 Lars Custers
Lars Custers Irene Paassen
Irene Paassen Jarno Drost
Jarno Drost- 1Princess Máxima Center for Pediatric Oncology, Utrecht, Netherlands
- 2Oncode Institute, Utrecht, Netherlands
A subset of pediatric tumors affects very young children and are thought to arise during fetal life. A common theme is that these embryonal tumors hijack developmental programs, causing a block in differentiation and, as a consequence, unrestricted proliferation. Embryonal tumors, therefore typically maintain an embryonic gene signature not found in their differentiated progeny. Still, the processes underpinning malignant transformation remain largely unknown, which is hampering therapeutic innovation. To gain more insight into these processes, in vitro and in vivo research models are indispensable. However, embryonic development is an extremely dynamic process with continuously changing cellular identities, making it challenging to define cells-of-origin. This is crucial for the development of representative models, as targeting the wrong cell or targeting a cell within an incorrect developmental time window can result in completely different phenotypes. Recent innovations in in vitro cell models may provide more versatile platforms to study embryonal tumors in a scalable manner. In this review, we outline different in vitro models that can be explored to study embryonal tumorigenesis and for therapy development.
Introduction
Cancer is the leading disease-related cause of death in children (Siegel et al., 2016; Cunningham et al., 2018). A significant subset of pediatric tumors occurs in early childhood, suggestive of an origin in prenatal life (Marshall et al., 2014). These so-called embryonal tumors are thought to develop as a consequence of aberrant development. However, for many embryonal tumors the processes driving tumorigenesis remain unknown. Whereas, adult cancers develop by a progressive accumulation of mutations over many years (Stratton et al., 2009), embryonal tumors are typically characterized by a relatively low mutational burden and only a few genetic events to drive tumorigenesis (Vogelstein et al., 2013; Gröbner et al., 2018; Rahal et al., 2018; Kattner et al., 2019). The few genetic alterations that do occur likely cause fetal cells to maintain a progenitor-like state and prohibit differentiation. This maturation block has been suggested to prime cells for malignant transformation (Chen et al., 2015; Puisieux et al., 2018; Rahal et al., 2018; Jessa et al., 2019). To better understand the processes underpinning embryonal tumorigenesis, a direct comparison between normal and tumor development is key. Gene expression profiling of fetal tissues with single cell resolution has provided more insights into the developmental trajectories driving embryogenesis. Comparison of such profiles with tumor gene expression signatures have defined the cellular identity of several embryonal tumors, possibly pointing to their cellular origin (Boeva et al., 2017; Young et al., 2018, 2020; Hovestadt et al., 2019; Jessa et al., 2019; Vladoiu et al., 2019). Yet, in many cases these studies are merely correlative and lack subsequent functional validation. To do so, representative in vitro and in vivo preclinical models are crucial.
Genetically engineered mouse models (GEMMs) have been the golden standard for finding the cellular origin of cancers, by introducing tumor driver events in putative tumor-initiating cells (Visvader, 2011; Marshall et al., 2014). Although GEMMs have provided important insights into tumorigenesis, several drawbacks limit their potential as a representative model of embryonal tumors. Embryonic development is an extremely dynamic process with continuously changing cellular identities, which makes it very challenging to target the right cell at the right time. For instance, homozygous loss of the Wilms tumor driver gene Wt1 was shown to be embryonically lethal in mice (Kreidberg et al., 1993), whereas a specific Wt1 ablation at E11.5 in a small fraction of nephron progenitor cells resulted in Wilms tumor formation (Hu et al., 2011; Berry et al., 2015; Huang et al., 2016). Moreover, GEMM generation is time consuming and mouse development does not fully recapitulate human embryogenesis (Navin et al., 2010, 2011; Blakeley et al., 2015; Theunissen and Jaenisch, 2017). The development of new in vitro cell models increasingly recapitulating the complexity of organogenesis will open new avenues for the development of novel, relevant embryonal tumor models. In this review, we discuss the currently available in vitro models to study embryonal tumorigenesis as well as the discovery of new therapeutic strategies.
Cell Lines of Fetal Origin
A broad range of cell lines has been established over the last decades. Cell lines are easy to maintain and typically do not consume many resources, which allows for fast and parallel modeling of multiple tumor driver events. This is particularly useful to interrogate the complex genetics underlying heterogeneous tumor phenotypes. One such tumor is neuroblastoma, which is characterized by a variety of driver events, including MYCN amplification and ALK mutations (Johnsen et al., 2019). To study neuroblastoma initiation, models of its embryonic origin, neural crest (Johnsen et al., 2019), are required. In vitro murine neural crest models can be generated by extraction of neural tubes from mouse embryos, which are subsequently placed in a culture dish to initiate the migration of neural crest cells onto the plate (Maurer et al., 2007; Olsen et al., 2017). The neural crest cells lose their multipotency over time in vitro (6–10 cell divisions) (Stemple and Anderson, 1992) and are, therefore only suitable for short-term experiments. However, multipotency can be maintained by exogenous c-Myc expression. Accordingly, Maurer et al. (2007) generated the JoMa1 neural crest cell line, which was established from mouse embryos carrying the inducible c-MycER transgene, enabling tamoxifen-inducible c-Myc expression and maintenance of multipotency. In both the JoMa1 cell line (Schulte et al., 2013) and non-genetically modified neural crest cells (Olsen et al., 2017), overexpression of MycN was proven sufficient to generate neuroblastoma upon transplantation in immune-deficient mice. Other murine neural crest-derived neuroblastoma models accommodate oncogenic variants of Alk or Phox2b, which was shown to impair neural crest development and inhibit sympathoadrenal differentiation processes (Reiff et al., 2010; Schulte et al., 2013; Montavon et al., 2014). However, murine neural crest development has been shown to be different from human in many aspects (O’Rahilly and Müller, 2007; Betters et al., 2010). Cohen et al. (2020), therefore developed a mouse-human chimera to study neuroblastoma formation in a human setting. Human iPSC-derived neural crest cells were injected in utero into gastrulating mouse embryos to form a human neural crest lineage in mice. For neuroblastoma modeling, the neural crest cells were subsequently genetically engineered with inducible expression constructs of MYCN and an oncogenic variant of ALK. Upon induction, mice developed tumors characteristic of patient neuroblastoma, and tumor transcriptomes resembled neuroblastoma patients more closely than GEMMs. Interestingly, injections subcutaneously lead to tumor formation but without expression of neuroblastoma markers (Cohen et al., 2020). These findings suggest that human neural crest cells serve as a more representative model than mouse, but only when generated in the appropriate developmental context and orthotopic environment.
Another embryonal tumor entity where differences between human and mouse models of tumorigenesis were observed is retinoblastoma. The common driver event of retinoblastoma is loss of RB1 during retinal development (Dimaras et al., 2015). Retinoblastoma modeling using GEMMs has proven challenging, as engineering of Rb1-deficient mice resulted in embryonic lethality (Lee et al., 1992; Wikenheiser-Brokamp, 2006) and retina-specific depletion of Rb1 was required. However, in contrast to human, mouse retinal cells were proven insensitive to Rb1 depletion and required additional knock-outs of tumor suppressors p107 or p130 for retinoblastoma development (Robanus-Maandag et al., 1998; Dannenberg et al., 2004; MacPherson et al., 2004). To generate human models of retinal development, Xu et al. (2014) isolated human fetal retinal cells post-fertilization retaining all retinal precursor cell types (RPCs) at distinct maturation states. Depletion of RB1 within the different RPCs indicated post-mitotic cone-precursors to be most prone to develop into retinoblastoma, based on its ability to form tumors with expression of retinoblastoma markers upon xenografting in mice (Xu et al., 2014). Furthermore, RB1 loss in matured retinal cells did not induce retinoblastoma, validating that tumor initiation is restricted to a specific cell within retinal development.
Overall, in vitro modeling of retinoblastoma and neuroblastoma in human and mouse fetal cell cultures uncovered that fundamental differences between mice and human development can impede representative modeling of embryonal tumors.
Pluripotent Stem Cell-Derived Cell Lines
Classical cell lines are typically composed of a single type of progenitor-like cell representing a specific germ layer (i.e., endoderm, ectoderm, mesoderm, neural crest). Culture models still capable of generating the different germ layers give the opportunity to model embryonal tumors of which it is not yet clear from which lineage they arise, or which seem to arise across the boundaries of the different germ layers. Current in vitro models capable of recapitulating these different developmental trajectories include pluripotent stem cells (PSCs) such as embryonic stem cells (ESCs) and induced PSCs (iPSCs), which can self-renew and be subjected to differentiation protocols that enforce all germ layers (Liu G. et al., 2020). PSCs can be stably maintained in culture and are permissive for genetic manipulation (Liu G. et al., 2020). With the development of effective differentiation protocols, PSCs can mirror embryonic development and therefore serve as a valuable model to study tumorigenesis. iPSCs are generated through the forced dedifferentiation of somatic cells, which thereby regain pluripotency. The molecular mechanisms that underly this reprogramming show significant similarities with the processes driving a subset of the embryonal germ cell tumors (GCTs) (Oosterhuis and Looijenga, 2019), including yolk sac tumors, embryonal carcinomas, and teratomas. GCTs encompass a diverse group of cancer entities that arise from cells of the early embryo or germ line (Oosterhuis and Looijenga, 2019). Interestingly, somatic mutations play a minor role as drivers of GCT development. Tumors are thought to arise by epigenetic deregulation of the cell-of-origin or aberrant stem cell niche factors (Oosterhuis and Looijenga, 2019). The developmental potency of the cell-of-origin can be reprogrammed through increased expression of well-known pluripotency factors, such as NANOG and OCT4 (De Jong and Looijenga, 2006; Thomas et al., 2011). Xenograft studies have shown that iPSCs and ESCs are intrinsically tumorigenic (Ben-David and Benvenisty, 2011). Upon xenografting, iPSCs develop into a benign GCT referred to as teratoma or in some cases more malignant GCTs, dependent on the reprogramming method applied (Lee et al., 2013). These findings indicate that maintaining an early embryonic cellular context is, by itself, sufficient for tumor initiation. Although PSC tumorigenicity is a limitation for its potential application in regenerative medicine, iPSCs and ESCs can on the other hand serve as in vitro models of GCTs.
A major class of genes mutated in childhood as well as adult cancers are subunits of the SWItch/Sucrose Non-Fermentable (SWI/SNF) chromatin remodeling complex (Wilson and Roberts, 2011; Shain and Pollack, 2013). The role of this complex in embryonal tumors is clearly exemplified in malignant rhabdoid tumors (MRT), which are characterized by the complete loss of SWI/SNF subunit SMARCB1 (95% of cases) or SMARCA4 (5% of cases) (Lee et al., 2012; Hasselblatt et al., 2014). To study MRT initiation, SMARCB1 was knocked down in hESCs using RNA interference (Langer et al., 2019). The differentiation capacity of hESCs was subsequently assessed, demonstrating that SMARCB1 inhibition specifically repressed neural induction, whereas mesodermal and endodermal lineage induction was not affected (Langer et al., 2019). In culture conditions inducing neural differentiation, SMARCB1 was shown to be essential for increased chromatin accessibility at neural differentiation genes and silencing of pluripotency-related super-enhancers (Wang et al., 2017; Langer et al., 2019). Furthermore, SMARCB1-null iPSCs that were transplanted into mice were able to generate MRT (Terada et al., 2019). Interestingly, iPSCs that had further progressed to neural progenitor cells (NPCs) generated tumors without rhabdoid features. These results show a lineage-specific role for SMARCB1 in vitro, validating recently developed MRT GEMMs wherein Smarcb1 loss-induced rhabdoid tumor development was demonstrated to be limited to a specific developmental time and lineage (Han et al., 2016; Vitte et al., 2017).
A different layer of epigenetic regulation affected in embryonal tumors is the post-translational modification of histone tails, which enables a rapid switch between active or repressive histone marks to dynamically regulate gene expression during development. Mutations in histones are specifically characterized in a subset of pediatric gliomas. In diffuse intrinsic pontine glioma (DIPG), nearly 80% of cases have a missense mutation in the histone 3.3 gene (H3F3A), causing a substitution of methionine for lysine 27 (H3K27M) (Khuong-Quang et al., 2012; Schwartzentruber et al., 2012; Wu et al., 2012). The origin of DIPG was indicated to lie in early neural development (Filbin et al., 2018; Sun et al., 2019), presumably making NPCs derived from ESCs a suitable model for tumor initiation. In line with this, overexpression of the H3K27M mutant in NPCs resulted in increased proliferation (Funato et al., 2014). Interestingly, introduction of the mutation was ineffective in uninduced ESCs or mature astrocytes. For a majority of DIPG cases, H3K27M mutations are typically co-occurring with amplification of PDGFRA and loss of TP53 (Khuong-Quang et al., 2012). Combined introduction of these three genetic events in NPCs induced more extensive neoplastic features, generating DIPG when transplanted in mice (Funato et al., 2014). This combination of mutations prohibited early NPCs to differentiate to astrocytes (Funato et al., 2014), explaining the observed maturation block in DIPG.
MRT and DIPG modeling approaches using ESCs and iPSCs have demonstrated that a specific cellular context is required for malignant transformation, meaning that tumorigenesis is restricted to a specific developmental time and fetal cell type.
Pluripotent Stem Cell-Derived Organoids
Recent innovations in three-dimensional (3D) culture technology, such as organoids, has opened new opportunities for generating additional representative models of embryonal tumors. Organoids can be derived from adult (ASC) or pluripotent stem cells. They typically capture the cellular and genetic heterogeneity of native tissue and recapitulate cellular hierarchy and dynamics to a large extent, which is most likely a consequence of their 3D architecture (Clevers, 2016). Therefore, 3D organoid cultures seem to better recapitulate organ morphogenesis (Clevers, 2016).
Following that rationale, 3D retinal organoids were established from hESCs or iPSCs, allowing for more comprehensive studies of retinoblastoma initiation in human cells (Zhong et al., 2014; Kuwahara et al., 2015). Loss of RB1 in retinal organoids showed a dysregulation of retinal maturation processes, impairing differentiation toward photoreceptors, ganglion, and bipolar cells (Zheng et al., 2020). However, the depletion of RB1 was not sufficient for retinoblastoma initiation as the organoids did not fully recapitulate the retinoblastoma cell phenotype. In addition, transplantation of RB1-null organoids into immune-deficient mice did not result in retinoblastoma formation (Zheng et al., 2020). In contrast, Liu H. et al. (2020) utilized an alternative hESC-derived retinal organoid model, in which RB1 depletion did successfully generate tumors upon xenografting and better resembled patient retinoblastoma. These findings illustrate that the finetuning of retinal organoid establishment can affect the outcome of RB1 depletion, possibly due to differences in cellular composition and the presence or absence of the cell-of-origin. These studies further highlight the specific cellular context required for retinoblastoma initiation and point out a possible limitation of PSC-derived models, as they may not be able to generate the full extent of cell-types found in vivo.
A frequent source of embryonal tumors is the embryonic brain. Human brain development can be mimicked by differentiation of PSCs to neural progenitor cells. In culture, they can self-organize into cerebral or cerebellar organoids containing different cell types in a polarized structure (Muguruma et al., 2015; Luo et al., 2016). Embryonic cerebellar organoids have been successfully used to model pediatric brain tumors, including medulloblastoma and rhabdoid tumors (Ballabio et al., 2020; Parisian et al., 2020). Organoid cultures can be utilized to introduce tumorigenic mutations in a systematic manner, as shown for cerebral organoids (Bian et al., 2018), demonstrating the potential for high-throughput in vitro tumor modeling. Furthermore, cerebellar organoids can be exploited to decipher tumor subtype-specific processes. Medulloblastoma, among other embryonal tumor entities, is classified into subtypes based on the oncogenic activation of specific signaling pathways (Cavalli et al., 2017). The medulloblastoma subgroup 3 (MYC amplified subgroup) was successfully modeled in cerebellar organoids by combination of MYC and OTX2 or GFI1 overexpression (Ballabio et al., 2020). The genetically modified cerebellar organoids showed increased proliferation and enrichment for progenitor cells, indicative of a differentiation block. Upon transplantation into mice, medulloblastomas developed resembling subgroup 3 tumors based on marker genes and DNA methylation patterns. Other medulloblastoma subtypes, likely arising from distinct neural differentiation trajectories, have not been modeled in vitro up to date (Gibson et al., 2010; Grammel et al., 2012; Hovestadt et al., 2019). To do so, tumor initiation models composed of different neural lineages may be required.
Overall, the development of embryonic organoid cultures has provided relevant models of embryonal tumorigenesis. By approaching in vivo physiology, human organoids may serve as a promising alternative for time- and labor-intensive in vivo studies.
Reverse Tumor Modeling and Differentiation Therapy
Relieving the differentiation block underpinning embryonal tumor development could potentially serve as a therapeutic approach (i.e., maturation therapy). To develop such therapies, the differentiation block must first be defined, which can be achieved through reverse tumor modeling by, for instance, reverting the oncogenic driver in cultured tumor cells. Following this principle, inhibition of N-MYC in MYCN-amplified neuroblastoma cell lines induced a differentiation morphology as well as upregulation of neural differentiation genes (Kang et al., 2006; Henriksen et al., 2011; Jiang et al., 2011; Westermark et al., 2011; Hossain et al., 2013). Differentiation phenotypes were also observed upon genetic manipulation of medulloblastoma models (Liu et al., 2017; Cheng et al., 2020; Zagozewski et al., 2020), and MRT models (Betz et al., 2002; Nakayama et al., 2017; Wang et al., 2017). These studies show that reversal of the genetic driver can transform tumor cells to a more mature cell state, possibly reflecting the matured cell type it would have become, had it not become cancerous. Genetic repair of driver genes is not feasible at present (Dunbar et al., 2018). An alternative strategy is to induce differentiation pharmacologically. For instance, experiments performed in MRT models with SMARCB1 re-expression identified EZH2 and BRD9 as promising therapeutic targets (Erkek et al., 2019; Wang et al., 2019). Moreover, aberrant epigenetic regulation is often causal of the malignant embryonic state of pediatric cancer cells (Lawlor and Thiele, 2012), potentially explaining the sensitivity of different embryonal tumors to drugs targeting epigenetic modifiers (Table 1). Treatment of in vitro pediatric tumor models with differentiation agents can recapitulate the effects achieved by driver reversal. However, a durable effect of differentiation therapy can only be acquired through induction of an irreversible growth arrest. As in vivo studies have shown, single agent treatment may not suffice to induce terminal differentiation and that combination therapy is required to do so (Hahn et al., 2008; Botrugno et al., 2009; Westerlund et al., 2017; Chen et al., 2018). A powerful tool to identify new (combinations of) drugs are high-throughput drug screens performed on in vitro tumor models. Organoids directly derived from patient tumor tissue could provide such models, as they have been shown to closely resemble its parental tissue (Drost and Clevers, 2018). Confirming their potential, an increasing number of reports described that tumor organoids are predictive for patient drug response (Tiriac et al., 2018; Vlachogiannis et al., 2018; Ganesh et al., 2019; Ooft et al., 2019; Yao et al., 2020). Recently, the organoid technology was also successfully applied to several pediatric cancers, including embryonal tumors such as MRT and Wilms tumors (Schutgens et al., 2019; Calandrini et al., 2020). The efficient establishment and cryopreservation of tumor organoid models from primary patient tissue allows for the generation of large patient cohorts stored in organoid biobanks. This is seemingly of particular interest for rare tumors, such as embryonal tumors, for which research material is scarce. In conclusion, the generation of novel and more representative in vitro embryonal tumor models is key for the improvement of differentiation therapeutics.

Table 1. In vitro embryonic tumor initiation models and differentiation therapies.
Discussion
In this review, we have attempted to outline the rapidly developing field of in vitro embryonal tumor models and discussed their added value to embryonal tumor research (Figure 1 and Table 1). Still, each model has its intrinsic limitations. For instance, fetal cells can be extracted and cultured from fetal tissues (Xu et al., 2014), but in many cases they do not represent the continuously changing cellular identities found during embryonic development. Alternatively, iPSCs or ESCs cell lines can be deployed to recapitulate these dynamics. Still, even though the spectrum of differentiation protocols is rapidly expanding, many embryonic cell types found in vivo cannot yet be captured in vitro. Additionally, in vitro cultures of ESCs or iPSCs have been shown to be susceptible to “spontaneous” malignant transformation, which can complicate the interpretation of modeling experiments (Ben-David and Benvenisty, 2011). Furthermore, 2D cultures do not capture 3D tissue architecture (Pampaloni et al., 2007). These limitations have been to some extent improved in 3D organoid cultures, which better capture the cell-cell interactions found during embryonic organogenesis (Clevers, 2016). The development of mouse-human chimeras has highlighted the role of the microenvironment in tumor progression (Cohen et al., 2020) and reveals a promising opportunity to bridge the gap of in vitro and in vivo tumor modeling, as mouse-human chimeras have the advantage of having human cells combined with an in vivo murine microenvironment. A good representation of patient tumor evolution remains challenging in in vitro models. In patients, tumors originate from a single tumor-initiating cell, wherein a genetic driver event induces aberrant signaling pathways that provide a cell with competitive advantages. Continuous selection of such cells (clonal selection) is thought to form the basis of tumor initiation, progression, and heterogeneity (Navin et al., 2010, 2011). In vitro models typically do not reflect the environmental conditions causing clonal selection, as culture conditions are only a simplified version of in vivo signaling complexity. Embryonal tumors maintain a fetal identity, which is no longer present in matured tissues (Orbach et al., 2013; Marshall et al., 2014). The characterization of developmental programs in embryonal tumors can therefore give crucial insights into the processes underpinning malignant growth. Single cell transcriptome profiling of tumors and developing tissues has proven to be a promising tool to reveal such processes, which could potentially serve as therapeutic targets (Filbin et al., 2018; Zhang et al., 2019). Similar methods can also be applied to in vitro models recapitulating embryonal tumorigenesis, as demonstrated for the retinoblastoma organoid model generated by Liu H. et al. (2020), which has the advantage that it allows for a direct comparison of normal and tumor development.
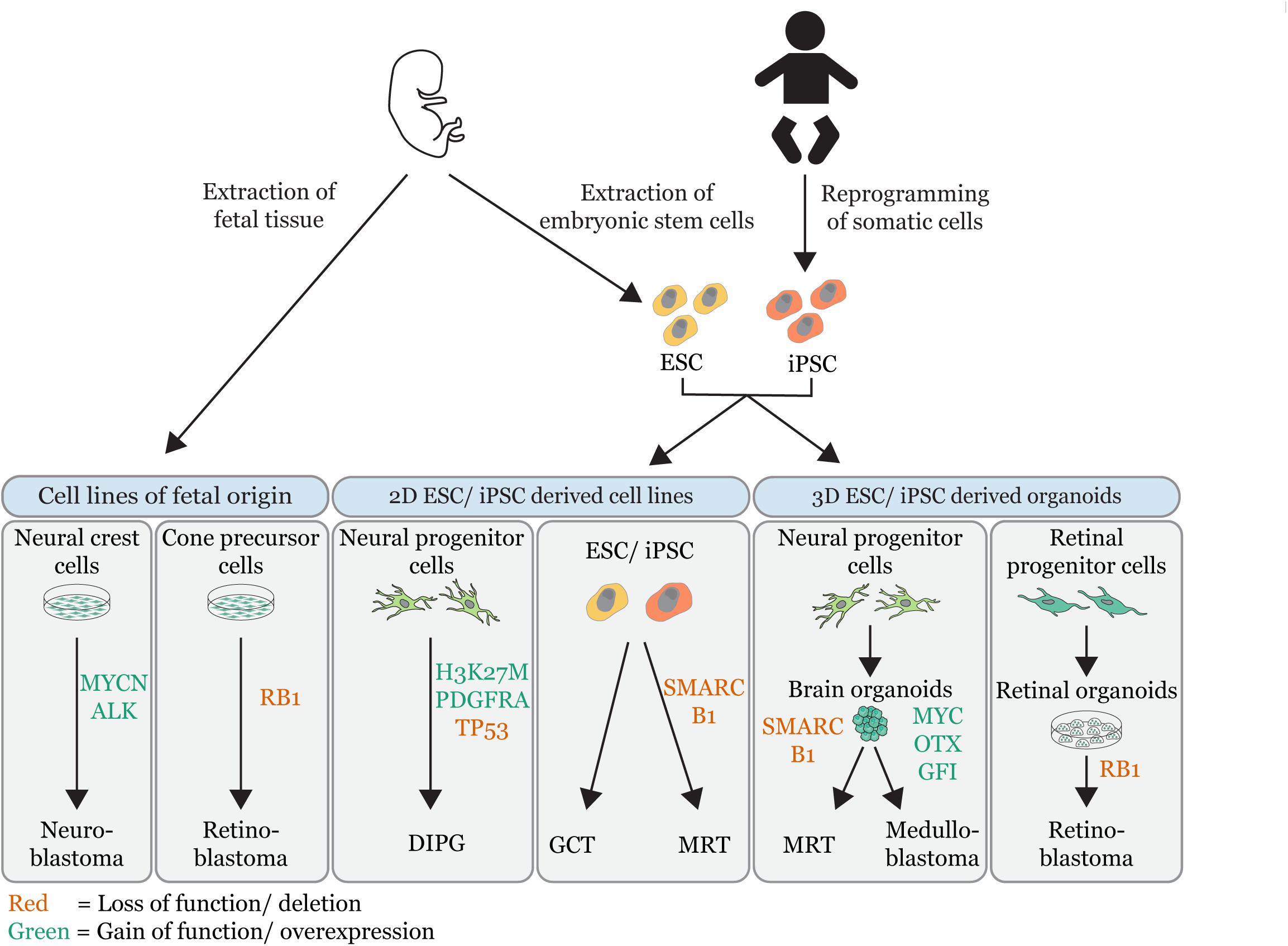
Figure 1. Overview of embryonal tumor modeling techniques. Illustration that summarizes the different in vitro approaches used to model embryonal tumors (MRT = malignant rhabdoid tumor; DIPG = diffuse intrinsic pontine glioma, GCT = germ cell tumor). In vitro tumor models are grouped by their source: cell lines of fetal origin, 2D embryonic stem cell (ESC) derived or induced pluripotent stem cell derived (iPSC) cell lines and 3D ESC/iPSC derived organoids. The gene-editing of tumor driver events is indicated (red = loss of function/deletions; green = gain of function/overexpression). Permission to reuse and Copyright: Medical illustrations used in in this figure were modified from Servier Medical Art, licensed under a Creative Commons Attribution 3.0 Generic License.
Although many in vitro embryonic cell-derived tumor models have been established over the years, the spectrum is biased toward ectoderm-derived tumors. It seems a matter of time before mesoderm- or endoderm-derived in vitro tumor models (e.g., Wilms tumor and hepatoblastoma) will be developed, as the number of culture systems for fetal tissues is rapidly expanding (Low et al., 2019; Ooms et al., 2020; Hendriks et al., 2021).
We are only just beginning to understand the complexity of embryonal tumor development. Although capturing this complexity in a single in vitro model might not be feasible, further development of representative in vitro cell models recapitulating at least part of it is crucial to gain further insight into the fundamental processes underpinning malignant growth and the development of new therapeutic strategies.
Author Contributions
LC, IP, and JD wrote the manuscript. JD supervised the work. All authors approved the manuscript for publication.
Funding
We are grateful for support from the European Research Council (ERC) starting Grant 850571 (JD), the Dutch Cancer Society (KWF)/Alpe d’HuZes Bas Mulder Award to JD (KWF/Alpe d’HuZes, 10218), and the Children Cancer-free Foundation (KiKa #338, LC; base funding, IP).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
ASC, adult stem cell; DIPG, diffuse intrinsic pontine glioma; ESC, embryonic stem cell; GCT, germ cell tumor; GEMM, genetically engineered mouse model; H3K27M, histone 3 methionine for lysine 27 substitution; iPSC, induced pluripotent stem cell; MRT, malignant rhabdoid tumor; NPC, neural progenitor cell; NCC, neural crest cell; RPC, retinal precursor cell; SWI/SNF, SWItch/Sucrose Non-Fermentable.
References
Anastas, J. N., Zee, B. M., Kalin, J. H., Kim, M., Guo, R., Alexandrescu, S., et al. (2019). Re-programing chromatin with a bifunctional LSD1/HDAC inhibitor induces therapeutic differentiation in DIPG. Cancer Cell 36, 528–544.e10. doi: 10.1016/j.ccell.2019.09.005
Ballabio, C., Anderle, M., Gianesello, M., Lago, C., Miele, E., Cardano, M., et al. (2020). Modeling medulloblastoma in vivo and with human cerebellar organoids. Nat. Commun. 11:583. doi: 10.1038/s41467-019-13989-3
Bandopadhayay, P., Piccioni, F., O’Rourke, R., Ho, P., Gonzalez, E. M., Buchan, G., et al. (2019). Neuronal differentiation and cell-cycle programs mediate response to BET-bromodomain inhibition in MYC-driven medulloblastoma. Nat. Commun. 10:2400. doi: 10.1038/s41467-019-10307-9
Ben-David, U., and Benvenisty, N. (2011). The tumorigenicity of human embryonic and induced pluripotent stem cells. Nat. Rev. Cancer 11, 268–277. doi: 10.1038/nrc3034
Berry, R. L., Ozdemir, D. D., Aronow, B., Lindström, N. O., Dudnakova, T., Thornburn, A., et al. (2015). Deducing the stage of origin of Wilms’ tumours from a developmental series of Wt1-mutant mice. DMM Dis. Models Mechan. 8, 903–917. doi: 10.1242/dmm.018523
Betters, E., Liu, Y., Kjaeldgaard, A., Sundström, E., and García-Castro, M. I. (2010). Analysis of early human neural crest development. Dev. Biol. 344, 578–592. doi: 10.1016/j.ydbio.2010.05.012
Betz, B. L., Strobeck, M. W., Reisman, D. N., Knudsen, E. S., and Weissman, B. E. (2002). Re-expression of hSNF5/INI1/BAF47 in pediatric tumor cells leads to G1 arrest associated with induction of p16ink4a and activation of RB. Oncogene 21, 5193–5203. doi: 10.1038/sj.onc.1205706
Bian, S., Repic, M., Guo, Z., Kavirayani, A., Burkard, T., Bagley, J. A., et al. (2018). Genetically engineered cerebral organoids model brain tumor formation. Nat. Methods 15, 631–639. doi: 10.1038/s41592-018-0070-7
Blakeley, P., Fogarty, N. M. E., Del Valle, I., Wamaitha, S. E., Hu, T. X., Elder, K., et al. (2015). Defining the three cell lineages of the human blastocyst by single-cell RNA-seq. Development (Cambridge) 142, 3151–3165. doi: 10.1242/dev.123547
Boeva, V., Louis-Brennetot, C., Peltier, A., Durand, S., Pierre-Eugène, C., Raynal, V., et al. (2017). Heterogeneity of neuroblastoma cell identity defined by transcriptional circuitries. Nat. Genet. 49, 1408–1413. doi: 10.1038/ng.3921
Botrugno, O. A., Santoro, F., and Minucci, S. (2009). Histone deacetylase inhibitors as a new weapon in the arsenal of differentiation therapies of cancer. Cancer Lett. 280, 134–144. doi: 10.1016/j.canlet.2009.02.027
Calandrini, C., Schutgens, F., Oka, R., Margaritis, T., Candelli, T., Mathijsen, L., et al. (2020). An organoid biobank for childhood kidney cancers that captures disease and tissue heterogeneity. Nat. Commun. 11:1310. doi: 10.1038/s41467-020-15155-6
Cavalli, F. M. G., Remke, M., Rampasek, L., Peacock, J., Shih, D. J. H., Luu, B., et al. (2017). Intertumoral heterogeneity within medulloblastoma subgroups. Cancer Cell 31, 737–754.e6. doi: 10.1016/j.ccell.2017.05.005
Chen, L., Alexe, G., Dharia, N. V., Ross, L., Iniguez, A. B., Conway, A. S., et al. (2018). CRISPR-Cas9 screen reveals a MYCN-amplified neuroblastoma dependency on EZH2. J. Clin. Invest. 128, 446–462. doi: 10.1172/JCI90793
Chen, X., Pappo, A., and Dyer, M. A. (2015). Pediatric solid tumor genomics and developmental pliancy. Oncogene 34, 5207–5215. doi: 10.1038/onc.2014.474
Cheng, Y., Liao, S., Xu, G., Hu, J., Guo, D., Du, F., et al. (2020). NeuroD1 dictates tumor cell differentiation in medulloblastoma. Cell Rep. 31:107782. doi: 10.1016/j.celrep.2020.107782
Clevers, H. (2016). Modeling development and disease with organoids. Cell 165, 1586–1597. doi: 10.1016/j.cell.2016.05.082
Cohen, M. A., Zhang, S., Sengupta, S., Ma, H., Bell, G. W., Horton, B., et al. (2020). Formation of human neuroblastoma in mouse-human neural crest chimeras. Cell Stem Cell 26, 579–592.e6. doi: 10.1016/j.stem.2020.02.001
Cunningham, R. M., Walton, M. A., and Carter, P. M. (2018). The major causes of death in children and adolescents in the United States. N. Eng. J. Med. 379, 2468–2475. doi: 10.1056/nejmsr1804754
Dannenberg, J. H., Schuijff, L., Dekker, M., Van Der Valk, M., and Te Riele, H. (2004). Tissue-specific tumor suppressor activity of retinoblastoma gene homologs p107 and p130. Genes Dev. 18, 2952–2962. doi: 10.1101/gad.322004
De Jong, J., and Looijenga, L. H. J. (2006). Stem cell marker OCT3/4 in tumor biology and germ cell tumor diagnostics: history and future. Crit. Rev. Oncogen. 12, 171–203. doi: 10.1615/CritRevOncog.v12.i3-4.10
Dimaras, H., Corson, T. W., Cobrinik, D., White, A., Zhao, J., Munier, F. L., et al. (2015). Retinoblastoma. Nat. Rev. Dis. Primers 1:1502. doi: 10.1038/nrdp.2015.21
Drost, J., and Clevers, H. (2018). Organoids in cancer research. Nat. Rev. Cancer 18, 407–418. doi: 10.1038/s41568-018-0007-6
Dunbar, C. E., High, K. A., Joung, J. K., Kohn, D. B., Ozawa, K., and Sadelain, M. (2018). Gene therapy comes of age. Science 359:eaan4672. doi: 10.1126/science.aan4672
Erkek, S., Johann, P. D., Finetti, M. A., Drosos, Y., Chou, H. C., Zapatka, M., et al. (2019). Comprehensive analysis of chromatin states in atypical teratoid/rhabdoid tumor identifies diverging roles for SWI/SNF and polycomb in gene regulation. Cancer Cell 35, 95–110.e8. doi: 10.1016/j.ccell.2018.11.014
Filbin, M. G., Tirosh, I., Hovestadt, V., Shaw, M. L., Escalante, L. E., Mathewson, N. D., et al. (2018). Developmental and oncogenic programs in H3K27M gliomas dissected by single-cell RNA-seq. Science 360, 331–335. doi: 10.1126/science.aao4750
Frumm, S. M., Fan, Z. P., Ross, K. N., Duvall, J. R., Gupta, S., Verplank, L., et al. (2013). Selective HDAC1/HDAC2 inhibitors induce neuroblastoma differentiation. Chem. Biol. 20, 713–725. doi: 10.1016/j.chembiol.2013.03.020
Funato, K., Major, T., Lewis, P. W., Allis, C. D., and Tabar, V. (2014). Use of human embryonic stem cells to model pediatric gliomas with H3.3K27M histone mutation. Science 346, 1529–1533. doi: 10.1126/science.1253799
Ganesh, K., Wu, C., O’Rourke, K. P., Szeglin, B. C., Zheng, Y., Sauvé, C. E. G., et al. (2019). A rectal cancer organoid platform to study individual responses to chemoradiation. Nat. Med. 25, 1607–1614. doi: 10.1038/s41591-019-0584-2
García-López, J., Wallace, K., Otero, J. H., Olsen, R., Wang, Y., and dong (2020). Large 1p36 deletions affecting arid1a locus facilitate mycn-driven oncogenesis in neuroblastoma. Cell Rep. 30, 454–464.e5. doi: 10.1016/j.celrep.2019.12.048
Gibson, P., Tong, Y., Robinson, G., Thompson, M. C., Currle, D. S., Eden, C., et al. (2010). Subtypes of medulloblastoma have distinct developmental origins. Nature 468, 1095–1099. doi: 10.1038/nature09587
Grammel, D., Warmuth-Metz, M., Von Bueren, A. O., Kool, M., Pietsch, T., Kretzschmar, H. A., et al. (2012). Sonic hedgehog-associated medullobla stoma arising from the cochlear nuclei of the brainstem. Acta Neuropathol. 123, 601–614. doi: 10.1007/s00401-012-0961-0
Gröbner, S. N., Worst, B. C., Weischenfeldt, J., Buchhalter, I., Kleinheinz, K., Rudneva, V. A., et al. (2018). The landscape of genomic alterations across childhood cancers. Nature 555, 321–327. doi: 10.1038/nature25480
Hahn, C. K., Ross, K. N., Warrington, I. M., Mazitschek, R., Kanegai, C. M., Wright, R. D., et al. (2008). Expression-based screening identifies the combination of histone deacetylase inhibitors and retinoids for neuroblastoma differentiation. Proc. Natl. Acad. Sci. U S A. 105, 9751–9756. doi: 10.1073/pnas.0710413105
Han, Z. Y., Richer, W., Fréneaux, P., Chauvin, C., Lucchesi, C., Guillemot, D., et al. (2016). The occurrence of intracranial rhabdoid tumours in mice depends on temporal control of Smarcb1 inactivation. Nat. Commun. 7:10421. doi: 10.1038/ncomms10421
Hasselblatt, M., Nagel, I., Oyen, F., Bartelheim, K., Russell, R. B., Schüller, U., et al. (2014). SMARCA4-mutated atypical teratoid/rhabdoid tumors are associated with inherited germline alterations and poor prognosis. Acta Neuropathol. 128, 453–456. doi: 10.1007/s00401-014-1323-x
Hendriks, D., Artegiani, B., Hu, H., Chuva, de Sousa Lopes, S., and Clevers, H. (2021). Establishment of human fetal hepatocyte organoids and CRISPR–Cas9-based gene knockin and knockout in organoid cultures from human liver. Nat. Protocols 16, 182–217. doi: 10.1038/s41596-020-00411-2
Henriksen, J. R., Haug, B. H., Buechner, J., Tømte, E., Løkke, C., Flaegstad, T., et al. (2011). Conditional expression of retrovirally delivered anti-MYCN shRNA as an in vitro model system to study neuronal differentiation in MYCN-amplified neuroblastoma. BMC Dev. Biol. 11:1. doi: 10.1186/1471-213X-11-1
Hoeman, C. M., Cordero, F. J., Hu, G., Misuraca, K., Romero, M. M., Cardona, H. J., et al. (2019). ACVR1 R206H cooperates with H3.1K27M in promoting diffuse intrinsic pontine glioma pathogenesis. Nat. Commun. 10:1023. doi: 10.1038/s41467-019-08823-9
Hossain, M. M., Banik, N. L., and Ray, S. K. (2013). N-Myc knockdown and apigenin treatment controlled growth of malignant neuroblastoma cells having N-Myc amplification. Gene 529, 27–36. doi: 10.1016/j.gene.2013.07.094
Hovestadt, V., Smith, K. S., Bihannic, L., Filbin, M. G., Shaw, M. K. L., Baumgartner, A., et al. (2019). Resolving medulloblastoma cellular architecture by single-cell genomics. Nature 572, 74–79. doi: 10.1038/s41586-019-1434-6
Hu, Q., Gao, F., Tian, W., Ruteshouser, E. C., Wang, Y., Lazar, A., et al. (2011). Wt1 ablation and Igf2 upregulation in mice result in wilms tumors with elevated ERK1/2 phosphorylation. J. Clin. Invest. 121, 174–183. doi: 10.1172/JCI43772
Huang, L., Mokkapati, S., Hu, Q., Ruteshouser, E. C., Hicks, M. J., and Huff, V. (2016). Nephron Progenitor but not stromal progenitor cells give rise to wilms tumors in mouse models with β-Catenin activation or Wt1 ablation and Igf2 upregulation. Neoplasia (United States) 18, 71–81. doi: 10.1016/j.neo.2015.12.001
Huang, M., Tailor, J., Zhen, Q., Gillmor, A. H., Miller, M. L., Weishaupt, H., et al. (2019). Engineering genetic predisposition in human neuroepithelial stem cells recapitulates medulloblastoma tumorigenesis. Cell Stem Cell 25, 433–446.e7. doi: 10.1016/j.stem.2019.05.013
Jessa, S., Blanchet-Cohen, A., Krug, B., Vladoiu, M., Coutelier, M., Faury, D., et al. (2019). Stalled developmental programs at the root of pediatric brain tumors. Nat. Genet. 51, 1702–1713. doi: 10.1038/s41588-019-0531-7
Jiang, R., Xue, S., and Jin, Z. (2011). Stable knockdown of MYCN by lentivirus-based RNAi inhibits human neuroblastoma cells growth in vitro and in vivo. Biochem. Biophys. Res. Commun. 410, 364–370. doi: 10.1016/j.bbrc.2011.06.020
Johnsen, J. I., Dyberg, C., and Wickström, M. (2019). Neuroblastoma—a neural crest derived embryonal malignancy. Front. Mol. Neurosci. 12:9. doi: 10.3389/fnmol.2019.00009
Kang, J. H., Rychahou, P. G., Ishola, T. A., Qiao, J., Evers, B. M., and Chung, D. H. (2006). MYCN silencing induces differentiation and apoptosis in human neuroblastoma cells. Biochem. Biophys. Res. Commun. 351, 192–197. doi: 10.1016/j.bbrc.2006.10.020
Kattner, P., Strobel, H., Khoshnevis, N., Grunert, M., Bartholomae, S., Pruss, M., et al. (2019). Compare and contrast: pediatric cancer versus adult malignancies. Cancer Metastasis Rev. 38, 673–682. doi: 10.1007/s10555-019-09836-y
Khuong-Quang, D. A., Buczkowicz, P., Rakopoulos, P., Liu, X. Y., Fontebasso, A. M., Bouffet, E., et al. (2012). K27M mutation in histone H3.3 defines clinically and biologically distinct subgroups of pediatric diffuse intrinsic pontine gliomas. Acta Neuropathol. 124, 439–447. doi: 10.1007/s00401-012-0998-0
Knutson, S. K., Warholic, N. M., Wigle, T. J., Klaus, C. R., Allain, C. J., Raimondi, A., et al. (2013). Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proc. Natl. Acad. Sci. U S A. 110, 7922–7927. doi: 10.1073/pnas.1303800110
Kreidberg, J. A., Sariola, H., Loring, J. M., Maeda, M., Pelletier, J., Housman, D., et al. (1993). WT-1 is required for early kidney development. Cell 74, 679–691. doi: 10.1016/0092-8674(93)90515-R
Kuwahara, A., Ozone, C., Nakano, T., Saito, K., Eiraku, M., and Sasai, Y. (2015). Generation of a ciliary margin-like stem cell niche from self-organizing human retinal tissue. Nat. Commun. 6:6286. doi: 10.1038/ncomms7286
Langer, L. F., Ward, J. M., and Archer, T. K. (2019). Tumor suppressor SMARCB1 suppresses super-enhancers to govern hESC lineage determination. eLife 8:e45672. doi: 10.7554/eLife.45672
Lawlor, E. R., and Thiele, C. J. (2012). Epigenetic changes in pediatric solid tumors: promising new targets. Clin. Cancer Res. 18, 2768–2779. doi: 10.1158/1078-0432.CCR-11-1921
Lee, A. S., Tang, C., Rao, M. S., Weissman, I. L., and Wu, J. C. (2013). Tumorigenicity as a clinical hurdle for pluripotent stem cell therapies. Nat. Med. 19, 998–1004. doi: 10.1038/nm.3267
Lee, E. Y. H. P., Chang, C. Y., Hu, N., Wang, Y. C. J., Lai, C. C., Herrup, K., et al. (1992). Mice deficient for Rb are nonviable and show defects in neurogenesis and haematopoiesis. Nature 359, 288–294. doi: 10.1038/359288a0
Lee, R. S., Stewart, C., Carter, S. L., Ambrogio, L., Cibulskis, K., Sougnez, C., et al. (2012). A remarkably simple genome underlies highly malignant pediatric rhabdoid cancers. J. Clin. Invest. 122, 2983–2988. doi: 10.1172/JCI64400
Liu, G., David, B. T., Trawczynski, M., and Fessler, R. G. (2020). Advances in pluripotent stem cells: history, mechanisms, technologies, and applications. Stem Cell Rev. Rep. 16, 3–32. doi: 10.1007/s12015-019-09935-x
Liu, H., Sun, Q., Sun, Y., Zhang, J., Yuan, H., Pang, S., et al. (2017). MELK and EZH2 cooperate to regulate medulloblastoma cancer stem-like cell proliferation and differentiation. Mol. Cancer Res. 15, 1275–1286. doi: 10.1158/1541-7786.MCR-17-0105
Liu, H., Zhang, Y., Zhang, Y.-Y., Li, Y.-P., Hua, Z.-Q., Zhang, C.-J., et al. (2020). Human embryonic stem cell-derived organoid retinoblastoma reveals a cancerous origin. Proc. Natl. Acad. Sci. 117, 33628–33638. doi: 10.1073/pnas.2011780117
Lone, A. M., Dar, N. J., Hamid, A., Shah, W. A., Ahmad, M., and Bhat, B. A. (2016). Promise of retinoic acid-triazolyl derivatives in promoting differentiation of neuroblastoma cells. ACS Chem. Neurosci. 7, 82–89. doi: 10.1021/acschemneuro.5b00267
Low, J. H., Li, P., Chew, E. G. Y., Zhou, B., Suzuki, K., Zhang, T., et al. (2019). Generation of human PSC-Derived kidney organoids with patterned nephron segments and a de novo vascular network. Cell Stem Cell 25, 373–387.e9. doi: 10.1016/j.stem.2019.06.009
Luo, C., Lancaster, M. A., Castanon, R., Nery, J. R., Knoblich, J. A., and Ecker, J. R. (2016). Cerebral organoids recapitulate epigenomic signatures of the human fetal brain. Cell Rep. 17, 3369–3384. doi: 10.1016/j.celrep.2016.12.001
MacPherson, D., Sage, J., Kim, T., Ho, D., McLaughlin, M. E., and Jacks, T. (2004). Cell type-specific effects of Rb deletion in the murine retina. Genes Dev. 18, 1681–1694. doi: 10.1101/gad.1203304
Marshall, G. M., Carter, D. R., Cheung, B. B., Liu, T., Mateos, M. K., Meyerowitz, J. G., et al. (2014). The prenatal origins of cancer. Nat. Rev. Cancer 14, 277–289. doi: 10.1038/nrc3679
Maurer, J., Fuchs, S., Jäger, R., Kurz, B., Sommer, L., and Schorle, H. (2007). Establishment and controlled differentiation of neural crest stem cell lines using conditional transgenesis. Differentiation 75, 580–591. doi: 10.1111/j.1432-0436.2007.00164.x
Mohammad, F., Weissmann, S., Leblanc, B., Pandey, D. P., Højfeldt, J. W., Comet, I., et al. (2017). EZH2 is a potential therapeutic target for H3K27M-mutant pediatric gliomas. Nat. Med. 23, 483–492. doi: 10.1038/nm.4293
Montavon, G., Jauquier, N., Coulon, A., Peuchmaur, M., Flahaut, M., Bourloud, K. B., et al. (2014). Wild-type ALK and activating ALK-R1275Q and ALK-F1174L mutations upregulate Myc and initiate tumor formation in murine neural crest progenitor cells. Oncotarget 5, 4452–4466. doi: 10.18632/oncotarget.2036
Muguruma, K., Nishiyama, A., Kawakami, H., Hashimoto, K., and Sasai, Y. (2015). Self-organization of polarized cerebellar tissue in 3D culture of human pluripotent stem cells. Cell Rep. 10, 537–550. doi: 10.1016/j.celrep.2014.12.051
Muscat, A., Popovski, D., Jayasekara, W. S. N., Rossello, F. J., Ferguson, M., Marini, K. D., et al. (2016). Low-dose histone deacetylase inhibitor treatment leads to tumor growth arrest and multi-lineage differentiation of malignant rhabdoid tumors. Clin. Cancer Res. 22, 3560–3570. doi: 10.1158/1078-0432.CCR-15-2260
Nakayama, R. T., Pulice, J. L., Valencia, A. M., McBride, M. J., McKenzie, Z. M., Gillespie, M. A., et al. (2017). SMARCB1 is required for widespread BAF complex-mediated activation of enhancers and bivalent promoters. Nat. Genet. 49, 1613–1623. doi: 10.1038/ng.3958
Navin, N., Kendall, J., Troge, J., Andrews, P., Rodgers, L., McIndoo, J., et al. (2011). Tumour evolution inferred by single-cell sequencing. Nature 472, 90–95. doi: 10.1038/nature09807
Navin, N., Krasnitz, A., Rodgers, L., Cook, K., Meth, J., Kendall, J., et al. (2010). Inferring tumor progression from genomic heterogeneity. Genome Res. 20, 68–80. doi: 10.1101/gr.099622.109
Ocasio, J., Babcock, B., Malawsky, D., Weir, S. J., Loo, L., Simon, J. M., et al. (2019). scRNA-seq in medulloblastoma shows cellular heterogeneity and lineage expansion support resistance to SHH inhibitor therapy. Nat. Commun. 10:5829. doi: 10.1038/s41467-019-13657-6
Olsen, R. R., Otero, J. H., García-López, J., Wallace, K., Finkelstein, D., Rehg, J. E., et al. (2017). MYCN induces neuroblastoma in primary neural crest cells. Oncogene 36, 5075–5082. doi: 10.1038/onc.2017.128
Ooft, S. N., Weeber, F., Dijkstra, K. K., McLean, C. M., Kaing, S., van Werkhoven, E., et al. (2019). Patient-derived organoids can predict response to chemotherapy in metastatic colorectal cancer patients. Sci. Trans. Med. 11:eaay2574. doi: 10.1126/scitranslmed.aay2574
Ooms, A. H. A. G., Calandrini, C., de Krijger, R. R., and Drost, J. (2020). Organoid models of childhood kidney tumours. Nat. Rev. Urol. 17, 311–313. doi: 10.1038/s41585-020-0315-y
Oosterhuis, J. W., and Looijenga, L. H. J. (2019). Human germ cell tumours from a developmental perspective. Nat. Rev. Cancer 19, 522–537. doi: 10.1038/s41568-019-0178-9
O’Rahilly, R., and Müller, F. (2007). The development of the neural crest in the human. J. Anatomy 211, 335–351. doi: 10.1111/j.1469-7580.2007.00773.x
Orbach, D., Sarnacki, S., Brisse, H. J., Gauthier-Villars, M., Jarreau, P. H., Tsatsaris, V., et al. (2013). Neonatal cancer. Lancet Oncol. 14, e609–e620. doi: 10.1016/S1470-2045(13)70236-5
Pampaloni, F., Reynaud, E. G., and Stelzer, E. H. K. (2007). The third dimension bridges the gap between cell culture and live tissue. Nat. Rev. Mol. Cell Biol. 8, 839–845. doi: 10.1038/nrm2236
Parisian, A. D., Koga, T., Miki, S., Johann, P. D., Kool, M., Crawford, J. R., et al. (2020). SMARCB1 loss interacts with neuronal differentiation state to block maturation and impact cell stability. Genes Dev. 34, 1316–1329. doi: 10.1101/gad.339978.120
Patties, I., Kortmann, R. D., Menzel, F., and Glasow, A. (2016). Enhanced inhibition of clonogenic survival of human medulloblastoma cells by multimodal treatment with ionizing irradiation, epigenetic modifiers, and differentiation-inducing drugs. J. Exp. Clin. Cancer Res. 35:1. doi: 10.1186/s13046-016-0376-1
Puisieux, A., Pommier, R. M., Morel, A. P., and Lavial, F. (2018). Cellular pliancy and the multistep process of tumorigenesis. Cancer Cell 33, 164–172. doi: 10.1016/j.ccell.2018.01.007
Rahal, Z., Abdulhai, F., Kadara, H., and Saab, R. (2018). Genomics of adult and pediatric solid tumors. Am. J. Cancer Res. 8, 1356–1386.
Reiff, T., Tsarovina, K., Majdazari, A., Schmidt, M., Del Pino, I., and Rohrer, H. (2010). Neuroblastoma Phox2b variants stimulate proliferation and dedifferentiation of immature sympathetic neurons. J. Neurosci. 30, 905–915. doi: 10.1523/JNEUROSCI.5368-09.2010
Robanus-Maandag, E., Dekker, M., Van Der Valk, M., Carrozza, M. L., Jeanny, J. C., Dannenberg, J. H., et al. (1998). p107 is a suppressor of retinoblastoma development in pRB-deficient mice. Genes Dev. 12, 1599–1609. doi: 10.1101/gad.12.11.1599
Schulte, J. H., Lindner, S., Bohrer, A., Maurer, J., De Preter, K., Lefever, S., et al. (2013). MYCN and ALKF1174L are sufficient to drive neuroblastoma development from neural crest progenitor cells. Oncogene 32, 1059–1065. doi: 10.1038/onc.2012.106
Schutgens, F., Rookmaaker, M. B., Margaritis, T., Rios, A., Ammerlaan, C., Jansen, J., et al. (2019). Tubuloids derived from human adult kidney and urine for personalized disease modeling. Nat. Biotechnol. 37, 303–313. doi: 10.1038/s41587-019-0048-8
Schwartzentruber, J., Korshunov, A., Liu, X. Y., Jones, D. T. W., Pfaff, E., Jacob, K., et al. (2012). Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 482, 226–231. doi: 10.1038/nature10833
Shain, A. H., and Pollack, J. R. (2013). The spectrum of SWI/SNF mutations, ubiquitous in human cancers. PLoS One 8:e55119. doi: 10.1371/journal.pone.0055119
Siegel, R. L., Miller, K. D., and Jemal, A. (2016). Cancer statistics, 2016. CA: Cancer J. Clin. 66, 7–30. doi: 10.3322/caac.21332
Stemple, D. L., and Anderson, D. J. (1992). Isolation of a stem cell for neurons and glia from the mammalian neural crest. Cell 71, 973–985. doi: 10.1016/0092-8674(92)90393-Q
Stratton, M. R., Campbell, P. J., and Futreal, P. A. (2009). The cancer genome. Nature 458, 719–724. doi: 10.1038/nature07943
Sun, Y., Xu, C., Pan, C., Chen, X., Geng, Y., Wu, Y., et al. (2019). Diffuse intrinsic pontine gliomas exhibit cell biological and molecular signatures of fetal hindbrain-derived neural progenitor cells. Neurosci. Bull. 35, 216–224. doi: 10.1007/s12264-018-00329-6
Terada, Y., Jo, N., Arakawa, Y., Sakakura, M., Yamada, Y., Ukai, T., et al. (2019). Human pluripotent stem cell-derived tumor model uncovers the embryonic stem cell signature as a key driver in atypical teratoid/rhabdoid tumor. Cell Rep. 26, 2608–2621.e6. doi: 10.1016/j.celrep.2019.02.009
Theunissen, T. W., and Jaenisch, R. (2017). Mechanisms of gene regulation in human embryos and pluripotent stem cells. Development (Cambridge) 144, 4496–4509. doi: 10.1242/dev.157404
Thomas, J., Adegboyega, P., Iloabachie, K., Mooring, J. W., and Lian, T. (2011). Sinonasal teratocarcinosarcoma with yolk sac elements: a neoplasm of somatic or germ cell origin? Annals Diagn. Pathol. 15, 135–139. doi: 10.1016/j.anndiagpath.2010.01.004
Tiriac, H., Belleau, P., Engle, D. D., Plenker, D., Deschênes, A., Somerville, T. D. D., et al. (2018). Organoid profiling identifies common responders to chemotherapy in pancreatic cancer. Cancer Discovery 8, 1112–1129. doi: 10.1158/2159-8290.CD-18-0349
Vitte, J., Gao, F., Coppola, G., Judkins, A. R., and Giovannini, M. (2017). Timing of Smarcb1 and Nf2 inactivation determines schwannoma versus rhabdoid tumor development. Nat. Commun. 8:300. doi: 10.1038/s41467-017-00346-5
Vlachogiannis, G., Hedayat, S., Vatsiou, A., Jamin, Y., Fernández-Mateos, J., Khan, K., et al. (2018). Patient-derived organoids model treatment response of metastatic gastrointestinal cancers. Science 359, 920–926. doi: 10.1126/science.aao2774
Vladoiu, M. C., El-Hamamy, I., Donovan, L. K., Farooq, H., Holgado, B. L., Sundaravadanam, Y., et al. (2019). Childhood cerebellar tumours mirror conserved fetal transcriptional programs. Nature 572, 67–73. doi: 10.1038/s41586-019-1158-7
Vogelstein, B., Papadopoulos, N., Velculescu, V. E., Zhou, S., Diaz, L. A., and Kinzler, K. W. (2013). Cancer genome landscapes. Science 340, 1546–1558. doi: 10.1126/science.1235122
Wang, X., Lee, R. S., Alver, B. H., Haswell, J. R., Wang, S., Mieczkowski, J., et al. (2017). SMARCB1-mediated SWI/SNF complex function is essential for enhancer regulation. Nat. Genet. 49, 289–295. doi: 10.1038/ng.3746
Wang, X., Wang, S., Troisi, E. C., Howard, T. P., Haswell, J. R., Wolf, B. K., et al. (2019). BRD9 defines a SWI/SNF sub-complex and constitutes a specific vulnerability in malignant rhabdoid tumors. Nat. Commun. 10:1881. doi: 10.1038/s41467-019-09891-7
Westerlund, I., Shi, Y., Toskas, K., Fell, S. M., Li, S., Surova, O., et al. (2017). Combined epigenetic and differentiation-based treatment inhibits neuroblastoma tumor growth and links HIF2α to tumor suppression. Proc. Natl. Acad. Sci. U S A. 114, E6137–E6146. doi: 10.1073/pnas.1700655114
Westermark, U. K., Wilhelm, M., Frenzel, A., and Henriksson, M. A. (2011). The MYCN oncogene and differentiation in neuroblastoma. Sem. Cancer Biol. 21, 256–266. doi: 10.1016/j.semcancer.2011.08.001
Wikenheiser-Brokamp, K. A. (2006). Retinoblastoma family proteins: insights gained through genetic manipulation of mice. Cell. Mol. Life Sci. 63, 767–780. doi: 10.1007/s00018-005-5487-3
Wilson, B. G., and Roberts, C. W. M. (2011). SWI/SNF nucleosome remodellers and cancer. Nat. Rev. Cancer 11, 481–492. doi: 10.1038/nrc3068
Wu, G., Broniscer, A., McEachron, T. A., Lu, C., Paugh, B. S., Becksfort, J., et al. (2012). Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat. Genet. 44, 251–253. doi: 10.1038/ng.1102
Xu, X. L., Singh, H. P., Wang, L., Qi, D. L., Poulos, B. K., Abramson, D. H., et al. (2014). Rb suppresses human cone-precursor-derived retinoblastoma tumours. Nature 514, 385–388. doi: 10.1038/nature13813
Yao, Y., Xu, X., Yang, L., Zhu, J., Wan, J., Shen, L., et al. (2020). Patient-Derived organoids predict chemoradiation responses of locally advanced rectal cancer. Cell Stem Cell 26, 17–26.e6. doi: 10.1016/j.stem.2019.10.010
Young, M. D., Mitchell, T. J., Custers, L., Margaritis, T., Morales, F., Kwakwa, K., et al. (2020). Single cell derived mRNA signals across human kidney tumors. BioRxiv [preprint] doi: 10.1101/2020.03.19.998815
Young, M. D., Mitchell, T. J., Vieira Braga, F. A., Tran, M. G. B., Stewart, B. J., Ferdinand, J. R., et al. (2018). Single-cell transcriptomes from human kidneys reveal the cellular identity of renal tumors. Science 361, 594–599. doi: 10.1126/science.aat1699
Zagozewski, J., Shahriary, G. M., Morrison, L. C., Saulnier, O., Stromecki, M., Fresnoza, A., et al. (2020). An OTX2-PAX3 signaling axis regulates Group 3 medulloblastoma cell fate. Nat. Commun. 11:3627. doi: 10.1038/s41467-020-17357-4
Zhang, L., He, X., Liu, X., Zhang, F., Huang, L. F., Potter, A. S., et al. (2019). Single-Cell transcriptomics in medulloblastoma reveals tumor-initiating progenitors and oncogenic cascades during tumorigenesis and relapse. Cancer Cell 36, 302–318.e7. doi: 10.1016/j.ccell.2019.07.009
Zheng, C., Schneider, J. W., and Hsieh, J. (2020). Role of RB1 in human embryonic stem cell-derived retinal organoids. Dev. Biol. 462, 197–207. doi: 10.1016/j.ydbio.2020.03.011
Keywords: pediatric cancer, embryonal tumors, in vitro models, organoids, therapy
Citation: Custers L, Paassen I and Drost J (2021) In vitro Modeling of Embryonal Tumors. Front. Cell Dev. Biol. 9:640633. doi: 10.3389/fcell.2021.640633
Received: 11 December 2020; Accepted: 25 January 2021;
Published: 26 February 2021.
Edited by:
Benedetta Artegiani, Princess Maxima Center for Pediatric Oncology, NetherlandsReviewed by:
Vasiliy Galat, Ann & Robert H. Lurie Children’s Hospital of Chicago, United StatesPatompon Wongtrakoongate, Mahidol University, Thailand
Copyright © 2021 Custers, Paassen and Drost. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jarno Drost, Si5Ecm9zdEBwcmluc2VzbWF4aW1hY2VudHJ1bS5ubA==
†These authors have contributed equally to this work