 Jixia Li1,2*
Jixia Li1,2* Maggie L. Kalev-Zylinska1,3*
Maggie L. Kalev-Zylinska1,3*- 1Blood and Cancer Biology Laboratory, Department of Molecular Medicine and Pathology, University of Auckland, Auckland, New Zealand
- 2Department of Laboratory Medicine, School of Medicine, Foshan University, Foshan, China
- 3Haematology Laboratory, Department of Pathology and Laboratory Medicine, Auckland City Hospital, Auckland, New Zealand
Acute megakaryoblastic leukemia (AMKL) is a rare subtype of acute myeloid leukemia (AML) in which leukemic blasts have megakaryocytic features. AMKL makes up 4%–15% of newly diagnosed pediatric AML, typically affecting young children (less than 2 years old). AMKL associated with Down syndrome (DS) shows GATA1 mutations and has a favorable prognosis. In contrast, AMKL in children without DS is often associated with recurrent and mutually exclusive chimeric fusion genes and has an unfavorable prognosis. This review mainly summarizes the unique features of pediatric non-DS AMKL and highlights the development of novel therapies for high-risk patients. Due to the rarity of pediatric AMKL, large-scale multi-center studies are needed to progress molecular characterization of this disease. Better disease models are also required to test leukemogenic mechanisms and emerging therapies.
1 Introduction
Acute megakaryoblastic leukemia (AMKL) is a rare but life-threatening hematological malignancy characterized by clonal megakaryoblastic proliferation and impaired differentiation of these cells. AMKL was first described in 1931 and included in the French-American-British (FAB) classification as acute myeloid leukemia (AML) the M7 subtype (De Marchi et al., 2019). In the 2016 World Health Organization (WHO) revision of myeloid neoplasms, AMKL in Down syndrome (DS) (also named as myeloid leukemia associated with DS, ML-DS) was included in myeloid proliferations associated with DS, while AMKL in non-Down syndrome (non-DS) patients was included in the category of acute myeloid leukemia not otherwise specified (AML NOS) (Arber et al., 2016). This categorization remains similar in the latest 2022 WHO and International Consensus classifications (ICC) (Arber et al., 2022; Khoury et al., 2022). However, cases with KMT2A (lysine methyltransferase 2A) and NUP98 (nucleoporin 98) rearrangements now form independent subgroups of AML with defining genetic abnormalities (that also include other non-AMKL phenotypes), and cases with CBFA2T3 (CBFA2/RUNX1 partner transcriptional co-repressor 3)::GLIS2 (GLIS family zinc finger 2) and RBM15 (RNA binding motif protein 15)::MKL1 (megakaryoblastic leukemia 1) fusions belong to a subgroup of AML with other defined genetic alterations. Other cases of AMKL lacking defined genetic alterations remain defined by differentiation, which requires blasts to express at least one of the platelet glycoproteins: CD41 (glycoprotein IIb), CD61 (glycoprotein IIIa), or CD42b (glycoprotein Ib). Myeloid proliferations associated with DS form a subtype of myeloid neoplasms associated with germline predisposition (Arber et al., 2022; Khoury et al., 2022).
AMKL has a bimodal age distribution, so it can be grouped into adult and pediatric. AMKL is very rare in adults accounting for only 1% of all AML (Wen et al., 2011), but in children, AMKL makes up 4%–15% of newly diagnosed AML cases (de Rooij et al., 2017). AMKL is most frequent in young children with DS (median age at diagnosis 1–1.8 years), accounting for the vast majority of AML in DS (∼70%) (Bhatnagar et al., 2016; Masetti et al., 2019b). By contrast, in non-DS patients, AMKL comprises 5%–10% of AML with a slightly older median age at diagnosis ranging from 1.6 to 1.8 years (de Rooij et al., 2016; Maarouf et al., 2019). Genomic studies highlighted the heterogeneity of pediatric AMKL with the specific alterations characterizing different disease subgroups. ML-DS is driven by overexpressed genes and micro-RNAs located on chromosome 21, GATA1 (GATA Binding Protein 1) mutations, and a range of other somatic mutations, in particular those affecting cohesin molecules, signaling pathways, epigenetic regulators, and hematopoietic transcription factors (Boucher et al., 2021; de Castro et al., 2021; Grimm et al., 2021). In contrast, non-DS AMKL is characterized by recurrent chromosomal translocations, complex karyotype, and DNA copy number abnormalities (de Rooij et al., 2017). Children with non-DS AMKL show more heterogeneity and more chromosomal abnormalities than ML-DS. Strikingly, recurrent oncogene fusions are found in more than 70% of non-DS pediatric AMKL, including CBFA2T3::GLIS2, KMT2A rearrangements, NUP98::KDM5A (lysine-specific demethylase 5A), RBM15::MKL1, and HOX (homeobox) gene rearrangements (de Rooij et al., 2016; de Rooij et al., 2017). For completeness, AMKL in adults is more often secondary, preceded by an antecedent hematologic disorder or exposure to chemotherapy or radiotherapy (Dastugue et al., 2002). Adults with AMKL have different cytogenetic and molecular features compared to pediatric patients, showing a diversity of chromosomal abnormalities, the lack of recurrent genetic fusions, and the presence of highly recurrent mutations in genes encoding TP53 (tumor protein p53), cohesins, splicing factors, and epigenetic regulators such as ASXL (additional sex combs-like), DNMT3A (DNA methyltransferase 3 alpha), and TET2 (tet methylcytosine dioxygenase 2) (Dastugue et al., 2002; de Rooij et al., 2017). Cytogenetic and molecular aberrations influence the outcomes of pediatric AMKL subgroups. ML-DS has an excellent prognosis even with low-dose induction chemotherapy, while the outcomes of non-DS AMKL remain unsatisfactory despite improved diagnostics, intensified treatment protocols, and advanced supportive care (de Rooij et al., 2017; Wang et al., 2021). Non-DS children carrying CBFA2T3::GLIS2, KMT2A rearrangements and NUP98::KDM5A have a very poor prognosis (de Rooij et al., 2016; de Rooij et al., 2017). Molecular features of ML-DS have been recently reviewed, including by our group (Boucher et al., 2021; de Castro et al., 2021; Grimm et al., 2021; Li and Kalev-Zylinska, 2022). Here, we provide an updated summary of molecular abnormalities and emerging novel therapeutic strategies in pediatric non-DS AMKL.
2 Molecular characterization of pediatric non-DS AMKL
Pediatric non-DS AMKL (also referred to in this review as non-DS AMKL for simplicity) is a highly molecularly heterogeneous disease characterized by recurrent and mutually exclusive genetic aberrations, including CBFA2T3::GLIS2 (18%–27%), KMT2A rearrangements (KMT2Ar) (7%–17%), NUP98::KDM5A (11%–12%), RBM15::MKL1 (10%–11%), and HOX rearrangements (HOXr) (14%–15%) (Figure 1A) (de Rooij et al., 2016; de Rooij et al., 2017; Masetti et al., 2019b). A further 4% of patients have non-recurrent fusions engaging hematopoietic transcription factors and epigenetic regulators, e.g., MN1 (meningioma [disrupted in balanced translocation] 1)::FLI1 (friend leukemia integration 1 transcription factor), BCR (breakpoint cluster region protein):ABL1 (ABL1, tyrosine-protein kinase ABL1), and MAP2K2 (mitogen-activated protein kinase 2)::MLLT10 (myeloid/lymphoid or mixed-lineage leukemia translocated to 10) (de Rooij et al., 2017). Molecular alterations are crucial for risk stratification and tailored treatment. Subgroups carrying CBFA2T3::GLIS2, KMT2Ar, and NUP98::KDM5A are associated with poor prognosis and a high risk of disease recurrence (Figure 1B). Thus, intensive chemotherapy and hematopoietic stem cell transplantation (HSCT) are often used in first remission for these patients (de Rooij et al., 2016; de Rooij et al., 2017; Masetti et al., 2019b). In contrast, cases with RBM15::MKL1, HOXr, non-recurrent fusions, and unknown drivers have an intermediate prognosis and are treated with standard chemotherapy (Figure 1B). This review compiled molecular changes reported in pediatric non-DS AMKL (Tables 1, 2). Ongoing molecular characterization of AMKL is needed to guide future mechanistic studies into disease pathogenesis and provide essential clues on developing new targeted therapies.
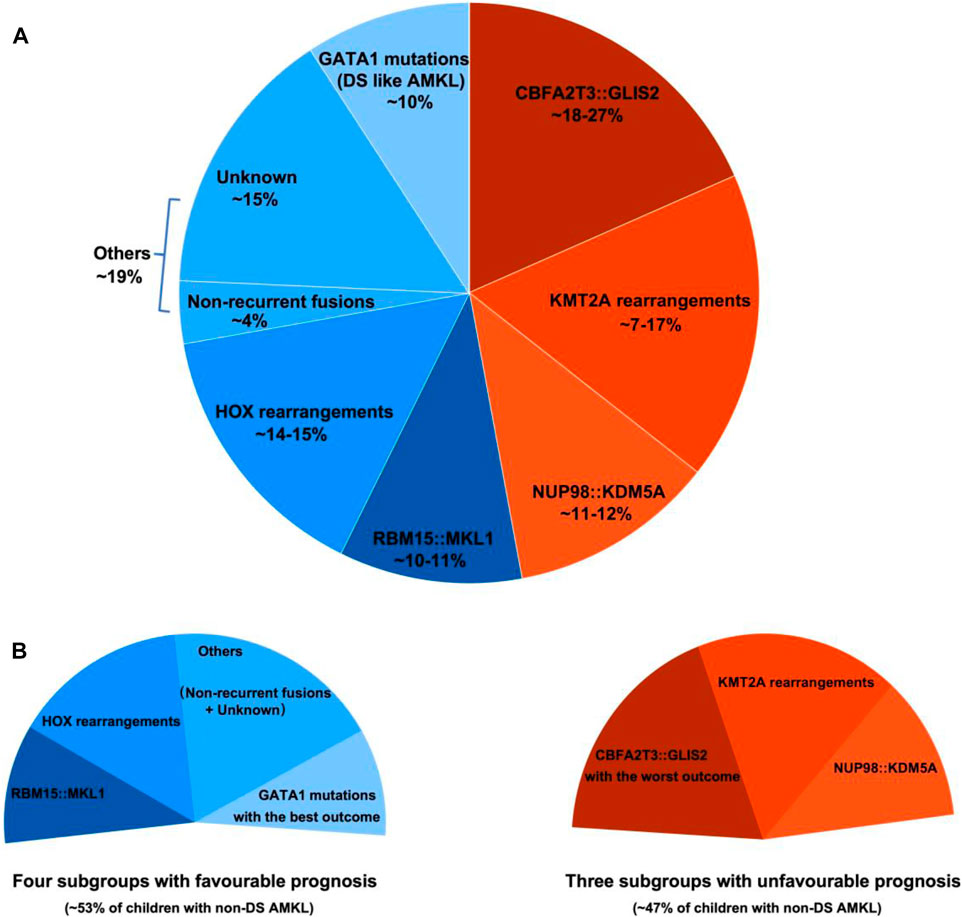
FIGURE 1. Distribution of distinct molecular subgroups and their prognostic impact in pediatric non-DS AMKL. (A) Distribution of distinct molecular subgroups in pediatric non-DS AMKL. Pediatric non-DS AMKL is characterized by recurrent and mutually exclusive chromosomal abnormalities, primarily including CBFA2T3::GLIS2 (18%–27%), KMT2A rearrangements (KMT2Ar) (7%–17%), NUP98::KDM5A (11%–12%), RBM15::MKL1 (10%–11%), and HOX rearrangements (HOXr) (14%–15%) (de Rooij et al., 2016; de Rooij et al., 2017; Hara et al., 2017; Lalonde et al., 2021). Further 4% of patients have non-recurrent fusions engaging hematopoietic transcription factors and epigenetic regulators (de Rooij et al., 2017). Other cases do not have known oncogenic fusions, but GATA1 mutations are detected in about half of this population (overall found in approximately 10% of pediatric non-DS AMKL) (de Rooij et al., 2017). (B) Prognosis of distinct molecular subgroups in pediatric non-DS AMKL. Based on molecular aberrations, three subgroups (CBFA2T3::GLIS2, KMT2Ar, and NUP98::KDM5A) have an unfavorable prognosis, amounting to nearly half of patients; other three subgroups (RBM15::MKL1, HOXr, non-recurrent fusions and unknown drivers) have an intermediate prognosis, accounting for approximately 40% of patients; and patients with GATA1 mutations represent an excellent prognostic group, making up for roughly 10% of children with non-DS AMKL (de Rooij et al., 2017). Among the unfavorable subsets, CBFA2T3::GLIS2 has the worst outcome, with the 5-year overall survival rate being 14% (de Rooij et al., 2017). In contrast, DS-like AMKL has the best outcome, with the 5-year overall survival rate being 100% (de Rooij et al., 2017).
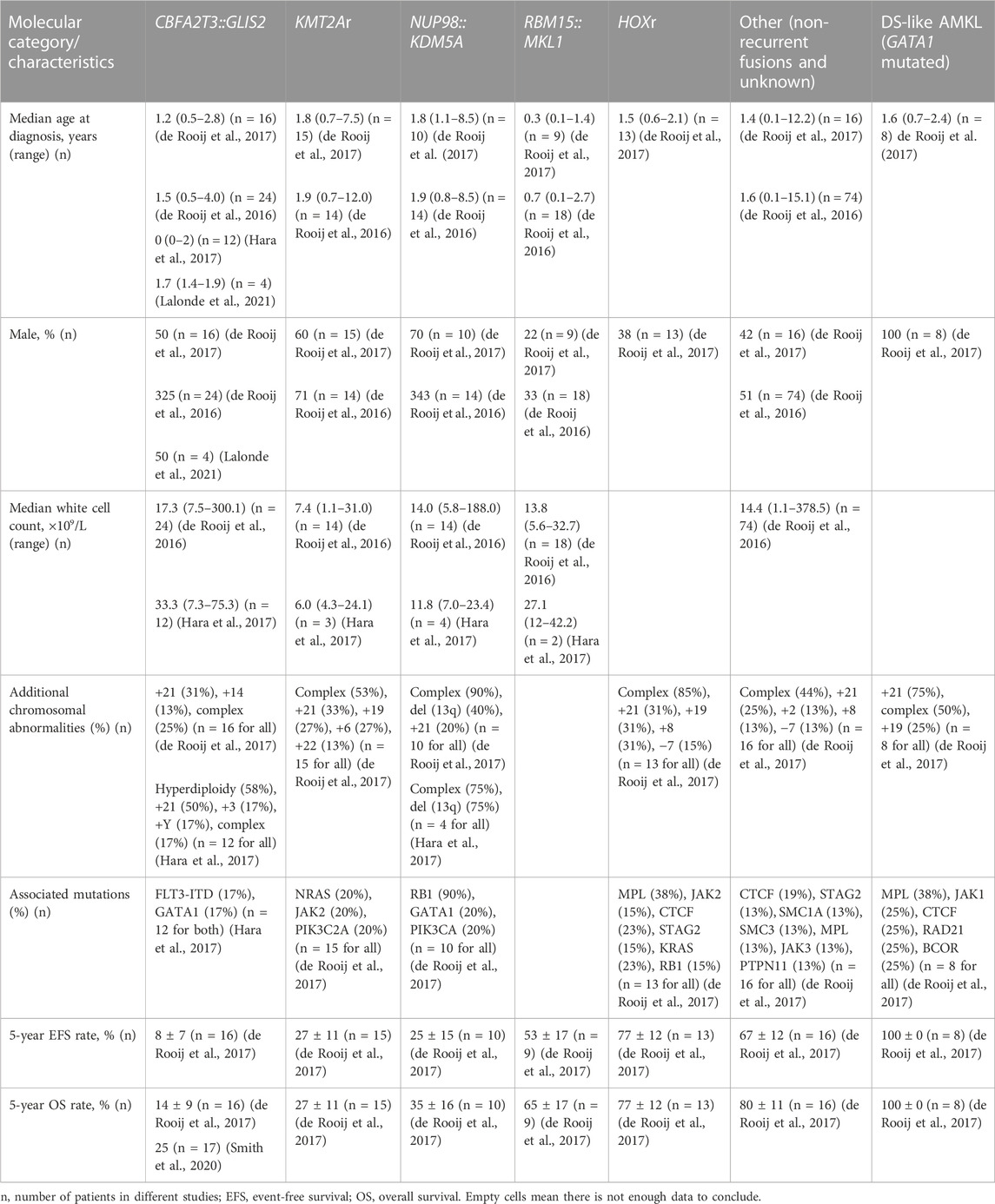
TABLE 1. Key features of distinct molecular subgroups of pediatric non-DS AMKL.
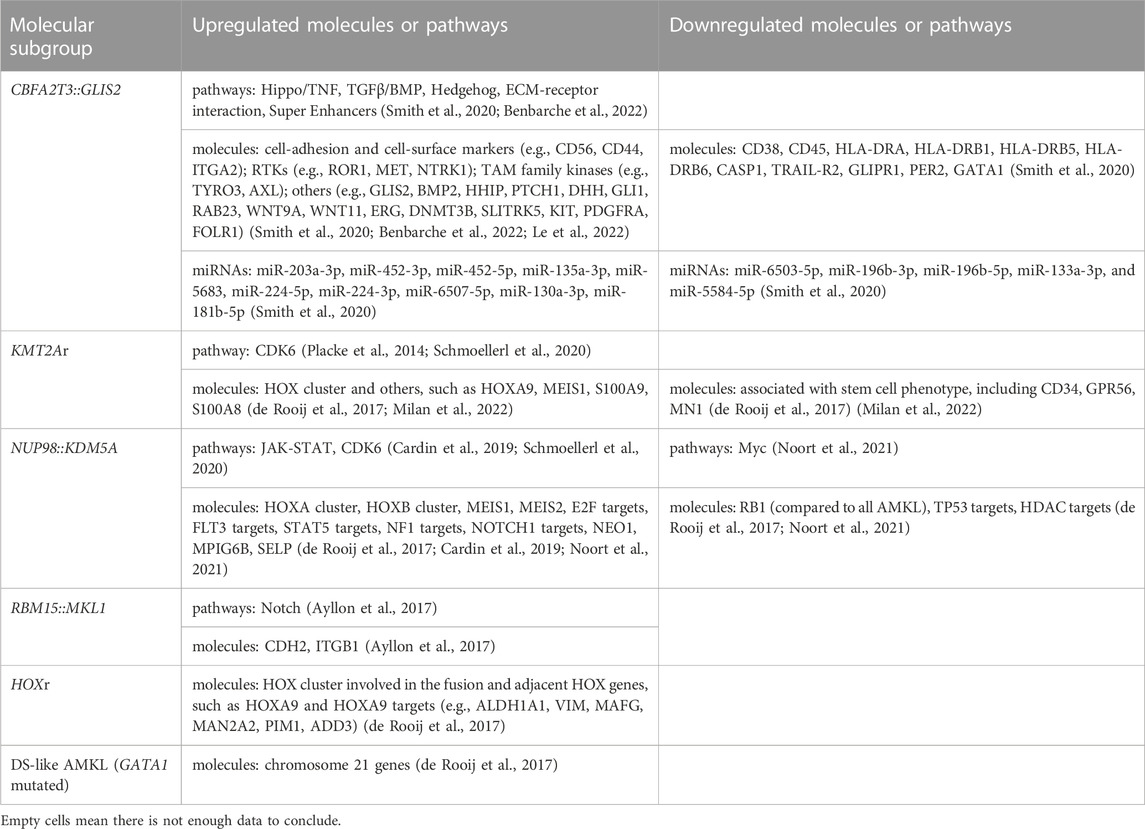
TABLE 2. Gene signatures associated with distinct molecular subgroups of pediatric non-DS AMKL.
2.1 CBFA2T3::GLIS2
The inversion inv(16)(p13.3q24.3) (generating the CBFA2T3::GLIS2 fusion) is the most frequently, but not exclusively detected in 18%–27% of pediatric non-DS AMKL (Figure 1) (de Rooij et al., 2016; de Rooij et al., 2017; Hara et al., 2017). A newer study suggested that this lesion may be even more common, as it was identified in 4 of 6 children with non-DS AMKL (Lalonde et al., 2021). CBFA2T3::GLIS2 is not specific to AMKL and occurs in diverse morphologic subtypes of de novo AML (except for acute promyelocytic leukemia (APL) and leukemia with erythroid differentiation), overall found in approximately 8.4% of cytogenetically normal AML cases (Masetti et al., 2013a; Smith et al., 2020). The inv(16)(p13.3q24.3) fuses the transcriptional repressor CBFA2T3 with the zinc finger DNA-binding transcription factor GLIS2. The prevalence of CBFA2T3::GLIS2 appears higher in African American children, accounting for about 29% of patients with this fusion in the AAML0531 and AAML1031 cohorts (Smith et al., 2020). CBFA2T3::GLIS2 characterizes an extremely aggressive leukemic subgroup with a grim prognosis across all differentiation subtypes (overall survival rates 14%–40%) (Gruber and Downing, 2015; de Rooij et al., 2016; de Rooij et al., 2017; Smith et al., 2020). Among CBFA2T3::GLIS2 AMKL, approximately 40% of patients also carry a DHH (desert hedgehog):RHEBL1 (Ras homologue enriched in brain like 1) fusion (Masetti et al., 2013b; Jetten, 2019). The co-occurrence of these two fusions has an even poorer prognosis than the CBFA2T3::GLIS2 alone (Masetti et al., 2013b; Jetten, 2019). Children with CBFA2T3::GLIS2 AML (including AMKL) tend to be younger (median age of onset ∼1.5 years) and display distinct clinical and laboratory features, including stronger expression of CD56 (neural cell adhesion molecule, NCAM) and a more frequent extramedullary involvement compared to other AML (Masetti et al., 2013a; de Rooij et al., 2016; Hara et al., 2017; Smith et al., 2020; Zangrando et al., 2021). Standard karyotyping cannot identify the CBFA2T3::GLIS2, because of its cryptic nature. Immunophenotypic features may suggest the presence of this fusion, as blasts carrying CBFA2T3::GLIS2 have bright CD56 expression with dim-to-negative HLA-DR (human leukocyte antigen DR isotype), CD38, and CD45 referred to as the RAM immunophenotype (Zangrando et al., 2021). The somatic mutational burden of CBFA2T3::GLIS2 subtype is much lower than in other AMKL subgroups (Gruber et al., 2012; Dang et al., 2017; Masetti et al., 2019a). However, mutations in tyrosine kinases such as FLT3 (fms-like tyrosine kinase 3), KIT (tyrosine-protein kinase KIT), RAS (Rat sarcoma), JAK/STAT (Janus kinases/signal transducer and activator of transcription), and transcription factor GATA1 can co-occur (Gruber et al., 2012; de Rooij et al., 2017; Hara et al., 2017). CBFA2T3::GLIS2 can also be accompanied by trisomies of chromosomes 3, 21 and 8, complex karyotype, or hyperdiploidy (Hara et al., 2017; Amano et al., 2020; Smith et al., 2020; Gillam et al., 2022).
CBFA2T3::GLIS2 fuses the 5′ portion of CBFA2T3 (also named as ETO2, MTG16, RUNX1T3 or ZMYND4) in frame with the 3’ region of GLIS2. The majority of chimeric CBFA2T3::GLIS2 fusions (80%) are between exon 11 of CBFA2T3 and exon 3 of GLIS2. Other rare fusion transcripts have also been reported, including between exons 9, 10 or 12 of CBFA2T3 and exons 2 or 3 of GLIS2 (Masetti et al., 2019b; De Marchi et al., 2019; Amano et al., 2020; Smith et al., 2020). The chimeric protein generated by the most common fusion retains most of the functional domains of both proteins with the loss of myeloid, nervy and DEAF-1 domain (MYND) that interacts with nuclear receptor corepressor complex in CBFA2T3, as well as the loss of transactivation domain (TAD) that mediates transcriptional activation of target genes in GLIS2 (Figure 2A). Both CBFA2T3 and GLIS2 participate in leukemic transformation driven by CBFA2T3::GLIS2 (Thirant et al., 2017a).
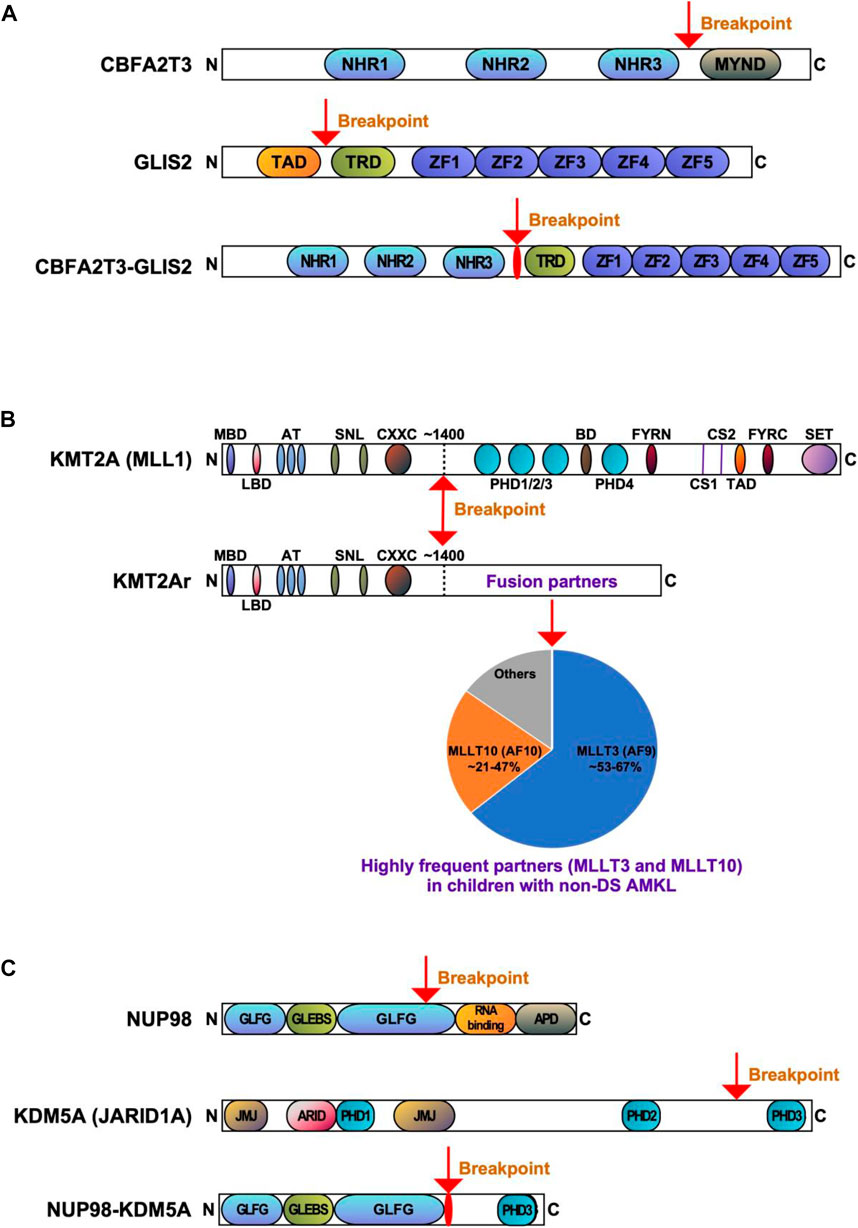
FIGURE 2. Recurrent gene fusions associated with poor outcomes in pediatric non-DS AMKL. Schematics display the functional domains but are not drawn to scale. The breakpoints are indicated by arrows. (A). Structure of CBFA2T3, GLIS2, and CBFA2T3-GLIS2 proteins. NHR, nervy homology region; MYND, myeloid, nervy, and DEAF-1; TAD, transactivation domain; TRD, transcriptional repression domain; ZF, zinc finger. (B) Structure of KMT2A (MLL) and KMT2Ar (chimeric) proteins. MBD, menin-binding domain; LBD, LEDGF-binding domain; AT, AT hooks; SNL, speckled nuclear localization domains; CXXC, CXXC domain; PHD1/2/3/4, plant homeodomain 1/2/3/4; BD, bromodomain. CS1 and CS2 are the taspase-1 cleavage sites, and FYRN and FYRC are the domains whereby KMT2A-N and KMT2A-C interact after cleavage. TAD, transactivation domain; SET, H3K4 histone methyltransferase domain. KMT2A fusion proteins are caused by chromosomal rearrangements leading to an in-frame fusion between the N-terminus of KMT2A and one of the multiple fusion partners. The breakpoints of KMT2A are located in the region of ∼1400 aa that contains the AT-hook DNA binding motifs and CXXC domains binding unmethylated CpG-containing DNA. Frequencies of the KMT2A fusion partner proteins are shown, with transcription cofactors MLLT3 (AF9) (found in 53%–67% of cases) and MLLT10 (AF10) (found in 21%–47% of cases) being the most common (de Rooij et al., 2016; de Rooij et al., 2017; Hara et al., 2017). (C) Structure of NUP98, KDM5A, and NUP98-KDM5A proteins. GLFG, GLFG repeats; GLEBS, Gle2-binding domain; RNA binding, RNA binding domain; APD, autoproteolytic domain; JMJ, Jumonji domain; ARID, AT-rich interactive domain; PHD1/2/3, plant homeodomains 1/2/3.
CBFA2T3 is a member of the RUNX1T1 (runt-related transcription factor 1, translocated) complex. CBFA2T3 belongs to the eight-twenty-one (ETO) family of chromatin-associated proteins, functioning as a master transcriptional coregulator and a Wnt (wingless/integrated) and Notch (neurogenic locus notch homolog protein) signaling suppressor in normal and malignant hematopoiesis (Steinauer et al., 2017; Masetti et al., 2019a; Steinauer et al., 2019; Steinauer et al., 2020; Jakobczyk et al., 2021). CBFA2T3 is expressed in all hematopoietic cells and contains three Nervy homology region (NHR) domains and a MYND domain (Figure 2A). CBFA2T3 participates in hemopoietic stem cell (HSC) self-renewal and differentiation, megakaryocyte-erythrocyte progenitor development, and leukemia stem cell (LSC) expansion (Steinauer et al., 2017; Masetti et al., 2019a; Steinauer et al., 2019; Steinauer et al., 2020; Jakobczyk et al., 2021). Physiological binding partners for CBFA2T3 include transcription factors and chromatin modifiers, and it is generally presumed that CBFA2T3 represses gene transcription through binding to multiple corepressors, such as E-proteins (e.g., E2A basic helix-loop-helix transcription factor), nuclear receptor corepressors (NCOR), and histone deacetylases (HDAC) (Steinauer et al., 2017). CBFA2T3 contributes to transcriptional repression via GATA1-SCL (stem cell leukemia, also known as TAL1) complex during megakaryopoiesis (Schuh et al., 2005; Hamlett et al., 2008). In the megakaryoblastic cell line L8057, CBFA2T3 knockdown enhances megakaryocytic differentiation via elevating gene expression associated with terminal megakaryocytic maturation (Hamlett et al., 2008); whilst CBFA2T3 deletion in a mouse model induces differentiation along the granulocytic-monocytic lineages at the expense of erythroid-megakaryocytic differentiation (Chyla et al., 2008).
GLIS2 is a member of the GLIS subfamily of Krüppel-like zinc finger transcription factors closely related to the GLI and ZIC subfamilies (Scoville et al., 2017). GLIS2 contains a TAD, a transcriptional repression domain (TRD), and a DNA binding domain (five zinc finger motifs) (Figure 2A). GLIS2 function is implicated in the processes of hematopoiesis and leukemogenesis (Jetten, 2019; Pinto and Chetty, 2020). Normally, GLIS2 is not expressed in the differentiating hematopoietic cells (Gruber et al., 2012; Thirant et al., 2017b); however, its expression contributes to HSC repopulation (Holmfeldt et al., 2016). Knockdown of GLIS2 in murine LSK (Lin-SCA1+c-KIT+) cells reduces HSC repopulation, suggesting GLIS2 could regulate HSC engraftment and hematopoietic reconstitution (Holmfeldt et al., 2016). GLIS2 overexpression in MOZ (monocytic leukemia zinc finger protein):TIF2 (transcriptional intermediary factor 2) leukemic cells promotes their differentiation into mature myeloid cells and delays AML development in mice, indicating that GLIS2 inhibits AML initiation by inducing LSC differentiation (Shima et al., 2018).
Although the molecular bases for AMKL transformation by CBFA2T3::GLIS2 are still far from being elucidated, distinct functional properties of this chimeric protein have been illustrated (Masetti et al., 2019a). CBFA2T3::GLIS2 AMKL has a unique gene expression pattern (Gruber et al., 2012; Thirant et al., 2017a; Benbarche et al., 2022). GATA1 downregulation, ERG (ETS-related gene) upregulation, and activation of super enhancer elements may impair megakaryocytic differentiation and increase abnormal self-renewal of leukemic cells in AMKL associated with CBFA2T3::GLIS2 (Thirant et al., 2017a; Benbarche et al., 2022). Super enhancers are clusters of regulatory elements characterized by high intensity of enhancer-related histone tail modifications (Benbarche et al., 2022). Ectopic expression of CBFA2T3::GLIS2 induces the formation of super enhancers, which controls the expression of KIT and platelet-derived growth factor receptor alpha (PDGFRA) involved in leukemic progression (Benbarche et al., 2022). CBFA2T3::GLIS2 positive cases also display higher expression of CD56, a CD56-interacting partner CACNB2 (calcium voltage-gated channel auxiliary subunit beta 2), GABRE (gamma-aminobutyric acid type A receptor subunit epsilon), miR-224 and miR-452 (Smith et al., 2020). Both miR-224 and miR-452 are intronic miRNAs transcribed from the GABRE locus and have numerous mRNA targets involved in immune responses, leukocyte activation, and leukocyte differentiation. Upregulation of other miRNAs (e.g., miR-181b-5p) may be responsible for the reduction in expression of apoptotic and tumor suppressor genes, and simultaneous downregulation of tumor suppressive miRNAs (e.g., miR-196a/b, miR-133a, and miR-199a/b) (Smith et al., 2020). The role of miRNA changes in leukemia initiation by CBFA2T3::GLIS2 is not well defined. Overexpression of miR-181b-5p downregulates apoptotic genes CASP1 (caspase 1) and TRAIL-R2 (TNF-related apoptosis-inducing ligand receptor 2), and the circadian rhythm gene PER2 (period circadian regulator 2), while loss of miR-196b is associated with ITGA2 (integrin subunit alpha 2) and DNMT3B (DNA methyltransferase 3 beta) overexpression (Smith et al., 2020). When highly expressed, ITGA2 and DNMT3B are independent indicators of poor prognosis in AML (Lamba et al., 2018; Lian et al., 2018). CBFA2T3::GLIS2 is also associated with an overexpression of several cell adhesion and cell surface molecules (e.g., extracellular matrix binding, cell adhesion, and integrin binding genes) and signaling pathways (e.g., Hippo, transforming growth factor beta (TGFβ), bone morphogenetic proteins (BMP), JAK/STAT and Hedgehog) (Gruber et al., 2012; Smith et al., 2020). Overall, multiple molecular alterations may contribute to the phenotype of CBFA2T3::GLIS2 leukemia.
Considering the lack of other recurrent mutations in patients carrying CBFA2T3::GLIS2, this fusion probably is the primary genomic alteration. However, the evidence is mixed on whether CBFA2T3::GLIS2 alone is sufficient for leukemic transformation. On the one hand, the introduction of CBFA2T3::GLIS2 into murine bone marrow is insufficient to induce overt leukemia in mice (Dang et al., 2017). On the other hand, the transduction of human CD34+ cord blood stem cells with a lentivirus carrying CBFA2T3::GLIS2 leads to increased proliferation, maturation arrest, and morphologic and immunophenotypic aberrations consistent with AMKL (Le et al., 2022). Further, an inducible transgenic mouse model demonstrated that CBFA2T3::GLIS2 expressed in fetal HSCs leads to a rapid and aggressive AMKL, whereas its expression in adult bone marrow HSCs results in AML (Lopez et al., 2019). In an endothelial cell coculture system, the expression of CBFA2T3::GLIS2 transforms human cord blood hematopoietic stem and progenitor cells (HSPCs) and leads to highly aggressive leukemia in mice (Le et al., 2022).
In summary, cytogenetically cryptic CBFA2T3::GLIS2 fusion is a potent oncogene contributing to malignant transformation and an extremely lethal and treatment-refractory AML. Disease expressing this fusion have unique molecular features with a specific gene expression signature, dysregulated expression of cell adhesion molecules, RTKs, and signaling pathways (e.g., Hippo, TGFβ, BMP, JAK/STAT and Hedgehog). Despite the use of intensive chemotherapy and HSCT, the outcomes of CBFA2T3::GLIS2 leukemia are poor. The mechanism of leukemogenesis associated with CBFA2T3::GLIS2 remains incompletely understood. Therefore, further mechanistic studies are required to inform the development of novel therapies.
2.2 KMT2A rearrangements
Alterations of the KMT2A gene (also known as mixed lineage leukemia 1, MLL1 or MLL gene) located at 11q23.3 are found in both acute lymphoblastic leukemia (ALL) and AML, making up ∼10% of all leukemia cases in all age groups (Winters and Bernt, 2017). KMT2A rearrangements (KMT2Ar) are one of the most common recurrent genetic aberrations found in 15%–25% of all newly diagnosed cases of pediatric AML (Meyer et al., 2006; Meyer et al., 2009; Meyer et al., 2013; Meyer et al., 2018; Hoffmeister et al., 2021; Quessada et al., 2021; Meyer et al., 2023; Yuen et al., 2023). KMT2Ar are particularly prevalent in infant AML, accounting for approximately 30% of children presenting below the age of 2 years. KMT2Ar correlate with monoblastic and monocytic AML subtypes (71.5%–73%) and are much less common in other subtypes (Meyer et al., 2023). In pediatric AMKL, KMT2Ar are reported in 7%–17.4% of patients, with the median age at diagnosis of 1.9 years (range 0.7–12 years) (Chen and Armstrong, 2015; de Rooij et al., 2016; de Rooij et al., 2017; Hara et al., 2017; Masetti et al., 2019b; Maarouf et al., 2019). Numerous KMT2A fusion partners have been identified in children with AMKL, including MLLT1 (myeloid/lymphoid or mixed-lineage leukemia translocated to 1, also known as ENL), AFF1 (AF4/FMR2 family member 1, previously known as AF4), MLLT3 (also known as AF9), MLLT6, MLLT9, MLLT10 (also known as AF10), and SEPT9 (septin 9) (Takita et al., 2009; de Rooij et al., 2016; de Rooij et al., 2017; Hara et al., 2017; Forlenza et al., 2018; Lalonde et al., 2021; Qiu et al., 2022). The two most common KMT2A translocations in AMKL are t(9;11)(p21;q23) (generating KMT2A::MLLT3 fusion) and t(10;11)(q12;q23) (generating KMT2A::MLLT10 fusion) (Figure 2B). Translocation t(9;11)(p21;q23) was found in 8 of 15 (53%), 9 of 14 (64%) and 2 of 3 (66.7%) of patients in three different studies (de Rooij et al., 2016; de Rooij et al., 2017; Hara et al., 2017). In the same studies, t(10;11)(q12;q23) was the second most common, detected in 7 of 15 (46.7%), 3 of 14 (21.4%), and 1 of 3 (33.3%) of patients, respectively (de Rooij et al., 2016; de Rooij et al., 2017; Hara et al., 2017). The prognosis of KMT2Ar AML varies, heavily relying on its translocation partner. However, in pediatric AMKL KMT2Ar are always regarded as high-risk fusion events linked to a greater risk of relapse and poorer overall survival (de Rooij et al., 2016; de Rooij et al., 2017). KMT2Ar subgroup of pediatric non-DS AMKL tends to have higher white cell count (WCC) and is more common in males, although these differences were not statistically significant in the study by de Rooij et al. (de Rooij et al. 2016). Pediatric KMT2Ar AML was shown to have a higher expression of CD33 compared to KMT2A wild-type AML (Pollard et al., 2021). However, it is unclear whether pediatric KMT2Ar AMKL has the same pattern, so further studies are required. Co-occurring mutations can be found in AMKL associated with KMT2Ar, including in molecules such as NRAS (neuroblastoma RAS viral oncogene homolog), KRAS (Ki-ras2 Kirsten rat sarcoma viral oncogene homolog), PTPN11 (tyrosine-protein phosphatase non-receptor type 11), NF1 (neurofibromatosis type 1), EPOR (erythropoietin receptor), MPL (thrombopoietin receptor), JAK1/2/3, PIK3C2A (phosphatidylinositol-4-phosphate 3-kinase catalytic subunit type 2 alpha), KIT, cohesin and epigenetic regulators, e.g., STAG3 (stromal antigen 3), SETD2 (SET domain containing 2, histone lysine methyltransferase), IDH1 (isocitrate dehydrogenase 1), CREBBP (cyclic adenosine monophosphate response element binding protein binding protein), transcription factors, e.g., GATA1, RNA splicing and regulatory proteins, e.g., U2AF1 (U2 small nuclear RNA auxiliary factor 1) and DDX3X (DEAD-Box helicase 3 X-linked) (de Rooij et al., 2017; Foster et al., 2019). KMT2Ar associated with KRAS mutations confer a particularly poor prognosis (Matsuo et al., 2020). Several additional chromosomal abnormalities can be observed in pediatric AMKL with KMT2Ar, such as trisomy 8, trisomy 21, hyperdiploidy, monosomy 15, and a complex karyotype (de Rooij et al., 2016; de Rooij et al., 2017; Hara et al., 2017). The clinical impact of these co-occurring chromosomal changes remains unclear.
KMT2A represents a key transcription factor and histone-H3 lysine-4 (H3K4) methyltransferase that serves as a master controller for the transcription of critical genes implicated in normal embryonic development and hematopoiesis (Li and Song, 2021; Sugeedha et al., 2021). KMT2A is a large protein (500 kDa, 3696 amino acids) and contains multiple conserved domains, including menin binding motif, LEDGF (lens epithelium-derived growth factor) binding domain, AT-hook (DNA binding) motifs, nuclear localization signals, CXXC domain (nonmethylated CpG DNA binding domain), PHD (plant homeodomain) fingers, bromodomain, FYRN (FY-rich domain N-terminal) domain, taspase 1 cleavage sites, TAD, FYRC (FY-rich domain C-terminal), and SET domains (Li and Song, 2021) (Figure 2B). Interestingly, its N-terminal ∼1400 residues, containing the AT-hook and CXXC domains, act as a transcription factor that recognizes and binds target genes such as HOXA9 and MEIS1 (Meis homeobox 1) (Muntean et al., 2010). The C-terminal SET domain functions as an H3K4 methyltransferase that mediates chromatin modifications associated with epigenetic transcriptional activation (Li and Song, 2021). An oncogenic fusion protein produced by KMT2Ar predominantly consists of the N-terminal DNA-interacting domains of KMT2A (residues 1–∼1400) fused in frame with one of over 100 fusion partners (Li and Song, 2021) (Figure 2B). KMT2A fusions act as oncoproteins in different leukemic cell and animal models, and expression of those fusions can promote proliferation and arrest myeloid differentiation of hematopoietic progenitors, resulting in their accumulation (Slany, 2016; Skucha et al., 2018). It appears that all forms of KMT2A fusion oncoproteins positively regulate the expression of HOX genes and the HOX cofactor MES1 critical for the leukemic transformation of hematopoietic progenitors (Muntean et al., 2010; Chen and Armstrong, 2015). The mechanism of leukemogenesis may therefore rely on the upregulation of target genes by KMT2A fusion proteins and other recruited proteins, e.g., super elongation complexes involving DOT1L (DOT1 like histone lysine methyltransferase) and H3K79 (lysine 79 of histone H3), or polycomb repressive complex 1 (Chen and Armstrong, 2015; Slany, 2016; Li and Song, 2021). KMT2A::MLLT3, the most common form of KMT2Ar in children with non-DS AMKL, is sufficient to induce a myeloproliferative disorder in mice and to generate leukemia in a mouse model using healthy CD34+ cord blood HSPCs (Mulloy et al., 2008; Wei et al., 2008; Stavropoulou et al., 2016). Wild-type MLLT3 is a positive regulator of early erythroid and megakaryocytic cell fates in primitive human cord blood cells, together with GATA1 (Pina et al., 2008). In a mouse model generated using human CD34+ cord blood HSPCs containing KMT2A::MLLT3, the development of a pronounced hypomethylation phenotype is an early event during leukemogenesis (Milan et al., 2022). Leukemia development in this model was associated with the loss of expression of stem-cell associated genes, gain of expression of HOXA9 and MEIS1, and increased expression ratio of S100 (S100 calcium-binding proteins) A8 to A9 proteins. KMT2A fusion proteins, particularly KMT2A::ENL, regulate only a small subset of genes recognized by wild-type KMT2A (e.g., HOXA9, MEIS1, and several other transcription factor genes, e.g., EYA1 (eyes absent homolog 1), SIX1 (SIX homeobox 1) and SIX4 (SIX homeobox 4), highlighting that the transforming capacity of KMT2A fusion may not be limited to HOXA/MEIS1 genes (Wang et al., 2011). Another study showed that IKAROS (IKAROS family zinc finger 1) acts as an essential regulator in KMT2Ar AML by influencing tumor suppressor pathways, immune dysregulation, and changes in cell differentiation (Aubrey et al., 2022). Cyclin-dependent kinase 6 (CDK6), a cell cycle regulator, was also shown to play a role in the development of KMT2Ar AML (Placke et al., 2014). Inhibition of CDK6 by shRNA or pharmacological inhibitor (e.g., PD-0332991) decreases leukemic cell growth and promotes myeloid cell differentiation in cell lines and primary human AML cells harboring different KMT2A translocations. In a mouse model with KMT2A::MLLT3, CDK6 inhibition increases cell differentiation and prolongs mice survival (Placke et al., 2014).
In summary, similar to CBFA2T3::GLIS2, KMT2Ar in pediatric non-DS AMKL drive disease with inferior outcomes, high incidence of treatment non-response and relapse. Children carrying KMT2Ar present with distinct clinical and molecular features, including monocytic differentiation, higher WCC, higher expression of CD33, fewer co-occurring mutations compared with KMT2A wild-type AML, and higher expression of HOXA/MEIS1 genes. However, despite improvements in the molecular characterization of KMT2Ar AML, leukemogenic mechanisms remain incompletely understood, and new therapeutic approaches are needed.
2.3 NUP98::KDM5A and other NUP98 rearrangements
The cytogenetically cryptic translocation t(11;12)(p15;q35) (resulting in NUP98::KDM5A fusion, also known as NUP98::JARID1A or NUP98:: RBP2) was initially identified in pediatric AMKL in 2006 (van Zutven et al., 2006). NUP98::KDM5A fuses NUP98 located on chromosome 11p15 with KDM5A located on the telomeric end of chromosome 12p13.3 (Masetti et al., 2019b). The fusion is found in 2% of pediatric AML overall, primarily in 8%–12% of non-DS AMKL and 12% of infant AML (de Rooij et al., 2013; de Rooij et al., 2016; de Rooij et al., 2017; Masetti et al., 2019b; Hara et al., 2020; Noort et al., 2021). NUP98::KDM5A AML patients have a median age at diagnosis of 3.2 years (ranging from 3 weeks to 18.5 years) (Noort et al., 2021). NUP98::KDM5A can be found in all morphologic subtypes of AML except for APL. Megakaryocytic differentiation was seen in 34% of NUP98::KDM5A AML, monocytic in 21%, and erythroid in 17% of these cases (Noort et al., 2021). The median age of AMKL patients carrying NUP98::KDM5A is lower than in AML overall (1.8–1.9 years, range: 0.7–12 years) (de Rooij et al., 2016; de Rooij et al., 2017; Noort et al., 2021). NUP98::KDM5A confers a dismal prognosis with an overall survival of 33%–36% due to a high incidence of induction failure or relapse (de Rooij et al., 2016; Hara et al., 2020; Noort et al., 2021). Additional chromosomal abnormalities can be found in association with NUP98::KDM5A, in particular trisomy 21, hyperdiploidy, −13/-13q and a complex karyotype (de Rooij et al., 2016).
NUP98 has many different partner genes (over 30) to produce a series of abnormal fusion proteins in several hematopoietic malignancies (including AML, myelodysplastic syndrome, T-ALL, and mixed-phenotype acute leukemia) (Michmerhuizen et al., 2020). KDM5A is the most common fusion partner of NUP98 in AML and AMKL (de Rooij et al., 2017; Cardin et al., 2019), and NSD1 (nuclear receptor) is the second most common (Mercher and Schwaller, 2019). NUP98::KDM5A and NUP98::NSD1 chimeric proteins fuse the N-terminus of NUP98 with the C-terminus of KDM5A (harboring PHD3) or NSD1 (harboring the PHD and SET domains), respectively (Wang et al., 2007; Wang et al., 2009; Hollink et al., 2011; Schmoellerl et al., 2020). NUP98::KDM5A patients have distinct clinical features compared with NUP98::NSD1, presenting at a younger age and showing a lower WCC (Noort et al., 2021). Additional mutations that associate with NUP98::NSD1, particularly those affecting RAS, WT1 and FLT3, are rarely found in association with NUP98::KDM5A (Noort et al., 2021). In contrast, loss of RB1 (Retinoblastoma 1) locus frequently associates with NUP98::KDM5A (90%, n = 9), and GATA1 mutations were present in 2 of 9 NUP98::KDM5A patients with the RB1 loss (de Rooij et al., 2017; Masetti et al., 2019b; Iacobucci et al., 2019; Michmerhuizen et al., 2020). Leukemic cells carrying NUP98::KDM5A show a strong upregulation of HOXA (HOXA5, HOXA9, HOXA10), and HOXB (HOXB2, HOXB3, HOXB4, HOXB5, HOXB6) genes compared with RBM::MKL1 and CBFA2T3::GLIS2 fusions (de Rooij et al., 2013; Noort et al., 2021). Intriguingly, this upregulation of HOX genes is shared with NUP98::NSD1, DEK (DEK proto-oncogene)::NUP214 (nucleoporin 214), and NPM1 (nucleophosmin 1) mutated cases, suggesting HOXA and HOXB overexpression is a common alteration in leukemia development (de Rooij et al., 2013; Noort et al., 2021). Additionally, both NUP98::KDM5A and NUP98::NSD1 AML show upregulation of targets of E2F (E2F transcription factor 1) and FLT3, and downregulation of targets of TP53 and HDAC (Noort et al., 2021). The upregulation of STAT5, NF1, and NOTCH1, and the downregulation of MYC targets were also identified in NUP98::KDM5A cases, but not in NUP98::NSD1 cases (Noort et al., 2021).
NUP98::KDM5A is undetectable with conventional karyotyping, whereas newly developed next-generation sequencing (NGS) technologies can detect it (Mercher and Schwaller, 2019). The most common in-frame fusion is between exon 13 of NUP98 and exon 27 of KDM5A, but an in-frame fusion involving exon 14 of NUP98 has also been described (de Rooij et al., 2013). NUP98:KDM5A protein contains the N-terminal glycine-leucine-phenylalanine-glycine (GLFG) repeats of NUP98 fused to the C-terminal PHD3 finger of KDM5A (Figure 2C) (Gough et al., 2011; Cardin et al., 2019). Both, the GLFC repeat of NUP98 and the PHD3 domain of KDM5A are thought to participate in leukemic transformation (Wang et al., 2009; Gough et al., 2011; Michmerhuizen et al., 2020).
NUP98 is a structural component of a nuclear pore complex responsible for transporting small ions, polypeptides and macromolecules (e.g., RNA and proteins) into and out of the nucleus (Gough et al., 2011; Michmerhuizen et al., 2020). NUP98 also functions as a transcriptional regulator and assists cell cycle progression (Gough et al., 2011). NUP98 protein consists of GLFG repeats, GLEBS (Gle2-binding sequence) binding domain, RNA-binding sites, and autoproteolytic cleavage site (Figure 2C). GLFG repeats are thought to function as docking sites for karyopherins (that support molecular trafficking), CREBBP, p300 (EP300), exportin 1 (XPO1), and the mRNA export factor TAP (Gough et al., 2011; Michmerhuizen et al., 2020). In hematopoietic progenitors, NUP98 regulates H3K4me3 (trimethylation of histone H3 at lysine 4) via binding to promoters adjacent to regions associated with H3K4me3 and via interaction with Wdr82-Set1A/COMPASS (complex of proteins associated with Set1) (Franks et al., 2017). The role of NUP98 in leukemia development depends on its interaction with the PHD3 domain in KDM5A (Wang et al., 2009; Cardin et al., 2019; Zhang et al., 2020).
KDM5A is a histone lysine demethylase that can remove methyl groups from histones H3K4me1/2/3, thus modulating transcriptional activation or repression (Yang et al., 2021). KDM5A is composed of a Jumonji (JMJ) N domain, a Bright/ARID DNA binding domain, a JMJ C domain, and three PHD domains (Figure 2C) (Yang et al., 2021). PHD3 finger is capable of binding to H3K4me1/2/3, with H3K4me3 being the preferred substrate (Yang et al., 2021). KDM5A overexpression in leukemia associates with a poor prognosis (Gale et al., 2016; Yang et al., 2021).
NUP98::KDM5A fusion is sufficient to induce leukemic transformation by altering proliferation, differentiation, maturation, and self-renewal in different cellular and animal models (Wang et al., 2009; Cardin et al., 2019; Domingo-Reines et al., 2022). The bone marrow-derived HSPCs transduced with NUP98::KDM5A fusion show myeloid differentiation arrest and sustained self-renewal (Wang et al., 2009). Overexpression of NUP98::KDM5A in human cord blood stem/progenitor cells results in maturation block and abnormal proliferation with short latency (Cardin et al., 2019). Similarly, in a human embryonic stem cell (hESC) model with doxycycline-regulated NUP98::KDM5A expression, inducible expression of the fusion protein affects progenitor cell production, accompanied by enhanced expression of HOXA gene cluster (Domingo-Reines et al., 2022). Mice transplanted with NUP98::KDM5A bone marrow progenitors develop CD34+CD117+ AML, characterized by transcriptional upregulation of lineage-specific factors (HOXA, GATA3, MEIS1, EYA, PBX1 (pre-B-cell leukemia transcription factor 1), and epigenetic activation of the HOXA gene cluster (Wang et al., 2009). Transfer of human cord blood HSPCs modified to express NUP98::KDM5A in mice results in AML, including AMKL (Cardin et al., 2019). Expression profiling of these synthetic AMKL xenografts closely matches those of AMKL patients (correlation coefficients >0.9). Prominent expression changes include transcriptional upregulation of HOXA, HOXB, MEIS1 and MEIS2, epigenetic activation of HOXB, and overactivation of STAT5A signaling (Cardin et al., 2019). Integrative analysis of transcriptomic and proteomic data from NUP98::KDM5A AMKL models identified cell membrane proteins SELP (selectin P), MPIG6B (megakaryocyte and platelet inhibitory receptor G6b), and NEO1 (neogenin) as novel disease biomarkers (Cardin et al., 2019). Upregulation of JAK-STAT signaling occurs in both synthetic AMKL xenografts and NUP98-rearranged AMKL patient-derived xenografts (Cardin et al., 2019). Like in KMT2Ar AML, CDK6 is highly expressed in NUP98 rearranged AML, representing a critical direct target of NUP98 fusion proteins (Schmoellerl et al., 2020). CDK6 expression is essential for initiating and maintaining AML driven by NUP98 fusion. CDK6 inhibitor palbociclib induces myeloid differentiation, apoptosis and cell cycle arrest in vitro and in vivo (Schmoellerl et al., 2020). Thus, CDK6 inhibition has been proposed as a rational strategy to target NUP98 fusion effects in AML (Schmoellerl et al., 2020).
Although the detailed molecular mechanism of leukemic transformation by NUP98::KDM5A is unclear, it appears that the upregulation of HOX, GATA3, MESI1 and PBX1 transcription factors are essential (Wang et al., 2009; Franks et al., 2017). The N-terminal domain is responsible for recruiting the WDR82-SET1A/B-COMPASS complex to promote H3K4me through which it upregulates gene expression (Franks et al., 2017). The C-terminal PHD3 domain that recognizes H3K4me3 also contributes (Wang et al., 2009). Cell transformation is dependent on the integrity of the PHD3 finger that specifically binds H3K4me2/3. Mutations affecting conserved residues in this domain abrogate H3K4me3 binding, which prevents the binding of NUP98:KDM5A protein to the HOXA9 promoter (Wang et al., 2009).
In summary, NUP98::KDM5A fusion identifies a high-risk group of patients with inferior outcomes. Leukemic cells harboring NUP98::KDM5A have unique molecular features. NUP98:KDM5A is recruited to the promoters of HOX genes associated with H3K4me3/2. This causes transcriptional upregulation of HOXA, GATA3, MEIS1, MEIS2, EYA, and PBX1, and epigenetic activation of the HOXA and HOXB gene clusters. Upregulation of these genes blocks differentiation and maturation, and sustains proliferation and self-renewal of leukemic cells. Nevertheless, the exact molecular mechanism of malignant transformation driven by NUP98::KDM5A is not fully understood. The pathogenetic significance of the RB1 loss in the presence of NUP98::KDM5A is unknown. Novel molecularly guided treatment options are required for this disease.
2.4 RBM15::MKL1
The t(1;22)(p13;q13) results in a fusion of the RBM15 (the RNA-binding motif protein 15) gene, also named as OTT (one twenty-two) located on chromosome 1p13 to the MKL1 (megakaryoblastic leukemia-1) gene, also named as MAL (megakaryocytic acute leukemia) or MRTFA (myocardin-related transcription factor A) located on chromosome 22q13 (Ma et al., 2001; Raffel et al., 2007). Although the translocation t(1;22)(p13;q13) is rare in pediatric AML (seen in approximately 0.3% of cases) (Quessada et al., 2021), it is almost exclusively seen in infants or young children (age <3 years) with AMKL (10%–15% of pediatric non-DS AMKL) (Carroll et al., 1991; de Rooij et al., 2016; Masetti et al., 2019b; Quessada et al., 2021). The t(1;22)(p13;q13) AMKL is associated with young patient age (median 5–8 months), female prevalence, and intermediate risk (Inaba et al., 2015; de Rooij et al., 2016; Masetti et al., 2019b). Because of the high selectivity of the t(1;22)(p13;q13) for infants with AMKL, the hypothesis was raised that this translocation may arise in utero, causing transformation of a unique developmental stage in HSPCs (Reed et al., 2021). Children with t(1;22)(p13;q13) younger than 6 months do not harbor other cytogenetic abnormalities, while children older than 6 months may have hyperdiploidy with the duplication of der(1)t(1;22) or gains of chromosomes 2, 6, 19, or 21 (de Rooij et al., 2016; Quessada et al., 2021). The RBM15::MKL1 chimeric protein contains all known functional domains of RBM15 and MKL1 (Figure 3A). The molecular mechanisms through which RBM15:MKL1 drives AMKL transformation still need to be better understood. Normal functions of RBM15 and MKL1 provide some clues to this fusion’s roles in the AMKL development.
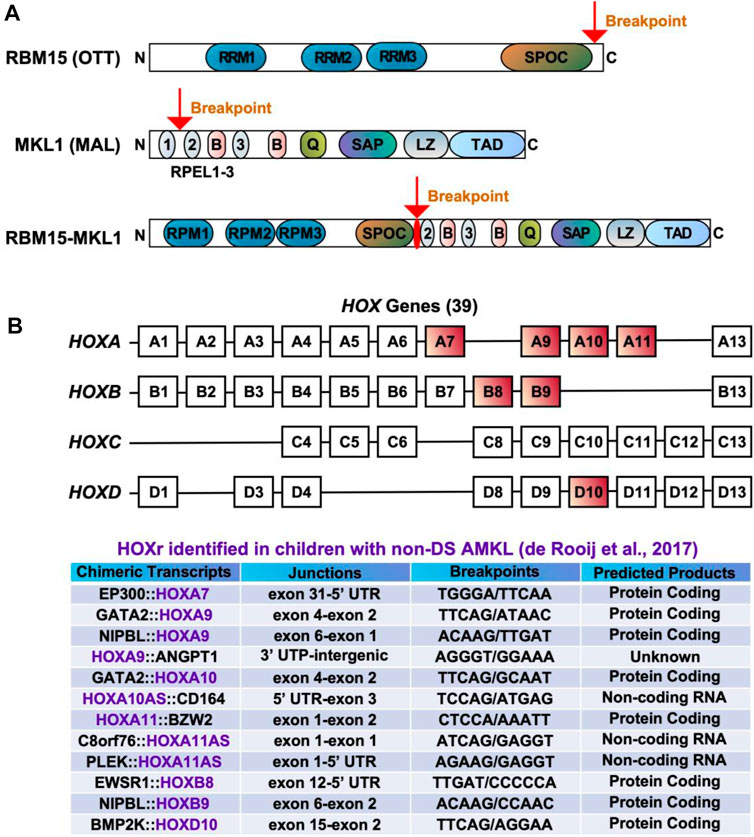
FIGURE 3. Chimeric fusions associated with favorable outcomes in pediatric non-DS AMKL. (A) Structure of RBM15 (OTT), MKL1 (MAL), and RBM15-MKL1 proteins. RRM, RNA recognition motif; SPOC, Spen paralogue and orthologue C-terminal domain; RPEL, actin-binding motifs with Arg-Pro-X-X-X- Glu-Leu core consensus; B, basic domains; Q, glutamine-rich domain; SAP, homology domain found in SAF-A/B, acinus, PIAS; LZ, leucine-zipper-like domain; TAD, transactivation domain. (B) HOX genes and HOXr identified in pediatric non-DS AMKL (de Rooij et al., 2017). There are 39 HOX genes clustered into four families: HOXA, HOXB, HOXC and HOXD. HOXA7, HOXA9, HOXA10, HOXA11, HOXB8, HOXB9 and HOXD10 are rearranged in AMKL, with HOXA9, HOXA10 and HOXA11 being the most frequently affected (de Rooij et al., 2017).
RBM15 is a crucial regulator of N6-methyladenosine methylation of RNA (Zhao et al., 2022). It contains three RNA recognition motifs (RRMs) and a SPEN paralogue and orthologue C-terminal (SPOC) domain (Figure 3A) (Raffel et al., 2007; Niu et al., 2009; Patil et al., 2016; Jin et al., 2018; Schuschel et al., 2020). RBM15 can regulate RNA splicing and histone modification of MPL critical for the HSC and megakaryocyte function (Xiao et al., 2015). RMB15 modulates several transcription factors associated with megakaryocytic differentiation, including RUNX1 (runt-related transcription factor 1), GATA1, and c-MYC (Niu et al., 2009; Zhang et al., 2015). The depletion of RBM15 in the human megakaryoblastic leukemia cell line Meg-01 enhances the formation of alternatively spliced isoforms of RUNX1a and GATA1s (Zhang et al., 2015). RUNX1a (also known as AML1a), a C-terminally truncated RUNX1 isoform, increases DNA binding and affects target gene transcription, with its overexpression increasing functional HSCs and decreasing hematopoietic differentiation (Davis et al., 2021). GATA1s, an N-terminally truncated GATA1 isoform, plays a major role in transient abnormal myelopoiesis and ML-DS development by promoting megakaryocytic progenitor expansion and disrupting megakaryocytic and erythroid differentiation (Wechsler et al., 2002; Shimizu et al., 2009; Chlon et al., 2015; Banno et al., 2016; Juban et al., 2021). The c-MYC proto-oncogene was discovered as a target of RBM15 during HSC and megakaryocyte development (Niu et al., 2009). RBM15 is also involved in regulating the activity of RBPJ (recombination signal binding protein for immunoglobulin kappa J region), which is an important player in the Notch pathway (Ma et al., 2007). RBM15 overexpression suppresses myeloid differentiation, while its knockdown enhances myeloid differentiation in the myeloid precursor cell line 32DWT18 (Ma et al., 2007). In human umbilical cord blood CD34+ cells, RBM15 knockdown inhibits the maturation of megakaryocytes (Jin et al., 2018). RBM15 deletion is lethal in embryonic mice, while conditional-knockout causes pleiotropic effects in stem cells and progenitors, including megakaryocytic expansion in the bone marrow and spleen (Raffel et al., 2007).
MKL1, a transcriptional coactivator of serum response factor (SRF), has two isoforms consisting of two or three N-terminal RPEL-repeats, basic regions, a glutamine-rich domain, a SAP domain (homology domain found in SAF-A/B, acinus, PIAS), a leucine zipper-like domain, and a TAD domain (Figure 3A) (Scharenberg et al., 2010; Kalita et al., 2012). MKL1 regulates cell morphology, adhesion, migration and differentiation in various cell types, including myeloid cells (Reed et al., 2021; Sprenkeler et al., 2021; Tabuchi and Ihara, 2021). Beyond being a coactivator for SRF, MKL1 modulates certain transcription factors (e.g., SMADs) and impacts histone modifications (Reed et al., 2021). MKL1 knockout mice show partial embryonic lethality and aberrant megakaryopoiesis characterized by increased progenitor numbers and reduced numbers of mature megakaryocytes (Sun et al., 2006; Cheng et al., 2009). The ability of MKL1 to promote megakaryocytic maturation largely depends on the SRF regulatory axis. Knockout of either MKL1 or SRF reduces megakaryocytic maturation in primary cultures, while MKL1 overexpression promotes megakaryopoiesis by augmenting both the genomic associations and activity of SRF (Rahman et al., 2018).
The RBM15::MKL1 fusion protein encompasses all putative functional domains encoded by both genes. Thus the fusion possesses the function of both proteins, including the ability to constitutively activate RBPJ, MKL1-and SRF- dependent target genes (Figure 3A) (Descot et al., 2008; Mercher et al., 2009). In a conditional knockin mouse model, RBM15::MKL1 causes abnormal megakaryopoiesis during embryonic and adult development but rarely generates AMKL (Mercher et al., 2009). However, in combination with a MPL mutation, RBM15::MKL1 leads to rapid cell transformation and a fatal disease with features similar to human AMKL (Mercher et al., 2009). This suggests that cooperating mutations are required to develop leukemia in the presence of RBM15::MKL1. One mechanism through which RBM15::MKL1 induces leukemia in vitro and in vivo is through aberrant binding and activation of RBPJ, which is essential for the differentiation bias toward megakaryocytes and proliferation of leukemic cells (Mercher et al., 2009). RBM15::MKL1 may also exert its oncogenic functions through dysregulation of MKL1 and SRF target genes caused by overactivation of MKL1-and SRF-dependent gene transcription (Descot et al., 2008). RBM15::MKL1 is associated with histone modifications, which may lead to the epigenetic deregulation of genes that control megakaryopoiesis (Lee and Skalnik, 2012). Levels of RBM15::MKL1 expression and its endogenous components may also contribute to leukemogenesis. For example, RBM15::MKL1 overexpression decreases endogenous RBM15 levels and increases endogenous MKL1 expression in the megakaryoblastic leukemia cell line 6133, while RBM15 overexpression reduces the fusion protein expression (Lee and Skalnik, 2012). It seems the N-terminal domain of RBM15 controls endogenous RBM15 expression, but the exact mechanism of this regulation is unknown (Lee and Skalnik, 2012). Relevant hPSC-based human models have been developed to study the impact of RBM15::MKL1. These models recreate AMKL features seen in patients, including overexpression of adhesion molecules CDH2 (cadherin 2) and ITGB1, and several components of Notch signaling (Ayllon et al., 2017).
In summary, RBM15::MKL1 is almost exclusively seen in infants or young children (age <3 years) with AMKL. RBM15::MKL1 possesses the function of both RBM15 and MKL1 proteins, including constitutive activation of RBPJ, MKL1-and SRF- dependent target genes. By itself, RBM15::MKL1 appears insufficient to drive AMKL. However, with a MPL mutation, RBM15::MKL1 causes rapid transformation to AMKL. It remains to be determined how RBM15::MKL1 alters megakaryopoiesis and the mechanism through which cooperating mutations drive leukemogenesis.
2.5 HOX rearrangements
HOX rearrangements (HOXr) are seen in 14.9% of pediatric non-DS AMKL and associate with better outcomes (5-year overall survival of 77%) than the aforementioned molecular subgroups (CBFA2T3::GLIS2, KMT2Ar, NUP98::KDM5A, and RBM15::MKL1) (de Rooij et al., 2017). The median age at diagnosis for these patients is 1.5 years (range of approximately 6 months to 2 years) (de Rooij et al., 2017; Masetti et al., 2019b). There are a variety of HOXr in AMKL, including GATA2::HOXA9, GATA2::HOXA10, NIPBL (Drosophila melanogaster Nipped-B)::HOXA9, NIPBL::HOXB9, GATA2::HOXA10, EWSR1 (Ewing Sarcoma breakpoint region 1)::HOXB8, PLEK (pleckstrin)::HOXA11AS (HOXA11 antisense RNA), BMP2K (BMP-2-inducible protein kinase)::HOXD10, EP300::HOXA7, C8orf76 (C8orf76, chromosome 8 open reading frame 76)::HOXA11AS, HOXA11::BZW2 (basic leucine zipper and W2 domains 2), HOXA9::ANGPT1 (angiopoietin 1) and HOXA10AS::CD164 (sialomucin core protein 24) (Figure 3B) (de Rooij et al., 2017; Masetti et al., 2019b). HOXr cause the upregulation of the HOX gene in the fusion and of the adjacent HOX genes (de Rooij et al., 2017). As highlighted in previous sections, overexpression of HOX genes is also associated with KMT2Ar and NUP98::KDM5A, so together, increased expression of HOX genes occurs in approximately half of pediatric non-DS AMKL (de Rooij et al., 2017). Multiple other mutations can be seen in association with HOXr in AMKL, including in NRAS, KRAS, MPL, JAK2, PI3K3R1, PTEN (phosphatase and tensin homolog deleted on chromosome 10), STAG2, CTCF (CCCTC-binding factor), and other genes (e.g., RB1, RUNX1, SETX (senataxin), ATM (ataxia-telangiesctasia mutated), SMARCA2 (SWI/SNF related, matrix associated, actin dependent regulator of chromatin, subfamily A, member 2), NSD1, and TP53) (de Rooij et al., 2017). HOXr AMKL is enriched in MPL mutations (41.7%, n = 12) (de Rooij et al., 2017). Murine bone marrow cells transfected with MPL p.W515L and GATA2::HOXA9 or NIPBL::HOXB9 show growth advantage and increased phosphorylation of JAK2 and STAT5 (de Rooij et al., 2017). Additional cytogenetic aberrations observed in HOXr AMKL include gains or losses of certain chromosomes, mostly +5, +6, +8, +10, +16, +18, +19, +21, −6, −7, −8, −16, −17, −19, −22, hyperdiploidy, or a complex karyotype (de Rooij et al., 2017). Given that HOXr AMKL is a rare and newly identified entity, little is known about the clinical and laboratory features of patients with this disease subgroup.
Homeobox genes encode a family of homeodomain-containing transcription factors that play diverse roles ranging from embryogenesis to carcinogenesis, including hematopoiesis and leukemogenesis (Bhatlekar et al., 2018; Collins and Thompson, 2018). In humans, a total of 39 HOX genes are found, situated in clusters on four chromosomes (7p15, 17q21.2, 12q13, and 2q31) (Bhatlekar et al., 2018; Collins and Thompson, 2018). HOX genes are grouped as HOXA, HOXB, HOXC, and HOXD (Figure 3B). Except for HOXD, HOX transcription factors play essential roles in the generation of blood cells (Bhatlekar et al., 2018; Collins and Thompson, 2018). For example, HOXA9, one of the most abundant HOX transcription factors in HSCs, impacts HSC proliferation, self-renewal, and myeloid and lymphoid differentiation (Bhatlekar et al., 2018; Collins and Thompson, 2018). HOXA9 is overexpressed in more than 50% of AML, which is associated with poor outcomes (Andreeff et al., 2008; Rio-Machin et al., 2017; Gavory et al., 2021). A recurrent fusion of NUP98 with HOXA9 via chromosome translocation t(7;11)(p15;p15) drives AML by deregulating expression of MEIS1, HOXA9, and PBX3, arresting differentiation and inducing long-term proliferation of human HSCs (Takeda et al., 2006; Rio-Machin et al., 2017). Nevertheless, this fusion has not been associated with the megakaryocytic phenotype. In contrast, GATA2::HOXA9 and NIPBL::HOXB9 have been demonstrated to efficiently generate AMKL in mice (Dang et al., 2017). Both fusions transfected into murine hematopoietic cells upregulate RUNX1, FLI1, and HOX target genes, increase the self-renewal capacity of HSCs and cause dysplastic megakaryopoiesis (Dang et al., 2017). HOXr are predicted to produce either in-frame functional fusion proteins or loss of function of the regulatory transcripts, highlighting the crucial roles of HOX genes in the pathogenesis of pediatric non-DS AMKL (de Rooij et al., 2017). However, more studies are required on the mechanism through which HOXr drive AMKL.
2.6 Non-recurrent gene fusions and unknown drivers
A few non-recurrent fusion genes have been identified in 3.5% of pediatric non-DS AMKL, including MN1::FLI1, GRB10 (growth factor receptor bound protein 10)::SDK1 (sidekick cell adhesion molecule 1), BCR::ABL1 and MAP2K2::AF10 (de Rooij et al., 2017). MN1::FLI1 induces AMKL in mice with a strong gene expression signature characteristic of megakaryocyte progenitors (Dang et al., 2017). Chimeric transcripts harboring STAG2 (e.g., STAG2::GPR119 (G protein-coupled receptor 119) and STAG2::LINCO1285) are detected in 3.5% of non-DS AMKL, and all are predicted to induce a truncated STAG2 protein (de Rooij et al., 2017). Because of the very low incidence of non-recurrent gene fusions in AMKL, little is known about their roles in leukemogenesis.
Monosomy 7 is detected in 5%–6% of pediatric AMKL and may confer poor risk (de Rooij et al., 2016; de Rooij et al., 2017) (Supplementary Table S1). Monosomy 7 is common in CBFA2T3::GLIS2, HOXr, and unknown driver subgroups (de Rooij et al., 2016; de Rooij et al., 2017). However, how monosomy 7 contributes to AMKL pathogenesis remains poorly understood.
Finally, the aetiology of approximately 15% of pediatric AMKL without any known genetic aberrations is unknown (de Rooij et al., 2017). Therefore, other molecular drivers are expected to be found.
2.7 GATA1 mutations
Although truncating mutations in GATA1 exon 2 or 3 are known to play essential roles in ML-DS, they can also be found in 9%–10% of pediatric non-DS AMKL (Figure 1A) (de Rooij et al., 2017). The non-DS AMKL patients with GATA1 mutations do not have germline trisomy 21 or any physical stigmata of DS, but GATA1 mutations and chromosome 21 amplifications are observed in major leukemic clones in these patients (de Rooij et al., 2017). Amplifications of chromosome 21 are one of the most highly recurrent copy number alterations found in approximately 39% of children with non-DS AMKL, possibly because certain chromosome 21 genes (e.g., DYRK1A [dual specificity tyrosine phosphorylation regulated kinase 1A] and ERG) act as critical players in megakaryopoiesis (de Rooij et al., 2017; Li and Kalev-Zylinska, 2022). Like ML-DS, GATA1 mutant cases of non-DS AMKL show significant overexpression of chromosome 21 genes and co-operating mutations in cohesins and the JAK pathway. Also similar to ML-DS, these patients have excellent outcomes (de Rooij et al., 2016; de Rooij et al., 2017; Masetti et al., 2019b) (Figure 1B). Because of these crucial similarities with ML-DS, non-DS AMKL cases with somatic GATA1 mutations are referred to as DS-like AMKL (de Rooij et al., 2017).
3 Novel molecularly targeted therapeutic strategies being developed for pediatric non-DS AMKL
In pediatric non-DS AMKL, high-risk subgroups (CBFA2T3::GLIS2, KMT2Ar, and NUP98::KDM5A) have inferior outcomes, mostly because of the primary refractoriness to chemotherapy and/or early relapse (de Rooij et al., 2016; de Rooij et al., 2017). Novel therapeutic strategies are urgently needed to improve the long-term survival of these patients. With the molecular abnormalities of pediatric non-DS AMKL being unravelled, some novel molecular targets are being considered, including driver fusions, their associated molecules, downstream targets or signaling pathways, and cooperating alterations or their related molecules (Table 3). Next, we present a brief overview of selected therapeutic targets being developed for AMKL.
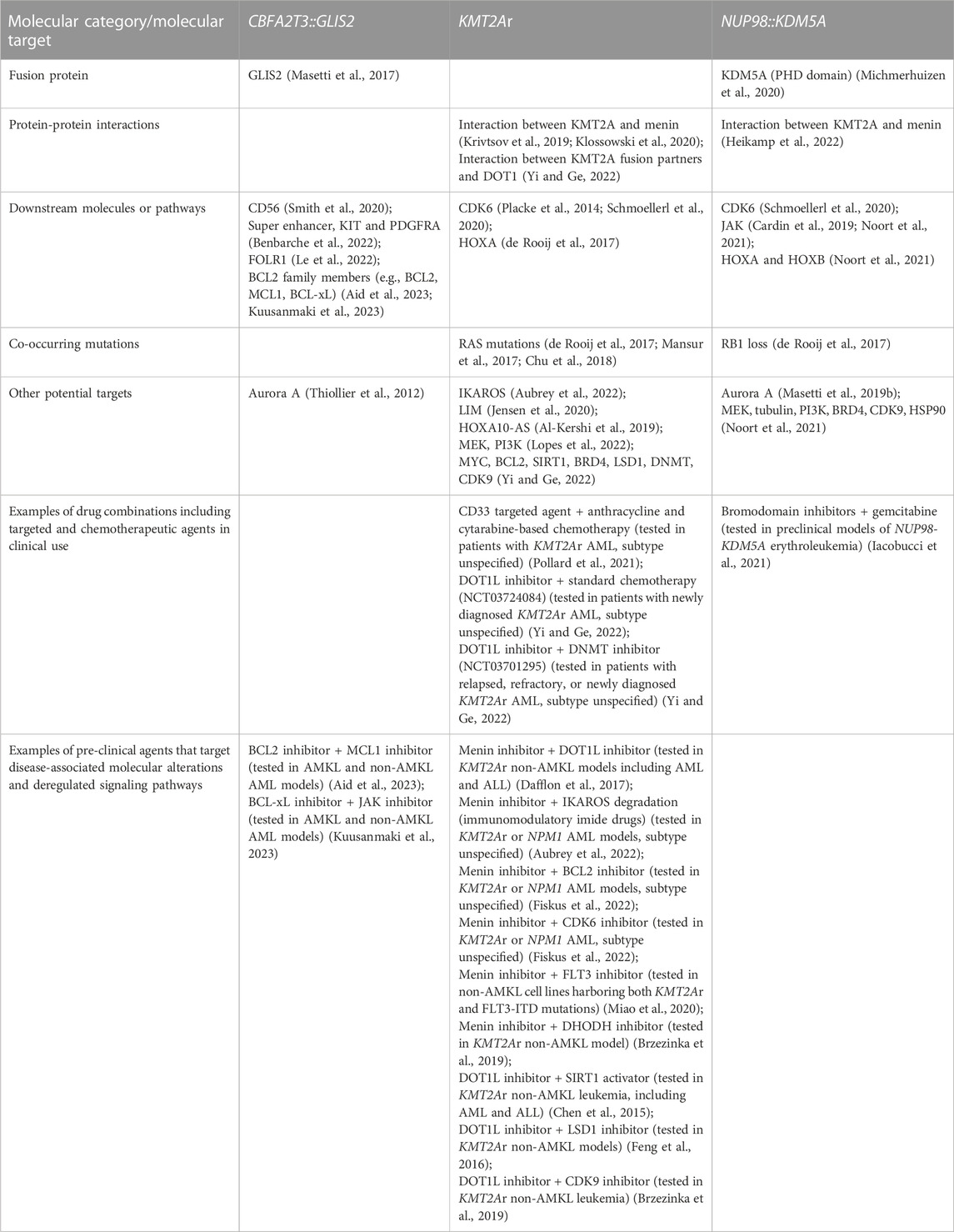
TABLE 3. Novel molecular targets/drugs being considered in high-risk pediatric non-DS AMKL.
The aberrant expression of fusion proteins in leukemic cells represents an opportunity for therapeutic targeting and is of significant research interest. For example, GANT61 is a small molecule that represses the DNA-binding and transcriptional activities of GLI proteins (Masetti et al., 2017; Lau et al., 2019). Since GLIS2 has a high homology of DNA-binding domain with other GLI proteins, GANT61 may target CBFA2T3:GLIS2 fusion in pediatric AML (Masetti et al., 2017; Lau et al., 2019). CBFA2T3::GLIS2 positive non-AMKL and AMKL cell lines (WSU-AML and M07e, respectively) and primary leukemic cells from patients are more sensitive to GANT61 than fusion-negative cells (Masetti et al., 2017). GANT61 induces apoptosis and G1 cell-cycle arrest, decreases the expression of GLIS2, CBFA2T3::GLIS2 and its target molecules (e.g., CD56, GATA3, CRISP3, and H2AFY) (Masetti et al., 2017). Nevertheless, further testing of GANT61 in mice models of CBFA2T3::GLIS2 leukemia is required.
Targeting interactions of the driver fusion with its associated molecules is another way to inhibit its leukemogenic drive. For instance, KMT2A directly binds to menin through the MBD domain and forms a menin-KMT2A complex. This complex is critical in regulating the HOX gene cluster, including the leukemogenic HOXA9 and its co-factor MEIS1 in myeloid stem and progenitor cells (Li and Song, 2021; Fiskus et al., 2022). Hence, disrupting the interaction between KMT2A and menin has emerged as a promising strategy. Menin inhibitors (e.g., VPT-50469, MI-3454, KO-539, and SNDX-5613) are capable of perturbing menin binding to KMT2A fusion, leading to a reduction in expression of its key targets, such as HOXA9, MEIS1, FLT3, and CDK6 (Krivtsov et al., 2019; Klossowski et al., 2020; Li and Song, 2021). Menin inhibitors inhibit proliferation and promote differentiation and apoptosis in non-AMKL cell lines expressing KMT2Ar, and achieve remission in KMT2Ar non-AMKL mouse models, including patient-derived xenografts (Krivtsov et al., 2019; Klossowski et al., 2020; Li and Song, 2021). Two inhibitors KO-539 and SNDX-5613 have entered phase I/II clinical trials for treating refractory/relapsed KMT2Ar AML (NCT04067336 and NCT04065399) (Li and Song, 2021). Leukemic cells with NUP98 fusions are also dependent on KMT2A, as KMT2A recruits a fusion onto the HOXA gene locus. Inhibition of interactions between KMT2A and menin by VTP50469 upregulates expression of molecules required for megakaryocytic and erythroid differentiation and downregulates expression of pro-leukemogenic genes (e.g., HOXA cluster) (Heikamp et al., 2022). VTP50469 prolongs the survival of mice engrafted with NUP98::KDM5A patient-derived cells (Heikamp et al., 2022). These findings highlight that targeting KMT2A-menin interactions may be helpful as a novel therapeutic approach. Likewise, targeting interactions between KMTA2 fusion partners and DOT1L emerged as a valid strategy against KMT2Ar leukemia. DOT1L peptide mimetics and/or small molecule inhibitors disrupting interactions between DOT1L and AF9/AF10/ENL are currently under investigation (Wu et al., 2021; Yi and Ge, 2022; Yuan et al., 2022). For example, a DOT1L peptide mimetic was synthesized to target DOT1L and AF9/ENL, and its use supressed growth of non-AMKL leukemic cell lines harboring KMT2Ar (Yuan et al., 2022). The most potent mimetic has similar anticancer activities to the DOT1L inhibitor EPZ5676 in KMT2Ar non-AMKL cell lines, demonstrating that inhibition of interactions between DOT1L and a KMT2A fusion is a promising approach (Yuan et al., 2022). DOT1L inhibitor EPZ5676 was trialled in pediatric and adult patients with KMT2Ar refractory or relapsed leukemia (NCT02141828 and NCT01684150 clinical trials) (Yi and Ge, 2022). The drug was well tolerated but had modest clinical activity due to drug resistance emerging upon long-term administration (Yi and Ge, 2022). It was concluded that EPZ5676 might be more efficacious when used in synergy with other anti-leukemic agents. For instance, a combination of DOT1L inhibitor with an inhibitor of the KMT2A-menin interactions was proposed to overcome resistance to a single agent (Dafflon et al., 2017).
Targeting downstream targets or signaling pathways of the driver fusion represents another rational approach to designing anti-leukemic treatment. For example, CBFA2T3::GLIS2 is associated with higher expression of CD56 and FOLR1, and activation of super enhancers, which provides highly plausible therapeutic targets (Benbarche et al., 2022). Surface expression of CD56 is high in patient-derived blasts, and an anti-CD56 antibody-drug conjugate (m906-PBD-ADC) exhibits a CD56-specific cell killing against primary leukemic blasts carrying this fusion (Smith et al., 2020). FOLR1 has also been validated as a valuable target in CBFA2T3::GLIS2 AMKL (Le et al., 2022). CAR T cells against FOLR1 showed pre-clinical efficacy in leukemic cell lines, patient-derived cells and xenografts of CBFA2T3::GLIS2 leukemia (Le et al., 2022). One super enhancer specific for leukemic cells carrying CBFA2T3::GLIS2 controls the expression of KIT and PDGFRA (Benbarche et al., 2022). Inhibition of this super enhancer combined with tyrosine kinase inhibitors specific for KIT and PDGFRA are able to impair leukemic progression in xenograft models, validating super enhancers, KIT and PDGFRA as useful therapeutic targets (Benbarche et al., 2022). Other targets are also being investigated. High expression of STAT5 in AMKL cell lines correlates with sensitivity to JAK inhibitor ruxolitinib (Drenberg et al., 2019). In three distinct murine models of AMKL carrying CBFA2T3::GLIS2 alone, in combination with JAK2 p.V617F, or with copy number alterations on chromosome 21, ruxolitinib significantly prolongs survival, justifying its therapeutic testing in pediatric AMKL (Drenberg et al., 2019). A new study showed that patient-derived CBFA2T3::GLIS2-positive AMKL cells express high levels of pro-apoptotic CASP3 and anti-apoptotic BCL2 (Aid et al., 2023). CBFA2T3::GLIS2-positive cell lines are dependent on BCL2 family members for cell survival. Combined targeting of BCL2 (using ABT199) and the myeloid cell lymphoma-1 (MCL1) protein (using S63845) inhibits the proliferation of CBFA2T3::GLIS2 cells in vitro and abrogates leukemia progression in mouse xenografts (Aid et al., 2023). Another pro-survival protein BCL-xL was also identified as a potential therapeutic vulnerability in erythroid and megakaryocytic lineage leukemias, including in the M07e AMKL cell line harboring CBFA2T3::GLIS2 (Kuusanmaki et al., 2023). The BCL-xL-specific inhibitor A-1331852 inhibits growth of erythroid and megakaryocytic blasts, patient-derived and cell lines, including cell lines resistant to BCL2 inhibitor venetoclax. The combination of A-1331852 with ruxolitinib eliminates growth of the erythroid (TF1 and HEL) and megakaryocytic (CMK) cell lines in long-term cultures spanning over a month (Kuusanmaki et al., 2023). These findings suggest new therapeutic targets for further testing in AMKL and reinforce the value of retaining the cell of origin information in AML classification (Arber et al., 2022; Khoury et al., 2022; Brown and Wei, 2023).
In NUP98::KDM5A-positive patient cells and mouse models, CDK6 and JAK-STAT pathways are upregulated (Cardin et al., 2019; Schmoellerl et al., 2020). Studies show that NUP98::KDM5A-positive cells are sensitive to CDK4/6 inhibitor (palbociclib) and JAK inhibitors (ruxolitinib and tofacitinib) (Cardin et al., 2019; Schmoellerl et al., 2020). A combination of menin inhibitor SNDX-50469 and CDK6 inhibitor abemaciclib has a synergistic activity in non-AMKL cell lines (MOLM13 and MV4-11) and patient-derived AML blasts harboring KMT2Ar (Fiskus et al., 2022).
Targeting cooperating alterations or their related molecules may assist in leukemia treatment. For example, MEK (mitogen-activated protein kinase) inhibitors (selumetinib and MEK162) have been proposed as potential options for KMT2Ar infants with ALL carrying RAS mutations (Kerstjens et al., 2017; Mansur et al., 2017; Chu et al., 2018). RAS mutations commonly occur in KMT2Ar AMKL (de Rooij et al., 2017); thus inhibition of the RAS pathway may be helpful in this disease, but this awaits experimental testing. Other possible therapeutic targets in three molecular subtypes of AMKL classified into high-risk category include Aurora A, PI3K, BRD4 (bromodomain-containing protein 4), CDK9 (cyclin-dependent kinase 9), HSP90 (heat shock protein 90), CD33, IKAROS, LIM (Lin11/Isl1/Mec3), HOXA10-AS, SIRT1 (sirtuin 1), LSD1 (lysine-specific demethylase 1), DNMT, c-MYC, BCL-2 (B Cell lymphoma 2), and ATM (Thiollier et al., 2012; Al-Kershi et al., 2019; Jensen et al., 2020; Smith et al., 2020; Noort et al., 2021; Pollard et al., 2021; Aubrey et al., 2022; Lopes et al., 2022; Yi and Ge, 2022). Foretinib (GSK1363089) is an oral multikinase inhibitor targeting MET (mesenchymal-epithelial transition factor), RON (recepteur d’origine nantais), AXL (AXL receptor tyrosine kinase), VEGFR (vascular endothelial growth factor receptor), c-KIT, FLT3, and PDGFR pathways. Patient-derived non-AMKL leukemic cells containing KMT2Ar and FLT3 mutations have a higher sensitivity to foretinib, suggesting this drug could benefit patients with multiple molecular aberrations (Lopes et al., 2022).
Rational combination therapies will be essential to improve treatment for high-risk AMKL patients. Combined therapies may include conventional cytotoxic drugs integrated with molecularly targeted agents or combinations of targeted agents informed by molecular alterations present in patients. The addition of anti-CD33 antibody gemtuzumab ozogamicin to conventional chemotherapy improves outcomes of children with KMT2Ar AML (Pollard et al., 2021). Another pre-clinical example is that menin inhibitor SNDX-5613 combined with BCL2 inhibitor venetoclax shows synergistic activity in patient-derived KMT2Ar AML cells and PDX mouse models (Fiskus et al., 2022). Table 3 contains examples of drug combinations tested in different experimental models and/or clinical trials against leukemias driven by CBFA2T3::GLIS2, NUP98::KDM5A, and KMT2Ar.
4 Conclusion
Pediatric non-DS AMKL is a rare, heterogeneous entity characterized by mostly poor outcomes, in contrast to excellent outcomes seen in ML-DS. Assisted by advanced genomic characterization, pediatric non-DS AMKL has been divided into distinct molecular subtypes. In the latest 2022 WHO and ICC classifications, cases with KMT2A and NUP98 rearrangements form independent AML subgroups, and cases with CBFA2T3::GLIS2 and RBM15::MKL1 belong to a subgroup of AML with other defined genetic alterations. Patients with CBFA2T3::GLIS2, KMT2Ar and NUP98::KDM5A have adverse risk, while other AMKL patients (e.g., with RBM15::MKL1 and HOXr) are considered an intermediate risk, except for DS-like AMKL that has an excellent prognosis. Different subgroups of AMKL share similarities and differences in gene and transcript changes, which offer new therapeutic targets. Novel drugs that interfere with driver fusions, downstream molecules, and cooperating alterations are being developed for these patients.
Progress in understanding AMKL pathogenesis has been immense in the last decade, but many challenges persist. We still need to improve our knowledge of this disease’s genetic and molecular landscape. Leukemogenic drivers are unknown in approximately 15% of non-DS AMKL, and the role of many known genetic alterations and cooperating events is unclear. Further progress in these areas will be critical to enable better insights into AMKL pathogenesis and improve patient outcomes.
Author contributions
JL drafted the manuscript. MK-Z provided supervision and guidance, and helped write the manuscript. All authors contributed to the article and approved the submitted version.
Funding
Auckland Medical Research Foundation (funder reference 1119009) and Leukaemia and Lymphoma Research Fund funded from donations by Anne Norman and Victoria Nicholls (UoA grant number 3715253).
Acknowledgments
We are very grateful to Taryn Green for her careful proofreading.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2023.1170622/full#supplementary-material
References
Aid, Z., Robert, E., Lopez, C. K., Bourgoin, M., Boudia, F., Le Mene, M., et al. (2023). High caspase 3 and vulnerability to dual BCL2 family inhibition define ETO2::GLIS2 pediatric leukemia. Leukemia 37 (3), 571–579. doi:10.1038/s41375-022-01800-0
Al-Kershi, S., Bhayadia, R., Ng, M., Verboon, L., Emmrich, S., Gack, L., et al. (2019). The stem cell-specific long noncoding RNA HOXA10-AS in the pathogenesis of KMT2A-rearranged leukemia. Blood Adv. 3 (24), 4252–4263. doi:10.1182/bloodadvances.2019032029
Amano, K., Takasugi, N., Kubota, Y., Mitani, Y., Sekiguchi, M., Watanabe, K., et al. (2020). CBFA2T3-GLIS2-positive acute megakaryoblastic leukemia in a patient with Down syndrome. Pediatr. Blood Cancer 67 (2), e28055. doi:10.1002/pbc.28055
Andreeff, M., Ruvolo, V., Gadgil, S., Zeng, C., Coombes, K., Chen, W., et al. (2008). HOX expression patterns identify a common signature for favorable AML. Leukemia 22 (11), 2041–2047. doi:10.1038/leu.2008.198
Arber, D. A., Orazi, A., Hasserjian, R. P., Borowitz, M. J., Calvo, K. R., Kvasnicka, H. M., et al. (2022). International consensus classification of myeloid neoplasms and acute leukemias: Integrating morphologic, clinical, and genomic data. Blood 140 (11), 1200–1228. doi:10.1182/blood.2022015850
Arber, D. A., Orazi, A., Hasserjian, R., Thiele, J., Borowitz, M. J., Le Beau, M. M., et al. (2016). The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 127 (20), 2391–2405. doi:10.1182/blood-2016-03-643544
Aubrey, B. J., Cutler, J. A., Bourgeois, W., Donovan, K. A., Gu, S., Hatton, C., et al. (2022). IKAROS and MENIN coordinate therapeutically actionable leukemogenic gene expression in MLL-r acute myeloid leukemia. Nat. Cancer 3 (5), 595–613. doi:10.1038/s43018-022-00366-1
Ayllon, V., Vogel-Gonzalez, M., Gonzalez-Pozas, F., Domingo-Reines, J., Montes, R., Morales-Cacho, L., et al. (2017). New hPSC-based human models to study pediatric Acute Megakaryoblastic Leukemia harboring the fusion oncogene RBM15-MKL1. Stem Cell. Res. 19, 1–5. doi:10.1016/j.scr.2016.12.019
Banno, K., Omori, S., Hirata, K., Nawa, N., Nakagawa, N., Nishimura, K., et al. (2016). Systematic cellular disease models reveal synergistic interaction of trisomy 21 and GATA1 mutations in hematopoietic abnormalities. Cell. Rep. 15 (6), 1228–1241. doi:10.1016/j.celrep.2016.04.031
Benbarche, S., Lopez, C. K., Salataj, E., Aid, Z., Thirant, C., Laiguillon, M. C., et al. (2022). Screening of ETO2-GLIS2-induced Super Enhancers identifies targetable cooperative dependencies in acute megakaryoblastic leukemia. Sci. Adv. 8 (6), 9455. doi:10.1126/sciadv.abg9455
Bhatlekar, S., Fields, J. Z., and Boman, B. M. (2018). Role of HOX genes in stem cell differentiation and cancer. Stem Cells Int. 2018, 3569493. doi:10.1155/2018/3569493
Bhatnagar, N., Nizery, L., Tunstall, O., Vyas, P., and Roberts, I. (2016). Transient abnormal myelopoiesis and AML in Down syndrome: An update. Curr. Hematol. Malig. Rep. 11 (5), 333–341. doi:10.1007/s11899-016-0338-x
Boucher, A. C., Caldwell, K. J., Crispino, J. D., and Flerlage, J. E. (2021). Clinical and biological aspects of myeloid leukemia in Down syndrome. Leukemia 35 (12), 3352–3360. doi:10.1038/s41375-021-01414-y
Brown, F. C., and Wei, A. H. (2023). Is BCL-xL the achilles' heel of AEL and AMKL? Blood 141 (13), 1505–1506. doi:10.1182/blood.2022019246
Brzezinka, K., Nevedomskaya, E., Lesche, R., Steckel, M., Eheim, A. L., Haegebarth, A., et al. (2019). Functional diversity of inhibitors tackling the differentiation blockage of MLL-rearranged leukemia. J. Hematol. Oncol. 12 (1), 66. doi:10.1186/s13045-019-0749-y
Cardin, S., Bilodeau, M., Roussy, M., Aubert, L., Milan, T., Jouan, L., et al. (2019). Human models of NUP98-KDM5A megakaryocytic leukemia in mice contribute to uncovering new biomarkers and therapeutic vulnerabilities. Blood Adv. 3 (21), 3307–3321. doi:10.1182/bloodadvances.2019030981
Carroll, A., Civin, C., Schneider, N., Dahl, G., Pappo, A., Bowman, P., et al. (1991). The t(1;22) (p13;q13) is nonrandom and restricted to infants with acute megakaryoblastic leukemia: A pediatric oncology group study. Blood 78 (3), 748–752.
Chen, C. W., and Armstrong, S. A. (2015). Targeting DOT1L and HOX gene expression in MLL-rearranged leukemia and beyond. Exp. Hematol. 43 (8), 673–684. doi:10.1016/j.exphem.2015.05.012
Chen, C. W., Koche, R. P., Sinha, A. U., Deshpande, A. J., Zhu, N., Eng, R., et al. (2015). DOT1L inhibits SIRT1-mediated epigenetic silencing to maintain leukemic gene expression in MLL-rearranged leukemia. Nat. Med. 21 (4), 335–343. doi:10.1038/nm.3832
Cheng, E. C., Luo, Q., Bruscia, E. M., Renda, M. J., Troy, J. A., Massaro, S. A., et al. (2009). Role for MKL1 in megakaryocytic maturation. Blood 113 (12), 2826–2834. doi:10.1182/blood-2008-09-180596
Chlon, T. M., McNulty, M., Goldenson, B., Rosinski, A., and Crispino, J. D. (2015). Global transcriptome and chromatin occupancy analysis reveal the short isoform of GATA1 is deficient for erythroid specification and gene expression. Haematologica 100 (5), 575–584. doi:10.3324/haematol.2014.112714
Chu, S. H., Song, E. J., Chabon, J. R., Minehart, J., Matovina, C. N., Makofske, J. L., et al. (2018). Inhibition of MEK and ATR is effective in a B-cell acute lymphoblastic leukemia model driven by Mll-Af4 and activated Ras. Blood Adv. 2 (19), 2478–2490. doi:10.1182/bloodadvances.2018021592
Chyla, B. J., Moreno-Miralles, I., Steapleton, M. A., Thompson, M. A., Bhaskara, S., Engel, M., et al. (2008). Deletion of Mtg16, a target of t(16;21), alters hematopoietic progenitor cell proliferation and lineage allocation. Mol. Cell. Biol. 28 (20), 6234–6247. doi:10.1128/MCB.00404-08
Collins, E. M., and Thompson, A. (2018). HOX genes in normal, engineered and malignant hematopoiesis. Int. J. Dev. Biol. 62 (11-12), 847–856. doi:10.1387/ijdb.180206at
Dafflon, C., Craig, V. J., Mereau, H., Grasel, J., Schacher Engstler, B., Hoffman, G., et al. (2017). Complementary activities of DOT1L and Menin inhibitors in MLL-rearranged leukemia. Leukemia 31 (6), 1269–1277. doi:10.1038/leu.2016.327
Dang, J., Nance, S., Ma, J., Cheng, J., Walsh, M. P., Vogel, P., et al. (2017). AMKL chimeric transcription factors are potent inducers of leukemia. Leukemia 31 (10), 2228–2234. doi:10.1038/leu.2017.51
Dastugue, N., Lafage-Pochitaloff, M., Pages, M. P., Radford, I., Bastard, C., Talmant, P., et al. (2002). Cytogenetic profile of childhood and adult megakaryoblastic leukemia (M7): A study of the groupe francais de Cytogenetique hematologique (GFCH). Blood 100 (2), 618–626. doi:10.1182/blood-2001-12-0241
Davis, A. G., Einstein, J. M., Zheng, D., Jayne, N. D., Fu, X. D., Tian, B., et al. (2021). A CRISPR RNA-binding protein screen reveals regulators of RUNX1 isoform generation. Blood Adv. 5 (5), 1310–1323. doi:10.1182/bloodadvances.2020002090
de Castro, C. P. M., Cadefau, M., and Cuartero, S. (2021). The mutational landscape of myeloid leukaemia in Down syndrome. Cancers (Basel) 13 (16). doi:10.3390/cancers13164144
De Marchi, F., Araki, M., and Komatsu, N. (2019). Molecular features, prognosis, and novel treatment options for pediatric acute megakaryoblastic leukemia. Expert Rev. Hematol. 12 (5), 285–293. doi:10.1080/17474086.2019.1609351
de Rooij, J. D., Branstetter, C., Ma, J., Li, Y., Walsh, M. P., Cheng, J., et al. (2017). Pediatric non-Down syndrome acute megakaryoblastic leukemia is characterized by distinct genomic subsets with varying outcomes. Nat. Genet. 49 (3), 451–456. doi:10.1038/ng.3772
de Rooij, J. D., Hollink, I. H., Arentsen-Peters, S. T., van Galen, J. F., Berna Beverloo, H., Baruchel, A., et al. (2013). NUP98/JARID1A is a novel recurrent abnormality in pediatric acute megakaryoblastic leukemia with a distinct HOX gene expression pattern. Leukemia 27 (12), 2280–2288. doi:10.1038/leu.2013.87
de Rooij, J. D., Masetti, R., van den Heuvel-Eibrink, M. M., Cayuela, J. M., Trka, J., Reinhardt, D., et al. (2016). Recurrent abnormalities can be used for risk group stratification in pediatric AMKL: A retrospective intergroup study. Blood 127 (26), 3424–3430. doi:10.1182/blood-2016-01-695551
Descot, A., Rex-Haffner, M., Courtois, G., Bluteau, D., Menssen, A., Mercher, T., et al. (2008). OTT-MAL is a deregulated activator of serum response factor-dependent gene expression. Mol. Cell. Biol. 28 (20), 6171–6181. doi:10.1128/MCB.00303-08
Domingo-Reines, J., Martinez-Navajas, G., Montes, R., Lamolda, M., Simon, I., Castano, J., et al. (2022). Generation of a H9 clonal cell line with inducible expression of NUP98-kdm5a fusion gene in the AAVS1 safe harbor locus. Front. Cell. Dev. Biol. 10, 846092. doi:10.3389/fcell.2022.846092
Drenberg, C. D., Shelat, A., Dang, J., Cotton, A., Orwick, S. J., Li, M., et al. (2019). A high-throughput screen indicates gemcitabine and JAK inhibitors may be useful for treating pediatric AML. Nat. Commun. 10 (1), 2189. doi:10.1038/s41467-019-09917-0
Feng, Z., Yao, Y., Zhou, C., Chen, F., Wu, F., Wei, L., et al. (2016). Pharmacological inhibition of LSD1 for the treatment of MLL-rearranged leukemia. J. Hematol. Oncol. 9, 24. doi:10.1186/s13045-016-0252-7
Fiskus, W., Boettcher, S., Daver, N., Mill, C. P., Sasaki, K., Birdwell, C. E., et al. (2022). Effective Menin inhibitor-based combinations against AML with MLL rearrangement or NPM1 mutation (NPM1c). Blood Cancer J. 12 (1), 5. doi:10.1038/s41408-021-00603-3
Forlenza, C. J., Zhang, Y., Yao, J., Benayed, R., Steinherz, P., Ramaswamy, K., et al. (2018). A case of KMT2A-SEPT9 fusion-associated acute megakaryoblastic leukemia. Cold Spring Harb. Mol. Case Stud. 4 (6). doi:10.1101/mcs.a003426
Foster, J. H., Williams, C. L., Elghetany, M. T., Liu, P., Krance, R. A., Bertuch, A. A., et al. (2019). Monozygotic twins with non-Down syndrome associated MLL-rearranged hematologic malignancy and megakaryoblastic differentiation. Leuk. Lymphoma 60 (4), 1083–1086. doi:10.1080/10428194.2018.1516883
Franks, T. M., McCloskey, A., Shokirev, M. N., Benner, C., Rathore, A., and Hetzer, M. W. (2017). Nup98 recruits the Wdr82-Set1A/COMPASS complex to promoters to regulate H3K4 trimethylation in hematopoietic progenitor cells. Genes. Dev. 31 (22), 2222–2234. doi:10.1101/gad.306753.117
Gale, M., Sayegh, J., Cao, J., Norcia, M., Gareiss, P., Hoyer, D., et al. (2016). Screen-identified selective inhibitor of lysine demethylase 5A blocks cancer cell growth and drug resistance. Oncotarget 7 (26), 39931–39944. doi:10.18632/oncotarget.9539
Gavory, G., Baril, C., Laberge, G., Bidla, G., Koonpaew, S., Sonea, T., et al. (2021). A genetic screen in Drosophila uncovers the multifaceted properties of the NUP98-HOXA9 oncogene. PLoS Genet. 17 (8), 1009730. doi:10.1371/journal.pgen.1009730
Gillam, J., Catic, A., Paulraj, P., Dalton, J., Lai, G., Jackson-Cook, C., et al. (2022). Acute megakaryoblastic leukemia with trisomy 3 and cbfa2t3::GLIS2: A case report. Genes. Chromosom. Cancer 61 (8), 491–496. doi:10.1002/gcc.23039
Gough, S. M., Slape, C. I., and Aplan, P. D. (2011). NUP98 gene fusions and hematopoietic malignancies: Common themes and new biologic insights. Blood 118 (24), 6247–6257. doi:10.1182/blood-2011-07-328880
Grimm, J., Heckl, D., and Klusmann, J. H. (2021). Molecular mechanisms of the genetic predisposition to acute megakaryoblastic leukemia in infants with Down syndrome. Front. Oncol. 11, 636633. doi:10.3389/fonc.2021.636633
Gruber, T. A., and Downing, J. R. (2015). The biology of pediatric acute megakaryoblastic leukemia. Blood 126 (8), 943–949. doi:10.1182/blood-2015-05-567859
Gruber, T. A., Larson Gedman, A., Zhang, J., Koss, C. S., Marada, S., Ta, H. Q., et al. (2012). An Inv(16)(p13.3q24.3)-encoded CBFA2T3-GLIS2 fusion protein defines an aggressive subtype of pediatric acute megakaryoblastic leukemia. Cancer Cell. 22 (5), 683–697. doi:10.1016/j.ccr.2012.10.007
Hamlett, I., Draper, J., Strouboulis, J., Iborra, F., Porcher, C., and Vyas, P. (2008). Characterization of megakaryocyte GATA1-interacting proteins: The corepressor ETO2 and GATA1 interact to regulate terminal megakaryocyte maturation. Blood 112 (7), 2738–2749. doi:10.1182/blood-2008-03-146605
Hara, Y., Shiba, N., Ohki, K., Tabuchi, K., Yamato, G., Park, M. J., et al. (2017). Prognostic impact of specific molecular profiles in pediatric acute megakaryoblastic leukemia in non-Down syndrome. Genes. Chromosom. Cancer 56 (5), 394–404. doi:10.1002/gcc.22444
Hara, Y., Shiba, N., Yamato, G., Ohki, K., Tabuchi, K., Sotomatsu, M., et al. (2020). Patients aged less than 3 years with acute myeloid leukaemia characterize a molecularly and clinically distinct subgroup. Br. J. Haematol. 188 (4), 528–539. doi:10.1111/bjh.16203
Heikamp, E. B., Henrich, J. A., Perner, F., Wong, E. M., Hatton, C., Wen, Y., et al. (2022). The menin-MLL1 interaction is a molecular dependency in NUP98-rearranged AML. Blood 139 (6), 894–906. doi:10.1182/blood.2021012806
Hoffmeister, L. M., Orhan, E., Walter, C., Niktoreh, N., Hanenberg, H., von Neuhoff, N., et al. (2021). Impact of KMT2A rearrangement and CSPG4 expression in pediatric acute myeloid leukemia. Cancers (Basel) 13 (19). doi:10.3390/cancers13194817
Hollink, I. H., van den Heuvel-Eibrink, M. M., Arentsen-Peters, S. T., Pratcorona, M., Abbas, S., Kuipers, J. E., et al. (2011). NUP98/NSD1 characterizes a novel poor prognostic group in acute myeloid leukemia with a distinct HOX gene expression pattern. Blood 118 (13), 3645–3656. doi:10.1182/blood-2011-04-346643
Holmfeldt, P., Ganuza, M., Marathe, H., He, B., Hall, T., Kang, G., et al. (2016). Functional screen identifies regulators of murine hematopoietic stem cell repopulation. J. Exp. Med. 213 (3), 433–449. doi:10.1084/jem.20150806
Iacobucci, I., Qu, C., Varotto, E., Janke, L. J., Yang, X., Seth, A., et al. (2021). Modeling and targeting of erythroleukemia by hematopoietic genome editing. Blood 137 (12), 1628–1640. doi:10.1182/blood.2020009103
Iacobucci, I., Wen, J., Meggendorfer, M., Choi, J. K., Shi, L., Pounds, S. B., et al. (2019). Genomic subtyping and therapeutic targeting of acute erythroleukemia. Nat. Genet. 51 (4), 694–704. doi:10.1038/s41588-019-0375-1
Inaba, H., Zhou, Y., Abla, O., Adachi, S., Auvrignon, A., Beverloo, H. B., et al. (2015). Heterogeneous cytogenetic subgroups and outcomes in childhood acute megakaryoblastic leukemia: A retrospective international study. Blood 126 (13), 1575–1584. doi:10.1182/blood-2015-02-629204
Jakobczyk, H., Debaize, L., Soubise, B., Avner, S., Rouger-Gaudichon, J., Commet, S., et al. (2021). Reduction of RUNX1 transcription factor activity by a cbfa2t3-mimicking peptide: application to B cell precursor acute lymphoblastic leukemia. J. Hematol. Oncol. 14 (1), 47. doi:10.1186/s13045-021-01051-z
Jensen, P., Carlet, M., Schlenk, R. F., Weber, A., Kress, J., Brunner, I., et al. (2020). Requirement for LIM kinases in acute myeloid leukemia. Leukemia 34 (12), 3173–3185. doi:10.1038/s41375-020-0943-5
Jetten, A. M. (2019). Emerging roles of GLI-similar kruppel-like zinc finger transcription factors in leukemia and other cancers. Trends Cancer 5 (9), 547–557. doi:10.1016/j.trecan.2019.07.005
Jin, S., Mi, Y., Song, J., Zhang, P., and Liu, Y. (2018). PRMT1-RBM15 axis regulates megakaryocytic differentiation of human umbilical cord blood CD34(+) cells. Exp. Ther. Med. 15 (3), 2563–2568. doi:10.3892/etm.2018.5693
Juban, G., Sakakini, N., Chagraoui, H., Cruz Hernandez, D., Cheng, Q., Soady, K., et al. (2021). Oncogenic Gata1 causes stage-specific megakaryocyte differentiation delay. Haematologica 106 (4), 1106–1119. doi:10.3324/haematol.2019.244541
Kalita, K., Kuzniewska, B., and Kaczmarek, L. (2012). MKLs: Co-factors of serum response factor (SRF) in neuronal responses. Int. J. Biochem. Cell. Biol. 44 (9), 1444–1447. doi:10.1016/j.biocel.2012.05.008
Kerstjens, M., Driessen, E. M., Willekes, M., Pinhancos, S. S., Schneider, P., Pieters, R., et al. (2017). MEK inhibition is a promising therapeutic strategy for MLL-rearranged infant acute lymphoblastic leukemia patients carrying RAS mutations. Oncotarget 8 (9), 14835–14846. doi:10.18632/oncotarget.11730
Khoury, J. D., Solary, E., Abla, O., Akkari, Y., Alaggio, R., Apperley, J. F., et al. (2022). The 5th edition of the World Health organization classification of haematolymphoid tumours: Myeloid and histiocytic/dendritic neoplasms. Leukemia 36 (7), 1703–1719. doi:10.1038/s41375-022-01613-1
Klossowski, S., Miao, H., Kempinska, K., Wu, T., Purohit, T., Kim, E., et al. (2020). Menin inhibitor MI-3454 induces remission in MLL1-rearranged and NPM1-mutated models of leukemia. J. Clin. Investig. 130 (2), 981–997. doi:10.1172/JCI129126
Krivtsov, A. V., Evans, K., Gadrey, J. Y., Eschle, B. K., Hatton, C., Uckelmann, H. J., et al. (2019). A menin-MLL inhibitor induces specific chromatin changes and eradicates disease in models of MLL-rearranged leukemia. Cancer Cell. 36 (6), 660–673 e611. doi:10.1016/j.ccell.2019.11.001
Kuusanmaki, H., Dufva, O., Vaha-Koskela, M., Leppa, A. M., Huuhtanen, J., Vanttinen, I., et al. (2023). Erythroid/megakaryocytic differentiation confers BCL-XL dependency and venetoclax resistance in acute myeloid leukemia. Blood 141 (13), 1610–1625. doi:10.1182/blood.2021011094
Lalonde, E., Rentas, S., Wertheim, G., Cao, K., Surrey, L. F., Lin, F., et al. (2021). Clinical impact of genomic characterization of 15 patients with acute megakaryoblastic leukemia-related malignancies. Cold Spring Harb. Mol. Case Stud. 7 (2). doi:10.1101/mcs.a005975
Lamba, J. K., Cao, X., Raimondi, S. C., Rafiee, R., Downing, J. R., Lei, S., et al. (2018). Integrated epigenetic and genetic analysis identifies markers of prognostic significance in pediatric acute myeloid leukemia. Oncotarget 9 (42), 26711–26723. doi:10.18632/oncotarget.25475
Lau, B. W., Huh, K., Madero-Marroquin, R., De Marchi, F., Lim, Y., Wang, Q., et al. (2019). Hedgehog/GLI1 activation leads to leukemic transformation of myelodysplastic syndrome in vivo and GLI1 inhibition results in antitumor activity. Oncogene 38 (5), 687–698. doi:10.1038/s41388-018-0431-9
Le, Q., Hadland, B., Smith, J. L., Leonti, A., Huang, B. J., Ries, R., et al. (2022). CBFA2T3-GLIS2 model of pediatric acute megakaryoblastic leukemia identifies FOLR1 as a CAR T cell target. J. Clin. Investig. 132 (22). doi:10.1172/JCI157101
Lee, J. H., and Skalnik, D. G. (2012). Rbm15-Mkl1 interacts with the Setd1b histone H3-Lys4 methyltransferase via a SPOC domain that is required for cytokine-independent proliferation. PLoS One 7 (8), e42965. doi:10.1371/journal.pone.0042965
Li, J., and Kalev-Zylinska, M. L. (2022). Advances in molecular characterization of myeloid proliferations associated with Down syndrome. Front. Genet. 13, 891214. doi:10.3389/fgene.2022.891214
Li, X., and Song, Y. (2021). Structure, function and inhibition of critical protein-protein interactions involving mixed lineage leukemia 1 and its fusion oncoproteins. J. Hematol. Oncol. 14 (1), 56. doi:10.1186/s13045-021-01057-7
Lian, X. Y., Zhang, W., Wu, D. H., Ma, J. C., Zhou, J. D., Zhang, Z. H., et al. (2018). Methylation-independent ITGA2 overexpression is associated with poor prognosis in de novo acute myeloid leukemia. J. Cell. Physiol. 233 (12), 9584–9593. doi:10.1002/jcp.26866
Lopes, B. A., Poubel, C. P., Teixeira, C. E., Caye-Eude, A., Cave, H., Meyer, C., et al. (2022). Novel diagnostic and therapeutic options for kmt2a-rearranged acute leukemias. Front. Pharmacol. 13, 749472. doi:10.3389/fphar.2022.749472
Lopez, C. K., Noguera, E., Stavropoulou, V., Robert, E., Aid, Z., Ballerini, P., et al. (2019). Ontogenic changes in hematopoietic hierarchy determine pediatric specificity and disease phenotype in fusion oncogene-driven myeloid leukemia. Cancer Discov. 9 (12), 1736–1753. doi:10.1158/2159-8290.CD-18-1463
Ma, X., Renda, M. J., Wang, L., Cheng, E. C., Niu, C., Morris, S. W., et al. (2007). Rbm15 modulates Notch-induced transcriptional activation and affects myeloid differentiation. Mol. Cell. Biol. 27 (8), 3056–3064. doi:10.1128/MCB.01339-06
Ma, Z., Morris, S. W., Valentine, V., Li, M., Herbrick, J. A., Cui, X., et al. (2001). Fusion of two novel genes, RBM15 and MKL1, in the t(1;22)(p13;q13) of acute megakaryoblastic leukemia. Nat. Genet. 28 (3), 220–221. doi:10.1038/90054
Maarouf, N., Mahmoud, S., Khedr, R., Lehmann, L., Shaaban, K., Ibrahim, S., et al. (2019). Outcome of childhood acute megakaryoblastic leukemia: Children's cancer hospital Egypt 57357 experience. Clin. Lymphoma Myeloma Leuk. 19 (3), e142–e152. doi:10.1016/j.clml.2018.12.011
Mansur, M. B., Ford, A. M., and Emerenciano, M. (2017). The role of RAS mutations in MLL-rearranged leukaemia: A path to intervention? Biochim. Biophys. Acta Rev. Cancer 1868 (2), 521–526. doi:10.1016/j.bbcan.2017.10.005
Masetti, R., Bertuccio, S. N., Astolfi, A., Chiarini, F., Lonetti, A., Indio, V., et al. (2017). Hh/Gli antagonist in acute myeloid leukemia with CBFA2T3-GLIS2 fusion gene. J. Hematol. Oncol. 10 (1), 26. doi:10.1186/s13045-017-0396-0
Masetti, R., Bertuccio, S. N., Pession, A., and Locatelli, F. (2019a). CBFA2T3-GLIS2-positive acute myeloid leukaemia. A peculiar paediatric entity. Br. J. Haematol. 184 (3), 337–347. doi:10.1111/bjh.15725
Masetti, R., Guidi, V., Ronchini, L., Bertuccio, N. S., Locatelli, F., and Pession, A. (2019b). The changing scenario of non-Down syndrome acute megakaryoblastic leukemia in children. Crit. Rev. Oncol. Hematol. 138, 132–138. doi:10.1016/j.critrevonc.2019.04.011
Masetti, R., Pigazzi, M., Togni, M., Astolfi, A., Indio, V., Manara, E., et al. (2013a). CBFA2T3-GLIS2 fusion transcript is a novel common feature in pediatric, cytogenetically normal AML, not restricted to FAB M7 subtype. Blood 121 (17), 3469–3472. doi:10.1182/blood-2012-11-469825
Masetti, R., Togni, M., Astolfi, A., Pigazzi, M., Manara, E., Indio, V., et al. (2013b). DHH-RHEBL1 fusion transcript: A novel recurrent feature in the new landscape of pediatric cbfa2t3-GLIS2-positive acute myeloid leukemia. Oncotarget 4 (10), 1712–1720. doi:10.18632/oncotarget.1280
Matsuo, H., Yoshida, K., Nakatani, K., Harata, Y., Higashitani, M., Ito, Y., et al. (2020). Fusion partner-specific mutation profiles and KRAS mutations as adverse prognostic factors in MLL-rearranged AML. Blood Adv. 4 (19), 4623–4631. doi:10.1182/bloodadvances.2020002457
Mercher, T., Raffel, G. D., Moore, S. A., Cornejo, M. G., Baudry-Bluteau, D., Cagnard, N., et al. (2009). The OTT-MAL fusion oncogene activates RBPJ-mediated transcription and induces acute megakaryoblastic leukemia in a knockin mouse model. J. Clin. Investig. 119 (4), 852–864. doi:10.1172/JCI35901
Mercher, T., and Schwaller, J. (2019). Pediatric acute myeloid leukemia (AML): From genes to models toward targeted therapeutic intervention. Front. Pediatr. 7, 401. doi:10.3389/fped.2019.00401
Meyer, C., Burmeister, T., Groger, D., Tsaur, G., Fechina, L., Renneville, A., et al. (2018). The MLL recombinome of acute leukemias in 2017. Leukemia 32 (2), 273–284. doi:10.1038/leu.2017.213
Meyer, C., Hofmann, J., Burmeister, T., Groger, D., Park, T. S., Emerenciano, M., et al. (2013). The MLL recombinome of acute leukemias in 2013. Leukemia 27 (11), 2165–2176. doi:10.1038/leu.2013.135
Meyer, C., Kowarz, E., Hofmann, J., Renneville, A., Zuna, J., Trka, J., et al. (2009). New insights to the MLL recombinome of acute leukemias. Leukemia 23 (8), 1490–1499. doi:10.1038/leu.2009.33
Meyer, C., Larghero, P., Almeida Lopes, B., Burmeister, T., Groger, D., Sutton, R., et al. (2023). Leukemia. The KMT2A recombinome of acute leukemias in 2023. doi:10.1038/s41375-023-01877-1
Meyer, C., Schneider, B., Jakob, S., Strehl, S., Attarbaschi, A., Schnittger, S., et al. (2006). The MLL recombinome of acute leukemias. Leukemia 20 (5), 777–784. doi:10.1038/sj.leu.2404150
Miao, H., Kim, E., Chen, D., Purohit, T., Kempinska, K., Ropa, J., et al. (2020). Combinatorial treatment with menin and FLT3 inhibitors induces complete remission in AML models with activating FLT3 mutations. Blood 136 (25), 2958–2963. doi:10.1182/blood.2020006575
Michmerhuizen, N. L., Klco, J. M., and Mullighan, C. G. (2020). Mechanistic insights and potential therapeutic approaches for NUP98-rearranged hematologic malignancies. Blood 136 (20), 2275–2289. doi:10.1182/blood.2020007093
Milan, T., Celton, M., Lagace, K., Roques, E., Safa-Tahar-Henni, S., Bresson, E., et al. (2022). Epigenetic changes in human model KMT2A leukemias highlight early events during leukemogenesis. Haematologica 107 (1), 86–99. doi:10.3324/haematol.2020.271619
Mulloy, J. C., Wunderlich, M., Zheng, Y., and Wei, J. (2008). Transforming human blood stem and progenitor cells: A new way forward in leukemia modeling. Cell. Cycle 7 (21), 3314–3319. doi:10.4161/cc.7.21.6951
Muntean, A. G., Tan, J., Sitwala, K., Huang, Y., Bronstein, J., Connelly, J. A., et al. (2010). The PAF complex synergizes with MLL fusion proteins at HOX loci to promote leukemogenesis. Cancer Cell. 17 (6), 609–621. doi:10.1016/j.ccr.2010.04.012
Niu, C., Zhang, J., Breslin, P., Onciu, M., Ma, Z., and Morris, S. W. (2009). c-Myc is a target of RNA-binding motif protein 15 in the regulation of adult hematopoietic stem cell and megakaryocyte development. Blood 114 (10), 2087–2096. doi:10.1182/blood-2009-01-197921
Noort, S., Wander, P., Alonzo, T. A., Smith, J., Ries, R. E., Gerbing, R. B., et al. (2021). The clinical and biological characteristics of NUP98-KDM5A in pediatric acute myeloid leukemia. Haematologica 106 (2), 630–634. doi:10.3324/haematol.2019.236745
Patil, D. P., Chen, C. K., Pickering, B. F., Chow, A., Jackson, C., Guttman, M., et al. (2016). m(6)A RNA methylation promotes XIST-mediated transcriptional repression. Nature 537 (7620), 369–373. doi:10.1038/nature19342
Pina, C., May, G., Soneji, S., Hong, D., and Enver, T. (2008). MLLT3 regulates early human erythroid and megakaryocytic cell fate. Cell. Stem Cell. 2 (3), 264–273. doi:10.1016/j.stem.2008.01.013
Pinto, K., and Chetty, R. (2020). Gene of the month: GLIS1-3. J. Clin. Pathol. 73 (9), 527–530. doi:10.1136/jclinpath-2020-206859
Placke, T., Faber, K., Nonami, A., Putwain, S. L., Salih, H. R., Heidel, F. H., et al. (2014). Requirement for CDK6 in MLL-rearranged acute myeloid leukemia. Blood 124 (1), 13–23. doi:10.1182/blood-2014-02-558114
Pollard, J. A., Guest, E., Alonzo, T. A., Gerbing, R. B., Loken, M. R., Brodersen, L. E., et al. (2021). Gemtuzumab ozogamicin improves event-free survival and reduces relapse in pediatric kmt2a-rearranged AML: Results from the phase III children's oncology group trial AAML0531. J. Clin. Oncol. 39 (28), 3149–3160. doi:10.1200/JCO.20.03048
Qiu, L., Nunez, C. A., Tang, G., Cuglievan, B., Issa, G. C., Wang, S. A., et al. (2022). A rare case of acute megakaryoblastic leukemia with t(11;17)(q23;q21) and KMT2A::MLLT6 fusion. Ann. Hematol. 101 (7), 1579–1581. doi:10.1007/s00277-022-04769-z
Quessada, J., Cuccuini, W., Saultier, P., Loosveld, M., Harrison, C. J., and Lafage-Pochitaloff, M. (2021). Cytogenetics of pediatric acute myeloid leukemia: A review of the current knowledge. Genes. (Basel) 12 (6). doi:10.3390/genes12060924
Raffel, G. D., Mercher, T., Shigematsu, H., Williams, I. R., Cullen, D. E., Akashi, K., et al. (2007). Ott1(Rbm15) has pleiotropic roles in hematopoietic development. Proc. Natl. Acad. Sci. U. S. A. 104 (14), 6001–6006. doi:10.1073/pnas.0609041104
Rahman, N. T., Schulz, V. P., Wang, L., Gallagher, P. G., Denisenko, O., Gualdrini, F., et al. (2018). MRTFA augments megakaryocyte maturation by enhancing the SRF regulatory axis. Blood Adv. 2 (20), 2691–2703. doi:10.1182/bloodadvances.2018019448
Reed, F., Larsuel, S. T., Mayday, M. Y., Scanlon, V., and Krause, D. S. (2021). Mrtfa: A critical protein in normal and malignant hematopoiesis and beyond. J. Biol. Chem. 296, 100543. doi:10.1016/j.jbc.2021.100543
Rio-Machin, A., Gomez-Lopez, G., Munoz, J., Garcia-Martinez, F., Maiques-Diaz, A., Alvarez, S., et al. (2017). The molecular pathogenesis of the NUP98-HOXA9 fusion protein in acute myeloid leukemia. Leukemia 31 (9), 2000–2005. doi:10.1038/leu.2017.194
Scharenberg, M. A., Chiquet-Ehrismann, R., and Asparuhova, M. B. (2010). Megakaryoblastic leukemia protein-1 (MKL1): Increasing evidence for an involvement in cancer progression and metastasis. Int. J. Biochem. Cell. Biol. 42 (12), 1911–1914. doi:10.1016/j.biocel.2010.08.014
Schmoellerl, J., Barbosa, I. A. M., Eder, T., Brandstoetter, T., Schmidt, L., Maurer, B., et al. (2020). CDK6 is an essential direct target of NUP98 fusion proteins in acute myeloid leukemia. Blood 136 (4), 387–400. doi:10.1182/blood.2019003267
Schuh, A. H., Tipping, A. J., Clark, A. J., Hamlett, I., Guyot, B., Iborra, F. J., et al. (2005). ETO-2 associates with SCL in erythroid cells and megakaryocytes and provides repressor functions in erythropoiesis. Mol. Cell. Biol. 25 (23), 10235–10250. doi:10.1128/MCB.25.23.10235-10250.2005
Schuschel, K., Helwig, M., Huttelmaier, S., Heckl, D., Klusmann, J. H., and Hoell, J. I. (2020). RNA-binding proteins in acute leukemias. Int. J. Mol. Sci. 21 (10). doi:10.3390/ijms21103409
Scoville, D. W., Kang, H. S., and Jetten, A. M. (2017). GLIS1-3: Emerging roles in reprogramming, stem and progenitor cell differentiation and maintenance. Stem Cell. Investig. 4, 80. doi:10.21037/sci.2017.09.01
Shima, H., Takamatsu-Ichihara, E., Shino, M., Yamagata, K., Katsumoto, T., Aikawa, Y., et al. (2018). Ring1A and Ring1B inhibit expression of Glis2 to maintain murine MOZ-TIF2 AML stem cells. Blood 131 (16), 1833–1845. doi:10.1182/blood-2017-05-787226
Shimizu, R., Kobayashi, E., Engel, J. D., and Yamamoto, M. (2009). Induction of hyperproliferative fetal megakaryopoiesis by an N-terminally truncated GATA1 mutant. Genes. cells. 14 (9), 1119–1131. doi:10.1111/j.1365-2443.2009.01338.x
Skucha, A., Ebner, J., Schmollerl, J., Roth, M., Eder, T., Cesar-Razquin, A., et al. (2018). MLL-fusion-driven leukemia requires SETD2 to safeguard genomic integrity. Nat. Commun. 9 (1), 1983. doi:10.1038/s41467-018-04329-y
Slany, R. K. (2016). The molecular mechanics of mixed lineage leukemia. Oncogene 35 (40), 5215–5223. doi:10.1038/onc.2016.30
Smith, J. L., Ries, R. E., Hylkema, T., Alonzo, T. A., Gerbing, R. B., Santaguida, M. T., et al. (2020). Comprehensive transcriptome profiling of cryptic cbfa2t3-GLIS2 fusion-positive AML defines novel therapeutic options: A cog and target pediatric AML study. Clin. Cancer Res. 26 (3), 726–737. doi:10.1158/1078-0432.CCR-19-1800
Sprenkeler, E. G. G., Guenther, C., Faisal, I., Kuijpers, T. W., and Fagerholm, S. C. (2021). Molecular mechanisms of leukocyte migration and its potential targeting-lessons learned from MKL1/SRF-related primary immunodeficiency diseases. Front. Immunol. 12, 615477. doi:10.3389/fimmu.2021.615477
Stavropoulou, V., Kaspar, S., Brault, L., Sanders, M. A., Juge, S., Morettini, S., et al. (2016). MLL-AF9 expression in hematopoietic stem cells drives a highly invasive AML expressing EMT-related genes linked to poor outcome. Cancer Cell. 30 (1), 43–58. doi:10.1016/j.ccell.2016.05.011
Steinauer, N., Guo, C., Huang, C., Wong, M., Tu, Y., Freter, C. E., et al. (2019). Myeloid translocation gene CBFA2T3 directs a relapse gene program and determines patient-specific outcomes in AML. Blood Adv. 3 (9), 1379–1393. doi:10.1182/bloodadvances.2018028514
Steinauer, N., Guo, C., and Zhang, J. (2017). Emerging roles of MTG16 in cell-fate control of hematopoietic stem cells and cancer. Stem Cells Int. 2017, 6301385. doi:10.1155/2017/6301385
Steinauer, N., Guo, C., and Zhang, J. (2020). The transcriptional corepressor CBFA2T3 inhibits all-trans-retinoic acid-induced myeloid gene expression and differentiation in acute myeloid leukemia. J. Biol. Chem. 295 (27), 8887–8900. doi:10.1074/jbc.RA120.013042
Sugeedha, J., Gautam, J., and Tyagi, S. (2021). SET1/MLL family of proteins: Functions beyond histone methylation. Epigenetics 16 (5), 469–487. doi:10.1080/15592294.2020.1809873
Sun, Y., Boyd, K., Xu, W., Ma, J., Jackson, C. W., Fu, A., et al. (2006). Acute myeloid leukemia-associated Mkl1 (Mrtf-a) is a key regulator of mammary gland function. Mol. Cell. Biol. 26 (15), 5809–5826. doi:10.1128/MCB.00024-06
Tabuchi, A., and Ihara, D. (2021). Regulation of dendritic synaptic morphology and transcription by the SRF cofactor MKL/MRTF. Front. Mol. Neurosci. 14, 767842. doi:10.3389/fnmol.2021.767842
Takeda, A., Goolsby, C., and Yaseen, N. R. (2006). NUP98-HOXA9 induces long-term proliferation and blocks differentiation of primary human CD34+ hematopoietic cells. Cancer Res. 66 (13), 6628–6637. doi:10.1158/0008-5472.CAN-06-0458
Takita, J., Motomura, A., Koh, K., Ida, K., Taki, T., Hayashi, Y., et al. (2009). Acute megakaryoblastic leukemia in a child with the MLL-AF4 fusion gene. Eur. J. Haematol. 83 (2), 149–153. doi:10.1111/j.1600-0609.2009.01275.x
Thiollier, C., Lopez, C. K., Gerby, B., Ignacimouttou, C., Poglio, S., Duffourd, Y., et al. (2012). Characterization of novel genomic alterations and therapeutic approaches using acute megakaryoblastic leukemia xenograft models. J. Exp. Med. 209 (11), 2017–2031. doi:10.1084/jem.20121343
Thirant, C., Ignacimouttou, C., Lopez, C. K., Diop, M., Le Mouel, L., Thiollier, C., et al. (2017a). ETO2-GLIS2 hijacks transcriptional complexes to drive cellular identity and self-renewal in pediatric acute megakaryoblastic leukemia. Cancer Cell. 31 (3), 452–465. doi:10.1016/j.ccell.2017.02.006
Thirant, C., Lopez, C., Malinge, S., and Mercher, T. (2017b). Molecular pathways driven by ETO2-GLIS2 in aggressive pediatric leukemia. Mol. Cell. Oncol. 4 (6), 1345351. doi:10.1080/23723556.2017.1345351
van Zutven, L. J., Onen, E., Velthuizen, S. C., van Drunen, E., von Bergh, A. R., van den Heuvel-Eibrink, M. M., et al. (2006). Identification of NUP98 abnormalities in acute leukemia: JARID1A (12p13) as a new partner gene. Genes. Chromosom. Cancer 45 (5), 437–446. doi:10.1002/gcc.20308
Wang, G. G., Cai, L., Pasillas, M. P., and Kamps, M. P. (2007). NUP98-NSD1 links H3K36 methylation to Hox-A gene activation and leukaemogenesis. Nat. Cell. Biol. 9 (7), 804–812. doi:10.1038/ncb1608
Wang, G. G., Song, J., Wang, Z., Dormann, H. L., Casadio, F., Li, H., et al. (2009). Haematopoietic malignancies caused by dysregulation of a chromatin-binding PHD finger. Nature 459 (7248), 847–851. doi:10.1038/nature08036
Wang, Q. F., Wu, G., Mi, S., He, F., Wu, J., Dong, J., et al. (2011). MLL fusion proteins preferentially regulate a subset of wild-type MLL target genes in the leukemic genome. Blood 117 (25), 6895–6905. doi:10.1182/blood-2010-12-324699
Wang, Y., Lu, A., Jia, Y., Zuo, Y., and Zhang, L. (2021). Outcome and prognostic features in pediatric acute megakaryoblastic leukemia without Down syndrome: A retrospective study in China. Clin. Lymphoma Myeloma Leuk. 21 (4), e301–e308. doi:10.1016/j.clml.2020.11.001
Wechsler, J., Greene, M., McDevitt, M. A., Anastasi, J., Karp, J. E., Le Beau, M. M., et al. (2002). Acquired mutations in GATA1 in the megakaryoblastic leukemia of Down syndrome. Nat. Genet. 32 (1), 148–152. doi:10.1038/ng955
Wei, J., Wunderlich, M., Fox, C., Alvarez, S., Cigudosa, J. C., Wilhelm, J. S., et al. (2008). Microenvironment determines lineage fate in a human model of MLL-AF9 leukemia. Cancer Cell. 13 (6), 483–495. doi:10.1016/j.ccr.2008.04.020
Wen, Q., Goldenson, B., and Crispino, J. D. (2011). Normal and malignant megakaryopoiesis. Expert Rev. Mol. Med. 13, e32. doi:10.1017/S1462399411002043
Winters, A. C., and Bernt, K. M. (2017). MLL-rearranged leukemias-an update on science and clinical approaches. Front. Pediatr. 5, 4. doi:10.3389/fped.2017.00004
Wu, F., Nie, S., Yao, Y., Huo, T., Li, X., Wu, X., et al. (2021). Small-molecule inhibitor of AF9/ENL-DOT1L/AF4/AFF4 interactions suppresses malignant gene expression and tumor growth. Theranostics 11 (17), 8172–8184. doi:10.7150/thno.56737
Xiao, N., Laha, S., Das, S. P., Morlock, K., Jesneck, J. L., and Raffel, G. D. (2015). Ott1 (Rbm15) regulates thrombopoietin response in hematopoietic stem cells through alternative splicing of c-Mpl. Blood 125 (6), 941–948. doi:10.1182/blood-2014-08-593392
Yang, G. J., Wu, J., Miao, L., Zhu, M. H., Zhou, Q. J., Lu, X. J., et al. (2021). Pharmacological inhibition of KDM5A for cancer treatment. Eur. J. Med. Chem. 226, 113855. doi:10.1016/j.ejmech.2021.113855
Yi, Y., and Ge, S. (2022). Targeting the histone H3 lysine 79 methyltransferase DOT1L in MLL-rearranged leukemias. J. Hematol. Oncol. 15 (1), 35. doi:10.1186/s13045-022-01251-1
Yuan, Y., Du, L., Tan, R., Yu, Y., Jiang, J., Yao, A., et al. (2022). Design, synthesis, and biological evaluations of DOT1L peptide mimetics targeting the protein-protein interactions between DOT1L and MLL-AF9/MLL-ENL. J. Med. Chem. 65 (11), 7770–7785. doi:10.1021/acs.jmedchem.2c00083
Yuen, K. Y., Liu, Y., Zhou, Y. Z., Wang, Y., Zhou, D. H., Fang, J. P., et al. (2023). Mutational landscape and clinical outcome of pediatric acute myeloid leukemia with 11q23/KMT2A rearrangements. Cancer Med. 12 (2), 1418–1430. doi:10.1002/cam4.5026
Zangrando, A., Cavagnero, F., Scarparo, P., Varotto, E., Francescato, S., Tregnago, C., et al. (2021). CD56, HLA-DR, and CD45 recognize a subtype of childhood AML harboring CBFA2T3-GLIS2 fusion transcript. Cytom. A 99 (8), 844–850. doi:10.1002/cyto.a.24339
Zhang, L., Tran, N. T., Su, H., Wang, R., Lu, Y., Tang, H., et al. (2015). Cross-talk between PRMT1-mediated methylation and ubiquitylation on RBM15 controls RNA splicing. Elife 4. doi:10.7554/eLife.07938
Zhang, Y., Guo, Y., Gough, S. M., Zhang, J., Vann, K. R., Li, K., et al. (2020). Mechanistic insights into chromatin targeting by leukemic NUP98-PHF23 fusion. Nat. Commun. 11 (1), 3339. doi:10.1038/s41467-020-17098-4
Keywords: acute myeloid leukemia, acute megakaryoblastic leukemia (AMKL), non-Down syndrome AMKL, chimeric fusions, cytogenetics, genomics, transcriptomics, therapeutic targets
Citation: Li J and Kalev-Zylinska ML (2023) Advances in molecular characterization of pediatric acute megakaryoblastic leukemia not associated with Down syndrome; impact on therapy development. Front. Cell Dev. Biol. 11:1170622. doi: 10.3389/fcell.2023.1170622
Received: 21 February 2023; Accepted: 19 May 2023;
Published: 01 June 2023.
Edited by:
Feng Zhu, Affiliated Hospital of Guilin Medical University, ChinaReviewed by:
Huimin Sun, Xiamen University, ChinaTomasz Szczepanski, Medical University of Silesia, Poland
Mélanie Bilodeau, University of Montreal, Canada
Copyright © 2023 Li and Kalev-Zylinska. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maggie L. Kalev-Zylinska, bS5rYWxldkBhdWNrbGFuZC5hYy5ueg==; Jixia Li, aml4aWFsaUBmb3N1LmVkdS5jbg==