 Ezekiel Uba Nwose1,2*
Ezekiel Uba Nwose1,2* Phillip Taderera Bwititi3
Phillip Taderera Bwititi3- 1School of Community Health, Charles Sturt University, Orange, NSW, Australia
- 2Department of Public and Community Health, Novena University, Kwale, Nigeria
- 3School of Biomedical Sciences, Charles Sturt University, Wagga Wagga, NSW, Australia
Introduction—Phenomenology
Autophagy or auto-phagocytosis is self-phagocytosis of tissues and cellular materials i.e., “self-maintenance.” While phagocytosis normally refers to the innate immune process of white blood cells engulfing foreign bodies such as infectious materials, autophagy is a regulatory phenomenon of dysfunctional cellular components being removed (Kobayashi, 2015). Autophagy is inseparable from inflammation and oxidative stress phenomena (Turkmen, 2017); which are intricately involved in pathophysiology of diabetes mellitus (DM) (Muriach et al., 2014). The relationships between phenomena is shown in Figure 1.
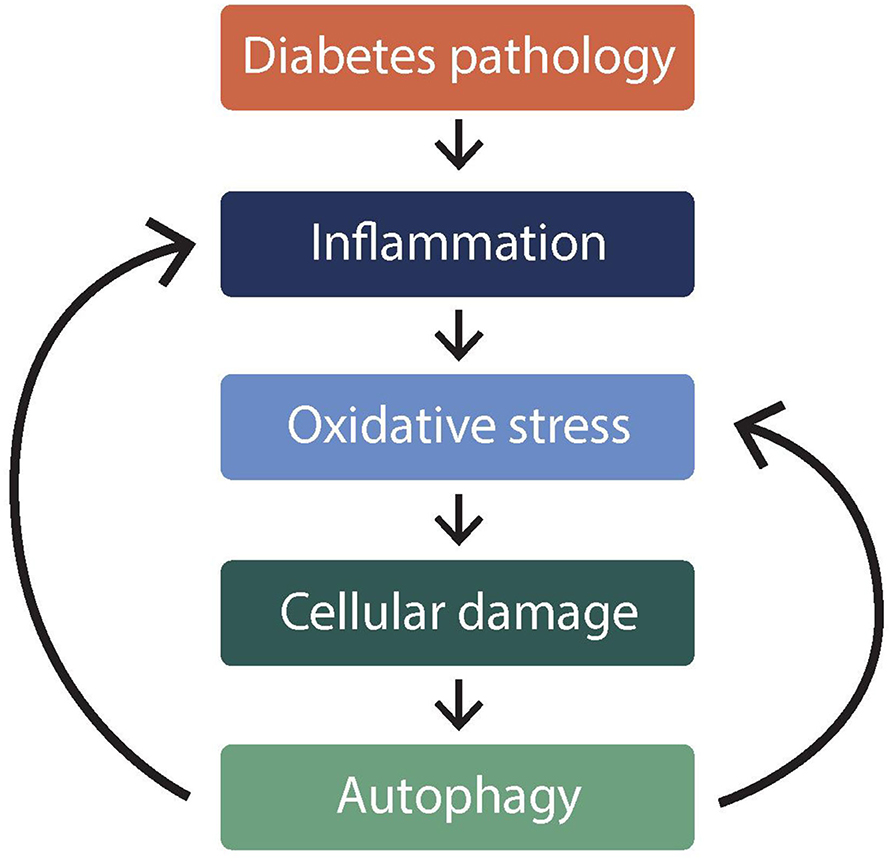
Figure 1. Illustration* of relationship between autophagy and oxidative stress in diabetes mellitus. *This figure illustrates how autophagy is inseparable from inflammation and oxidative stress phenomena. Figure shows that autophagy is caused by (downward arrows), but it also exacerbates (backward arrows), inflammation, and oxidative stress in diabetes.
Autophagy in association with oxidative stress is involved in pathophysiology. For instance, in the non-modifiable aging process, autophagy is involved in the associated oxidative stress and this can be assessed by glycation end-products as well as indices of lipid oxidation such as malondialdehyde (Moldogazieva et al., 2019). What is yet to be articulated for clinical translation in terms of laboratory assessment of autophagy is the concept of oxidative stress screening. A recent review highlights cell culture and electron microscopy methods (Yoshii and Mizushima, 2017) but not blood tests for oxidative stress indices. Thus, the gap between knowledge and practice are the apparent lack of acknowledgment of oxidative damage interplay between autophagy and metabolic diseases.
The objective of this paper is to bring to the fore the way in which clinical laboratory tests for oxidative stress panel can be used to assess autophagy in metabolic syndrome, especially the relevance of tests from different thematic sub-panels to establish cellular damage in metabolic syndrome. In this objective, cognizance is taken that metabolic syndrome is a constellation of diabetes and its cardiovascular complications including dyslipidaemia factor. For instance, abnormal cholesterol can exacerbate oxidative stress to increase autophagy in diabetes.
This opinion paper is organized in four sections. First three sections cover “causes, consequences, and therapeutic challenges” in terms of oxidative stress, effects on vascular physiology, and implications for management by laboratory methods, respectively. A brief fourth section is on availability of clinical laboratory tests for oxidative stress panel and how to interpret the results in terms of autophagy-inflammation interplay.
Causes of Autophagy in Diabetes—Erythrocyte Oxidative Stress (EOS) Perspective
Cellular oxidative stress can induce mitochondrial damage, which then requires autophagy to maintain homeostasis (Lee et al., 2012). Oxidative stress is a disturbance of the physiological control of oxidant/antioxidant (redox) balance, in which oxidants become dominant. It is a state in which a cell experiences alteration of cellular components, due to exposure to free radicals and other reactive oxygen species (ROS) beyond its antioxidant capacity (Sies, 1991). Redox reactions are essential for cellular functions such as the utilization of chemical energy from nutrients for the production of adenosine triphosphate. However, excessive oxidants expose cells including erythrocytes to oxidative stress (Kuhn et al., 2017).
EOS is a type of cellular oxidative stress, which arises from over-exposure of the cellular components of red blood cells to various ROS (Richards et al., 1998). It is a situation whereby the erythrocyte's functional mechanisms are impaired or overwhelmed by alteration in the normal metabolic and/or physiological activities that generate ROS (Taniyama and Griendling, 2003). Although the red cell has an efficient antioxidant system for the normal levels of oxidants generated in its membranes, the oxidant challenge can exceed the capacity of the antioxidant system (Fung and Zhang, 1990).
There is a tendency for imperfect reduction of oxygen in the mitochondrial electron transport systems e.g., the leak of superoxide radicals (Maxwell and Lip, 1997). The cascade reaction induced by the superoxide radicals involves antioxidant function of “reduced” glutathione (GSH) that leads to reduction in concentration and exacerbates EOS (McMullin, 1999; Boada et al., 2000; Ulusu et al., 2003). Thus, there are three possible sources of ROS that predispose the erythrocytes to oxidative stress:
• There exist special channels on the membrane by which superoxide radicals permeate the erythrocyte from the mitochondria of other cells (Richards et al., 1998). This is more so during hyperglycaemia or dyslipidaemia (Taniyama and Griendling, 2003). A discussed how EOS is strongly implicated in diabetes and its cardiovascular complications (Nwose et al., 2007a). Laboratory-based investigations have also reported on erythrocyte morphology or oxidative stress being associated with oxidative stress (Parthiban et al., 1995; Nwose et al., 2009; Gyawali et al., 2015). What is being advanced here is the EOS interplay with autophagy in metabolic syndrome (Figure 1).
• Secondly, the erythrocyte can paradoxically become oxidatively stressed from normal physiological processes (Kuhn et al., 2017). Due to the role of erythrocytes in oxygen transport and the presence of redox-active hemoglobin molecules, they generate pro-oxidant radicals by the Fenton reaction.
• Thirdly, there is hyperglycaemia-induced oxidative stress. Besides glycolysis being associated with oxidative stress in diabetic cardiovascular physiology (Zinman, 2003; Brownlee, 2005), there are points of pro-oxidant production when the erythrocyte is utilizing glucose to generate energy in the pentose phosphate pathway (Nwose et al., 2007a).
• Cellular oxidative stress can induce mitochondrial damage, which then requires autophagy to maintain homeostasis.
Therefore, point of emphasize is that fragments of damaged red blood cell materials are removed from the system by the phagocytosis function of the spleen—the basic cleaning function of the blood by spleen. In the context of splenectomy, red blood cells tend to acquire autophagic vacuoles (Holroyde and Gardner, 1970). Current research is yet to translate the basic science that EOS is followed by splenic autophagy of the damaged red blood cells. Therefore, what is being brought to the fore is a measurable perspective of autophagy in terms of EOS that is integral to diabetes and associated metabolic syndrome indices.
Consequences—Potential Implications of Autophagy-EOS Interplay in Diabetes
There can be autophagy of pancreatic beta-cells (Marasco and Linnemann, 2018), and this has implications, but is not the focus of this discussion. Mitochondrial oxidative stress and autophagy are implicated in diabetes (Muriach et al., 2014); and although red blood cells lack mitochondria, there are potential effects on glucose metabolic pathways. Whether in glycolysis or pentose phosphate pathway, the physiology to meet the cellular need of energy in the erythrocyte is associated with the propensity to deplete GSH content, which in turn leads to EOS. The aberrant state of EOS is of clinical importance in diabetes (Nwose et al., 2007a), especially because hyperglycaemia exacerbates oxidative stress (Yano et al., 2004).
In the diabetes pathophysiology, GSH level is depleted as it is converted to oxidized glutathione. This leads to reduced erythrocyte antioxidant capacity including impaired vitamin E regeneration system (Nwose et al., 2008a), which feedforward to constitutes a possible cause of EOS (Nwose et al., 2007a). Methaemoglobin reductase activity is the other pathway, which impairs GSH functions and potentially complicates the entire vitamin E recycling system (Nwose et al., 2008a).
Thus, diabetes is associated with a decrease of erythrocyte GSH level, which translates to antioxidant imbalance of the cell. Prolonged impairment of the vitamin E recycling in the red blood cell membrane amounts to EOS that damages the cell (Nwose et al., 2008a). What is being brought to the fore is that hyperglycaemia-induced EOS can cause autophagy and may complicate a cause-and-effect phenomenon in diabetes (Figure 1). Therefore, the laboratory perspective is to view autophagy and associated challenges as follows:
• Causes—oxidative stress
• Consequences—effects on cardiovascular physiology
• Therapeutic challenges—implications for management by oxidative stress panel screening.
Therapeutic Challenges
Implications for Management by Laboratory Methods
For over two decades, research has demonstrated that changes in erythrocyte antioxidant and haem components in DM lead to complications such as cardiovascular diseases (Dominguez et al., 1998; Dumaswala et al., 2001; Memisogullari et al., 2003), but the question is how EOS is involved in macrovascular complications of DM. As illustrated (Nwose et al., 2007a), EOS may effect macrovascular events including increased blood viscosity, hypercoagulation, and endothelial dysfunction. It is noteworthy that these vascular events constitute Virchow's triad, which has been a subject of research (Makin et al., 2002; Lowe, 2003; Nwose et al., 2014); and shrouded in discussion (Bagot and Arya, 2008; Dickson, 2009; Malone and Agutter, 2009). There is also the effect of tissue hypoxia as discussed that can lead to high blood pressure hence exacerbate metabolic syndrome.
Endothelial Dysfunction Exacerbated by Metabolic Syndrome
There is knowledge of hyperglycaemia-induced endothelial dysfunction (De Vriese et al., 2000; Zinman, 2003); and that diabetic dyslipidaemia (component of metabolic syndrome) initiates a chronic inflammatory reaction that results in endothelial damage, which culminates in endothelial dysfunction such as atherosclerosis and coronary artery disease (Gonzalez and Selwyn, 2003). It is established that oxidative stress results in endothelial dysfunction, which has plasma homocysteine levels as clinical index (Dumaswala et al., 2001; Nwose et al., 2007a).
Blood Viscosity
This is an intrinsic resistance of blood flow in the vascular system (Lowe et al., 1997). Normally, erythrocyte membrane deformability is a physical property that enables cells to change shape and flow with little or no aggregation/friction. When EOS occurs through lipid peroxidation within the membrane, the cell membrane becomes more rigid and less adaptable (Suda et al., 1980). This makes the blood more viscous, which leads to the development of vascular abnormalities including atherothrombosis and endothelial dysfunction that are associated with coronary artery disease (Solans et al., 2000), as well as tissue hypoxia (El-Sayed et al., 2005). The implication in diabetes and dyslipidaemia has been highlighted (Nwose, 2010, 2013; Richards and Nwose, 2010; Nwose et al., 2014). The point advanced here is the potential, as part of oxidative damage indices for screening splenic autophagy of red blood cellular materials after EOS.
Imbalance of Coagulation and Fibrinolysis
There are several theories surrounding hypercoagulation in DM. For instance, hypo-fibrinolysis occurring as thrombomodulin-thrombin complex—which is formed on intact vascular endothelium—may activate thrombin-activatable fibrinolysis inhibitor (Yano, 2003). This suggestion is supported by the observation that hyperglycaemia and insulin enhance the synthesis and secretion of plasminogen activator inhibitor type 1 (Kohler and Grant, 2000). These findings imply that fibrinolysis and therefore the generation of D-dimer are reduced in DM. A seemingly opposing theory is that EOS leads to enhancement of events such as increased production of procoagulant tissue factors at the gene level (Brownlee, 2005), which imply that D-dimer changes in diabetes have not been adequately explained.
It has been reported that some coagulation markers such as D-dimer and fibrinogen are elevated in DM (Sommeijer et al., 2004). Preliminary reports have also shown increased D-dimer levels in DM (Nwose et al., 2007b). Therefore, it is advanced that D-dimer constitutes a potential option for oxidative damage screening (Table 1).
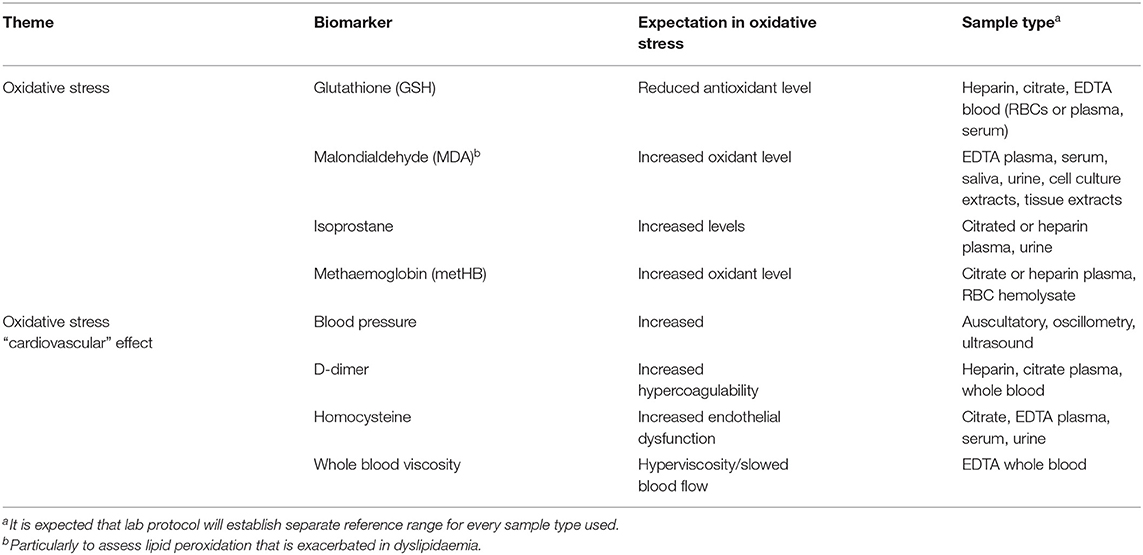
Table 1. Oxidative damage panel useable in the laboratory as oxidative stress screening.
EOS-Induced Haemolytic Anemia Exacerbating High Blood Pressure
There is a likelihood that hyperglycaemia-depleted GSH occurs via either reduced regeneration due to deficient pentose phosphate pathway (McMullin, 1999), or enhanced hexosamine pathway flux (Brownlee, 2005). The effect is EOS, which leads to a sequence of membrane rigidity and lysis (Fung and Zhang, 1990). The impacts are both hyperviscosity and anemia, respectively. That is, disruption of the normal rheological properties (El-Sayed et al., 2005), which leads to a sequence of anemia, reduced blood/O2 supply, ischaemia and subsequently angina, chronic ischaemic heart disease, myocardial infarction or sudden death (McCance et al., 2002). It is noteworthy that the homeostatic response to reduced blood/O2 supply involves increase in cardiac output, which may lead to high blood pressure. Hence, evaluation of fluctuations in blood pressure can support oxidative stress panel.
Indeed, anemia is associated with an increased risk of diabetic macrovascular disease or metabolic syndrome per se (Thomas et al., 2006). Autophagy is implicated in anemia (Grosso et al., 2017), but what has not been given adequate attention is that:
• EOS compromises the free radical basis of anemia (Dumaswala et al., 2001)
• Anemia is an effect of autophagy-related EOS
• To date, there are no defined signs and symptoms of autophagy for clinical decision making.
Oxidative Stress Panel Indices for Autophagy—Screening Suggestion
There are limited laboratory methods for autophagy evaluation (Klionsky et al., 2016), especially in diabetes research and practice. Thus, the justification of this paper is to advance the role of oxidative stress panel indices for the screening of autophagy in people living with diabetes. For instance, this could be used to monitor therapeutic management of cardiovascular complications of diabetes.
Diagnostic laboratory markers include the traditional risk evaluation markers such as cholesterol and glucose profiles. Emerging biomarkers include C-reactive protein, D-dimer, and homocysteine (Tracy, 2003; Ridker et al., 2004), as well as oxidative stress indices that include GSH and MDA (Tsimikas, 2006; Gyawali et al., 2015; Nwose et al., 2018; Lubrano et al., 2019). Uric acid and albumin levels have been considered (Bwititi et al., 2012). Pending clear definition of the signs and symptoms of autophagy, it is recommendable that the common laboratory principle applies whereby more than three positive results from this suite of biomarkers may indicate high levels of autophagy, be adopted.
It is becoming more clear that pharmacological agents to promote autophagy work by mediating oxidative stress (Wu et al., 2020). This implies that autophagy status is associated with degree of oxidative stress. The contribution being made in this paper is that methods for clinical evaluation of oxidative stress used in research can be integrated into clinical practice.
Further, it has been established that oxidative damage must involve reduction in antioxidant status, increase in oxidant levels and evidence of cardiovascular effects such as hypercoagulation, endothelial dysfunction, and blood viscosity that could easily lead to diseases (Nwose et al., 2008b). Thus, laboratory screening of oxidative damage includes indices of oxidative stress indices and their effects (Table 1). Oxidative stress markers listed here are just examples of what is readily available. There are a whole lot of others (Keaney et al., 2003; Frijhoff et al., 2015).
Perhaps, it is pertinent to emphasize that although assessment of EOS and its role in autophagy would be a potential therapeutic screening method, what needs to be addressed is “if it will be an effective indicator of overall metabolic dysregulation.” For this reason, it is pertinent to point out the concept of high cholesterol (indicated by lipid profile) being a factor that can exacerbate lipid peroxidation (i.e., oxidative stress) to increase autophagy in diabetes. Also, oxidative damage panel screening will show cardiovascular effect such as increased blood viscosity (Nwose, 2013). Thus, beyond e.g., blood glucose and cholesterol dysregulation, there are effects or subclinical pathophysiological indices.
This perspective recommendation advances the knowledge that oxidative stress induces autophagy (Hariharan et al., 2011; Turkmen, 2017; Gao, 2019; Moldogazieva et al., 2019). This therefore justifies carrying out laboratory tests for oxidative stress. The theme of this special issue, therapeutic challenges in the management of autophagy implies the need for monitoring of treatment using tests that assess therapeutic efficacy. It has recently been highlighted that targeting autophagy is a means to counteract oxidative stress in obesity (Pietrocola and Bravo-San Pedro, 2021). Another advantage is therefore identification of need for change in treatment regimen. For instance, there is indication of possible simultaneous induction of oxidative stress and autophagy in diabetes as well as obese patients (Klionsky et al., 2016). What this opinion paper contributes is the need to look at a panel of tests and the availability of the test methods.
Conclusion
Current considerations of autophagy acknowledge oxidative stress in the cause-and-effect physiology as well as therapeutic modes of action. However, monitoring by laboratory methods is yet to integrate the available and validated oxidative stress parameters. What this opinion paper brings to fore is that tests for oxidative stress panel can be used to assess autophagy in metabolic syndrome, including diabetes and its cardiovascular complications, at no major cost.
Author Contributions
EN and PB conceptualized the opinion. EN did the initial draft of main text. PB did the revision as well as the abstract and conclusion. Both authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This opinion paper was predominantly based on, and represents a translational update of, several pieces of doctoral research work done more than 13 years ago. Kay Skinner from Charles Sturt University has kindly edited the manuscript to improve the English language and is hereby appreciated.
Abbreviations
DM, diabetes mellitus; EOS, erythrocyte oxidative stress; GSH, reduced glutathione; MDA, Malondialdehyde; ROS, reactive oxygen species.
References
Bagot, C. N., and Arya, R. (2008). Virchow's triad: a question of attribution. Br. J. Haematol. 143, 180–190. doi: 10.1111/j.1365-2141.2008.07323.x
Boada, J., Riog, T., Xavier, P., Gamez, A., Bartrons, R., Cascante, M., et al. (2000). Cells overexpressing fructose-2-6-bisphosphatase showed enhanced pentose phosphate pathway flux and resistance to oxidative stress. FEBS Lett. 480:261. doi: 10.1016/S0014-5793(00)01950-5
Brownlee, M. (2005). The pathobiology of diabetic complications: a unifying mechanism. Diabetes 54, 1615–1625. doi: 10.2337/diabetes.54.6.1615
Bwititi, P., Nwose, U., Nielsen, S., Ruven, H., Kalle, W., Richards, R., et al. (2012). Serum uric acid and albumin levels and estimated glomerular filtration rate: oxidative stress considerations. Aust. J. Med. Sci. 33, 82–87.
De Vriese, A. S., Verbeuren, T. J., Van de Voorde, J., Lameire, N. H., and Vanhoutte, P. M. (2000). Endothelial dysfuntion in diabetes. Br. J. Pharmacol. 130, 963–974. doi: 10.1038/sj.bjp.0703393
Dickson, B. C. (2009). Virchow's triad. Br. J. Haematol. 145:433. doi: 10.1111/j.1365-2141.2009.07617.x
Dominguez, C., Ruiz, E., Gussinye, M., and Carrascosa, A. (1998). Oxidative stress at onset and in early stages of type I diabetes in children and adolescents. Diabetes Care 21, 1736–1742. doi: 10.2337/diacare.21.10.1736
Dumaswala, U. J., Zhuo, L., Mahajan, S., Nair, P. N. M., Shertzer, H. G., Dibello, P., et al. (2001). Glutathione protects chemokine-scavenging and antioxidative defense functions in human RBC. Am. J. Physiol. Cell Physiol. 280, C867–C873. doi: 10.1152/ajpcell.2001.280.4.C867
El-Sayed, M. S., Ali, N., and El-Sayed, A. Z. (2005). Haemorheology in exercise and training. Sports Med. 35, 649–670. doi: 10.2165/00007256-200535080-00001
Frijhoff, J., Winyard, P. G., Zarkovic, N., Davies, S. S., Stocker, R., Cheng, D., et al. (2015). Clinical relevance of biomarkers of oxidative stress. Antioxid. Redox Signal. 23, 1144–1170. doi: 10.1089/ars.2015.6317
Fung, L. W., and Zhang, Y. (1990). A method to evaluate the antioxidant system for radicals in erythrocyte membranes. Free Radic. Biol. Med. 9, 289–298. doi: 10.1016/0891-5849(90)90003-2
Gao, Q. (2019). Oxidative stress and autophagy. Adv. Exp. Med. Biol. 1206, 179–198. doi: 10.1007/978-981-15-0602-4_9
Gonzalez, M. A., and Selwyn, A. P. (2003). Endothelial dysfunction, inflammation, and prognosis in cardiovascular disease. Am. J. Med. 115, 99S−106S. doi: 10.1016/j.amjmed.2003.09.016
Grosso, R., Fader, C. M., and Colombo, M. I. (2017). Autophagy: a necessary event during erythropoiesis. Blood Rev. 31, 300–305. doi: 10.1016/j.blre.2017.04.001
Gyawali, P., Richards, R. S., Bwititi, P. T., and Nwose, E. U. (2015). Association of abnormal erythrocyte morphology with oxidative stress and inflammation in metabolic syndrome. Blood Cells Mol. Dis. 54, 360–363. doi: 10.1016/j.bcmd.2015.01.005
Hariharan, N., Zhai, P., and Sadoshima, J. (2011). Oxidative stress stimulates autophagic flux during ischemia/reperfusion. Antioxid. Redox Signal. 14, 2179–2190. doi: 10.1089/ars.2010.3488
Holroyde, C. P., and Gardner, F. H. (1970). Acquisition of autophagic vacuoles by human erythrocytes physiological role of the spleen. Blood 36, 566–575. doi: 10.1182/blood.V36.5.566.566
Keaney, J. F., Larson, M. G., Vasan, R. S., Wilson, P. W. F., Lipinska, I., Corey, D., et al. (2003). Obesity and systemic oxidative stress: clinical correlates of oxidative stress in the Framingham study. Arterioscler. Thromb. Vasc. Biol. 23, 434–439. doi: 10.1161/01.ATV.0000058402.34138.11
Klionsky, D. J., Abdelmohsen, K., Abe, A., Abedin, M. J., Abeliovich, H., Acevedo Arozena, A., et al. (2016). Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 12, 1–222. doi: 10.1080/15548627.2015.1100356
Kobayashi, S. (2015). Choose delicately and reuse adequately: the newly revealed process of autophagy. Biol. Pharm. Bull. 38, 1098–1103. doi: 10.1248/bpb.b15-00096
Kohler, H. P., and Grant, P. J. (2000). Plasminogen-activator inhibitor type 1 and coronary artery disease. New Engl. J. Med. 342, 1792–1801. doi: 10.1056/NEJM200006153422406
Kuhn, V., Diederich, L., Keller, T. C. S., Kramer, C. M., Lückstädt, W., et al. (2017). Red blood cell function and dysfunction: redox regulation, nitric oxide metabolism, anemia. Antioxid. Redox Signal. 26, 718–742. doi: 10.1089/ars.2016.6954
Lee, J., Giordano, S., and Zhang, J. (2012). Autophagy, mitochondria, and oxidative stress: cross-talk and redox signalling. Biochem. J. 441, 523–540. doi: 10.1042/BJ20111451
Lowe, G. D. (2003). Virchow's triad revisited: abnormal flow. Pathophysiol. Haemost. Thromb. 33, 455–457. doi: 10.1159/000083845
Lowe, G. D., Lee, A. J., Rumley, A., Price, J. F., and Fowkes, F. G. (1997). Blood viscosity and risk of cardiovascular events: the Edinburgh artery study. Br. J. Haematol. 96, 168–173. doi: 10.1046/j.1365-2141.1997.8532481.x
Lubrano, V., Pingitore, A., Traghella, I., Storti, S., Parri, S., Berti, S., et al. (2019). Emerging biomarkers of oxidative stress in acute and stable coronary artery disease: levels and determinants. Antioxidants (Basel) 8:115. doi: 10.3390/antiox8050115
Makin, A., Silverman, S. H., and Lip, G. Y. H. (2002). Peripheral vascular disease and Virchow's triad for thrombogenesis. QJM 95, 199–210. doi: 10.1093/qjmed/95.4.199
Malone, P. C., and Agutter, P. S. (2009). Is 'Virchow's triad' useful? Br. J. Haematol. 145:839. doi: 10.1111/j.1365-2141.2009.07685.x
Marasco, M. R., and Linnemann, A. K. (2018). β-Cell autophagy in diabetes pathogenesis. Endocrinology 159, 2127–2141. doi: 10.1210/en.2017-03273
Maxwell, S. R. J., and Lip, G. Y. H. (1997). Free radicals and antioxidants in cardiovascular disease. Br. J. Clin. Pharmacol. 44, 307–317. doi: 10.1046/j.1365-2125.1997.t01-1-00594.x
McCance, K. L., Grey, T. C., and Huether, S. E. (2002). “Altered cellular and tissue biology,” in Pathophysiology: The Biologic Basis for Disease in Adults and Children, 4th Edn, ed K. L. McCance (Maryland Heights, MO: Mosby), 48–51.
McMullin, M. F. (1999). The molecular basis of disorders of red cell enzymes. J. Clin. Pathol. 52, 241–244. doi: 10.1136/jcp.52.4.241
Memisogullari, R., Taysi, S., Bakan, E., and Capoglu, I. (2003). Antioxidant status and lipid peroxidation in type II diabetes mellitus. Cell Biochem. Funct. 21, 291–296. doi: 10.1002/cbf.1025
Moldogazieva, N. T., Mokhosoev, I. M., Mel'nikova, T. I., Porozov, Y. B., and Terentiev, A. A. (2019). Oxidative stress and advanced lipoxidation and glycation end products (ALEs and AGEs) in aging and age-related diseases. Oxid. Med. Cell. Longev. 2019:3085756. doi: 10.1155/2019/3085756
Muriach, M., Flores-Bellver, M., Romero, F. J., and Barcia, J. M. (2014). Diabetes and the brain: oxidative stress, inflammation, and autophagy. Oxid. Med. Cell. Longev. 2014:102158. doi: 10.1155/2014/102158
Nwose, E. U. (2010). Whole blood viscosity assessment issues V: prevalence in hypercreatinaemia, hyperglycaemia, and hyperlipidaemia. North Am. J. Med. Sci. 2, 403–408. doi: 10.4297/najms.2010.2408
Nwose, E. U. (2013). Blood viscosity, lipid profile, and lipid peroxidation in type-1 diabetic patients with good and poor glycemic control: the promise and reality. North Am. J. Med. Sci. 5, 567–568. doi: 10.4103/1947-2714.118926
Nwose, E. U., Bwititi, P. T., and Chalada, M. J. (2018). Influence of anticoagulants on determination of H2O2 levels in blood: comparison of citrate and EDTA. Int. J. Pathol. Clin. Res. 4:083. doi: 10.23937/2469-5807/1510083
Nwose, E. U., Jelinek, H. F., Richards, R. S., and Kerr, P. G. (2007a). Erythrocyte oxidative stress in clinical management of diabetes and its cardiovascular complication. Br. J. Biomed. Sci. 64, 35–43. doi: 10.1080/09674845.2007.11732754
Nwose, E. U., Jelinek, H. F., Richards, R. S., and Kerr, P. G. (2008a). The “vitamin E regeneration system” (VERS) and an algorithm to justify antioxidant supplementation in diabetes–a hypothesis. Med. Hypotheses 70, 1002–1008. doi: 10.1016/j.mehy.2007.07.048
Nwose, E. U., Jelinek, H. F., Richards, R. S., Tinley, P., and Kerr, P. G. (2009). Atherothrombosis and oxidative stress: the connection and correlation in diabetes. Redox Rep. 14, 55–60. doi: 10.1179/135100009X392458
Nwose, E. U., Richards, R. S., and Bwititi, P. T. (2014). Cardiovascular risks in prediabetes: preliminary data on “vasculopathy triad.” North Am. J. Med. Sci. 6, 328–332. doi: 10.4103/1947-2714.136913
Nwose, E. U., Richards, R. S., Jelinek, H. F., and Kerr, P. G. (2007b). D-dimer identifies stages in the progression of diabetes mellitus from family history of diabetes to cardiovascular complications. Pathology 39, 252–257. doi: 10.1080/00313020701230658
Nwose, E. U., Richards, R. S., Kerr, P. G., Tinley, R., and Jelinek, H. F. (2008b). Oxidative damage indices for the assessment of subclinical diabetic macrovascular complications. Br. J. Biomed. Sci. 65, 136–141. doi: 10.1080/09674845.2008.11732817
Parthiban, A., Vijayalingam, S., Shanmugasundaram, K. R., and Mohan, R. (1995). Oxidative stress and the development of diabetic complications–antioxidants and lipid peroxidation in erythrocytes and cell membrane. Cell Biol. Int. 19, 987–993. doi: 10.1006/cbir.1995.1040
Pietrocola, F., and Bravo-San Pedro, J. M. (2021). Targeting autophagy to counteract obesity-associated oxidative stress. Antioxidants (Basel) 10:102. doi: 10.3390/antiox10010102
Richards, R. S., and Nwose, E. U. (2010). Blood viscosity at different stages of diabetes pathogenesis. Br. J. Biomed. Sci. 67, 67–70. doi: 10.1080/09674845.2010.11730293
Richards, R. S., Roberts, T. K., McGregor, N. R., Dunstan, R. H., and Butt, H. L. (1998). Erythrocyte antioxidant systems protect cultured endothelial cells against oxidant damage. Biochem. Mol. Biol. Int. 46, 857–865. doi: 10.1080/15216549800204402
Ridker, P. M., Brown, N. J., Vaughan, D. E., Harrison, D. G., and Mehta, J. L. (2004). Established and emerging plasma biomarkers in the prediction of first atherothrombotic events. Circulation 109(25 Suppl. 1), IV6–IV19. doi: 10.1161/01.CIR.0000133444.17867.56
Sies, H. (1991). Oxidative stress: from basic research to clinical application. Am. J. Med. 91, S31–S38. doi: 10.1016/0002-9343(91)90281-2
Solans, R., Motta, C., Sol,á, R., La Ville, A. E., Lima, J., Simeón, P., et al. (2000). Abnormalities of erythrocyte membrane fluidity, lipid composition, and lipid peroxidation in systemic sclerosis: evidence of free radical-mediated injury. Arthritis Rheum. 43, 894–900. doi: 10.1002/1529-0131(200004)43:4<894::AID-ANR22>3.0.CO;2-4
Sommeijer, D. W., MacGillavry, M. R., Meijers, J. C. M., Zanten, A. P., Reitsma, P. H., and Ten Cate, H. (2004). Anti-inflammatory and anticoagulant effects of pravastatin in patients with type 2 diabetes. Diabetes Care 27, 468–474. doi: 10.2337/diacare.27.2.468
Suda, T., Maeda, N., and Shiga, T. (1980). Effect of cholesterol on human erythrocyte membrane. A spin label study. J. Biochem. (Tokyo) 87, 1703–1713. doi: 10.1093/oxfordjournals.jbchem.a132914
Taniyama, Y., and Griendling, K. K. (2003). Reactive oxygen species in the vasculature: molecular and cellular mechanisms. Hypertension 42, 1075–1081. doi: 10.1161/01.HYP.0000100443.09293.4F
Thomas, M. C., Cooper, M. E., Rossing, K., and Parving, H. H. (2006). Anaemia in diabetes: is there a rationale to TREAT? Diabetologia 49, 1151–1157. doi: 10.1007/s00125-006-0215-6
Tracy, R. P. (2003). Thrombin, inflammation, and cardiovascular disease: an epidemiologic perspective. Chest 124(3 Suppl.), 49S−57S. doi: 10.1378/chest.124.3_suppl.49S
Tsimikas, S. (2006). Measures of oxidative stress. Clin. Lab. Med. 26, 571–590. doi: 10.1016/j.cll.2006.06.004
Turkmen, K. (2017). Inflammation, oxidative stress, apoptosis, and autophagy in diabetes mellitus and diabetic kidney disease: the Four Horsemen of the Apocalypse. Int. Urol. Nephrol. 49, 837–844. doi: 10.1007/s11255-016-1488-4
Ulusu, N. N., Sahilli, M., Avci, A., Canbolat, O., Ozansoy, G., Ari, N., et al. (2003). Pentose phosphate pathway, glutathione-dependent enzymes, and antioxidant defense during oxidative stress in diabetic rodent brain and peripheral organs: effects of stobadine and vitamin E. Neurochem. Res. 28, 815–823. doi: 10.1023/A:1023202805255
Wu, D., Zhong, P., Wang, Y., Zhang, Q., Li, J., Liu, Z., et al. (2020). Hydrogen sulfide attenuates high-fat diet-induced non-alcoholic fatty liver disease by inhibiting apoptosis and promoting autophagy via reactive oxygen species/phosphatidylinositol 3-kinase/AKT/mammalian target of rapamycin signaling pathway. Front. Pharmacol. 11:585860. doi: 10.3389/fphar.2020.585860
Yano, M., Hasegawa, G., Ishii, M., Yamasaki, M., Fukuri, M., Nakamura, N., et al. (2004). Short-term exposure of high glucose concentration induces generation of reactive oxygen species in endothelial cells: implication for the oxidative stress associated with postprandial hyperglycaemia. Redox Rep. 9, 111–116. doi: 10.1179/135100004225004779
Yano, Y. (2003). “Diabetes pathogenesis: thrombin-activatable fibrinolysis inhibitor implicated in vascular damage,” in Diabetes Week (Atlanta), 18.
Yoshii, S. R., and Mizushima, N. (2017). Monitoring and measuring autophagy. Int. J. Mol. Sci. 18:1865. doi: 10.3390/ijms18091865
Zinman, B. (2003). Pathways Leading to Diabetic Microvascular Complications and the Latest Therapies CME. Available online at: http://www.medscape.com/viewprogram/2636?src=search (accessed November 29, 2020).
Keywords: biomarkers, cause-and-effect, erythrocyte, laboratory tests, oxidative stress
Citation: Nwose EU and Bwititi PT (2021) Autophagy in Diabetes Pathophysiology: Oxidative Damage Screening as Potential for Therapeutic Management by Clinical Laboratory Methods. Front. Cell Dev. Biol. 9:651776. doi: 10.3389/fcell.2021.651776
Received: 11 January 2021; Accepted: 31 March 2021;
Published: 27 April 2021.
Edited by:
Joilson O. Martins, University of São Paulo, BrazilReviewed by:
Fabiola Castro, University of São Paulo, BrazilMariana Davanso, University of Tuiuti Do Paraná, Brazil
Kalhara Menikdiwela, Texas Tech University, United States
Copyright © 2021 Nwose and Bwititi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ezekiel Uba Nwose, ZW53b3NlQGNzdS5lZHUuYXU=