Abstract
Autophagy is a stable self-sustaining process in eukaryotic cells. In this process, pathogens, abnormal proteins, and organelles are encapsulated by a bilayer membrane to form autophagosomes, which are then transferred to lysosomes for degradation. Autophagy is involved in many physiological and pathological processes. Nucleotide-binding oligomerization domain-like receptor protein 3 (NLRP3) inflammasome, containing NLRP3, apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC) and pro-caspase-1, can activate caspase-1 to induce pyroptosis and lead to the maturation and secretion of interleukin-1 β (IL-1 β) and IL-18. NLRP3 inflammasome is related to many diseases. In recent years, autophagy has been reported to play a vital role by regulating the NLRP3 inflammasome in inflammatory nervous system diseases. However, the related mechanisms are not completely clarified. In this review, we sum up recent research about the role of the effects of autophagy on NLRP3 inflammasome in Alzheimer’s disease, chronic cerebral hypoperfusion, Parkinson’s disease, depression, cerebral ischemia/reperfusion injury, early brain injury after subarachnoid hemorrhage, and experimental autoimmune encephalomyelitis and analyzed the related mechanism to provide theoretical reference for the future research of inflammatory neurological diseases.
Introduction
Autophagy, which is a closely coordinated process, isolates aged/damaged organelles and misfolded and mutated proteins into bilayer membrane vesicles named autophagosomes and then fuses into lysosomes, leading to the degradation of isolated components (Lv et al., 2021). Autophagy can be divided into macroautophagy, microautophagy, and chaperone-mediated autophagy according to the inducing signal, action time, target type, and transport pathway into lysosomes. Macroautophagy involves the formation of a double membranous vesicle that isolates the cytoplasm. The complete vesicles, called autophagosomes, then fuse with lysosomes for subsequent degradation (Wang et al., 2019a; Zhu et al., 2019). In microautophagy, the substances destined for degradation reach lysosome cavity through invagination of lysosome or endoplasmic membrane (Sahu et al., 2011). Chaperone-mediated autophagy occurs only in mammalian cells, allowing selective degradation of proteins with specific amino acid sequences (Figure 1; Kaushik and Cuervo, 2012). Among these three autophagy processes, macroautophagy, referred to as autophagy, is the most active form and has been widely studied in diseases (Ueno and Komatsu, 2017; Galluzzi and Green, 2019). Beclin1, LC3, P62, and other conserved proteins participate in the autophagy process and are regarded as autophagy-related proteins (Wang et al., 2019a). Among them, LC3, a ubiquitin-like protein, promotes autophagosome formation (Fujita et al., 2008; Pyo et al., 2012). It regulates the elongation and closure of the autophagic membrane by binding with phosphatidylethanolamine (Ichimiya et al., 2020). Autophagy is affected by many factors, such as endoplasmic reticulum stress (ERS), immune or inflammatory stimulation, nutritional deficiency, Ca2+ concentration and accumulation of organelle damage (Tooze and Yoshimori, 2010; Mizushima et al., 2011). Autophagy is usually maintained at the basic level under physiological conditions. In the pathological state, the upregulated autophagy can eliminate the dysfunctional proteins in cells and help them survive (Glick et al., 2010). Autophagy is a double-edged sword because, if autophagy is maintained at a high level, autophagy leads to cell death (Liu and Levine, 2015; Garcia-Huerta et al., 2016). Many studies find that autophagy played an important role in neurodegenerative diseases (Menzies et al., 2015), cardiovascular diseases (Shirakabe et al., 2016), infection, and immunity (Deretic et al., 2013). In particular, the role of autophagy in inflammatory nervous system diseases is reported by many researchers; for example, MiR-124 inhibits the secretion of proinflammatory mediators by promoting autophagy in Parkinson’s disease (PD) (Yao et al., 2019), and the upregulation of autophagy of hippocampal cells improved memory impairment led by ethanol through an anti-inflammatory mechanism (Liu et al., 2019). The mechanism about autophagy in inflammatory nervous system diseases needs to be further studied.
FIGURE 1
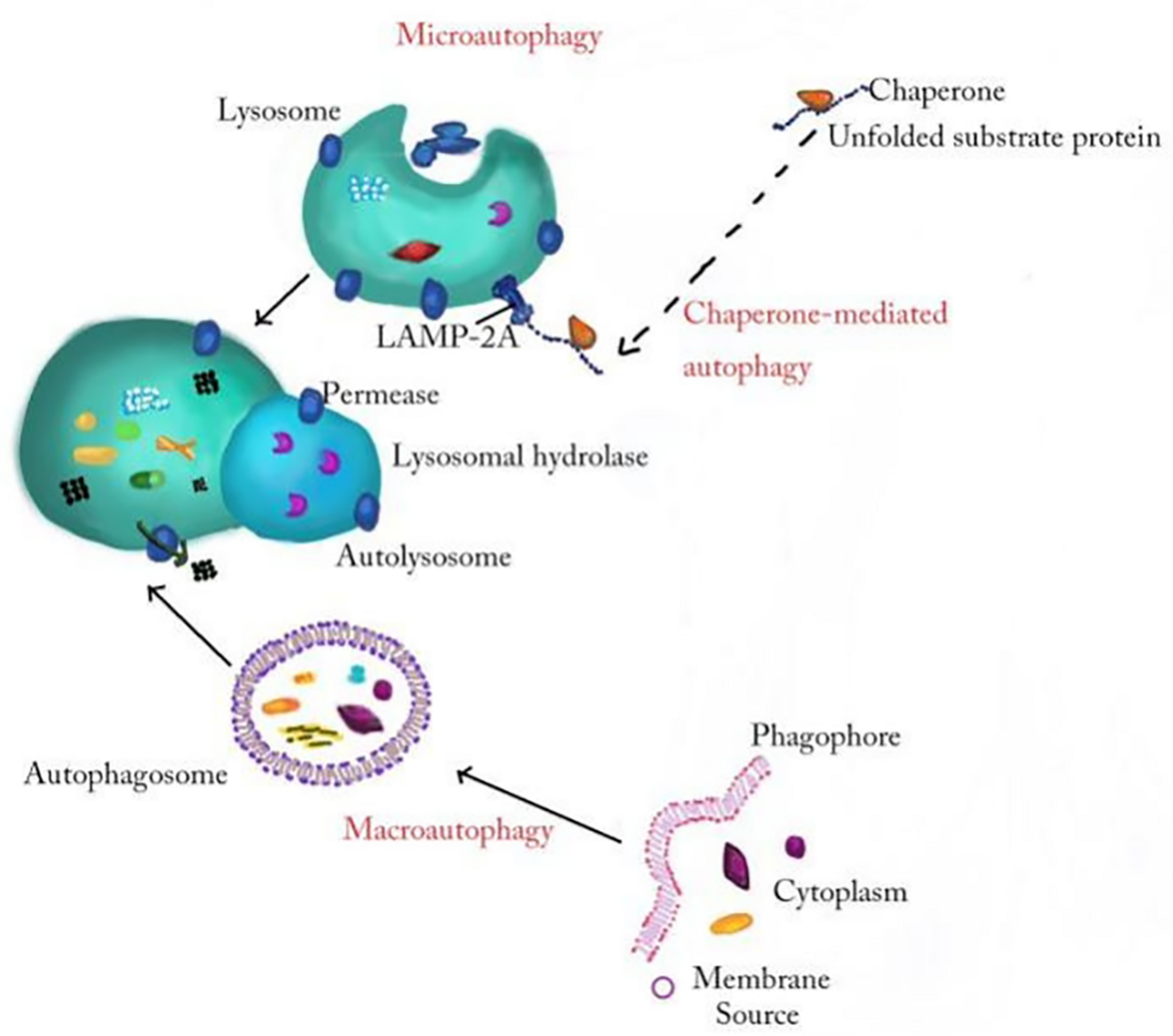
The general processes of macroautophagy, microautophagy, and chaperone-mediated autophagy. In the process of macroautophagy, the inclusion is wrapped by a double membrane structure to form an autophagosome and then fuses with a lysosome to degrade. Microautophagy refers to the direct invagination of the lysosomal membrane and the encapsulation of the cell contents. In chaperone-mediated autophagy, cytoplasmic proteins combine with chaperones, and are transported to lysosomal chambers and then digested by lysosomal enzymes.
Inflammasomes, first proposed by Martinon and coworkers in 2002 (Martinon et al., 2002), are a kind of protein cytoplasmic complex, which can activate the effective inflammatory mediators. As a part of the innate immune response against invading pathogens, inflammasomes are activated by cell infection or pressure stimulation and induce the expression, maturation, and release of a variety of proinflammatory cytokines; therefore, triggering a series of inflammatory reactions (Schroder and Tschopp, 2010; Yaribeygi et al., 2019). The nucleotide-binding oligomerization domain-like receptor (NLR) family can be divided into three subfamilies: the NLRPs (NLRP1-14), the NODs (NOD1-2, NOD3/NLRC3, NOD4/NLRC5, NOD5/NLRX1, and CIITA), and the IPAF subfamily, including NAIP and IPAF (Schroder and Tschopp, 2010). The NLRP3 inflammasome is the most extensively studied one and contains NLRP3, pro-caspase-1, and apoptosis-associated speck-like protein (ASC). The NLRP3 inflammasome can be activated by different stimuli, including damage-associated molecular patterns (DAMPS) and pathogen-associated molecular patterns (PAMPs). The first stimulation is mediated by pro-inflammatory pathways, such as toll like receptor (TLR)-mediated activation of nuclear factor kB (NF-kB), which upregulates the protein expressions of NLRP3 and pro-IL-1β (Munoz-Planillo et al., 2013; Lu et al., 2014; Abais et al., 2015; Toldo and Abbate, 2018) and reduces the activation threshold of NLRP3 through additional post-translational modifications (Swanson et al., 2019; Yang et al., 2019). The second stimulation includes Ca2+ signaling disturbance, K+ efflux, ROS production, mitochondrial dysfunction, and lysosomal rupture, which promotes the assembly of inflammasome and activates caspase-1, thus catalyzing the conversion of pro-IL-1β to active IL-1β (Figure 2; Schroder and Tschopp, 2010; He et al., 2016). Activated caspase-1 also cleaves gasdermin D to trigger a specific cell death form named pyroptosis (Shi et al., 2015). Pyroptosis is a new form of pro-inflammatory cell death program and characterized by the pore formation induced by the Gasdermin family and subsequently cellular lysis as well as the release of several pro-inflammatory intracellular cytokines. Two signaling pathways participate in pyroptosis, including caspase-4/5/11 and caspase-1mediated pathways (Shi et al., 2015). The NLRP3 inflammasome is involved in the pathogenesis of many complex diseases, including type 2 diabetes (Hong et al., 2018), atherosclerosis (Grebe et al., 2018), obesity, and gout (Kim et al., 2014). It is reported that the NLRP3 inflammasome also played a vital role in central nervous system (CNS) diseases (Song et al., 2017), including Alzheimer’s disease (AD) (Ising et al., 2019) PD (Saresella et al., 2016; Wang et al., 2019b), and HIV-associated neurocognitive disorders (Walsh et al., 2014). The main neurotoxicity of NLRP3 is the release of IL-1β. The neuroinflammation mediated by IL-1β plays a vital role in CNS diseases, including AD, stroke, multiple dementia, and sclerosis. IL-1β, which is a pleiotropic cytokine, activates microglia and astrocytes to induce the synthesis of other pro-inflammatory and chemotactic mediators in the CNS. Peripherally, IL-1β can induce the expansion of brain-derived T cells (Mendiola and Cardona, 2018).
FIGURE 2
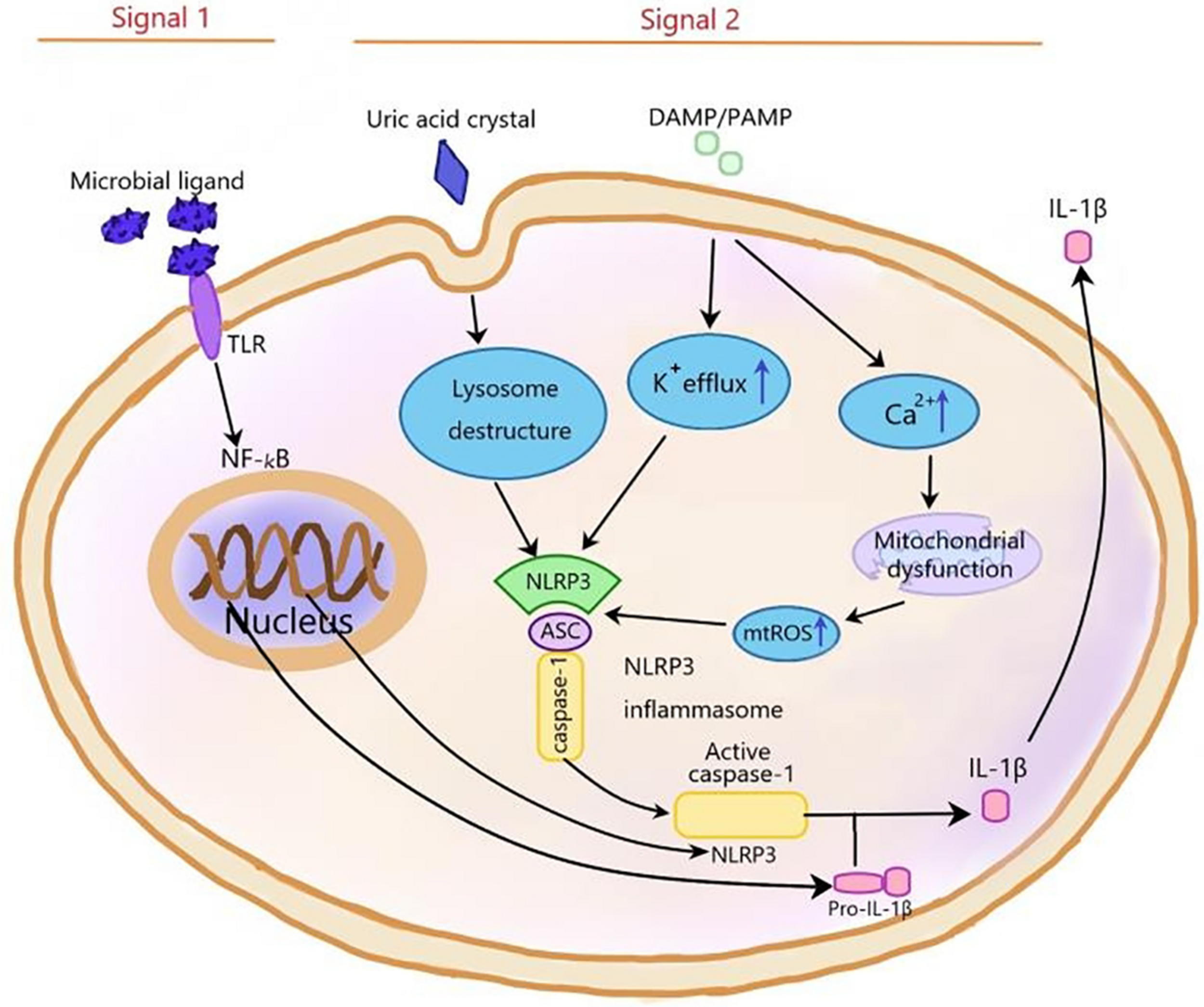
The NLRP3 inflammasome is activated by signal 1 and signal 2. Signal 1, which is mediated by microbial ligands recognized by TLR, activates NF-KB pathway to upregulate pro-IL-1β and NLRP3 expression. Signal 2, which consists of three ways, induces the assembly of NLRP3 inflammasome complex. (1) In the noninfectious condition, K+ efflux activates the NLRP3 inflammasome. (2) The endogenous and exogenous particles, such as uric acid crystallization, induce lysosome damage to activate NLRP3 inflammasome. (3) The increase of intracellular Ca2+ concentration increases mtROS level to activate NLRP3 inflammasome. NF-kB, nuclear factor kappa-light-chain-enhancer of activated B cells; ASC, apoptosis-associated speck-like protein containing a C-terminal caspase recruitment domain; ROS, reactive oxygen species; TLR, toll-like receptor.
Autophagy can negatively regulate the activation of the NLRP3 inflammasome by scavenging endogenous activators of inflammasome, including reactive oxygen species (ROS) of damaged mitochondria, inflammatory components, and cytokines. In contrast, NLRP3 inflammasome activation can inhibit autophagy by cleaving signal molecule Toll/IL-1R domain-containing adaptor-inducing IFN-β (TRIF) with caspase-1. The decrease of signal molecule TRIF inhibits autophagy induced by the TLR4-TRIF signaling pathway. The inflammasome activation can also suppress mitophagy in macrophages in a caspase-1-dependent manner, which leads to mitochondrial dysfunction. The interaction between inflammasome and autophagy is necessary to the balance host defense against inflammation and prevent excessive inflammation (Cao et al., 2019; Biasizzo and Kopitar-Jerala, 2020).
It is reported that autophagy plays an important role by influencing the NLRP3 inflammasome in many diseases, including nephropathy, inflammatory bowel disease, inflammatory lung disease, and sepsis (Cao et al., 2019); however, the relevant mechanism has not been fully explained. To study the mechanism of the effects of autophagy on the NLRP3 inflammasome in inflammatory nervous system diseases may provide a new strategy for the treatment of diseases. Therefore, in this review, we are the first to sum up the recent studies about the effects of autophagy on the NLRP3 inflammasome in inflammatory nervous system diseases including AD, Parkinson’s disease, Chronic cerebral hypoperfusion (CCH), depression, cerebral ischemia/reperfusion injury, Early brain injury (EBI) after Subarachnoid hemorrhage (SAH), and Experimental autoimmune encephalomyelitis (EAE) and analyzed the related mechanism to provide reference for future research.
The Effects of Autophagy on NLRP3 Inflammasome in Alzheimer’s Disease
Alzheimer’s disease is a destructive neurodegenerative disease characterized by extensive loss of neurons and synapsis and gradual loss of memory. The main pathological features of AD are amyloid plaques and neurofibrillary tangles consisting of hyperphosphorylated filaments of the microtubule-associated protein tau. The extracellular accumulation of amyloid beta (β-amyloid) in senile plaques is the main cause of neurodegeneration (Lane et al., 2018). β-amyloid activates the NLRP3 inflammasome to release proinflammatory cytokine IL-1β in microglia. The activated NLRP3 inflammasome leads to neuroinflammation in the brains of AD patients (Bodles and Barger, 2004; Heneka et al., 2013). ASC, a NLRP3 inflammasome component, is elevated in the blood of AD patients and may serve as a biomarker of AD (Scott et al., 2020). Cho et al. (2014) speculate that the autophagy of microglia might be involved in the degradation of extracellular amyloid fibers and play an important role in regulating β-amyloid fiber-mediated inflammatory response. Their research shows that autophagy was induced in microglia by extracellular β-amyloid fibers and involved in the degradation of extracellular β-amyloid fibers. The NLRP3 inflammasome was upregulated in microglia induced by β-amyloid fibers. In addition, the inhibited autophagy enhanced the activation of the NLRP3 inflammasome, indicating that autophagy negatively regulates the NLRP3 inflammasome induced by β-amyloid fibers (Cho et al., 2014). Collectively, it can be deduced that autophagy can eliminate β-amyloid fibers and inhibit the β-amyloid fiber-induced NLRP3 inflammasome to ameliorate AD. Heavy metals, including manganese, are involved in the etiology of neurodegenerative diseases. Manganese (Mn) is an important trace element, which is widely distributed in the earth’s crust. Excessive intake of Mn can lead to neurodegenerative diseases (Iregren, 1999; Erikson and Aschner, 2003). In Mn-induced microglia within the hippocampus of mice, Mn induced NLRP3 inflammasome-mediated inflammation by increasing the protein expression level of the NLRP3 inflammasome and the levels of caspase-1 and IL-1β. Mn treatment also dramatically inhibited autophagy by impairing autophagic lysosomal degradation activity, which mediated the activation of the NLRP3 inflammasome. In conclusion, Mn could induce NLRP3 inflammasome-mediated neuroinflammatory injury in the hippocampus of mice through inhibiting autophagy (Wang et al., 2017). Autophagy-NLRP3 inflammasome is a potential target for the treatment of neurotoxicity caused by heavy metals.
Eicosapentaenoic acid (EPA), mainly in the form of eicosapentaenoic acid-enriched phosphatidylcholine (EPA-PC), is mainly in marine products, including antarctic krill and marine cucumber (Burri et al., 2012). EPA-PC upregulated β-amyloid-suppressed autophagy through promoting the ratio of LC3II/LC3I and autophagosome formation and suppressed β-amyloid-induced the NLRP3 inflammasome to mitigate AD (Wen et al., 2019). Autophagy can clear β-amyloid; therefore, the protective effect of EPA-PC on AD is through inducing autophagy to eliminate β-amyloid-induced NLRP3 inflammasome. Another study shows that the reduction of Beclin 1 and the impairment of autophagy promoted IL-1β and IL-18 release from microglia (Houtman et al., 2019). In addition, microglia isolated from the brains of AD patients showed reduced level of Beclin 1, suggesting that microglial autophagy was impaired in the brains of AD patients (Lucin et al., 2013). Progesterone (PG) is an endogenous neurosteroid, which plays a neuroprotective role in several neurodegenerative disease models (Borowicz et al., 2011; Espinosa-Garcia et al., 2014). It is found that PG can improve the cognitive impairment of AD (Liu et al., 2013; Qin et al., 2015). To elucidate the molecular mechanism of PG neuroprotection, Yang Hong et al. treated astrocytes with PG and found that PG could significantly inhibit β-amyloid-induced neuroinflammatory response and regulated the function of astrocytes by inhibiting ERS and activating autophagy (Hong et al., 2016, 2018). β-amyloid activated NLRP3 inflammasome-mediated inflammation and inhibited autophagy in astrocytes, and PG inhibited β-amyloid-induced activation of NLRP3 inflammasome-mediated inflammation by upregulating astrocyte autophagy (Hong et al., 2019). In conclusion, enhancing autophagy to suppress an NLRP3 inflammasome-mediated neuroinflammatory response is a new strategy for the treatment of AD.
The Effects of Autophagy on the NLRP3 Inflammasome in Parkinson’s Disease
Parkinson’s disease is the second most common neurodegenerative disease and currently still incurable. It is characterized by major motor dysfunction and other nonmotor symptoms, such as cognitive changes, autonomic nervous dysfunction, sleep disorders. The pathological property of Parkinson’s disease is the progressive loss of dopaminergic neurons and the accumulation of Lewy bodies (LBS) in the neuron with fiber α-synuclein aggregates as the main protein component (Hou et al., 2020; Schwab et al., 2020). New evidence suggests that the low-grade systemic inflammation contributes to the development of degenerative changes in the brain in PD (Wang et al., 2014; Goldberg and Dixit, 2015; Poewe et al., 2017). Therefore, the precise control of excessive neuroinflammation can ameliorate PD. The NLRP3 inflammasome can cause chronic low-degree inflammation (Lu et al., 2014) and is activated in several PD models (Wang et al., 2019b; Haque et al., 2020). Zhou et al. (2016) found that the levels of mature IL-1β and activated caspase-1 in the plasma of PD patients were increased, indicating the inflammation was activated in the pathological process of PD. Consistent with the clinical data, the same results were confirmed in the serum of PD mice. Kaempferol (Ka), a natural polyphenolic small molecule, inhibited neurodegeneration by relieving neuroinflammation in a PD mouse model induced by lipopolysaccharide (LPS). Ka also decreased the protein expression level of the NLRP3 inflammasome and the levels of caspase-1 and IL-1β induced by LPS. The inhibition of Ka disappeared completely in the primary microglia from the NLRP3 knockout PD mouse model, indicating that the NLRP3 inflammasome mediated the neuroprotective effect of Ka. Further mechanism studies show that Ka could counteract LPS-induced inhibition of autophagy and inhibit LPS-induced NLRP3 inflammasome expression by inducing NLRP3 degradation, which were reversed by the autophagy inhibitor (3-MA). Therefore, it can be inferred that Ka inhibits the NLRP3 inflammasome through promoting autophagy. In addition, Ka can promote the ubiquitination of NLRP3. In summary, by inducing NLRP3 ubiquitination-modified degradation, Ka improves LPS-induced NLRP3 inflammasome-related neurodegeneration via promoting NLRP3 degradation through autophagy (Han et al., 2019). Whether Ka can suppress the NLRP3 inflammasome through autophagy via the other ways, such as scavenging reactive oxygen (ROS), needs further study. With the NLRP3 inflammasome and autophagy as targets, Ka has a potential therapeutic effect on PD.
The Effects of Autophagy on the NLRP3 Inflammasome in Chronic Cerebral Hypoperfusion
Chronic cerebral hypoperfusion, which is a state of chronic cerebral blood flow reduction, is associated with some cerebrovascular and neurodegenerative diseases, such as Alzheimer’s disease (AD) and carotid artery stenosis (Hainsworth and Markus, 2008; Arsava et al., 2018; Choi et al., 2019; Shang et al., 2019). CCH is reported to increase the levels of NLRP3, caspase-1, and IL-1β in the hippocampus and thalamus of AD mice (Shang et al., 2019; Matsuyama et al., 2020). However, the effects of autophagy on the NLRP3 inflammasome in CCH has not been studied. To determine whether autophagy is involved in the activation of the NLRP3 inflammasome and its possible mechanism in CCH, Shao-Hua Su et al. conducted a series of studies and found that CCH caused proinflammatory cytokine release, lysosome dysfunction, and autolysosome accumulation, resulting abnormal autophagy (Su et al., 2017, 2018). Mechanism research revealed that, in rat hippocampus, CCH activated the NLRP3 inflammasome and impaired autophagy, which was significantly attenuated by URB597 (the fatty acid amide hydrolase inhibitor). The autophagy inhibitor 3-MA and lysosome inhibitor CQ could neutralize the effects of URB597 on the CCH-induced NLRP3 inflammasome, suggesting that URB597 alleviated CCH-induced NLRP3 inflammasome activation through promoting restoring CCH-inhibited lysosomal function of autophagy. URB597 could promote CCH-induced defective autophagy by preventing ROS accumulation, and ROS could activate the NLRP3 inflammasome, suggesting that URB597 inhibited CCH-induced NLRP3 inflammasome partly via clearing ROS. In conclusion, URB597 alleviated inflammatory injury by suppressing the CCH-induced NLRP3 inflammasome through promoting autophagy and inhibiting ROS accumulation. Autophagy consisted of three steps, including autophagy formation, transport to lysosome, and degradation in lysosome (Su et al., 2019). Whether the first two participated in CCH-induced NLRP3 inflammasome needs further study.
The Effects of Autophagy on NLRP3 Inflammasome in Depression
Depression is a chronic recurrent and debilitating mental disease characterized by depression, loss of pleasure, inferiority complex, poor sleep or appetite, and inattention. Depression seriously impairs one’s ability to work or study and even has adverse effects on one’s daily life. In developed countries, depression is the main cause of disability (Smith, 2014; Wang et al., 2020). Increased inflammation is involved in the progression of depression, and the activation of NLRP3 inflammasome in microglia is an important feature of CNS inflammation under chronic stress (Pan et al., 2014; Pariante, 2017). Studies show that the NLRP3 inflammasome level in peripheral blood mononuclear cells is increased in depressive patients (Alcocer-Gomez et al., 2014). In rodents, depression induced by LPS is associated with activation of NLRP3 inflammasome in the brain (Pan et al., 2014). Inflammatory inhibitors have therapeutic effects on depression (Raison et al., 2013). Andrographolide is a diterpenoid lactone with anti-inflammatory and antitumor activities, and it exerts potential neuroprotective effects in diseases of the CNS (Wang et al., 2016; Islam et al., 2018). Andrographolide significantly inhibited inflammatory response in the prefrontal cortex of chronic unpredictable mild stress (CUMS)-induced mice to alleviate depression. In the prefrontal cortex of CUMS mice, Andrographolide inhibited NLRP3 inflammasome-mediated inflammation by decreasing the assembly of the NLRP3 inflammasome and induced autophagy. CQ, an autophagy flux blocker, could attenuate the antidepressant and anti-inflammatory effect of andrographolide, suggesting that andrographolide might ameliorate depression by inhibiting NLRP3 inflammasome-mediated inflammatory injury through inducing autophagy in CUMS mice, which needs further study (Geng et al., 2019). The underlying mechanism of andrographolide in improving depression by affecting autophagy and the NLRP3 inflammasome, including how andrographolide induced autophagy, remains to be elucidated. Autophagy and NLRP3 are potential targets for the treatment of depression.
The Effects of Autophagy on NLRP3 Inflammasome in Cerebral Ischemia/Reperfusion Injury
Ischemic stroke is still the leading cause of acquired disability and death in adults worldwide. Reperfusion is the main method for the treatment of ischemic stroke, but it can cause serious secondary brain tissue injury, which is called cerebral I/R injury (Donnan et al., 2008; Zhang et al., 2020). Much evidence suggests that inflammation plays a vital role in the occurrence and development of ischemic stroke (Dirnagl et al., 1999). The NLRP3 inflammasome participates in cerebral I/R injury after stroke (Wang et al., 2015; Qiu et al., 2016). Qingkailing alleviates cerebral I/R injury by inhibiting AMPK-mediated activation of NLRP3 inflammation (Ma et al., 2019). Edebenone improves cerebral I/R injury by inhibiting the activity of the NLRP3 inflammasome (Peng et al., 2020). Autophagy and the NLRP3 inflammasome are proven to be related to cerebral I/R injury (Qiu et al., 2016; Sun et al., 2020), but its mechanism is not fully clear. Resveratrol (3,4,5-trihydroxy-trans-stilbene, RSV), a natural polyphenolic compound, is shown to have protective effects in cerebral I/R injury (Burns et al., 2002; Shrikanta et al., 2015; Lu et al., 2020). The results of He et al. (2017) show that resveratrol could improve rat cerebral I/R injury by reducing brain water content and cerebral infarct volume and increasing neurological scores. The mechanism research revealed that RSV suppressed NLRP3 inflammasome-mediated inflammation through decreasing the levels of NLRP3 inflammasome, caspase-1, IL-1β, and IL-18 induced by cerebral I/R injury. Moreover, RSV upregulated Sirt1 expression and promoted autophagy in rat cerebral I/R injury, and 3-MA (an autophagy inhibitor) inhibited autophagy and NLRP3 inflammasome, suggesting that RSV suppressed NLRP3 inflammasome activation through autophagy promotion. Sirt1 siRNA downregulated Sirt1 expression and abolished the effects of RSV on autophagy and NLRP3 inflammasome. Given these results, it can be deduced that RSV ameliorates cerebral I/R injury by inhibiting the NLRP3 inflammasome through autophagy induction via increasing Sirt1expression (He et al., 2017). The Sirt1-AMPK pathway plays a protective role in ischemic stroke (Wang et al., 2011), so whether RSV ameliorates cerebral I/R injury by inhibiting the NLRP3 inflammasome through autophagy induction via Sirt1-AMPK pathway is worth studying. GSK3β is a serine/threonine kinase, which participates in the signal pathway through a phosphorylation-mediated signaling cascade, is activated by phosphorylation. Inactivation of GSK-3β promotes neuronal survival (Zhou et al., 2011; Chen et al., 2016; Chien et al., 2018). The expression of p-GSK-3β was increased in the 24 h following reperfusion after middle cerebral artery occlusion (Chen et al., 2016). GSK-3β siRNA and GSK-3β inhibitor alleviated cerebral I/R injury in rats, demonstrating that inhibition of GSK-3β could alleviate cerebral ischemia/reperfusion injury. The NLRP3 inflammasome played a vital role in cerebral I/R injury, and I/R significantly elevated the levels of the NLRP3 inflammasome, cleaved-caspase-1, IL-1β, and IL-18, which was abrogated by treatment with GSK-3β inhibitor or GSK-3β siRNA, indicating that the inhibition of GSK-3β alleviated I/R-induced brain injury by inhibiting NLRP3 inflammasome-mediated inflammation. The inhibiting GSK-3β could enhance autophagic activity under I/R stimulation, and the autophagy inhibitor could abrogate the effects of the inhibition of GSK-3β on I/R-induced brain injury, which suggested that the suppression of GSK-3β ameliorated cerebral I/R injury in rats by suppressing NLRP3 inflammasome activation via promoting autophagy (Wang et al., 2019c). The signaling pathways of GSK-3β regulating autophagy and NLRP3 are still to be elucidated.
The Effects of Autophagy on NLRP3 Inflammasome in Early Brain Injury After Subarachnoid Hemorrhage
Subarachnoid hemorrhage is a stroke with high mortality and a high incidence rate (Ciurea et al., 2013). Early brain injury occurs by increasing intracranial pressure and then decreasing cerebral perfusion at the moment of hemorrhage (Schneider et al., 2018). Recent studies show that EBI plays an important role in the poor prognosis of SAH patients. In the past decade, more and more evidence has shown that NLRP3 inflammasome-mediated neuroinflammation promotes the progression of EBI (Sercombe et al., 2002; Dumont et al., 2003; Ostrowski et al., 2006; Chen et al., 2013, 2016; Fann et al., 2013; Cho et al., 2014). Mitophagy is a selective form of autophagy that specifically scavenges damaged mitochondria and can negatively regulate NLRP3 inflammasome (Bienert et al., 2007; Youle and Narendra, 2011). It has been reported that melatonin could improve brain edema by reducing brain water content and attenuated neurological dysfunction by increasing the neurological scores after SAH. Mechanism studies show that melatonin could enhance mitophagy by increasing the LC3-II/LC3-I ratio and the expression levels of mitophagy-associated proteins (PINK1/Parkin) and Atg5, and inhibit NLRP3 inflammasome-mediated inflammation by decreasing the levels of ROS generation, NLRP3 inflammasome, and pro-inflammatory cytokine secretion, and microglial activation induced by SAH. Although 3-mA pretreatment reversed the above effects of melatonin. Moreover, melatonin treatment significantly reduced neuronal cell death induced by SAH. From all above, it could be inferred that melatonin-induced mitophagy protected EBI after SAH by inhibiting the activation of NLRP3 inflammasome (Cao et al., 2017). The relationship between mitophagy and reduced ROS generation needs to be further studied. Whether inflammasome activation affects mitophagy is still to be elucidated.
The Effects of Autophagy on NLRP3 Inflammasome in Experimental Autoimmune Encephalomyelitis
Multiple sclerosis (MS) is a chronic inflammatory autoimmune disease characterized by immune-mediated demyelination and neurodegeneration of the CNS. EAE is an animal model of MS that has been widely studied in recent years (Zepp et al., 2011). Cannabinoid receptor 1 (CB2R) is mainly expressed in immune cells intimately and has been reported to be related to the inflammation in MS. A large number of studies show that CB2R and the NLRP3 inflammasome play an important role in the development of EAE (Rossi et al., 2011; Lou et al., 2012; Inoue and Shinohara, 2013). Activating CB2R could ameliorate clinical symptoms and leukocyte infiltration in EAE. CB2R-deficiency notably increased NLRP3 expression and the secretion of IL-1β and activated Casp-1 activation in EAE, and HU-308 (CB2R agonist) had the opposite effects, indicating that CB2R inhibited NLRP3 inflammasome-mediated inflammation. CB2R-deficiency also decreased the levels of LC3-II/LC3-I ratio and Beclin 1 in EAE, and HU-308 had the opposite effects, suggesting that CB2R promoted autophagy. Inhibition of autophagy with ATG5 siRNA attenuated the inhibitory effect of HU-308 on the NLRP3 inflammasome, suggesting that the induction of autophagy mediates, at least partly, the inhibitory effect of CB2R on NLRP3 inflammasome formation. Collectively, activation of CB2R can improve EAE through suppression of NLRP3 inflammasome via upregulating autophagy, which provides a good strategy to treat MS (Shao et al., 2014). The mechanism of autophagy inhibiting NLRP3 inflammasome in EAE remains to be studied. Contrary to the above conclusion, there has been a report that inhibition of autophagy of dendritic cells attenuated inflammatory infiltration in EAE mice (Bhattacharya et al., 2014), which might be due to the different species of target cells.
Conclusion
In conclusion, autophagy plays an important role in the development and treatment of many nervous system diseases by affecting NLRP3 inflammasome. The relevant mechanism is very complex and needs to be further clarified. At present, most studies confirm that autophagy attenuates inflammatory injury by inhibiting the NLRP3 inflammasome. Autophagy inhibits the NLRP3 inflammasome by reducing ASC, phosphorylating NLRP3, and scavenging ROS (Figure 3; Cao et al., 2019). In contrast, autophagy also promotes the NLRP3 inflammasome in yeast cells (Dupont et al., 2011). Whether autophagy can promote the NLRP3 inflammasome in mammalian cells needs to be further explored. Although autophagy plays an important role by regulating the NLRP3 inflammasome in neuroinflammatory injury, there are still many problems to be solved. For example, can autophagy affect NLRP3 inflammasome through other pathways? Are there side effects of promoting autophagy to inhibit inflammatory injury? With the continuous progress of research, targeting autophagy and NLRP3 inflammation may offer a new way for the treatment of inflammatory nervous system diseases.
FIGURE 3
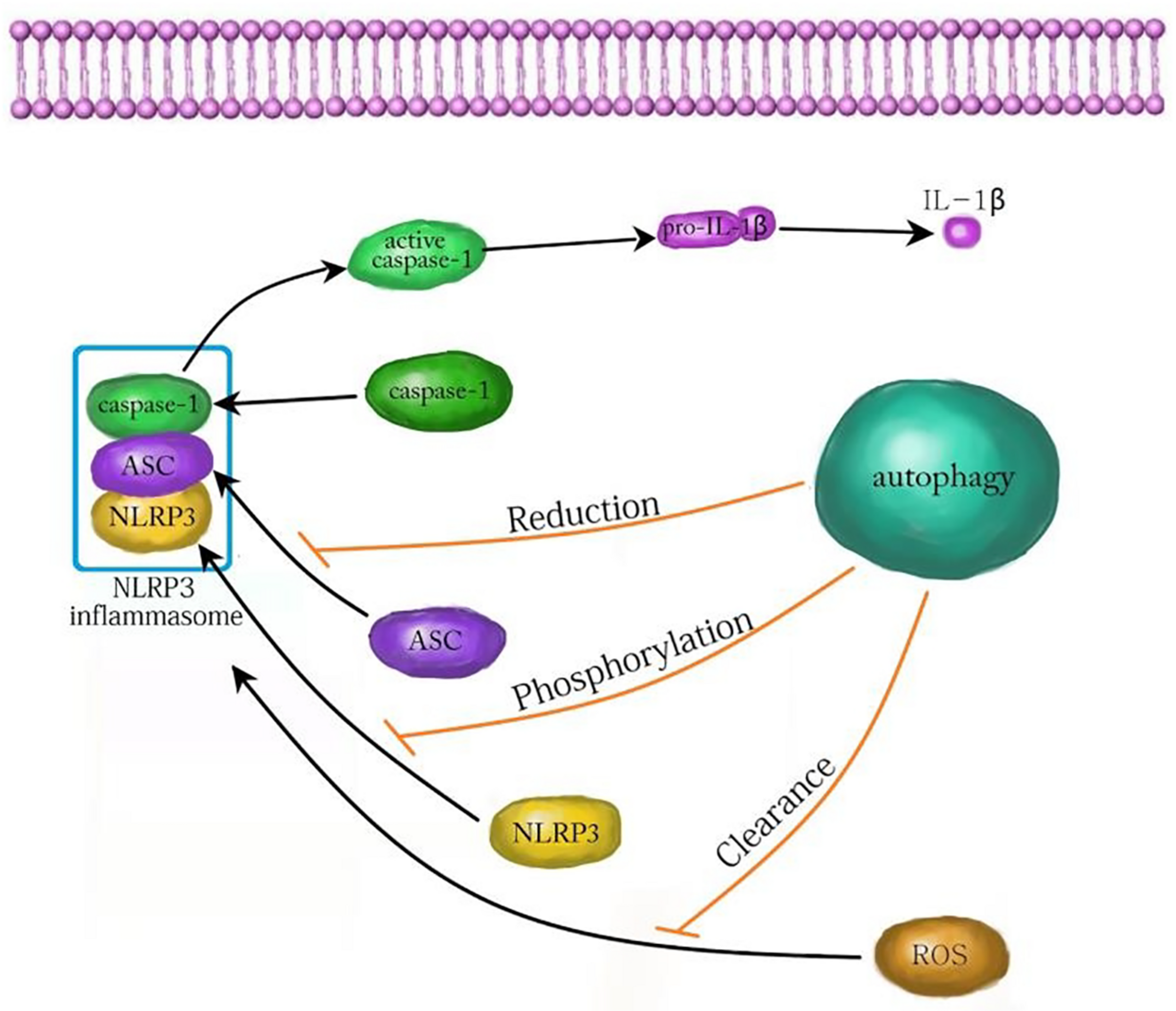
The mechanism of autophagy inhibiting NLRP3 inflammasome. Autophagy can inhibit the activation of the NLRP3 inflammasome by decreasing ASC, increasing NLRP3 phosphorylation and clearing ROS. ASC, apoptosis associated speck like protein; ROS, reactive oxygen species.
Statements
Author contributions
HW: devising, writing, and funding this manuscript. SZ: writing and funding this manuscript. XL: drawing. JW: writing. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by grants from the key scientific and technological projects in Henan Province, China (Grant No. 202102310153) and the National Natural Science Foundation of China (Grant No. 81903444).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1
Abais J. M. Xia M. Zhang Y. Boini K. M. Li P.-L. (2015). Redox regulation of NLRP3 inflammasomes: ROS as trigger or effector?Antioxid. Redox Signal.221111–1129. 10.1089/ars.2014.5994
2
Alcocer-Gomez E. de Miguel M. Casas-Barquero N. Nunez-Vasco J. Antonio Sanchez-Alcazar J. Fernandez-Rodriguez A. et al (2014). NLRP3 inflammasome is activated in mononuclear blood cells from patients with major depressive disorder.Brain Behav. Immun.36111–117. 10.1016/j.bbi.2013.10.017
3
Arsava E. M. Hansen M. B. Kaplan B. Peker A. Gocmen R. Arat A. et al (2018). The effect of carotid artery stenting on capillary transit time heterogeneity in patients with carotid artery stenosis.Eur. Stroke J.3263–271. 10.1177/2396987318772686
4
Bhattacharya A. Prakash Y. S. Eissa N. T. (2014). Secretory function of autophagy in innate immune cells.Cell. Microbiol.161637–1645. 10.1111/cmi.12365
5
Biasizzo M. Kopitar-Jerala N. (2020). Interplay between NLRP3 inflammasome and autophagy.Front. Immunol.11:591803. 10.3389/fimmu.2020.591803
6
Bienert G. P. Moller A. L. B. Kristiansen K. A. Schulz A. Moller I. M. Schjoerring J. K. et al (2007). Specific aquaporins facilitate the diffusion of hydrogen peroxide across membranes.J. Biol. Chem.2821183–1192. 10.1074/jbc.M603761200
7
Bodles A. M. Barger S. W. (2004). Cytokines and the aging brain – what we don’t know might help us.Trends Neurosci.27621–626. 10.1016/j.tins.2004.07.011
8
Borowicz K. K. Piskorska B. Banach M. Czuczwar S. J. (2011). Neuroprotective actions of neurosteroids.Front. Endocrinol.2:50. 10.3389/fendo.2011.00050
9
Burns J. Yokota T. Ashihara H. Lean M. E. J. Crozier A. (2002). Plant foods and herbal sources of resveratrol.J. Agric. Food Chem.503337–3340. 10.1021/jf0112973
10
Burri L. Hoem N. Banni S. Berge K. (2012). Marine omega-3 phospholipids: metabolism and biological activities.Int. J. Mol. Sci.1315401–15419. 10.3390/ijms131115401
11
Cao S. Shrestha S. Li J. Yu X. Chen J. Yan F. et al (2017). Melatonin-mediated mitophagy protects against early brain injury after subarachnoid hemorrhage through inhibition of NLRP3 inflammasome activation.Sci. Rep.7:2417. 10.1038/s41598-017-02679-z
12
Cao Z. Wang Y. Long Z. He G. (2019). Interaction between autophagy and the NLRP3 inflammasome.Acta Biochim. Biophys. Sin.511087–1095. 10.1093/abbs/gmz098
13
Chen S. Ma Q. Krafft P. R. Hu Q. Rolland W. II Sherchan P. et al (2013). P2X7R/cryopyrin inflammasome axis inhibition reduces neuroinflammation after SAH.Neurobiol. Dis.58296–307. 10.1016/j.nbd.2013.06.011
14
Chen X. Liu Y. Zhu J. Lei S. Dong Y. Li L. et al (2016). GSK-3 beta downregulates Nrf2 in cultured cortical neurons and in a rat model of cerebral ischemia-reperfusion.Sci. Rep.6:20196. 10.1038/srep20196
15
Chien T. Weng Y.-T. Chang S.-Y. Lai H.-L. Chiu F.-L. Kuo H.-C. et al (2018). GSK3 beta negatively regulates TRAX, a scaffold protein implicated in mental disorders, for NHEJ-mediated DNA repair in neurons.Mol. Psychiatry232375–2390. 10.1038/s41380-017-0007-z
16
Cho M.-H. Cho K. Kang H.-J. Jeon E.-Y. Kim H.-S. Kwon H.-J. et al (2014). Autophagy in microglia degrades extracellular beta-amyloid fibrils and regulates the NLRP3 inflammasome.Autophagy101761–1775. 10.4161/auto.29647
17
Choi S. A. Chong S. Kwak P. A. Moon Y. J. Jangra A. Phi J. H. et al (2019). Impaired functional recovery of endothelial colony-forming cells from moyamoya disease in a chronic cerebral hypoperfusion rat model.J. Neurosurg. Pediatr.23204–213. 10.3171/2018.8.PEDS1883
18
Ciurea A. V. Palade C. Voinescu D. Nica D. A. (2013). Subarachnoid hemorrhage and cerebral vasospasm – literature review.J. Med. Life6120–125.
19
Deretic V. Saitoh T. Akira S. (2013). Autophagy in infection, inflammation and immunity.Nat. Rev. Immunol.13722–737. 10.1038/nri3532
20
Dirnagl U. Iadecola C. Moskowitz M. A. (1999). Pathobiology of ischaemic stroke: an integrated view.Trends Neurosci.22391–397. 10.1016/S0166-2236(99)01401-0
21
Donnan G. A. Fisher M. Macleod M. Davis S. M. (2008). Stroke.Lancet3711612–1623. 10.1016/S0140-6736(08)60694-7
22
Dumont A. S. Dumont R. J. Chow M. M. Lin C. L. Calisaneller T. Ley K. F. et al (2003). Cerebral vasospasm after subarachnoid hemorrhage: putative role of inflammation.Neurosurgery53123–133. 10.1227/01.NEU.0000068863.37133.9E
23
Dupont N. Jiang S. Pilli M. Ornatowski W. Bhattacharya D. Deretic V. (2011). Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1 beta.EMBO J.304701–4711. 10.1038/emboj.2011.398
24
Erikson K. M. Aschner M. (2003). Manganese neurotoxicity and glutamate-GABA interaction.Neurochem. Int.43475–480. 10.1016/S0197-0186(03)00037-8
25
Espinosa-Garcia C. Aguilar-Hernandez A. Ceruantes M. Morali G. (2014). Effects of progesterone on neurite growth inhibitors in the hippocampus following global cerebral ischemia.Brain Res.154523–34. 10.1016/j.brainres.2013.11.030
26
Fann D. Y.-W. Lee S. Y. Manzanero S. Tang S. C. Gelderblom M. Chunduri P. et al (2013). Intravenous immunoglobulin suppresses NLRP1 and NLRP3 inflammasome-mediated neuronal death in ischemic stroke.Cell Death Dis.4:e790. 10.1038/cddis.2013.326
27
Fujita N. Itoh T. Omori H. Fukuda M. Noda T. Yoshimori T. (2008). The Atg16L complex specifies the site of LC3 lipidation for membrane biogenesis in autophagy.Mol. Biol. Cell192092–2100. 10.1091/mbc.e07-12-1257
28
Galluzzi L. Green D. R. (2019). Autophagy-independent functions of the autophagy machinery.Cell1771682–1699. 10.1016/j.cell.2019.05.026
29
Garcia-Huerta P. Troncoso-Escudero P. Jerez C. Hetz C. Vidal R. L. (2016). The intersection between growth factors, autophagy and ER stress: a new target to treat neurodegenerative diseases?Brain Res.1649173–180. 10.1016/j.brainres.2016.02.052
30
Geng J. Liu J. Yuan X. Liu W. Guo W. (2019). Andrographolide triggers autophagy-mediated inflammation inhibition and attenuates chronic unpredictable mild stress (CUMS)-induced depressive-like behavior in mice.Toxicol. Appl. Pharmacol.379:114688. 10.1016/j.taap.2019.114688
31
Glick D. Barth S. Macleod K. F. (2010). Autophagy: cellular and molecular mechanisms.J. Pathol.221:3–12. 10.1002/path.2697
32
Goldberg E. L. Dixit V. D. (2015). Drivers of age-related inflammation and strategies for healthspan extension.Immunol. Rev.26563–74. 10.1111/imr.12295
33
Grebe A. Hoss F. Latz E. (2018). NLRP3 inflammasome and the IL-1 pathway in atherosclerosis.Circ. Res.1221722–1740. 10.1161/CIRCRESAHA.118.311362
34
Hainsworth A. H. Markus H. S. (2008). Do in vivo experimental models reflect human cerebral small vessel disease? A systematic review.J. Cereb. Blood Flow Metab.281877–1891. 10.1038/jcbfm.2008.91
35
Han X. Sun S. Sun Y. Song Q. Zhu J. Song N. et al (2019). Small molecule-driven NLRP3 inflammation inhibition via interplay between ubiquitination and autophagy: implications for Parkinson disease.Autophagy151860–1881. 10.1080/15548627.2019.1596481
36
Haque M. E. Akther M. Jakaria M. Kim I.-S. Azam S. Choi D.-K. (2020). Targeting the microglial NLRP3 inflammasome and its role in Parkinson’s disease.Mov. Disord.3520–33. 10.1002/mds.27874
37
He Q. Li Z. Wang Y. Hou Y. Li L. Zhao J. (2017). Resveratrol alleviates cerebral ischemia/reperfusion injury in rats by inhibiting NLRP3 inflammasome activation through Sirt1-dependent autophagy induction.Int. Immunopharmacol.50208–215. 10.1016/j.intimp.2017.06.029
38
He Y. Hara H. Nunez G. (2016). Mechanism and regulation of NLRP3 inflammasome activation.Trends Biochem. Sci.411012–1021. 10.1016/j.tibs.2016.09.002
39
Heneka M. T. Kummer M. P. Stutz A. Delekate A. Schwartz S. Vieira-Saecker A. et al (2013). NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice.Nature493674–678. 10.1038/nature11729
40
Hong Y. Liu Y. Yu D. Wang M. Hou Y. (2019). The neuroprotection of progesterone against A beta-induced NLRP3-Caspase-1 inflammasome activation via enhancing autophagy in astrocytes.Int. Immunopharmacol.74:105669. 10.1016/j.intimp.2019.05.054
41
Hong Y. Liu Y. Zhang G. Wu H. Hou Y. (2018). Progesterone suppresses A beta(42)-induced neuroinflammation by enhancing autophagy in astrocytes.Int. Immunopharmacol.54336–343. 10.1016/j.intimp.2017.11.044
42
Hong Y. Wang X. Sun S. Xue G. Li J. Hou Y. (2016). Progesterone exerts neuroprotective effects against A beta-induced neuroinflammation by attenuating ER stress in astrocytes.Int. Immunopharmacol.3383–89. 10.1016/j.intimp.2016.02.002
43
Hou X. Watzlawik J. O. Fiesel F. C. Springer W. (2020). Autophagy in Parkinson’s disease.J. Mol. Biol.4322651–2672. 10.1016/j.jmb.2020.01.037
44
Houtman J. Freitag K. Gimber N. Schmoranzer J. Heppner F. L. Jendrach M. (2019). Beclin1-driven autophagy modulates the inflammatory response of microglia via NLRP3.EMBO J.38:e99430. 10.15252/embj.201899430
45
Ichimiya T. Yamakawa T. Hirano T. Yokoyama Y. Hayashi Y. Hirayama D. et al (2020). Autophagy and autophagy-related diseases: a review.Int. J. Mol. Sci.21:8974. 10.3390/ijms21238974
46
Inoue M. Shinohara M. L. (2013). NLRP3 inflammasome and MS/EAE.Autoimmune Dis.2013:859145. 10.1155/2013/859145
47
Iregren A. (1999). Manganese neurotoxicity in industrial exposures: proof of effects, critical exposure level, and sensitive tests.Neurotoxicology20315–323.
48
Ising C. Venegas C. Zhang S. Scheiblich H. Schmidt S. V. Vieira-Saecker A. et al (2019). NLRP3 inflammasome activation drives tau pathology.Nature575669–673. 10.1038/s41586-019-1769-z
49
Islam M. T. Ali E. S. Uddin S. J. Islam M. A. Shaw S. Khan I. N. et al (2018). Andrographolide, a diterpene lactone from Andrographis paniculata and its therapeutic promises in cancer.Cancer Lett.420129–145. 10.1016/j.canlet.2018.01.074
50
Kaushik S. Cuervo A. M. (2012). Chaperone-mediated autophagy: a unique way to enter the lysosome world.Trends Cell Biol.22407–417. 10.1016/j.tcb.2012.05.006
51
Kim H. Y. Lee H. J. Chang Y.-J. Pichavant M. Shore S. A. Fitzgerald K. A. et al (2014). Interleukin-17-producing innate lymphoid cells and the NLRP3 inflammasome facilitate obesity-associated airway hyperreactivity.Nat. Med.2054–61. 10.1038/nm.3423
52
Lane C. A. Hardy J. Schott J. M. (2018). Alzheimer’s disease.Eur. J. Neurol.2559–70. 10.1111/ene.13439
53
Liu S. Wu H. Xue G. Ma X. Wu J. Qin Y. et al (2013). Metabolic alteration of neuroactive steroids and protective effect of progesterone in Alzheimer’s disease-like rats.Neural Regen. Res.82800–2810.
54
Liu Y. Levine B. (2015). Autosis and autophagic cell death: the dark side of autophagy.Cell Death Differ.22367–376. 10.1038/cdd.2014.143
55
Liu Y. Zhang Y. Peng J. Wang H. Li X. Li X. et al (2019). Autophagy alleviates ethanol-induced memory impairment in association with anti-apoptotic and anti-inflammatory pathways.Brain Behav. Immun.8263–75. 10.1016/j.bbi.2019.07.033
56
Lou Z.-Y. Zhao C.-B. Xiao B.-G. (2012). Immunoregulation of experimental autoimmune encephalomyelitis by the selective CB1 receptor antagonist.J. Neurosci. Res.9084–95. 10.1002/jnr.22721
57
Lu A. Magupalli V. G. Ruan J. Yin Q. Atianand M. K. Vos M. R. et al (2014). Unified polymerization mechanism for the assembly of ASC-dependent inflammasomes.Cell1561193–1206. 10.1016/j.cell.2014.02.008
58
Lu X. Dong J. Zheng D. Li X. Ding D. Xu H. (2020). Reperfusion combined with intraarterial administration of resveratrol-loaded nanoparticles improved cerebral ischemia-reperfusion injury in rats.Nanomedicine28:102208. 10.1016/j.nano.2020.102208
59
Lucin K. M. O’brien C. E. Bieri G. Czirr E. Mosher K. I. Abbey R. J. et al (2013). Microglial beclin 1 regulates retromer trafficking and phagocytosis and is impaired in Alzheimer’s disease.Neuron79873–886. 10.1016/j.neuron.2013.06.046
60
Lv S. Wang H. Li X. (2021). The role of the interplay between autophagy and NLRP3 inflammasome in metabolic disorders.Front. Cell Dev. Biol.9:634118. 10.3389/fcell.2021.634118
61
Ma C. Wang X. Xu T. Yu X. Zhang S. Liu S. et al (2019). Qingkailing injection ameliorates cerebral ischemia-reperfusion injury and modulates the AMPK/NLRP3 inflammasome signalling pathway.BMC Complement. Altern. Med.19:320. 10.1186/s12906-019-2703-5
62
Martinon F. Burns K. Tschopp J. (2002). The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta.Mol. Cell10417–426. 10.1016/S1097-2765(02)00599-3
63
Matsuyama H. Shindo A. Shimada T. Yata K. Wakita H. Takahashi R. et al (2020). Chronic cerebral hypoperfusion activates AIM2 and NLRP3 inflammasome.Brain Res.1736:146779. 10.1016/j.brainres.2020.146779
64
Mendiola A. S. Cardona A. E. (2018). The IL-1 beta phenomena in neuroinflammatory diseases.J. Neural Transm.125781–795. 10.1007/s00702-017-1732-9
65
Menzies F. M. Fleming A. Rubinsztein D. C. (2015). Compromised autophagy and neurodegenerative diseases.Nat. Rev. Neurosci.16345–357. 10.1038/nrn3961
66
Mizushima N. Yoshimori T. Ohsumi Y. (2011). “The role of Atg proteins in autophagosome formation,” in Annual Review Of Cell And Developmental Biology, Vol. 27edsSchekmanR.GoldsteinL.LehmannR. (Palo Alto, CA: Annual reviews), 107–132. 10.1146/annurev-cellbio-092910-154005
67
Munoz-Planillo R. Kuffa P. Martinez-Colon G. Smith B. L. Rajendiran T. M. Nunez G. (2013). K+ efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter.Immunity381142–1153. 10.1016/j.immuni.2013.05.016
68
Ostrowski R. P. Colohan A. R. Zhang J. H. (2006). Molecular mechanisms of early brain injury after subarachnoid hemorrhage.Neurol. Res.28399–414. 10.1179/016164106X115008
69
Pan Y. Chen X.-Y. Zhang Q.-Y. Kong L.-D. (2014). Microglial NLRP3 inflammasome activation mediates IL-1 beta-related inflammation in prefrontal cortex of depressive rats.Brain Behav. Immun.4190–100. 10.1016/j.bbi.2014.04.007
70
Pariante C. M. (2017). Why are depressed patients inflamed? A reflection on 20 years of research on depression,glucocorticoid resistance and inflammation.Eur. Neuropsychopharmacol.27554–559. 10.1016/j.euroneuro.2017.04.001
71
Peng J. Wang H. Gong Z. Li X. He L. Shen Q. et al (2020). Idebenone attenuates cerebral inflammatory injury in ischemia and reperfusion via dampening NLRP3 inflammasome activity.Mol. Immunol.12374–87. 10.1016/j.molimm.2020.04.013
72
Poewe W. Seppi K. Tanner C. M. Halliday G. M. Brundin P. Volkmann J. et al (2017). Parkinson disease.Nat. Rev. Dis. Primers3:17013. 10.1038/nrdp.2017.13
73
Pyo J.-O. Nah J. Jung Y.-K. (2012). Molecules and their functions in autophagy.Exp. Mol. Med.4473–80. 10.3858/emm.2012.44.2.029
74
Qin Y. Chen Z. Han X. Wu H. Yu Y. Wu J. et al (2015). Progesterone attenuates A beta(25-35)-induced neuronal toxicity via JNK inactivation and progesterone receptor membrane component 1-dependent inhibition of mitochondrial apoptotic pathway.J. Steroid Biochem. Mol. Biol.154302–311. 10.1016/j.jsbmb.2015.01.002
75
Qiu J. Wang M. Zhang J. Cai Q. Lu D. Li Y. et al (2016). The neuroprotection of Sinomenine against ischemic stroke in mice by suppressing NLRP3 inflammasome via AMPK signaling.Int. Immunopharmacol.40492–500. 10.1016/j.intimp.2016.09.024
76
Raison C. L. Rutherford R. E. Woolwine B. J. Shuo C. Schettler P. Drake D. F. et al (2013). A randomized controlled trial of the tumor necrosis factor antagonist infliximab for treatment-resistant depression.JAMA Psychiatry7031–41. 10.1001/2013.jamapsychiatry.4
77
Rossi S. Furlan R. de Chiara V. Muzio L. Musella A. Motta C. et al (2011). Cannabinoid CB1 receptors regulate neuronal TNF-alpha effects in experimental autoimmune encephalomyelitis.Brain Behav. Immun.251242–1248. 10.1016/j.bbi.2011.03.017
78
Sahu R. Kaushik S. Clement C. C. Cannizzo E. S. Scharf B. Follenzi A. et al (2011). Microautophagy of cytosolic proteins by late endosomes.Dev. Cell20131–139. 10.1016/j.devcel.2010.12.003
79
Saresella M. La Rosa F. Piancone F. Zoppis M. Marventano I. Calabrese E. et al (2016). The NLRP3 and NLRP1 inflammasomes are activated in Alzheimer’s disease.Mol. Neurodegener.11:23. 10.1186/s13024-016-0088-1
80
Schneider U. C. Xu R. Vajkoczy P. (2018). Inflammatory events following subarachnoid hemorrhage (SAH).Curr. Neuropharmacol.161385–1395. 10.2174/1570159X16666180412110919
81
Schroder K. Tschopp J. (2010). The inflammasomes.Cell140821–832. 10.1016/j.cell.2010.01.040
82
Schwab A. D. Thurston M. J. Machhi J. Olson K. E. Namminga K. L. Gendelman H. E. et al (2020). Immunotherapy for Parkinson’s disease.Neurobiol. Dis.137:104760. 10.1016/j.nbd.2020.104760
83
Scott X. O. Stephens M. E. Desir M. C. Dietrich W. D. Keane R. W. de Rivero Vaccari J. P. (2020). The inflammasome adaptor protein ASC in mild cognitive impairment and Alzheimer’s disease.Int. J. Mol. Sci.21:4674. 10.3390/ijms21134674
84
Sercombe R. Dinh Y. R. T. Gomis P. (2002). Cerebrovascular inflammation following subarachnoid hemorrhage.Jpn. J. Pharmacol.88227–249. 10.1254/jjp.88.227
85
Shang J. Yamashita T. Zhai Y. Nakano Y. Morihara R. Li X. et al (2019). Acceleration of NLRP3 inflammasome by chronic cerebral hypoperfusion in Alzheimer’s disease model mouse.Neurosci. Res.14361–70. 10.1016/j.neures.2018.06.002
86
Shao B.-Z. Wei W. Ke P. Xu Z.-Q. Zhou J.-X. Liu C. (2014). Activating cannabinoid receptor 2 alleviates pathogenesis of experimental autoimmune encephalomyelitis via activation of autophagy and inhibiting NLRP3 inflammasome.CNS Neurosci. Ther.201021–1028. 10.1111/cns.12349
87
Shi J. Zhao Y. Wang K. Shi X. Wang Y. Huang H. et al (2015). Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death.Nature526660–665. 10.1038/nature15514
88
Shirakabe A. Ikeda Y. Sciarretta S. Zablocki D. K. Sadoshima J. (2016). Aging and autophagy in the heart.Circ. Res.1181563–1576. 10.1161/CIRCRESAHA.116.307474
89
Shrikanta A. Kumar A. Govindaswamy V. (2015). Resveratrol content and antioxidant properties of underutilized fruits.J. Food Sci. Technol.52383–390. 10.1007/s13197-013-0993-z
90
Smith K. (2014). Mental health: a world of depression.Nature515:181. 10.1038/515180a
91
Song L. Pei L. Yao S. Wu Y. Shang Y. (2017). NLRP3 inflammasome in neurological diseases, from functions to therapies.Front. Cell. Neurosci.11:63. 10.3389/fncel.2017.00063
92
Su S.-H. Wu Y.-F. Lin Q. Hai J. (2017). Cannabinoid receptor agonist WIN55,212-2 and fatty acid amide hydrolase inhibitor URB597 ameliorate neuroinflammatory responses in chronic cerebral hypoperfusion model by blocking NF-kappa B pathways.Naunyn Schmiedebergs Arch. Pharmacol.3901189–1200. 10.1007/s00210-017-1417-9
93
Su S.-H. Wu Y.-F. Lin Q. Wang D.-P. Hai J. (2019). URB597 protects against NLRP3 inflammasome activation by inhibiting autophagy dysfunction in a rat model of chronic cerebral hypoperfusion.J. Neuroinflammation16:260. 10.1186/s12974-019-1668-0
94
Su S.-H. Wu Y.-F. Wang D.-P. Hai J. (2018). Inhibition of excessive autophagy and mitophagy mediates neuroprotective effects of URB597 against chronic cerebral hypoperfusion.Cell Death Dis.9:733. 10.1038/s41419-018-0755-y
95
Sun X. Liu H. Sun Z. Zhang B. Wang X. Liu T. et al (2020). Acupuncture protects against cerebral ischemia-reperfusion injury via suppressing endoplasmic reticulum stress-mediated autophagy and apoptosis.Mol. Med.26:105. 10.1186/s10020-020-00236-5
96
Swanson K. V. Deng M. Ting J. P. Y. (2019). The NLRP3 inflammasome: molecular activation and regulation to therapeutics.Nat. Rev. Immunol.19477–489. 10.1038/s41577-019-0165-0
97
Toldo S. Abbate A. (2018). The NLRP3 inflammasome in acute myocardial infarction.Nat. Rev. Cardiol.15203–214. 10.1038/nrcardio.2017.161
98
Tooze S. A. Yoshimori T. (2010). The origin of the autophagosomal membrane.Nat. Cell Biol.12831–835. 10.1038/ncb0910-831
99
Ueno T. Komatsu M. (2017). Autophagy in the liver: functions in health and disease.Nat. Rev. Gastroenterol. Hepatol.14170–184. 10.1038/nrgastro.2016.185
100
Walsh J. G. Reinke S. N. Mamik M. K. Mckenzie B. A. Maingat F. Branton W. G. et al (2014). Rapid inflammasome activation in microglia contributes to brain disease in HIV/AIDS.Retrovirology11:35. 10.1186/1742-4690-11-35
101
Wang D. Wang H. Gao H. Zhang H. Zhang H. Wang Q. et al (2020). P2X7 receptor mediates NLRP3 inflammasome activation in depression and diabetes.Cell Biosci.10:28. 10.1186/s13578-020-00388-1
102
Wang D. Zhang J. Jiang W. Cao Z. Zhao F. Cai T. et al (2017). The role of NLRP3-CASP1 in inflammasome-mediated neuroinflammation and autophagy dysfunction in manganese-induced, hippocampal-dependent impairment of learning and memory ability.Autophagy13914–927. 10.1080/15548627.2017.1293766
103
Wang J. Wu D. Wang H. (2019a). Hydrogen sulfide plays an important protective role by influencing autophagy in diseases.Physiol. Res.68335–345. 10.33549/physiolres.933996
104
Wang L. Zhai Y.-Q. Xu L.-L. Qiao C. Sun X.-L. Ding J.-H. et al (2014). Metabolic inflammation exacerbates dopaminergic neuronal degeneration in response to acute MPTP challenge in type 2 diabetes mice.Exp. Neurol.25122–29. 10.1016/j.expneurol.2013.11.001
105
Wang P. Xu T.-Y. Guan Y.-F. Tian W.-W. Viollet B. Rui Y.-C. et al (2011). Nicotinamide phosphoribosyltransferase protects against ischemic stroke through SIRT1-dependent adenosine monophosphate-activated kinase pathway.Ann. Neurol.69360–374. 10.1002/ana.22236
106
Wang S. Yuan Y.-H. Chen N.-H. Wang H.-B. (2019b). The mechanisms of NLRP3 inflammasome/pyroptosis activation and their role in Parkinson’s disease.Int. Immunopharmacol.67458–464. 10.1016/j.intimp.2018.12.019
107
Wang W. Guo W. Li L. Fu Z. Liu W. Gao J. et al (2016). Andrographolide reversed 5-FU resistance in human colorectal cancer by elevating BAX expression.Biochem. Pharmacol.1218–17. 10.1016/j.bcp.2016.09.024
108
Wang X. Li R. Wang X. Fu Q. Ma S. (2015). Umbelliferone ameliorates cerebral ischemia-reperfusion injury via upregulating the PPAR gamma expression and suppressing TXNIP/NLRP3 inflammasome.Neurosci. Lett.600182–187. 10.1016/j.neulet.2015.06.016
109
Wang Y. Meng C. Zhang J. Wu J. Zhao J. (2019c). Inhibition of GSK-3 beta alleviates cerebral ischemia/reperfusion injury in rats by suppressing NLRP3 inflammasome activation through autophagy.Int. Immunopharmacol.68234–241. 10.1016/j.intimp.2018.12.042
110
Wen M. Ding L. Zhang L. Zhang T. Teruyosh Y. Wang Y. et al (2019). Eicosapentaenoic acid-enriched phosphatidylcholine mitigated Abeta-42-induced neurotoxicity via autophagy-inflammasome pathway.J. Agric. Food Chem.6713767–13774. 10.1021/acs.jafc.9b05947
111
Yang Y. Wang H. Kouadir M. Song H. Shi F. (2019). Recent advances in the mechanisms of NLRP3 inflammasome activation and its inhibitors.Cell Death Dis.10:128. 10.1038/s41419-019-1413-8
112
Yao L. Zhu Z. Wu J. Zhang Y. Zhang H. Sun X. et al (2019). MicroRNA-124 regulates the expression of p62/p38 and promotes autophagy in the inflammatory pathogenesis of Parkinson’s disease.FASEB J.338648–8665. 10.1096/fj.201900363R
113
Yaribeygi H. Katsiki N. Butler A. E. Sahebkar A. (2019). Effects of antidiabetic drugs on NLRP3 inflammasome activity, with a focus on diabetic kidneys.Drug Discov. Today24256–262. 10.1016/j.drudis.2018.08.005
114
Youle R. J. Narendra D. P. (2011). Mechanisms of mitophagy.Nat. Rev. Mol. Cell Biol.129–14. 10.1038/nrm3028
115
Zepp J. Wu L. Li X. (2011). IL-17 receptor signaling and T helper 17-mediated autoimmune demyelinating disease.Trends Immunol.32232–239. 10.1016/j.it.2011.02.007
116
Zhang Y.-M. Qu X.-Y. Tao L.-N. Zhai J.-H. Gao H. Song Y.-Q. et al (2020). XingNaoJing injection ameliorates cerebral ischaemia/reperfusion injury via SIRT1-mediated inflammatory response inhibition.Pharm. Biol.5816–24. 10.1080/13880209.2019.1698619
117
Zhou X. Zhou J. Li X. Guo C. A. Fang T. Chen Z. (2011). GSK-3 beta inhibitors suppressed neuroinflammation in rat cortex by activating autophagy in ischemic brain injury.Biochem. Biophys. Res. Commun.411271–275. 10.1016/j.bbrc.2011.06.117
118
Zhou Y. Lu M. Du R.-H. Qiao C. Jiang C.-Y. Zhang K.-Z. et al (2016). MicroRNA-7 targets nod-like receptor protein 3 inflammasome to modulate neuroinflammation in the pathogenesis of Parkinson’s disease.Mol. Neurodegener.11:28. 10.1186/s13024-016-0094-3
119
Zhu Y. Yin Q. Wei D. Yang Z. Du Y. Ma Y. (2019). Autophagy in male reproduction.Syst. Biol. Reprod. Med.65265–272. 10.1080/19396368.2019.1606361
Summary
Keywords
autophagy, NLRP3 inflammasome, Alzheimer’s disease, Parkinson’s disease, depression
Citation
Zhao S, Li X, Wang j and Wang H (2021) The Role of the Effects of Autophagy on NLRP3 Inflammasome in Inflammatory Nervous System Diseases. Front. Cell Dev. Biol. 9:657478. doi: 10.3389/fcell.2021.657478
Received
25 January 2021
Accepted
08 April 2021
Published
17 May 2021
Volume
9 - 2021
Edited by
Adelaide Fernandes, University of Lisbon, Portugal
Reviewed by
Claudia Alexandra Valente, University of Lisbon, Portugal; Juan Pablo de Rivero Vaccari, University of Miami, United States
Updates
Copyright
© 2021 Zhao, Li, Wang and Wang.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Honggang Wang, whg197167@vip.henu.edu.cn; whg1975316@sina.com
This article was submitted to Molecular Medicine, a section of the journal Frontiers in Cell and Developmental Biology
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.