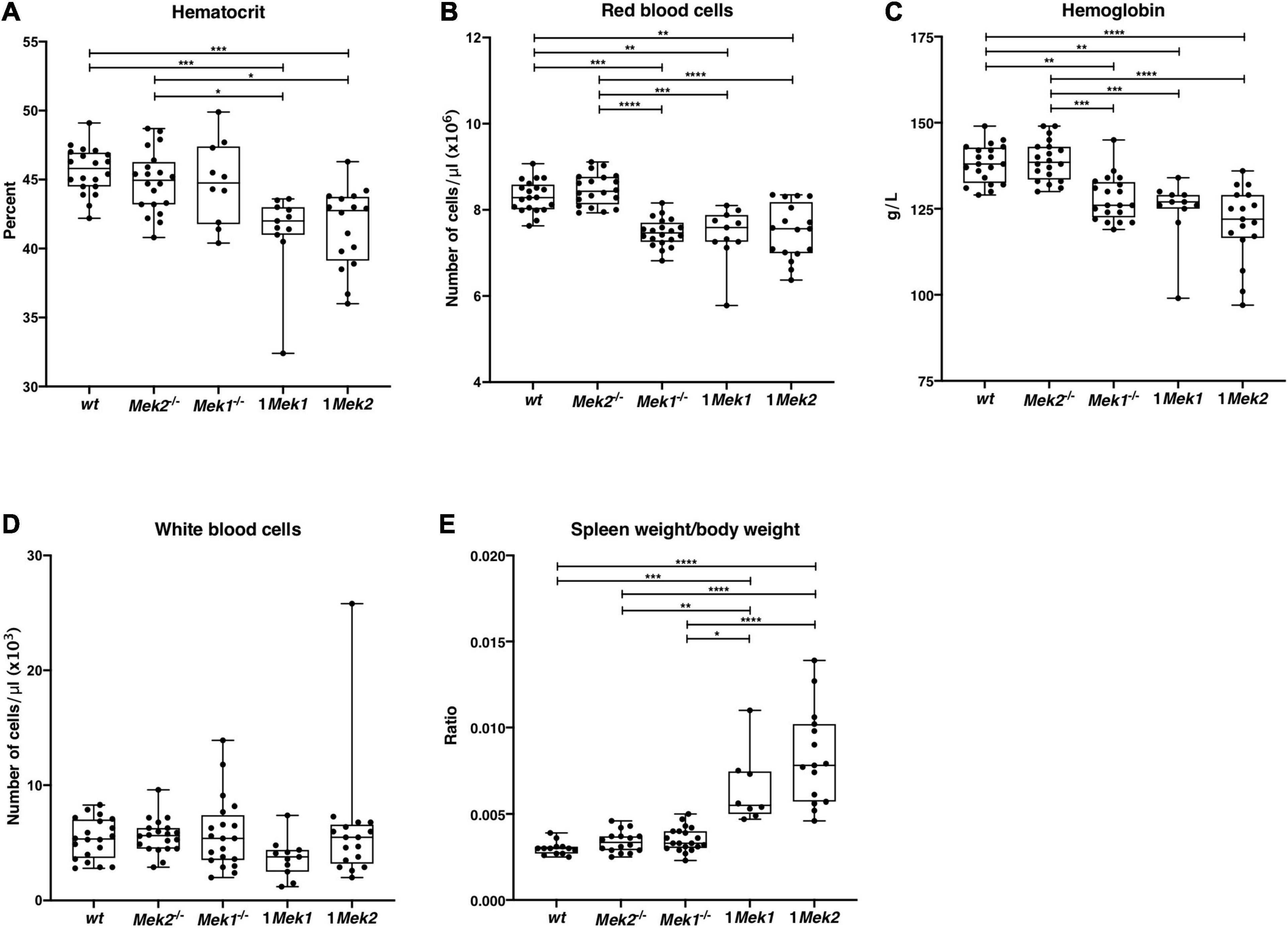
Figure 4. 1Mek1 and 1Mek2 mutants present a splenomegaly and a trend to anemia. Measurement of the hematologic parameters was performed on wt, Mek2–/–, Mek1–/–, 1Mek1, and 1Mek2 mutant mice at 2–4 months of age. Hematocrit (A), red blood cell count (B), hemoglobin concentration (C), and white blood cell count (D) are presented. (E) The spleen weight–to–body weight ratio was significantly reduced in 1Mek1 and 1Mek2 compared to controls, indicating splenomegaly. The results are presented as whiskers-and-boxes graph, and each dot represents an individual mouse. ANOVA with Kruskal–Wallis multiple-comparisons test was performed. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Normal Response to Erythropoietic Stress in 1Mek1 and 1Mek2 Mutants
Fewer circulating RBCs could be explained either by a defective generation or by a decrease in erythrocyte survival. Reduction in ERK signaling may impair the ability of erythropoietic progenitors to correctly respond to proliferative and differentiation signals. This was directly addressed in vivo by testing the ability of 1Mek1 and 1Mek2 mutants to recover from a hemolytic anemia induced by PHZ (Paulson et al., 2011). PHZ reacts with hemoglobin leading to its degradation, to the lysis of erythrocytes, and to a decreased hematocrit inducing an anemic condition. After injection of PHZ, hematocrit and reticulocyte counts were monitored over a period of 14 days. Hematocrit dropped sharply to around 25% 2 days after PHZ treatment in wt, 1Mek1, and 1Mek2 mice (Figure 5A). After this initial drop, hematocrit levels increased. 1Mek2 mutants showed an accelerated response at day 4, whereas 1Mek1mutants presented a delay in recovery. However, all genotypes returned to normal levels after 14 days. The proportion of peripheral blood reticulocytes was also evaluated by flow cytometry and used as an indicator of the erythropoietic proliferation response (Figure 5B). The basal reticulocyte count was less than 2.5% in wt, 1Mek1, and 1Mek2 animals. After PHZ treatment, erythropoiesis was induced, and reticulocyte counts increased for all genotypes to reach maximal levels at day 6. The reticulocyte counts were slightly higher in 1Mek2 mutants at day 6 and lower in 1Mek1 mutants between days 6 and 10. No significant difference was observed at day 14 between wt, 1Mek1, and 1Mek2 animals.
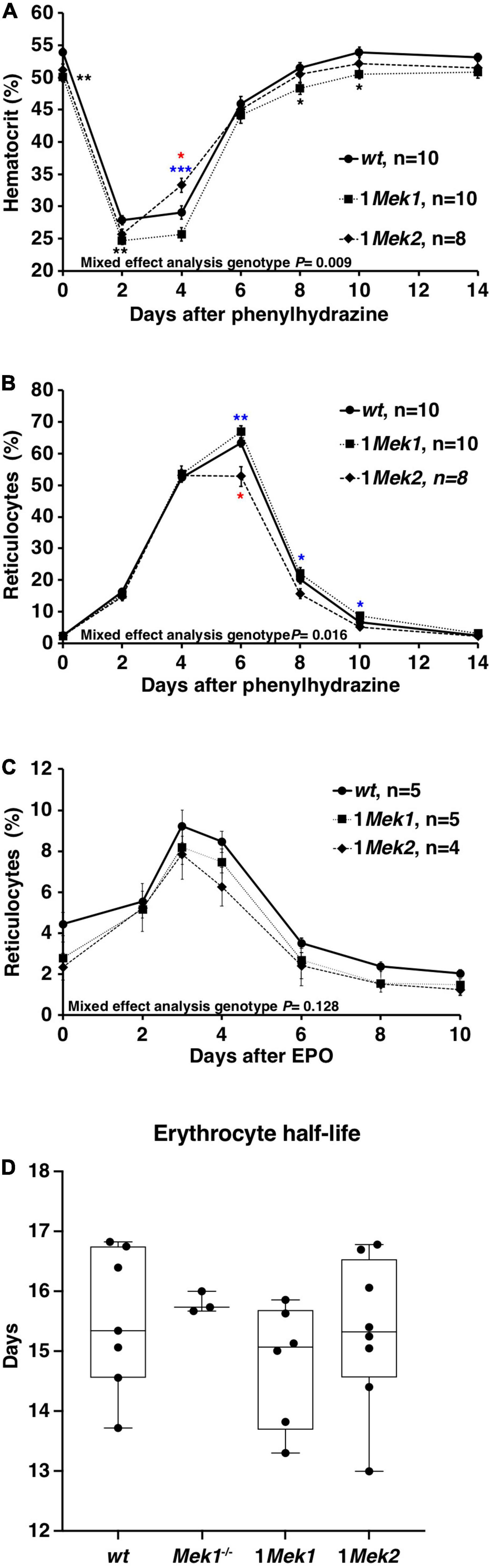
Figure 5. Normal erythropoiesis in 1Mek1 and 1Mek2. (A,B) wt, 1Mek1, and 1Mek2 mutant mice were injected with PHZ on days 0 and 1. Hematocrit (A) and reticulocyte count (B) were assessed every 2 days for 2 weeks. A normal response to this erythropoietic stress was observed in mutants. (C) The effect of a single subcutaneous injection of EPO on reticulocyte counts was monitored in controls and 1Mek1 and 1Mek2 mutants. Blood samples were collected every 2 days for reticulocyte count by flow cytometry. Response to EPO was unchanged in mutants. (D) Biotin-reactive agent was injected in tail vein to label RBCs and monitor their survival. The disappearance rate of biotinylated RBCs with time was used to calculate the RBC half-life. No statistical difference was observed between controls and 1Mek1 and 1Mek2 mutants. Boxplots depict data distribution; each dot represents one individual. Data are presented as the mean ± SEM (A–C). Mixed-effects analysis with Tukey multiple-comparisons test (A–C) and ANOVA (D) was performed. *P < 0.05, **P < 0.01, ***P < 0.001. Black: wt versus 1Mek1; red: wt versus 1Mek1; blue: 1Mek1 versus 1Mek2.
Another approach to test whether proliferation response during erythropoiesis was affected in 1Mek1 and 1Mek2 mutants was to treat mice with EPO and to assess if it causes an increase in reticulocyte cell counts. One subcutaneous injection of 50 U of EPO was performed, and reticulocyte counts were monitored by flow cytometry over a period of 10 days. wt, 1Mek1, and 1Mek2 mice showed a similar response indicating that the reaction to EPO was not significantly modified in 1Mek1 and 1Mek2 mutants versus wt animals (Figure 5C). Therefore, no major difference in the hematologic responses to PHZ and EPO was observed in 1Mek1 and 1Mek2 mutants that could explain the anemia observed in these mutants.
Alternatively, fewer circulating RBCs can be explained by a decrease in erythrocyte survival. To assess the half-life of circulating RBCs in 2- to 4-month-old 1Mek1 and 1Mek2 mutants, we measured the turnover of biotin-labeled RBCs by flow cytometry (Figure 5D). 1Mek1 and 1Mek2 RBCs showed a half-life similar to that of wt and Mek1–/– mutants indicating that RBC survival was normal.
Hematopoietic Stem and Progenitor Cells Are Altered in 1Mek1 Mutants
To verify whether the anemia in 1Mek1 and 1Mek2 resulted from a defect in a specific hematopoietic progenitor population, colony-forming assays in methylcellulose were performed with bone marrow and spleen from 10-week-old wt, Mek1–/–, 1Mek1, and 1Mek2 mice. While Mek1–/– and 1Mek2 mice behaved like wt controls in bone marrow, 1Mek1 mice displayed altered early hematopoiesis. Indeed, in 1Mek1 mutants, the numbers of multipotent CFU-GEMM and myeloid CFU-GM progenitors were significantly decreased (Figure 6A).
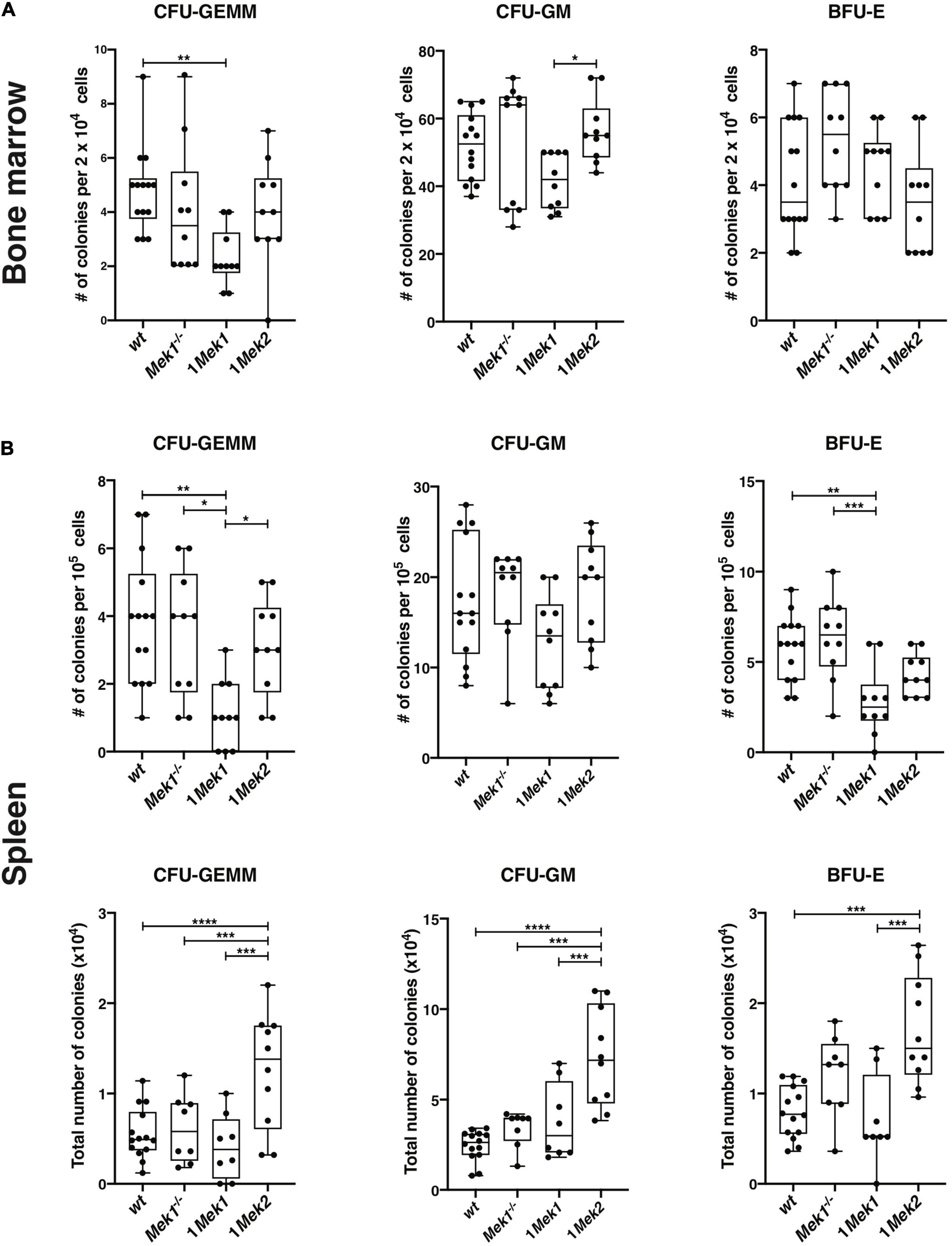
Figure 6. Counts of hematopoietic progenitors in bone marrow and spleen from 1Mek1 and 1Mek2 mutants. CFU-GEMM, CFU-GM, and BFU-E from wt and Mek mutant bone marrow (A) and spleen (B) were measured after 7 days in methylcellulose with cytokines. A decrease in the CFU-GEMM and CFU-GM populations was observed in bone marrow of 1Mek1 mutants, whereas an increase in the total number of progenitor populations was detected in spleen from 1Mek2 mutants. The results are presented as whiskers-and-boxes graph, and each dot represents an individual mouse. ANOVA with Kruskal–Wallis multiple-comparisons test was performed. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
HSCs generate mature leukocytes through successive differentiation steps of increasingly committed HSC progenitors (Freitas et al., 2017; Zhang et al., 2018; Singh and Cancelas, 2020). To determine whether impaired HSC maturation contributed to the 1Mek1 phenotype, global hematopoietic development was assessed in controls and 1Mek1 mice following the gating strategy described in Supplementary Figure 1 (Freitas et al., 2017). Flow cytometry analyses with the appropriate combination of cell markers were performed on bone marrow and spleen from 10-week-old mice to resolve each progenitor population (Figure 7A). Bone marrow from 1Mek1 mutants showed a two times reduction in the number of immature HSC between stages LT-HSC and LMPP when compared to wt specimens (Figures 7B–I). A trend for a decrease was also observed for the common lymphoid progenitor (CLP) population (Figure 7K), whereas no difference was measured for common myeloid progenitors (CMPs; Figure 7J), megakaryocyte-erythroid progenitors (MEPs; Figure 7L), and granulocyte–macrophage progenitors (GMPs; Figure 7M). Thus, in bone marrow from 1Mek1 mutants, multipotent progenitors were less abundant, but the number of committed progenitors recovered at later stages indicated that one functional Mek1 allele was sufficient to maintain hematopoietic progenitor homeostasis in accordance with the normal levels of mature myeloid cells observed (Figure 3).
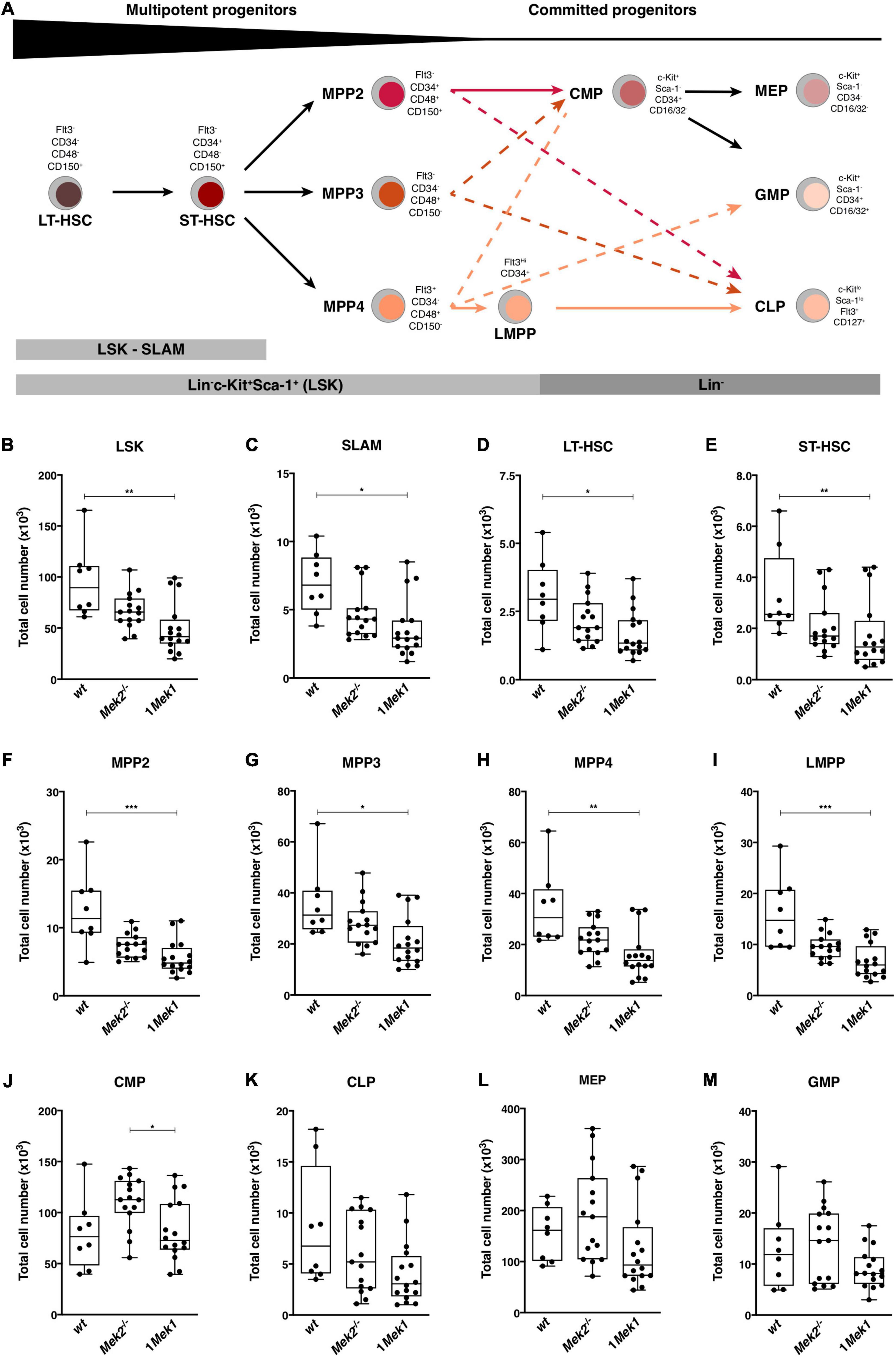
Figure 7. Reduced numbers of HSCs and multipotent HSC progenitors in bone marrow from 1Mek1 mutants. (A) Schematic representation of lineage determination in the mouse hematopoietic system with the flow cytometric gating strategy used to monitor the major classes of mouse hematopoietic stem and progenitor cells. (B–M) 1Mek1 mutant mice displayed a cell autonomous reduction in multipotent progenitors and lymphoid-biased progenitors in the bone marrow. Absolute numbers of LSK (B, Lin–c-Kit+Sca-1+), SLAM (C, Lin–c-Kit+Sca-1+CD48–CD150+), LT-HSC (D, Lin–c-Kit+Sca-1+CD48–CD150+Flt3–CD34–), ST-HSC (E, Lin–c-Kit+Sca-1+CD48–CD150–Flt3–CD34–), MPP2 (F, Lin–Sca-1+c-Kit+Flt3–CD34–CD48+CD150+), MPP3 (G, Lin–Sca-1+c-Kit+Flt3–CD34–CD48+ CD150–), MPP4 (H, Lin–Sca-1+c-Kit+Flt3+CD34–CD48+CD150–), LMPP (I, Lin–c-Kit+Sca-1+ Flt3highCD34+), CMP (J, Lin–c-Kit+Sca-1–CD34+CD16/32–), CLP (K, Lin–c-KitlowSca-1lowFlt3+CD127+), MEP (L, Lin–c-Kit+Sca-1+CD34–CD16/32–), and GMP (M, Lin–c-Kit+Sca-1–CD34+CD16/32+) in bone marrow of wt, Mek2–/–, and 1Mek1 mutants are presented. Data are from at least four independent experiments. The results are presented as whiskers-and-boxes graph, and each dot represents an individual mouse. ANOVA with Kruskal–Wallis multiple-comparisons test was performed. *P < 0.05, **P < 0.01, ***P < 0.001.
In the spleen, the proportions of CFU-GEMM and BFU-E progenitors were reduced in 1Mek1 mutants, but their total numbers in the organ were not significantly diminished compared to controls. Moreover, spleen from 1Mek1 mutants presented no significant change in HSC maturation when compared to wt and Mek2–/– controls (Supplementary Figure 2). In contrast, the total numbers of CFU-GM, CFU-GEMM, and BFU-E progenitors were significantly increased in 1Mek2 mutants, which might contribute to the expansion of the spleen in 1Mek2 mutants (Figure 6B).
To determine whether erythropoietic cell differentiation defects were associated with reduced Mek function, ProE, Ery.A, Ery.B, and Ery.C cell populations were assessed in bone marrow and spleen from wt, Mek2–/–, Mek1–/–, 1Mek1, and 1Mek2 adults. No significant difference was observed in bone marrow and spleen (Figure 8). However, Ery.A, Ery.B, and Ery.C cell populations showed a tendency to increase in 1Mek2 mutant spleens, suggesting that a distinct differentiation response may exist between 1Mek1 and 1Mek2 mutants (Figure 8B).
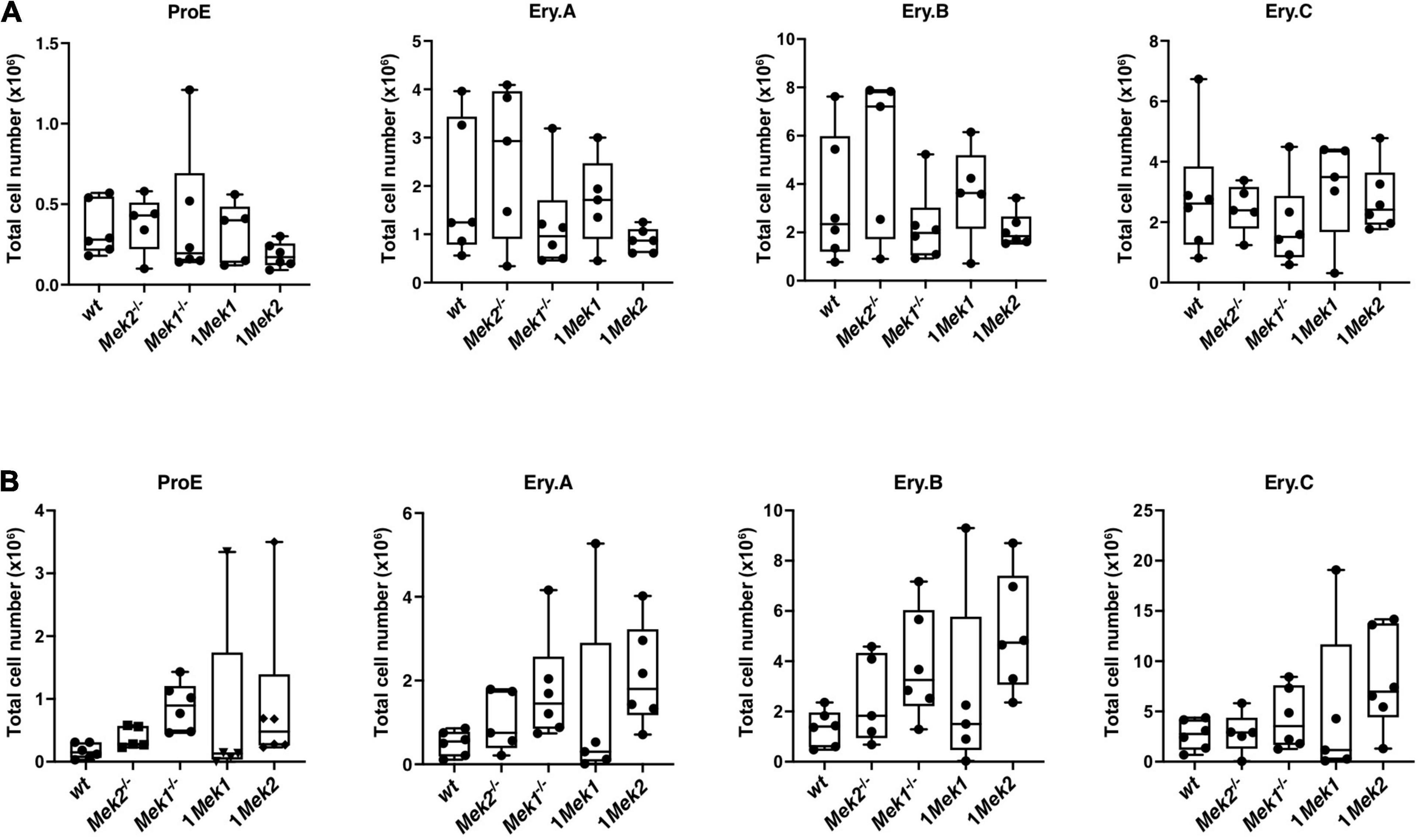
Figure 8. Erythroid subsets in bone marrow and spleen of adult Mek mutants. Erythroid subset analysis by flow cytometry on bone marrow (A) and spleen (B) from wt and Mek mutant adults. Frequency of Ter119medCD71highFSChigh (ProE), Ter119highCD71highFSChigh (Ery.A), Ter119highCD71highFSClow (Ery.B), and Ter119highCD71lowFSClow (Ery.C) are presented. The results are presented as whiskers-and-boxes graph, and each dot represents an individual mouse. ANOVA with Tukey multiple-comparisons test was performed. No significant difference was observed.
The impact of MEK dosage versus MEK isoform on the erythroid phenotype was assessed by quantifying the levels of MEK proteins by flow cytometry in CD71+ erythroid precursors using an antibody that cross-reacts with MEK1 and MEK2 (Figure 9A; Aoidi et al., 2016). Cell size was determined by the forward scatter: the larger cells correspond to ProE and Ery.A cell populations, whereas cells with intermediate and small sizes correspond to the more differentiated Ery.B cells. High levels of MEK proteins were detected in ProE and Ery.A erythroid precursors from wt specimens, whereas 5–10 times lower levels of MEK protein were measured in the more differentiated Ery.B erythroid populations (Figure 9B). Quantification of MEK protein levels in the allelic series of Mek mutants was performed for ProE and Ery.A erythroid precursors. Levels of MEK protein were maximal in wt specimens, and they gradually decreased in Mek2–/–, Mek1–/–, and 1Mek1, to finally be the lowest in 1Mek2 mutants (Figure 9C). The similar levels of MEK observed in Mek1–/– (two functional alleles of Mek2) and 1Mek1 (one functional allele of Mek1) specimens and the lowest MEK levels in 1Mek2 (one functional allele of Mek2) established that the Mek1 allele generates roughly twice the amount of MEK protein than the Mek2 allele. This suggests that MEK dosage rather than MEK isoform might be responsible for the different phenotypes.
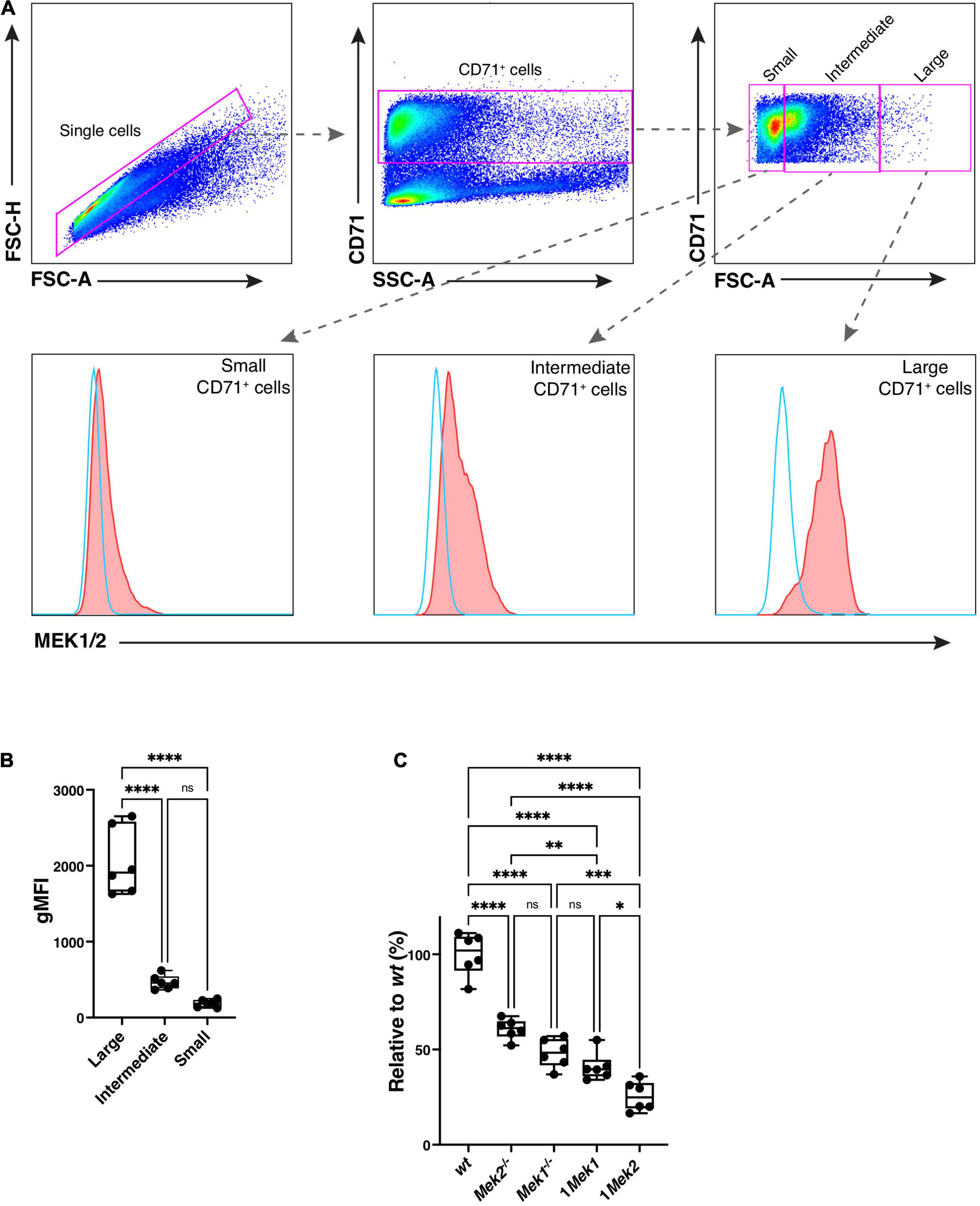
Figure 9. Loss of MEK Protein Expression During the Erythropoietic Cell Differentiation. (A) Representative dot plots show the flow cytometry gating strategy used to quantitate the levels of MEK1 and MEK2 proteins in CD71+ erythroid precursors isolated from bone marrow of wt and Mek mutants. MEK protein levels in wt specimens were measured in CD71+ cells of different sizes using the forward scatter. The larger cells correspond to the ProE and Ery.A cells, whereas the cells with the intermediate and small sizes correspond to more differentiated Ery.B cells. (B) The geometric mean of the fluorescence intensity (gMFI) for intracellular staining of MEK1/2 was used to quantify the levels of MEK protein. High levels of MEK protein were detected in ProE and Ery.A precursors (no antibody in blue and with anti-MEK1/2 in red). (C) Quantification of MEK protein levels in the allelic series of Mek mutants relative to the levels in wt was performed for the large CD71+ cells. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Discussion
Our previous characterization of the Mek1 Mek2 compound mutant mice has provided genetic evidence establishing the crucial and unique roles played by MEK1 and MEK2 in signal transduction during development. Using a Mek1 conditional allele and lineage-specific Cre mouse lines in a Mek2 null background, we showed that deletion of both Mek1 and Mek2 genes is necessary to generate phenotypes during gliogenesis, respiratory track development, skin formation, kidney branching, and male gonad ontogeny, revealing MEK functional redundancy in several embryonic processes (Scholl et al., 2007; Newbern et al., 2008; Yamashita et al., 2011; Li et al., 2012; Boucherat et al., 2014; Ihermann-Hella et al., 2014; Boucherat et al., 2017). During placenta formation, MEK1 and MEK2 can also substitute for each other, but a minimum threshold of MEK proteins is required for the proper development of extraembryonic structures and embryo survival (Nadeau and Charron, 2014; Aoidi et al., 2016). Here, we showed that the complete absence of MEK function in hematopoietic cell lineages in Mekhema null mutants causes neonatal death, confirming the essential role of the ERK pathway in hematopoiesis. Conversely, mice that retained one functional allele of either Mek1 (1Mek1) or Mek2 (1Mek2) gene in hematopoietic lineages can survive until adulthood. This establishes that MEK1 and MEK2 are partly interchangeable during early hematopoiesis as previously shown for other developmental processes.
Mekhema null mutants died shortly after birth from a severe anemia, even though Vav1-icre expression occurs much earlier, around E11.5, in definitive HSCs (dHSCs), which are known to be involved in the generation of the definitive erythroid, myeloid, and lymphoid lineages (de Boer et al., 2003; Medvinsky et al., 2011). In vertebrates, there are two main waves of hematopoiesis. First, the embryonic wave starts around E7.5 in the yolk sac and then migrates into AGM and placenta (Orkin and Zon, 2008). Embryonic hematopoiesis is responsible for the expansion of transient populations of nucleated primitive erythrocytes and some myeloid cells. Second, the definitive hematopoiesis starts at E10.5 in the fetal liver, which then becomes the major hematopoietic organ of the fetus (Bustelo et al., 1993; Ogilvy et al., 1999). Early production of RBCs is essential during development to sustain the rapid growth of the embryo after gastrulation. Our data indicated that embryonic hematopoiesis and few days of definitive hematopoiesis in Mekhema null mutants are sufficient to sustain the normal development of the embryo until birth. Inactivation of the two alleles of both Mek1 and Mek2 genes in dHSCs at E11.5 jeopardized the renewal of erythroid populations contributing to the anemia at birth. The anemia observed in Mekhema null newborns is most likely the cause of their death. However, the severe reduction in myeloid and lymphoid cell populations seen in the spleen of Mekhema null newborns raised the possibility that the HSC pools are affected and that a compromised immune system contributes to septicemia causing the death of Mekhema null mutants. For instance, it was reported that the mutation of the Spi1 gene, which encodes the PU.1 transcription factor specific to hematopoietic lineages, affects lymphoid development and causes septicemia and death at birth (Mckercher et al., 1996).
Mice carrying only one functional Mek allele in hematopoietic cell lineages survived after birth. Characterization of the hematologic parameters of 1Mek1 and 1Mek2 mutant mice revealed small but significant reductions of hematocrit, RBC numbers, and hemoglobin concentration, suggesting defective erythropoiesis. In bone marrow, 1Mek1 mutant mice presented altered early myelopoiesis, whereas 1Mek2 mutant mice behaved like control animals. In contrast, in the spleen, no significant differences in total hematopoietic progenitors CFU-GEMM, CFU-GM, and BFU-E were detected in 1Mek1 mutants, whereas 1Mek2 mutants showed a significant increase (Figure 6B). The 1Mek2 phenotype is evocative of the increased accumulation of erythroid progenitors in the spleen of Erk1–/– mutants (Guihard et al., 2010). In contrast to the augmented erythropoiesis in Erk1–/– mutants, 1Mek2 mutants showed a mild anemia. In fact, both 1Mek1 and 1Mek2 mutants presented similar levels of anemia even if 1Mek2 mice showed higher levels of hematopoietic progenitors and a stronger erythropoietic stress response compared to wt and 1Mek1 animals. However, 1Mek1 and 1Mek2 mutants showed normal erythroid precursor populations in bone marrow and spleen at late stages of erythroid differentiation (Figure 8). The exact cause of anemia in 1Mek1 and 1Mek2 mutants remains unresolved, but the return to normal levels of erythroid precursors suggests that the defect occurs at early stages of erythropoiesis with recovery later on.
We have previously shown that the Mek1 gene produces twice the amount of MEK protein and activity than Mek2. Analysis of Mek1 Mek2 allelic series has demonstrated that the severity of the placenta phenotype correlated with the amount of MEK protein and activity independently of the MEK isoform produced (Aoidi et al., 2016). Accordingly, 1Mek1 animals produce roughly twice the amount of MEK protein than 1Mek2 mice in early erythroid precursor cell (Figure 9C). A gradient of MEK protein from the highest level to the lowest in the wt, Mek2–/–, Mek1–/–, 1Mek1, and 1Mek2 was observed. This gradient of MEK protein should lead to a gradient of ERK activation allowing threshold levels of ERK signaling to be attained in 1Mek1 mice but not in 1Mek2 mutants. A minimal activity of the ERK pathway might be required to restrain the hematopoietic splenic progenitor expansion. Moreover, the reduction in MEK proteins at late stages of erythroid cell differentiation indicates that the ERK pathway acts mainly at earlier stages.
The reduction in the total numbers of HSC and CFU-GEMM and CFU-GM progenitors was specific to the bone marrow of 1Mek1 mutants. This phenotype is reminiscent of the HSC homing and maintenance defects previously described in Erk1–/– mice, which was attributed to osteopetrosis due to abnormal myeloid lineage progenitors and impaired osteoclastogenesis and osteoclast function (Saulnier et al., 2012). The impact of the 1Mek1 mutation on bone remodeling, HSC homing, and maintenance remains to be determined to establish if these factors can contribute to the decreased HSC populations in the bone marrow.
Finally, in 1Mek1 and 1Mek2 mutants, the erythrocyte half-life and the erythropoiesis response to hemolytic stress and to EPO remained unaffected, suggesting a relatively normal erythropoietic response despite a defective erythroid homeostasis.
In summary, the conditional deletion of Mek function in hematopoietic cell lineages unveiled the essential role of the ERK pathway in erythroid cell commitment and differentiation and the lack of ERK signaling causing a block at the last steps of erythroid cell differentiation. The characterization of our allelic series also revealed the specific contribution of each Mek gene in erythropoiesis during gestation despite a minimal impact on the process. The splenomegaly observed in mutants cannot be explained by the almost normal number of erythroid committed progenitors and precursors, which suggests that other hematopoietic cell lineages may also be affected in Mek mutants.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics Statement
The animal study was reviewed and approved by the Université Laval’s Animal Protection Committee (CPAUL) and C2EA-26, Animal Care and Use Committee, Villejuif, France.
Author Contributions
LB, S-PF-B, ME, and JC: conceptualization. LB, S-PF-B, VR, SR, and NH: investigation. JC: writing – original draft. LB, VR, NH, KB, ME, and JC: writing – review and editing. KB, ME, and JC: funding acquisition and resources. ME, KB, and JC: supervision. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by CIHR (MOP-97801 to JC), CRSNG (RGPIN-2016-05837 to JC), Concours Merck Sharpe and Dohme—Faculteì de Meìdecine, Université Laval (to JC), Fondation Arthritis (to ME), IDEX Université Paris-Saclay program “Jean d’Alembert” (to JC and ME), ANR (@RAction grant ANR-14-ACHN-0008 to ME), and ANR (PRC grant ANR-17-CE14-0019 to KB). VR was supported by a Ph.D. fellowship from the FRM and a 4th year Ph.D. fellowship from the Ligue Nationale Contre le Cancer.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank Lucie Jeannotte for critical comments and D. Dimitris Kioussis for the Vav1-icre mouse line. We thank M.-L. Aknin, Dr. H. Gary, B. Lecomte, and S. Mendez (IPSIT, technical facilities PLAIMMO and AnimEx, Clamart) and L. Poncet for their technical assistance.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2021.639022/full#supplementary-material
Supplementary Figure 1 | Flow cytometry gating strategy for delineating the major classes of mouse hematopoietic stem and progenitor cells. Representative dot plots show the flow cytometry gating strategy used for LSK (Lin–c-Kit+Sca-1+), SLAM (LSK CD48–CD150+), LT-HSC (LSK CD48–CD150+Flt3–CD34–), ST-HSC (LSK CD48–CD150+Flt3–CD34+), MPP2 (LSK Flt3–CD48+CD150+), MPP3 (LSK Flt3–CD48+CD150–), MPP4 (LSK Flt3+CD48+/–CD150–), LMPP (LSK Flt3highCD34+), CMP (LSK CD34+CD16/32–), CLP (Lin–c-KitlowSca-1lowFlt3+CD127+), MEP (Lin–c-Kit+Sca-1–CD34–CD16/32–), and GMP (Lin–c-Kit+Sca-1–CD34+CD16/32+) in the mouse bone marrow (BM).
Supplementary Figure 2 | Numbers of HSCs and multipotent HSC progenitors in spleen from 1Mek1 mutants. (A–L) Absolute numbers of LSK (A, Lin–c-Kit+Sca-1+), SLAM (B, Lin–c-Kit+Sca-1+CD48–CD150+), LT-HSC (C, Lin–c-Kit+Sca-1+CD48–CD150+Flt3–CD34–), ST-HSC (D, Lin–c-Kit+Sca-1+CD48–CD150–Flt3–CD34–), MPP2 (E, Lin–Sca-1+c-Kit+Flt3–CD34–CD48+CD150+), MPP3 (F, Lin–Sca-1+c-Kit+Flt3–CD34–CD48+ CD150–), MPP4 (G, Lin–Sca-1+c-Kit+Flt3+CD34–CD48+CD150–), LMPP (H, Lin–c-Kit+Sca-1+ Flt3highCD34+), CMP (I, Lin–c-Kit+Sca-1–CD34+CD16/32–), CLP (J, Lin–c-KitlowSca-1lowFlt3+CD127+), MEP (K, Lin–c-Kit+Sca-1+CD34–CD16/32–), and GMP (L, Lin–c-Kit+Sca-1–CD34+CD16/32+) in spleen of wt, Mek2–/–, and 1Mek1 mutants are presented. Data are from at least four independent experiments. The results are presented as whiskers-and-boxes graph, and each dot represents an individual mouse. ANOVA with Kruskal–Wallis multiple-comparisons test was performed. ∗P < 0.05.
References
Alberola-Ila, J., and Hernandez-Hoyos, G. (2003). The Ras/MAPK cascade and the control of positive selection. Immunol. Rev. 191, 79–96. doi: 10.1034/j.1600-065x.2003.00012.x
Aoidi, R., Maltais, A., and Charron, J. (2016). Functional redundancy of the kinases MEK1 and MEK2: rescue of the Mek1 mutant phenotype by Mek2 knock-in reveals a protein threshold effect. Sci. Signal 9:ra9. doi: 10.1126/scisignal.aad5658
Bélanger, L.-F., Roy, S., Tremblay, M., Brott, B., Steff, A.-M., Mourad, W., et al. (2003). Mek2 is dispensable for mouse growth and development. Mol. Cell Biol. 23, 4778–4787. doi: 10.1128/mcb.23.14.4778-4787.2003
Bissonauth, V., Roy, S., Gravel, M., Guillemette, S., and Charron, J. (2006). Requirement for Map2k1 (Mek1) in extra-embryonic ectoderm during placentogenesis. Development 133, 3429–3440. doi: 10.1242/dev.02526
Boucherat, O., Landry-Truchon, K., Aoidi, R., Houde, N., Nadeau, V., Charron, J., et al. (2017). Lung development requires an active ERK/MAPK pathway in the lung mesenchyme. Dev. Dyn. 246, 72–82. doi: 10.1002/dvdy.24464
Boucherat, O., Nadeau, V., Berube-Simard, F. A., Charron, J., and Jeannotte, L. (2014). Crucial requirement of ERK/MAPK signaling in respiratory tract development. Development 141, 3197–3211. doi: 10.1242/dev.110254
Bustelo, X. R., Rubin, S. D., Suen, K. L., Carrasco, D., and Barbacid, M. (1993). Developmental expression of the vav protooncogene. Cell Growth Differ. 4, 297–308.
Caunt, C. J., Sale, M. J., Smith, P. D., and Cook, S. J. (2015). MEK1 and MEK2 inhibitors and cancer therapy: the long and winding road. Nat. Rev. Cancer 15, 577–592. doi: 10.1038/nrc4000
de Boer, J., Williams, A., Skavdis, G., Harker, N., Coles, M., Tolaini, M., et al. (2003). Transgenic mice with hematopoietic and lymphoid specific expression of Cre. Eur. J. Immunol. 33, 314–325. doi: 10.1002/immu.200310005
Espeli, M., Bokers, S., Giannico, G., Dickinson, H. A., Bardsley, V., Fogo, A. B., et al. (2011). Local renal autoantibody production in lupus nephritis. J. Am. Soc. Nephrol. 22, 296–305. doi: 10.1681/asn.2010050515
Fischer, A. M., Katayama, C. D., Pages, G., Pouyssegur, J., and Hedrick, S. M. (2005). The role of erk1 and erk2 in multiple stages of T cell development. Immunity 23, 431–443. doi: 10.1016/j.immuni.2005.08.013
Freitas, C., Wittner, M., Nguyen, J., Rondeau, V., Biajoux, V., Aknin, M. L., et al. (2017). Lymphoid differentiation of hematopoietic stem cells requires efficient Cxcr4 desensitization. J. Exp. Med. 214, 2023–2040. doi: 10.1084/jem.20160806
Geest, C. R., Buitenhuis, M., Groot Koerkamp, M. J., Holstege, F. C., Vellenga, E., and Coffer, P. J. (2009). Tight control of MEK-ERK activation is essential in regulating proliferation, survival, and cytokine production of CD34+-derived neutrophil progenitors. Blood 114, 3402–3412. doi: 10.1182/blood-2008-08-175141
Geest, C. R., and Coffer, P. J. (2009). MAPK signaling pathways in the regulation of hematopoiesis. J. Leukoc. Biol. 86, 237–250. doi: 10.1189/jlb.0209097
Gifford, S. C., Yoshida, T., Shevkoplyas, S. S., and Bitensky, M. W. (2006). A high-resolution, double-labeling method for the study of in vivo red blood cell aging. Transfusion 46, 578–588. doi: 10.1111/j.1537-2995.2006.00776.x
Guihard, S., Clay, D., Cocault, L., Saulnier, N., Opolon, P., Souyri, M., et al. (2010). The MAPK ERK1 is a negative regulator of the adult steady-state splenic erythropoiesis. Blood 115, 3686–3694. doi: 10.1182/blood-2009-09-242487
Hayashi, S., Lewis, P., Pevny, L., and Mcmahon, A. P. (2002). Efficient gene modulation in mouse epiblast using a Sox2Cre transgenic mouse strain. Gene Expr. Patterns 2, 93–97. doi: 10.1016/s0925-4773(02)00292-7
Hsu, C. L., Kikuchi, K., and Kondo, M. (2007). Activation of mitogen-activated protein kinase kinase (MEK)/extracellular signal regulated kinase (ERK) signaling pathway is involved in myeloid lineage commitment. Blood 110, 1420–1428. doi: 10.1182/blood-2007-02-071761
Ihermann-Hella, A., Lume, M., Miinalainen, I. J., Pirttiniemi, A., Gui, Y., Peranen, J., et al. (2014). Mitogen-activated protein kinase (MAPK) pathway regulates branching by remodeling epithelial cell adhesion. PLoS Genet. 10:e1004193. doi: 10.1371/journal.pgen.1004193
Kamata, T., Kang, J., Lee, T. H., Wojnowski, L., Pritchard, C. A., and Leavitt, A. D. (2005). A critical function for B-Raf at multiple stages of myelopoiesis. Blood 106, 833–840. doi: 10.1182/blood-2004-11-4458
Kollmann, K., Warsch, W., Gonzalez-Arias, C., Nice, F. L., Avezov, E., Milburn, J., et al. (2017). A novel signalling screen demonstrates that CALR mutations activate essential MAPK signalling and facilitate megakaryocyte differentiation. Leukemia 31, 934–944. doi: 10.1038/leu.2016.280
Koulnis, M., Pop, R., Porpiglia, E., Shearstone, J. R., Hidalgo, D., and Socolovsky, M. (2011). Identification and analysis of mouse erythroid progenitors using the CD71/TER119 Flow-cytometric assay. J. Vis. Exp. 5:e2809.
Li, X., Newbern, J. M., Wu, Y., Morgan-Smith, M., Zhong, J., Charron, J., et al. (2012). MEK Is a key regulator of gliogenesis in the developing brain. Neuron 75, 1035–1050. doi: 10.1016/j.neuron.2012.08.031
Malynn, B. A., Demengeot, J., Stewart, V., Charron, J., and Alt, F. W. (1995). Generation of normal lymphocytes derived from N-myc-deficient embryonic stem cells. Int. Immunol. 7, 1637–1647. doi: 10.1093/intimm/7.10.1637
Matsuzaki, T., Aisaki, K., Yamamura, Y., Noda, M., and Ikawa, Y. (2000). Induction of erythroid differentiation by inhibition of Ras/ERK pathway in a friend murine leukemia cell line. Oncogene 19, 1500–1508. doi: 10.1038/sj.onc.1203461
Mckercher, S. R., Torbett, B. E., Anderson, K. L., Henkel, G. W., Vestal, D. J., Baribault, H., et al. (1996). Targeted disruption of the PU.1 gene results in multiple hematopoietic abnormalities. EMBO J. 15, 5647–5658. doi: 10.1002/j.1460-2075.1996.tb00949.x
Medvinsky, A., Rybtsov, S., and Taoudi, S. (2011). Embryonic origin of the adult hematopoietic system: advances and questions. Development 138, 1017–1031. doi: 10.1242/dev.040998
Mori, M., Uchida, M., Watanabe, T., Kirito, K., Hatake, K., Ozawa, K., et al. (2003). Activation of extracellular signal-regulated kinases ERK1 and ERK2 induces Bcl-xL up-regulation via inhibition of caspase activities in erythropoietin signaling. J. Cell Physiol. 195, 290–297. doi: 10.1002/jcp.10245
Nadeau, V., and Charron, J. (2014). Essential role of the ERK/MAPK pathway in blood-placental barrier formation. Development 141, 2825–2837. doi: 10.1242/dev.107409
Nadeau, V., Guillemette, S., Bélanger, L. F., Jacob, O., Roy, S., and Charron, J. (2009). Map2k1 and Map2k2 genes contribute to the normal development of syncytiotrophoblasts during placentation. Development 136, 1363–1374. doi: 10.1242/dev.031872
Newbern, J., Zhong, J., Wickramasinghe, R. S., Li, X., Wu, Y., Samuels, I., et al. (2008). Mouse and human phenotypes indicate a critical conserved role for ERK2 signaling in neural crest development. Proc. Natl. Acad. Sci. U.S.A. 105, 17115–17120. doi: 10.1073/pnas.0805239105
Ogilvy, S., Metcalf, D., Gibson, L., Bath, M. L., Harris, A. W., and Adams, J. M. (1999). Promoter elements of vav drive transgene expression in vivo throughout the hematopoietic compartment. Blood 94, 1855–1863. doi: 10.1182/blood.v94.6.1855
Orkin, S. H., and Zon, L. I. (2008). Hematopoiesis: an evolving paradigm for stem cell biology. Cell 132, 631–644. doi: 10.1016/j.cell.2008.01.025
Paulson, R. F., Shi, L., and Wu, D. C. (2011). Stress erythropoiesis: new signals and new stress progenitor cells. Curr. Opin. Hematol. 18, 139–145. doi: 10.1097/moh.0b013e32834521c8
Roskoski, R. Jr. (2012a). ERK1/2 MAP kinases: structure, function, and regulation. Pharmacol. Res. 66, 105–143. doi: 10.1016/j.phrs.2012.04.005
Roskoski, R. Jr. (2012b). MEK1/2 dual-specificity protein kinases: structure and regulation. Biochem. Biophys. Res. Commun. 417, 5–10. doi: 10.1016/j.bbrc.2011.11.145
Saulnier, N., Guihard, S., Holy, X., Decembre, E., Jurdic, P., Clay, D., et al. (2012). ERK1 regulates the hematopoietic stem cell niches. PLoS One 7:e30788.
Scholl, F. A., Dumesic, P. A., Barragan, D. I., Harada, K., Bissonauth, V., Charron, J., et al. (2007). Mek1/2 MAPK kinases are essential for Mammalian development, homeostasis, and raf-induced hyperplasia. Dev. Cell 12, 615–629. doi: 10.1016/j.devcel.2007.03.009
Shaul, Y. D., and Seger, R. (2007). The MEK/ERK cascade: from signaling specificity to diverse functions. Biochim. Biophys. Acta 1773, 1213–1226. doi: 10.1016/j.bbamcr.2006.10.005
Shim, J. H., Greenblatt, M. B., Zou, W., Huang, Z., Wein, M. N., Brady, N., et al. (2013). Schnurri-3 regulates ERK downstream of WNT signaling in osteoblasts. J. Clin. Invest. 123, 4010–4022. doi: 10.1172/jci69443
Siekmeier, R., Bierlich, A., and Jaross, W. (2000). Determination of reticulocytes: three methods compared. Clin. Chem. Lab. Med. 38, 245–249.
Singh, A. K., and Cancelas, J. A. (2020). gap junctions in the bone marrow lympho-hematopoietic stem cell niche, leukemia progression, and chemoresistance. Int. J. Mol. Sci. 21:796. doi: 10.3390/ijms21030796
Sun, Y., Liu, W. Z., Liu, T., Feng, X., Yang, N., and Zhou, H. F. (2015). Signaling pathway of MAPK/ERK in cell proliferation, differentiation, migration, senescence and apoptosis. J. Recept. Signal. Transduct. Res. 35, 600–604. doi: 10.3109/10799893.2015.1030412
Uchida, M., Kirito, K., Shimizu, R., Miura, Y., Ozawa, K., and Komatsu, N. (2001). A functional role of mitogen-activated protein kinases, erk1 and erk2, in the differentiation of a human leukemia cell line, UT-7/GM: a possible key factor for cell fate determination toward erythroid and megakaryocytic lineages. Int. J. Hematol. 73, 78–83. doi: 10.1007/bf02981906
Wortzel, I., and Seger, R. (2011). The ERK cascade: distinct functions within various subcellular organelles. Genes Cancer 2, 195–209. doi: 10.1177/1947601911407328
Yamashita, S., Tai, P., Charron, J., Ko, C., and Ascoli, M. (2011). The Leydig cell MEK/ERK pathway is critical for maintaining a functional population of adult Leydig cells and for fertility. Mol. Endocrinol. 25, 1211–1222. doi: 10.1210/me.2011-0059
Zhang, Y., Gao, S., Xia, J., and Liu, F. (2018). Hematopoietic hierarchy - an updated roadmap. Trends Cell Biol. 28, 976–986. doi: 10.1016/j.tcb.2018.06.001
Keywords: ERK/MAP kinase pathway, Mek genes, hematopoiesis, erythropoiesis, gene inactivation
Citation: Beuret L, Fortier-Beaulieu S-P, Rondeau V, Roy S, Houde N, Balabanian K, Espéli M and Charron J (2021) Mek1 and Mek2 Functional Redundancy in Erythropoiesis. Front. Cell Dev. Biol. 9:639022. doi: 10.3389/fcell.2021.639022
Received: 08 December 2020; Accepted: 21 June 2021;
Published: 27 July 2021.
Edited by:
Serge Roche, UMR 5237 Centre de Recherche en Biologie Cellulaire de Montpellier (CRBM), FranceReviewed by:
Philippe Lenormand, INSERM U1081 Institut de Recherche sur le Cancer et le Vieillissement, FranceIsabelle Plo, Inserm U1287, France
Copyright © 2021 Beuret, Fortier-Beaulieu, Rondeau, Roy, Houde, Balabanian, Espéli and Charron. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jean Charron, amVhbi5jaGFycm9uQGNyY2h1ZGVxdWViZWMudWxhdmFsLmNh