 Juan I. Young
Juan I. Young Susan Slifer1
Susan Slifer1 Jacqueline T. Hecht
Jacqueline T. Hecht Susan H. Blanton
Susan H. Blanton- 1John P. Hussman Institute for Human Genomics, Dr. John T. Macdonald Foundation Department of Human Genetics, Miller School of Medicine, University of Miami, Miami, FL, United States
- 2McGovern Medical School, University of Texas Health Science Center, Houston, TX, United States
Non-syndromic cleft lip with or without cleft palate (NSCLP) is the most common craniofacial birth defect. The etiology of NSCLP is complex with multiple genes and environmental factors playing causal roles. Although studies have identified numerous genetic markers associated with NSCLP, the role of epigenetic variation remains relatively unexplored. Because of their identical DNA sequences, monozygotic (MZ) twins discordant for NSCLP are an ideal model for examining the potential contribution of DNA methylation to non-syndromic orofacial clefting. In this study, we compared the patterns of whole genome DNA methylation in six MZ twin pairs discordant for NSCLP. Differentially methylated positions (DMPs) and regions (DMRs) were identified in NSCLP candidate genes, including differential methylation in MAFB and ZEB2 in two independent MZ twin pairs. In addition to DNA methylation differences in NSCLP candidate genes, we found common differential methylation in genes belonging to the Hippo signaling pathway, implicating this mechanosensory pathway in the etiology of NSCLP. The results of this novel approach using MZ twins discordant for NSCLP suggests that differential methylation is one mechanism contributing to NSCLP, meriting future studies on the role of DNA methylation in familial and sporadic NSCLP.
Introduction
Non-syndromic cleft lip with and without cleft palate (NSCLP) is a common birth defect occurring in 1/700-1000 livebirths, affecting 135,000 newborns worldwide. The severity varies based on laterality (bi- versus unilateral involvement) and extensiveness (involves only the lip, the primary palate and/or the secondary palate). The defect affects feeding initially and, in the long term, speech articulation, hearing, dental hygiene, dentition and overall well-being as there is always residual scarring. The cost of multiple surgical interventions exact a large emotional burden for patients and families, and high financial burden for families and society.
Despite many decades of research, only limited environmental agents and genetic causes of NSCLP are known (Chiquet et al., 2011; Letra et al., 2012, 2014; Cvjetkovic et al., 2015; Leslie et al., 2015, 2016a,b; Fish et al., 2017; Ludwig et al., 2017; Cox et al., 2018). While candidate gene and genome-wide studies of case-control and family-based datasets have identified causal/susceptibility loci and variants, the majority of the genetic causes remain elusive. Based on the analysis of multiplex families, in which there are multiple, but not necessarily first degree, affected relatives, there are likely extra-genic factors affecting penetrance and/or expression of the genetic liability. For example, MZ twins have a concordance rate of » 25–50% for NSCLP (compared to 1–3% in DZ twins) with a heritability estimate of approximately 70%, indicating that other factors play a role (Christensen, 1999; Grosen et al., 2011). It is likely that variation in multiple genes is etiologically necessary, with extra-genic factors contributing with a threshold effect.
Epigenetics is well-recognized as a critical mediator of the interplay between genes and the environment and influences phenotypic outcomes. DNA methylation, a major substrate of epigenetic information, regulates genome activity and provides an alternative mechanism for modulating cellular physiology compared to those arising solely from genetic changes (Esteller, 2008). Thus, a potential mechanism by which environmental factors could elicit altered tissue morphogenesis is variation in gene methylation. Support for this hypothesis comes from the observation that maternal use of the methyl-group donor folic acid may decrease the risk for NSCLP (Butali et al., 2013; Wallenstein et al., 2013).
Methylation differences within normal MZ twin pairs are well documented (Kaminsky et al., 2009; Czyz et al., 2012; Ollikainen et al., 2015). Importantly, methylation differences have been identified in MZ twin pairs discordant for a variety of congenital defects, including Beckwith-Wiedemann syndrome (OMIM 130650), congenital cataracts, congenital renal agenesis, cryptorchidism, Mayer-Rokitansky-Kuster-Hauser syndrome (OMIM 277000) and congenital heart disease (OMIM 217095), yielding insights into the underlying pathology (Weksberg et al., 2002; Rall et al., 2011; Tierling et al., 2011; Jin et al., 2014; Wei et al., 2015; Lu et al., 2017; Lyu et al., 2018). Given the relevance of DNA methylation in neural crest development, which plays a critical role in craniofacial development (Hu et al., 2014; Seelan et al., 2019), and the potential influence of epigenetics in the etiology of NSCLP, we compared genomic DNA methylation patterns between six pairs of monozygotic twins discordant for NSCLP.
Materials and Methods
Editorial Policies and Ethical Considerations
All DNA samples were collected and studied following informed and signed written consent from a parent or guardian. This study was approved by the University of Texas Health Science Center Committee for Protection of Human Subjects (HSC-MS-03-090) and the University of Miami Human Subject Research Office (IRB #20100505).
Dataset
Six monozygotic twin pairs discordant for NSCLP were ascertained and characterized as part of the larger genetic study of NSCLP that has been previously described (Chiquet et al., 2007; Morris et al., 2020). Briefly, participants were recruited at the craniofacial clinics affiliated with the UTHealth Science Center at Houston under IRB-approved protocols and written informed consent was obtained from parents and assents from the children. All participants were evaluated and syndromic forms of NSCLP were excluded. Study inclusion was irrespective of ethnicity and/or sex. Family history and a minimum of a three-generation pedigree were obtained from all families. The characteristics of the six pairs of MZ twins and their samples are listed in Table 1. All affected individuals were post-surgical repair. Saliva samples were collected using the Oragene® Saliva Collection Kit or the Oragene® Saliva Self−Collection Kit for Young Children (Sponge Kit) according to manufacturer’s protocol. Genomic DNA was isolated from saliva samples obtained from these male MZ twins discordant for NSCLP and from their available parents.
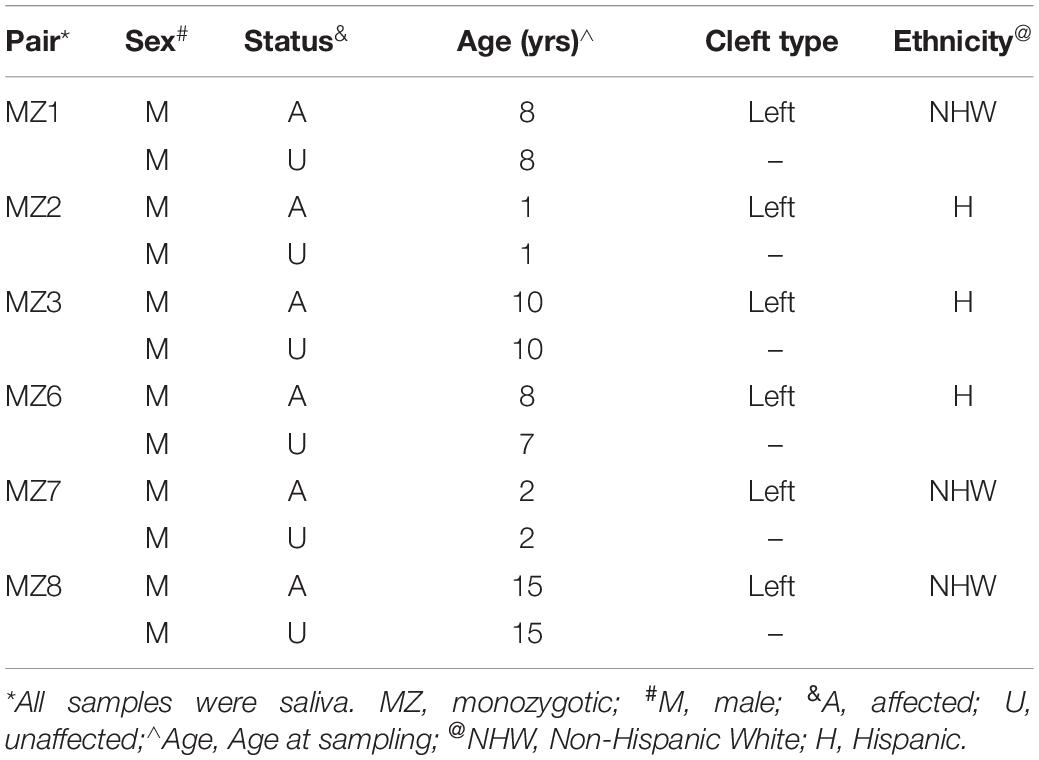
Table 1. Description of twin pairs.
Candidate Gene Assembly
A list of candidate genes for NSCLP was assembled from a thorough review of the literature of orofacial clefting including human association and sequencing studies and animal models. Genes associated with non-syndromic cleft lip with or without cleft palate, cleft palate only, and syndromic forms of clefting were included (Supplementary Table 1).
Whole Genome Sample Preparation and Sequencing
gDNA samples were evaluated for concentration by Qubit® fluorometric assay or by using Picogreen®. The integrity of the gDNA was assessed by loading approximately 100 ng per sample on a 0.75% agarose gel and comparing size distribution to a size marker (Quick-Load® 1 kb Extend DNA Ladder, NEB). 0.6–1 μg of gDNA was fragmented using the Covaris L-series or E-series instrument and sheared to a final insert size of approximately 400 bp. The purified fragmented DNA was repaired by the phosphorylation of the 5′ end and the conversion of overhangs to blunt ends. One “A” nucleotide was added to each 3′ end of the blunt fragments. The indexing-specific paired-end adaptors were ligated to the ends of the DNA fragments and the adaptor-ligated library was PCR amplified. Quality and quantity of the purified ligated products were verified using the Caliper LabChip® GX. Libraries were pooled and sequenced according to manufacturer’s instructions via the Illumina HiSeq X platform. Multiplexing 6 whole human genomes per flow cell generated an average depth of 30× per sample, exceeding read depth recommendations for Whole genome bisulfite sequencing (WGBS) analysis (Ziller et al., 2015).
Comparison of the whole genome sequencing (WGS, Table 2) between the affected and unaffected co-twins did not identify a pathogenic point mutation, insertion/deletion, or structural alteration in any of the known CLP-associated genes listed in Supplementary Table 1.

Table 2. Whole genome DNA sequencing statistics.
Whole Genome Bisulfite Sequencing
Whole genome bisulfite sequencing gDNA library preparations were carried out using the TruSeq DNA Sample Prep Kit v2 (Illumina Inc.) combined with sodium bisulfite conversion. Briefly, 1 μg of gDNA spiked with 5 ng unmethylated λ DNA (Promega) was fragmented to 200–300 bp peak size (Covaris, Woburn, MA, United States). Fragment size was analyzed on a Bioanalyzer DNA 1000 Chip (Agilent). End repair, sample purification with AMPure beads (Agencourt Bioscience Corp.), adenylation of 3′ ends, and adaptor ligation were carried out following the Illumina standard protocol. Ligation DNA was purified with AMPure, analyzed on a Bioanalyzer and subjected to bisulfite conversion using the MethylCode Bisulfite Conversion Kit (ThermoFisher) according to manufacturer’s instructions. Adaptor-ligated DNA was amplified through five cycles of PCR using the Hifi Uracil + DNA polymerase (Kapa Biosystems, Woburn, MA, United States) according to manufacturer’s protocol. Library quality was monitored using the Bioanalyzer (Agilent) and the concentration of fragments carrying adapters at both ends determined by quantitative PCR using the Library Quantification Kit from KAPA Biosystem. Paired-end DNA sequencing (124 bp each) using the Illumina Hi-Seq 2500 was performed.
Generated sequencing reads (average read length 120.9) were trimmed using Trim Galore1 and aligned to the bisulfite-converted human reference genome (GRCh37/hg19) using Burrows–Wheeler alignment. Methylation states were called using Bismark v0.10.0 (Krueger and Andrews, 2011). Confirmed clonal reads and reads with low mapping quality score were removed (Johnson et al., 2012), as well as, reads aligning to regions of the reference genome identified as troublesome for high- throughput sequencing aligners [DAC Blacklisted Regions (DBR) and Duke Excluded Regions (DER)2].
After data preprocessing, »54.3% of the resulting reads were uniquely mapped to the human reference genome. The median bisulfite conversion efficiency of CpGs was determined to be ∼98%. CpGs not covered by at least 20 reads were excluded, which resulted in a minimum of two reads per strand across all twelve samples. Reads on opposite strands with an absolute strand difference in methylation > 20% were excluded, leaving on average 2.4 × 106 unique and high-confidence CpGs per sample. Most CpG sites retained were highly methylated (over 50%, Supplementary Figure 1).
Array-Based Methylation Assay
Array-based methylation profiling was performed using the MethylationEPIC BeadChip (Illumina, San Diego, CA, United States) assay. Briefly, 300 ng gDNA per sample were bisulfite converted using the EZ-96 DNA Methylation kit (Zymo Research, Irvine, CA, United States) and processed as described by Illumina. The MethylationEPIC BeadChip measures over 850,000 CpG loci across a diverse set of functionally relevant genomic regions that include CpG islands (CGI) and promoters as well as CGI shores, gene body and intergenic CpGs. BeadChip images were scanned and the data analyzed using the R software (version 3.2.43). Raw intensity files (.idat) were obtained and processed using RnBeads package (Assenov et al., 2014). For quality control, we selected probes that had a detection p-value < 0.01 for all samples. The detection p-value represents the quality of the probes compared to background noise. Because all samples were male, we did not filter probes associated with sex. Methylated and unmethylated signal intensities were quantile normalized for each probe type separately and both beta and M values were estimated. The beta values are the ratio of the methylated signal intensity to the sum of both methylated and unmethylated signals after background subtraction (beta values range from 0 to 1, corresponding to completely unmethylated and fully methylated sites, respectively). M values are logit transformation of the beta values, which is M = log (beta/1-beta). M values were used as outcome variables for subsequent analysis because they have been shown to have better statistical properties such as homoscedasticity (Du et al., 2017). For the analysis of individual CpGs, an absolute delta beta > 0.30 was used to detect differential methylation between twins.
Identification of Differentially Methylated Positions Within Twin Pairs
From the strand-overlapping sites with > 20 reads, we selected differentially methylated sites based on the following criteria: (1) intra-twin pair difference in methylation levels of at least 30% (delta methylation > | 0.3|) and (2) a coefficient of variation in methylation levels of the unaffected individuals of less than 0.25. This strategy of restricting the search on differential methylation to sites in which the variation in unaffected twins is <25% is based on the idea that sites that exhibit large inter-individual variation in methylation levels have less regulatory potential than those sites in which the methylation levels are more tightly preserved between unaffected individuals.
DMR Analysis
Differentially methylated regions (DMR) analysis used a Bayesian hierarchical model (Wu et al., 2015) to detect regional methylation differences with ≥ 3 contiguous sites each with a methylation difference > 20% within a 2 kb distance with unidirectional methylation changes and a region-wise average methylation difference > 25%.
DNA Bisulfite Conversion and Bisulfite Specific PCR
Flanking primers (methylation-unbiased nested PCR primers) were designed for a subset of differentially methylated CpG sites and quantitative levels of methylation for each CpG dinucleotide were evaluated using the BiQ Analyzer software. Bisulfite-PCR was followed by cloning and Sanger sequencing of multiple clones for short-range mapping of methylation levels in selected loci. Genomic DNA (0.25 μg) was bisulfite converted using the EpiTect Kit (Qiagen) following the manufacturer’s protocol. Converted DNA was amplified with primers designed in MethPrimer. Optimized PCR reactions were followed by cloning using the TOPO TA Kit (Invitrogen). Multiple clones were sequenced. Bisulfite-cloning validated 70% of the DMS (Supplementary Figure 2).
Pyrosequencing Validation
Locus-specific pyrosequencing was conducted to validate the methylation data at selected loci. Pyrosequencing assays were designed and implemented by EpigenDx (Worcester, MA, United States) following the manufacturer’s recommended protocol. The correlation between the WGBS and pyrosequencing data was evaluated using the Spearman rank order correlation test. Pyrosequencing validated 73% of the DMS. Interestingly, the majority of validated DMSs had contiguous CpGs that also showed differential methylation in the same direction and with a similar magnitude to the targeted CpG.
Results
DNA Methylation Profiles Are Different in Monozygotic Twins Discordant for NSCLP
Methylome analysis was performed on saliva samples from six sets of male MZ twins discordant for NSCLP using whole-genome bisulfite sequencing (WGBS). Globally, DNA methylation profiles generated by WGBS were highly correlated across all individuals, indicating inter-individual conservation in methylation levels. The correlation of methylation patterns of the co-twins was significantly higher compared to unrelated individuals, indicative of low within-pair DNA methylation variation (average r = 0.84 for twins and r = 0.75 for unrelated individuals at 30X coverage; Figure 1A and Supplementary Figure 3). Removal of areas of extreme variability across individuals (variably methylated regions, VMRs) (Feinberg et al., 2010) from the analysis of the 30X coverage increased the correlation for intra-twin comparisons (average r = 0.93), but decreased it for unrelated samples (average r = 0.65) (Figure 1B). This pattern of correlation change suggests that the polymorphic nature of these VMRs is affected to a larger extent by environmental rather than genetic influences.
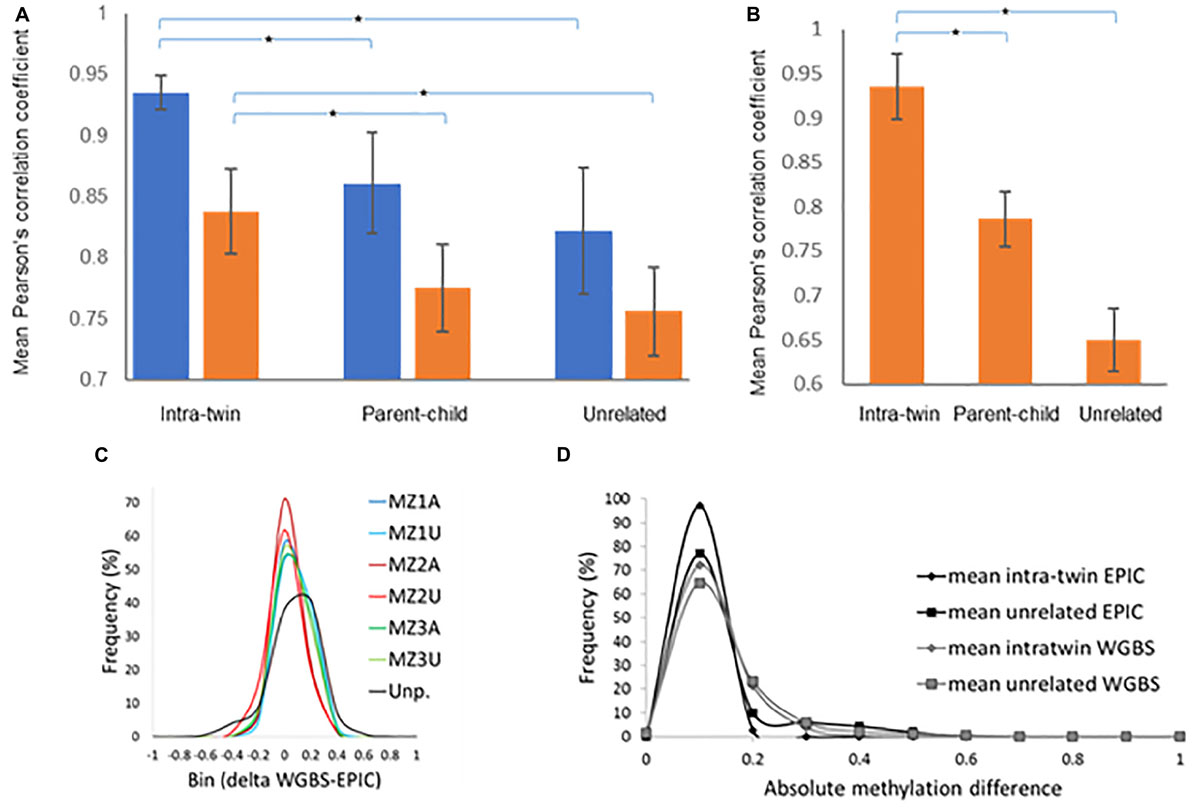
Figure 1. MZ co-twins are highly correlated for DNA methylation but have site-specific differences. (A) Pearson’s correlation coefficients derived from comparing intra-twin, parent-child and parent to parent (unrelated) methylation levels at 100X (blue) or 30X (brown) genome coverage. (B) Correlation coefficients using only variable CpGs as identified by Feinberg et al. (2010) at 30X genome coverage. (C) Comparison between DNA methylation values for 16,920 individual CpG loci generated by MethylationEPIC microarray and whole-genome bisulfite sequencing. Methylation values generated by Rnbeads for the MethylationEPIC microarray were subtracted from methylation levels calculated from the sequencing data for the same sample (MZ1A to MZ3U). Averaged comparisons contrasting EPIC and WGBS data from unpaired (Unp.) samples is also included (black line). (D) Distribution of average absolute differences in DNA methylation within the MZ twins as well as comparisons of unrelated individuals. A significantly (P<1.2e-10 for WGBS and P<1.9E-12 for EPIC, Kolmogorov–Smirnov test) smaller number of CpG sites with a large average within-twin differences in methylation level was observed in within twin comparisons as contrasted with comparisons of unrelated individuals (unaffected-unaffected, NSCLP-unaffected (inter-twins) or NSCLP-NSCLP).
The WGBS data was validated globally by subjecting DNA from three of the MZ pairs to the MethylationEPIC microarray (Illumina) assay. This assay interrogates the DNA methylation status of ∼865,000 CpG sites, which account for only ∼3% of all human autosomal CpGs. The methylation values obtained from the microarray were correlated with those from WGBS. WGBS data (prefiltered as described in the Methods section) were further filtered to include only CpGs present in the MethylationEPIC microarray, resulting in a total of 16,920 loci for comparison (Figure 1C). The mean methylation level correlation between WGBS and the microarray determinations for the six samples was r = 0.90 and, on average, 97.63% of loci have delta methylation levels < | 0.2|, indicating that the methylation values derived from the sequencing and the array methods are consistent across commonly interrogated loci.
To identify CpGs displaying differential methylation in any of the discordant twin pairs, the differences in CpG methylation between any two paired samples were estimated. Figure 1D shows the distribution of the mean absolute differences in DNA methylation levels per individual CpG within twins, as well as between unrelated samples. The overall distribution of average intra-twin DNA methylation differences showed a significant skew to the left as compared to the distribution of the average unrelated pair differences, with fewer CpG sites having large average differences in DNA methylation (Figure 1D). However, there is still variability in DNA methylation levels at individual CpG sites within the pairs of MZ twins.
DNA Methylation Differences in MZ Twin Pairs
The differentially methylated CpG sites (DMSs) identified within twin pairs is presented in Supplementary Table 2. The number of DMS per MZ pair varies from 7 (MZ2) to 42(MZ1), for a total of 151 DMSs associated with 147 genes. No single CpG is differentially methylated in more than one twin pair. This lack of overlap in DMS per pair is also seen at the gene level; there are no shared genes containing DMSs in the different MZ pairs. Table 3 lists the five genes with the most differential methylation per twin pair.
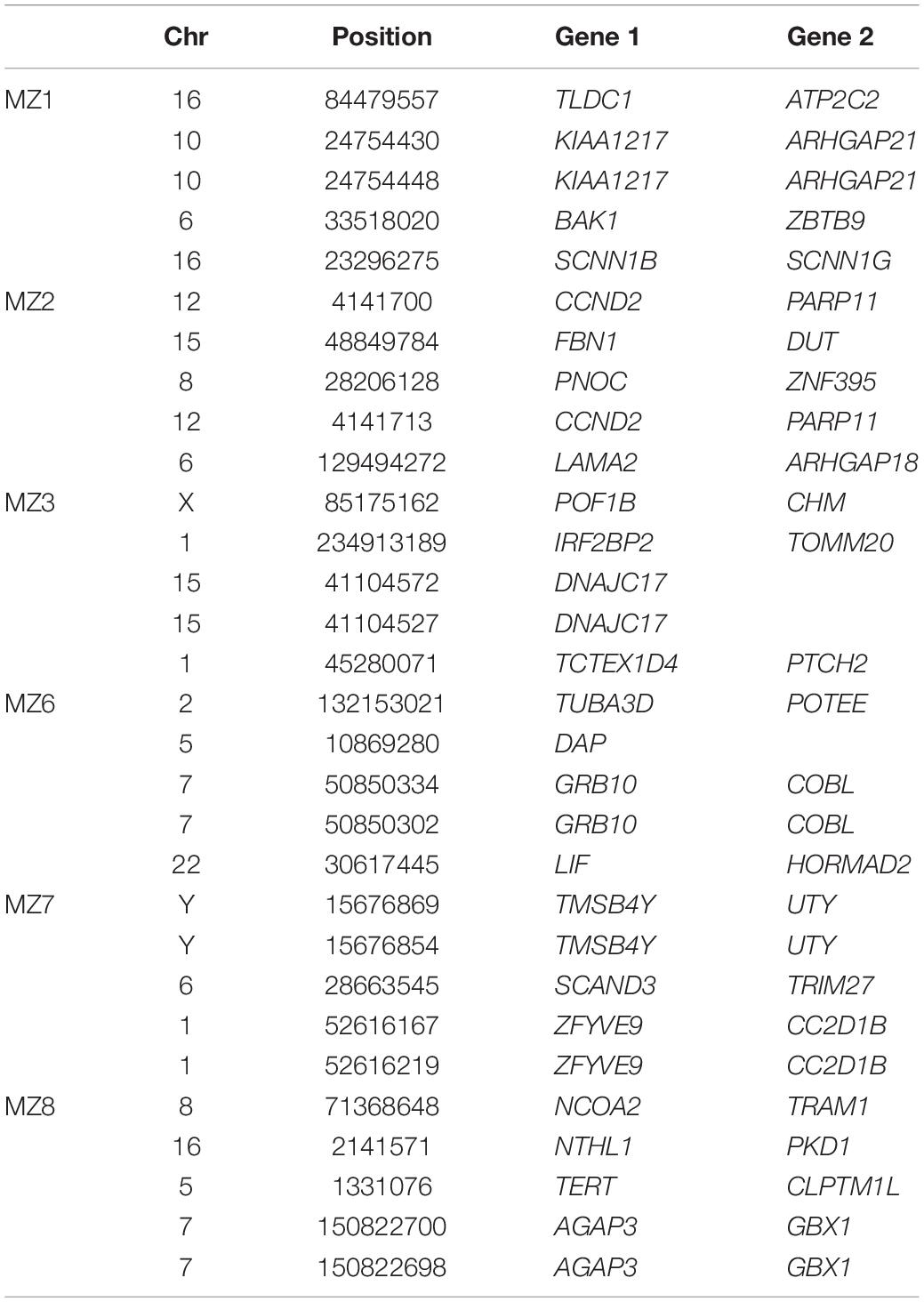
Table 3. Five most differentially methylated CpGs for each NSCLP twin pair.
To evaluate epigenetic involvement of candidate NSCLP genes, we tested whether any of the identified DMSs in the discordant twins were located in known or suspected NSCLP genes (Supplementary Table 1). Evidence of aberrant methylation in single CpGs was observed in seven NSCLP genes, four in MZ1, two in MZ2 and one in MZ6 (Table 4). Note that the number of DMSs in candidate genes is more than expected by chance in MZ1 (p-value < 0.003) and in MZ2 (p-value < 0.0001).
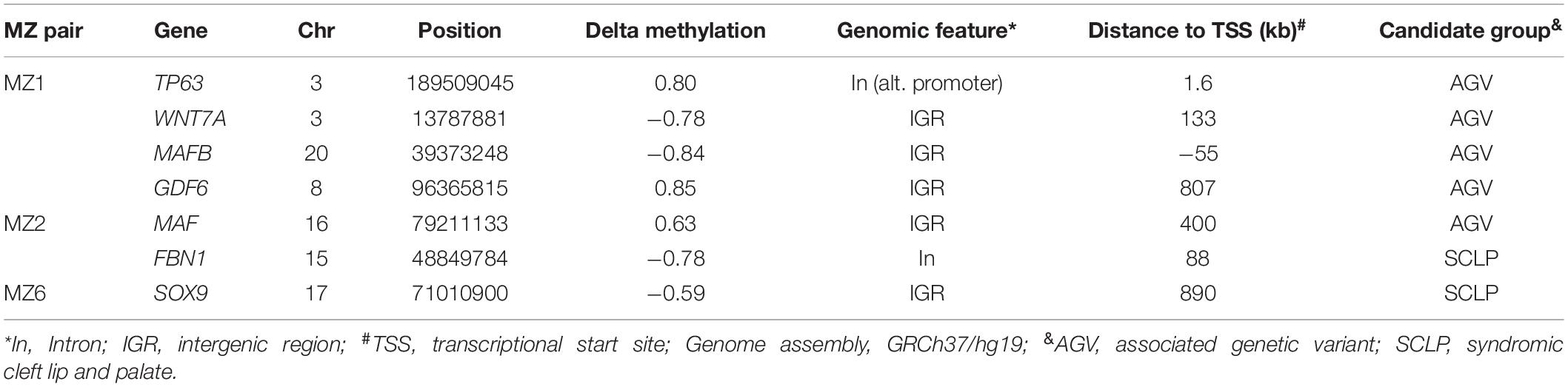
Table 4. Differentially methylated positions in candidate genes identified in three NSCLP twin pairs.
DNA methylation levels are strongly correlated among neighboring sites across the genome, and identification of clusters of differentially methylated positions rather than single CpGs usually predicts functional relevance (Eckhardt et al., 2006). Regional analysis is also less prone to be affected by the technical artifacts associated with individual sites. Thus, we performed systematic regional analyses of the WGBS data to identify differentially methylated regions (DMRs) in the MZ NSCLP discordant twins. Amongst the six MZ pairs, we identified a total of 768 DMRs, varying in numbers from 61 in MZ2 to 349 in MZ1 (Supplementary Table 2). Table 5 lists the 10 most differentially methylated DMRs per MZ twin pair. Candidate genes containing DMRs are listed in Table 6. While each twin pair had DMRs in one or more candidate genes, only twin pairs MZ1 MZ2, and MZ3 had an overrepresentation of candidate genes among all their respective DMRs (MZ1 p < 0.004, MZ2 p < 0.02, and MZ3 p < 0.0006). A gene-based overlap analysis detected only two candidate genes containing DMRs in more than a single MZ pair, MAFB (MZ6 and MZ8) and ZEB2 (MZ3 and MZ8). The DMRs in ZEB2 are in two different areas of intron 2 (AB011141) while the DMRs in MAFB are overlapping and located in the proximal upstream region.
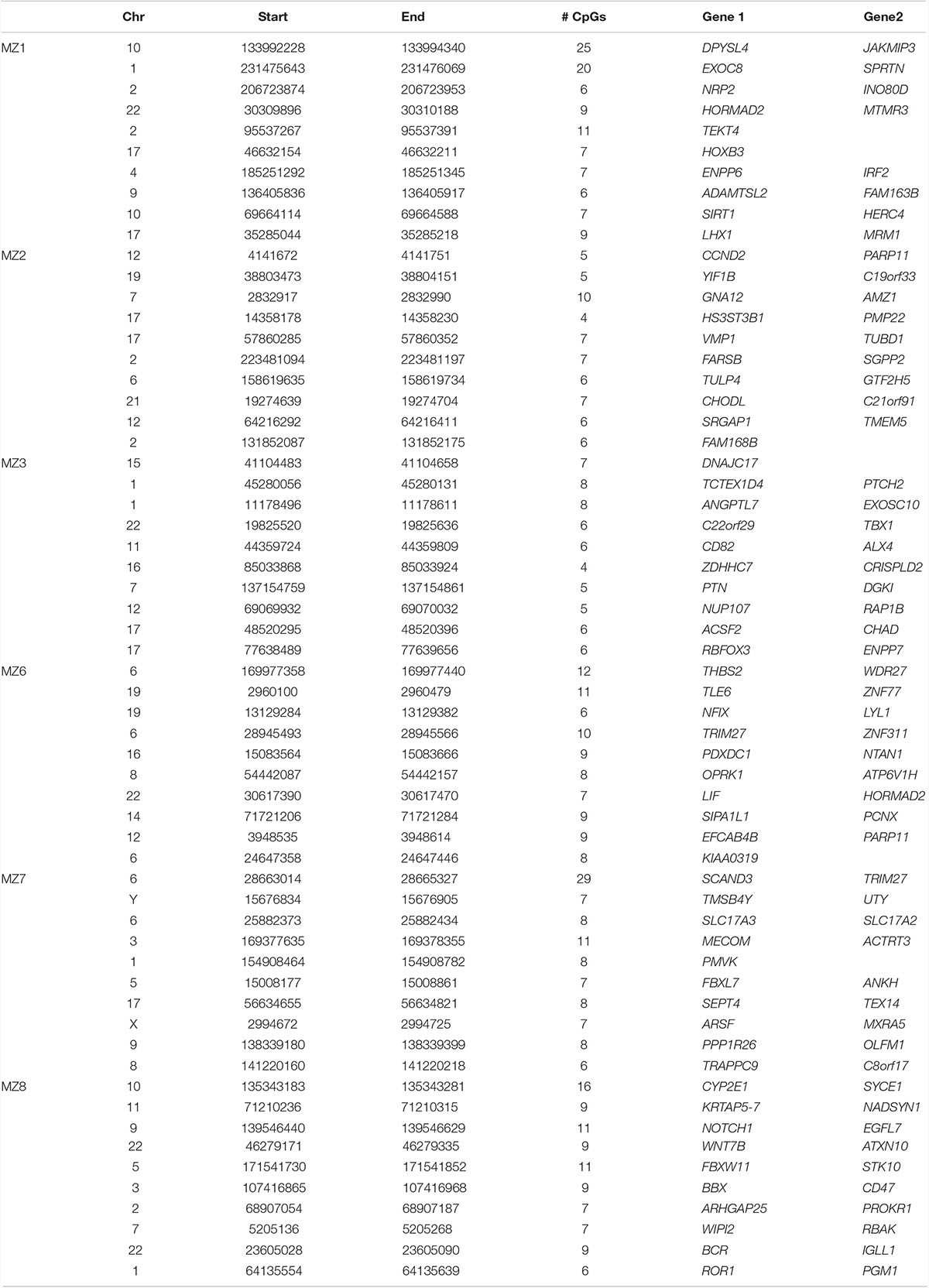
Table 5. Top ten differentially methylated regions in each NSCLP twin pair.
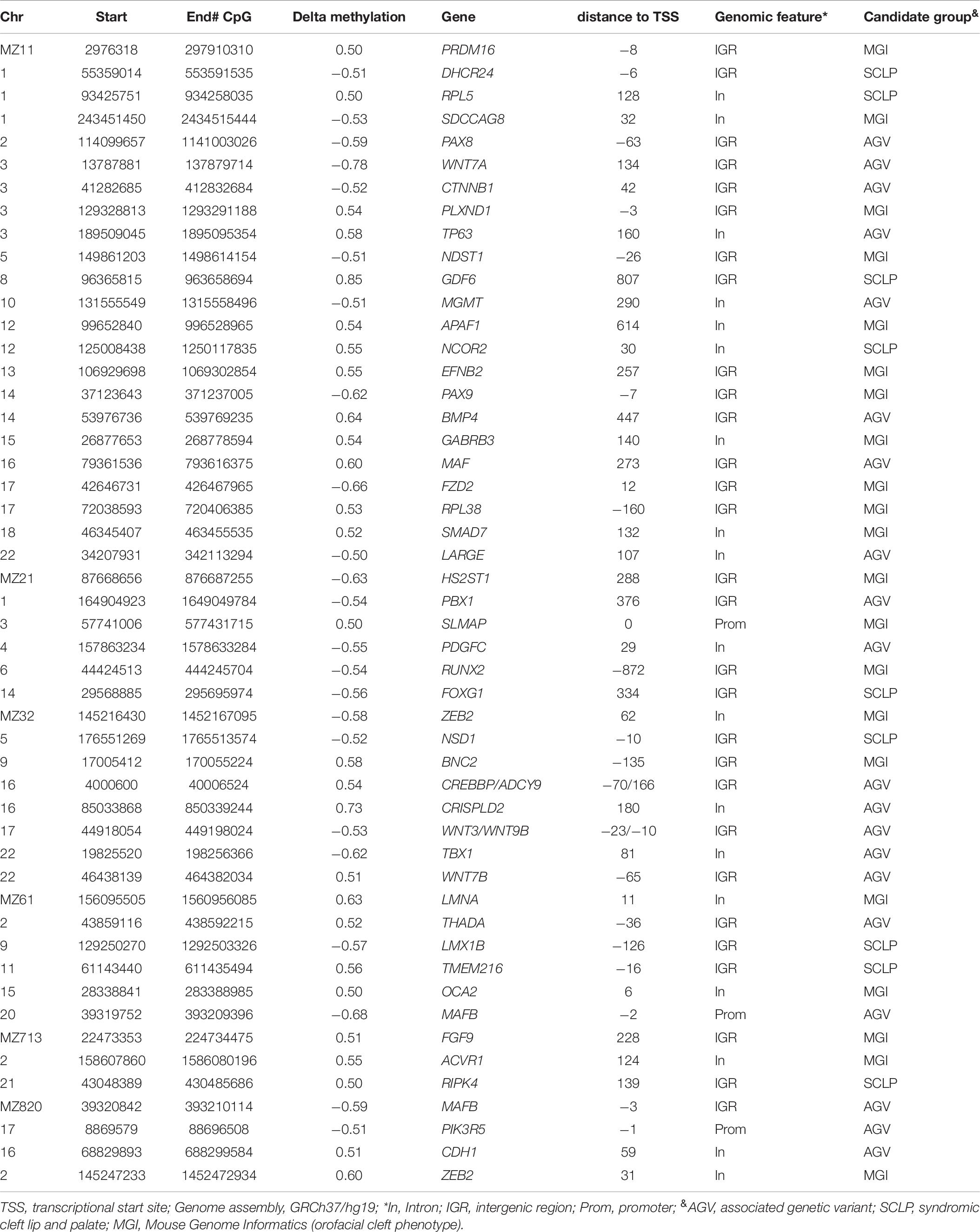
Table 6. Differentially methylated regions identified in candidate genes.
Enrichment of Identified DMRs in Shared Disease-Relevant Pathways
To identify enriched molecular pathways, EnrichR (Chen et al., 2013; Kuleshov et al., 2016) was used to analyze the DMR-gene set from each twin pair (Table 7). Across the six twin pairs, comparison of the top 10 significantly enriched Kegg pathways from all differentially methylated genes identified common enrichment of the “Hippo Signaling Pathway” in three pairs (MZ1, MZ3 and MZ8) and “Signaling pathways regulating pluripotency of stem cells” in two pairs (MZ1 and MZ3). Interestingly, pathway analysis using only our candidate genes identified enrichment in Hippo signaling in MZ1 (BMP4, FZD2, WNT7A, CTNNB1, GDF6, and SMAD7) and Wnt signaling in MZ3 (CREBBP, WNT9B, and WNT3). Additionally, we found that the combined set of genes identified as differentially methylated in any twin pair (set of 1,234 genes) was significantly enriched in the terms Osteoblast differentiation (p < 6.6E-8), and Neural crest migration (p < 4.1E-4) (Supplementary Figure 4).
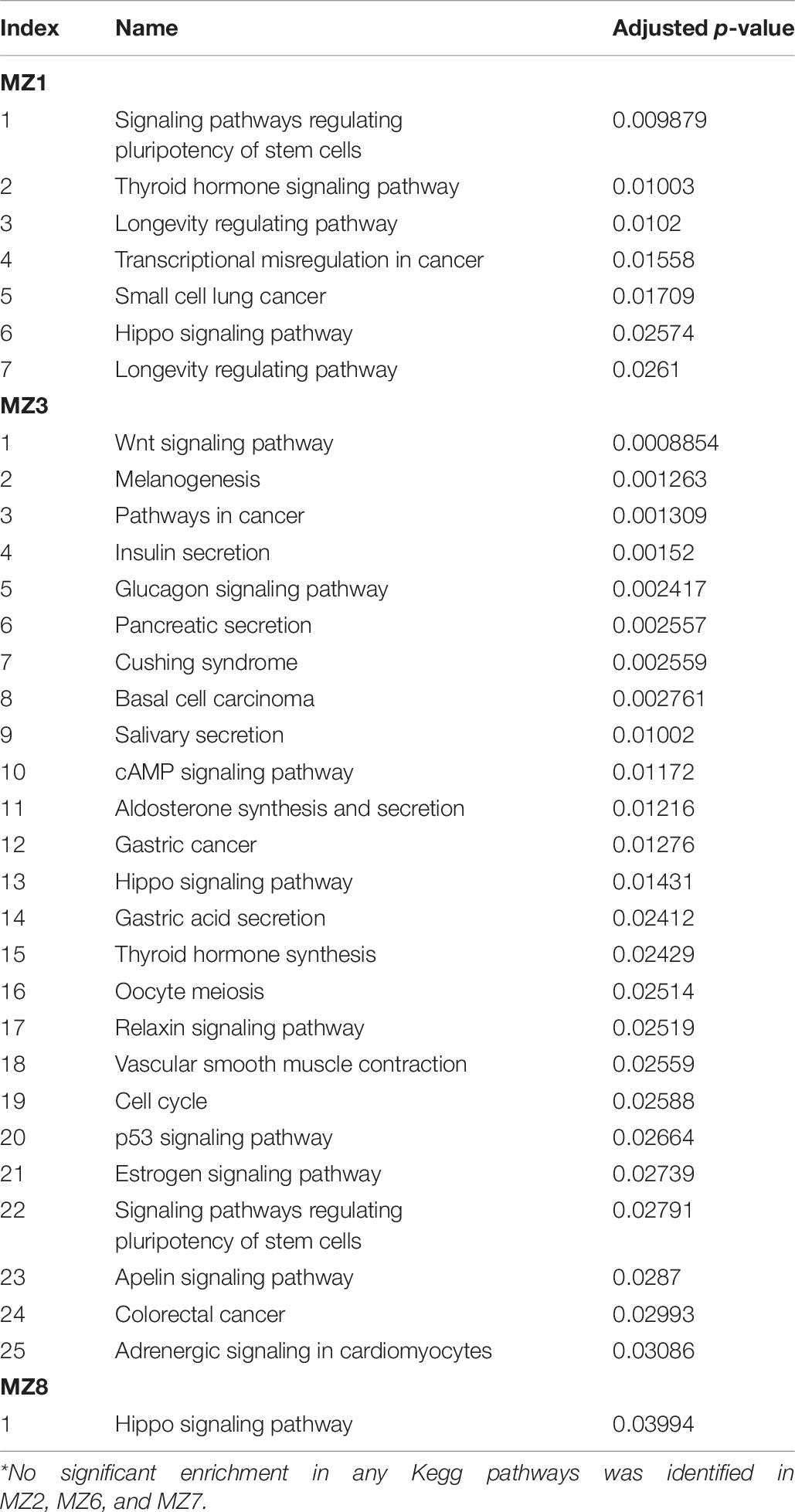
Table 7. Pathways enriched for differentially methylated genes*.
Between-Group Analyses Identified Additional DMRs
Group level DNA methylation differences between NSCLP cases and controls were also examined. Unlike the within-pair discordant MZ twin design, between-group DNA methylation differences can be attributed to both genetic and epigenetic factors. This analysis identified significantly different methylation regions only in the genes KCNAB3, NWD1, IFLTD1, and TRBJ2-3. No DMRs in NSCLP candidate genes were identified in this analysis.
Discussion
This is the first comprehensive study of epigenetic differences in MZ twins discordant for NSCLP. This epigenome-wide study was performed using saliva DNA from six male MZ twin pairs discordant for NSCLP to determine if differences in DNA methylation could explain the discordant NSCLP phenotype. Results from this study provide novel and exciting findings which point to the importance of methylation in craniofacial development.
We found regional methylation differences in a number of genes in each twin pair (range 61–349). Less than 3% (37 out of 1,338) of these genes were differentially methylated in more than one twin pair. A single gene, TRIM27, was differentially methylated in three pairs. TRIM27 has not been previously associated with clefting or craniofacial development but has been shown to be differentially methylated in prostate and lung cancers (Ji et al., 2020; Wang et al., 2020). Interestingly, differential methylation of TRIM27 was reported in a study comparing cleft lip only to cleft palate only (Sharp et al., 2017).
When we restricted our analysis to NSCLP candidate genes, evidence of methylation differences was found, suggesting their possible contribution to NSCLP through epigenetic variation. Of the 49 candidate genes which exhibited differential methylation, only two, MAFB and ZEB2, were differentially methylated in more than one twin pair. Importantly, the changes were all in the same direction, suggesting that these two genes are epigenetically important in orofacial development. This provides additional support for the previously reported association between NSCLP and MAFB, which is expressed in palatal shelves (Beaty et al., 2010). ZEB2 is involved in embryonic development of facial features (Ghoumid et al., 2013; Funato and Nakamura, 2017), and Zeb2 mutant mice exhibit alterations in neural crest migration and midfacial clefting (Van de Putte et al., 2003; Miyoshi et al., 2006). The lack of significant gene overlap points to the underlying heterogeneity of NSCLP.
Interestingly, although the majority of the identified DMRs do not lie in genes previously associated with NSCLP, pathway enrichment analysis revealed overrepresentation of the Hippo pathway in three of the six twin pairs. This signaling pathway was initially discovered as a key regulator of tissue growth in flies and regulates organ size and tissue repair by controlling cell proliferation, survival, mobility, stemness, and differentiation. The Hippo pathway is tightly regulated by both intrinsic and extrinsic signals, such as mechanical force, cell–cell contact, polarity, energy status, stress, and many diffusible hormonal factors. Studies have shown evidence of crosstalk between the Hippo pathway and other key signaling pathways, such as Wnt signaling (Li et al., 2019), and has recently been associated with craniofacial development (Wang et al., 2016; Wang and Martin, 2017; Sun et al., 2018) and mouse secondary palate formation (Goodwin et al., 2020). The common enrichment in this pathway is not due to alterations in a small subset of similar genes in all pairs (see Supplementary Figure 5). Rather, there is pathway convergence through alterations in MZ pair-specific methylation in different genes. A comparison of our results with a recent orofacial clefting case-control report revealed a little overlap (Alvizi et al., 2017). Interestingly, this overlap includes the NSCLP candidate gene MAFB (Supplementary Table 3). The different findings between these studies provides further evidence for complex genetic and epigenetics involvement in the etiology of NSCLP.
It is well known that genetic variation in a single gene can cause syndromic cases of orofacial clefting. It is therefore possible that extreme epigenetic variation in a few key genes could produce a clefting phenotype. However, it is more likely that the effects of most DNA methylation changes are subtle, similar to some non-coding genetic variants, and that multiple changes act in concert to ultimately contribute to non-sydromic orofacial clefting, as discussed above. The observation of DMPs in genes previously implicated in NSCLP, such as TP63, TBX1, CRISPLD2, CREBBP, and BMP4, suggest that de novo epimutations, which could imitate genetic mutations (Martin et al., 2005), are likely involved in the pathogenesis of NSCLP. Since we found methylation changes in several NSCLP candidate genes in each affected twin, we propose that an “epigenetic load” mechanism is involved in the dysregulation of orofacial morphogenesis, rather than a single dysfunctional NSCLP gene. This is consistent with the accepted multifactorial inheritance of NSCLP (Carter, 1969; Hecht et al., 1991; Howe et al., 2018). Based on the important role of epigenetic mechanisms in regulating gene expression, it is likely that methylomic variation could mediate disease susceptibility by cumulative altered gene expression, similar to genetic variation in regulatory regions of the genome.
The use of saliva as a proxy for the lip and palatal tissues is justified by their common developmental origin (Patel and Hoffman, 2014). Salivary glands, lip, and palate all arise from neural crest cells, making cells from saliva a developmentally appropriate proxy for these tissues. In addition, saliva is a good source of high quality DNA for use in epigenomics (Thompson et al., 2013; Smith et al., 2015) and has been deemed more informative than blood for DNA methylation analysis (Lowe et al., 2013) in spite of the possibility of bacterial contamination resulting in non-human reads in human sequencing data (Samson et al., 2020).
Our sample size, while small, exceeds the number of twins in most epigenetic studies of birth defects with most being based on a single discordant twin pair (Weksberg et al., 2002; Rall et al., 2011; Tierling et al., 2011; Jin et al., 2014; Wei et al., 2015; Lu et al., 2017; Lyu et al., 2018). NSCLP has an estimated frequency of 1 in 700; thus, discordant MZ twin pairs occur infrequently but are rich source of information.
The stringent parameters used in this study, together with the validation of selected DMPs by either bisulfite sequencing, pyrosequencing, or methylation arrays, suggest that these are likely real differences rather than reflecting experimental noise. Amongst the identified methylation differences between co-twins, we propose that many of them underlie the discordance in NSCLP but recognize that there will be some that may reflect individual exposure to environmental factors unrelated to NSCLP, or perhaps non-craniofacial phenotypic differences. However, identification of DMRs in NSCLP candidate genes and the pathway analyses of the unbiased genes containing DMRs suggest that these DMRs play an etiologic role in NSCLP.
In summary, we show for the first time that MZ twins discordant for NSCLP have differential methylation. The findings from this novel study demonstrate that methylation may play a significant role in the etiology of NSCLP and, more importantly, differential methylation could account for the “reduced penetrance” seen in many multiplex families (Hecht et al., 1991; Hecht and Blanton, 1998). Since the discordant twins share age, sex and socioeconomic status and do not exhibit CLP-related genotype variability (as analyzed by WGS), our data highlights the role of non-shared environmental and stochastic factors in the etiology of epigenetic differences and resulting discordant NSCLP phenotypes. Importantly, we find methylation differences in genes previously implicated by NSCLP genetic studies. Moreover, our data suggest that although DNA methylation at some CpG sites is consistently altered in affected twins, most differences are individual-specific, revealing epigenetic heterogeneity between NSCLP individuals. This suggests that each affected individual/family may have their own unique epigenetic signature making etiologic identification complex. However, the differences converge at the level of potentially affected pathways including the Hippo signaling pathway. Our findings complement a recent NSCLP case-control study suggesting that epigenetic mechanisms underlie NSCLP (Alvizi et al., 2017). Future studies are aimed at defining epigenetic changes in sporadic cases, as well as in multiplex NSCLP families in which DNA methylation patterns can be traced through multiple generations.
Data Availability Statement
The data presented in the study are deposited in the Gene Expression Omnibus (GEO) repository, accession number GSE173211.
Ethics Statement
The studies involving human participants were reviewed and approved by this study was approved by the University of Texas Health Science Center Committee for Protection of Human Subjects (HSC-MS-03-090) and the University of Miami Human Subject Research Office (IRB #20100505). Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
Author Contributions
JY, JH, and SB: conceptualization, investigation, writing—original draft, and writing—review and editing. JY and SS: formal analysis and methodology. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by National Institute of Dental and Craniofacial Research Grant R01DE011931-17 (JH and SB).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Anastasia Vedenko, Ian Ergui, Sathesh Sivasankaran, and Derek Van Booven for their research and analytical assistance.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2021.656865/full#supplementary-material
Footnotes
- ^ https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/
- ^ http://hgwdev.cse.ucsc.edu/cgi-bin/hgFileUi?db=hg19&g=wgEncodeMapability
- ^ https://www.r-project.org/
References
Alvizi, L., Ke, X., Brito, L. A., Seselgyte, R., Moore, G. E., Stanier, P., et al. (2017). Differential methylation is associated with non-syndromic cleft lip and palate and contributes to penetrance effects. Sci. Rep. 7:2441. doi: 10.1038/s41598-017-02721-0
Assenov, Y., Muller, F., Lutsik, P., Walter, J., Lengauer, T., and Bock, C. (2014). Comprehensive analysis of DNA methylation data with RnBeads. Nat. Methods 11, 1138–1140. doi: 10.1038/nmeth.3115
Beaty, T. H., Murray, J. C., Marazita, M. L., Munger, R. G., Ruczinski, I., Hetmanski, J. B., et al. (2010). A genome-wide association study of cleft lip with and without cleft palate identifies risk variants near MAFB and ABCA4. Nat. Genet. 42, 525–529. doi: 10.1038/ng.580
Butali, A., Little, J., Chevrier, C., Cordier, S., Steegers-Theunissen, R., Jugessur, A., et al. (2013). Folic acid supplementation use and the MTHFR C677T polymorphism in orofacial clefts etiology: an individual participant data pooled-analysis. Birth Defects Res. A Clin. Mol. Teratol. 97, 509–514. doi: 10.1002/bdra.23133
Carter, C. O. (1969). Genetics of common disorders. Br. Med. Bull. 25, 52–57. doi: 10.1093/oxfordjournals.bmb.a070671
Chen, C. P., Lin, S. P., Chern, S. R., Wu, P. S., Su, J. W., and Wang, W. (2013). “A boy with cleft palate, hearing impairment, microcephaly, micrognathia and psychomotor retardation and a microdeletion in 6p25.3 involving the DUSP22 gene”, in Genetic Counseling, Vol. 1, Geneva, 243–246.
Chiquet, B. T., Lidral, A. C., Stal, S., Mulliken, J. B., Moreno, L. M., Arcos-Burgos, M., et al. (2007). CRISPLD2: a novel NSCLP candidate gene. Hum. Mol. Genet. 16, 2241–2248. doi: 10.1093/hmg/ddm176
Chiquet, B. T., Henry, R., Burt, A., Mulliken, J. B., Stal, S., Blanton, S. H., et al. (2011). Nonsyndromic cleft lip and palate: CRISPLD genes and the folate gene pathway connection. Birth Defects Res. A Clin. Mol. Teratol. 91, 44–49. doi: 10.1002/bdra.20737
Christensen, K. (1999). The 20th century danish facial cleft population–epidemiological and genetic-epidemiological studies. Cleft Palate Craniofac. J. 36, 96–104. doi: 10.1597/1545-1569_1999_036_0096_tcdfcp_2.3.co_2
Cox, L. L., Cox, T. C., Moreno Uribe, L. M., Zhu, Y., Richter, C. T., Nidey, N., et al. (2018). Mutations in the epithelial cadherin-p120-catenin complex cause mendelian non-syndromic cleft lip with or without cleft palate. Am. J. Hum. Genet. 102, 1143–1157. doi: 10.1016/j.ajhg.2018.04.009
Cvjetkovic, N., Maili, L., Weymouth, K. S., Hashmi, S. S., Mulliken, J. B., Topczewski, J., et al. (2015). Regulatory variant in FZD6 gene contributes to nonsyndromic cleft lip and palate in an african-american family. Mol. Genet. Genomic Med. 3, 440–451. doi: 10.1002/mgg3.155
Czyz, W., Morahan, J. M., Ebers, G. C., and Ramagopalan, S. V. (2012). Genetic, environmental and stochastic factors in monozygotic twin discordance with a focus on epigenetic differences. BMC Med. 10:93. doi: 10.1186/1741-7015-10-93
Du, W., He, D., Wang, Y., Liu, H., Liao, C., Fei, W., et al. (2017). Upper airway changes after mandibular setback and/or advancement genioplasty in obese patients. J. Oral. Maxillofac. Surg. 75, 2202–2210. doi: 10.1016/j.joms.2017.04.012
Eckhardt, F., Lewin, J., Cortese, R., Rakyan, V. K., Attwood, J., Burger, M., et al. (2006). DNA methylation profiling of human chromosomes 6, 20 and 22. Nat. Genet. 38, 1378–1385. doi: 10.1038/ng1909
Esteller, M. (2008). Epigenetics in cancer. N. Engl. J. Med. 358, 1148–1159. doi: 10.1056/NEJMra072067
Feinberg, A. P., Irizarry, R. A., Fradin, D., Aryee, M. J., Murakami, P., Aspelund, T., et al. (2010). Personalized epigenomic signatures that are stable over time and covary with body mass index. Sci. Transl. Med. 2:49ra67. doi: 10.1126/scitranslmed.3001262
Fish, J. L., Albertson, C., Harris, M. P., Lozanoff, S., Marcucio, R. S., Richtsmeier, J. T., et al. (2017). The society for craniofacial genetics and developmental biology 39th annual meeting. Am. J. Med. Genet. A 173, 985–1006. doi: 10.1002/ajmg.a.38096
Funato, N., and Nakamura, M. (2017). Identification of shared and unique gene families associated with oral clefts. Int. J. Oral Sci. 9, 104–109. doi: 10.1038/ijos.2016.56
Ghoumid, J., Drevillon, L., Alavi-Naini, S. M., Bondurand, N., Rio, M., Briand-Suleau, A., et al. (2013). ZEB2 zinc-finger missense mutations lead to hypomorphic alleles and a mild Mowat-Wilson syndrome. Hum. Mol. Genet. 22, 2652–2661. doi: 10.1093/hmg/ddt114
Goodwin, A. F., Chen, C. P., Vo, N. T., Bush, J. O., and Klein, O. D. (2020). YAP/TAZ regulate elevation and bone formation of the mouse secondary palate. J. Dent. Res. 99, 1387–1396. doi: 10.1177/0022034520935372
Grosen, D., Bille, C., Petersen, I., Skytthe, A., Hjelmborg, J., Pedersen, J. K., et al. (2011). Risk of oral clefts in twins. Epidemiology 22, 313–319. doi: 10.1097/EDE.0b013e3182125f9c
Hecht, J. T., Yang, P., Michels, V. V., and Buetow, K. H. (1991). Complex segregation analysis of nonsyndromic cleft lip and palate. Am. J. Hum. Genet. 49, 674–681.
Hecht, J. T., and Blanton, S. H. (1998). “Orofacial clefting,” in Principles of Molecular Medicine, ed. J. L. Jameson (Totowa, NJ: Humana Press Inc.). doi: 10.1007/978-1-59259-726-0_121
Howe, L. J., Lee, M. K., Sharp, G. C., Davey Smith, G., St Pourcain, B., Shaffer, J. R., et al. (2018). Investigating the shared genetics of non-syndromic cleft lip/palate and facial morphology. PLoS Genet. 14:e1007501. doi: 10.1371/journal.pgen.1007501
Hu, N., Strobl-Mazzulla, P. H., and Bronner, M. E. (2014). Epigenetic regulation in neural crest development. Dev. Biol. 396, 159–168. doi: 10.1016/j.ydbio.2014.09.034
Ji, X., Lin, L., Shen, S., Dong, X., Chen, C., Li, Y., et al. (2020). Epigenetic–smoking interaction reveals histologically heterogeneous effects of TRIM27 DNA methylation on overall survival among early-stage NSCLC patients. Mol. Oncol. 14, 2759–2774. doi: 10.1002/1878-0261.12785
Jin, M., Zhu, S., Hu, P., Liu, D., Li, Q., Li, Z., et al. (2014). Genomic and epigenomic analyses of monozygotic twins discordant for congenital renal agenesis. Am. J. Kidney Dis. 64, 119–122. doi: 10.1053/j.ajkd.2014.01.423
Johnson, M. D., Mueller, M., Game, L., and Aitman, T. J. (2012). Single nucleotide analysis of cytosine methylation by whole-genome shotgun bisulfite sequencing. Curr. Protoc. Mol. Biol. 99, 21.23.1–21.23.28. doi: 10.1002/0471142727.mb2123s99
Kaminsky, Z. A., Tang, T., Wang, S. C., Ptak, C., Oh, G. H., Wong, A. H., et al. (2009). DNA methylation profiles in monozygotic and dizygotic twins. Nat. Genet. 41, 240–245. doi: 10.1038/ng.286
Krueger, F., and Andrews, S. R. (2011). Bismark: a flexible aligner and methylation caller for bisulfite-seq applications. Bioinformatics 27, 1571–1572. doi: 10.1093/bioinformatics/btr167
Kuleshov, M. V., Jones, M. R., Rouillard, A. D., Fernandez, N. F., Duan, Q., Wang, Z., et al. (2016). Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 44, W90–W97. doi: 10.1093/nar/gkw377
Leslie, E. J., Carlson, J. C., Shaffer, J. R., Feingold, E., Wehby, G., Laurie, C. A., et al. (2016a). A multi-ethnic genome-wide association study identifies novel loci for non-syndromic cleft lip with or without cleft palate on 2p24.2, 17q23 and 19q13. Hum. Mol. Genet. 25, 2862–2872.
Leslie, E. J., Liu, H., Carlson, J. C., Shaffer, J. R., Feingold, E., Wehby, G., et al. (2016b). A genome-wide association study of nonsyndromic cleft palate identifies an etiologic missense variant in GRHL3. Am. J. Hum. Genet. 98, 744–754. doi: 10.1016/j.ajhg.2016.02.014
Leslie, E. J., Taub, M. A., Liu, H., Steinberg, K. M., Koboldt, D. C., Zhang, Q., et al. (2015). Identification of functional variants for cleft lip with or without cleft palate in or near PAX7, FGFR2, and NOG by targeted sequencing of GWAS loci. Am. J. Hum. Genet. 96, 397–411. doi: 10.1016/j.ajhg.2015.01.004
Letra, A., Maili, L., Mulliken, J. B., Buchanan, E., Blanton, S. H., and Hecht, J. T. (2014). Further evidence suggesting a role for variation in ARHGAP29 variants in nonsyndromic cleft lip/palate. Birth Defects Res. A Clin. Mol. Teratol. 100, 679–685. doi: 10.1002/bdra.23286
Letra, A., Silva, R. M., Motta, L. G., Blanton, S. H., Hecht, J. T., Granjeirol, J. M., et al. (2012). Association of MMP3 and TIMP2 promoter polymorphisms with nonsyndromic oral clefts. Birth Defects Res. A Clin. Mol. Teratol. 94, 540–548. doi: 10.1002/bdra.23026
Li, N., Lu, N., and Xie, C. (2019). The Hippo and Wnt signalling pathways: crosstalk during neoplastic progression in gastrointestinal tissue. FEBS J. 286, 3745–3756. doi: 10.1111/febs.15017
Lowe, R., Gemma, C., Beyan, H., Hawa, M. I., Bazeos, A., Leslie, R. D., et al. (2013). Buccals are likely to be a more informative surrogate tissue than blood for epigenome-wide association studies. Epigenetics 8, 445–454. doi: 10.4161/epi.24362
Lu, P., Wang, P., Li, L., Xu, C., Liu, J. C., Guo, X., et al. (2017). Exomic and epigenomic analyses in a pair of monozygotic twins discordant for cryptorchidism. Twin Res. Hum. Genet. 20, 349–354. doi: 10.1017/thg.2017.33
Ludwig, K. U., Bohmer, A. C., Bowes, J., Nikolic, M., Ishorst, N., Wyatt, N., et al. (2017). Imputation of orofacial clefting data identifies novel risk loci and sheds light on the genetic background of cleft lip +/- cleft palate and cleft palate only. Hum. Mol. Genet. 26, 829–842. doi: 10.1093/hmg/ddx012
Lyu, G., Zhang, C., Ling, T., Liu, R., Zong, L., Guan, Y., et al. (2018). Genome and epigenome analysis of monozygotic twins discordant for congenital heart disease. BMC Genomics 19:428. doi: 10.1186/s12864-018-4814-7
Martin, D. I., Ward, R., and Suter, C. M. (2005). Germline epimutation: a basis for epigenetic disease in humans. Ann. N. Y. Acad. Sci. 1054, 68–77. doi: 10.1196/annals.1345.009
Miyoshi, T., Maruhashi, M., Van De Putte, T., Kondoh, H., Huylebroeck, D., and Higashi, Y. (2006). Complementary expression pattern of Zfhx1 genes Sip1 and deltaEF1 in the mouse embryo and their genetic interaction revealed by compound mutants. Dev. Dyn. 235, 1941–1952. doi: 10.1002/dvdy.20799
Morris, V. E., Hashmi, S. S., Zhu, L., Maili, L., Urbina, C., Blackwell, S., et al. (2020). Evidence for craniofacial enhancer variation underlying nonsyndromic cleft lip and palate. Hum. Genet. 139, 1261–1272. doi: 10.1007/s00439-020-02169-9
Ollikainen, M., Ismail, K., Gervin, K., Kyllonen, A., Hakkarainen, A., Lundbom, J., et al. (2015). Genome-wide blood DNA methylation alterations at regulatory elements and heterochromatic regions in monozygotic twins discordant for obesity and liver fat. Clin. Epigenetics 7:39. doi: 10.1186/s13148-015-0073-5
Patel, V. N., and Hoffman, M. P. (2014). Salivary gland development: a template for regeneration. Semin. Cell Dev. Biol. 2, 52–60. doi: 10.1016/j.semcdb.2013.12.001
Rall, K., Barresi, G., Walter, M., Poths, S., Haebig, K., Schaeferhoff, K., et al. (2011). A combination of transcriptome and methylation analyses reveals embryologically-relevant candidate genes in MRKH patients. Orphanet J. Rare Dis. 6:32. doi: 10.1186/1750-1172-6-32
Samson, C. A., Whitford, W., Snell, R. G., Jacobsen, J. C., and Lehnert, K. (2020). Contaminating DNA in human saliva alters the detection of variants from whole genome sequencing. Sci. Rep. 10:19255. doi: 10.1038/s41598-020-76022-4
Seelan, R. S., Pisano, M., and Greene, R. M. (2019). Nucleic acid methylation and orofacial morphogenesis. Birth Defects Res. 111, 1593–1610. doi: 10.1002/bdr2.1564
Sharp, G. C., Ho, K., Davies, A., Stergiakouli, E., Humphries, K., McArdle, W., et al. (2017). Distinct DNA methylation profiles in subtypes of orofacial cleft. Clin. Epigenetics 9:63. doi: 10.1186/s13148-017-0362-2
Smith, A. K., Kilaru, V., Klengel, T., Mercer, K. B., Bradley, B., Conneely, K. N., et al. (2015). DNA extracted from saliva for methylation studies of psychiatric traits: evidence tissue specificity and relatedness to brain. Am. J. Med. Genet. B Neuropsychiatr. Genet. 168B, 36–44. doi: 10.1002/ajmg.b.32278
Sun, Z., da Fontoura, C. S. G., Moreno, M., Holton, N. E., Sweat, M., Sweat, Y., et al. (2018). FoxO6 regulates Hippo signaling and growth of the craniofacial complex. PLoS Genet. 14:e1007675. doi: 10.1371/journal.pgen.1007675
Thompson, T. M., Sharfi, D., Lee, M., Yrigollen, C. M., Naumova, O. Y., and Grigorenko, E. L. (2013). Comparison of whole-genome DNA methylation patterns in whole blood, saliva, and lymphoblastoid cell lines. Behav. Genet. 43, 168–176. doi: 10.1007/s10519-012-9579-1
Tierling, S., Souren, N. Y., Reither, S., Zang, K. D., Meng-Hentschel, J., Leitner, D., et al. (2011). DNA methylation studies on imprinted loci in a male monozygotic twin pair discordant for Beckwith-Wiedemann syndrome. Clin. Genet. 79, 546–553. doi: 10.1111/j.1399-0004.2010.01482.x
Van de Putte, T., Maruhashi, M., Francis, A., Nelles, L., Kondoh, H., Huylebroeck, D., et al. (2003). Mice lacking ZFHX1B, the gene that codes for Smad-interacting protein-1, reveal a role for multiple neural crest cell defects in the etiology of Hirschsprung disease-mental retardation syndrome. Am. J. Hum. Genet. 72, 465–470. doi: 10.1086/346092
Wallenstein, M. B., Shaw, G. M., Yang, W., and Carmichael, S. L. (2013). Periconceptional nutrient intakes and risks of orofacial clefts in California. Pediatr. Res. 74, 457–465. doi: 10.1038/pr.2013.115
Wang, J., and Martin, J. F. (2017). Hippo pathway: an emerging regulator of craniofacial and dental development. J. Dent. Res. 96, 1229–1237. doi: 10.1177/0022034517719886
Wang, J., Xiao, Y., Hsu, C. W., Martinez-Traverso, I. M., Zhang, M., Bai, Y., et al. (2016). Yap and Taz play a crucial role in neural crest-derived craniofacial development. Development 143, 504–515. doi: 10.1242/dev.126920
Wang, L., Wang, B., and Quan, Z. (2020). Identification of aberrantly methylated differentially expressed genes and gene ontology in prostate cancer. Mol. Med. Rep. 21, 744–758. doi: 10.3892/mmr.2019.10876
Wei, T., Sun, H., Hu, B., Yang, J., Qiao, C., and Yan, M. (2015). Exome sequencing and epigenetic analysis of twins who are discordant for congenital cataract. Twin Res. Hum. Genet. 18, 393–398. doi: 10.1017/thg.2015.34
Weksberg, R., Shuman, C., Caluseriu, O., Smith, A. C., Fei, Y. L., Nishikawa, J., et al. (2002). Discordant KCNQ1OT1 imprinting in sets of monozygotic twins discordant for Beckwith-Wiedemann syndrome. Hum. Mol. Genet. 11, 1317–1325. doi: 10.1093/hmg/11.11.1317
Wu, H., Xu, T., Feng, H., Chen, L., Li, B., Yao, B., et al. (2015). Detection of differentially methylated regions from whole-genome bisulfite sequencing data without replicates. Nucleic Acids Res. 43:e141. doi: 10.1093/nar/gkv715
Keywords: methylation, NSCLP, non-syndromic cleft lip and cleft palate, twins, whole genome bisulfite sequencing
Citation: Young JI, Slifer S, Hecht JT and Blanton SH (2021) DNA Methylation Variation Is Identified in Monozygotic Twins Discordant for Non-syndromic Cleft Lip and Palate. Front. Cell Dev. Biol. 9:656865. doi: 10.3389/fcell.2021.656865
Received: 21 January 2021; Accepted: 22 March 2021;
Published: 12 May 2021.
Edited by:
Miguel Constancia, University of Cambridge, United KingdomReviewed by:
Michele Rubini, University of Ferrara, ItalyMichelle Holland, King’s College London, United Kingdom
Copyright © 2021 Young, Slifer, Hecht and Blanton. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Juan I. Young, anlvdW5nM0BtZWQubWlhbWkuZWR1
†These authors have contributed equally to this work