 Ziran Yang1†Xuehong Zhou1†Enrun Zheng1Yizhou Wang1
Ziran Yang1†Xuehong Zhou1†Enrun Zheng1Yizhou Wang1 Xinhua Liu2Yue Wang1,2Yanpu Wang3
Xinhua Liu2Yue Wang1,2Yanpu Wang3 Zhaofei Liu3
Zhaofei Liu3 Fei Pei4Yue Zhang1Jie Ren1
Fei Pei4Yue Zhang1Jie Ren1 Yunchao Huang1Lu Xia1Sudun Guan1Sen Qin1Feiya Suo1
Yunchao Huang1Lu Xia1Sudun Guan1Sen Qin1Feiya Suo1 Jie Shi5Lijing Wang6
Jie Shi5Lijing Wang6 Lin He1,5*
Lin He1,5* Luyang Sun1,7*
Luyang Sun1,7*- 1Department of Biochemistry and Molecular Biology, School of Basic Medical Sciences, Key Laboratory of Carcinogenesis and Translational Research (Ministry of Education), Peking University Health Science Center, Beijing, China
- 2Department of Biochemistry and Molecular Biology, School of Basic Medical Sciences, Hangzhou Normal University, Hangzhou, China
- 3Medical Isotopes Research Center and Department of Radiation Medicine, School of Basic Medical Sciences, Peking University Health Science Center, Beijing, China
- 4Department of Pathology, Peking University Third Hospital, School of Basic Medical Sciences, Peking University Health Science Center, Beijing, China
- 5National Institute on Drug Dependence, Peking University, Beijing, China
- 6Vascular Biology Research Institute, Guangdong Pharmaceutical University, Guangzhou, China
- 7Department of Integration of Chinese and Western Medicine, School of Basic Medical Sciences, Peking University Health Science Center, Beijing, China
Many carcinomas feature hypoxia, a condition has long been associated with tumor progression and poor prognosis, as well as resistance to chemoradiotherapy. Here, we report that the F-box protein JFK promotes mammary tumor initiation and progression in MMTV-PyMT murine model of spontaneous breast cancer. We find that JFK is inducible under hypoxic conditions, in which hypoxia-inducible factor HIF-1α binds to and transcriptionally activates JFK in breast cancer cells. Consistently, analysis of public clinical datasets reveals that the mRNA level of JFK is positively correlated with that of HIF-1α in breast cancer. We show that JFK deficiency leads to a decrease in HIF-1α-induced glycolysis in breast cancer and sensitizes hypoxic breast cancer cells to ionizing radiation and chemotherapeutic treatment. These results indicate that JFK is an important player in hypoxic response, supporting the pursuit of JFK as a potential therapeutic target for breast cancer intervention.
Introduction
Among females, breast cancer is the most commonly diagnosed cancer and the leading cause of cancer death (Bray et al., 2018). Although the development of comprehensive breast cancer treatment strategies has supported a downward trend in breast cancer mortality, there are still no means of definitively identifying patients who will be resistance to radiotherapy and chemotherapy and relapse (Akram et al., 2017). Evidence is mounting that the oxygen (O2) content of tumor tissue is an important determinant of metastasis, and tumor hypoxia has been associated with increased malignancy, poor prognosis, and resistance to both radiotherapy and chemotherapy (Keith et al., 2011). Thus, particular interest has been focused on the mechanisms by which hypoxic tumor cells alter their transcriptional profiles to modulate processes including initiation, progression, and metastasis to persist under conditions of hypoxic stress.
Aerobic glycolysis, also termed the Warburg effect, is a general feature of glucose metabolism in tumor cells (Hanahan and Weinberg, 2011; Chen et al., 2019). Tumor cells predominantly utilize glycolysis for energy production mechanisms and have higher rates of glycolysis, which increases biosynthesis, inhibits apoptosis, and generates signaling metabolites to enhance tumor cell survival under difficult conditions including hypoxia (Pavlova and Thompson, 2016; Wu et al., 2020). It is well established that hypoxia can lead to high rates of glycolysis through stabilization of hypoxia-inducible factors (HIFs) (Wu et al., 2020). HIFs are heterodimeric complexes composed of basic helix-loop-helix/PAS (bHLH-PAS) proteins including an O2-labile alpha subunit (HIF-1α, HIF-2α, or HIF-3α) and a stable beta subunit (HIF-1β) (Iommarini et al., 2017). HIF transcription factors are master regulators of the cellular response to hypoxia and coordinate a transcriptional program that ensures optimal functional, metabolic, and vascular adaptation to oxygen shortages (Jurikova et al., 2016). In breast cancer, HIF-1α transcriptionally activates the expression of genes with known functions in cell survival, angiogenesis, metabolic reprogramming, invasion, and metastasis, through binding to hypoxia response elements (HREs) in the promoters of its target genes (Masoud and Li, 2015). However, it remains to be investigated the mechanism(s) underlying how cancer cells response to hypoxic conditions through HIF-1α-mediated transcriptional regulation.
To date, the best-characterized mammalian multi-subunit RING-finger type of E3 ligases is the SCF (SKP1-CUL1-F-box) complex, comprising SKP1, RBX1, CUL1, and a flexible F-box protein (FBP) that acts as a receptor for substrate recognition (Jia and Sun, 2011). In previous studies, we reported that JFK is the only Kelch domain-containing F-box protein in humans, and targets p53 and ING4 for degradation through the assembly of a SCF complex (Sun et al., 2009, 2011; Yan et al., 2015). We showed that JFK destabilizes p53, contributing to reduced cell apoptosis and sensitizing cells to ionizing radiation-induced cell death. Additionally, JFK-mediated ING4 proteasomal degradation promotes angiogenesis and metastasis of breast cancer. JFK is highly expressed in breast cancer, supporting that it could be used as a target molecule for the design of new breast cancer therapies. However, the upstream activators (and cellular contexts) which drive elevated JFK levels in breast cancers are currently unknown.
In the present study, using MMTV-PyMT model of spontaneous breast cancer, we report that JFK promotes mammary tumorigenesis. HIF-1α binds to and transcriptionally activates JFK in hypoxic breast cancer cells, and analyses of public clinical datasets support the positive correlation between mRNA expression of JFK and HIF-1α in breast cancer. We showed that JFK knockdown leads to a decrease in hypoxia-enhanced glycolysis and overcomes chemo-radiotherapeutic resistance in hypoxic breast cancer cells. We demonstrated that JFK is a hypoxia-induced oncogene, and explored the clinicopathological significance of the HIF-1α-JFK axis in breast cancer progression and intervention.
Materials and Methods
Antibodies
The antibodies used were αTubulin from Sigma; αHIF-1α from Abcam; αp53, αING4, αKi-67, αPCNA, αE2F1, αFOXP3, and αMYC from Santa Cruz. Polyclonal antibodies against JFK were raised against the C-terminal epitope of the JFK protein (CYPKTNALYFVRAKR) in rabbits.
JFK Transgenic Mice
JFK transgenic mice were generated by Cyagen Biosciences on the C57BL/6 background. ORF of human JFK with a FLAG tag was cloned into the mammalian expression vector pRP (Exp)-EF1A. After digestion, linearized DNA was used for microinjection into the pro-nuclei of fertilized oocytes from hormonally superovulated C57BL/6 female mice under a microscope. The injected fertilized eggs were transplanted into the oviducts of pseudo-pregnant mice. The genomic DNA were extracted from mouse tail tips for molecular genotyping. The primers specific for the transgene JFK (forward: 5′-GCTTTTGGAGTACGTCGTCT-3′; reverse: 5′-GGCTCCTCATCTTGATCCAT-3′) were used to amplify a 334 bp fragment, and the primers for internal control (forward: 5′-CAACCACTTACAAGAGACCCGTA-3′; reverse: 5′-GAGCCCTTAGAAATAACGTTCACC-3′) were used to amplify a 632 bp fragment.
MMTV-PyMT Mouse Model
FVB/N-Tg (MMTV-PyMT) mice were crossed with wild-type mice (C57BL/6 strain) for six generations to backcross into the C57BL/6 strain. Genotyping analyses were performed by PCR with genomic DNA extracted from tail tips. The primers specific for the transgene PyMT (forward: 5′-GGAAGCAAGTACTTCACAAGGG-3′; reverse: 5′-GGAAAGTCACTAGGAGCAGGG-3′) were used to amplify a 556 bp fragment. Animal handling and procedures were approved by the Institutional Animal Care of Peking University Health Center.
Small-Animal SPECT/CT Imaging of 99mTc-3PRGD2
The integrin αvβ3-targeting radiotracer 99mTc-3PRGD2 was synthesized by labeling the precursor HYNIC-3PRGD2 with Na99mTcO4 as previously described (Jia et al., 2011). 99mTc-3PRGD2 was prepared with the radiochemical purity of >98% and was then used for in vivo imaging studies. For small-animal SPECT/CT, MMTV-PyMT mice were intravenously injected with 37 MBq of 99mTc-3PRGD2. At 30 min postinjection, SPECT/CT scanning was performed using a NanoScan SPECT/CT Imaging System (Mediso, Budapest, Hungary) as previously described (Liu et al., 2012, 2014).
Cell Culture and Transfection
MCF-7 and T47D cells were maintained according to the ATCC’s recommendation. Transfections of expression plasmids were carried out using polyethyleneimine (PEI, Polysciences), and siRNA oligonucleotides (GeneChem Inc.) were transfected into cells using RNAiMAX (Invitrogen) according to the manufacturer’s instructions. The sequences of siRNAs were: HIF-1α siRNA, 5′-UUCAAGUUGGAAUUGGUAG-3′; JFK siRNA, 5′-GGUGUAGCCCAUCAGUGUU-3′; and control siRNA, 5′-UUCUCCGAACGUGUCACGU-3′.
Western Blotting
Cellular lysates were prepared by incubating the cells in lysis buffer (50 mM Tris–HCl, pH 7.5, 150 mM NaCl, 0.5% NP-40, and 2 mM EDTA) containing protease inhibitor cocktail and phosphatase inhibitor for 20 min at 4°C, followed by centrifugation at 14,000 g for 15 min at 4°C. The protein concentration of the lysates was determined using the BCA protein assay kit (Pierce) according to the manufacturer’s protocol. For western blotting, the samples were resolved on SDS-PAGE gels and transferred onto nitrocellulose membranes. The membranes were incubated with appropriate antibodies overnight at 4°C followed by incubation with a secondary antibody. Immunoreactive bands were visualized using western blotting Luminol reagent (Santa Cruz) according to the manufacturer’s recommendation. The dilutions of primary antibodies were: 1: 1000 with αJFK, αING4, αKi-67, αPCNA, or αHIF-1α; 1: 2000 with αp53 or αTubulin.
Real-Time Reverse Transcription PCR
Total mRNAs were isolated with Trizol reagents (Invitrogen) for cDNA synthesis. Real-time reverse transcriptase PCR was performed using the ABI PRISM 7500 system (Applied Biosystems) that measures real-time SYBR green fluorescence and then calculated by means of the comparative Ct method (2-ΔΔCt) with the expression of GAPDH as an internal control. The primers used were: JFK: 5′-AGCCCCACCCAGTATTGGA-3′ (forward) and 5′-CCTGATACGGTGAGAGAAAGGA-3′ (reverse); GAPDH: 5′-GAAGGTGAAGGTCGGAGTC-3′ (forward) and 5′-GAAGATGGTGATGGGATTTC-3′ (reverse).
ChIP and qChIP
MCF-7 or T47D cells treated with hypoxia (1% O2) for 24 h were crosslinked with 1% formaldehyde for 10 min at room temperature and quenched by the addition of glycine to a final concentration of 125 mM for 5 min. The fixed cells were resuspended in lysis buffer (1% SDS, 5 mM EDTA, and 50 mM Tris–HCl, pH 8.1) containing protease inhibitors, then subjected to 30 cycles (30 s on and off) of sonication (Bioruptor, Diagenode) to generate chromatin fragments of ∼300 bp in length. Lysates were diluted in buffer (1% Triton X-100, 2 mM EDTA, 150 mM NaCl, and 20 mM Tris–HCl, pH 8.1) containing protease inhibitors. For immunoprecipitation, the diluted chromatin was incubated with normal IgG (control) or HIF-1α antibodies overnight at 4°C with constant rotation, followed by incubation with 50 μl of 50% (v/v) protein A/G Sepharose beads for an additional 2 h. Beads were successively washed with the following buffers: TSE I (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 150 mM NaCl, and 20 mM Tris–HCl, pH 8.0); TSE II (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 500 mM NaCl, and 20 mM Tris–HCl, pH 8.0); TSE III (0.25 M LiCl, 1% Nonidet P-40, 1% sodium deoxycholate, 1 mM EDTA, and 10 mM Tris–HCl, pH 8.0). The pulled-down chromatin complex eluted by TE (1 mM EDTA and 10 mM Tris–HCl, pH 8.0) and input were de-crosslinked at 55°C for 12 h in elution buffer (1% SDS and 0.1 M NaHCO3) (Li et al., 2016). The DNA was purified with the QIAquick PCR Purification Kit (QIAGEN). qChIPs were performed using Power SYBR Green PCR Master Mix and an ABI PRISM 7500 system (Applied Biosystems, Foster City, CA, United States). The primers used were: JFK: 5′-TGAGCACATTTGTGTGCCG-3′ (forward) and 5′-TGGACACTACTAGGCGGTCA-3′ (reverse).
Luciferase Reporter Assay
MCF-7 or T47D cells in 24-well plates were infected with lentiviruses carrying luciferase reporter, renilla, and indicated expression constructs. The amount of DNA in each transfection was kept constant by addition of empty vector. Twenty-four hours after transfection, the firefly and renilla luciferase were assayed according to the manufacturer’s protocol (Promega), and the firefly luciferase activity was normalized to that of renilla luciferase. Each experiment was performed in triplicate and repeated at last three times.
Measurement of Glucose Uptake and Lactate Production
The extracellular lactate was measured using the cell culture medium with Lactate Colorimetric Assay Kit (BioVision) according to the manufacturer’s instruction. Intracellular glucose was measured using cell lysates with Glucose Colorimetric/Fluorometric Assay Kit (BioVision) according to the manufacturer’s instruction.
Histologic Analysis
Mammary tumors or livers of female mice were excised, fixed with 10% neutral buffered formalin for 24 h. After paraffin embedding and sectioning (5 μm), tissues were stained with hematoxylin and eosin (H&E). In tissue immunohistochemical staining, the antigen was retrieved by high pressure and incubation in 0.01 M sodium citrate buffer. Then the samples were blocked in 10% normal goat serum in PBS, and incubated with primary antibodies at 4°C overnight in primary antibody solution. After being washed with PBS buffer, the samples were incubated with polymer HRP goat anti-rabbit (DAKO, Agilent) for 30 min at room temperature, developed with DAB (3,3′-diaminobenzidine tetrahydrochloride), and counterstained with hematoxylin (Zhongshan Golden Bridge Biotechnology Company). All specimens were evaluated by two pathologists without knowledge of the experimental group.
Cell Viability Assay
MCF-7 cells with control or JFK-depleted were cultured in normoxia or hypoxia for 48 h, and were subsequently treated with X-ray irradiation, tamoxifen, or fulvestrant before growing for 5 days. The cell viability rate of MCF-7 cells was evaluated using Cell Counting Kit (CCK, TransGen Biotech) according to the manufacturer’s instruction.
Statistical Analysis
Results are reported as mean ± SD for triplicate experiments or mean ± SEM unless otherwise noted. SPSS V.19.0 was used for statistical analysis. Comparisons between two groups were performed using two-tailed paired Student’s t test. Comparisons among three groups were performed using one-way ANOVA followed by Tukey’s honestly significant difference (HSD) post hoc test.
Results
JFK Promotes Mammary Tumor Initiation and Metastasis in Mice
It has been reported that JFK is highly expressed in tumor tissues including breast, kidney, and pancreatic cancer, with the highest JFK expression level reported in breast cancer (Yan et al., 2015). To further investigate the potential function of JFK in breast cancer development, we first generated JFK transgenic (JFKTG) mice by nuclear transplantation. Briefly, the open reading frame (ORF) of human JFK (with a FLAG tag) was cloned into the mammalian expression vector pRP(Exp)-EF1A. After digestion, linearized DNA was micro-injected into the pronuclei of fertilized zygotes derived from C57BL/6 female mice, and the embryos were transplanted onto pseudo-pregnant mice to breed JFKTG founders.
The expression of mRNA and protein of JFK in JFKTG mice was verified by real-time reverse transcriptase PCR (qPCR) and western blotting, and these both indicated that JFK transgenic mice were obtained successfully (Figure 1A). We then crossed the JFKTG mice with mouse mammary tumor virus-polyoma virus middle T-antigen (MMTV-PyMT) transgenic mice (Murai, 2015) – a widely utilized preclinical mouse model that recapitulates human breast cancer progression from early hyperplasia to malignant breast carcinoma – to obtain female JFKWT/PyMTTG and JFKTG/PyMTTG mice. We observed that JFKTG/PyMTTG mice developed palpable tumors at the average age of 12-week-old, 2 weeks earlier than JFKWT/PyMTTG mice. Moreover, the number of subcutaneous tumors was significantly greater, and the volume of subcutaneous tumors was significantly larger in JFKTG/PyMTTG mice compared with JFKWT/PyMTTG mice (Figure 1B).
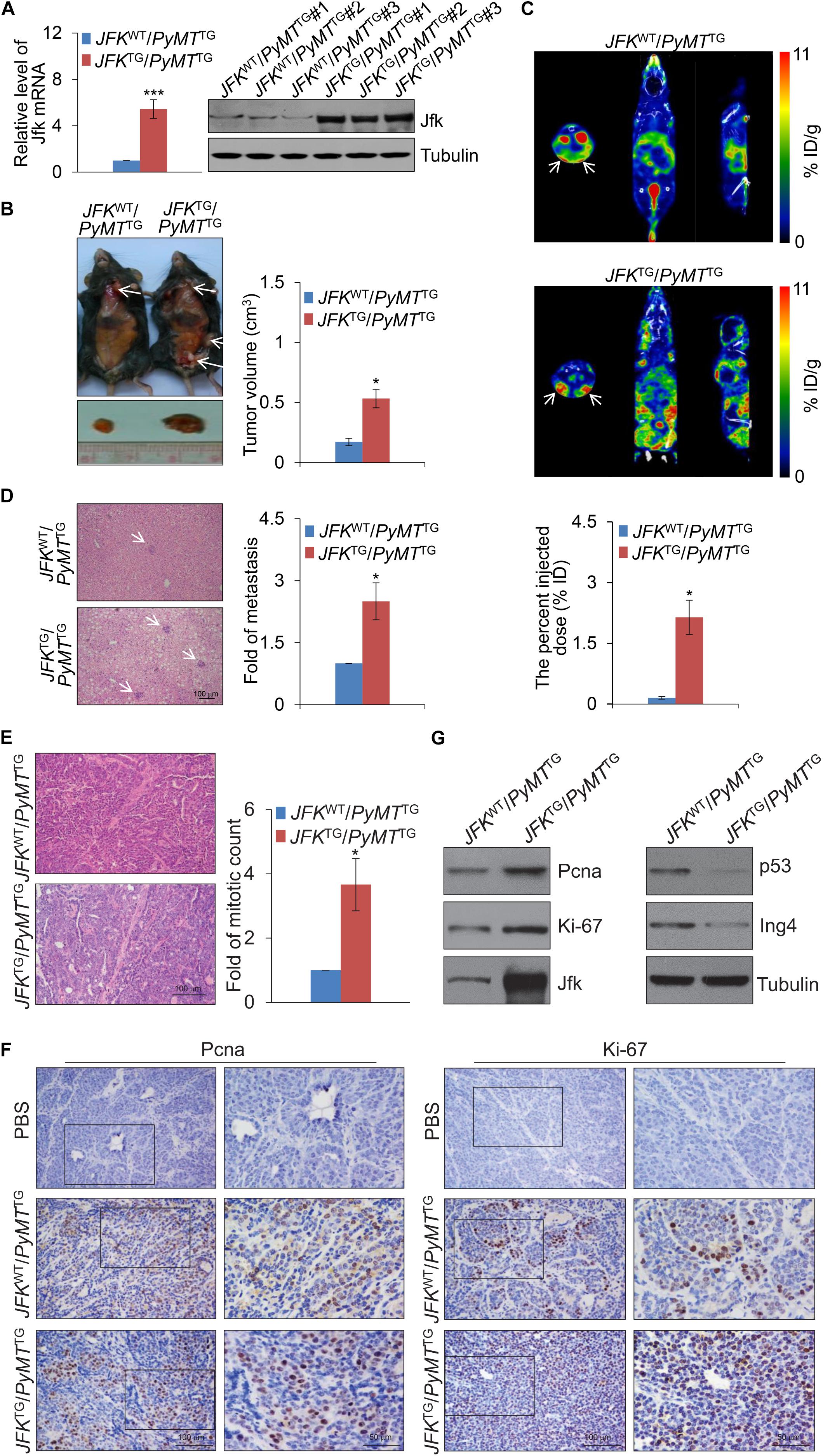
Figure 1. JFK promotes mammary tumor initiation and metastasis in mice. (A) Total RNAs or proteins from the tail tip of JFKWT/PyMTTG or JFKTG/PyMTTG mice were extracted, and respectively, analyzed for Jfk expression by qPCR or western blotting with the indicated antibodies. Error bars represent the mean ± SD for triplicate experiments (***p < 0.001). (B) Representative images of tumors resected from 14-week-old JFKWT/PyMTTG or JFKTG/PyMTTG mice on autopsy. Arrows indicate the subcutaneous breast tumors, and tumor size was measured using digital calipers. Error bars represent the mean ± SEM (n = 6, *p < 0.05). (C) Representative SPECT/CT images of JFKWT/PyMTTG or JFKTG/PyMTTG mice at 30 min after injection of 99mTc-3PRGD2 (37 MBq) via the tail vein. Error bars represent the mean ± SEM (n = 6, *p < 0.05). (D) Representative images of liver sections from JFKWT/PyMTTG or JFKTG/PyMTTG mice stained with hematoxylin and eosin (H&E) are shown. Bar, 100 μm. Error bars represent the mean ± SEM (n = 6, *p < 0.05). (E) Representative images of tumor sections from 14-week-old JFKWT/PyMTTG and JFKTG/PyMTTG mice stained with H&E. Bar, 100 μm. Error bars represent the mean ± SEM (n = 6, *p < 0.05). (F) Immunohistochemical staining of mammary tumors with from 14-week-old JFKWT/PyMTTG and JFKTG/PyMTTG mice for the expression of Ki-67 and Pcna. (G) Total protein extracts were prepared from mammary tumor tissues from 14-week-old JFKWT/PyMTTG and JFKTG/PyMTTG mice and analyzed via western blotting with the indicated antibodies.
Previous studies reported that integrin αvβ3 is expressed at high levels on the surface of a variety of solid tumor cells and on the tumor vasculature (Liu et al., 2011). To further explore the effect of JFK on breast carcinogenesis, we used an integrin αvβ3-targeting radiotracer, 99mTc-3PRGD2 (Zhu et al., 2012), to conduct single photon emission computed tomography (SPECT)/computed tomography (CT) in vivo imaging. To this end, 99mTc-3PRGD2 was injected into the 14-week-old JFKWT/PyMTTG and JFKTG/PyMTTG mice via the tail vein. SPECT/CT of JFKWT/PyMTTG and JFKTG/PyMTTG mice at 30 min after injection showed that there was remarkable accumulation of 99mTc-3PRGD2 in mammary tumors at 30 min post-injection, and JFKTG/PyMTTG mice developed more multifocal palpable tumors compared with JFKWT/PyMTTG mice (Figure 1C and Supplementary Movies 1, 2). The quantification analysis of total tumor volumes revealed that JFKTG/PyMTTG mice exhibited increased tumorigenesis compared with JFKWT/PyMTTG mice (Figure 1C). Hematoxylin-eosin (H&E) staining analysis of liver tissues also revealed that the JFKTG/PyMTTG mice had significantly elevated numbers of liver metastases compared to JFKWT/PyMTTG mice (Figure 1D). Collectively, these in vivo results support that JFK overexpression somehow promotes mammary tumor initiation and metastasis.
To further explore the role of JFK in promoting breast cancer, we collected breast tumors from 14-week-old JFKWT/PyMTTG and JFKTG/PyMTTG mice and performed histological analysis. H&E staining of tumor sections showed that abnormal mitoses in mammary tumor tissue of JFKTG/PyMTTG mice are about 3-fold greater in number than in JFKWT/PyMTTG mice (Figure 1E), clearly suggesting that JFK overexpression somehow promotes cell division of breast cancer cells in vivo. Pursuing this, we performed immunohistochemical analysis of breast tumor tissues of JFKWT/PyMTTG and JFKTG/PyMTTG mice for expression of Ki-67 and Pcna, two well-documented markers for cellular proliferation (Jurikova et al., 2016). JFKTG/PyMTTG mice exhibited substantially more Ki-67-positive and Pcna-positive nuclei than JFKWT/PyMTTG mice (Figure 1F), indicating that JFK promotes cell proliferation in vivo. Western blotting analysis showed that overexpression of JFK was accompanied by increased accumulation of the Ki-67 and Pcna proteins but reduced levels of the p53 and Ing4 proteins, findings consistent with previous studies showing that JFK targets p53 and ING4 proteins for ubiquitination and degradation (Sun et al., 2009; Yan et al., 2015; Figure 1G) and supporting the aforementioned hypothesis that JFK impacts tumor progression through its function in SCFJFK in destabilizing tumor suppressor genes. Together, these in vivo results show that JFK functions in mammary tumorigenesis in the MMTV-PyMT mouse model by promoting cell proliferation.
Genome-Wide Identification of Transcriptional Targets of HIF-1α
Given that JFK is expressed at aberrantly high levels in breast cancer at both the mRNA and protein levels, we next explored mechanism(s) underlying JFK dysregulation in diverse samples by adopting a phenolyzer approach (Yang H. et al., 2015). Specifically, to predict any transcription factors likely to bind at the JFK promoter in breast cancer, we analyzed data from PROMO1 using tools for the phenotype-based gene analyzer2. We initially focused on transcription factors with reported links to breast cancer, and found that 16 transcription factors including HIF-1α, MYC, FOXP3, and E2F1 were predicted to target the JFK locus (Figure 2A). Soluble chromatin from MCF-7 cells was prepared for ChIP assays, and the results showed that HIF-1α, FOXP3, and E2F1 were enriched at JFK promoter (Figure 2B). Briefly, HIF-1α overexpression, but not FOXP3 nor E2F1, has been reported to be closely related to high histological grade, lymph node metastasis, large tumor size, and increased angiogenesis in breast cancer (Qin et al., 2006; Yamamoto et al., 2008; Li et al., 2018), which is consistent with the function of JFK. In light of the common view that tumor microenvironment is often hypoxic and HIF-1α is a hypoxia-inducible factor (Barker et al., 2015), we focused on the effect of HIF-1α on transcriptional regulation of JFK.
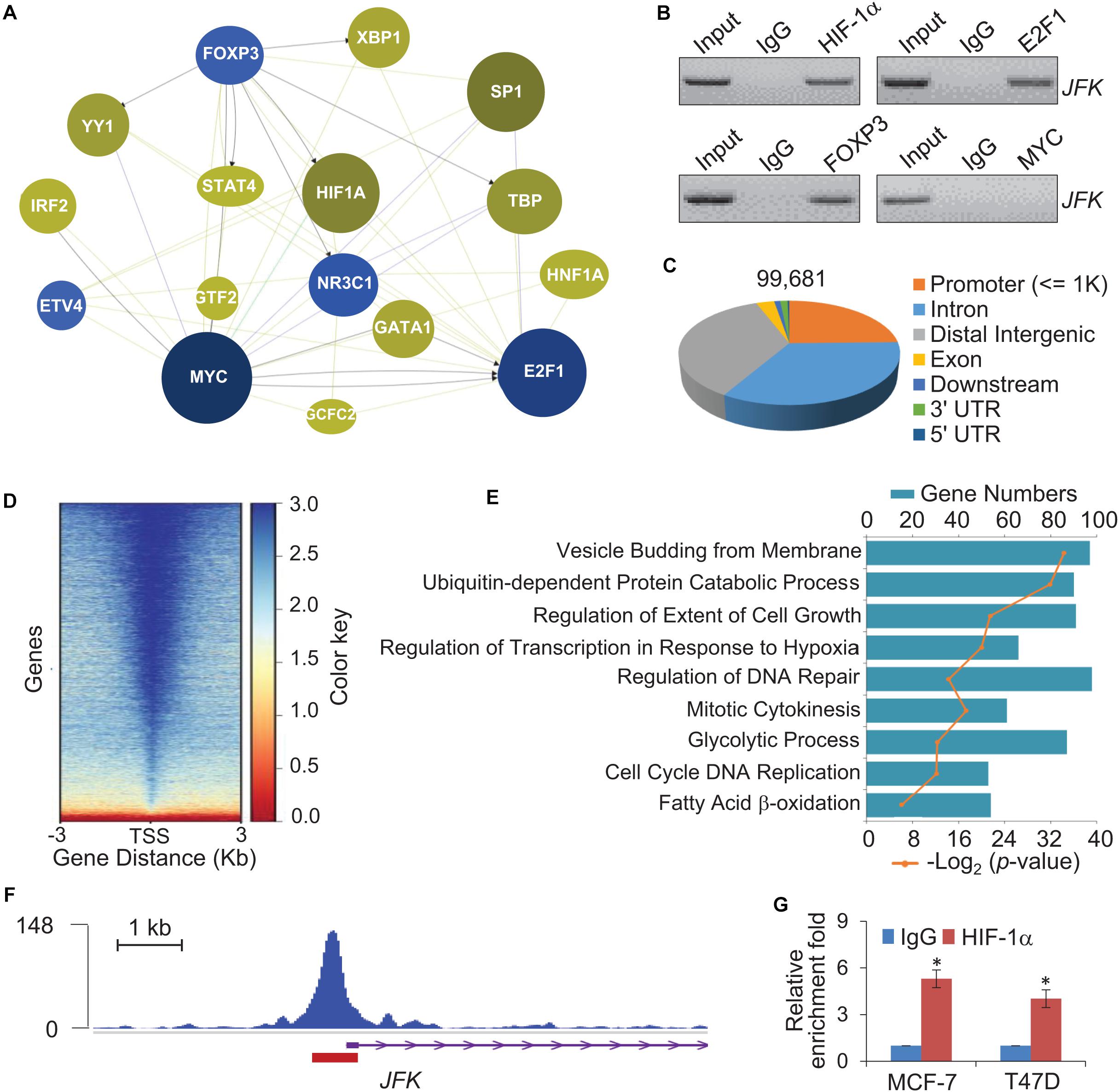
Figure 2. Genome-wide identification of transcriptional targets for HIF-1α. (A) Breast cancer associated transcription factors which bind at the promoter of JFK were assessed using a Phenolyzer approach. The size and shade of each circle represent the strength of correlation between a given transcription factor that was predicted to bind at the promoter of JFK and breast cancer. (B) Soluble chromatin from MCF-7 cells was prepared for ChIP assays with antibodies against the indicated proteins. (C) Genomic distribution of HIF-1α determined by ChIP-seq analysis. ChIP was performed in hypoxia-treated T47D cells with antibodies against HIF-1α, followed by deep sequencing. The raw data were analyzed by MACS2 with a cutoff q value < 1e-2. (D) Binding density of HIF-1α across its specific binding sites, visualized by deepTools. The heatmap shows the normalized ChIP-seq tag counts ordered by signal strength. (E) GO enrichment (biological processes) analysis of potential target genes. (F) Tracks visualizing the ChIP signal for HIF-1α binding at representative locus. The red rectangle indicates the peak region/binding site of HIF-1α at the JFK promoter. (G) qChIP-based verification of HIF-1α binding at the JFK locus under hypoxia. Soluble chromatin from MCF-7 or T47D cells exposed with hypoxia (1% O2) for 24 h was prepared, and qChIP analysis was performed for the JFK promoter using the antibody against HIF-1α. Error bars represent the mean ± SD for triplicate experiments. Statistical significance of differences is indicated as *p < 0.05.
We further analyzed published chromatin immunoprecipitation-coupled massive parallel DNA sequencing (ChIP-seq) data for the genome-wide transcriptional profile of HIF-1α and to identify candidate direct transcriptional activation targets of HIF-1α in breast cancer (GSE59935) (Zhang et al., 2015). In these experiments, ChIP was performed in hypoxia-treated T47D cells using antibodies against HIF-1α (Zhang et al., 2015). After processing with the alignment to the unmasked human reference genome (GRCh37, hg19) using Bowtie2, we performed MACS2 (Model-based Analysis for ChIP-Seq) analysis using a cutoff of q < 1e-2. After filtering, a total of 99,681 HIF-1α-specific binding peaks were called, with the majority of the peaks located at promoter sequences (23.92%) and the remainder at intronic (32.71%) or intergenic (34.75%) regions (Figure 2C).
The genomic landscape of HIF-1α binding peaks at promoter sequences (≤3 K) were analyzed by deepTools (Figure 2D). Using R and Bioconductor, further GO enrichment analysis of the promoter regions (≤500 bp) with enriched HIF-1α occupancy revealed enrichment for functional annotations including mitotic cytokinesis, glycolytic process, cell cycle, and fatty acid β-oxidation, which were related to cell proliferation and metabolism (Figure 2E). Consistent with above prediction, tracks visualizing the ChIP signal analysis indicated that HIF-1α binds at the JFK promoter (Figure 2F). Quantitative ChIP (qChIP) analysis with antibodies against HIF-1α and JFK-specific primers of MCF-7 cells or T47D cells cultured in hypoxia (1% O2) conditions validated that the HIF-1α protein is highly enriched at JFK promoter (Figure 2G). Together, these observations suggest that JFK is a potential target gene of HIF-1α.
JFK Is Transcriptionally Activated by HIF-1α
To further investigate the expression regulation of JFK, we next cloned a 600 bp fragment from the JFK promoter (Chr 1: 16,679,286–16,679,886) and constructed a JFK promoter-driven luciferase reporter (JFK-Luc). In these experiments, MCF-7 or T47D cells were co-transfected with HIF-1α expression plasmids together with the wild-type JFK-Luc promoter or a mutant JFK-Luc promoter lacking the predicted HIF-1α binding motif. The results showed that HIF-1α was able to activate transcription from the JFK promoter, but only when the CACGTG motif was present (Figure 3A), demonstrating that the HIF-1α protein can directly bind to the JFK promoter.
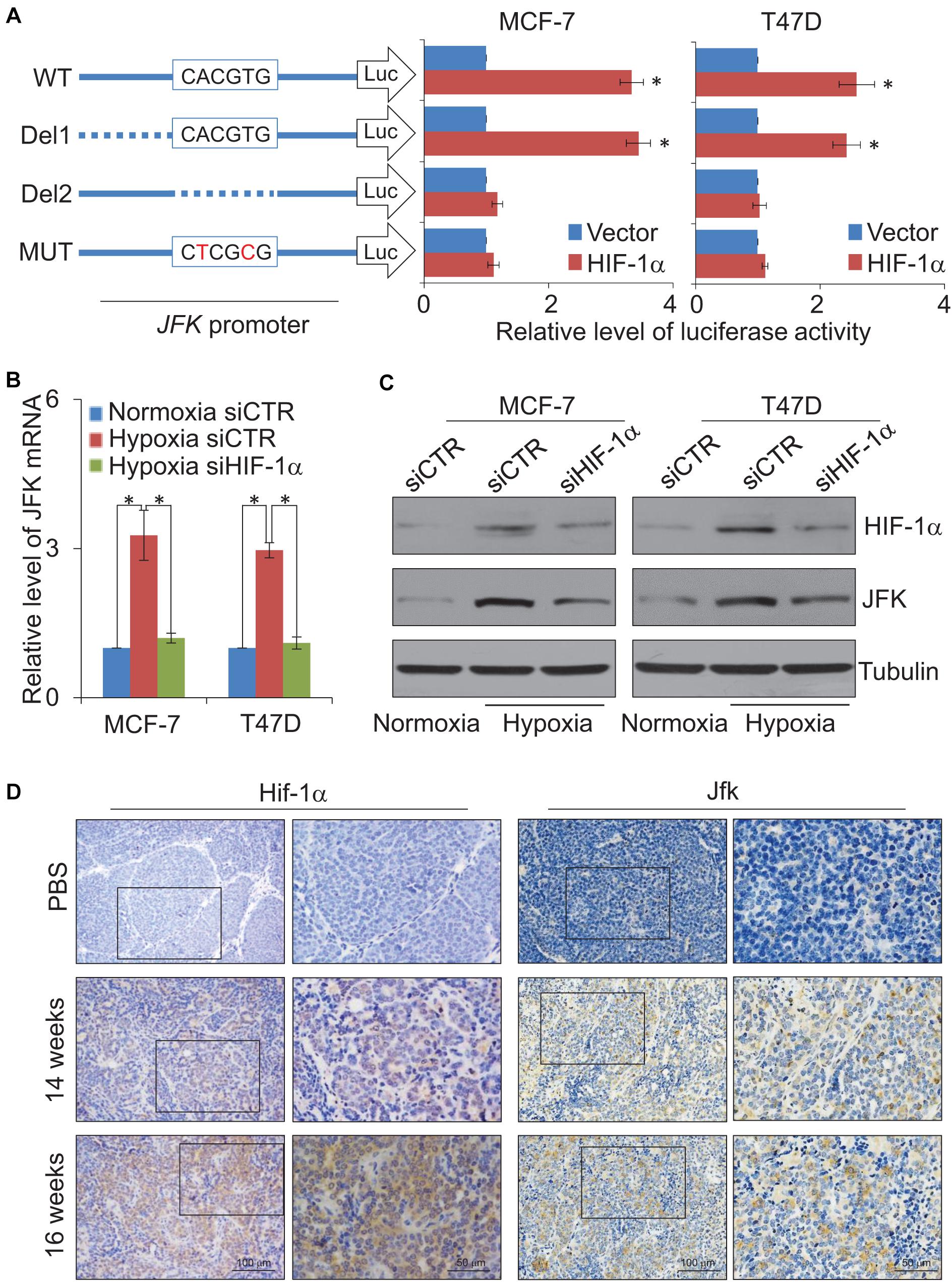
Figure 3. JFK is transcriptionally activated by HIF-1α. (A) MCF-7 or T47D cells infected with lentiviruses carrying JFK-Luc wild-type, deletion, or mutant variants, as well as control vector or HIF-1α expression constructs, were harvested, lysed, and assayed for firefly and Renilla luciferase activity using the dual-luciferase reporter assay system. Error bars represent the mean ± SD for triplicate experiments. (B) qPCR measurement of the JFK expression in MCF-7 or T47D cells treated with control or HIF-1α siRNAs, under normoxia or hypoxia (1% O2) for 24 h. Error bars represent the mean ± SD for triplicate experiments. (C) Western blotting analysis of cellular extracts from MCF-7 or T47D cells treated with control or HIF-1α siRNAs, under normoxia or hypoxia (1% O2) for 24 h. (D) Immunohistochemical staining of breast tumors from 14-week-old or 16-week-old JFKWT/PyMTTG mice to assess the expression of Hif-1α and Jfk. Statistical significance of differences is indicated as *p < 0.05.
Both qPCR and western blotting were then performed in MCF-7 or T47D cells treated with control or HIF-1α siRNAs, under normoxia or hypoxia treatment for 24 h. The results showed that hypoxic conditions led to significantly increased expression of JFK (Figures 3B,C). No such increase in JFK mRNA or protein accumulation was observed upon HIF-1α knockdown in hypoxic condition (Figures 3B,C). These results demonstrate a direct functional impact of HIF-1α in the observed hypoxia-induced transcriptional activation of JFK in breast cancer cells.
Extending our investigation to the MMTV-PyMT mouse model, we harvested tumors from 14-week-old or 16-week-old JFKWT/PyMTTG mice. Immunohistochemical staining showed that, compared to 14-week-old JFKWT/PyMTTG mice, the Hif-1α and Jfk protein levels were clearly up-regulated in the mammary tumors of the 16-week-old JFKWT/PyMTTG mice (Figure 3D). Collectively, these results support that the hypoxia-mediated induction of JFK is mediated by HIF-1α binding and transcriptional activation and, further, defines an HIF-1α-JFK axis which apparently functions in promoting breast carcinogenesis.
JFK Promotes HIF-1α-Induced Glycolysis
It is known that HIF-1α is a metabolic regulator that plays an important role in glycolysis metabolism (Wu et al., 2020), we further investigated whether JFK functions in hypoxia-induced glycolysis of breast cancer cells. To this end, MCF-7 cells treated with control or JFK siRNAs were cultured in normoxia or hypoxia conditions, or co-transfected with vector or HIF-1α, and we performed glucose colorimetric/fluorometric or lactate colorimetric assay to detect glucose uptake or lactate production. The results showed that JFK knockdown significantly dampened hypoxia or HIF-1α-induced glycolysis in breast cancer cells (Figures 4A,B).
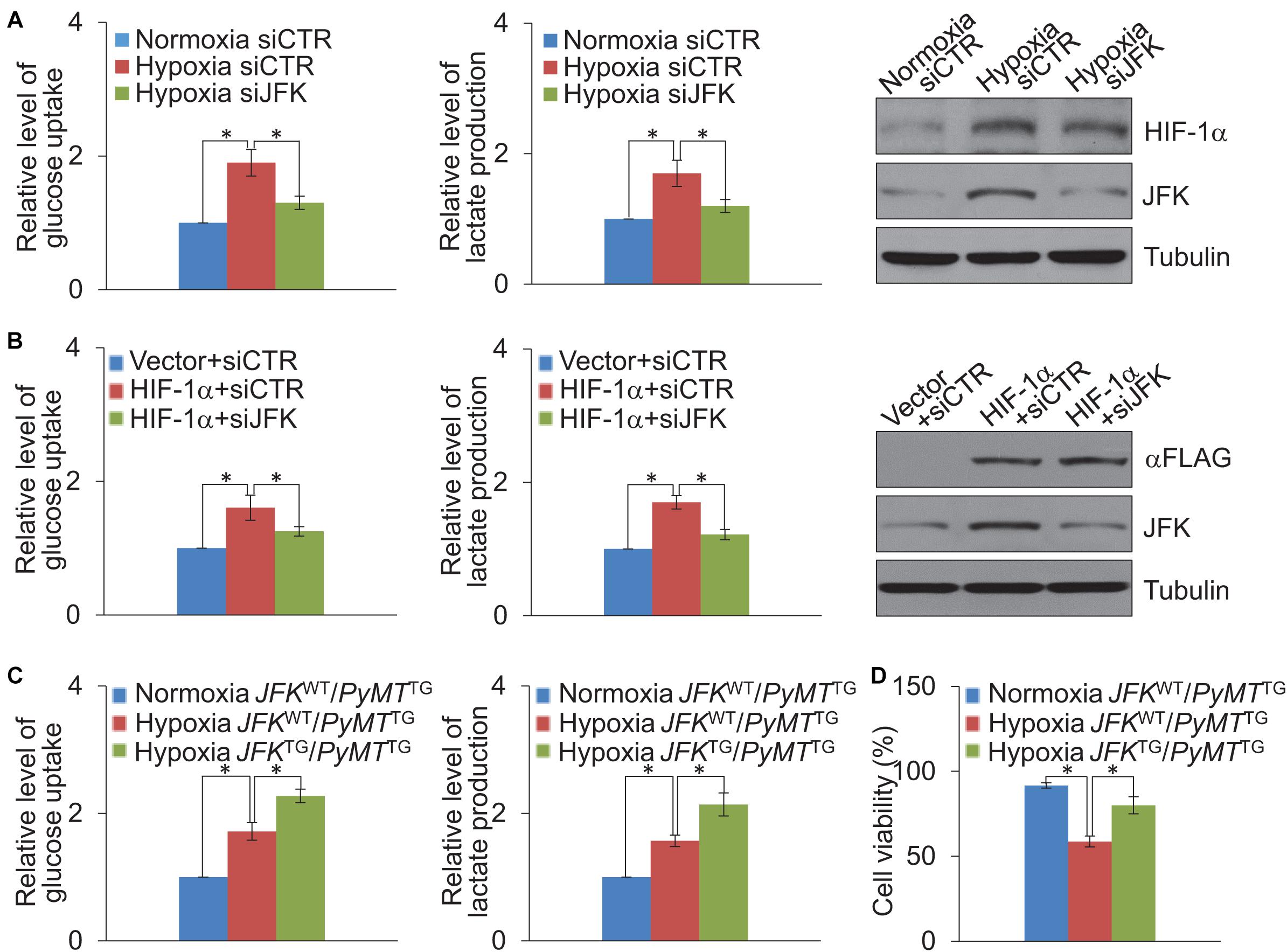
Figure 4. JFK promotes HIF-1α-induced glycolysis. (A) Cellular extracts or cell culture medium of MCF-7 cells treated with control or JFK siRNAs and cultured in hypoxia (1% O2) conditions for 24 h were prepared for measurement of intracellular glucose or extracellular lactate. Error bars represent the mean ± SD for triplicate experiments. Cellular extracts were prepared for western blotting analysis with the indicated antibodies. (B) Cellular extracts or cell culture medium of MCF-7 cells treated with control or JFK siRNAs and transfected with vector or HIF-1α were prepared for measurement of intracellular glucose or extracellular lactate. Error bars represent the mean ± SD for triplicate experiments. Cellular extracts were prepared for western blotting analysis with the indicated antibodies. (C) Cellular extracts or cell culture medium of primary tumor cells from JFKWT/PyMTTG or JFKTG/PyMTTG mice cultured in normoxia or hypoxia (1% O2) conditions for 24 h were prepared for measurement of intracellular glucose or extracellular lactate. Error bars represent the mean ± SD for triplicate experiments. (D) Primary tumor cells from JFKWT/PyMTTG or JFKTG/PyMTTG mice cultured in normoxia or hypoxia (0.2% O2) conditions for 72 h, followed by cell viability assessment. Error bars represent the mean ± SD for three independent experiments. Statistical significance of differences is indicated as *p < 0.05.
To further extend our investigation to the MMTV-PyMT mouse model, we digested tumor tissues from JFKWT/PyMTTG or JFKTG/PyMTTG mice, and the isolated cells were put into primary culture. Isolated cells cultured in normoxia or hypoxia conditions were performed glucose colorimetric/fluorometric or lactate colorimetric assay to detect glucose uptake or lactate production. The results revealed that JFK overexpression significantly strengthened hypoxia-induced glycolysis in breast cancer (Figure 4C). Furthermore, primary tumor cells were cultured in normoxia or hypoxia conditions for 72 h, and cell viability analysis showed that the JFK overexpression significantly weakened the decreased cell viability under hypoxia (Figure 4D). Together, these results suggest that JFK promotes HIF-1α-induced glycolysis thus elevating cellular tolerance to hypoxia in breast cancer.
JFK Deficiency Sensitizes Hypoxic Breast Cancer Cells to Chemo-Radiotherapeutic Treatment
Considering that the high expression of HIF-1α has been related to resistance to radiotherapy and chemotherapy in breast cancer (Yang J. et al., 2015), we next investigated the role of JFK in breast cancer cell sensitivity to ionizing radiation (IR) and chemotherapeutic agents under hypoxia. To this end, MCF-7 cells treated with control or JFK siRNAs under normoxia or hypoxia were exposed to X-ray irradiation or treated with chemotherapeutic agents (tamoxifen or fulvestrant). Cell viability analysis showed that JFK deficiency significantly weakened the increased cell viability in response to hypoxia-induced resistance to IR or chemotherapeutic agents of MCF-7 cells (Figures 5A,B). These observations suggest that JFK protected cells from toxic insults and promoted cell survival under hypoxia.
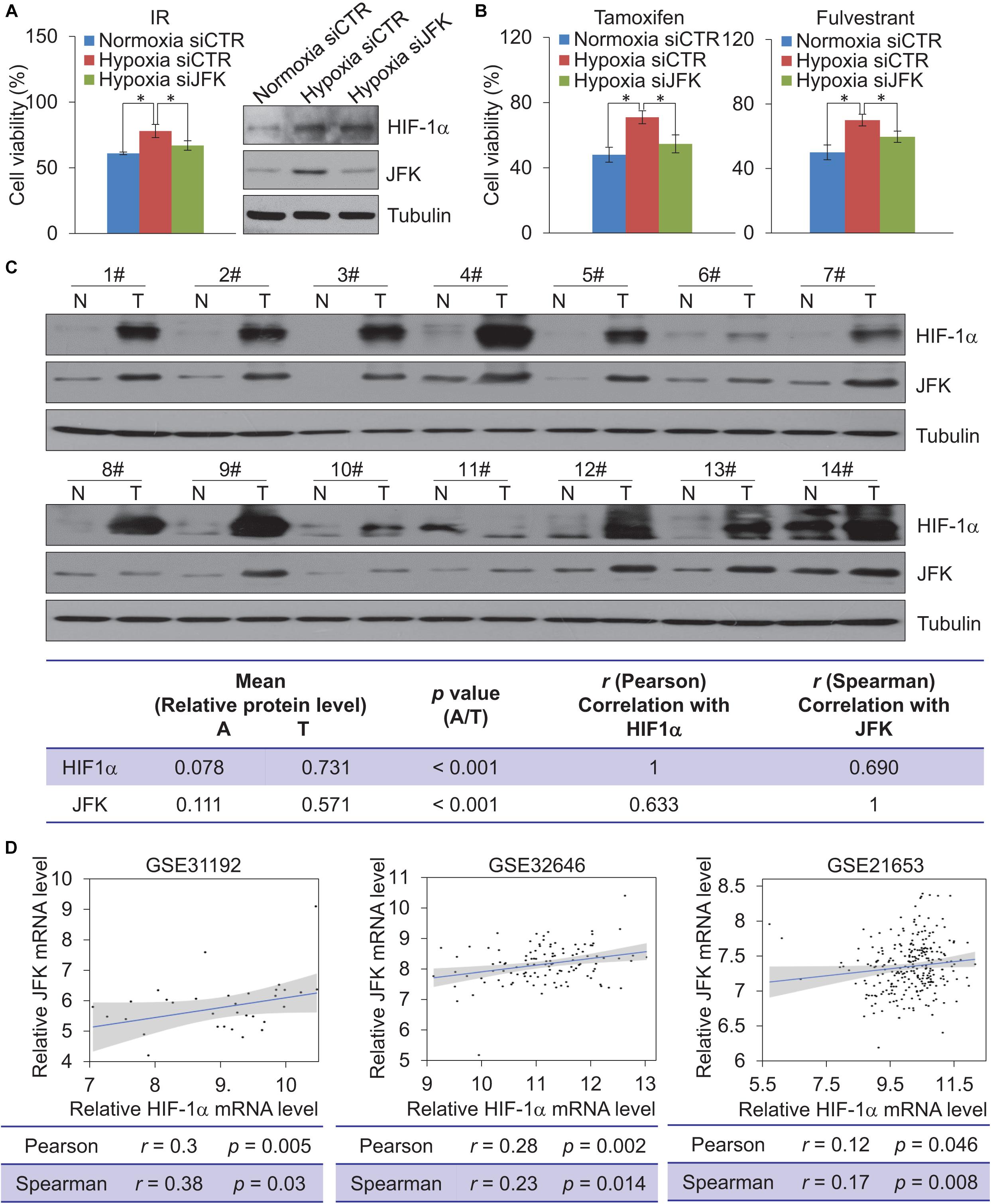
Figure 5. JFK deficiency sensitizes hypoxic breast cancer cells to chemo-radiotherapeutic treatment. (A) MCF-7 cells treated with control or JFK siRNAs were cultured in normoxia or hypoxia (1% O2) conditions, and treated with IR (5 Gy) and subjected to cell viability assessment. Error bars represent the mean ± SD for triplicate experiments. Cellular extracts were prepared for western blotting analysis with the indicated antibodies. (B) MCF-7 cells treated with control or JFK siRNAs were cultured in normoxia or hypoxia (1% O2) conditions, and treated with tamoxifen (0.5 μM) or fulvestrant (0.5 μM) and subjected to cell viability assessment. Error bars represent the mean ± SD for triplicate experiments. (C) Western blotting analysis in 14 breast tumors (T) paired with adjacent normal tissues (N) from breast carcinoma patients for the expression of HIF-1α and JFK. Quantitation was performed based on densitometry using Image J, and the values are expressed after normalization to the level of tubulin. The results from correlation analysis between HIF-1α and JFK expression are shown. (D) The level of JFK mRNA expression was plotted against the level of HIF-1α mRNA expression in published clinical datasets (GSE31992, GSE32646, and GSE21653); the line presents a fitted linear model, and the shading represents the 95% confidence interval. Statistical significance of differences is indicated as *p < 0.05.
To gain further support for the impacts of this HIF-1α-JFK axis on breast cancer, and seeking to add clinicopathologically relevant data to our observations, we collected samples from resected breast carcinomas – paired with adjacent normal mammary tissues – from 14 breast cancer patients. Western blotting analysis showed that both HIF-1α and JFK protein levels were significantly upregulated in the cancer tissue compared to the normal tissue (Figure 5C). Additionally, and consistent with HIF-1α as a transcription activator of JFK, quantitation and statistical analysis revealed that the relative level of JFK protein was significantly and positively correlated with that of HIF-1α (Spearman’s rank correlation coefficient, 0.690; p < 0.001) (Figure 5C).
We also analyzed public data to evaluate the HIF-1α and JFK mRNA expression levels in breast cancers. Analysis of published clinical datasets (GSE31192, GSE32646, and GSE21653) revealed a positive correlation between HIF-1α and JFK mRNA expression detected for all of the examined datasets (Figure 5D). At minimum, these analyses of previously published breast cancer datasets and our own analysis of tissues from 14 breast cancer patients support our identification of the HIF-1α-JFK axis. Collectively, these findings support a notion that HIF-1α-JFK axis is an important regulator that acts to elevate cellular tolerance to hypoxia and contribute to cell survival in promoting breast carcinogenesis.
Discussion
Elevated accumulation of the HIF-1α protein has been observed in a broad array of human cancer cell types including breast cancer cells, in which elevated levels of HIF-1α are associated with poor prognosis (Jun et al., 2017). Specifically, knockout of HIF-1α in the MMTV-PyMT mouse model was reported to suppress both primary tumor growth and lung metastasis (Schwab et al., 2012), which is consistent with our observation that JFK overexpression promotes mammary tumor initiation and metastasis in the MMTV-PyMT mouse model. Together these findings support our hypothesis that the HIF-1α-JFK axis contributes to breast cancer cell survival. Our observation of selective (i.e., hypoxia-specific) transcriptional activation of JFK by HIF-1α is notable because JFK dysregulation/overexpression has been reported to be a frequent event in breast cancer and has been associated with especially aggressive clinical features and worse survival in breast cancer patients (Yan et al., 2015).
The HREs bound by HIF-1α and HIF-2α have both been shown to contain a conserved RCGTG core sequence, and there are reports that they share transcription target genes including IL-6, FILAG, and GLUT1, which are involved in the adaptation of tumor cells to hypoxia (Keith et al., 2011); thus, it is possible that HIF-2α may also bind to the HRE in the JFK promoter. A previous study reported that the HIF-2α protein is stabilized under 5% O2 (resembling end capillary oxygen conditions) and – in contrast to the low transcription activation activity of HIF-1α at this oxygen level – HIF-2α strongly promotes the transcription of genes such as VEGF (Holmquist-Mengelbier et al., 2006). It is known that untreated breast cancer tumors can be hypoxic (∼1.3% oxygen); this level is >5 times lower than oxygen levels found in the normal breast tissues (Wang et al., 2015; Rankin and Giaccia, 2016). Given that O2 levels vary widely across sub-domains of solid tumors [owing, for example, to rapid cell division and aberrant tumor angiogenesis and blood flow (Bertout et al., 2008)], it will be interesting to explore whether JFK is a co-target of both HIF-1α and HIF-2α, perhaps under different severities of hypoxia, in different tumor sub-domains, or at different stages of breast cancer development. Clarifying these points should deepen our understanding of the roles of hypoxia generally, and JFK and HIF-1α/HIF-2α specifically, in breast cancer.
Among breast cancer tumors, basal-like tumors are the most aggressive and are associated with the highest rates of metastasis and recurrence (Alluri and Newman, 2014). Clinical dataset analyses in our previous study revealed that higher JFK expression levels were associated with worse overall survival in basal-like breast cancer patients (Yan et al., 2015), so it will be interesting to further investigate whether JFK levels in, for example, diagnostic tumor biopsies may be useful for identifying basal-like breast cancers in patients, which are at increased risk of developing metastasis. Consistently, HIF-1α has been identified as one of five signature markers predictive of disease outcomes among node-negative breast cancer patients (Charpin et al., 2012). In addition, it is known that basal-like tumors frequently show morphologic evidence of hypoxia (central fibrosis and necrosis) (Livasy et al., 2006); conventional chemotherapy is not always effective on these tumors and a systemic relapse is often observed, potentially due to the development of drug resistance (Boichuk et al., 2017). Our study revealed that JFK deficiency sensitizes breast cancer cells to chemo-radiotherapeutic treatment under hypoxia, indicating that hypoxia-induced JFK upregulation in basal-like tumors might contribute to drug resistance.
Obesity has been proposed to be a highly significant risk factor for breast carcinogenesis and aggressiveness (Perez-Hernandez et al., 2014), as excessive adipose tissue in obese patients not only alters the local microenvironment by remodeling the extracellular matrix but also provides both energy and niches for cancer cell proliferation and invasion (Seo et al., 2015; Gao et al., 2020). In this regard, drugs (e.g., metformin) targeting signaling pathways that are deregulated in obesity might help to prevent carcinogenesis in obese women (Oppong et al., 2014; Gao et al., 2020). Intriguingly, our recent study demonstrated that JFK suppresses hepatic lipid catabolism and exacerbates the development of obesity and non-alcoholic fatty liver disease (NAFLD) by destabilizing ING5. Moreover, JFK is up-regulated in liver tissues from NAFLD patients (n = 206) as compared with healthy controls (n = 10) according to a recently published public dataset (GSE135251) (Govaere et al., 2020). In the present study, we show that JFK promotes mammary progression in mice, and that the protein level of JFK is up-regulated in resected breast tumors compared with the adjacent normal tissues from breast cancer patients. Although the molecular mechanisms underlying the strong association between obesity and poor breast cancer outcomes are not well understood, it seems plausible that JFK acts as a regulatory hub that coordinates obesity and breast carcinogenesis. Considering that HIF-1α controls oxygen delivery into tumors via angiogenesis and metabolic adaptation of tumors to hypoxia via glycolysis (Semenza, 2002), the HIF-1α-JFK axis we identified in the present study may regulate processes (including glycolysis and fatty-acid β-oxidation) with broad impacts on tumor initiation and metastasis. Moreover, further exploratory screens for interaction partners or degradation substrates of JFK under several conditions including hypoxia, dysregulated energy metabolism, and lipid abnormalities to characterize how JFK functions in apparently diverse cellular processes would likely be scientifically informative.
Our previous studies also revealed that JFK depletion suppresses proliferation and angiogenesis of breast cancer cells through accumulation of tumor suppressors p53 and ING4 (Sun et al., 2009, 2011; Yan et al., 2015). In this study, we showed that JFK depletion reduces cellular tolerance to hypoxia by suppressing glycolysis, and thus contributes to cell survival in breast cancer, which deepens the mechanistic understanding of cancer cell responses to normoxic versus hypoxic conditions. The fact that cancer cells can become addicted to specific metabolic pathways has led to the recent development of novel drugs that target these metabolic vulnerabilities (Tennant et al., 2010). Beyond defining the hypoxia-sensitive HIF-1α-SCFJFK axis, which promotes breast carcinogenesis, we extended these molecular and cell-context specific insights into clinical relevance when targeting HIF-1α is not effective, supporting the hypothesis that JFK is a vulnerable target for the development of innovative therapeutic interventions against breast cancer.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Ethics Statement
The animal study was reviewed and approved by the Institutional Animal Care and Use Committee at Peking University.
Author Contributions
ZY, LH, and LS designed the research. ZY, XZ, and LH conducted the experiments. ZY, XZ, LH, YpW, EZ, and YZ performed the animal experiments and analyzed the data. YzW, XL, YW, ZL, FP, JR, YH, LX, SG, SQ, FS, JS, and LW provided technical assistance. ZY, XZ, LH, and LS wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by a grant (2016YFC1302304 to LS) from the Ministry of Science and Technology of China, and grants (81874158 to LS and 82002992 to LH) from the National Natural Science Foundation of China, a grant (QNBJ2020-2 to LS) from National Program for Support of Top-notch Young Professionals, a grant (Z200020 to LS) from Beijing Natural Science Foundation of China, and a grant (2019M650385 to LH) from China Postdoctoral Science Foundation.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the National Center for Protein Sciences at Peking University (Beijing, China) for providing technical support.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2021.686737/full#supplementary-material
Supplementary Movie 1 | Three-dimensional SPECT/CT image of JFKWT/PyMTTG at 30 min after intravenous injection of 99mTc-3PRGD2.
Supplementary Movie 2 | Three-dimensional SPECT/CT image of JFKTG/PyMTTG at 30 min after intravenous injection of 99mTc-3PRGD2.
Footnotes
- ^ http://alggen.lsi.upc.es/cgi-bin/promo_v3/promo/promoinit.cgi?dirDB=TF_8.3
- ^ http://phenolyzer.wglab.org/
References
Akram, M., Iqbal, M., Daniyal, M., and Khan, A. U. (2017). Awareness and current knowledge of breast cancer. Biol. Res. 50:33. doi: 10.1186/s40659-017-0140-9
Alluri, P., and Newman, L. A. (2014). Basal-like and triple-negative breast cancers: searching for positives among many negatives. Surg. Oncol. Clin. N. Am. 23, 567–577. doi: 10.1016/j.soc.2014.03.003
Barker, H. E., Paget, J. T., Khan, A. A., and Harrington, K. J. (2015). The tumour microenvironment after radiotherapy: mechanisms of resistance and recurrence. Nat. Rev. Cancer 15, 409–425. doi: 10.1038/nrc3958
Bertout, J. A., Patel, S. A., and Simon, M. C. (2008). The impact of O2 availability on human cancer. Nat. Rev. Cancer 8, 967–975. doi: 10.1038/nrc2540
Boichuk, S., Galembikova, A., Sitenkov, A., Khusnutdinov, R., Dunaev, P., Valeeva, E., et al. (2017). Establishment and characterization of a triple negative basal-like breast cancer cell line with multi-drug resistance. Oncol. Lett. 14, 5039–5045. doi: 10.3892/ol.2017.6795
Bray, F., Ferlay, J., Soerjomataram, I., Siegel, R. L., Torre, L. A., and Jemal, A. (2018). Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 68, 394–424. doi: 10.3322/caac.21492
Charpin, C., Tavassoli, F., Secq, V., Giusiano, S., Villeret, J., Garcia, S., et al. (2012). Validation of an immunohistochemical signature predictive of 8-year outcome for patients with breast carcinoma. Int. J. Cancer 131, E236–E243. doi: 10.1002/ijc.27371
Chen, F., Chen, J., Yang, L., Liu, J., Zhang, X., Zhang, Y., et al. (2019). Extracellular vesicle-packaged HIF-1alpha-stabilizing lncRNA from tumour-associated macrophages regulates aerobic glycolysis of breast cancer cells. Nat. Cell Biol. 21, 498–510. doi: 10.1038/s41556-019-0299-0
Gao, Y., Chen, X., He, Q., Gimple, R. C., Liao, Y., Wang, L., et al. (2020). Adipocytes promote breast tumorigenesis through TAZ-dependent secretion of Resistin. Proc. Natl. Acad. Sci. U.S.A. 117, 33295–33304. doi: 10.1073/pnas.2005950117
Govaere, O., Cockell, S., Tiniakos, D., Queen, R., Younes, R., Vacca, M., et al. (2020). Transcriptomic profiling across the nonalcoholic fatty liver disease spectrum reveals gene signatures for steatohepatitis and fibrosis. Sci. Transl. Med. 12:eaba4448. doi: 10.1126/scitranslmed.aba4448
Hanahan, D., and Weinberg, R. A. (2011). Hallmarks of cancer: the next generation. Cell 144, 646–674. doi: 10.1016/j.cell.2011.02.013
Holmquist-Mengelbier, L., Fredlund, E., Lofstedt, T., Noguera, R., Navarro, S., Nilsson, H., et al. (2006). Recruitment of HIF-1alpha and HIF-2alpha to common target genes is differentially regulated in neuroblastoma: HIF-2alpha promotes an aggressive phenotype. Cancer Cell 10, 413–423. doi: 10.1016/j.ccr.2006.08.026
Iommarini, L., Porcelli, A. M., Gasparre, G., and Kurelac, I. (2017). Non-canonical mechanisms regulating hypoxia-inducible factor 1 Alpha in Cancer. Front. Oncol. 7:286. doi: 10.3389/fonc.2017.00286
Jia, B., Liu, Z., Zhu, Z., Shi, J., Jin, X., Zhao, H., et al. (2011). Blood clearance kinetics, biodistribution, and radiation dosimetry of a kit-formulated integrin alphavbeta3-selective radiotracer 99mTc-3PRGD 2 in non-human primates. Mol. Imaging Biol. 13, 730–736. doi: 10.1007/s11307-010-0385-y
Jia, L., and Sun, Y. (2011). SCF E3 ubiquitin ligases as anticancer targets. Curr. Cancer Drug Targets 11, 347–356. doi: 10.2174/156800911794519734
Jun, J. C., Rathore, A., Younas, H., Gilkes, D., and Polotsky, V. Y. (2017). Hypoxia-inducible factors and Cancer. Curr. Sleep Med. Rep. 3, 1–10. doi: 10.1007/s40675-017-0062-7
Jurikova, M., Danihel, L., Polak, S., and Varga, I. (2016). Ki67, PCNA, and MCM proteins: markers of proliferation in the diagnosis of breast cancer. Acta Histochem. 118, 544–552. doi: 10.1016/j.acthis.2016.05.002
Keith, B., Johnson, R. S., and Simon, M. C. (2011). HIF1alpha and HIF2alpha: sibling rivalry in hypoxic tumour growth and progression. Nat. Rev. Cancer 12, 9–22. doi: 10.1038/nrc3183
Li, L., Shi, L., Yang, S., Yan, R., Zhang, D., Yang, J., et al. (2016). SIRT7 is a histone desuccinylase that functionally links to chromatin compaction and genome stability. Nat. Commun. 7:12235. doi: 10.1038/ncomms12235
Li, X., Gao, Y., Li, J., Zhang, K., Han, J., Li, W., et al. (2018). FOXP3 inhibits angiogenesis by downregulating VEGF in breast cancer. Cell Death Dis. 9:744. doi: 10.1038/s41419-018-0790-8
Liu, Z., Huang, J., Dong, C., Cui, L., Jin, X., Jia, B., et al. (2012). 99mTc-labeled RGD-BBN peptide for small-animal SPECT/CT of lung carcinoma. Mol. Pharm. 9, 1409–1417. doi: 10.1021/mp200661t
Liu, Z., Liu, H., Ma, T., Sun, X., Shi, J., Jia, B., et al. (2014). Integrin alphavbeta(6)-Targeted SPECT imaging for pancreatic cancer detection. J. Nucl Med. 55, 989–994. doi: 10.2967/jnumed.113.132969
Liu, Z., Wang, F., and Chen, X. (2011). Integrin targeted delivery of radiotherapeutics. Theranostics 1, 201–210. doi: 10.7150/thno/v01p0201
Livasy, C. A., Karaca, G., Nanda, R., Tretiakova, M. S., Olopade, O. I., Moore, D. T., et al. (2006). Phenotypic evaluation of the basal-like subtype of invasive breast carcinoma. Mod. Pathol. 19, 264–271. doi: 10.1038/modpathol.3800528
Masoud, G. N., and Li, W. (2015). HIF-1alpha pathway: role, regulation and intervention for cancer therapy. Acta Pharm. Sin. B 5, 378–389. doi: 10.1016/j.apsb.2015.05.007
Murai, T. (2015). Cholesterol lowering: role in cancer prevention and treatment. Biol. Chem. 396, 1–11. doi: 10.1515/hsz-2014-0194
Oppong, B. A., Pharmer, L. A., Oskar, S., Eaton, A., Stempel, M., Patil, S., et al. (2014). The effect of metformin on breast cancer outcomes in patients with type 2 diabetes. Cancer Med. 3, 1025–1034. doi: 10.1002/cam4.259
Pavlova, N. N., and Thompson, C. B. (2016). The emerging hallmarks of cancer metabolism. Cell Metab. 23, 27–47. doi: 10.1016/j.cmet.2015.12.006
Perez-Hernandez, A. I., Catalan, V., Gomez-Ambrosi, J., Rodriguez, A., and Fruhbeck, G. (2014). Mechanisms linking excess adiposity and carcinogenesis promotion. Front. Endocrinol. 5:65. doi: 10.3389/fendo.2014.00065
Qin, G., Kishore, R., Dolan, C. M., Silver, M., Wecker, A., Luedemann, C. N., et al. (2006). Cell cycle regulator E2F1 modulates angiogenesis via p53-dependent transcriptional control of VEGF. Proc. Natl. Acad. Sci. U.S.A. 103, 11015–11020. doi: 10.1073/pnas.0509533103
Rankin, E. B., and Giaccia, A. J. (2016). Hypoxic control of metastasis. Science 352, 175–180. doi: 10.1126/science.aaf4405
Schwab, L. P., Peacock, D. L., Majumdar, D., Ingels, J. F., Jensen, L. C., Smith, K. D., et al. (2012). Hypoxia-inducible factor 1alpha promotes primary tumor growth and tumor-initiating cell activity in breast cancer. Breast Cancer Res. 14:R6. doi: 10.1186/bcr3087
Semenza, G. L. (2002). HIF-1 and tumor progression: pathophysiology and therapeutics. Trends Mol. Med. 8(4 Suppl.), S62–S67. doi: 10.1016/s1471-4914(02)02317-1
Seo, B. R., Bhardwaj, P., Choi, S., Gonzalez, J., Andresen Eguiluz, R. C., Wang, K., et al. (2015). Obesity-dependent changes in interstitial ECM mechanics promote breast tumorigenesis. Sci. Transl. Med. 7:301ra130. doi: 10.1126/scitranslmed.3010467
Sun, L., Shi, L., Li, W., Yu, W., Liang, J., Zhang, H., et al. (2009). JFK, a Kelch domain-containing F-box protein, links the SCF complex to p53 regulation. Proc. Natl. Acad. Sci. U.S.A. 106, 10195–10200. doi: 10.1073/pnas.0901864106
Sun, L., Shi, L., Wang, F., Huangyang, P., Si, W., Yang, J., et al. (2011). Substrate phosphorylation and feedback regulation in JFK-promoted p53 destabilization. J. Biol. Chem. 286, 4226–4235. doi: 10.1074/jbc.M110.195115
Tennant, D. A., Duran, R. V., and Gottlieb, E. (2010). Targeting metabolic transformation for cancer therapy. Nat. Rev. Cancer 10, 267–277. doi: 10.1038/nrc2817
Wang, Z., Dabrosin, C., Yin, X., Fuster, M. M., Arreola, A., Rathmell, W. K., et al. (2015). Broad targeting of angiogenesis for cancer prevention and therapy. Semin. Cancer Biol. 35(Suppl.), S224–S243. doi: 10.1016/j.semcancer.2015.01.001
Wu, Z., Wu, J., Zhao, Q., Fu, S., and Jin, J. (2020). Emerging roles of aerobic glycolysis in breast cancer. Clin. Transl. Oncol. 22, 631–646. doi: 10.1007/s12094-019-02187-8
Yamamoto, Y., Ibusuki, M., Okumura, Y., Kawasoe, T., Kai, K., Iyama, K., et al. (2008). Hypoxia-inducible factor 1alpha is closely linked to an aggressive phenotype in breast cancer. Breast Cancer Res. Treat. 110, 465–475. doi: 10.1007/s10549-007-9742-1
Yan, R., He, L., Li, Z., Han, X., Liang, J., Si, W., et al. (2015). SCF(JFK) is a bona fide E3 ligase for ING4 and a potent promoter of the angiogenesis and metastasis of breast cancer. Genes Dev. 29, 672–685. doi: 10.1101/gad.254292.114
Yang, H., Robinson, P. N., and Wang, K. (2015). Phenolyzer: phenotype-based prioritization of candidate genes for human diseases. Nat. Methods 12, 841–843. doi: 10.1038/nmeth.3484
Yang, J., AlTahan, A., Jones, D. T., Buffa, F. M., Bridges, E., Interiano, R. B., et al. (2015). Estrogen receptor-alpha directly regulates the hypoxia-inducible factor 1 pathway associated with antiestrogen response in breast cancer. Proc. Natl. Acad. Sci. U.S.A. 112, 15172–15177. doi: 10.1073/pnas.1422015112
Zhang, J., Wang, C., Chen, X., Takada, M., Fan, C., Zheng, X., et al. (2015). EglN2 associates with the NRF1-PGC1alpha complex and controls mitochondrial function in breast cancer. EMBO J. 34, 2953–2970. doi: 10.15252/embj.201591437
Keywords: breast carcinogenesis, transcriptional regulation, cancer metabolism, hypoxia, JFK
Citation: Yang Z, Zhou X, Zheng E, Wang Y, Liu X, Wang Y, Wang Y, Liu Z, Pei F, Zhang Y, Ren J, Huang Y, Xia L, Guan S, Qin S, Suo F, Shi J, Wang L, He L and Sun L (2021) JFK Is a Hypoxia-Inducible Gene That Functions to Promote Breast Carcinogenesis. Front. Cell Dev. Biol. 9:686737. doi: 10.3389/fcell.2021.686737
Received: 27 March 2021; Accepted: 21 June 2021;
Published: 15 July 2021.
Edited by:
Qinong Ye, Institute of Biotechnology (CAAS), ChinaReviewed by:
Binghui Li, Capital Medical University, ChinaShan Gao, Suzhou Institute of Biomedical Engineering and Technology (CAS), China
Copyright © 2021 Yang, Zhou, Zheng, Wang, Liu, Wang, Wang, Liu, Pei, Zhang, Ren, Huang, Xia, Guan, Qin, Suo, Shi, Wang, He and Sun. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lin He, bGluaGVAYmptdS5lZHUuY24=; Luyang Sun, bHV5YW5nX3N1bkBoc2MucGt1LmVkdS5jbg==
†These authors have contributed equally to this work